

eToc:
Collins et al. describe the role of centriole number and microtubules during the developmental process of radial intercalation in the skin of Xenopus embryos. Increasing the number of centrioles andƒor microtubules within an intercalating cell drives early apical insertion, whereas decreasing centrioles and microtubules delays insertion.
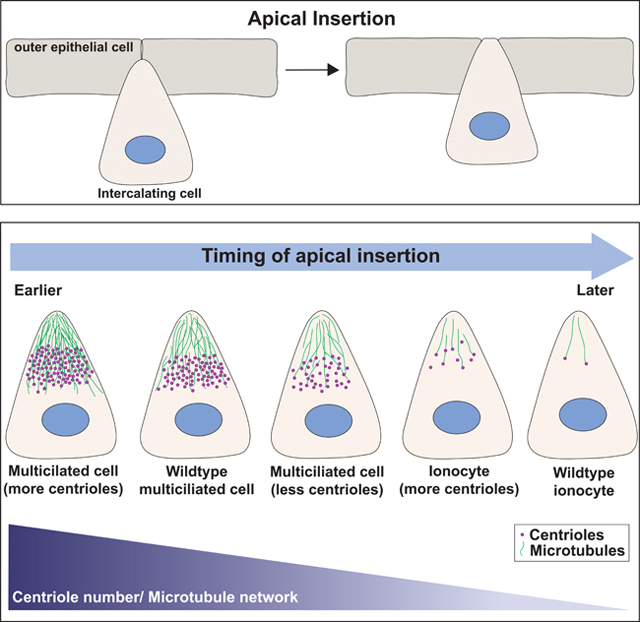
Summary
Centrioles are microtubule (MT)-based structures that provide important functions during cell migration, cell division and cell signaling [1]. Modulating centriole number in 3D cell cultures has been shown to influence protrusive behavior [2–5]. Here we address, in vivo, the role of centrioles and the accumulation of MTs on the protrusive behavior required during the initiation of radial intercalation. Radial intercalation is an important developmental process whereby cells undergo polarized movements and interdigitate into a more superficial layer [6, 7]. It is commonly employed during metamorphic events, such as the tissue thinning coupled with expansion or during the introduction of different cell types into an epithelium. During radial intercalation, cells emerge from a basal layer by undergoing a process of apical migration, apical insertion, and expansion [8]. In Xenopus skin, multiciliated cells (MCCs), which contain ~150 centrioles, and ionocytes (ICs), which contain two centrioles, differentiate during the same developmental window, but MCCs complete intercalation prior to ICs. Here we utilize this difference in timing to create a quantifiable assay for insertion and find that the timing of insertion is modulated by changes in centriole number and the accumulation of acetylated MTs. Additionally, centrioles align between the nucleus and the leading edge creating an axis of migration with apically oriented (+) ends. Using the MT (−) end protein CAMSAP1 fused to the apically positioned Par6 protein, we have artificially reversed the orientation of MTs and find that the accumulation of MTs in either orientation is sufficient to promote apical insertion.
Results and Discussion
In the Xenopus skin, multiciliated cells (MCCs) and ionocytes (ICs) undergo radial intercalation. Intercalating cells must first apically migrate from a basal layer towards the outer epithelium (Figure 1A, left). Once they encounter the outer cells, they protrude between cell vertices in a process of apical emergence, whereby they apically insert into the epithelium (Figure 1A, middle) and expand their apical surface until they achieve their final size (Figure 1A, right) [7, 8]. Recently published single cell RNAseq data indicates that both MCCs and ICs differentiate at developmental stage (ST) 13, several hours prior to the onset of radial intercalation [9, 10]. Importantly, these distinct cell types have different numbers of centrioles, with ICs containing two centrioles and MCCs containing approximately 150. Given the ability of centrioles to influence migration and protrusive behavior, we hypothesize that the difference in centriole number will result in a quantifiable difference in their efficiency to undergo intercalation. To follow cells during intercalation, we injected 2–4 cell embryos with DNA expressing fluorescent proteins via the tubulin a1a (Tub) promoter which drives expression specifically in MCCs or with the pendrin (Pen) promoter which drives expression specifically in a subset of ICs [7, 11]. It has previously been proposed that ICs intercalate later than MCCs [12]. Here, we performed a detailed time course where we analyzed the size of the apical surface of intercalating MCCs and ICs once they reached the outer epithelial layer at each developmental ST between ST17 and ST28 as a percentage of the average final apical size (Figure 1B–C). This analysis was done independently for each cell type, as MCCs have a larger final apical size (233 μm2) compared to ICs (95 μm2). In Wild Type (WT) embryos at ST17, neither MCCs nor ICs have initiated intercalation. MCCs begin intercalating at ST19 and continue to increase their apical size until ST24, by which time most cells are completely intercalated and have reached their final apical area. In striking contrast, ICs only begin intercalating around ST22/23 and do not complete intercalation until ST28. Importantly, the slope of these two growth curves appears to be nearly identical, which indicates that the rate of apical expansion is not dramatically different between the two cell types (Figure 1C). However, these results suggest that the initiation of apical insertion of ICs is dramatically delayed compared to MCCs by approximately 3–4 hours (at 18°C). To better quantify the initial apical insertion specifically, we scored the percentage of cells at each developmental ST which had achieved a small apical domain of 35 μm2, a size that represents the first observable insertion in either MCCs or ICs (Figure S1). Using this analysis, we see a similar separation with MCCs beginning to insert at ST19 and ICs beginning to insert at ST24 (Figure 1D). At ST21, nearly half of MCCs have initiated insertion compared to essentially no ICs. MCCs have completed insertion by ST24 at which point only 45% of ICs have inserted.
Figure 1. MCCs and ICs undergo intercalation at different developmental stages.

A, (top) Schematic of the progression of intercalation during development and (bottom) representative Z-projections and zoomed in side projections of embryos injected with Tub-GFP (to mark MCCs) or Pen-GFP (to mark ICs) during intercalation. B, Developmental time-course showing Z-projections of Xenopus embryos injected with Tub-GFP or Pen-GFP and stained with phalloidin. Scale bar, 10um. C, Quantification of MCC or IC apical area (as a percentage of final cell area) to measure intercalation progress. 100% indicates complete intercalation and incorporation into the outer epithelium. D, Quantification of the percentage of MCCs or ICs that have apically inserted (defined as an apical area >35μm2, see Figure S1) throughout the radial intercalation process. For graphs, dots (C) and bars (D) represent the mean, error bars indicate SD, and *p<0.05, **p<0.01. The n’s for each experiment are indicated in in Table S1. See also Figure S1 and Table S1.
MCCs and ICs represent distinct cell types with vastly different transcriptional profiles and cellular functions. However, the process of apical insertion appears quite similar in these cells suggesting that centriole number could be the underlying difference that drives the delay in the rate of IC insertion. To test this hypothesis, we modulated centriole number in MCCs and quantified the initiation of insertion. First, we injected embryos with either a control Morpholino (MO; Gene-Tools Inc.) or a MO targeting CCDC78 which encodes a protein previously shown to regulate centriole biogenesis [13]. Depletion of CCDC78 leads to an approximately 50% decrease in centrioles (82 ± 15 compared to 165 ± 18 with control MO, p=0.04; Figure 2A–B) [13]. In WT MCCs, there is a substantial accumulation of post-translationally modified acetylated tubulin in cells undergoing intercalation [14]. Significantly, in CCDC78 MO MCCs with decreased centriole numbers, we see a corresponding 40% decrease in acetylated tubulin (relative fluorescent intensity of 0.6 compared to control MO) (Figure 2C–E) which reflects a decrease in the overall MT network (Figure S2A–B). We measured the apical insertion under these conditions and found that in control MO injected embryos, MCCs initiate intercalation with similar timing to WT (Figure 1B,D and 2F–G). In contrast, we found that in CCDC78 morphant MCCs apical insertion was significantly delayed relative to controls (Figure 2F–G). Importantly, an analysis of the apical size over time indicates that the delay is specific to apical insertion as apical expansion or growth appears relatively normal (Figure S2C–D). Additionally, when we analyzed ST30 embryos we found that similar numbers of MCCs had succeeded intercalating in both control (24.8 ± 4.3 per field) and CCDC78 MO embryos (25.0 ± 4.9), indicating that MCCs in CCDC78 morphants are delayed in apically inserting rather than dying off or delaminating. To substantiate our observations, we repeated the centriole decrease experiment using CRISPR/Cas9 mediated gene editing of CCDC78. Using this approach, we see a decrease in centriole number similar to the MO data. Furthermore, while the F0 embryos are blindly mosaic for editing making quantification imprecise, we find a significant decrease in the percentage of MCCs that have apically inserted at ST21 in embryos with CRISPRƒCas9 editing (Figure S2E–F).
Figure 2. Decreasing MCC centriole number results in decreased MT density and a delay in apical insertion.

A, MCCs injected with Control MO or CCDC78 MO and GFP-Sas6 mRNA stained with phalloidin. B, Quantification of centriole number in Control and CCDC78 MO MCCs. C-D, Z-projections of MCCs injected with Control MO (C) or CCDC78 MO (D) with dextran blue as a tracer and GFP-Sas6 and stained with α-acetyl. tub. and phalloidin with side projections (C’, D’). E, Quantification of acetyl. tub. staining in intercalating Control and CCDC78 MO MCCs. Fluorescence was normalized relative to the mean Control MO fluorescence for each independent experiment. F, Time-course of Z-projections displaying progression of MCC intercalation in Control and CCDC78 MO embryos injected with GFP-Sas6 and membrane (mem)-RFP as a tracer and stained with phalloidin. G, Quantification of the percentage of MCCs apically inserted throughout the radial intercalation process. For all bar graphs, bars represent the mean, error bars indicate SD, and *p<0.05, **p<0.01. Scale bar in A, C, C’, D, D’ is 5μm and in F is 10μm. The n’s for each experiment are indicated in Table S1. See also Figure S2 and Table S1.
As shown above, decreasing centriole number in MCCs delays, but does not block, insertion. Next, we tested if increasing centriole number above normal levels would impact apical insertion. Cep152 and Plk4 are critical regulators of centriole biogenesis [15]. In cycling cells, overexpression (OE) of either protein leads to an over production of centrioles. To promote increased centriole amplification, we drove OE of both Cep152 and Plk4 in MCCs using the Tub promoter and found a significant increase in the number of centrioles generated from 168 ± 11 to 229 ± 14 (Figure 3A–B). This increase in centriole number leads to a corresponding increase in acetylated tubulin (Figure 3C–E) and overall MTs (Figure S3A–B). Strikingly, these cells now apically insert precociously, with 35% having inserted at ST17 (compared to 2% control MCC, p=0.01) and 65% inserted at ST19 (compared to 20% Control, p=0.005) (Figure 3F–G). Recent work has indicated that recruitment of Plk4 to the cell cortex can also promote protrusive behavior independent of centriole number [16]. Given this potential non–centriolar effect, we repeated these experiments using only OE of Cep152. Similarly, this led to an increase in centriole number (albeit lesser than OE of both Plk4 and Cep152, Figure S3C–D) and precocious apical insertion (Figure S3E–F). While we can’t rule out a non–centriolar role for Plk4 in driving precocious insertion, the data indicate that increasing centriole number independent of Plk4 expression is sufficient to drive precocious apical insertion (Figure S3F). Our results reveal that increasing the number of centrioles in MCCs leads to early insertion, while decreasing the number of centrioles leads to delayed insertion.
Figure 3. Increasing the number of centrioles in MCCs or ICs results in increased MT accumulation and precocious apical insertion.

A, MCCs expressing RFP-centrin or OE Tub-GFP-Plk4 and Tub-RFP-Cep152 stained with phalloidin. B, Quantification of centriole number in Control or Plk4+Cep152 OE MCCs. C-D, Side projections of intercalating control (C) and Plk4+Cep152 OE (D) MCCs expressing mem-RFP stained with α-acetyl. tub. E, Quantification of acetyl. tub. staining. Fluorescence was normalized relative to the mean control fluorescence for each independent experiment. F, Z-projections displaying progression of MCC apical insertion in Control and Plk4 + Cep152 OE embryos. G, Quantification of the percentage of MCCs apically inserted at each stage. H, ICs expressing RFP-centrin or OE Pen-GFP-Plk4 and Pen-GFP-Cep152 stained with phalloidin. I, Quantification of centriole number in Control or Plk4+Cep152 OE ICs. J-K, Side projections of intercalating control (J) and Plk4+Cep152 OE (K) ICs stained with α-acetyl. tub. and phalloidin. L, Quantification of acetyl. tub. staining. Fluorescence was normalized relative to the control (uninjected) IC in mosaic embryos for each experiment. M, Time-course of embryos injected with Pen-GFP or Pen-GFP-Plk4 + Pen-GFP-Cep152 fixed and stained with phalloidin to assay apical insertion. N, Quantification of the percentage of ICs apically inserted at each stage. For all bar graphs, bars represent the mean, error bars indicate SD, and *p<0.05, **p<0.01, ***p<0.001. Scale bar in C, D, J, K is 5μm and in A, F, H, and M is 10μm. The n’s for each experiment are indicated in Table S1. See also Figure S3 and Table S1.
We next tested if this relationship between centriole number and timing of apical insertion is conserved in ICs. Ectopic expression of the MCC inducing factor Multicilin (MCIDAS) leads to an amplification of centrioles as cells are converted into MCCs [17]. We drove expression of MCIDAS in ICs using the Pen promoter. It is difficult in this context to distinguish between WT MCCs and ectopic ICs converted to MCCs. However, we found that in Pen–MCIDAS embryos there were more MCCs per field at ST21 (compared to control embryos), consistent with the interpretation that ICs were converted to MCCs and furthermore had inserted precociously (Figure S3G). Since the Pen promoter turns on during IC differentiation this result confirms that the timing of differentiation is not what delays IC insertion. These cells have an increase in centriole number and precociously insert. However, since MCIDAS causes a cell type conversion, one cannot rule out the possibility that some other MCC specific factor is responsible for the effect. To address the specific role for centrioles we drove OE of both Cep152 and Plk4 in ICs using the Pen promoter and found an increase in centrioles from the normal two to 16 centrioles (Figure 3H–I). Additionally, with the increase in centriole number we see a concomitant increase in relative acetylated tubulin intensity approximately 1.5–fold above control ICs (Figure 3J–L) as well as an overall MT increase (Figure S3H–I). Similar to MCCs, OE of Cep152 and Plk4 in ICs also leads to precocious apical insertion (Figure 3M–N). We found that 26% of these OE ICs had inserted at ST20 (relative to 0% Control cells, p<0.001) and that 43% had inserted at ST22 (compared to only 16% Control ICs, p=0.005). Similar to MCCs, OE of Cep152 alone in ICs also shows an increase in centriole number (Figure S3J–K) and precocious intercalation independent of Plk4 (Figure S3L–M). These results indicate that similar to MCCs, centriole number influences the timing of apical insertion of ICs.
Centrioles within the centrosome are part of the MT organizing center of many cells. During intercalation we see an accumulation of MTs, and importantly, we see corresponding changes to MT accumulation relative to changes in centriole number in all of our experimental conditions suggesting that these centrioles are actively promoting MT nucleation. Additionally, we previously showed that MTs, as well as the MT stabilizing protein CLAMP, are essential to intercalation [14]. One possibility is that MTs could facilitate trafficking of vesicles or signaling molecules such as Rac1 towards the leading edge [5]. Alternatively, an increase in MTs at the leading edge could provide the structural strength and rigidity required to facilitate penetration through cell vertices. We reasoned that if trafficking is the primary function, then there would be a preferential directionality to the MTs. We therefore performed localization experiments in intercalating MCCs using both a MT (+) end protein GFP–EB3 and a MT (−) end protein GFP–CAMSAP2 [18–20]. There is a marked accumulation of the (+) end EB3 polarized towards the apical edge of intercalating MCCs and enriched above the cluster of centrioles (Figure 4A). In contrast, while CAMSAP2–GFP localizes between the nucleus and the leading edge near the centrioles, we do not see an enrichment at the apical surface (Figure 4B). These observations suggest that while MTs can nucleate in all directions, there is a preferential accumulation of MTs nucleated towards the leading edge (i.e. apical tip) of intercalating cells. This directionality of MTs is consistent with a role in directional trafficking of signaling molecules.
Figure 4. MT function during intercalation.

A-B, Side projections of intercalating MCCs expressing GFP-EB3 (A) or GFP-CAMSAP2 (B) and RFP-Sas6 stained with phalloidin and DAPI. C, Side projection of intercalating control and GFP-CAMSAP1-Par6 ICs stained with α-acetyl. tub. and phalloidin. D, Quantification of acetyl. tub. in control and GFP-CAMSAP1-Par6 ICs. Fluorescence was normalized relative to the control (uninjected) IC in mosaic embryos for each experiment. E, Side projection of embryos injected with EGFP-ninein to mark MT (−) ends and Pen-RFP or RFP-CAMSAP1-Par6 and stained with phalloidin and DAPI. F, Embryos mosaically injected with Pen-RFP and GFP-CAMSAP1-Par6 mRNA and stained with phalloidin to assay apical insertion. G, Quantification of the percentage of ICs apically inserted at each stage. For all bar graphs, bars represent the average, error bars indicate SD, and *p<0.05, **p<0.01. Scale bars in A–D are 5μm and in E is 10μm. The n’s for each experiment are indicated in Table S1. See also Figure S4 and Table S1.
CAMSAP proteins localize and stabilize the (−) ends of MTs and have been associated with the accumulation of non–centrosomal MTs [20]. To further test the possibility that MT orientation is important for radial intercalation, we mislocalized CAMSAP1 to the leading edge of intercalating cells by generating a fusion protein that combines CAMSAP1 with Par6, a component of the apical basal polarity “Par” complex that localizes to the apical side of intercalating cells [14]. mRNA injection of this construct leads to an apical accumulation of GFP–CAMSAP1–Par6 and importantly a marked increase in MTs (Figure 4C–D and S4A–B). We confirmed that the MT (−) ends were apically positioned by localizing an independent (−) end protein, ninein, which becomes apically enriched in the presence of the CAMSAP1–Par6 fusion protein (Figure 4E) [21]. We addressed the importance of MT orientation by expressing GFP–CAMSAP1–Par6 in ICs with normal centriole numbers (two) that have a paucity of MT (+) ends at the apical surface. In mosaic embryos, we compared the timing of Pen–RFP IC apical insertion alone or in cells co-expressing Pen–RFP and GFP–CAMSAP1–Par6 and, surprisingly, found that at each time point more GFP–CAMSAP1–Par6 cells had inserted than Control cells (Figure 4F–G), indicating precocious insertion. Importantly, expression of GFP–Par6 alone had no impact on the timing of insertion (Figure S4C–D). These results suggest that an apical accumulation of MTs, independent of orientation, is sufficient to promote apical insertion. While we cannot rule out the potential benefit of MT–based trafficking, our results suggest that the structural rigidity of MTs, in either orientation, can facilitate the process of breaking through an epithelial barrier.
The process of radial intercalation occurs during a developmental window spanning approximately 10 hours. While there is some overlap in the timing for individual cells, as a population MCCs intercalate several hours before ICs. This difference in timing has provided us a robust assay to quantifiably address the role of centriole number on apical insertion. Here we have tested the hypothesis that this difference in timing is due to differences in centriole number and the corresponding accumulation of MTs. By modulating centriole numbers in both cell types, we have directly addressed the role of centrioles on the timing of apical insertion. Decreasing centriole number delays insertion whereas increasing centriole number promotes early insertion. These data provide quantifiable in vivo evidence that centrioles promote protrusive behavior during apical insertion.
Radial intercalation is a complex phenomenon that involves coordination of various cytoskeletal elements and signaling pathways. Indeed, apical expansion of MCCs has been well characterized and is known to require RhoA and actin–based pushing forces [8, 22]. MCCs are known to create an elaborate actin network via interactions between basal bodies and WDR5 that ultimately facilitates spacing, polarization and metachronal synchrony of the motile cilia [23–25]. Furthermore, in any in vivo context, protrusive behavior involves dynamic and reciprocal changes in both the invading cell, as well as the surrounding neighboring cells. As such, radial intercalation in Xenopus skin is a useful model, as we can manipulate cellular protrusive and migratory properties in a complex 3D tissue environment. While we do not refute the potential importance of trafficking, signaling, and secretion of pro-invasive cytokines, our data provides mechanistic insight into the relationship between centrioles, the accumulation of MTs, and the protrusive behavior that occurs during radial intercalation [2–5]. Increasing MTs, independent of centrioles or independent of MT orientation, promotes apical insertion. Intercalating cells undoubtedly experience resistance as they meet and insert in between outer epithelial cells. Functionally, we propose that increasing MTs provides structural stability, and confers mechanical resilience to an intercalating cell. While individual MTs are ultimately dynamic, an overall increase in the pool of MTs would provide an increase in the collective stability of the MT network that facilitates the penetration between outer cells.
Our results contribute to the collective understanding of how cytoskeletal networks influence radial intercalation. We favor a model in which intercalating cells contain actin-based protrusions that identify vertices in the epithelial layer. Furthermore, the ability of cells to break through these vertices benefits not only from the dynamic pools of actin in the protrusions but also from a more stable pool of MTs. The larger the pool of MTs, the easier it is for the cells to penetrate through cell-cell junctions. In this model, the accumulation of MTs in either direction provides a structurally important pool that facilitates apical insertion.
STAR Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Brian Mitchell (brian-mitchell@northwestern.edu). All unique/stable reagents generated in this study are available without restriction.
Experimental Models and Subject Details
Xenopus laevis were used and maintained in accordance with standards established by the Northwestern University Institutional Animal Care and Use Committee. Mature X. laevis frogs were obtained from NASCO (Fort Atkinson, WI). Frog were housed in a recirculating tank system with regularly monitored temperature and water quality (pH, conductivity, and nitrate/nitrite levels) at a temperature of 16–18°C and were fed frog brittle.
Method Details
Embryo injections/CRISPR
All Xenopus experiments were performed using previously described techniques [26]. In brief, Xenopus embryos were obtained by in vitro fertilization using standard protocols [27] approved by the Northwestern University Institutional Animal Care and Use Committee. Embryos were injected at the two- or four-cell stage with 40–250 pg mRNA or 10–20 pg of plasmid DNA. To create mosaic embryos, only one or two blastomeres were injected at the four-cell stage. Morpholino antisense oligonucleotides (GeneTools) were injected into four-cell stage embryos to inhibit the expression of Xenopus CCDC78 (Unigene Xl.4890). A morpholino was used to target CCDC78 at the initiation site, as previously described [13]: CCDC78 MO: 5’-CATCAGTGTTACTAGGATAGGCAGG-3’; Control MO: 5’-CCTCTTACCTCAGTTACAATTTATA-3’. Morpholinos were injected into one blastomere at the four-cell stage with membrane RFP or dextran blue as a tracer. CCDC78 mutant cells were generated in F0 animals using CRISPR/Cas9-mediated genome editing. A gRNA (GGGCGAGTTACAGGACAAGATGG) was made that targeted both alleles of Xenopus CCDC78 at exon 2. Fertilized eggs were injected at the one-cell stage with 500 pg Cas9 protein (PNAbio, CP01-250) and 300 pg of sgRNA, and 40 pg GFP-Sas6 mRNA.
Plasmids/mRNA
pCS2+ plasmids containing an N-terminal GFP or RFP as a tracer containing the α-tubulin promoter (TUBA1A-B on Scaffold 127187, pCS2tub) [7] were used to drive expression of some constructs specifically in MCCs. pCS2+ plasmids containing the Pendrin (Pen) promoter [11] with an N-terminal GFP or RFP were used to drive expression of some constructs specifically in ionocytes. The membrane(mem)-RFP construct and GFP-Par6 constructs were previously described [14] and the RFP-centrin construct was described [25]. The Tub-RFP-Cep152, Tub-GFP-Plk4, GFP-Sas6, and RFP-Sas6 constructs were previously described [13]. To generate the Pen-GFP-Plk4 construct, Plk4 was excised from the Tub-GFP-Plk4 construct with SacI and NotI and cloned into the pCS2+ vector containing the Pen promoter and an N-terminal GFP. The Pen-GFP-Cep152 construct was generated by PCR amplification of Cep152 (Xl.13956) with BamHI and StuI sites added. The following primers were used, with BamHI (forward) and StuI (reverse) sites underlined: Cep152 (forward) - 5’-GCGGATCCATGTCTATCGACTTTGATAGTGGA-3’ Cep152 (reverse) - 5’-GGAGGCCTTTAGTTGAAGTTATTTAAGTTGGGAAATG-3’. The PCR amplified fragment was cloned into the pCS2+ vector containing the Pen promoter and an N-terminal GFP. GFP-tagged EB3 and human CAMSAP2 with an N-terminal GFP were used to track MT (+) ends and (−) ends, respectively [28]. The GFP-CAMSAP1-Par6 construct was generated by PCR amplification of CAMSAP1 from a pcDNA4TO-CAMSAP1L1-24xGCN4-IRES-puro (Addgene #60912) with BamHI and ClaI sites added. The following primers were used, with BamHI (forward) and ClaI (reverse) sites underlined: CAMSAP1(forward)-5’-GCGGATCCATGGGGGATGCTGCAGAC-3’; CAMSAP1 (reverse) - 5’-GCATCGATTGCCTTAGTGGGTAAAAGTTTTTTGG-3’. The PCR fragment was then ligated into pCS2 with an N-terminal GFP. Par6 (Xl.626) was then ligated downstream of the CAMSAP1 sequence with EcoRI and XhoI to create the fusion protein. The RFP-CAMSAP1-Par6 construct was generated by replacing the N-terminal GFP with RFP. The Pen-MCIDAS construct was generated by replacing the CMV promoter of a CS2-MT-MCIDAS plasmid [17] with the Pen promoter. All cloning was verified by sequencing. The EGFP-ninein construct was a gift from Michel Bornens (Addgene plasmid #73519) [21]. mRNA was generated using the Sp6 in vitro mRNA transcription kit (Ambion) following linearization of plasmid DNA with NotI. Capped mRNA was isolated using an RNA isolation kit (Qiagen). For some experiments, dextran cascade-blue (10,000 MW, ThermoFisher) was co-injected with RNA or morpholino as a tracer.
Immunofluorescence
For antibody staining, embryos were fixed with 3% PFA in PBS, blocked in 10% goat serum, and primary and secondary antibody solutions were prepared in 5% goat serum. Mouse anti-acetylated α-tubulin (T7451; Sigma-Aldrich) was used at a 1:500 dilution, mouse anti-beta tubulin (DHSB;E7) supernatant was used at a 1:10 dilution. E7 (anti-tubulin) was deposited to the DSHB by Klymkowsky, M. Cy-2-, Cy-3-, or Cy-5-conjugated goat anti-mouse secondary antibodies were used at the manufacturers’ recommended dilution. Phalloidin 650 (1:600, Invitrogen) and Alexa Fluor Plus 405 Phalloidin (1:40, Invitrogen) were used to visualize actin. DAPI nuclear counterstain (ThermoFisher, #62248) was used at a dilution of 1:300. Embryos were mounted between two coverslips using Fluoro-Gel (Electron Microscopy Sciences).
Microscopy
All microscopy was performed on a laser-scanning confocal microscope (A1R; Nikon) using a 60× oil Plan-Apochromat objective lens with a 1.4 NA. Nikon Elements Software was used for all acquisition and image processing. For all images, multiple z planes were visualized every 0.2–0.4 μm. Images are maximum intensity projections of z stacks. Images were processed in Nikon Elements Software.
Quantification and Statistical Analysis
Quantification of apical area and centriole numbers were performed manually in ImageJ Software [29]. Apical area of intercalating cells was measured at each stage (based on phalloidin staining). For analysis of apical area as a percentage of final area (Figure 1C), the mean apical area of MCCs and ICs at ST28 was defined as the ‘final apical area.’ MCC and IC apical area were determined independently due to the difference in size between cell types. The mean apical area at each stage was divided by the final apical area to define area as a percentage. For apical insertion analysis, an apical domain area of 35μm2 was set as a threshold to determine apical insertion. This area was chosen based the average apical area of MCCs and ICs at the stage when we first observed an increase in the apical area during intercalation (MCCs - ST19, ICs - ST24; see Figure S1). The percentage of MCCs or ICs apically inserted (as opposed to still below the surface) at each stage represent the percentage of cells measured at the indicated stage that have an area >35μm2 and is independent of measurements taken at other stages. For all apical insertion analyses, cells below the surface of the outer epithelium were included in the analysis and were given an apical area of 0μm2. For acetylated and beta tubulin intensity measurements, a z-projection of the intercalating cell was created and the fluorescence intensity was measured in ImageJ by outlining the cell of interest and only measuring anti-acetylated or anti-beta tubulin fluorescence within the outline. Tubulin intensities were normalized relative to the mean fluorescence intensity measured for the ‘control’ condition for each experiment. For figure presentations, acetylated tubulin and beta tubulin images were deconvolved in Nikon Elements Software. Some images were smoothed and processed for figure presentation only. Raw images were used for all analyses. Significance was determined with a student’s t-test. Number of cells and embryos quantified for each analysis are indicated in Table S1. For all bar graphs, bars represent the mean and error bars indicate the standard deviation (SD). For all statistical analyses, *p<0.05, **p<0.01, and ***p<0.001.
Supplementary Material
Highlights:
Radial intercalation of multiciliated cells and ionocytes is temporally separable
Timing of apical insertion correlates with centriole number and microtubules
Modulating centriole numbers influences the timing of apical insertion
Ectopic accumulation of microtubules accelerates apical insertion
Acknowledgments
This work was supported by NIH/NIGMS (R01GM119322). We would like to thank Jen Mitchell for helpful comments during preparation of this manuscript. We would like to thank the National Xenopus Resource and the Marine Biological Laboratories for technical support and reagents.
Footnotes
Declaration of Interests
The authors declare no competing interests.
Data and Code Availability
This study did not generate any unique datasets or code.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Loncarek J, and Bettencourt-Dias M. (2018). Building the right centriole for each cell type. J Cell Biol 217, 823–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnandis T, Monteiro P, Adams SD, Bridgeman VL, Rajeeve V, Gadaleta E, Marzec J, Chelala C, Malanchi I, Cutillas PR, et al. (2018). Oxidative Stress in Cells with Extra Centrosomes Drives Non-Cell-Autonomous Invasion. Dev Cell 47, 409–424 e409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Godinho SA, Kwon M, and Pellman D. (2009). Centrosomes and cancer: how cancer cells divide with too many centrosomes. Cancer Metastasis Rev 28, 85–98. [DOI] [PubMed] [Google Scholar]
- 4.Godinho SA, and Pellman D. (2014). Causes and consequences of centrosome abnormalities in cancer. Philos Trans R Soc Lond B Biol Sci 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Godinho SA, Picone R, Burute M, Dagher R, Su Y, Leung CT, Polyak K, Brugge JS, Thery M, and Pellman D. (2014). Oncogene-like induction of cellular invasion from centrosome amplification. Nature 510, 167–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drysdale TA, and Elinson RP (1992). Cell Migration and Induction in the Development of the Surface Ectodermal Pattern of the Xenopus laevis Tadpole Dev Growth Differ 34, 51–59. [DOI] [PubMed] [Google Scholar]
- 7.Stubbs JL, Davidson L, Keller R, and Kintner C. (2006). Radial intercalation of ciliated cells during Xenopus skin development. Development 133, 2507–2515. [DOI] [PubMed] [Google Scholar]
- 8.Sedzinski J, Hannezo E, Tu F, Biro M, and Wallingford JB (2016). Emergence of an Apical Epithelial Cell Surface In Vivo. Dev Cell 36, 24–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Briggs JA, Weinreb C, Wagner DE, Megason S, Peshkin L, Kirschner MW, and Klein AM (2018). The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science 360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nieuwkoop PDF, J. (1994). Normal table of Xenopus laevis (Daudin), (New York, New York: Garland Publishing Inc; ). [Google Scholar]
- 11.Quigley IK, Stubbs JL, and Kintner C. (2011). Specification of ion transport cells in the Xenopus larval skin. Development 138, 705–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubaissi E, Rousseau K, Lea R, Soto X, Nardeosingh S, Schweickert A, Amaya E, Thornton DJ, and Papalopulu N. (2014). A secretory cell type develops alongside multiciliated cells, ionocytes and goblet cells, and provides a protective, anti-infective function in the frog embryonic mucociliary epidermis. Development 141, 1514–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Klos Dehring DA, Vladar EK, Werner ME, Mitchell JW, Hwang P, and Mitchell BJ (2013). Deuterosome-mediated centriole biogenesis. Dev Cell 27, 103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Werner ME, Mitchell JW, Putzbach W, Bacon E, Kim SK, and Mitchell BJ (2014). Radial intercalation is regulated by the Par complex and the microtubule-stabilizing protein CLAMPƒSpef1. J Cell Biol 206, 367–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Avidor-Reiss T, and Gopalakrishnan J. (2013). Building a centriole. Curr Opin Cell Biol 25, 72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luo Y, Barrios-Rodiles M, Gupta GD, Zhang YY, Ogunjimi AA, Bashkurov M, Tkach JM, Underhill AQ, Zhang L, Bourmoum M, et al. (2019). Atypical function of a centrosomal module in WNT signalling drives contextual cancer cell motility. Nat Commun 10, 2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stubbs JL, Vladar EK, Axelrod JD, and Kintner C. (2012). Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Nat Cell Biol 14, 140–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hendershott MC, and Vale RD (2014). Regulation of microtubule minus-end dynamics by CAMSAPs and Patronin. Proc Natl Acad Sci U S A 111, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stepanova T, Slemmer J, Hoogenraad CC, Lansbergen G, Dortland B, De Zeeuw CI, Grosveld F, van Cappellen G, Akhmanova A, and Galjart N. (2003). Visualization of microtubule growth in cultured neurons via the use of EB3-GFP (end-binding protein 3-green fluorescent protein). J Neurosci 23, 2655–2664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang K, Hua S, Mohan R, Grigoriev I, Yau KW, Liu Q, Katrukha EA, Altelaar AF, Heck AJ, Hoogenraad CC, et al. (2014). Microtubule minus-end stabilization by polymerization-driven CAMSAP deposition. Dev Cell 28, 295–309. [DOI] [PubMed] [Google Scholar]
- 21.Delgehyr N, Sillibourne J, and Bornens M. (2005). Microtubule nucleation and anchoring at the centrosome are independent processes linked by ninein function. J Cell Sci 118, 1565–1575. [DOI] [PubMed] [Google Scholar]
- 22.Sedzinski J, Hannezo E, Tu F, Biro M, and Wallingford JB (2017). RhoA regulates actin network dynamics during apical surface emergence in multiciliated epithelial cells. J Cell Sci 130, 420–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kulkarni SS, Griffin JN, Date PP, Liem KF Jr., and Khokha MK (2018). WDR5 Stabilizes Actin Architecture to Promote Multiciliated Cell Formation. Dev Cell 46, 595–610 e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahuzier A, Shihavuddin A, Fournier C, Lansade P, Faucourt M, Menezes N, Meunier A, Garfa-Traore M, Carlier MF, Voituriez R, et al. (2018). Ependymal cilia beating induces an actin network to protect centrioles against shear stress. Nat Commun 9, 2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Werner ME, Hwang P, Huisman F, Taborek P, Yu CC, and Mitchell BJ (2011). Actin and microtubules drive differential aspects of planar cell polarity in multiciliated cells. J Cell Biol 195, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Werner ME, and Mitchell BJ (2013). Using Xenopus Skin to Study Cilia Development and Function In Methods in Enzymology, Volume 525: Cilia, Part B, Volume 525, Marshall WF, ed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sive HL, Grainger RM, and Harland RM (2007). Xenopus laevis In Vitro Fertilization and Natural Mating Methods. CSH protocols 2007, pdb prot4737. [DOI] [PubMed] [Google Scholar]
- 28.Kim SK, Zhang S, Werner ME, Brotslaw EJ, Mitchell JW, Altabbaa MM, and Mitchell BJ (2018). CLAMPƒSpef1 regulates planar cell polarity signaling and asymmetric microtubule accumulation in the Xenopus ciliated epithelia. J Cell Biol 217, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.