

Abstract
Next-generation sequencing and other genomic technologies are transforming prenatal and reproductive screening and testing for fetal genetic disorders at an unprecedented pace. Original approaches of screening and testing for fetal genetic and genomic disorders were focused on a few more prevalent conditions that were easily diagnosable with pre-genomic era diagnostic tools. First, chromosomal microarray analysis and then next-generation sequencing brought technology capable of more detailed genomic evaluation to prenatal genetic screening and diagnosis. This has facilitated parallel introduction of a variety of new tests on maternal blood samples, including expanded carrier screening and cell-free DNA-based non-invasive screening for fetal aneuploidy, selected copy number variants, and single gene disorders. Genomic tests on fetal DNA samples, obtained primarily through amniocentesis or chorionic villus sampling, include chromosomal microarray analysis and gene panel and exome sequencing. All these form the diagnostic pillar of the emerging field of fetal precision medicine, but their implementation is associated with ethical, counseling and healthcare resource utilization challenges. We discuss where in the reproductive and prenatal care continuum these exciting new technologies are integrated, along with associated challenges. We propose areas of priority for research to gain the data in support of their responsible implementation into clinical reproductive and prenatal care.
Keywords: prenatal diagnosis, carrier screening, exome sequencing, genome sequencing, fetal phenotypes, non-invasive prenatal testing
Introduction
Until the early 2000’s, in the “pre-genomic” era of prenatal genetic testing and screening (Fig. 1a), the main goal was to identify women at increased risk for children with common aneuploidies, trisomies 13, 18 and 21. Screening methods included various combinations of family history, maternal age, levels of specific maternal serum analytes and findings on prenatal ultrasound (ACOG 2016b). Women at increased risk were offered either chorionic villus sampling (CVS) or amniocentesis with a karyotype, and in some cases with fluorescence in situ hybridization (FISH) for rapid detection of common trisomies. Although much less frequently, if indicated based on high suspicion or a specific condition, a locus-specific FISH for a known deletion or a Sanger-sequencing based or other molecular test for a specific single gene defect could be added. Although paternal age was well recognized as a risk factor for diseases caused by de novo pathogenic single nucleotide variants (SNVs), no specific screening or testing addressing paternal age was available (ACOG 2016a; Friedman 1981; Kong et al. 2012; Toriello et al. 2008).
Fig 1. Prenatal Genetic Testing and Screening Timeline.
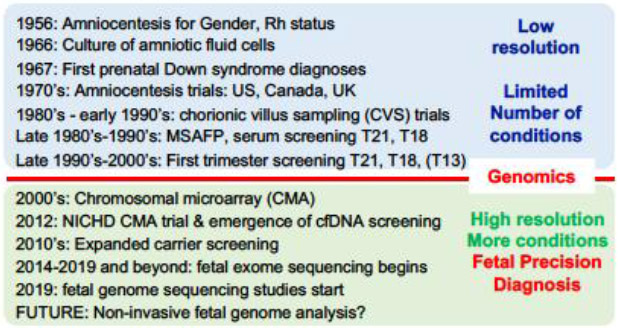
a. Pre-genomic era; b. Genomic era.
Thus, the methods mentioned above were the only option available until the early 2000’s when chromosomal microarray analysis (CMA), which can identify unbalanced numerical chromosomal abnormalities (aneuploidies) and (sub)microscopic structural chromosomal defects (deletions and duplications), ushered in the “genomic era” in prenatal genetic testing (Fig. 1b). A definitive multicenter trial that included more than 4000 women, along with other large series, demonstrated that prenatal CMA on DNA from CVS or amniotic fluid (AF) samples detects clinically significant copy number variants (CNVs) in 1-1.7% of fetuses when amniocentesis was performed for standard indications and in 6-7% of fetuses with congenital anomalies identified by prenatal ultrasound imaging (Hillman et al. 2013; Wapner et al. 2012) and also has a better diagnostic yield than karyotypes for stillbirths and miscarriages (Dhillon et al. 2014; Reddy et al. 2012). These results demonstrated that more widely offering CVS or amniocentesis to all pregnant women would significantly improve the ability to prenatally detect clinically significant chromosomal abnormalities (ACOG 2016a; Evans et al. 2016). While this was a major advance, in particular for pregnancies complicated by fetal structural anomalies, the combination of karyotype analysis and CMA still leaves at least 60-70% of fetuses with prenatally detected congenital anomalies without a genetic diagnosis (Best et al. 2018). A substantial proportion of these are expected to be caused by deleterious sequence variants in single genes, consistent with the high burden of Mendelian disease in newborns (Baird et al. 1988; Meng et al. 2017).
Slightly later, the transformative technology of high-throughput next-generation sequencing (NGS) matured and became cheaper. This resulted in an exponential rise in the use of exome sequencing for research to identify new disease genes and for clinical genetic diagnosis in children and adults with suspected genetic disorders where exome sequencing has an incremental diagnostic rate of 25-40% when CMA, karyotype and targeted testing cannot reveal a diagnosis (Clark et al. 2018; Yang et al. 2014). In this population, it has become the primary diagnostic sequencing method, replacing Sanger sequencing, which is now mostly used for confirmation of putative pathogenic sequence variants identified through NGS or for testing known familial variants.
The versatility of NGS has resulted in development and rapid introduction into the clinic of a variety of NGS-based tests for prenatal screening and diagnosis of Mendelian and chromosomal disorders. Among those, non-invasive prenatal screening (NIPS) for fetal chromosomal abnormalities by analysis of cell-free DNA (cfDNA) in the maternal plasma, now offered to millions of women worldwide each year, has been characterized as a disruptive innovation (Rifai et al. 2015) that has profoundly changed the practice of prenatal testing and screening for chromosomal and other genetic disorders (Bianchi and Chiu 2018). Other NGS-based genetic tests that are changing the practice of reproductive and prenatal genetics include expanded carrier screening (Gregg and Edwards 2018) and exome sequencing on fetal samples obtained through amniocentesis and CVS (Best et al. 2018; Lord et al. 2019; Normand et al. 2018a; Petrovski et al. 2019; Vora and Hui 2018). More recently, NGS-based assays on cfDNA in maternal plasma for non-invasive prenatal detection of mutations in single genes or small gene panels have been introduced (Hayward and Chitty 2018; Zhang et al. 2019). Furthermore, proof of concept has been demonstrated that single nucleotide variants (SNVs) across the fetal genome can be interrogated from cfDNA in maternal plasma (Fan et al. 2012; Kitzman et al. 2012; Lo et al. 2010). Lastly, NGS is now also becoming the research method of choice to identify fetal aneuploidy and CNVs in DNA from intact fetal cells isolated from the blood of pregnant women (Vossaert et al. 2018).
NGS is thus becoming the diagnostic pillar of Fetal Precision Medicine, by transforming prenatal genetic diagnosis testing from a broad screening approach for few conditions to a more precise individualized evaluation for a large number of genetic disorders. Here, we review where in the reproductive and prenatal care continuum these exciting new technologies are integrated, along with their pitfalls, challenges, and we propose research priorities for their responsible implementation in prenatal and reproductive care (Table 1).
Table 1:
Selected research priorities for NGS-based prenatal precision diagnosis
| NGS Technology | Research Priorities |
|---|---|
| General implementation research for all technologies | Development and evaluation of counseling and education tools |
| Public and provider education | |
| Cost and healthcare resource utilization | |
| Ethical and social issues | |
| Expanded Carrier screening | Size and content of panels |
| Best time for implementation | |
| Best approach and timing of partner screening | |
| Non-invasive prenatal testing and screening | Technological improvement of cfDNA testing for chromosomal abnormalities, focused on improving specificity and sensitivity, and increasing resolution |
| Technological improvement of cfDNA testing and screening for single gene disorders and larger clinical validation | |
| Technological improvement of cell-based non-invasive testing and clinical validation | |
| Exome and genome sequencing | Effective pre- and post-test counseling approaches |
| Indications for exome or genome sequencing | |
| Phenotype-genotype correlations, registries and database tools | |
| Healthcare resource use analysis of exomes, genomes and panels, and incremental benefit assessment compared to other testing for various indications |
Next-generation sequencing (NGS) principles and their implications for prenatal diagnostic applications.
For the most commonly used form of NGS, massively parallel sequencing, extracted genomic DNA is first fragmented and adaptors are ligated to those fragments to prepare the sequencing library. For gene panel and exome sequencing, the library is captured by hybridization to biotinylated probes or baits, bound to streptavidin-coated magnetic beads to enrich for the regions of interest. The final sequencing library is then immobilized on a solid substrate for cluster formation through bridge amplification, and sequencing by synthesis to obtain multiple copies of the nucleotide sequence of each overlapping fragment. The resulting sequence is computationally aligned to the reference genome, during which any discrepancy between sample and reference is marked as a putative genomic variant for further interpretation of potential pathogenicity (Normand et al. 2018a). A more detailed description of NGS can be found elsewhere (Biesecker and Green 2014), but several key features are important when we consider downstream applications. These include the length of the sequenced fragments and the fidelity and error rate of the sequencing process that vary between methods and the sequencing depth of coverage or average number of times a single base is read during a sequencing run. If identifying copy-number variation across a large region is the goal of genome-wide sequencing, lower depths are acceptable but more uniform coverage is desired. If the goal is to identify specific single-nucleotide variants, deeper sequence (larger number of overlapping reads) at targeted sites is required. The American College of Medical Genetics and Genomics (ACMG), along with the Association for Molecular Pathology (AMP) have issued guidelines for depth of coverage of diagnostic exome sequencing (Richards et al. 2015) and criteria for interpretation of pathogenicity of an identified variant. These include known association with a genetic condition relevant to the indication for sequencing, the variant’s frequency in the population, its segregation in the family consistent with the inheritance pattern of the phenotype, its predicted impact on protein function determined by bioinformatic functional predication models and, if available, experimental functional data from human studies and model systems.
There are additional technical considerations unique to prenatal applications of NGS (Abou Tayoun et al. 2018; Best et al. 2018; Normand et al. 2018a). For amniotic fluid or CVS samples, an assay to determine evidence of maternal cell contamination (MCC) is required. Depending on the volume of the obtained sample, the amount of DNA can be small. In addition, assays for MCC also use some of the limited amount of DNA and cell culture may be required to obtain enough DNA for sequencing. This not only lengthens the time to obtain a diagnosis, but replication errors in cell culture could potentially introduce SNVs resulting from culture artefacts that must be considered in data analysis. For CVS, DNA extracted from chorionic villi is primarily from trophoblast cells, whereas cell cultures represent the mesenchymal core of the villi. Because decisions about pregnancy management are made with sequencing results, a short turn-around time is critical. In order to accommodate smaller amounts of DNA and the need for a rapid turn-around time, the sequencing library preparation may include more amplification steps. The DNA amplification process can also introduce artefactual sequence errors. In addition, prenatal sequencing is often done as a trio, whereby parental and fetal DNAs are sequenced in parallel, allowing faster interpretation of de novo dominant and recessive variants. Interpretation of the pathogenicity of sequence variants requires effective communication of precise phenotypic information from the clinic to the lab. Yet, accurate phenotyping using consistent nomenclature is particularly challenging prenatally, as phenotypes can be incompletely defined due to imaging limitations and also evolve as pregnancy progresses (Best et al. 2018; Gray et al. 2019). Furthermore, the general knowledge of which fetal phenotypes are associated with specific genes and gene variants is more limited than postnatally. Prenatally lethal phenotypes are especially underrepresented in the current available databases that can be queried to help support variant interpretation (Gray et al. 2019).
Expanded carrier screening (ECS)
Parental carrier screening is an important component of prenatal and preconception reproductive genetic risk assessment (Gregg and Edwards 2018). Its primary goal is to identify carrier couples for recessive conditions and carrier mothers of X-linked conditions (Edwards et al. 2015; Henneman et al. 2016). For years, carrier screening was focused on a limited number of higher prevalence moderate to severe recessive conditions, with ethnicity-based screening for disorders that are more common in specific populations. Recognized disadvantages of this strategy are the growing number of individuals of mixed ethnic backgrounds, high inaccuracy of self-declared ethnicity, and lack of screening for the majority of rare autosomal recessive and X-linked disorders (Langlois et al. 2015). With NGS-based sequencing, different expanded carrier screening panels containing up to hundreds of genetic disorders have been developed and introduced into the clinic. There is no strict guidance on panel content and variant interpretation, and though various ECS panels overlap, they have important differences, mostly in the number of genes included (Antonarakis 2019; Chokoshvili et al. 2017). With panels of >400 or more genes, >85% of screened individuals carry ≥1 or more pathogenic variants (Bell et al. 2011; Martin et al. 2015), but the identification of “at risk” carrier couples, ~1-3% for most panels with >100 genes and 5% with a panel of 549 genes (Martin et al. 2015), does not increase dramatically, because each included disorder is rare in the population. When couples are screened sequentially, a positive result in the first screened partner (usually the pregnant woman) can cause anxiety and increases healthcare resource utilization to address additional genetic counseling and coordination of partner testing or screening. Turn-around time to the final “couple carrier status” result is increased and partners don’t always pursue the follow-up testing. With more genes on carrier screening panels, sequential screening may become impractical because of the large proportion of screened individuals that are identified as carriers. Thus, concurrent couple screening will be more efficient, but its benefits, counseling aspects, costs and healthcare utilization burden should be prospectively investigated.
Variant reporting for ECS is appropriately limited to well described pathogenic and likely pathogenic variants. Yet, neonatal and prenatal exome sequencing have already identified cases where a pathogenic or likely pathogenic variant in one allele and a VUS in trans or two high-risk VUS in trans are interpreted as likely causing the proband’s phenotype. In these cases, couple carrier status in genes on ECS panels would not be reported until they are reclassified. This highlights the importance of depositing all sequence data and associated phenotypes in public databases (see below). How to address new information on variant pathogenicity in ECS programs by either rescreening on a regular basis or reinterpretation of existing results is to our knowledge incompletely addressed. Integrating this is in reproductive health care also carries counseling and ethical challenges which should be formally studied.
Lastly, for some diseases, SNVs are not the only possible mutation mechanism and this should be conveyed in pre-test counseling, along with information about included methods in the ECS panel for detection of other mutation types for some genes; e.g. copy numbers of exons of DMD (X-linked Duchene Muscular Dystrophy gene), and copy number of SMN1 (spinal muscular atrophy gene) and its neighboring SMN2, or of HBA (alpha-thalassemia).
Cell-free DNA sequencing for non-invasive assessment of chromosomal aneuploidy and copy number variants.
The discovery of fetal cfDNA in maternal plasma in 1997 (Lo et al. 1997) was followed by demonstration in 2008 that massively parallel sequencing of circulating cfDNA fragments coupled with counting of fragments that map to each chromosome can provide information about fetal chromosomal aneuploidy (Chiu et al. 2008; Fan et al. 2008). The subsequent commercial introduction of cfDNA-based NIPS had an unprecedented impact on the practice of prenatal screening for trisomies 13, 18 and 21 for which it has superior positive and negative predictive values compared to standard multiple marker screening (Gil et al. 2015). However, this coincided with the demonstration of the contribution to fetal morbidity of clinically significant CNVs, detectable by CMA on DNA from CVS and AF samples but not by NIPS. Although this expanded the indications for genetic amniocenteses or CVS (Evans et al. 2016; Wapner et al. 2012), with the rapid commercialization of NIPS, the number of these procedures declined significantly (Williams et al. 2015), despite evidence that their associated risk of pregnancy loss ~1:909 for amniocentesis and ~1:450 for CVS (Akolekar et al. 2015; Eddleman et al. 2003) is also lower than previously thought, with data supporting that risks of both procedures are identical if performed by experienced providers (Wulff et al. 2016). The predicted consequence of the reduction in amniocenteses and CVS is that many diagnosable disorders, which in aggregate are more common than Down syndrome and other common trisomies, will not be identified prenatally (Evans et al. 2016). Although cfDNA screening for sex chromosome aneuploidy, rarer autosomal aneuploidies and selected clinically significant copy number variants, including a genome-wide screen for CNVs larger than 7 Mb (Lefkowitz et al. 2016), are increasingly offered, these are not yet recommended by professional societies because of the low PPVs and high false positive rates of these expanded NIPS tests (ACOG 2016b). These limitations of NIPS are not optimally communicated by primary prenatal care providers to patients, with a tendency to emphasize the risk of diagnostic procedures.
Efforts to develop and clinically evaluate the benefit of new tools and strategies for educating providers and stakeholders are a priority. In parallel, development and thorough validation of improved non-invasive tests that can accurately diagnose the same genetic disorders as those identifiable by amniocentesis or CVS, should be prioritized. New approaches to cell-free DNA analysis that take into account size differences between fetal and maternal cfDNA fragments, along with recognition of preferred fragment ends that differ between mother and fetus, offer opportunities for improvements (Sun et al. 2018). Ultimately, a long-term goal for the field should be to offer all prenatal genetic testing non-invasively. While this may eventually be attainable, to date, such precision prenatal genetic diagnosis still requires a diagnostic procedure.
Sequencing of gene panels, exomes and genomes for prenatal diagnosis
Once a pathogenic CNV or aneuploidy are ruled out in the setting of ultrasound-detected fetal anomalies, causative sequence variants in single genes should be considered. The ability to sequence gene panels, exomes or genomes to identify these variants is another area where NGS technology is changing prenatal genetic diagnosis. Initially, smaller studies and series showed that prenatal exome sequencing can identify genetic causes for fetal birth defects in ongoing pregnancies and stillbirths, that were unexplained after standard testing, but with variable detection rates, ranging from 6 to >80% (Best et al. 2018). Results from one diagnostic lab, which reflects clinical practice of prenatal exome sequencing on selected cases, showed a diagnostic rate of 32% (Normand et al. 2018b), while recent larger studies on unselected cases with one or more anomalies or enlarged nuchal translucency, showed detection rates between 8.5 and 10% (Lord et al. 2019; Petrovski et al. 2019). Data thus far indicate higher yields for CNS defects, skeletal phenotypes, and for fetuses with multiple versus single isolated congenital anomalies. The table in Online Resource 1 lists key published prenatal exome sequencing experience. When they are reported there is, not surprisingly, a relatively high rate of VUS and what is labeled by one group as “bioinformatic signatures” of plausible new causative genes or variants in known disease genes for which evidence of pathogenicity is too limited to unequivocally attribute causation (Drury et al. 2015; Lord et al. 2019; Petrovski et al. 2019; Vora et al. 2017). This highlights the importance of detailed fetal phenotyping along with a concerted international effort to collect and catalogue information that can support sequence variant interpretation for fetal phenotypes (Filges and Friedman 2015; Gray et al. 2019; Meier et al. 2019).
The importance of the fetal phenotype
Detailed and accurate information is critical for interpretation of the pathogenicity of sequence variants. Postnatally this derives from a combination of the external dysmorphological exam, imaging results of internal organ systems and the skeleton, and information on functional impairments, including muscular strength and tone, growth, neurodevelopmental and behavioral assessments, and the results of metabolic and other laboratory tests. Of these, growth and major structural defects can be assessed prenatally, but functional assessments are more limited and different from those obtained during a postnatal evaluation. They include gross fetal movements and tone, bladder and stomach filling and emptying, amniotic fluid levels, placental and umbilical cord morphology, and Doppler measurements of blood flow dynamics in fetal heart and fetal, uterine and placental vessels. In addition, the fetus is still developing and depending on the gestational age at assessment not all organs are fully formed or grown. Emerging exome data are teaching us that prenatal phenotypes of known genetic disorders can diverge from those seen postnatally and have even revealed different and unsuspected prenatal phenotypes of known disease genes. Examples include, pleural effusions and increased nuchal translucency or cystic hygroma as the predominant prenatal presentation of Noonan syndrome, while postnatally congenital heart defects are the predominant findings (Gray et al. 2019), de novo mutations in KMT2D, which postnatally are associated with Kabuki syndrome, in fetuses with congenital heart effects, renal and other congenital anomalies (Lord et al. 2019; Normand et al. 2018b; Petrovski et al. 2019), and RYR1 mutations in fetal akinesia (McKie et al. 2014; Suzumori et al. 2018). These examples illustrate that some prenatal phenotypes, in particular lethal ones, are underrepresented in genotype-phenotype databases which are biased towards postnatal presentations of genetic diseases or syndromes. Thus, systematic international multicenter efforts are needed to include prenatal phenotype-genotype data using standardized nomenclature in commonly used databases and registries and align prenatal imaging reports with this nomenclature.
Considerations for variant annotation on prenatal samples
The same principles for variant annotation and reporting are followed for prenatal (fetal) sequencing as in other clinical scenarios, but require additional considerations. In most cases prenatal exome sequencing is performed as a trio, and hence results relevant to the parents can be obtained. This can complicate interpretation and reporting of actionable incidental findings. The most complex are the 59 “actionable” genes for which the ACMG recommends reporting pathogenic variants because measures can be taken to improve health if they are identified (Green et al. 2013), with the ability of patients to opt out of receiving the information (ACMG 2015; Kalia et al. 2017). These guidelines exclude fetal samples (Green et al. 2013), but if these variants are identified in one of the parents with trio sequencing, there is a 50% chance the fetus has the same finding, and vice versa. The medical actions typically recommended for conditions associated with these 59 genes do not involve prenatal care, but if reported in the fetus, a decision to terminate a pregnancy could be taken by parents for potentially treatable conditions that may not present until later in life. While many choose not to report such secondary findings prenatally, approaches vary and their handling with appropriate consent procedures must be addressed in pre-test counseling. Other findings that are ethically complex prenatally include incidental detection of other adult-onset conditions not on this list of 59 genes, carrier status, and variants of uncertain significance. More research on the ethical and counseling aspects surrounding these is needed.
Cell-free DNA sequencing for non-invasive assessment of single gene disorders
The ability to isolate and sequence fetal cfDNA has led to exciting new developments for non-invasive genetic diagnosis of single gene disorders. Various research labs have developed and evaluated tests for specific de novo dominant mutations (for example in COL1A1, COL1A2, FGFR2, and FGFR3 associated with skeletal dysplasias) and for selected recessive mutations (Hayward and Chitty 2018). In the United Kingdom, cfDNA-based single gene tests and small multiplex panels have been approved as clinical tests that can be offered to selected patients, most frequently for fetal skeletal dysplasias (Drury et al. 2016). In the United States, a screening test with 30 genes has been introduced with excellent technical validation results (Zhang et al. 2019). One professional society currently recommends against its use based on concern for incomplete information on clinical utility (ACOG 2019). However, current data indicates that issues like confined placental mosaicism, which complicates aneuploidy assessment are of much less concern for cfDNA-based single-gene testing (Zhang et al. 2019). Thus, these tests have value when there is advanced paternal age or when the fetus is suspected to have a disease caused by variants in included genes and amniocentesis or CVS are declined or not possible. Larger studies and more accumulated clinical use data will be needed before more widespread adoption. The growing experience with diagnostic prenatal and neonatal exome sequencing can guide future adjustments in gene content of such panels.
Use of NGS for cell-based non-invasive prenatal fetal genome assessment
A final application, that now benefits from NGS is the analysis of the genome of intact fetal cells isolated from maternal blood. This is an area of intense ongoing research with recent significant improvements in the isolation of these rare cells (Beaudet 2016; Rezaei et al. 2018; Vestergaard et al. 2017). The analysis of the genome of these purified cells is now shifting from single-cell CMA to sequencing. It has been demonstrated that low-coverage genome-wide sequencing allows determination of aneuploidy and CNVs as small as 1 Mb from a single circulating trophoblast cell. Early data also show that by analyzing sequence data of individual cells separately, apoptosis artefacts can be better accounted for than when isolated cells are pooled (Vossaert et al. 2018). This is a promising non-invasive prenatal diagnosis method that deserves further validation in larger studies and such work is ongoing.
A pathway to prenatal precision diagnosis
We envision a future wherein all individuals are offered some form of ECS when they reach young adulthood, or become sexually active. We propose here that carrier screening be offered earlier, uncoupled from preconception and prenatal care if the goal is wide implementation of carrier screening before pregnancy, allowing more opportunities for reproductive planning. The optimal content of screening panels and the types of variants reported is currently under debate. Some argue for smaller panels to balance cost, equal access, and burden on counseling and healthcare utilization, with the benefits of detecting risks for a larger number of diseases. Others argue that more comprehensive panels, including “carrier exomes” are the ultimate goal. This debate may end if, as part of their healthcare, individuals will in the future more commonly have their exomes or genomes sequenced as children or young adults. Carrier status information could then be queried when needed from stored exome results in an individual’s health record and be interpreted based on knowledge that is current at the time of carrier data query.
The high detection rate of clinically significant CNVs by prenatal CMA and the relatively low risk of amniocentesis and CVS validate offering amniocentesis or CVS with CMA to all pregnant women who desire comprehensive genetic assessment of their pregnancy. Yet, cfDNA-based NIPS marketing strategies focus on the benefits of screening for common fetal aneuploidies, while underemphasizing the impact of rare CNVs as contributors to fetal and childhood diseases and overemphasizing the risk of CVS and amniocentesis. When women who desire genetic risk assessment for their pregnancies present for genetic counseling, they often already favor NIPS based on information from peers, online sources, or primary providers. Universal formal genetic counseling to objectively review all genetic screening and testing options is not currently possible because of limitations in access to genetics trained providers, while primary care providers have time constraints and are not as extensively trained in genetic counseling. There is thus a need for development and testing of better education tools, such as easy-to-use self-directed electronic tools for providers and patients with standardized information on the benefits, indications and pitfalls of the different prenatal genetic testing strategies.
Prenatal exome sequencing is currently either conducted in research studies or for highly selected cases (Best et al. 2018; Vora and Hui 2018), because of our still incomplete understanding of pathogenicity of prenatally detected sequence variants. Yet, important data on incremental diagnostic rate of prenatal exome sequencing in the setting of fetal anomalies, and in particular on the contribution of de novo dominant mutations are emerging (Lord et al. 2019; Normand et al. 2018b; Petrovski et al. 2019). Together with the growing knowledge about variant classification and fetal phenotype annotation, this indicates that exome sequencing for prenatal diagnosis is likely to become more widespread. Clinical utility and cost-benefit analysis studies should continue to evaluate where this technology best fits in the prenatal diagnostic continuum (ISPD et al. 2018). At least one trial is ongoing in the United States to evaluate the clinical utility of genome sequencing, which has already entered clinical care in other settings, for prenatal genetic evaluation of fetuses with congenital anomalies. With further experience and technological refinement, genome sequencing has the potential to replace exome sequencing as the genetic test of choice on prenatal samples. Its advantages over exomes include the more even coverage of the genome, the improved ability to detect copy number variation, the potential ability to detect balanced structural rearrangements at higher resolution than a karyotype, and the ability to detect non-coding clinically significant variants in a single assay, but those results are more complex to interpret and its overall cost is higher. Although this remains to be tested and validated, with prenatal genome sequencing it may become possible to avoid the lengthier stepwise diagnostic odyssey of karyotype and CMA first, followed by panel sequencing or exome sequencing, with the potential for a more favorable total turn-around time. Ultimately, although still more remote, if cfDNA-based analysis or intact circulating fetal or trophoblast cell analysis continues to improve, we may be able to do the same on non-invasively obtained samples. This would not only eliminate the very small risk of amniocentesis and CVS, it would also provide access to precision diagnosis for patients who cannot easily reach specialized care by experts who perform amniocentesis and CVS. Ongoing research and already existing technological advances suggest that this will likely become technically possible, but the fundamental challenge will be to address the associated counseling needs and ethical, societal and healthcare utilization aspects of these emerging technologies.
Finally, improved early detection of single gene disorders can pave the way for prenatal precision therapies that target the genetic defect or its consequences on the protein product or downstream pathways or are aimed at stem-cell replacement (Larson and Cohen 2000; Chitty et al. 2016; Sagar et al. 2018; Schneider et al. 2018). Although gene therapy holds promise for treating genetic diseases, a major hurdle postnatally is rejection by the body’s immune system. In utero gene or cell therapy carries the advantage of a window of opportunity where the fetal immune system is quiescent and therefore gene insertion can be considered. (Larson and Cohen 2000). Successful prenatal protein replacement for X-linked hypohydrotic ectodermal dysplasia has been reported (Schneider et al. 2018) and a European multicenter study BOOSTB4 (Boost Brittle Bones Before Birth) is aimed at evaluating the prenatal transplantation of fetal mesenchymal cells in severe viable forms of osteogenesis imperfecta (Chitty et al. 2016; Sagar et al. 2018). A study to prenatally treat alpha-thalassemia major with in utero stem cell therapy is also underway. In addition, postnatal targeted therapy can be initiated immediately after birth if prenatally diagnosed, for example for conditions like the severe X-linked metabolic disorder, ornithine transcarbamylase deficiency. With further advances in prenatal diagnosis these approaches can potentially be expanded in the future for the treatment of other fetal genetic disorders.
Supplementary Material
Acknowledgments
Funding: This work is supported in part by the administrative core of the Baylor College of Medicine Intellectual and Developmental disabilities Research Center, National Institutes of Health (NIH) grant U54HD083092. IVdV also receives support for research on prenatal genome sequencing from NIH grant HD055651. The content is solely the responsibility of the authors and does not represent the official views of the Eunice Kennedy Shriver National Institute of Child Health and Human Development or the National Institutes of Health.
Footnotes
Conflict of interest disclosures: RS has no conflicts to declare. IVdV conducts research on prenatal genome sequencing research that receives support from Illumina.
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- Abou Tayoun AN, Spinner NB, Rehm HL, Green RC, Bianchi DW (2018) Prenatal DNA Sequencing: Clinical, Counseling, and Diagnostic Laboratory Considerations Prenat Diagn 38:26–32 doi: 10.1002/pd.5038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American College of Medical Genetics and Genomics (ACMG) Board of Directors (2015) ACMG policy statement: updated recommendations regarding analysis and reporting of secondary findings in clinical genome-scale sequencing Genet Med 17:68–69 doi: 10.1038/gim.2014.151 [DOI] [PubMed] [Google Scholar]
- American College of Obstetrics and Gynecology (ACOG) (2019). Practice Advisory_ Cell-free DNA to Screen for Single-Gene Disorders (https://www.acog.org/Clinical-Guidance-and-Publications/Practice-Advisories/Cell-free-DNA-to-Screen-for-Single-Gene-Disorders; February 21, 2019)
- American College of Obstetrics and Gynecology (ACOG) Committee on Genetics (2016a) Practice Bulletin No. 162 Summary: Prenatal Diagnostic Testing for Genetic Disorders Obstet Gynecol 127:976–978 doi: 10.1097/AOG.0000000000001438 [DOI] [PubMed] [Google Scholar]
- American College of Obstetrics and Gynecology (ACOG) Committee on Genetics (2016b) Practice Bulletin No. 163 Summary: Screening for Fetal Aneuploidy Obstet Gynecol 127:979–981 doi: 10.1097/AOG.0000000000001439 [DOI] [PubMed] [Google Scholar]
- Akolekar R, Beta J, Picciarelli G, Ogilvie C, D'Antonio F (2015) Procedure-related risk of miscarriage following amniocentesis and chorionic villus sampling: a systematic review and meta-analysis Ultrasound Obstet Gynecol 45:16–26 doi: 10.1002/uog.14636 [DOI] [PubMed] [Google Scholar]
- Antonarakis SE (2019) Carrier screening for recessive disorders Nature reviewsGenetics 20:549–561 doi: 10.1038/s41576-019-0134-2 [DOI] [PubMed] [Google Scholar]
- Baird PA, Anderson TW, Newcombe HB, Lowry RB (1988) Genetic disorders in children and young adults: a population study Am J Hum Genet 42:677–693 [PMC free article] [PubMed] [Google Scholar]
- Beaudet AL (2016) Using fetal cells for prenatal diagnosis: History and recent progress American journal of medical genetics Part C, Seminars in medical genetics 172:123–127 doi: 10.1002/ajmg.c.31487 [DOI] [PubMed] [Google Scholar]
- Bell CJ et al. (2011) Carrier testing for severe childhood recessive diseases by next-generation sequencing Science translational medicine 3:65ra64 doi: 10.1126/scitranslmed.3001756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best S, Wou K, Vora N, Van der Veyver IB, Wapner R, Chitty LS (2018) Promises, pitfalls and practicalities of prenatal whole exome sequencing Prenat Diagn 38:10–19 doi: 10.1002/pd.5102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi DW, Chiu RWK (2018) Sequencing of Circulating Cell-free DNA during Pregnancy N Engl J Med 379:464–473 doi: 10.1056/NEJMra1705345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biesecker LG, Green RC (2014) Diagnostic clinical genome and exome sequencing N Engl J Med 371:1170 doi: 10.1056/NEJMc1408914 [DOI] [PubMed] [Google Scholar]
- Chitty LS, David AL, Gottschalk I, Oepkes D, Westgren M, Götherström C, Consortium O (2016) EP21.04. BOOSTB4: a clinical study to determine safety and efficacy of pre-and/ or postnatal stem cell transplantation for treatment of osteogenesis imperfecta. Ultrasound Obstet Gynecol 48S1:356 doi: 10.1002/uog.17084 [DOI] [Google Scholar]
- Chiu RW et al. (2008) Noninvasive prenatal diagnosis of fetal chromosomal aneuploidy by massively parallel genomic sequencing of DNA in maternal plasma Proc Natl Acad Sci U S A 105:20458–20463 doi: 10.1073/pnas.0810641105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chokoshvili D, Borry P, Vears DF (2017) A systematic analysis of online marketing materials used by providers of expanded carrier screening Genet Med doi: 10.1038/gim.2017.222 [DOI] [PubMed] [Google Scholar]
- Clark MM, Stark Z, Farnaes L, Tan TY, White SM, Dimmock D, Kingsmore SF (2018) Meta-analysis of the diagnostic and clinical utility of genome and exome sequencing and chromosomal microarray in children with suspected genetic diseases NPJ Genom Med 3:16 doi: 10.1038/s41525-018-0053-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon RK, Hillman SC, Morris RK, McMullan D, Williams D, Coomarasamy A, Kilby MD (2014) Additional information from chromosomal microarray analysis (CMA) over conventional karyotyping when diagnosing chromosomal abnormalities in miscarriage: a systematic review and meta-analysis BJOG 121:11–21 doi: 10.1111/1471-0528.12382 [DOI] [PubMed] [Google Scholar]
- Drury S, Mason S, McKay F, Lo K, Boustred C, Jenkins L, Chitty LS (2016) Implementing Non-Invasive Prenatal Diagnosis (NIPD) in a National Health Service Laboratory; From Dominant to Recessive Disorders Adv Exp Med Biol 924:71–75 doi: 10.1007/978-3-319-42044-8_14 [DOI] [PubMed] [Google Scholar]
- Drury S et al. (2015) Exome sequencing for prenatal diagnosis of fetuses with sonographic abnormalities Prenat Diagn 35:1010–1017 doi: 10.1002/pd.4675 [DOI] [PubMed] [Google Scholar]
- Eddleman K et al. (2003) Pregnancy loss rates after midtrimester amniocentesis: the faster trial American Journal of Obstetrics and Gynecology 189:S111 [Google Scholar]
- Edwards JG et al. (2015) Expanded carrier screening in reproductive medicine-points to consider: a joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal-Fetal Medicine Obstet Gynecol 125:653–662 doi: 10.1097/AOG.0000000000000666 [DOI] [PubMed] [Google Scholar]
- Evans MI, Wapner RJ, Berkowitz RL (2016) Noninvasive prenatal screening or advanced diagnostic testing: caveat emptor Am J Obstet Gynecol 215:298–305 doi: 10.1016/j.ajog.2016.04.029 [DOI] [PubMed] [Google Scholar]
- Fan HC, Blumenfeld YJ, Chitkara U, Hudgins L, Quake SR (2008) Noninvasive diagnosis of fetal aneuploidy by shotgun sequencing DNA from maternal blood Proc Natl Acad Sci U S A 105:16266–16271 doi: 10.1073/pnas.0808319105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan HC, Gu W, Wang J, Blumenfeld YJ, El-Sayed YY, Quake SR (2012) Non-invasive prenatal measurement of the fetal genome Nature 487:320–324 doi: 10.1038/nature11251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filges I, Friedman JM (2015) Exome sequencing for gene discovery in lethal fetal disorders - harnessing the value of extreme phenotypes Prenat Diagn 35:1005–1009 doi: 10.1002/pd.4464 [DOI] [PubMed] [Google Scholar]
- Friedman JM (1981) Genetic disease in the offspring of older fathers Obstet Gynecol 57:745–749 [PubMed] [Google Scholar]
- Gil MM, Quezada MS, Revello R, Akolekar R, Nicolaides KH (2015) Analysis of cell-free DNA in maternal blood in screening for fetal aneuploidies: updated meta-analysis Ultrasound Obstet Gynecol 45:249–266 doi: 10.1002/uog.14791 [DOI] [PubMed] [Google Scholar]
- Gray KJ, Wilkins-Haug LE, Herrig NJ, Vora NL (2019) Fetal phenotypes emerge as genetic technologies become robust Prenat Diagn doi: 10.1002/pd.5532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green RC et al. (2013) ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing Genet Med 15:565–574 doi: 10.1038/gim.2013.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg AR, Edwards JG (2018) Prenatal genetic carrier screening in the genomic age Seminars in perinatology 42:303–306 doi: 10.1053/j.semperi.2018.07.019 [DOI] [PubMed] [Google Scholar]
- Hayward J, Chitty LS (2018) Beyond screening for chromosomal abnormalities: Advances in non-invasive diagnosis of single gene disorders and fetal exome sequencing Semin Fetal Neonatal Med 23:94–101 doi: 10.1016/j.siny.2017.12.002 [DOI] [PubMed] [Google Scholar]
- Henneman L et al. (2016) Responsible implementation of expanded carrier screening Eur J Hum Genet 24:e1–e12 doi: 10.1038/ejhg.2015.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman SC et al. (2013) Use of prenatal chromosomal microarray: prospective cohort study and systematic review and meta-analysis Ultrasound Obstet Gynecol 41:610–620 doi: 10.1002/uog.12464 [DOI] [PubMed] [Google Scholar]
- ISPD, SMFM, PQF (2018) Joint Position Statement from the International Society for Prenatal Diagnosis (ISPD), the Society for Maternal Fetal Medicine (SMFM), and the Perinatal Quality Foundation (PQF) on the use of genome-wide sequencing for fetal diagnosis Prenat Diagn 38:6–9 doi: 10.1002/pd.5195 [DOI] [PubMed] [Google Scholar]
- Larson JE, Cohen JC (2000) In utero gene therapy Ochsner J 2:107–110 [PMC free article] [PubMed] [Google Scholar]
- Kalia SS et al. (2017) Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics Genet Med 19:249–255 doi: 10.1038/gim.2016.190 [DOI] [PubMed] [Google Scholar]
- Kitzman JO et al. (2012) Noninvasive whole-genome sequencing of a human fetus Science translational medicine 4:137ra176 doi: 10.1126/scitranslmed.3004323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong A et al. (2012) Rate of de novo mutations and the importance of father's age to disease risk Nature 488:471–475 doi: 10.1038/nature11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langlois S, Benn P, Wilkins-Haug L (2015) Current controversies in prenatal diagnosis 4: pre-conception expanded carrier screening should replace all current prenatal screening for specific single gene disorders Prenat Diagn 35:23–28 doi: 10.1002/pd.4532 [DOI] [PubMed] [Google Scholar]
- Lefkowitz RB et al. (2016) Clinical Validation of a Non-Invasive Prenatal Test for Genome-Wide Detection of Fetal Copy Number Variants Am J Obstet Gynecol 215:227.e221–227.e216. doi: 10.1016/j.ajog.2016.02.030 [DOI] [PubMed] [Google Scholar]
- Lo YM et al. (2010) Maternal plasma DNA sequencing reveals the genome-wide genetic and mutational profile of the fetus Science translational medicine 2:61ra91 doi: 10.1126/scitranslmed.3001720 [DOI] [PubMed] [Google Scholar]
- Lo YM, Corbetta N, Chamberlain PF, Rai V, Sargent IL, Redman CW, Wainscoat JS (1997) Presence of fetal DNA in maternal plasma and serum Lancet 350:485–487 [DOI] [PubMed] [Google Scholar]
- Lord J et al. (2019) Prenatal exome sequencing analysis in fetal structural anomalies detected by ultrasonography (PAGE): a cohort study Lancet 393:747–757 doi: 10.1016/s0140-6736(18)31940-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin J et al. (2015) Comprehensive carrier genetic test using next-generation deoxyribonucleic acid sequencing in infertile couples wishing to conceive through assisted reproductive technology Fertil Steril 104:1286–1293 doi: 10.1016/j.fertnstert.2015.07.1166 [DOI] [PubMed] [Google Scholar]
- McKie AB et al. (2014) Germline mutations in RYR1 are associated with foetal akinesia deformation sequence/lethal multiple pterygium syndrome Acta Neuropathol Commun 2:148 doi: 10.1186/s40478-014-0148-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier N et al. (2019) Exome sequencing of fetal anomaly syndromes: novel phenotype-genotype discoveries Eur J Hum Genet 27:730–737 doi: 10.1038/s41431-018-0324-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng L et al. (2017) Use of Exome Sequencing for Infants in Intensive Care Units: Ascertainment of Severe Single-Gene Disorders and Effect on Medical Management JAMA Pediatr:e173438 doi: 10.1001/jamapediatrics.2017.3438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Normand EA, Alaimo JT, Van den Veyver IB (2018a) Exome and genome sequencing in reproductive medicine Fertil Steril 109:213–220 doi: 10.1016/j.fertnstert.2017.12.010 [DOI] [PubMed] [Google Scholar]
- Normand EA et al. (2018b) Clinical exome sequencing for fetuses with ultrasound abnormalities and a suspected Mendelian disorder Genome medicine 10:74 doi: 10.1186/s13073-018-0582-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovski S et al. (2019) Whole-exome sequencing in the evaluation of fetal structural anomalies: a prospective cohort study Lancet 393:758–767 doi: 10.1016/S0140-6736(18)32042-7 [DOI] [PubMed] [Google Scholar]
- Reddy UM et al. (2012) Karyotype versus microarray testing for genetic abnormalities after stillbirth N Engl J Med 367:2185–2193 doi: 10.1056/NEJMoa1201569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezaei M et al. (2018) A Reappraisal of Circulating Fetal Cell Noninvasive Prenatal Testing Trends in biotechnology doi: 10.1016/j.tibtech.2018.11.001 [DOI] [PubMed] [Google Scholar]
- Richards S et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Genet Med 17:405–424 doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifai N, Topol E, Chan E, Lo YM, Wittwer CT (2015) Disruptive Innovation in Laboratory Medicine Clinical chemistry 61:1129–1132 doi: 10.1373/clinchem.2015.243667 [DOI] [PubMed] [Google Scholar]
- Sagar R, Walther-Jallow L, David AL, Götherström C, Westgren M (2018) Fetal Mesenchymal Stromal Cells: an Opportunity for Prenatal Cellular Therapy Curr Stem Cell Rep 4:61–68 doi: 10.1007/s40778-018-0118-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider H et al. (2018) Prenatal Correction of X-Linked Hypohidrotic Ectodermal Dysplasia N Engl J Med 378:1604–1610 doi: 10.1056/NEJMoa1714322 [DOI] [PubMed] [Google Scholar]
- Sun K et al. (2018) Size-tagged preferred ends in maternal plasma DNA shed light on the production mechanism and show utility in noninvasive prenatal testing Proc Natl Acad Sci U S A doi: 10.1073/pnas.1804134115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzumori N et al. (2018) Compound heterozygous RYR1 mutations by whole exome sequencing in a family with three repeated affected fetuses with fetal akinesia Eur J Obstet Gynecol Reprod Biol 230:200–202 doi: 10.1016/j.ejogrb.2018.09.013 [DOI] [PubMed] [Google Scholar]
- Toriello HV, Meck JM, Professional P, Guidelines C (2008) Statement on guidance for genetic counseling in advanced paternal age Genet Med 10:457–460 doi: 10.1097/GIM.0b013e318176fabb [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vestergaard EM et al. (2017) On the road to replacing invasive testing with cell-based NIPT: five clinical cases with aneuploidies, microduplication, unbalanced structural rearrangement or mosaicism Prenat Diagn 37:1120–1124 doi: 10.1002/pd.5150 [DOI] [PubMed] [Google Scholar]
- Vora NL, Hui L (2018) Next-generation sequencing and prenatal 'omics: advanced diagnostics and new insights into human development Genet Med 20:791–799 doi: 10.1038/s41436-018-0087-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vora NL et al. (2017) Prenatal exome sequencing in anomalous fetuses: new opportunities and challenges Genet Med 19:1207–1216 doi: 10.1038/gim.2017.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossaert L et al. (2018) Reliable detection of subchromosomal deletions and duplications using cell-based noninvasive prenatal testing Prenat Diagn 38:1069–1078 doi: 10.1002/pd.5377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapner RJ et al. (2012) Chromosomal microarray versus karyotyping for prenatal diagnosis N Engl J Med 367:2175–2184 doi: 10.1056/NEJMoa1203382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J 3rd, Rad S, Beauchamp S, Ratousi D, Subramaniam V, Farivar S, Pisarska MD (2015) Utilization of noninvasive prenatal testing: impact on referrals for diagnostic testing Am J Obstet Gynecol 213:102 e101–106 doi: 10.1016/j.ajog.2015.04.005 [DOI] [PubMed] [Google Scholar]
- Wulff CB, Gerds TA, Rode L, Ekelund CK, Petersen OB, Tabor A, Danish Fetal Medicine Study G (2016) Risk of fetal loss associated with invasive testing following combined first-trimester screening for Down syndrome: a national cohort of 147,987 singleton pregnancies Ultrasound Obstet Gynecol 47:38–44 doi: 10.1002/uog.15820 [DOI] [PubMed] [Google Scholar]
- Yang Y et al. (2014) Molecular Findings Among Patients Referred for Clinical Whole-Exome Sequencing Jama 312:1870–1879 doi: 10.1001/jama.2014.14601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J et al. (2019) Non-invasive prenatal sequencing for multiple Mendelian monogenic disorders using circulating cell-free fetal DNA Nat Med 25:439–447 doi: 10.1038/s41591-018-0334-x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.