

Abstract
Nicotinic acetylcholine receptors (nAChRs) are known to play a role in cognitive functions of the hippocampus, such as memory consolidation. Given that they conduct Ca2+ and are capable of regulating the release of glutamate and γ-aminobutyric acid (GABA) within the hippocampus, thereby shifting the excitatory-inhibitory ratio, we hypothesized that the activation of nAChRs will result in the potentiation of hippocampal networks and alter synchronization. We used nicotine as a tool to investigate the impact of activation of nAChRs on neuronal network dynamics in primary embryonic rat hippocampal cultures prepared from timed-pregnant Sprague-Dawley rats. We perturbed cultured hippocampal networks with increasing concentrations of bath-applied nicotine and performed network extracellular recordings of action potentials using a microelectrode array (MEA). We found that nicotine modulated network dynamics in a concentration-dependent manner; it enhanced firing of action potentials as well as facilitated bursting activity. In addition, we used pharmacological agents to determine the contributions of discrete nAChR subtypes to the observed network dynamics. We found that β4-containing nAChRs are necessary for the observed increases in spiking, bursting and synchrony, while the activation of α7 nAChRs augments nicotine-mediated network potentiation but is not necessary for its manifestation. We also observed that antagonists of N-methyl-D-aspartate receptors (NMDARs) and group I metabotropic glutamate receptors (mGluRs) partially blocked the effects of nicotine. Furthermore, nicotine exposure promoted autophosphorylation of Ca2+/calmodulin-dependent kinase II (CaMKII) and serine 831 phosphorylation of the α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPAR) subunit GluA1. These results suggest that nicotinic receptors induce potentiation and synchronization of hippocampal networks and glutamatergic synaptic transmission. Findings from this work highlight the impact of cholinergic signaling in generating network-wide potentiation in the form of enhanced spiking and bursting dynamics that coincide with molecular correlates of memory such as increased phosphorylation of CaMKII and GluA1.
Keywords: Nicotine, nicotinic acetylcholine receptors, in vitro hippocampal networks, neural network dynamics, micro-electrode arrays
Graphical abstract
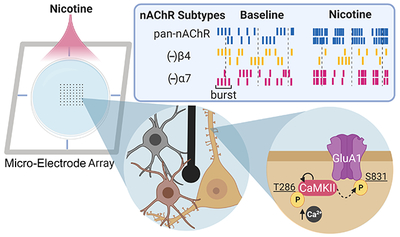
Nicotinic acetylcholine receptor (nAChR) activation within the hippocampus is capable of shifting the excitatory-inhibitory ratio. We found that pan-activation of nAChRs using nicotine potentiates cultured hippocampal networks through β4-containing nAChRs, and this effect is augmented by activation of α7 nAChRs. Furthermore, nicotine exposure promotes threonine 286 autophosphorylation of CaMKII and serine 831 phosphorylation of the AMPA receptor subunit GluA1. These findings highlight the impact of cholinergic signaling in generating network-wide potentiation in the form of enhanced spiking and bursting dynamics as well as elevated synchrony that coincide with molecular correlates of memory such as increased phosphorylation of CaMKII and GluA1.
Introduction
The neuromodulator acetylcholine acts through a variety of nAChRs to modulate many aspects of central nervous system (CNS) physiology including synaptic plasticity, neurogenesis, and neuroprotection (Mudo et al. 2007; Belluardo et al. 2000; Harrist et al. 2004), and orchestrates learning and memory-related circuits in the brain (Luchicchi et al. 2014). Neuronal nAChRs are abundant in the CNS, and in the hippocampus, the most highly expressed nAChR subtypes are those containing α7 and α4β2 (Alkondon and Albuquerque 2004; Dani 2015; Alkondon and Albuquerque 1993). Studies have shown that both α7 and α4β2 nAChRs localize on the soma of inhibitory interneurons (Zarei et al. 1999; Alkondon and Albuquerque 2001). Activation of α7 and α4β2 nAChRs on interneurons within the hippocampus can inhibit or disinhibit excitatory pyramidal neurons (Alkondon and Albuquerque 2001; Ji and Dani 2000) dependent on the locations of the receptors. For example, presynaptic α7 nAChRs directly enhance the release of glutamate or GABA when localized on glutamatergic or GABAergic terminals, respectively (Wonnacott et al. 2006; Wonnacott 1997; Schicker et al. 2008; Sher et al. 2004; MacDermott et al. 1999; Marchi and Grilli 2010; Radcliffe et al. 1999; Fabian-Fine et al. 2001). Interestingly, α3β4 nAChRs may also be implicated in modulating network dynamics due to their action on inhibitory transmission, despite being less predominant. These receptors can localize on perisomatic-targeting parvalbumin-containing GABAergic interneurons, and their activation can initiate a prolonged quantal GABA release from nerve terminals by activating axonal T-Type (Cav3) Ca2+ channels and Ca2+ release from stores (Tang et al. 2011). Furthermore, the activation of a non-α7 nAChR, likely α3β4, enhances inhibition by increasing excitatory input to hippocampal CA1 GABAergic interneurons (Alkondon and Albuquerque 2002).
As a consequence of their activation, nAChRs modulate both excitation and inhibition, thereby altering the excitatory-inhibitory ratio (Cobb et al. 1999; Griguoli and Cherubini 2012; Somogyi and Klausberger 2005). Precise regulation of the excitatory-inhibitory ratio is crucial for the occurrence of collective phenomena that arise from synchronous bursting within the neuronal circuitry. Several of these phenomena, such as hippocampal network oscillations (Freund and Buzsáki 1996; Miles et al. 1996; Cobb et al. 1995) and sharp-wave ripples (Buzsáki 1986), are believed to support hippocampus-dependent cognitive tasks (Feder and Ranck 1973; Otto et al. 1991; Singer 1993; Buzsaki and Draguhn 2004; Buzsaki and Watson 2012). Disruption of these crucial network dynamics has been observed in several disease models in which nAChRs have been implicated, such as Alzheimer’s disease (Scott et al. 2012; Goutagny et al. 2013; Hajos et al. 2005).
While the impact of nicotine on nAChRs has been well studied at the receptor level (Nelson et al. 2003; Dani 2015; Mansvelder and McGehee 2000; Alkondon et al. 1994; Lester et al. 2009; Clarke et al. 1985; Gray et al. 1996) as well as in vivo (Rezvani and Levin 2001; Fujii and Sumikawa 2001; Hulihan-Giblin et al. 1990; Mudo et al. 2007; Harrist et al. 2004; Barrass et al. 1969; Kellar and Wonnacott 1990; Miner and Collins 1988), much less attention has been directed towards the effects of nicotine and the activation of nAChRs on neuronal networks (Cobb et al. 1999; Cobb and Davies 2005; Palop et al. 2006; Ruivo and Mellor 2013). Investigations within this mesoscopic spatial scale are essential for the study of collective phenomena as it is not possible to extrapolate to emergent dynamics from single electrode studies. Given the important roles of nAChRs in maintaining proper brain function (Leiser et al. 2009; Belluardo et al. 2000; Levin 2013; Luchicchi et al. 2014) and the observations that aberrant nicotinic transmission results in various neurological disorders (Uhlhaas and Singer 2010; Levin 2013; Scott et al. 2012; Leiser et al. 2009; Schmitz et al. 2016), elucidating the effects of nicotine on the integrated response from neuronal networks may inform the development of more effective drugs for treatments.
Based on this rationale, we used a network of cultured hippocampal neurons in order to understand how nicotine modulates network spiking, bursting, and rhythmicity. This model will facilitate determination of the effects of nAChR activation without the confounding contribution of receptor desensitization resulting from cholinergic tone. Hippocampal cultures lack endogenous cholinergic tone due to the absence of the septum, the major source of cholinergic input (Feder and Ranck 1973; Mesulam et al. 1983; Freund and Buzsáki 1996), and there is little consensus regarding the existence of intrinsic cholinergic neurons within the intact hippocampus proper (Frotscher et al. 1986; Lauterborn et al. 1993; Blaker et al. 1988). Additionally, cholinergic neurons do not exhibit high rates of survival in culture without growth factor supplementation (Culmsee et al. 2002; Hartikka and Hefti 1988). Here, we analyzed such cultures using a microelectrode array (MEA) to investigate network dynamics, allowing us to perform simultaneous extracellular recordings of action potentials from multiple sites within the networks. MEAs enable both acute and chronic characterization of network activity from slices (Novak and Wheeler 1988; Boppart et al. 1992; Gonzalez-Sulser et al. 2012) as well as from cultured cell preparations (Potter and DeMarse 2001; Chen and Dzakpasu 2010). In addition, the use of pharmacological manipulations with the MEA permits the study of collective, rhythmic network activity (Wagenaar et al. 2006; Segev et al. 2001; Gandolfo et al. 2010; Niedringhaus et al. 2012). Although intact anatomical connections between discrete brain areas are essential to the generation of some types of in vivo network activity, primary cultured hippocampal networks facilitate the study of neuronal network dynamics including increased bursting activity that may, in turn, impact the likelihood of synchronous activity. Lastly, oscillatory activity has been shown to develop in cultured hippocampal networks despite the absence of inputs from other brain regions (Boehler et al. 2012).
In the present study, we tested whether nicotine exhibits a concentration-dependent impact on network activity, and if so, which nAChR subtypes and what molecular pathways may mediate this effect. We found that nicotine can modulate network dynamics in a concentration-dependent manner, increasing the firing of action potentials (spikes) and facilitating bursting activity. To determine the contribution of the various nAChR subtypes present in the hippocampus to the observed network activity, prior to nicotine stimulation we introduced a suite of pharmacological agents that block conductances of distinct nAChR subtypes. Interestingly, we found that β4-containing nAChRs were required for increases in network spiking, bursting, and synchrony, whereas α7 nAChRs play a more nuanced role in mediating the effects of nicotine. To address the role of synaptic NMDARs and group I mGluRs in the observed dynamics, we blocked either NMDARs or group I mGluRs, then stimulated the network with nicotine. Finally, we examined a potential molecular mechanism that may underlie the observed potentiation via a CaMKII-AMPAR signaling pathway. We report on the emergent neural network dynamics and discuss the implications of our findings.
Experimental Procedures
Animals
All experimental procedures were approved (IACUC protocol #: 15-026-100232) and performed in accordance with the Georgetown University Animal Care and Use Committee (GUACUC) regulations and recommendations. A total of 30 pregnant mothers were used for these studies in which each group included cultures from at least two animals. The following study was not pre-registered as it does not include any human subject data.
Cell culture
Timed-pregnant Sprague-Dawley rats (8-10 week old females, RRID: RGD_734476) were obtained from Charles River (Raleigh, NC). Pregnant rats were singly housed in individually ventilated cages for 2 days to acclimate to the animal facility, with ad libitum access to food and water. At embryonic day 18 (E18), pregnant rats were euthanized using a carbon dioxide chamber with a flow meter regulator to minimize asphyxiation distress. No anesthetics were used as these agents may interfere with the proper growth of neurons in culture (Mintz et al. 2012). Euthanasia was verified by lack of response to toe pinch and decapitation was used as secondary means of assuring death. No other procedures involved animal pain, suffering or distress. Immediately following euthanasia, embryos were removed by laparotomy and hippocampal tissue was harvested using a protocol modified from a previously published study (Pak et al. 2001). Briefly, neural tissue was digested with 0.1% trypsin and by mechanical trituration. Cells were plated onto microelectrode arrays (MEA, Multi Channel Systems MCS GmbH, Reutlingen, Germany), 12-well tissue culture plates (Fisher Scientific, Waltham, MA) or acid-washed borosilicate cover glass that were previously treated with poly-D-lysine and laminin (Sigma, St. Louis, MO) at an approximate density of 700 cells/mm2, 1000 cells/ mm2, or 150 cells/ mm2, respectively. Hippocampal cultures were continuously maintained in Neurobasal medium with B27 (Invitrogen, Carlsbad, CA) and stored in a 5% CO2 and 95% O2 humidified incubator at 37°C. Experiments were performed on cultures at 14 days in vitro (DIV).
Antibodies
Anti-CaMKII pT286 (1:500, cat. #V1111, Promega, RRID: AB_430841), CaMKIIα monoclonal 6G9 (1:5000, cat. #MA1-048, ThermoFisher, RRID: AB_325403), goat anti-rabbit (1:1000, Millipore Sigma, RRID: AB_257896) and anti-mouse HRP-conjugated IgG secondary antibodies (1:1000, Millipore Sigma, RRID: AB_258431) were used for immunoblotting. GluA1 pS831 monoclonal N453 (1:500, cat. #04-823, Millipore Sigma, RRID: AB_1977218), chicken MAP2 (1:1000, cat. #AB15452, Millipore Sigma, RRID: AB_805385), mouse GFAP monoclonal (GA-5) (1:1000, cat. # IF03L, Millipore Sigma, RRID: AB_2294571), donkey anti-rabbit Alexa Fluor 555 (1:150, cat. #A31572, ThermoFisher, RRID: AB_162543) and goat anti-chicken Alexa Fluor 647 (1:250, cat. #A21449, ThermoFisher, RRID: AB_1500594) were used for immunocytochemistry.
Western blot assay
Cells were lysed in 2-mercaptoethanol (BioRad) containing NuPAGE LDS sample buffer (Thermo Fisher Scientific). Equal amounts of each sample were separated by 10% SDS-PAGE gels and transferred to nitrocellulose membranes. Blots were then blocked with TBST containing 5% nonfat dried milk and incubated with primary antibodies overnight at 4°C. Following three washes in TBST, blots were incubated in horseradish peroxidase-conjugated secondary antibodies, then washed thoroughly before visualization using KwikQuant Ultra Digital-ECL Substrate Solution (Kindle Biosciences, LLC). Samples for Figure 6 were run contiguously on the same gel (no APV on one gel, and +APV in another gel). Phospho-specific antibody blots were stripped and reprobed for total CaMKII.
Figure 6.

Nicotine induces NMDAR-dependent CaMKII phosphorylation in primary cultured hippocampal neurons. (a,d) Neurons (DIV14) plated on tissue culture dishes were pre-incubated with vehicle (a) or APV (50 μM, 15 min) (d) and subsequently treated with nicotine (90 μM for 1, 5, or 15 min). Lysates were immunoblotted for CaMKII-α/β phosphothreonine 286 (pT286) and total CaMKIIα as a loading control. (b-c, e-f) Quantification of average intensity of CaMKIIα/β pT286. (n=3 independent wells from a single culture for each condition; *p < 0.05, **p < 0.01; ns = not significant; one-way ANOVA with Holm-Sidak post-hoc correction). Error bars represent SEM. Full uncropped blots are shown in Supplemental Figure S7.
Immunocytochemistry and quantification of immunofluorescence intensity
For immunocytochemistry, primary cultured hippocampal neurons (14 DIV) were fixed in 1% paraformaldehyde (PFA)/4% sucrose, followed by methanol (−20°C). Primary antibodies were incubated in GDB buffer (30 mM phosphate buffer, pH 7.4, containing 0.1% gelatin, 0.3% Triton X-100, 450 mM NaCl). Alexa 647- and Alexa 555-conjugated secondary antibodies (Invitrogen) were used for double immunolabeling. For intensity measurement, images were acquired using an Axiovert 200M microscope (Zeiss) for epifluorescence. The experimenter was blinded to treatments while acquiring and analyzing images. To identify puncta for control and treated groups, images were flattened and thresholded at a constant value (2x background), resulting in the selection of regions with intense staining, which were primarily soma and puncta. Regions of interest were selected automatically using the ROI tool in Metamorph software (Universal Imaging), leading to the selection of puncta and the soma; the soma was then excluded from quantification. This method allowed for unbiased identification of puncta in each cell. The intensity of signal for each GluA1 pS831-positive cluster and the total number of puncta were measured using Metamorph software.
MEA recordings
Spontaneous extracellular electrical activity was recorded using an MEA (MEA2100, Multi Channel Systems MCS GmbH, Reutlingen, Germany). The MEA is an 8x8 square array composed of 59 titanium nitride electrodes and one reference electrode, accompanied by four auxiliary analog channels. Each electrode is 30 μm in diameter, and the inter-electrode distance is 200 μm. Upon plating, cells adhere to the poly-D-lysine- and laminin-treated silicon nitride substrate of the MEA, and spontaneous electrical activity is detected after seven days. MEAs were covered with a hydrophobic membrane that is permeable to CO2 and O2 to reduce osmolarity and pH changes (Potter and DeMarse 2001). Recordings were performed on a heated stage at 37°C at DIV14. In order to allow for the detection of spikes, the time series of amplified electrical activity was sampled at a rate of 10 kHz. Data were digitized and stored for offline analysis.
Drugs
Reagents were obtained from the following sources: (−) Nicotine hydrogen tartrate salt [nAChR agonist] (N5260, Millipore Sigma, 2013), (+)-MK-801 Maleate [NMDAR antagonist] (0924, Ascent Scientific, 2009), and APV [NMDAR antagonist] (A5282, Millipore Sigma, 2012); MLA [α7 nAChR antagonist] (1029, Tocris Biosciences, 2015), MPEP [mGluR5 antagonist] (1212, Tocris Biosciences, 2015), and 3-MATIDA [mGluR1 antagonist] (2196, Tocris Biosciences, 2015), Gabazine [GABAAR antagonist] (1262, Tocris Biosciences, 2009). Sazetidine-A (SAZ-A) tartrate [α4β2 nAChR partial agonist] was synthesized by Drs. Milton L. Brown, Mikell A. Paige, and Brian E. McDowell (Georgetown University, Washington, DC) and kindly provided by Dr. Kenneth Kellar (Georgetown University, 2015) (Xiao et al. 2006). AT-1001 [α3β4 nAChR partial agonist] was a gift from Astraea Therapeutics (Mountain View, CA, USA, 2012) (Zaveri et al. 2010).
Drug treatment
Each drug was dissolved in ultrapure water to prepare a 10 mM stock solution. Conditioned media (500 μL) was removed from each MEA, mixed with an aliquot of the stock drug solution to the desired final concentration, then added back to the MEA. In the experiments using MLA, SAZ-A, AT-1001, APV, MK-801 or MPEP and 3-MATIDA, each MEA was first incubated in the respective drug(s) for 15 minutes, stimulated with nicotine, then recorded for network activity. Unless otherwise indicated, all MEA data represents the last 3 minutes of the 15-minute drug incubation and the last 3 minutes of the 15-minute recording after nicotine stimulation.
MEA data analysis
MEA traces were high-pass filtered at 200 Hz to remove low-frequency components. Recorded spikes from these traces were detected using Offline Sorter (Plexon Inc., Dallas TX) and thresholded at 4.5σ of the mean biological noise for each channel, as this study investigated responses from the network rather than from individual units. The z-score threshold of 4.5σ was established by extensively sampling high-pass filtered data at various σ values using a threshold algorithm from Offline Sorter and identifying optimal signal-to-noise ratio in which the ground channel noise (spiking within channel #15) was minimized. Since the signal from each electrode represents a collective response, we did not sort spikes by electrode. Network activity was analyzed with custom software written in MATLAB (MathWorks, Natick, MA). First, to investigate changes in overall network activity, we measured the total number of spikes over a 3-minute window for each electrode within the MEA. Next, we isolated bursts, which are a common temporal feature of cultured networks and can occur across the entire network. Each electrode had a resulting spike train, τST(t), expressed as:
where N is the total number of spikes, tn is the time of the nth spike and δt is a delta function that indicates a spike taking place at time t = tn. The inter-spike interval (ISI) between spike n and spike n-1 (n > 1) is:
For all experimental groups, a burst recorded from each electrode was defined as a cluster of activity containing no fewer than four spikes with a maximum ISI of 100 ms. This value was selected because it represents the temporal boundary between a distribution of ISIs thought to be within bursts and the intervals between bursts within our networks (Niedringhaus et al. 2012). The number of bursts was quantified in a 3-minute window between the 13-15th minute time points. The fraction of spikes within bursts was calculated as the total number of spikes within bursts divided by the total number of spikes within a 3-minute window between the 13-15th minute time points (see Fig. S1 for experimental design). We calculated the correlation coefficient as a measure of synchronized activity between pairs of electrodes, using a sample of 15 electrodes. We used a 1 kHz Butterworth low pass filter to eliminate noise and sporadic, low amplitude spikes but preserve bursts and large amplitude voltage transients. All MEA data analysis was performed blind and in automated fashion to minimize experimenter bias.
Statistics
Statistical tests were performed using GraphPad Prism 7 (GraphPad Software, San Diego, CA). Normality was determined using the Shapiro-Wilk test. Data are expressed as means ± SEM, and differences were considered significant at p<0.05. No sample size calculation was performed and no randomization was performed to allocate subjects for this study. No test for outliers was performed and no data points were excluded.
Results
Concentration-dependent effects of nicotine on network activity
We cultured embryonic hippocampal neurons on MEAs to study the effects of nicotine on neuronal network activity. By DIV7, processes can be visualized, and cell bodies are frequently proximal to electrodes (Fig. 1a). By DIV14, robust, spontaneous network-wide activity was evident across all electrodes (Fig. 1b). A rich mixture of spiking and bursting activity was apparent as seen from representative individual electrodes (Fig. 1c–d, Fig. S2), consistent with previous reports that networks at this developmental time point display well-established connectivity (Wagenaar et al. 2006) and vigorous spontaneous electrical activity comprised of spikes super-imposed on paroxysmal depolarizations (Köller et al. 1993).
Figure 1.

Spontaneous activity from rat hippocampal neural networks. (a) An MEA (left) and differential interference contrast (DIC) images of a DIV7 culture of hippocampal neurons plated on the MEA at low (center) and high (right) magnification. (b) Network electrical activity from all 60 electrodes of the MEA at DIV14. Each box corresponds to one second of activity (x-axis) with a voltage range of ± 100 μV (y-axis). (c) Representative 3-ms spike traces from a single electrode during baseline. (d) Three representative 1-second traces of filtered activity from an MEA. Within a single electrode, a wide range of different types of bursts is observed, including those with long, intermediate or short duration.
To determine how nicotine impacts basal network activity, we quantified changes in action potential firing after nicotine application. Five concentrations of nicotine, ranging from 0.1 μM to 90 μM, were applied to naïve cultured hippocampal networks. Concentrations from 50 to 90 μM nicotine significantly increased spiking as well as bursting activity (Fig. 2a–b) compared to pretreatment baseline. Most significantly, this range of nicotine concentration reorganized the pattern of action potential firing by increasing the number of spikes within each burst (Fig. 2c and Fig. S3). Bursts are thought to facilitate information transmission within hippocampal networks due to the increased probability of inducing postsynaptic neurons to fire action potentials (Lisman 1997; Izhikevich et al. 2003; Csicsvari et al. 1998; Miles and Wong 1987; Thomson 2000) as well as an increased likelihood of synaptic potentiation (Pike et al. 1999; Paulsen and Sejnowski 2000). Thus, we refer to our observed phenomena within these cultured networks as nicotine-mediated network potentiation (Fig. 2b–c). No significant change in bursting activity or spikes within bursts was observed in 0.1–10 μM nicotine and vehicle-treated networks (Fig. 2b–c).
Figure 2.

Nicotine potentiates network-wide spiking and bursting, and reorganizes network activity in a concentration-dependent manner. (a-c) MEAs treated with nicotine concentrations of 50 μM and greater show a significant increase in (a) spikes, (b) bursts and (c) spikes within bursts during a 3-minute recording. Data expressed as percent change normalized to baseline. (n=5 MEAs for each treatment from two independent cultures; *p<0.05, **p<0.01, ****p<0.0001; one-way ANOVA and Dunnett post hoc correction). (d-f) Representative spatial heat maps of correlation coefficients between active electrodes of the network (d) during baseline, (e) 1 minute, and (f) 15 minutes after applying 50 μM nicotine. (g) Quantification of correlation coefficients between active electrodes of the network after 1 and 15-minute application of 50 μM nicotine. (n=5 MEAs for each treatment; *p<0.05, ns = not significant; repeated-measures ANOVA with Tukey’s post hoc correction). Error bars represent SEM. (h-j) Representative spatial heat maps of correlation coefficients between active electrodes of the network (h) during baseline, (i) 1 minute, and (j) 15 minutes after applying 90 μM nicotine. (k) Quantification of correlation coefficients between active electrodes of the network after 1 and 15-minute application of 90 μM nicotine. (n=5 MEAs for each treatment from two independent cultures; **p<0.01, ns = not significant; repeated-measures ANOVA with Tukey’s post hoc correction). Error bars represent SEM.
Synchronous firing of converging afferents is necessary for proper signal transmission to postsynaptic neurons (Singer 1993). Given that concentrations of nicotine within the range of 50–90 μM enhanced network parameters that give rise to more efficient information transmission, specifically increased bursting activity as well as the number of spikes within bursts, we hypothesized that concentrations of nicotine within this range would also increase network synchrony. To test this idea, we used the correlation coefficient as a direct measure of synchrony. Pairwise correlations were calculated between the times of spikes of active electrodes within the network and shown as representative spatial heat maps of correlation coefficients (Fig. 2d–f, h–j). We observed a significant increase in correlation coefficients after treatment of 50 and 90 μM nicotine that was sustained for at least 15 minutes (quantified in Fig. 2g, k), demonstrating the emergence of persistent synchronous activity. There was no significant increase in correlation after vehicle (Fig. S4a–d) or 10 μM nicotine (Fig. S4e–h) application.
Contribution of specific nAChR subtypes to nicotine-mediated network potentiation and synchrony
Since nicotine acts at a variety of CNS nAChRs, we investigated which nAChR subtypes might be involved in facilitating nicotine-mediated network potentiation. The hippocampus has been shown to contain α7-, α4β2-, and α3β4-containing receptors, with α7 and α4β2 being the most prevalent subtypes (Alkondon and Albuquerque 2004; Alkondon and Albuquerque 1993; Alkondon et al. 1994; Zarei et al. 1999; Gasparini et al. 1999; Moroni et al. 2002). We first treated hippocampal networks with 30 nM of MLA, a highly selective and potent competitive antagonist of α7 nAChRs. After a 15-minute incubation, MLA by itself did not significantly change spiking, bursting, or spikes within bursts. However, in contrast to the robust network potentiation produced by 90 μM nicotine on naïve networks, MLA attenuated the nicotine-induced increase in spikes (Fig. 3a), bursts (Fig. 3b), and spikes within bursts (Fig. 3c). These data suggest that α7 nAChR activation contributes to nicotine-mediated network potentiation, particularly to the number of spikes within bursts.
Figure 3.

Contributions of nAChR subtypes to nicotine-mediated network potentiation. Subtypes antagonized are indicated at top of graphs. (a-c) Effects of α7 nAChR antagonist MLA (before and during stimulation with nicotine) on (a) spikes, (b) bursts, and (c) spikes within bursts during a 3-minute recording. (d-f) Effects of α4β2 nAChR partial agonist SAZ-A (before and during stimulation with nicotine) on (d) spikes, (e) bursts, and (f) spikes within bursts during a 3-minute recording. (g-i) Effects of α3β4 nAChR partial agonist AT-1001 (before and during stimulation with nicotine) on (g) spikes, (h) bursts, and (i) spikes within bursts during a 3-minute recording. Statistical significance was calculated by using one-way ANOVA followed by a Holm-Bonferroni post-hoc correction, which allows for pairwise comparisons and preserves pairing where pairing is appropriate (antagonist(s) alone vs. antagonist(s)+nicotine) (n=5 MEAs for each treatment from two independent cultures; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001; ns = not significant). Data expressed as percent change normalized to baseline. Error bars represent SEM.
There is a dearth of antagonists specific to the α4β2 and α3β4 nAChR receptor subtypes. High concentrations of the α4β2 antagonist DhβE that are needed to block their activation with 90 μM nicotine can also block α3β4 (Harvey et al. 1996). Consequently, to distinguish the contributions of α4β2 and α3β4 nAChRs to network activity, we used sazatidine-A (SAZ-A), a partial agonist at rat α4β2 nAChRs (Tuan et al. 2015; DeDominicis et al. 2017) which potently desensitizes rat α4β2 but not rat α3β4 nAChRs (Xiao et al. 2006). We also used AT-1001, a partial agonist at rat α3β4 nAChRs that is known to desensitize rat α3β4 and human α4β2 nAChRs (Tuan et al. 2015).
The concentrations of SAZ-A and AT-1001 were chosen based on the maximal desensitization each drug would cause at its target receptor (Tuan et al. 2015; Xiao et al. 2006), which coincides with the maximal activation of each drug. As they are partial agonists, the long application time will desensitize the target nAChR receptor, essentially giving rise to a “time-averaged antagonism” (Hulihan-Giblin et al. 1990). Thus, to assess the role of α4β2 nAChRs, we applied 1 μM of SAZ-A onto hippocampal networks for 15 minutes to activate α4β2, then stimulated the network with 90 μM of nicotine in the continued presence of SAZ-A during which the α4β2 receptors were presumably desensitized (Fig. 3d–f). The initial activation of α4β2 nAChRs with SAZ-A was not sufficient to increase spiking or spikes within bursts, nor was the desensitization of α4β2 nAChRs effective in blocking the effect of nicotine on spiking activity or spikes within bursts (Fig. 3d, f). Interestingly, the activation and subsequent desensitization of α4β2 nAChRs did cause an increase in bursting that occluded nicotine’s effects on bursts (Fig. 3e), suggesting that the activation of α4β2 nAChRs was sufficient to increase bursting within the network.
Blocking α7 nAChRs with MLA did not entirely block the network-potentiation parameters mediated by nicotine. Additionally, the desensitization of α4β2 nAChRs due to pre-incubation in SAZ-A did not fully attenuate nicotine’s effects; activation of α4β2 nAChRs was only able to occlude the impact of nicotine on bursting activity. These data suggest that other nAChR subtypes may play a role in nicotine-mediated network potentiation. To address this possibility, we used AT-1001 to investigate whether α3β4 nAChRs - reportedly the least prevalent subtype within the hippocampus (Alkondon and Albuquerque 1993; Alkondon et al. 1994) - might be involved. During AT-1001 treatment by itself, approximately one minute of continued robust network activity was followed by a long-lasting decrease in activity. The time-averaged effect of the activation of β4-containing receptors with 20 μM AT-1001 resulted in a sharp decrease in spiking (Fig. 3g), bursting (Fig. 3h), and spikes within bursts (Fig. 3i). Desensitization of β4-containing receptors by AT-1001 also blocked the impact of 90 μM nicotine on spiking, bursting, and the fraction of spikes within bursts, suggesting that β4-containing nAChRs are necessary for the effects of nicotine on network potentiation.
We next sought to assess the role of nAChR subtypes in nicotine-mediated synchrony. To delineate the role of α7nAChRs, we calculated correlation coefficients in MLA-treated networks before and after stimulation with 90 μM of nicotine (Fig. 4a). We found that 30 nM of MLA by itself did not significantly affect network synchrony, nor did it attenuate nicotine-mediated network synchronization, suggesting that α7 nAChRs may not be necessary for nicotine to enhance network synchrony.
Figure 4.

Effects of nAChR ligands on nicotine-mediated network synchrony. (a-c) Quantification of correlation coefficients measured at baseline, during the 1st and 15th minute, and during co-stimulation with 90 μM nicotine (15th minute) upon application of (a) 30 nM MLA, (b) 1 μM SAZ-A, and (c) 20 μM AT-1001. (n=5 MEAs for each treatment from two independent cultures; *p<0.05, **p<0.01; ns = not significant; repeated-measures ANOVA with a Tukey’s post hoc correction). Error bars represent SEM.
Since activation of α4β2 nAChRs caused an increase in bursting, we asked whether increased bursting without a change in the fraction of spikes within bursts would be sufficient to promote network synchrony. We, therefore, calculated the correlation coefficients of SAZ-A treated networks and found that SAZ-A did not cause a significant change in network synchrony (Fig. 4b). Moreover, after the activation and subsequent desensitization of α4β2 nAChRs with prolonged SAZ-A, 90 μM nicotine was still capable of increasing network synchrony, albeit more modestly (Fig. 4b). These results suggest that α4β2 nAChRs are also not essential for the nicotine-mediated increase in network synchrony and that increased bursting alone is not sufficient to enhance network synchrony.
With regard to α3β4 nAChRs, correlation coefficients for the AT-1001 treated networks showed that after one minute of incubation, synchrony was maintained at basal levels. However, during the fifteenth minute of AT-1001 application, there was a marked de-synchronization within the network, and subsequent stimulation with 90 μM nicotine did not rescue the effect (Fig. 4c). These data suggest that β4-containing nAChRs are necessary for nicotine-mediated network synchrony. Activation of α3β4 nAChRs within the hippocampus has been shown to support inhibitory transmission (Tang et al. 2011). We hypothesized that the observed desynchronization might be a result of enhanced GABAergic transmission. To test this idea, we applied 10 μM of gabazine (GBZN), a GABAA receptor antagonist, and observed the return of synchrony within the networks (in the continued presence of both AT-1001 and nicotine) (Fig. 4c). This observation suggests that the activation of β4-containing nAChRs may enhance GABAergic transmission.
Due to the presence of nicotine throughout the recording, the contribution of steady-state stochastic activation of nAChRs (Papke 2014; Campling et al. 2013) to the maintenance of nicotine-mediated network potentiation was investigated. We tested whether an increase in spiking that is mediated by nicotine can be attenuated or blocked by specific nAChR antagonists. Briefly, after recording baseline activity for 15 minutes, we applied nicotine to increase network activity and recorded network spiking for 12 minutes. We followed with either MLA, DHβE (which preferentially blocks α4β2 nAChRs at the concentration administered), or vehicle and recorded spiking activity for an additional 6 minutes. If steady-state activation of α7 or α4β2 nAChRs contributes to the increase in network spiking during stimulation with nicotine, then we would expect a decrease in spiking during the application of MLA or DHβE. We found that the application of MLA (Fig. S5) or DHβE (Fig. S6) did not cause a significant change in spiking during the first or the last 3 minutes of application. These results suggest that neither α7 nor α4β2 nAChRs are stochastically activated.
A component of nicotine-mediated network potentiation is independent of NMDAR activation
To assess whether nicotine-mediated network potentiation is dependent on NMDARs, we blocked synaptic NMDARs with 10 μM MK-801 and challenged the network with 90 μM nicotine. The blockade of synaptic NMDARs by itself resulted in a network depotentiation characterized by reduced spiking, bursting, and the fraction of spikes within bursts (Fig. 5a–c). Interestingly, blocking synaptic NMDARs failed to prevent the nicotine-induced increase in these parameters (Fig. 5a–c). However, the impact of nicotine on spiking (Fig. 5a) and bursting (Fig. 5b) was partially diminished when NMDARs were blocked, suggesting the existence of an NMDAR-dependent component to the nicotine-mediated network potentiation.
Figure 5.

Contribution of synaptic NMDARs and group I mGluRs to nicotine-mediated network potentiation. (a-c) Effects of NMDAR channel blocker MK-801 (before and during stimulation with nicotine) on (a) spikes, (b) bursts, and (c) spikes within bursts during a 3-minute recording. (d-f) Effects of group I mGluR antagonists MPEP (for mGluR1) and 3-MATIDA (for mGluR5) (before and during stimulation with nicotine) on (d) spikes, (e) bursts and (f) spikes within bursts during a 3-minute recording. Statistical significance was calculated by one-way ANOVA followed by a Holm-Bonferroni post-hoc correction which allows for pairwise comparisons and preserves pairing where pairing is appropriate (antagonist(s) alone vs. antagonist(s)+nicotine) (n=4 MEAs for each treatment from two independent cultures; *p<0.05, **p<0.01, ***p<0.001; ns = not significant). Data expressed as percent change normalized to baseline. Error bars represent SEM.
Nicotine has also been reported to enhance long-term potentiation (LTP) in a group I mGluR-dependent manner (Welsby et al. 2006). We assessed the role of group I mGluRs in nicotine-mediated network potentiation with the co-application of specific antagonists, MPEP and 3-MATIDA, at concentrations that would result in the maximal blockade of their respective receptors, mGluR1 and mGluR5 (Gasparini et al. 1999; Moroni et al. 2002). As with blocking NMDARs, blocking group I mGluRs resulted in a deceased excitatory-inhibitory ratio as exhibited by decreased spiking, bursting, and the fraction of spikes within bursts (Fig. 5d–f). In contrast to NMDARs, however, blocking group I mGluRs largely failed to prevent nicotine-mediated network potentiation, suggesting that group I mGluRs are not necessary for the effects of nicotine on the network, but did modestly reduce the fraction of spikes within bursts (Fig. 5f).
Nicotine induces the phosphorylation of proteins involved in molecular memory and potentiation of synaptic plasticity
Our observed nicotine-induced network activity – lasting at least 15 minutes - is significantly longer than the time needed for nAChRs to become desensitized, which is on the order of tens of milliseconds (Quick and Lester 2002). The initial effects are likely due to the influx of cations contributing to the depolarization of neurons resulting from the activation of nAChRs (Dani and Bertrand 2007). As the receptors desensitize rapidly after activation, their contribution to the sustained activity becomes less clear. Thus, we sought to investigate the molecular determinants of the persistent nicotine-mediated network potentiation that outlasts nAChR activation. Nicotine has been reported to support LTP within the hippocampus in an NMDAR-dependent manner (Mann and Greenfield 2003) and to induce AMPAR phosphorylation via CaMKII within layer I of the cortex (Tang et al. 2015). Interestingly, we found that the application of nicotine resulted in a time-dependent increase in the activation of both CaMKII-α and -β isoforms via threonine-286 (T286) autophosphorylation (Fig. 6a–c). Given that the phosphorylation of CaMKII is known to be NMDAR-dependent (Lisman et al. 2002), we asked whether this nicotine-mediated phosphorylation is also NMDAR-dependent. MK-801 is an open channel blocker that requires sufficient baseline activity in order to block NMDARs. Since it is difficult to assess whether sufficient baseline activity is present in cultures not plated on MEAs, we opted to use the competitive antagonist APV because it does not require the channel to be opened and does not rely on baseline receptor activity for inhibition. Indeed, we found that the nicotine-mediated phosphorylation of CaMKII-α and -β T286 was fully abrogated by the pre-incubation of cultures with the NMDAR antagonist APV (Fig. 6d–f).
Finally, we sought to determine whether the activation of CaMKII results in a functionally relevant change in excitatory glutamate receptors involved in synaptic potentiation. We found that activation of nAChRs with nicotine elevated levels of AMPAR GluA1 phosphoserine-831 (pS831), a CaMKII-targeted residue necessary for LTP (Diering et al. 2016) (Fig. 7a–f). A significant increase was observed in both the relative intensity and the number of GluA1 pS831 puncta (Fig. 7g,h). These data suggest that nicotine-mediated network potentiation shares molecular components with NMDAR-dependent LTP.
Figure 7.

Nicotine induces GluA1 phosphorylation in primary cultured hippocampal neurons. (a-f) Cultures (DIV14) grown on glass coverslips were treated with vehicle (a-c) or nicotine (90 μM, 15 min) (d-f) and immunolabeled for GluA1 phosphoserine 831 (pS831) (b,e) and MAP2, a marker of neuronal dendrites (a, d), with merged images shown (c,f). Scale bar, 20 μm. Insets at bottom show higher magnification of dendrites indicated by white boxes above. Scale bar, 10 μm. (g) Quantification of average intensity of GluA1 pS831 puncta in spines. (h) Quantification of number of GluA1 pS831 puncta. (n=30 neurons for control and 33 for nicotine-treated; *p<0.05; unpaired two-tailed Student’s t-test). Error bars represent SEM.
Discussion
Within the hippocampus, nAChRs are located at both synaptic and non-synaptic sites, and they are capable of influencing synaptic plasticity due to their ability to conduct calcium (Lena et al. 1993). Due to the fact that kinetics of nAChR desensitization occur on a millisecond timescale, the precise time point at which activation of nAChRs takes place or desensitization begins was not analyzed. We focused here on the long-lasting downstream plasticity outcomes induced by the initial activation, as these effects better reflect a sustained response of network potentiation. To this end, we showed that nicotine can potentiate in vitro hippocampal network dynamics in a concentration-dependent manner. Increasing concentrations of nicotine not only enhanced action potential firing but importantly, reorganized the temporal pattern of spiking towards more bursts that contain a larger fraction of spikes. We propose that this reorganization of spikes is integral to the manifestation of potentiation, based on the observation that a burst of action potentials of 4 or more spikes is more likely to give rise to LTP than one, two or a burst of 3 spikes (Lisman 1997). Furthermore, spikes that do not participate in a burst, i.e., errant spikes, tend to “veto” successive spikes and bursts from occurring (Harris et al. 2001). Therefore, when spiking activity reorganizes a larger fraction of spikes into bursting epochs, this “veto effect” of errant spikes is decreased and the “burstiness” of the network increases, facilitating information transmission (Lisman 1997; Buzsaki et al. 2002). Although spiking, bursting, and spikes per burst can be interdependent, this is not necessarily so. For example, one could have an increase in spikes that are not within bursts, and this would have much different computational effect than an increase in spikes that are within bursts, e.g., pacemaker cell activity. Collectively, this increase in bursts, as well as spikes within bursts, promotes network dynamics that enhance encoding and transmission of information and can, therefore, be considered a form of potentiation.
The fact that these emergent phenomena described above are accompanied by the phosphorylation of two proteins (CaMKII and GluA1) at sites that are necessary for the manifestation of LTP further supports the notion of nicotine-dependent potentiation. The observed increase in active CaMKII is likely due to membrane depolarization resulting from the gating of nAChRs, leading to sustained network potentiation via recruitment of NMDARs and subsequent activation of Ca2+-dependent synaptic plasticity-related proteins, such as CaMKII. The enhancement in active CaMKII during nicotine application is accompanied by increased intensity and number of puncta containing phosphoS831 GluA1. This observation is consistent with findings by others that show CaMKIIα phosphorylates the GluA1 subunit of AMPARs at S831, which increases GluA1 conductance and is a hallmark of NMDAR-dependent LTP in the hippocampus (Benke et al. 1998; Lisman et al. 2002). The phosphorylation of AMPARs during nicotine stimulation and at a time point during which nicotine promotes the activation of CaMKII further strengthens the idea that nicotine is inducing excitatory potentiation within the network. Moreover, CaMKII β regulates dendritic morphology and synapse formation (Fink et al. 2003), and such structural remodeling could further contribute to persistent changes in activity. Taken together, these results suggest that NMDAR-dependent alterations in glutamatergic synaptic strength contribute to nicotine-mediated network potentiation.
We examined the role of NMDARs in nicotine-mediated network potentiation with a well-established procedure that uses MK-801 to test the requirement for NMDARs in a particular pathway (Hardingham et al. 2001; Hardingham and Bading 2002; Ivanov et al. 2006; Liu et al. 2007; Stanika et al. 2009; Bordji et al. 2010; Stark and Bazan 2011; Zhang et al. 2011; Arnold et al. 2005). This treatment impacts baseline network activity by reducing bursts and spikes within bursts due to the requirement of calcium influx through synaptic NMDARs for the initiation of recurrent synchronous bursting (Arnold et al. 2005). With regard to network potentiation in our experiments, two components were diminished due to NMDAR inhibition by MK-801: the number of spikes and bursts. The impact of NMDAR blockade on the spikes within bursts is less clear (Fig. 5c), as these results did not reach significance, and therefore we cannot rule out that blocking NMDARs may have a minor effect on the nicotine-mediated enhancement of spikes within bursts. Surprisingly, when NMDARs were blocked, the activation of nAChRs within the network was still capable of inducing nicotine-mediated network potentiation. The observation that nicotine can act, at least in part, independently of NMDARs to increase network activity might have implications for disease states with known NMDAR hypofunction, such as schizophrenia.
Given that there was an NMDAR-independent component to our findings, we sought to investigate whether group I mGluRs - which are known to also play a role in hippocampal LTP (Lu et al. 1997; Balschun et al. 1999) - contribute to nicotine-mediated network potentiation. As with blocking NMDARs, inhibition of group I mGluRs was not able to abrogate nicotine-mediated network potentiation completely but resulted in a diminished fraction of spikes within bursts. Although the pharmacological and molecular findings support a role for NMDAR and mGluR signaling, it is unknown why blockade of these receptors impaired nicotine-mediated potentiation. As others have reported (Suresh et al. 2016; Lanneau et al. 2002; Arnold et al. 2005), ionotropic and metabotropic glutamate receptor tone seems to be necessary for baseline synaptic transmission and spiking activity, and thus we cannot exclude the possibility of an indirect effect on basic membrane properties of cultured neurons with NMDAR/mGluR antagonists. Interestingly, however, NMDARs and group I mGluRs appeared to contribute to different and complementary components of nicotine-mediated network dynamics. Thus, the activation of both types of glutamate receptors during stimulation with nicotine support nicotine-mediated network potentiation, which may reflect signal integration and plasticity (Sharma and Vijayaraghavan 2003; Zarei et al. 1999; Dannenberg et al. 2017). Additionally, studies suggest a direct coupling of α7 nAChRs to G protein signaling cascades that enable a downstream calcium response to persist past the expected time course of channel activation, which may maintain the long-lasting effects of nicotine. This G protein coupling process depends on the binding of Gαq at the G protein-binding cluster located in the M3-M4 loop of the α7 receptor (King et al. 2015). Although not yet established, the same study proposes the existence of a G protein-binding cluster in other nAChR subunits based on sequence homology of the M3-M4 loops (King et al. 2015), which could also contribute to a G protein-mediated maintenance of nicotine’s effects.
To delineate the nicotinic receptor subtypes that are involved in the effect of nicotine on network potentiation, we used various pharmacological tools. Surprisingly, we observed that the time-averaged effect of activation of α3β4 nAChR is network de-potentiation that is not recoverable via stimulation with nicotine but is recoverable via blocking GABARs. This result is consistent with evidence that shows that these receptors are found on the axonal terminals of GABAergic interneurons and that the release of GABA via this mechanism is slow but long-lasting (Tang et al. 2011). Moreover, AT-1001, which selectively activates β4-containing receptors, was recently shown to have anxiolytic properties (Cippitelli et al. 2015; Yuan et al. 2017), further suggesting a role for GABA in mediating effects of AT-1001.
We also showed that the pretreatment of networks with the α7 nAChR antagonist MLA attenuated the effects of nicotine on network potentiation. This finding is consistent with a predominance of α7 nAChR expression within the hippocampus, as well as a previously described role of α7 nAChRs in supporting increased hippocampal action potential firing (Sharma and Vijayaraghavan 2003). Additionally, we observed that the activation of α4β2 nAChRs was sufficient to enhance bursting. This effect is meaningful in view of observations that postsynaptic bursts are crucial for proper information transmission within hippocampal networks (Lisman 1997; Izhikevich et al. 2003), the occurrence Hebbian synaptic plasticity in vitro (Pike et al. 1999), and for homeostatic maintenance of network excitability in vivo (Buzsaki et al. 2002). Given the role of bursts in enhancing information transmission (Lisman 1997; Izhikevich et al. 2003), this action of α4β2 nAChRs could potentially play a significant role in the ability of nicotine to act as a cognitive enhancer.
Synchronization of neural activity is necessary for various normal and pathological functions that rely on oscillatory activity (Buzsaki and Watson 2012; Buzsaki and Draguhn 2004; Uhlhaas and Singer 2010; Singer 1993; Jefferys et al. 1996; Traub and Jefferys 1994). Studies have shown that cholinergic innervation plays a role in both physiological oscillatory states such as the hippocampal theta rhythm, which is necessary for learning and memory (Kramis et al. 1975; Bland and Colom 1993; Williams and Kauer 1997; Buzsáki 2002), as well as in hypersynchrony (Williams and Kauer 1997; Bui et al. 2015; Traub and Jefferys 1994). We hypothesized that the activation of nAChRs that leads to increased bursting detected at the single electrode level from small subpopulations of neurons could increase the likelihood of synchronous bursting across the network. To assess this possibility, we analyzed correlation coefficients as a direct measure of synchrony and found that increased bursting alone (e.g. via the activation of α4β2 nAChRs) was not sufficient to promote network synchrony. The degree of synchrony varied in the baseline activity as the correlation coefficients ranged from 0.2 to 0.4. While we do not know the precise reason for this variability, there are many possibilities, including cell density, connectivity, and culture viability. Despite this variation, we found that high concentrations of nicotine capable of generating network potentiation (50 and 90 μM) were able to significantly increase synchronized activity regardless of the initial baseline correlation. In agreement with the observation that activation of nAChRs within the hippocampus augments the power of pre-existing oscillatory states (Fig. S8) (Williams and Kauer 1997; Cobb and Davies 2005; Cobb et al. 1999; Griguoli and Cherubini 2012; Griguoli et al. 2009), we found that networks entering a desynchronized state after β4-containing nAChR activation were incapable of being subsequently synchronized by nicotine.
We highlight the fact that our studies regarding nicotine-induced synchrony were performed in a network of cultured hippocampal neurons; this is a fundamental first step in elucidating network dynamics but obvious anatomical differences between cultured networks and slice preparations preclude a direct comparison of results. For example, Wang et al. (2015) report that concentrations of nicotine in the range of 0.1 to 10 μM increases gamma power, whereas 100 μM nicotine decreases gamma power. Their studies involved the use of a slice preparation consisting only of the CA3 region of the hippocampus and importantly, they investigated the effects of nicotine on induced rather than spontaneous firing activity. Indeed, an advantage to using a cultured network system is the presence of robust, spontaneous activity. Furthermore, others have shown that nicotine attenuates carbachol-induced gamma oscillations in the prefrontal cortex (Mansvelder et al. 2006). Examples from these rich and diverse studies serve to underscore that the actions of nicotine on synchrony are complex; they depend upon the type of activity being modulated as well as the tissue preparation utilized (Mansvelder et al. 2006).
While these investigations focused on the impact of nicotine on neuronal networks, given the emerging role of astrocytes in synaptic regulation and information processing (Santello et al. 2019), we cannot rule out possible effects of nicotine on astroglial cells, which account for approximately 40% of the cells within our cultures (Fig. S9). Furthermore, hippocampal excitatory neurons in culture exhibit a heterogeneous composition containing pyramidal and granule cells from the different regions of the hippocampus (Lee et al. 2013). Our studies aimed to characterize the collective dynamical impacts and not those due to specific neuronal cell types. Since spike-sorting has been shown to not discriminate between individual excitatory and inhibitory neurons in vitro (Weir et al. 2015), future studies will be required to parse out which of the different cell types are most affected by nicotine.
In summary, we showed that nicotine significantly potentiates basal network activity in a concentration-dependent manner. This action of nicotine involved multiple nAChR subtypes, including α7, α4β2, and α3β4. While performed in a reduced preparation, these studies contribute to a better understanding of the role of nAChRs as modulators of information transmission within hippocampal networks and are a stepping stone to exploring how these mechanisms might regulate network properties in the intact brain.
--Human subjects --
Involves human subjects:
If yes: Informed consent & ethics approval achieved:
=> if yes, please ensure that the info “Informed consent was achieved for all subjects, and the experiments were approved by the local ethics committee.” is included in the Methods.
ARRIVE guidelines have been followed:
Yes
=> if it is a Review or Editorial, skip complete sentence => if No, include a statement: “ARRIVE guidelines were not followed for the following reason:
”
(edit phrasing to form a complete sentence as necessary).
=> if Yes, insert “All experiments were conducted in compliance with the ARRIVE guidelines.” unless it is a Review or Editorial
Supplementary Material
Acknowledgments
This research was supported by the National Science Foundation (PHY-1205919 and IOS-1755033), National Institutes of Health (RF1 AG056603-01) and the Georgetown-MedStar CERSI Scholars program. We thank Dr. Stefano Vicini for his generosity with the MEA2100, Dr. Kenneth Kellar and Astraea Therapeutics LLC for the donation of AT-1001 and Dr. Barbara Wroblewska for her assistance in mGluR pharmacology. Finally, we thank Drs. Kenneth Kellar, Robert Yasuda, Stefano Vicini, Gerard Ahern, and the late Dr. Barry Wolfe for their helpful discussions.
Abbreviations used
- nAChRs
nicotinic acetylcholine receptors
- NMDARs
N-methyl-D-aspartate receptors
- mGluRs
metabotropic glutamate receptors
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- MEA
microelectrode array
- CaMKII
Ca2+/calmodulin-dependent kinase II
- GABA
γ-aminobutyric acid
- RRID
research resource identifier
- LTP
long-term potentiation
- CNS
central nervous system
Footnotes
Open Science Badges
This article has received a badge for *Open Materials* because it provided all relevant information to reproduce the study in the manuscript. More information about the Open Practices badges can be found at https://cos.io/our-services/open-science-badges/
Conflict of Interest
The authors declare no conflict of interest.
References
- Alkondon M, Albuquerque EX (1993) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J. Pharmacol. Exp. Ther 265, 1455–1473. [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX (2001) Nicotinic acetylcholine receptor alpha7 and alpha4beta2 subtypes differentially control GABAergic input to CA1 neurons in rat hippocampus. J. Neurophysiol 86, 3043–3055. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX (2002) A non-alpha7 nicotinic acetylcholine receptor modulates excitatory input to hippocampal CA1 interneurons. J. Neurophysiol 87, 1651–1654. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX (2004) The nicotinic acetylcholine receptor subtypes and their function in the hippocampus and cerebral cortex. Prog. Brain Res 145, 109–120. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Reinhardt S, Lobron C, Hermsen B, Maelicke A, Albuquerque EX (1994) Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. II. The rundown and inward rectification of agonist-elicited whole-cell currents and identification of receptor subunits by in situ hybridization. J. Pharmacol. Exp. Ther 271, 494–506. [PubMed] [Google Scholar]
- Arnold FJL, Hoffmann F, Bengtson CP, Wittmann M, Vanhoutte P, Bading H (2005) Microelectrode array recordings of cultured hippocampal networks reveal a simple model for transcription and protein synthesis-dependent plasticity. J. Physiol 564, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balschun D, Manahan-Vaughan D, Wagner T, Behnisch T, Reymann KG, Wetzel W (1999) A specific role for group I mGluRs in hippocampal LTP and hippocampus-dependent spatial learning. Learn. Mem 6, 138–152. [PMC free article] [PubMed] [Google Scholar]
- Barrass BC, Blackburn JW, Brimblecombe RW, Rich P (1969) Modification of nicotine toxicity by pretreatment with different drugs. Biochem. Pharmacol 18, 2145–2152. [DOI] [PubMed] [Google Scholar]
- Belluardo N, Mudò G, Blum M, Fuxe K (2000) Central nicotinic receptors, neurotrophic factors and neuroprotection. Behav. Brain Res 113, 21–34. [DOI] [PubMed] [Google Scholar]
- Benke TA, Lüthi A, Isaac JTR, Collingridge GL (1998) Modulation of AMPA receptor unitary conductance by synaptic activity. Nature 393, 793–797. [DOI] [PubMed] [Google Scholar]
- Blaker SN, Armstrong DM, Gage FH (1988) Cholinergic neurons within the rat hippocampus: Response to fimbria-fornix transection. J. Comp. Neurol 272, 127–138. [DOI] [PubMed] [Google Scholar]
- Bland BH, Colom LV (1993) Extrinsic and intrinsic properties underlying oscillation and synchrony in limbic cortex. Prog. Neurobiol 41, 157–208. [DOI] [PubMed] [Google Scholar]
- Boehler MD, Leondopulos SS, Wheeler BC, Brewer GJ (2012) Hippocampal networks on reliable patterned substrates. J. Neurosci. Methods 203, 344–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boppart SA, Wheeler BC, Wallace CS (1992) A flexible perforated microelectrode array for extended neural recordings. IEEE Trans. Biomed. Eng 39, 37–42. [DOI] [PubMed] [Google Scholar]
- Bordji K, Becerril-Ortega J, Nicole O, Buisson A (2010) Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J. Neurosci 30, 15927–15942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui A, Kim HK, Maroso M, Soltesz I (2015) Microcircuits in Epilepsy: Heterogeneity and Hub Cells in Network Synchronization. Cold Spring Harb. Perspect. Med 5, 10.1101/cshperspect.a022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G (1986) Hippocampal sharp waves: their origin and significance. Brain Res 398, 242–252. [DOI] [PubMed] [Google Scholar]
- Buzsáki G (2002) Theta oscillations in the hippocampus. Neuron 33, 325–340. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Csicsvari J, Dragoi G, Harris K, Henze D, Hirase H (2002) Homeostatic maintenance of neuronal excitability by burst discharges in vivo. Cereb. cortex (New York, N.Y. 1991) 12, 893–899. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Draguhn A (2004) Neuronal oscillations in cortical networks. Science 304, 1926–1929. [DOI] [PubMed] [Google Scholar]
- Buzsaki G, Watson BO (2012) Brain rhythms and neural syntax: implications for efficient coding of cognitive content and neuropsychiatric disease. Dialogues Clin. Neurosci 14, 345–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campling BG, Kuryatov A, Lindstrom J (2013) Acute activation, desensitization and smoldering activation of human acetylcholine receptors. PLoS One 8, e79653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Dzakpasu R (2010) Observed network dynamics from altering the balance between excitatory and inhibitory neurons in cultured networks. Phys. Rev. E 82, 31907. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Brunori G, Gaiolini KA, Zaveri NT, Toll L (2015) Pharmacological stress is required for the anti-alcohol effect of the α3β4* nAChR partial agonist AT-1001. Neuropharmacology 93, 229–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A (1985) Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-alpha-bungarotoxin. J. Neurosci 5, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Buhl EH, Halasy K, Paulsen O, Somogyi P (1995) Synchronization of neuronal activity in hippocampus by individual GABAergic interneurons. Nature 378, 75–78. [DOI] [PubMed] [Google Scholar]
- Cobb SR, Bulters DO, Suchak S, Riedel G, Morris RGM, Davies CH (1999) Activation of nicotinic acetylcholine receptors patterns network activity in the rodent hippocampus. J. Physiol 518, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Davies CH (2005) Cholinergic modulation of hippocampal cells and circuits. J. Physiol 562, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csicsvari J, Hirase H, Czurko A, Buzsáki G (1998) Reliability and state dependence of pyramidal cell–interneuron synapses in the hippocampus: an ensemble approach in the behaving rat. Neuron 21, 179–189. [DOI] [PubMed] [Google Scholar]
- Culmsee C, Gerling N, Lehmann M, Nikolova-Karakashian M, Prehn JHM, Mattson MP, Krieglstein J (2002) Nerve growth factor survival signaling in cultured hippocampal neurons is mediated through TrkA and requires the common neurotrophin receptor P75. Neuroscience 115, 1089–1108. [DOI] [PubMed] [Google Scholar]
- Dani JA (2015) Chapter One-Neuronal Nicotinic Acetylcholine Receptor Structure and Function and Response to Nicotine. Int. Rev. Neurobiol 124, 3–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani JA, Bertrand D (2007) Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu.Rev.Pharmacol.Toxicol 47, 699–729. [DOI] [PubMed] [Google Scholar]
- Dannenberg H, Young K, Hasselmo M (2017) Modulation of hippocampal circuits by muscarinic and nicotinic receptors. Front. Neural Circuits 11, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeDominicis KE, Sahibzada N, Olson TT, Xiao Y, Wolfe BB, Kellar KJ, Yasuda RP (2017) The (alpha4)3(beta2)2 Stoichiometry of the Nicotinic Acetylcholine Receptor Predominates in the Rat Motor Cortex. Mol. Pharmacol 92, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Heo S, Hussain NK, Liu B, Huganir RL (2016) Extensive phosphorylation of AMPA receptors in neurons. Proc. Natl. Acad. Sci 113, E4920–E4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabian-Fine R, Skehel P, Errington ML, Davies HA, Sher E, Stewart MG, Fine A (2001) Ultrastructural distribution of the alpha7 nicotinic acetylcholine receptor subunit in rat hippocampus. J. Neurosci 21, 7993–8003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feder R, Ranck JB (1973) Studies on single neurons in dorsal hippocampal formation and septum in unrestrained rats: Part II. Hippocampal slow waves and theta cell firing during bar pressing and other behaviors. Exp. Neurol 41, 532–555. [DOI] [PubMed] [Google Scholar]
- Fink CC, Bayer K-U, Myers JW, Ferrell JE, Schulman H, Meyer T (2003) Selective regulation of neurite extension and synapse formation by the beta but not the alpha isoform of CaMKII. Neuron 39, 283–97. [DOI] [PubMed] [Google Scholar]
- Freund TF, Buzsáki GY (1996) Interneurons of the hippocampus. Hippocampus 6, 347–470. [DOI] [PubMed] [Google Scholar]
- Frotscher M, Schlander M, Léránth C (1986) Cholinergic neurons in the hippocampus. Cell Tissue Res 246, 293–301. [DOI] [PubMed] [Google Scholar]
- Fujii S, Sumikawa K (2001) Acute and chronic nicotine exposure reverse age-related declines in the induction of long-term potentiation in the rat hippocampus. Brain Res 894, 347–353. [DOI] [PubMed] [Google Scholar]
- Gandolfo M, Maccione A, Tedesco M, Martinoia S, Berdondini L (2010) Tracking burst patterns in hippocampal cultures with high-density CMOS-MEAs. J. Neural Eng 7, 56001. [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38, 1493–1503. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sulser A, Wang J, Queenan BN, Avoli M, Vicini S, Dzakpasu R (2012) Hippocampal neuron firing and local field potentials in the in vitro 4-aminopyridine epilepsy model. J. Neurophysiol 108, 2568–2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutagny R, Gu N, Cavanagh C, Jackson J, Chabot J, Quirion R, Krantic S, Williams S (2013) Alterations in hippocampal network oscillations and theta–gamma coupling arise before Aβ overproduction in a mouse model of Alzheimer’s disease. Eur. J. Neurosci 37, 1896–1902. [DOI] [PubMed] [Google Scholar]
- Gray R, Rajan AS, Radcliffe KA, Yakehiro M, Dani JA (1996) Hippocampal synaptic transmission enhanced by low concentrations of nicotine. Nature 383, 713. [DOI] [PubMed] [Google Scholar]
- Griguoli M, Cherubini E (2012) Regulation of hippocampal inhibitory circuits by nicotinic acetylcholine receptors. J. Physiol 590, 655–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griguoli M, Scuri R, Ragozzino D, Cherubini E (2009) Activation of nicotinic acetylcholine receptors enhances a slow calcium-dependent potassium conductance and reduces the firing of stratum oriens interneurons. Eur. J. Neurosci 30, 1011–1022. [DOI] [PubMed] [Google Scholar]
- Hajos M, Hurst RS, Hoffmann WE, Krause M, Wall TM, Higdon NR, Groppi VE (2005) The selective alpha7 nicotinic acetylcholine receptor agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide hydrochloride] enhances GABAergic synaptic activity in brain slices and restores auditory gating deficits in anesthetized rats. J. Pharmacol. Exp. Ther 312, 1213–1222. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJL, Bading H (2001) A calcium microdomain near NMDA receptors: On switch for ERK-dependent synapse-to-nucleus communication. Nat. Neurosci 4, 565–566. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2002) Coupling of extrasynaptic NMDA receptors to a CREB shut-off pathway is developmentally regulated, in Biochim. Biophys. Acta - Proteins Proteomics, Vol. 1600, pp. 148–153. [DOI] [PubMed] [Google Scholar]
- Harris KD, Hirase H, Leinekugel X, Henze DA, Buzsáki G (2001) Temporal Interaction between Single Spikes and Complex Spike Bursts in Hippocampal Pyramidal Cells. Neuron 32, 141–149. [DOI] [PubMed] [Google Scholar]
- Harrist A, Beech RD, King SL, Zanardi A, Cleary MA, Caldarone BJ, Eisch A, Zoli M, Picciotto MR (2004) Alteration of hippocampal cell proliferation in mice lacking the β2 subunit of the neuronal nicotinic acetylcholine receptor. Synapse 54, 200–206. [DOI] [PubMed] [Google Scholar]
- Hartikka J, Hefti F (1988) Development of septal cholinergic neurons in culture: plating density and glial cells modulate effects of NGF on survival, fiber growth, and expression of transmitter-specific enzymes. J.neurosci 8, 2967–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey SC, Maddox FN, Luetje CW (1996) Multiple Determinants of Dihydro-β-Erythroidine Sensitivity on Rat Neuronal Nicotinic Receptor α Subunits. J. Neurochem 67, 1953–1959. [DOI] [PubMed] [Google Scholar]
- Hulihan-Giblin BA, Lumpkin MD, Kellar KJ (1990) Acute effects of nicotine on prolactin release in the rat: agonist and antagonist effects of a single injection of nicotine. J. Pharmacol. Exp. Ther 252, 15–20. [PubMed] [Google Scholar]
- Ivanov A, Pellegrino C, Rama S, Dumalska I, Salyha Y, Ben-Ari Y, Medina I (2006) Opposing role of synaptic and extrasynaptic NMDA receptors in regulation of the extracellular signal-regulated kinases (ERK) activity in cultured rat hippocampal neurons. J. Physiol 572, 789–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izhikevich EM, Desai NS, Walcott EC, Hoppensteadt FC (2003) Bursts as a unit of neural information: selective communication via resonance. Trends Neurosci 26, 161–167. [DOI] [PubMed] [Google Scholar]
- Jefferys JGR, Traub RD, Whittington MA (1996) Neuronal networks for induced ‘40 Hz’rhythms. Trends Neurosci 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Ji D, Dani JA (2000) Inhibition and disinhibition of pyramidal neurons by activation of nicotinic receptors on hippocampal interneurons. J. Neurophysiol 83, 2682–2690. [DOI] [PubMed] [Google Scholar]
- Kellar KJ, Wonnacott S (1990) Nicotinic cholinergic receptors in Alzheimer’s disease, pp. 341–373. Oxford University Press, Oxford, UK. [Google Scholar]
- King JR, Nordman JC, Bridges SP, Lin MK, Kabbani N (2015) Identification and Characterization of a G Protein-binding Cluster in alpha7 Nicotinic Acetylcholine Receptors. J. Biol. Chem 290, 20060–20070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köller H, Siebler M, Müller HW (1993) Paroxysmal long-lasting depolarizations in cultured hippocampal neurons are generated by activation of NMDA and non-NMDA receptors. Synapse 14, 214–20. [DOI] [PubMed] [Google Scholar]
- Kramis R, Vanderwolf CH, Bland BH (1975) Two types of hippocampal rhythmical slow activity in both the rabbit and the rat: relations to behavior and effects of atropine, diethyl ether, urethane, and pentobarbital. Exp. Neurol 49, 58–85. [DOI] [PubMed] [Google Scholar]
- Lanneau C, Harries MH, Ray AM, Cobb SR, Randall A, Davies CH (2002) Complex interactions between mGluR1 and mGluR5 shape neuronal network activity in the rat hippocampus. Neuropharmacology 43, 131–140. [DOI] [PubMed] [Google Scholar]
- Lauterborn JC, Isackson PJ, Montalvo R, Gall CM (1993) In situ hybridization localization of choline acetyltransferase mRNA in adult rat brain and spinal cord. Mol. Brain Res 17, 59–69. [DOI] [PubMed] [Google Scholar]
- Lee KJ, Queenan BN, Rozeboom AM, Bellmore R, Lim ST, Vicini S, Pak DTS (2013) Mossy fiber-CA3 synapses mediate homeostatic plasticity in mature hippocampal neurons. Neuron 77, 99–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser SC, Bowlby MR, Comery TA, Dunlop J (2009) A cog in cognition: how the α7 nicotinic acetylcholine receptor is geared towards improving cognitive deficits. Pharmacol. Ther 122, 302–311. [DOI] [PubMed] [Google Scholar]
- Lena C, Changeux JP, Mulle C (1993) Evidence for “preterminal” nicotinic receptors on GABAergic axons in the rat interpeduncular nucleus. J. Neurosci 13, 2680–2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester HA, Xiao C, Srinivasan R, Son CD, Miwa J, Pantoja R, Banghart MR, Dougherty DA, Goate AM, Wang JC (2009) Nicotine is a selective pharmacological chaperone of acetylcholine receptor number and stoichiometry. Implications for drug discovery. AAPS J. 11, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED (2013) Complex relationships of nicotinic receptor actions and cognitive functions. Biochem. Pharmacol 86, 1145–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE (1997) Bursts as a unit of neural information: making unreliable synapses reliable. Trends Neurosci 20, 38–43. [DOI] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H (2002) The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci 3, 175. [DOI] [PubMed] [Google Scholar]
- Liu Y, Tak PW, Aarts M, Rooyakkers A, Liu L, Ted WL, Dong CW, et al. (2007) NMDA receptor subunits have differential roles in mediating excitotoxic neuronal death both in vitro and in vivo. J. Neurosci 27, 2846–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Jia Z, Janus C, Henderson JT, Gerlai R, Wojtowicz JM, Roder JC (1997) Mice lacking metabotropic glutamate receptor 5 show impaired learning and reduced CA1 long-term potentiation (LTP) but normal CA3 LTP. J. Neurosci 17, 5196–5205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchicchi A, Bloem B, Viana JN, Mansvelder HD, Role LW (2014) Illuminating the role of cholinergic signaling in circuits of attention and emotionally salient behaviors. Front. Synaptic Neurosci 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDermott AB, Role LW, Siegelbaum SA (1999) Presynaptic ionotropic receptors and the control of transmitter release. Annu. Rev. Neurosci 22, 443–485. [DOI] [PubMed] [Google Scholar]
- Mann EO, Greenfield SA (2003) Novel modulatory mechanisms revealed by the sustained application of nicotine in the guinea-pig hippocampus in vitro. J. Physiol 551, 539–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, Aerde KI van, Couey JJ, Brussaard AB (2006) Nicotinic modulation of neuronal networks: from receptors to cognition. Psychopharmacology (Berl). 184, 292–305. [DOI] [PubMed] [Google Scholar]
- Mansvelder HD, McGehee DS (2000) Long-term potentiation of excitatory inputs to brain reward areas by nicotine. Neuron 27, 349–357. [DOI] [PubMed] [Google Scholar]
- Marchi M, Grilli M (2010) Presynaptic nicotinic receptors modulating neurotransmitter release in the central nervous system: functional interactions with other coexisting receptors. Prog. Neurobiol 92, 105–111. [DOI] [PubMed] [Google Scholar]
- Mesulam M, Mufson EJ, Levey AI, Wainer BH (1983) Cholinergic innervation of cortex by the basal forebrain: cytochemistry and cortical connections of the septal area, diagonal band nuclei, nucleus basalis (substantia innominata), and hypothalamus in the rhesus monkey. J. Comp. Neurol 214, 170–197. [DOI] [PubMed] [Google Scholar]
- Miles R, Tóth K, Gulyás AI, Hájos N, Freund TF (1996) Differences between somatic and dendritic inhibition in the hippocampus. Neuron 16, 815–823. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RK (1987) Inhibitory control of local excitatory circuits in the guinea-pig hippocampus. J. Physiol 388, 611–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner LL, Collins AC (1988) The effect of chronic nicotine treatment on nicotine-induced seizures. Psychopharmacology (Berl). 95, 52–55. [DOI] [PubMed] [Google Scholar]
- Mintz CD, Smith SC, Barrett KMS, Benson DL (2012) Anesthetics interfere with the polarization of developing cortical neurons. J. Neurosurg. Anesthesiol 24, 368–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni F, Attucci S, Cozzi A, Meli E, Picca R, Scheideler MA, Pellicciari R, Noe C, Sarichelou I, Pellegrini-Giampietro DE (2002) The novel and systemically active metabotropic glutamate 1 (mGlu1) receptor antagonist 3-MATIDA reduces post-ischemic neuronal death. Neuropharmacology 42, 741–751. [DOI] [PubMed] [Google Scholar]
- Mudo G, Belluardo N, Fuxe K (2007) Nicotinic receptor agonists as neuroprotective/neurotrophic drugs. Progress in molecular mechanisms. J. Neural Transm 114, 135–147. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Kuryatov A, Choi CH, Zhou Y, Lindstrom J (2003) Alternate stoichiometries of alpha4beta2 nicotinic acetylcholine receptors. Mol. Pharmacol 63, 332–341. [DOI] [PubMed] [Google Scholar]
- Niedringhaus M, Chen X, Dzakpasu R, Conant K (2012) MMPs and soluble ICAM-5 increase neuronal excitability within in vitro networks of hippocampal neurons. PLoS One 7, e42631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak JL, Wheeler BC (1988) Multisite hippocampal slice recording and stimulation using a 32 element microelectrode array. J. Neurosci. Methods 23, 149–159. [DOI] [PubMed] [Google Scholar]
- Otto T, Eichenbaum H, Wible CG, Wiener SI (1991) Learning-related patterns of CA1 spike trains parallel stimulation parameters optimal for inducing hippocampal long-term potentiation. Hippocampus 1, 181–192. [DOI] [PubMed] [Google Scholar]
- Pak DTS, Yang S, Rudolph-Correia S, Kim E, Sheng M (2001) Regulation of dendritic spine morphology by SPAR, a PSD-95-associated RapGAP. Neuron 31, 289–303. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Mucke L (2006) A network dysfunction perspective on neurodegenerative diseases. Nature 443, 768–773. [DOI] [PubMed] [Google Scholar]
- Papke RL (2014) Merging old and new perspectives on nicotinic acetylcholine receptors. Biochem. Pharmacol 89, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen O, Sejnowski TJ (2000) Natural patterns of activity and long-term synaptic plasticity. Curr. Opin. Neurobiol 10, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike FG, Meredith RM, Olding AWA, Paulsen O (1999) Postsynaptic bursting is essential for ‘Hebbian’induction of associative long-term potentiation at excitatory synapses in rat hippocampus. J. Physiol 518, 571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Potter SM, DeMarse TB (2001) A new approach to neural cell culture for long-term studies. J. Neurosci. Methods 110, 17–24. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester RAJ (2002) Desensitization of neuronal nicotinic receptors. J. Neurobiol 53, 457–478. [DOI] [PubMed] [Google Scholar]
- Radcliffe KA, Fisher JL, Gray R, Dani JA (1999) Nicotinic modulation of glutamate and GABA synaptic transmission in hippocampal neurons. Ann. N. Y. Acad. Sci 868, 591–610. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Levin ED (2001) Cognitive effects of nicotine. Biol. Psychiatry 49, 258–267. [DOI] [PubMed] [Google Scholar]
- Ruivo LMT-G, Mellor JR (2013) Cholinergic modulation of hippocampal network function. Front. Synaptic Neurosci 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santello M, Toni N, Volterra A (2019) Astrocyte function from information processing to cognition and cognitive impairment. Nature Publishing Group. [DOI] [PubMed] [Google Scholar]
- Schicker KW, Dorostkar MM, Boehm S (2008) Modulation of transmitter release via presynaptic ligand-gated ion channels. Curr. Mol. Pharmacol 1, 106–129. [DOI] [PubMed] [Google Scholar]
- Schmitz TW, Nathan Spreng R, Weiner MW, Aisen P, Petersen R, Jack CR, Jagust W, et al. (2016) Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology. Nat. Commun 7, 13249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott L, Feng J, Kiss T, Needle E, Atchison K, Kawabe TT, Milici AJ, Hajós-Korcsok É, Riddell D, Hajós M (2012) Age-dependent disruption in hippocampal theta oscillation in amyloid-β overproducing transgenic mice. Neurobiol. Aging 33, 1481.e13–1481.e23. [DOI] [PubMed] [Google Scholar]
- Segev R, Shapira Y, Benveniste M, Ben-Jacob E (2001) Observations and modeling of synchronized bursting in two-dimensional neural networks. Phys. Rev. E 64, 11920. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S (2003) Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron 38, 929–939. [DOI] [PubMed] [Google Scholar]
- Sher E, Chen Y, Sharples TJW, Broad LM, Benedetti G, Zwart R, McPhie GI, Pearson KH, Baldwinson T, Filippi G De (2004) Physiological roles of neuronal nicotinic receptors subtypes: new insights on the nicotinic modulation of neurotransmitter release, synaptic transmission and plasticity. Curr. Top. Med. Chem 4, 283–297. [DOI] [PubMed] [Google Scholar]
- Singer W (1993) Synchronization of cortical activity and its putative role in information processing and learning. Annu. Rev. Physiol 55, 349–374. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Klausberger T (2005) Defined types of cortical interneurone structure space and spike timing in the hippocampus. J. Physiol 562, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanika RI, Pivovarova NB, Brantner CA, Watts CA, Winters CA, Andrews SB (2009) Coupling diverse routes of calcium entry to mitochondrial dysfunction and glutamate excitotoxicity. Proc. Natl. Acad. Sci. U. S. A 106, 9854–9859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark DT, Bazan NG (2011) Synaptic and extrasynaptic nmda receptors differentially modulate neuronal cyclooxygenase-2 function, lipid peroxidation, and neuroprotection. J. Neurosci 31, 13710–13721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suresh J, Radojicic M, Pesce LL, Bhansali A, Wang J, Tryba AK, Marks JD, Drongelen W van (2016) Network burst activity in hippocampal neuronal cultures: the role of synaptic and intrinsic currents. J. Neurophysiol 115, 3073–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Karson MA, Nagode DA, McIntosh JM, Uebele VN, Renger JJ, Klugmann M, Milner TA, Alger BE (2011) Nerve terminal nicotinic acetylcholine receptors initiate quantal GABA release from perisomatic interneurons by activating axonal T-type (Cav3) Ca(2)(+) channels and Ca(2)(+) release from stores. J. Neurosci 31, 13546–13561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang B, Luo D, Yang J, Xu X-Y, Zhu B-L, Wang X-F, Yan Z, Chen G-J (2015) Modulation of AMPA receptor mediated current by nicotinic acetylcholine receptor in layer I neurons of rat prefrontal cortex. Sci. Rep 5, 14099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson AM (2000) Facilitation, augmentation and potentiation at central synapses. Trends Neurosci 23, 305–312. [DOI] [PubMed] [Google Scholar]
- Traub RD, Jefferys JGR (1994) Simulations of epileptiform activity in the hippocampal CA3 region in vitro. Hippocampus 4, 281–285. [DOI] [PubMed] [Google Scholar]
- Tuan EW, Horti AG, Olson TT, Gao Y, Stockmeier CA, Al-Muhtasib N, Dalley CB, et al. (2015) AT-1001 Is a Partial Agonist with High Affinity and Selectivity at Human and Rat alpha3beta4 Nicotinic Cholinergic Receptors. Mol. Pharmacol 88, 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlhaas PJ, Singer W (2010) Abnormal neural oscillations and synchrony in schizophrenia. Nat. Rev. Neurosci 11, 100–113. [DOI] [PubMed] [Google Scholar]
- Wagenaar DA, Pine J, Potter SM (2006) An extremely rich repertoire of bursting patterns during the development of cortical cultures. BMC Neurosci 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir K, Blanquie O, Kilb W, Luhmann HJ, Sinning A (2015) Comparison of spike parameters from optically identified GABAergic and glutamatergic neurons in sparse cortical cultures. Front. Cell. Neurosci 8, 460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welsby P, Rowan M, Anwyl R (2006) Nicotinic receptor-mediated enhancement of long-term potentiation involves activation of metabotropic glutamate receptors and ryanodine-sensitive calcium stores in the dentate gyrus. Eur. J. Neurosci 24, 3109–3118. [DOI] [PubMed] [Google Scholar]
- Williams JH, Kauer JA (1997) Properties of carbachol-induced oscillatory activity in rat hippocampus. J. Neurophysiol 78, 2631–2640. [DOI] [PubMed] [Google Scholar]
- Wonnacott S (1997) Presynaptic nicotinic ACh receptors. Trends Neurosci 20, 92–98. [DOI] [PubMed] [Google Scholar]
- Wonnacott S, Barik J, Dickinson J, Jones IW (2006) Nicotinic receptors modulate transmitter cross talk in the CNS. J. Mol. Neurosci 30, 137–140. [DOI] [PubMed] [Google Scholar]
- Xiao Y, Fan H, Musachio JL, Wei ZL, Chellappan SK, Kozikowski AP, Kellar KJ (2006) Sazetidine-A, a novel ligand that desensitizes alpha4beta2 nicotinic acetylcholine receptors without activating them. Mol. Pharmacol 70, 1454–1460. [DOI] [PubMed] [Google Scholar]
- Yuan M, Malagon AM, Yasuda D, Belluzzi JD, Leslie FM, Zaveri NT (2017) The α3β4 nAChR partial agonist AT-1001 attenuates stress-induced reinstatement of nicotine seeking in a rat model of relapse and induces minimal withdrawal in dependent rats. Behav. Brain Res 333, 251–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarei MM, Radcliffe KA, Chen D, Patrick JW, Dani JA (1999) Distributions of nicotinic acetylcholine receptor α7 and β2 subunits on cultured hippocampal neurons. Neuroscience 88, 755–764. [DOI] [PubMed] [Google Scholar]
- Zaveri N, Jiang F, Olsen C, Polgar W, Toll L (2010) Novel α3β4 Nicotinic Acetylcholine Receptor-Selective Ligands. Discovery, Structure−Activity Studies, and Pharmacological Evaluation. J. Med. Chem 53, 8187–8191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang SJ, Buchthal B, Lau D, Hayer S, Dick O, Schwaninger M, Veltkamp R, Zou M, Weiss U, Bading H (2011) A signaling cascade of nuclear calcium-CREB-ATF3 activated by synaptic NMDA receptors defines a gene repression module that protects against extrasynaptic NMDA receptor-induced neuronal cell death and ischemic brain damage. J. Neurosci 31, 4978–4990. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.