

Abstract
Human papillomavirus (HPV) insertions in cancer genomes have been linked to various forms of focal genomic instability and altered expression of neighboring genes. Here we tested the hypothesis that investigation of HPV insertions in a head and neck cancer squamous cell carcinoma (HNSCC) cell line would identify targetable driver genes contributing to oncogenesis of other HNSCC. In the cell line UPCI:SCC090, HPV16 integration amplified PIM1 serine/threonine kinase gene ~16-fold, thereby increasing transcript and protein levels. We used genetic and pharmacological approaches to inhibit PIM kinases in this and other HNSCC cell lines. Knockdown of PIM1 transcripts by transfected short hairpin RNAs reduced UPCI:SCC090 viability. CRISPR/Cas9-mediated mutagenesis of PIM1 caused cell cycle arrest and apoptosis. Pharmacological inhibition of PIM family kinases decreased growth of UPCI:SCC090 and additional HNSCC cell lines in vitro and a xenograft UPCI:SCC090 model in vivo. Based on established interactions between intracellular signaling pathways and relatively high levels of gene expression in almost all HNSCC, we also evaluated combinations of PIM kinase and epidermal growth factor receptor (EGFR) inhibitors. Dual inhibition of these pathways resulted in supra-additive cell death. These data support clinical testing of PIM inhibitors alone or in combination in HNSCC.
Keywords: head and neck squamous cell carcinoma, human papillomavirus (HPV), PIM serine/threonine kinases, molecular therapeutics, virus insertional mutagenesis
1. INTRODUCTION
HNSCC ranks eighth in cancer incidence worldwide [1]. While most cases are attributable to tobacco and alcohol use, a subset of oropharyngeal cancers caused by HPV infection is increasing markedly in incidence in numerous developed countries [2]. The transforming ability of HPV partly has been linked to myriad functions of viral oncoproteins E6 and E7, which functionally inactivate tumor suppressor proteins p53 and pRb, respectively. While E6 and E7 expression is sufficient for keratinocyte immortalization, secondary genetic events also are necessary for cellular transformation and formation of cancer [3].
We previously reported a direct association between HPV insertions and various forms of flanking host genomic instability, including amplifications, rearrangements, deletions and translocations in human cancer cell lines and primary tumors [4]. Viral insertions frequently were accompanied by disruption in expression of neighboring host genes. We hypothesized that HPV-mediated alterations in gene expression or structure could serve as critical secondary genetic events in the pathogenesis of HPV-associated cancers [4]. For example, HPV insertions with flanking genomic rearrangements altered MYC expression in HeLa cells [5], GUMC395, a cell line derived from a cervical neuroendocrine cancer [6], and other cancer cell lines [7]. Marked alterations of MYC expression in these tumors may have contributed profoundly to their formation.
In studying the HNSCC cell line UPCI:SCC090 using comprehensive genomics methods including whole genome sequencing (WGS) and RNA sequencing (RNA-Seq), we discovered HPV insertions flanking a 16-fold somatic amplification of PIM1 (Proviral insertion site for Moloney murine leukemia virus MuLV), a serine/threonine kinase proto-oncogene located on chromosome 6p21 (Fig. 1) [4]. This HPV insertion-mediated increase in PIM1 genomic copy number was accompanied by marked increases of PIM1 transcripts, which were not associated with expression of chimeric HPV-PIM1 fusion transcripts.
Figure 1. HPV-mediated genomic amplification and expression of PIM1 in HNSCC cell lines.
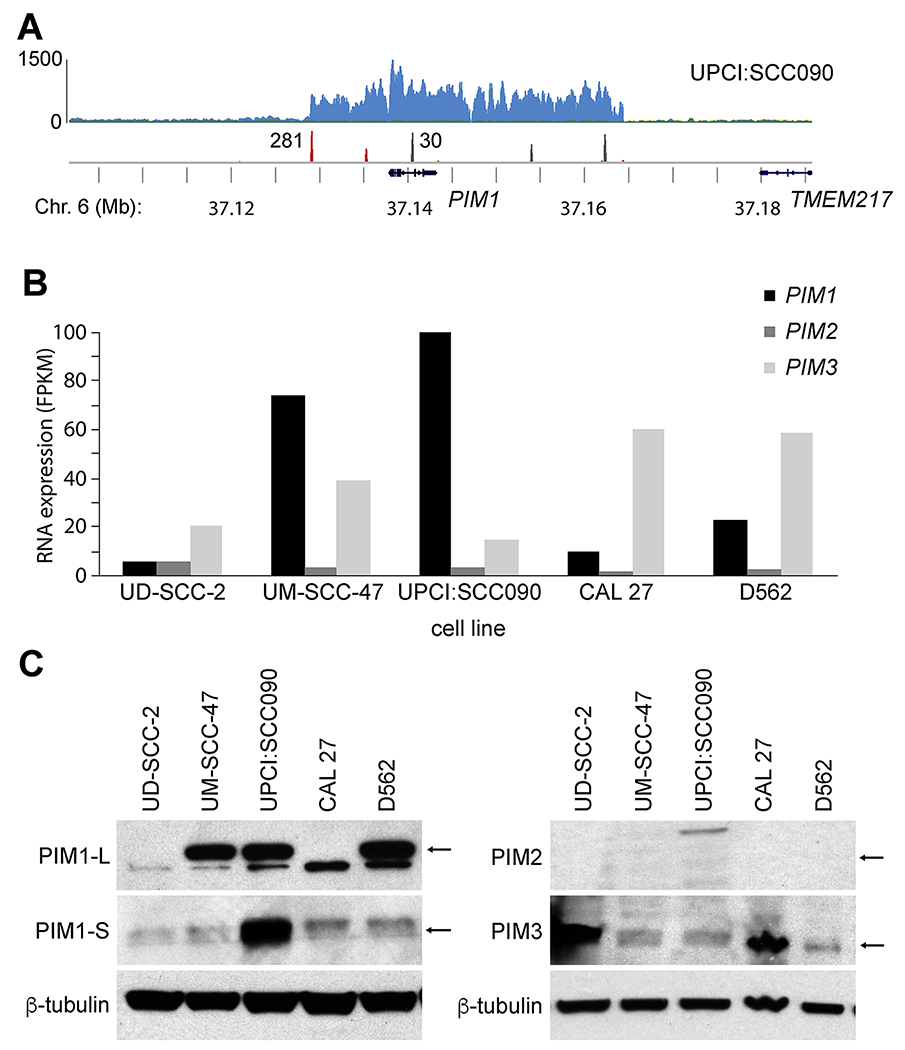
(A) HPV integrant-mediated genomic amplification of the PIM1 locus in UPCI:SCC090 HNSCC cells. (Top) Histograms show (y-axis) depth of WGS coverage by (blue) well-aligned sequence reads, and (middle) counts of (red) HPV insertional and (gray) host-host breakpoint reads in UPCI:SCC090 cells, mapped to the (bottom) reference human genome (hg19) at (x-axis) the PIM1 locus on Chr. 6 (bottom, gene schematics, chromosomal coordinates in Mb). Reproduced from Akagi et al. with permission [4]. See also Supp. Fig. S1. (B) RNA-seq expression levels (y-axis; FPKM) of PIM1, PIM2 and PIM3 transcripts expressed in HNSCC cell lines (i.e. UD-SCC-2, UM-SCC-47, UPCI:SCC090, CAL 27, D562). (C) Western blot analysis of PIM1 (L, long and S, short isoforms), PIM2, and PIM3 expressed in panel of HNSCC cell lines, with beta-tubulin as loading control. Arrows, expected protein mass.
The PIM kinase family (PIM1, PIM2, and PIM3) encodes constitutively active serine/threonine kinases that regulate cell cycle progression and apoptosis [8]. Pim1 originally was identified as a recurrent provirus integration site for the Moloney murine leukemia virus (Mo-MLV), resulting in mouse T cell lymphomas [9]. These viral insertions resulted in transcriptional upregulation of the gene, identifying it as a targetable cancer-causing driver gene. Subsequently, Pim2 was identified as another common viral insertion site in transplanted mouse T cell lymphomas, suggesting its important role in cancer progression [10]. The orthologous human gene, PIM1, also has been reported to be overexpressed in HNSCC [11]. Therefore, to confirm that HPV insertion-mediated alterations at PIM1 were required for the HNSCC cancer phenotype in UPCI:SCC090 cells, we investigated the antiproliferative and biochemical effects of genetic knockdown and small molecule inhibition of PIM kinases. To test the hypothesis that PIM kinases also contribute to HNSCC cancer formation more broadly, as candidate cancer-causing driver genes, we extended these experiments in additional HNSCC cell lines.
2. MATERIALS AND METHODS
2.1. Human HNSCC cancer cell lines and primary HNSCC tumor /normal pairs
Human HNSCC cell lines FaDu (HPV-negative), SCC-4 (HPV-negative), SCC-9 (HPV-negative), SCC-15 (HPV-negative), CAL 27 (HPV-negative), Detroit 562 (hereafter called D562, HPV-negative), and SCC-25 (HPV-negative) were purchased from American Type Culture Collection (ATCC) [12–15]; UM-SCC-47 (HPV-positive) and UM-SCC-104 (HPV-positive) [15], kindly provided by Dr. Thomas Carey, University of Michigan; UPCI:SCC090 (HPV-positive) [16], Dr. Susanne M. Gollin, University of Pittsburgh; UD-SCC-2 (HPV-positive) [17], Dr. Henning Bier, University of Dusseldorf; and HMS001 (HPV-positive) [4], Dr. James Rocco, Ohio State University. Cell lines were cultured according to instructions from ATCC, and as described previously [4] and in Supplementary Methods.
Patients with newly diagnosed oral cavity or oropharyngeal squamous cell carcinoma presenting at Ohio State University from 2011-2016 provided written, informed consent to participate in genomics studies [3], approved by Institutional Review Boards at Ohio State University and at University of Texas MD Anderson Cancer Center. WGS and RNA-Seq were performed on 101 HPV-positive HNSCC tumor/normal (T/N) pairs, including 84 in the Ohio cohort and 17 downloaded from the TCGA website at https://gdc.cancer.gov/, and 50 HPV-negative OSCC T/N pairs including 26 from our Ohio cohort and 24 from TCGA [3].
2.2. Quantitative realtime PCR assays for PIM1 and PIM3 expression
TaqMan assays to quantify PIM1 and PIM3 transcript levels were performed as described in Supplementary Methods.
2.3. Lentiviral shRNA
Knockdown of PIM1, PIM3 or FOXE1 was conducted using recombinant lentivirus to express specific or control scrambled shRNA sequences in the HNSCC cell lines UD-SCC-2, UM-SCC-47, UPCI:SCC090, CAL 27, and D562, as described in Supplementary Methods.
2.4. Generation of PIM1 knockout clones using CRISPR/Cas9 gene editing
CRISPR/Cas9-mediated genome editing was used to construct PIM1 knockout clones derived from UPCI:SCC090 parental cells. A lentiviral construct, LentiCRISPR-PIM1, was engineered to express a custom PIM1-specific guide RNA sequence, 5’-GTGGCGTGCAGGTCGTTGCA-3’, and functional effects of this genetic knockdown were assessed as described in Supplementary Methods.
2.5. Analysis of protein expression and phosphorylation by Western blotting
Expression and phosphorylation levels of proteins including PIM family kinases, downstream PIM kinase substrates and proteins upstream of the PIM signaling pathway were evaluated in HNSCC cell lines and CRISPR knockout clones by Western blot analysis, as described in Supplementary Methods.
2.6. Cell cycle and apoptosis analysis
Effects of PIM inhibition on the cell cycle and in inducing apoptosis were quantified using propidium iodide (Abcam) in flow cytometry and with the AlexaFluor 488 Annexin V dead cell apoptosis kit (Invitrogen Life Technologies), respectively, as described in Supplementary Methods.
2.7. Mouse xenograft model
Xenograft models in nude mice were generated by injection of UPCI:SCC090 cells. Female nude mice (Charles River) were maintained in compliance with a protocol approved by the Institutional Animal Care and Use Committee (Ohio State University). Mice were injected in the flank with 1.5x106 UPCI:SCC090 cells in a 1:1 ratio of Matrigel (Corning). Twenty days after inoculation of cells, when tumor volumes reached ~150 to 200 mm3, mice (n = 7) were arbitrarily assigned to treatment twice daily via oral gavage either with 100mg/kg INCB053914 in 5% dimethylacetamide and 0.5% methylcellulose, or vehicle. Tumor volumes were measured every 2 days until euthanasia due to tumor growth and/or toxicity.
2.8. Dual inhibition of PIM and EGFR pathways
To assay effects of PIM pathway inhibition on cell viability, cell lines were treated with pan-PIM inhibitor INCB053914 (Incyte Corp.) [18], or the PIM1 inhibitors Quercetagetin (Santa Cruz Technologies) or PIM1 Inhibitor 2 (Santa Cruz Technologies), or a negative control, DMSO, as described in Supplementary Methods. For dual inhibitor studies of the PIM and EGFR pathways, cells were incubated with INCB053914 [18] and either erlotinib or afatinib (Selleckchem), individually or in combination, as described in Supplementary Methods.
2.9. Informatics analysis of HNSCC samples
Informatics analysis of PIM gene copy numbers, RNA-Seq data and associations between copy numbers and gene expression levels in primary HNSCC tumors was performed as described in Supplementary Methods.
3. RESULTS
3.1. HPV integration upregulates PIM1 expression in UPCI:SCC090 cells
We conducted WGS and RNA-Seq of UPCI:SCC090 cells and other HPV-positive and negative HNSCC cell lines, and of primary HNSCC tumor/ normal (T/N) pairs [3, 4]. In the HNSCC cell line UPCI:SCC090, we identified ~200 copies of HPV16 genomic DNA per cell, and found 33 HPV-host insertional breakpoints located on Chromosomes 3p12, 6p21, and 9q22 [4]. HPV integrants on Chr. 6p21 flanked an ~16-fold amplification of PIM1, a serine/threonine kinase gene (Fig. 1A). On Chr. 9q22, HPV integrants flanked an ~7 fold amplification of FOXE1, a member of the forkhead family of transcription factors [4]. Amplification of PIM1 was associated with high levels of PIM1 transcript and protein expression, as assessed by RNA-Seq and Western blot (Fig. 1B, C). We detected only minimal numbers of chimeric HPV-PIM1 fusion transcripts, insufficient to account for the strong upregulation of PIM1 transcripts (Supp. Fig. S1). Both isoforms of PIM1 protein (i.e. PIM1-L [large], 44kDa; and PIM1-S [small], 33kDa) were highly expressed.
3.2. PIM gene family expression varies widely in HNSCC cell lines
We measured expression of all 3 members of the PIM family of kinases in UPCI:SCC090 and additional HNSCC cell lines, including HPV-positive (i.e. UD-SCC-2 and UM-SCC-47) and HPV-negative (i.e. CAL 27 and D562) lines. Considerable variation was detected in PIM1 mRNA expression (i.e. ranging from ~10 to 120 fragments per thousand transcripts per million mapped reads, FPKM) and in PIM3 expression (i.e. 25-60 FPKM) based on RNA-seq data. By contrast, PIM2 expression was consistently low across these cell lines (Fig. 1B). Quantitation of PIM1, PIM2 and PIM3 expression by quantitative reverse transcriptase mediated polymerase chain reaction (qRT-PCR) assays confirmed high expression of PIM1 (in UPCI:SCC090, UM-SCC-47 and D562 cells) and PIM3 (in UD-SCC-2 and CAL 27 cells) (Supp. Fig. S2).
Consistent with the known regulation of PIM1 expression at a transcriptional level [8, 19], PIM1 protein levels measured by Western blotting correlated closely with transcript levels as measured by RNA-Seq (Fig. 1C). By contrast, encoded PIM3 protein expression levels were not as closely associated with PIM3 transcript levels. For example, based on PIM3 transcript levels, PIM3 protein levels were higher than predicted in UD-SCC-2 cells, while they were lower than predicted in D562 cells (Fig. 1B, C). We surveyed expression of PIM1 and PIM3 proteins in additional HNSCC cell lines, again highlighting wide-ranging variability in expression (Supp. Fig. S3).
3.3. PIM knockdown reduces cell viability
We hypothesized that HPV-mediated amplification of PIM1 and FOXE1 could contribute to cancer formation of UPCI:SCC090 cells. Therefore, we examined a potential relationship between expression levels of these genes and UPCI:SCC090 cell viability. To knockdown these genes, we obtained lentivirus vectors to express shRNA directed against PIM1, FOXE1, or a scrambled shRNA sequence as a control. After anti-PIM1 shRNA was introduced, UPCI:SCC090 cell viability was reduced by 82% as quantified by cell counting with Trypan blue exclusion. By comparison, treatment with anti-FOXE1 shRNA reduced cell viability by 38%. As a control, treatment with scrambled shRNA reduced viability by 21% (Fig. 2A).
Figure 2. Anti-PIM1 shRNA compromises cell viability.
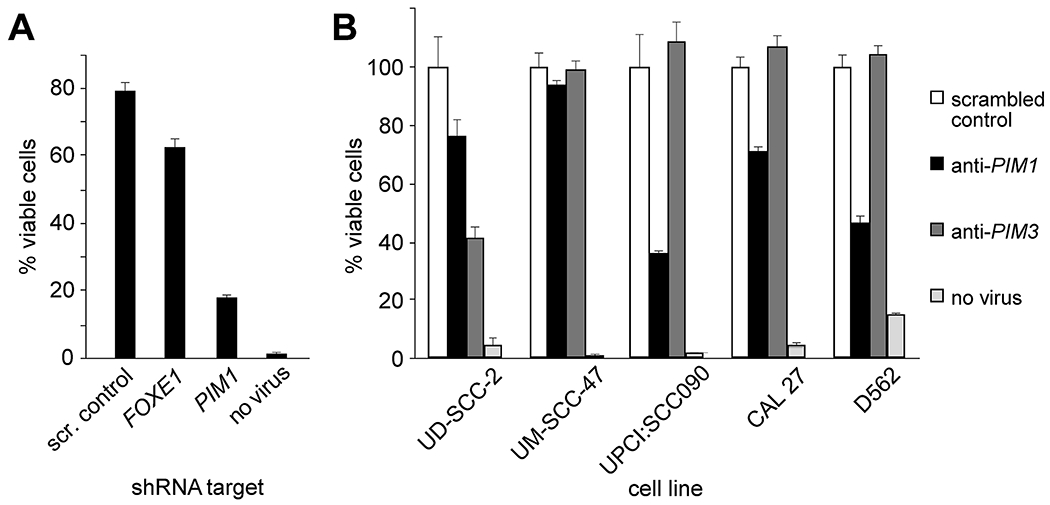
(A) UPCI:SCC090 cells were transduced with lentivirus expressing shRNAs with indicated targets of scrambled control (scr. control), FOXE1 or PIM1, or no lentivirus. Cell viability was determined by trypan blue exclusion using a hemacytometer in light microscopy. Barplots, mean of duplicate cell counts; error bars, range of data. (B) HPV-positive (UD-SCC-2, UM-SCC-47, UPCI:SCC090) and HPV negative (CAL 27, D562) OSCC cell lines were transduced with lentivirus expressing shRNAs with indicated targets of scrambled control, PIM1 or PIM3, or no lentivirus. Barplots, mean of duplicate cell counts; error bars, range.
To evaluate impacts of PIM1 or PIM3 knockdown on viability of additional cell lines, we transduced UD-SCC-2, UM-SCC-47, UPCI:SCC090, CAL 27, and D562 cells each with recombinant lentiviruses to express the respective knockdown shRNAs or scrambled control. Knockdown of PIM1 in D562 cells, which express high levels of PIM1 mRNA and protein, resulted in ~60% reduction in cell viability. By contrast, D562 cell viability was unaffected by PIM3 knockdown (Fig. 2B). The converse was true in UD-SCC-2 cells, which express high levels of PIM3 but low PIM1. By contrast, viability of UM-SCC-47 and CAL 27 cells was unaffected by PIM1 or PIM3 shRNA knockdown, despite high expression levels of PIM1 and PIM3, respectively (Fig. 2B), suggesting potential compensatory driver genes or pathways in them.
3.4. Genetic knockout of PIM1 reduces UPCI:SCC090 proliferation
Because genetic knockdown experiments involving shRNAs can produce off-target and transient effects on phenotypes[20], we also generated stable PIM1 genetic knockout subclones using CRISPR/Cas9 gene editing. This facilitated a comparison of their proliferation vs. that of wildtype cells at various timepoints in culture. We designed guide RNAs to target the first and fourth coding exons of PIM1 (Supp. Fig. S4). Five PIM1 knockout subclones (i.e. C9, C2, C5, C4, C11) were generated from parental UPCI:SCC090 cells by introduction of insertion-deletion (indel) and frameshift mutations in PIM1. Each was confirmed by PCR amplification and Sanger sequencing of the targeted region, which demonstrated that each subclonal line harbored both mutated and wildtype PIM1 sequences (Supp. Fig. S5). A possible explanation for the remaining wildtype allele is that CRISPR/Cas9 did not mutagenize all genomic copies of PIM1 per cell. Since the gene is amplified 16-fold in a concatenated array together with integrated HPV16 in parental UPCI:SCC090 cells (Supp. Fig. S1) [4], some wildtype alleles may have survived CRISPR-mediated gene editing in resulting cell clones. An alternative explanation is that subclonal cells harboring wildtype PIM1 persisted in mosaicism within each culture population, although each subclone was grown up from isolated cells that were considered to be singletons. Despite persistence of the wildtype allele in cloned lines, a 70 to 100% reduction in PIM1 protein expression was confirmed by Western blot (Fig. 3A and Supp. Fig. S6).
Figure 3. PIM1 knockout in UPCI:SCC090 cells reveals genetic driver of cell viability and proliferation.
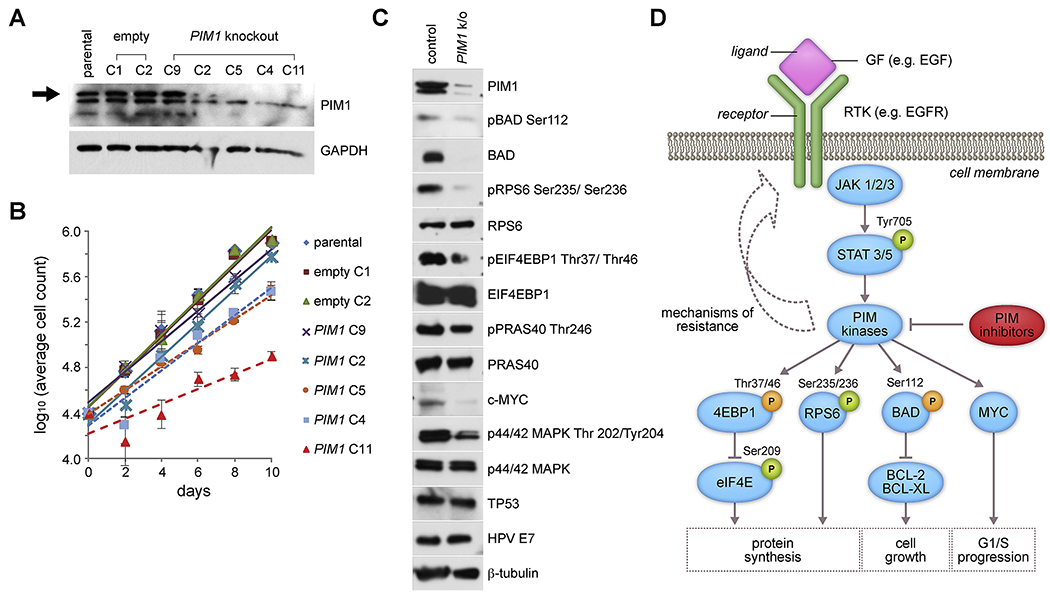
(A) Western blot analysis of PIM1 expression and (B) cell proliferation curves (y-axis, Log10 average cell count) of parental UPCI:SCC090 cell line, 2 negative controls (empty vector mock-edited by CRISPR, clones C1, C2), and 5 PIM1 knockout clones (gene edited by CRISPR, clones C9, C2, C5, C4, C11) cultured over time (x-axis, days). Key: colors, symbols: cell clones. Slopes of linear regression lines indicative of growth rates are: parental, 0.157; empty C1,0.156; empty C2, 0.157; PIM1 C9, 0.135; PIM1 C2, 0.148; PIM1 C5, 0.104; PIM1 C4, 0.121; and PIM1 C11, 0.066. Error bars, range of cell counts in duplicate wells. (C) Western blot analysis of downstream proteins and phosphorylation targets of PIM1, in a selected PIM1 knockout clone derived from UPCI:SCC090 cells by CRISPR gene editing, compared with parental UPCI:SCC090 control cells. (D) Schematic of possible mechanism of resistance in response to PIM inhibition in HNSCC cells such as CAL 27 cells, through feedback loops involving increased expression of PIM protein through STAT activation. Downstream targets of PIM kinases are displayed showing activating (green) and inactivating (orange) phosphorylation, and biological responses associated with these signaling interactions. GF, growth factor ligand, e.g. EGF, epidermal growth factor; RTK, receptor tyrosine kinase, e.g. EGFR, epidermal growth factor receptor; STAT, signal transducer and activator of transcription. See also Supp. Figs. S6 and S11.
These reductions in PIM1 protein expression, due to CRISPR-mediated genetic knockout in UPCI:SCC090 subclones, correlated with reductions in their proliferation rates (Fig. 3B). A nearly complete reduction of PIM1 expression in clone C11 was associated with a ~58% reduction in proliferation rate when compared to control cells (Fig. 3B). By contrast, minimal or more modest reductions of PIM1 expression in PIM clones C9 and C2 (Fig. 3A) were associated with proliferation rates that were essentially unchanged when compared with parental control cells (Fig. 3B). Upon serial passage of the CRISPR knockout clones, we observed a gradual recovery in PIM1 protein expression accompanied by an accelerated proliferation rate, again revealing an association between PIM1 expression levels and the cells’ proliferation rates (data not shown).
Despite genetic knockdown of PIM1, no change in HPV E7 oncoprotein was detected (Fig. 3). Since only minimal numbers of RNA-Seq reads were identified that documented expression of chimeric HPV-host transcripts (Supp. Fig. S1), this result confirmed our prediction that PIM1 disruption would not affect the autonomously expressed, independent E7 oncogene.
3.5. Downstream consequences of PIM1 genetic knockout
To explore potential mechanisms for reduced cell proliferation of PIM1 knockout cells, we examined phosphorylation levels of PIM1 kinase substrates in knockout clone C11. PIM1 has been shown to inhibit apoptosis through phosphorylation of Ser112 of the pro-apoptotic BCL2-associated agonist of death promoter (BAD) protein, thereby inducing anti-apoptotic activity of BCL2/BCL-xL and increasing cell survival [21]. Western blotting confirmed marked decreases in PIM1, pBAD Ser112 phosphorylation and total BAD levels in the knockout cells (Fig. 3 and Supp. Fig. S6).
PIM1 also has been shown to regulate protein synthesis downstream of the phosphatidylinositol-4,5-bisphosphonate 3-kinase catalytic subunit alpha, AKT serine/threonine kinase, mammalian target of rapamycin (PIK3CA/AKT/mTOR) signaling pathway. It activates ribosomal protein S6 (RPS6, also termed S6) via phosphorylation of Ser235/236, and inhibits eukaryotic translation initiation factor 4E-binding protein (EIF4EBP1) via phosphorylation of Thr37 and Thr46, thereby promoting cap-dependent translation and cell growth [22, 23]. Upon knockdown of PIM1 in UPCI:SCC090 cells, we observed decreased phosphorylation of pRPS6 and of EIF4EBP1 (i.e. pEIF4EBP1), respectively, while overall RPS6 and EIF4EBP1 protein levels remained unchanged (Figs. 3C and 3D). However, upon knockdown of PIM1, we did not observe decreases in the PIM1 phosphorylation target AKT1 substrate 1 (AKT1S1) at Thr246 or in total AKT1S1 levels (not shown), in contrast to treated leukemia cells [24].
PIM1 protein reportedly stabilizes MYC protein and promotes prostate cancer tumorigenesis through activation of mitogen-activated protein kinase 1 (MAPK1, also termed ERK) [25]. Decreased levels of MYC protein and phosphorylated MAPK1 (termed pMAPK1, phosphorylated at Thr 202/Tyr204) were observed in the CRISPR-generated PIM1 knockout clone C11 when compared with control (Figs. 3C and 3D). Similarly, reduced levels of MYC and phosphorylated MYC were observed in another CRISPR knockout clone (Supp. Fig. S6). By contrast, both HPV16 E7 and p53 protein expression levels were unaffected by PIM1 knockdown, arguing against an explanation for reduced cellular proliferation that involves off-target effects on HPV oncoproteins (Fig. 3C). We conclude that reduced proliferation and increased cell death upon PIM1 knockdown may be attributed to a combination of factors including inhibition of protein synthesis, generation of pro-apoptotic signals, and cell cycle arrest due to reduction in MYC function.
3.6. Pan-PIM Inhibitors inhibit HNSCC cell line growth
Genetic knockdown or knockout of PIM1 reduced UPCI-SCC090 cells’ viability and proliferation. These observations prompted us to investigate the effect of small molecule inhibitors of PIM kinase activity upon cell viability in our panel of HNSCC cell lines. Several PIM kinase inhibitors have been developed for potential therapeutic uses. We evaluated the anti-proliferative effects of INCB053914 [18], PIM1 Inhibitor 2 [26], and quercetagetin [27], using neutral red and WST-1 cell viability assays (Fig. 4A, Supp. Fig. S7). INCB053914 is a pan-PIM inhibitor, while PIM1 inhibitor 2 and quercetagetin are competitive inhibitors of ATP binding to the PIM1 active site.
Figure 4. Sensitivity of HNSCC cell lines to PIM inhibitors, in vitro and in vivo.

(A-C) Various cell lines (legend, color symbols) were treated with (A) INCB053914; (B) PIM1 inhibitor 2; or (C) quercetagetin at indicated concentrations (x-axis) for 72 h. Cell viability was assessed using neutral red assay (y-axis, normalized to DMSO control). Data represent averages of triplicate wells normalized to DMSO control. Error bars, standard deviation. (D) Nude mice injected with UPCI:SCC090 cells (day 0) were treated twice daily with either vehicle or INCB053914 via oral gavage, starting on day 20. Mean tumor volume (y-axis) was assessed for 8 days (x-axis); error bars, standard deviation (n = 7 mice).
Consistent with shRNA knockdown experiments, UPCI:SCC090, UD-SCC-2 and D562 cells each were sensitive to treatments with the pan-PIM inhibitor INCB053914 at low doses. The doses at which growth was half-maximally inhibited (EC50) ranged from approximately 0.045 to 8 μM (Fig. 4A). By contrast, UM-SCC-47 and CAL 27 cells were more resistant to this drug (Fig. 4A). Similar findings were obtained upon fitting drug inhibition curves according to a non-linear model (not shown). Treatment of seven additional HNSCC cell lines (i.e. SCC-9, SCC15, SCC-25, HMS-001, UM-SCC-104, SCC4, and FaDu cells) with INCB053914 revealed EC50 < 8 μM in SCC-9 and SCC15 cells (Supp. Fig. S8, Supp. Table S1). In total, 5 of 12 HNSCC cell lines were at least modestly sensitive to the pan-PIM inhibitor INCB053914, as defined by EC50 <8 μM. Similar results also were observed with other PIM kinase inhibitors tested, i.e. PIM1 Inhibitor 2 and quercetagetin (Fig. 4B, C).
3.7. In vivo effects of PIM inhibition in UPCI:SCC090 xenograft model
To further validate our in vitro findings that PIM1 amplification and upregulation contributed as a driver to the malignant phenotype of UPCI:SCC090 cells, we established a mouse xenograft tumor model to assess functional inhibition by INCB053914 in vivo. UPCI:SCC090 cells were injected bilaterally into flanks of nude mice and grown until tumors reached 150 – 200 mm3 prior to starting drug treatments. Administration of INCB053914, given twice daily at 100mg/kg via oral gavage for 8 days, resulted in approximately 60% tumor growth inhibition, corresponding to an approximately 2.5-fold decrease in mean tumor volume, relative to vehicle control (Fig. 4D). Results of this in vivo xenograft tumor model are consistent with inhibitory doses in xenograft models of lymphoma [18], and provide further evidence that PIM1 is a driver of UPCI:SCC090’s viability and growth as a tumor in vivo.
3.8. INCB053914 induces cell cycle arrest and apoptosis
We sought to evaluate the effect of pan-PIM inhibition on cell cycle progression, using flow cytometry to assay cell lines that were either sensitive (i.e. UPCI:SCC090, UD-SCC-2) or resistant (i.e. UM-SCC-47) to INCB053914. Treatment of UPCI:SCC090 cells with 1.25μM INCB053914 (i.e. twice the estimated EC50 dose) for 48 hours (h) caused cell cycle arrest. In comparison to DMSO vehicle control, the proportion of cells treated with INCB053914 that were in G0/G1 increased from 58.4% to 74.8%. Similarly, the proportion of cells in the sub-G1 phase also increased from 0.7% to 2.8% (Supp. Table S2). The proportion of UPCI:SCC090 cells in late apoptosis was 0.9% in the negative control, and 18.5% after treatment with 2.5μM INCB053914 for 48 h. The proportion of dead cells was 1.9% in the negative control, and 16% of cells treated with INCB053914 (Supp. Fig. S9). Similar findings were observed for the drug-sensitive cell line UD-SCC-2 (Supp. Fig. S9). By contrast, similar treatments of a resistant cell line UM-SCC-47 resulted in minimal alterations in cell cycle or apoptosis (Supp. Fig. S9, Supp. Table S2).
To elucidate possible mechanisms of sensitivity or resistance to INCB053914, we evaluated alterations in phosphorylation of PIM1 substrates by Western blot. In sensitive cell lines such as UPCI:SCC090, UD-SCC-2, and D562, we observed a consistent, dose-dependent decrease in pRPS6 phosphorylation at serine residues 235/236 (Supp. Fig. S10). By contrast, levels of pRPS6 phosphorylation were essentially unchanged in resistant cells such as UM-SCC-47 treated with a wide range of doses of INCB053914 (Supp. Fig. S11).
A functional deficiency of phosphorylated pRPS6 has been linked to reduced cell size [22, 28]. Consistent with this report, UPCI:SCC090 and UD-SCC-2 cells displayed 44% and 29% reductions in mean cell sizes, respectively, following 48 hours of exposure to 1.25μM INCB053914. The cell sizes were measured by flow cytometry as mean forward scatter height (FSC-H) of the gated G1 cell population. By contrast, UM-SCC-47 cells, resistant to INCB053914, displayed only a 7% decrease in cell size upon such treatment (Supp. Fig. S9).
When assessing phosphorylation of other PIM substrates after treatment with INCB053914, we observed reductions in phosphorylation of BAD serine 112 in drug-sensitive UPCI:SCC090 and D562 cells (Supp. Fig. S11). In the former cells, reductions in BAD phosphorylation were modest, whereas they were more robust in D562 cells. By contrast, no such inhibition was observed in drug-sensitive UD-SCC-2 cells. This indicated that modulation of phosphorylated pBAD is not required for growth inhibition. We also did not observe altered phosphorylation of AKT1S1 at Thr246 as previously reported [24]. Protein levels of MYC varied with INCB053914 treatment for each cell line (Supp. Fig. S11). Western blot analysis of UM-SCC-47 cell lysates showed an increase in inactivating phosphorylation of EIF4EBP1 at Thr37 and Thr46, which negatively regulates cap-dependent translation. In addition, increased activating phosphorylation of eIF4E protein at Ser209 was observed (Supp. Fig. S11). These modifications promote protein synthesis and could contribute to modest reductions in size of UM-SCC-47 cells upon treatment with INCB053914 (Supp. Fig. S9).
CAL 27 cells display high levels of PIM3 expression, but their viability was not affected either by pan-PIM inhibitor treatment or by genetic knockdown (Fig. 4A). Nevertheless, we observed modest, dose-dependent reductions in phosphorylation of pRPS6 at Ser235/236 (Supp. Fig. S12) and more robust decreases in phosphorylation of BAD at Ser112, phosphorylation of EIF4EBP1 at Thr37 and Thr46, and phosphorylation of eIF4E at Ser209, in response to treatment by pan-PIM inhibitor INCB053914 (Supp. Fig. S12). However, expression levels of activated, phosphorylated signal transducer and activator of transcription 3 (STAT3) at Tyr705 and of PIM3 were increased upon treatment with INCB053914 (Supp. Fig. S12). Since PIM expression has been reported to be regulated at the level of transcription by STAT proteins, we hypothesized that CAL 27 resistance to PIM inhibition could involve up-regulation of PIM3 by interacting signaling feedback loops involving STAT3 activation (Supp. Fig. S12).
3.9. Supra-additive inhibition of growth by pan-PIM and EGFR inhibitors
The oncogenic pathways driven by EGFR are interconnected with the complex network of PIM signaling pathways. A recent study showed that EGF and TGF-alpha signaling through EGFR can induce increased expression and nuclear translocation of PIM1 in 5 HNSCC cell lines, contributing to radiation resistance [29]. We noted that cell lines resistant to pan-PIM inhibitors (i.e. UM-SCC-47 and CAL 27) harbored ~10-fold amplification of EGFR copy number as shown by WGS [4], but sensitive cell lines had no such amplification (Supp. Table S4).
To test the hypothesis that combined inhibition of both upstream EGFR and downstream PIM1 could demonstrate supra-additivity, we assayed viability of the HNSCC cell lines UPCI:SCC090, UD-SCC-2, UM-SCC-47, CAL 27 and D562 after 72 h exposure to various concentrations of both INCB053914 and a small molecule EGFR inhibitor, i.e. afatinib or erlotinib. As indicated by a combination index (CI) <1, supra-additivity was observed with dual inhibitor treatments at nanomolar concentrations in CAL 27 cells (Fig. 5, Supp. Table S3). For example, the EC50 of INCB053914 for CAL 27 decreased from 20μM to less than 1.5μM upon addition of 0.05μM of afatinib. Supra-additivity also was observed in sensitive cell lines (i.e. UPCI:SCC090 and UD-SCC-2), and to a lesser extent in resistant D562 and UM-SCC-47 cells (Fig. 5, Supp. Fig. S13, Supp. Table S3). The EC50 of INCB053914 for UM-SCC-47 decreased from 30μM to less than 1.5μM upon addition of 0.3125μM of afatinib (Supp. Fig. S13, Supp. Table S3). These data support the hypothesis that combined inhibition of the EGFR and PIM signaling pathways may overcome resistance mechanisms that thwart single agent treatments.
Figure 5. Synergistic cell death upon targeting EGFR and PIM kinases.
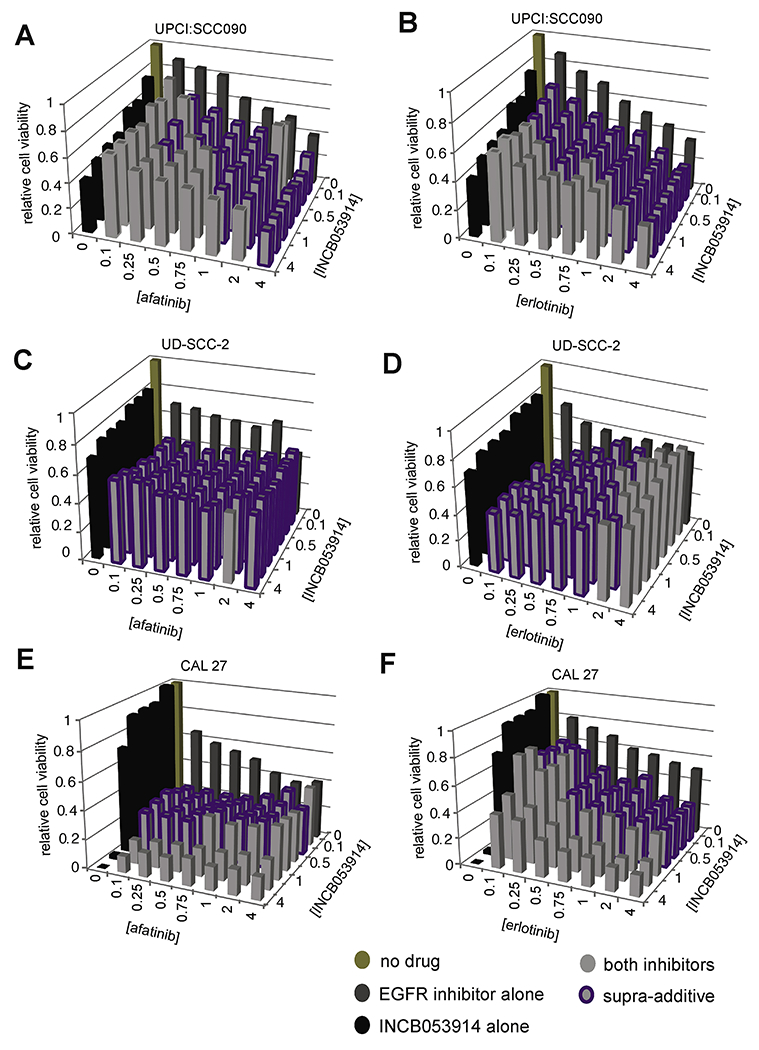
Cells were treated with INCB053914 (y-axis) and EGFR inhibitor (x-axis) alone or in combination at established effective dilutions (ED). Relative cell viability (z-axis) after 72 hours was normalized to DMSO control. Dual inhibitor treatments that display synergy (combination index (CI) <1 ) are highlighted, purple. Data represent average of triplicate wells normalized to DMSO control. Concentrations of inhibitor at each Drug ED are provided in Supp. Table S3. (A, B) UPCI:SCC090, (C, D) UD-SCC-2 and (E, F) CAL 27 cells were treated with (A) afatinib or (B) erlotinib.
3.10. PIM expression in primary tumors
We identified mutations, copy number changes, and expression levels of PIM family genes and EGFR in HPV-positive (n = 101) and HPV-negative (n = 50) primary oral and oropharyngeal squamous cell carcinomas by analyzing WGS and RNA-seq data from samples in our Ohio cohort or downloaded from the Cancer Genome Atlas project (TCGA) [3]. Mutation analysis did not reveal any mutations in the PIM family genes. However, PIM family gene amplification (defined as copy number > 2.5) was identified in 22 (14.6%) of 151 primary HNSCC tumors (Supp. Table S5). These were identified more frequently in 101 HPV-positive HNSCC (PIM1, n=6; PIM2, n=11; PIM3, n=1; total 17.8%) than in 50 HPV-negative tumors (all PIM family members, n=4; total 8%). We also observed EGFR amplification in 2 (2.0%) of 101 HPV-positive tumors and in 14 (28%) of 50 HPV-negative tumors (Supp. Table S5).
Mean PIM1 mRNA expression levels were significantly higher in tumors with PIM1 copy number amplification (expressed as log2(transcript fragments per kilobase million), TPM), Supp. Fig. S14). However, similar associations were not observed for PIM2 or PIM3 (Supp. Fig. S15). Mean EGFR expression levels were significantly higher in tumors with EGFR amplification (Supp. Fig. S14), as previously reported [30, 31]. However, EGFR copy number amplification was not associated with PIM gene expression in our panel of HNSCC cell lines (Supp. Fig. S16).
RNA-seq analysis of the 151 primary HNSCC tumors revealed high levels of expression (defined as TPM ≥ 30) of PIM1, PIM2, and PIM3 in 146 (96.7%), 75 (49.7%) and 151 (100%) of the samples, respectively (Fig. 6, Supp. Table S6). When compared with expression of all annotated genes across these primary HNSCC tumors, median PIM1 transcript levels ranked in the top 6.5th percentile, PIM2 ranked in the 21.4th percentile, PIM3 ranked in the top 1.3rd percentile and EGFR ranked in the 7.7th percentile (Fig. 6). By comparison, based on the same definition, only ~23% of 18,640 RefSeq genes were expressed at these high median levels across the 151 HNSCC samples (Fig. 6). While high levels of PIM1 and PIM3 expression were detected in almost all of the HPV-positive and HPV-negative HNSCC tumors, high levels of PIM2 expression were identified in a majority of only the HPV-positive tumors (Supp. Table S6). EGFR was highly expressed (TPM ≥ 30) in 134 (88.7%) of the HNSCC tumors studied.
Figure 6. The PIM kinase genes and EGFR are highly expressed in HNSCC.

The histogram shows the distribution of median gene expression levels in 151 primary human HNSCC tumors for each of 18,640 genes [3]. After normalization and batch correction of RNA-seq data, genes expressed in at least in one sample (log2(TPM + 1) > 0) were compared. Gencode v18 genes were used as a reference set of annotated genes for comparison of expression. X-axis, median of transcript levels for each gene across all HNSCC samples (transformed as log2 TPM+1), ranging from 0.014 to 13.7; y-axis, number of genes. Blue vertical lines, log2 transformed median expression levels of PIM1 = 6.51 (top 6.5th percentile), PIM2 =4.94 (21.4th percentile), PIM3 =8.43 (1.3rd percentile) and EGFR = 6.29 (7.7th percentile).
4. DISCUSSION
The transforming ability of HPV has been attributed primarily to inactivation of host p53 and pRb family members by viral E6 and E7 oncoproteins, respectively [32]. While expression of E6 and E7 is essential to the viral life cycle and sufficient for cellular immortalization, secondary genetic events are necessary for development of cancer. Analyses of genomic landscapes of HNSCC [3, 33, 34] and cervical cancers [35] by next generation DNA sequencing have identified numerous recurrent somatic mutations and genomic structural rearrangements. We and others have reported that HPV can act as an insertional mutagen, wherein HPV integration is directly associated with alterations in host genome structure and expression [4, 5, 7, 36]. We hypothesize that resulting alterations can drive clonal selection and are necessary for the malignant phenotype, and the data presented here support this hypothesis. As reported here, the study of HPV integration has led to the identification of a candidate driver gene (i.e. PIM1 kinase) that is a rational target for therapeutic agent development in HNSCC. Notably, groundbreaking studies of Mo-MuLV insertions in mouse lymphomas led to the initial identification of members of the same family of serine/threonine kinase genes [9, 10], activated by genomic insertions of a distinct virus in a different species and tissue type, again driving cancer formation or progression.
We previously showed that HPV insertion at PIM1 in the HNSCC cell line UPCI:SCC090 was associated with flanking genomic amplification and markedly increased expression of that serine/threonine kinase gene [4]. The 16-fold amplification of the PIM1 gene, its marked overexpression, and its well-documented role as a candidate oncogene in other cancers [37, 38] prompted us to investigate the hypothesis that the large-scale upregulation of PIM1 in UPCI:SCC090 cells, attributable to virus integration, could have contributed to the malignant phenotype and formation of this particular HNSCC [39]. We detected very low expression levels of chimeric HPV-PIM1 fusion transcripts, which we concluded were insufficient to account for the strong upregulation of PIM1. We experimentally inhibited PIM1 in UPCI:SCC090 cells at the DNA, RNA, and protein levels, using CRISPR-mediated genome editing, shRNA inhibition, and several pharmacological inhibitors. Resulting reductions of PIM1 serine/threonine kinase activity consistently were associated with decreased UPCI:SCC090 cell viability and proliferation, and also resulted in well-documented changes in phosphorylation of PIM substrates. Treatment with the pan-PIM inhibitor INCB053914 significantly decreased both UPCI:SCC090 cell growth in vitro and tumor volume in a mouse xenograft model in vivo. The growth of additional HNSCC cell lines also was inhibited by similar experimental treatments.
Subclonal cells derived from UPCI:SCC090 that underwent CRISPR/Cas9-mediated genetic mutagenesis of PIM1 displayed no changes in HPV E7 expression (Fig. 3). This result was expected, since minimal chimeric HPV-PIM1 fusion transcripts were detected in parental cells (Supp. Fig. S1). These proof-of-principle data support the hypothesis that HPV-mediated genetic rearrangements could alter expression, structure and function of candidate cancer-causing driver genes, and thereby contribute directly to cancer formation independent of viral oncogene expression. Comparable evidence was obtained in HPV-positive cervical neuroendocrine cancer GUMC395 cells, where experimental knockdown of MYC adjacent to sites of HPV integration resulted in decreased cell viability. These effects again were independent of the viral E6 and E7 transcripts [6].
PIM1 overexpression has been identified in HNSCC compared with matched normal controls, and has been associated with poor survival [11, 29, 40]. Here, we identified high levels of expression of at least one member of the PIM kinase family in almost all primary HNSCC (Fig. 6, Supp. Table S6). We attributed this finding in part to genomic copy number gains at these genes, found in ~13% of HNSCC. Overexpression of PIM family kinases also has been reported in numerous hematological malignancies and other solid tumors, including Hodgkin lymphoma [41], multiple myeloma [42], prostate [43], pancreatic [44] and triple negative breast cancers [45]. PIM kinase overexpression may contribute to chemotherapy resistance [46] and cellular survival by blocking apoptotic cell death in the context of oncogene-induced stress [47] and hypoxia [48].
In the case of the UPCI:SCC090 cell line, PIM1 overexpression was attributable to HPV-integrant mediated amplification, but we observed PIM family kinases to be overexpressed in other cell lines. This is likely because PIM1 kinase is downstream of several cellular signaling pathways frequently activated by host genetic alterations in HNSCC, including the PIK3CA/AKT/mTOR, EGFR and NFKB signaling pathways [3, 49]. PIM1 also increases the stability and transcriptional activity of MYC [50], and the PIM kinases and MYC are recognized oncogenic collaborators in cellular transformation [8]. The substrates of PIM kinase phosphorylation in turn regulate several critical cellular processes, including cell cycle progression (e.g. p21, p27), cell growth (e.g. eIF4E, 4EPB2), and cell death (e.g. BAD, BCL2; Fig. 3D) [8]. As a regulator of cell metabolism and growth, PIM1 phosphorylates several substrates in the PIK3CA/AKT/mTOR pathway. PIM1 inactivates negative regulators of this pathway (i.e. TSC2 and AKT1S1) and promotes pathway activation through eIF4B phosphorylation. As shown here, sensitivity to INCB053914 was linked to reduced levels of phosphorylated pRPS6 protein.
In several experimental models of other solid tumors, overexpression of PIM kinases has been implicated in resistance to PIK3CA/AKT/mTOR inhibitors. For example, in breast cancer cells treated with PI3K inhibitors, upregulation of PIM1 serves as a secondary resistance mechanism [51]. Similarly, resistance to PI3K-AKT inhibition in a prostate cancer model is mediated by PIM1 kinase [52]. In this latter model, PIM1 increased translation of the transcription factor NFR2 and downstream regulators of ROS scavengers and metabolism, promoting cell survival [53]. Treatment with a combination of PI3K/AKT and PIM kinase inhibitors overcame this resistance [52].
Several HNSCC cell lines with high PIM expression demonstrated sensitivity to PIM inhibitors. However, we also found that a few cell lines with high levels of PIM1 and PIM3 expression, respectively, were nevertheless resistant to PIM inhibition (i.e. UM-SCC-47 and CAL 27), in agreement with a previous report about PIM in other cancers [46]. Further analysis of factors upstream and downstream of PIM suggested potential mechanisms of resistance. Treatment of CAL 27 cells with INCB053914 induced compensatory STAT3 activation and increased PIM3 expression. By contrast, treatment of UM-SCC-47 cells resulted in increased phosphorylation of EIF4EBP1 and eIF4E proteins, suggesting alternative mechanisms of mTOR pathway activation. Both cell lines harbor EGFR gene amplification [4], raising the possibility that increased signaling through the activated EGFR pathway might promote PIM inhibitor resistance.
Based on these findings, we inferred that frequent, aberrant activation of signaling pathways upstream of PIM1 (including the EGFR and PIK3CA/AKT/mTOR pathways) in HNSCC could result in decreased sensitivity to PIM inhibition (Fig. 3D). Therefore, we tested effects of dual PIM and EGFR inhibition. We found strong supra-additivity in cell lines treated with various combined doses of inhibitors, regardless of their sensitivity to PIM inhibition alone. These data demonstrating supra-additivity in HNSCC are consistent with recently demonstrated synergy between PIM inhibitors and PI3K inhibitors in prostate cancer [54], MET inhibitors in lung cancer[55], mTOR inhibitors in leukemia [56] and Janus kinase (JAK) inhibitors in myeloproliferative neoplasms [57, 58].
We note that multiple mechanisms of resistance to EGFR inhibitors may limit their efficacy in the clinic, including acquisition of secondary EGFR mutations, synchronous activation of redundant receptor tyrosine kinases (e.g. c-Met or G-protein-coupled receptors), and aberrant activation of bypass pathways including the MAPK, RAS and PI3K/AKT pathways which maintain mitogenic signaling [59, 60]. Several proteins in these pathways activate PIM family kinases.
In summary, HPV insertion-mediated amplification of PIM1 in an HNSCC cell line prompted us to investigate that kinase gene’s functional roles in cancer cell viability and growth. The preclinical data shown here strongly suggest that the use of PIM inhibitors in HNSCC could provide a promising new therapeutic approach. In addition, since PIM kinases are involved in several signaling pathways dysregulated in cancer, including the EGFR pathway, the future clinical development of PIM inhibitors could be enhanced by studies of synergistic combinations of therapeutic agents that inhibit multiple targets in such pathways.
Supplementary Material
HIGHLIGHTS.
By studying targets of HPV integration in UPCI:SCC090 HNSCC cells, we identified the PIM family of serine/threonine kinases as targetable cancer driver genes
Expression of PIM1 and/or PIM family members was experimentally decreased using several genetic and pharmacological approaches in this and additional HNSCC cell lines, resulting in cell death in vitro and in xenograft models in vivo.
PIM family kinases were found to be highly expressed in almost all head and neck cancers.
Synergistic cell death occurred when EGFR inhibitors were combined with PIM kinase inhibitors.
These findings provide a strong rationale for clinical trials of combined EGFR and PIM kinase inhibitors for recurrent metastatic HNSCC.
ACKNOWLEDGMENTS
We thank the patients with oropharyngeal and oral cavity cancers at Ohio State University who enrolled in our genomics study. We thank Dr. Holly Koblish at Incyte Corporation; Dr. Faye M. Johnson at MD Anderson Cancer Center; members of Dr. Broutian’s doctoral thesis advisory committee including Drs. Gillison, Quintin Pan, Dawn Chandler and Nyla Heerema; and members of the Gillison and Symer laboratories for insightful comments at various stages of this study; TCGA for access to WGS and RNA-seq data; and Jordan Pietz and Jeff Flasik (MDA) for help preparing graphical figures. We gratefully acknowledge Incyte Corporation for providing the pan-PIM inhibitor INCB053914.
Financial support: This study was funded by the Cancer Prevention Research Institute of Texas (RR170005; MLG), University of Texas MD Anderson Cancer Center (DES, MLG), the Ohio State University Comprehensive Cancer Center (DES, MLG), the Ohio Supercomputer Center (PAS0425; DES), the Oral Cancer Foundation (MLG), and National Cancer Institute grant R50CA211533 (K.A.). Dr. Gillison is a CPRIT Scholar. We acknowledge the Analytical Cytometry Shared Resource and Genomics Shared Resource at OSUCCC, supported by NCI CCSG P30CA016058. We gratefully acknowledge Incyte Corporation for providing the pan-PIM inhibitor INCB053914.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data access: The whole genome sequencing and RNA-seq datasets from the Ohio cohort are archived at the European Genome-phenome Archive (EGA; https://ega-archive.org/) [3].
Conflict of interest disclosure: MLG has been a consultant for Eli Lilly and Company, Merck & Co. Inc, Bristol-Myers Squibb, Bayer, Amgen, AstraZeneca, Celgene Corporation, Genocea, GlaxoSmithKline, Rakuten Medical (Rakuten Aspyrian), Roche, and Aduro Biotech. The authors declare no other potential conflicts of interest.
REFERENCES
- [1].Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A, Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries, CA Cancer J Clin, 68 (2018) 394–424. [DOI] [PubMed] [Google Scholar]
- [2].Chaturvedi AK, Anderson WF, Lortet-Tieulent J, Curado MP, Ferlay J, Franceschi S, Rosenberg PS, Bray F, Gillison ML, Worldwide trends in incidence rates for oral cavity and oropharyngeal cancers, J Clin Oncol, 31 (2013) 4550–4559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gillison ML, Akagi K, Xiao W, Jiang B, Pickard RKL, Li J, Swanson BJ, Agrawal AD, Zucker M, Stache-Crain B, Emde AK, Geiger HM, Robine N, Coombes KR, Symer DE, Human papillomavirus and the landscape of secondary genetic alterations in oral cancers, Genome Res, 29 (2019) 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Akagi K, Li J, Broutian TR, Padilla-Nash H, Xiao W, Jiang B, Rocco JW, Teknos TN, Kumar B, Wangsa D, He D, Ried T, Symer DE, Gillison ML, Genome-wide analysis of HPV integration in human cancers reveals recurrent, focal genomic instability, Genome Res, 24 (2014) 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Adey A, Burton JN, Kitzman JO, Hiatt JB, Lewis AP, Martin BK, Qiu R, Lee C, Shendure J, The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line, Nature, 500 (2013) 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Yuan H, Krawczyk E, Blancato J, Albanese C, Zhou D, Wang N, Paul S, Alkhilaiwi F, Palechor-Ceron N, Dakic A, Fang S, Choudhary S, Hou TW, Zheng YL, Haddad BR, Usuda Y, Hartmann D, Symer D, Gillison M, Agarwal S, Wangsa D, Ried T, Liu X, Schlegel R, HPV positive neuroendocrine cervical cancer cells are dependent on Myc but not E6/E7 viral oncogenes, Sci Rep, 7 (2017) 45617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Durst M, Croce CM, Gissmann L, Schwarz E, Huebner K, Papillomavirus sequences integrate near cellular oncogenes in some cervical carcinomas, Proc Natl Acad Sci U S A, 84 (1987) 1070–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nawijn MC, Alendar A, Berns A, For better or for worse: the role of Pim oncogenes in tumorigenesis, Nat Rev Cancer, 11 (2011) 23–34. [DOI] [PubMed] [Google Scholar]
- [9].Cuypers HT, Selten G, Quint W, Zijlstra M, Maandag ER, Boelens W, van Wezenbeek P, Melief C, Berns A, Murine leukemia virus-induced T-cell lymphomagenesis: integration of proviruses in a distinct chromosomal region, Cell, 37 (1984) 141–150. [DOI] [PubMed] [Google Scholar]
- [10].Breuer ML, Cuypers HT, Berns A, Evidence for the involvement of pim-2, a new common proviral insertion site, in progression of lymphomas, EMBO J, 8 (1989) 743–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Beier UH, Weise JB, Laudien M, Sauerwein H, Gorogh T, Overexpression of Pim-1 in head and neck squamous cell carcinomas, Int J Oncol, 30 (2007) 1381–1387. [PubMed] [Google Scholar]
- [12].Rheinwald JG, Beckett MA, Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas, Cancer Res, 41 (1981) 1657–1663. [PubMed] [Google Scholar]
- [13].Gioanni J, Fischel JL, Lambert JC, Demard F, Mazeau C, Zanghellini E, Ettore F, Formento P, Chauvel P, Lalanne CM, et al. , Two new human tumor cell lines derived from squamous cell carcinomas of the tongue: establishment, characterization and response to cytotoxic treatment, Eur J Cancer Clin Oncol, 24 (1988) 1445–1455. [DOI] [PubMed] [Google Scholar]
- [14].Peterson WD Jr., Stulberg CS, Simpson WF, A permanent heteroploid human cell line with type B glucose-6-phosphate dehydrogenase, Proc Soc Exp Biol Med, 136 (1971) 1187–1191. [DOI] [PubMed] [Google Scholar]
- [15].Cooper T, Biron VL, Fast D, Tam R, Carey T, Shmulevitz M, Seikaly H, Oncolytic activity of reovirus in HPV positive and negative head and neck squamous cell carcinoma, J Otolaryngol Head Neck Surg, 44 (2015) 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].White JS, Weissfeld JL, Ragin CC, Rossie KM, Martin CL, Shuster M, Ishwad CS, Law JC, Myers EN, Johnson JT, Gollin SM, The influence of clinical and demographic risk factors on the establishment of head and neck squamous cell carcinoma cell lines, Oral Oncol, 43 (2007) 701–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lin CJ, Grandis JR, Carey TE, Gollin SM, Whiteside TL, Koch WM, Ferris RL, Lai SY, Head and neck squamous cell carcinoma cell lines: established models and rationale for selection, Head Neck, 29 (2007) 163–188. [DOI] [PubMed] [Google Scholar]
- [18].Koblish H, Li YL, Shin N, Hall L, Wang Q, Wang K, Covington M, Marando C, Bowman K, Boer J, Burke K, Wynn R, Margulis A, Reuther GW, Lambert QT, Dostalik Roman V, Zhang K, Feng H, Xue CB, Diamond S, Hollis G, Yeleswaram S, Yao W, Huber R, Vaddi K, Scherle P, Preclinical characterization of INCB053914, a novel pan-PIM kinase inhibitor, alone and in combination with anticancer agents, in models of hematologic malignancies, PLoS One, 13 (2018) e0199108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Saurabh K, Scherzer MT, Shah PP, Mims AS, Lockwood WW, Kraft AS, Beverly LJ, The PIM family of oncoproteins: small kinases with huge implications in myeloid leukemogenesis and as therapeutic targets, Oncotarget, 5 (2014) 8503–8514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Lin A, Giuliano CJ, Palladino A, John KM, Abramowicz C, Yuan ML, Sausville EL, Lukow DA, Liu L, Chait AR, Galluzzo ZC, Tucker C, Sheltzer JM, Off-target toxicity is a common mechanism of action of cancer drugs undergoing clinical trials, Sci Transl Med, 11 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Macdonald A, Campbell DG, Toth R, McLauchlan H, Hastie CJ, Arthur JS, Pim kinases phosphorylate multiple sites on Bad and promote 14–3-3 binding and dissociation from Bcl-XL, BMC Cell Biol, 7 (2006) 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O, Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis, Genes Dev, 19 (2005) 2199–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Schatz JH, Oricchio E, Wolfe AL, Jiang M, Linkov I, Maragulia J, Shi W, Zhang Z, Rajasekhar VK, Pagano NC, Porco JA Jr., Teruya-Feldstein J, Rosen N, Zelenetz AD, Pelletier J, Wendel HG, Targeting cap-dependent translation blocks converging survival signals by AKT and PIM kinases in lymphoma, J Exp Med, 208 (2011) 1799–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang F, Beharry ZM, Harris TE, Lilly MB, Smith CD, Mahajan S, Kraft AS, PIM1 protein kinase regulates PRAS40 phosphorylation and mTOR activity in FDCP1 cells, Cancer Biol Ther, 8 (2009) 846–853. [DOI] [PubMed] [Google Scholar]
- [25].Wang J, Kim J, Roh M, Franco OE, Hayward SW, Wills ML, Abdulkadir SA, Pim1 kinase synergizes with c-MYC to induce advanced prostate carcinoma, Oncogene, 29 (2010) 2477–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Pierce AC, Jacobs M, Stuver-Moody C, Docking study yields four novel inhibitors of the protooncogene Pim-1 kinase, J Med Chem, 51 (2008) 1972–1975. [DOI] [PubMed] [Google Scholar]
- [27].Holder S, Zemskova M, Zhang C, Tabrizizad M, Bremer R, Neidigh JW, Lilly MB, Characterization of a potent and selective small-molecule inhibitor of the PIM1 kinase, Mol Cancer Ther, 6 (2007) 163–172. [DOI] [PubMed] [Google Scholar]
- [28].Fingar DC, Salama S, Tsou C, Harlow E, Blenis J, Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E, Genes Dev, 16 (2002) 1472–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Peltola K, Hollmen M, Maula SM, Rainio E, Ristamaki R, Luukkaa M, Sandholm J, Sundvall M, Elenius K, Koskinen PJ, Grenman R, Jalkanen S, Pim-1 kinase expression predicts radiation response in squamocellular carcinoma of head and neck and is under the control of epidermal growth factor receptor, Neoplasia, 11 (2009) 629–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sheu JJ, Hua CH, Wan L, Lin YJ, Lai MT, Tseng HC, Jinawath N, Tsai MH, Chang NW, Lin CF, Lin CC, Hsieh LJ, Wang TL, Shih Ie M, Tsai FJ, Functional genomic analysis identified epidermal growth factor receptor activation as the most common genetic event in oral squamous cell carcinoma, Cancer Res, 69 (2009) 2568–2576. [DOI] [PubMed] [Google Scholar]
- [31].Leemans CR, Braakhuis BJ, Brakenhoff RH, The molecular biology of head and neck cancer, Nat Rev Cancer, 11 (2011) 9–22. [DOI] [PubMed] [Google Scholar]
- [32].Munger K, Baldwin A, Edwards KM, Hayakawa H, Nguyen CL, Owens M, Grace M, Huh K, Mechanisms of human papillomavirus-induced oncogenesis, J Virol, 78 (2004) 11451–11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cancer Genome Atlas Network, Comprehensive genomic characterization of head and neck squamous cell carcinomas, Nature, 517 (2015) 576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A, Shefler E, Ramos AH, Stojanov P, Carter SL, Voet D, Cortes ML, Auclair D, Berger MF, Saksena G, Guiducci C, Onofrio RC, Parkin M, Romkes M, Weissfeld JL, Seethala RR, Wang L, Rangel-Escareno C, Fernandez-Lopez JC, Hidalgo-Miranda A, Melendez-Zajgla J, Winckler W, Ardlie K, Gabriel SB, Meyerson M, Lander ES, Getz G, Golub TR, Garraway LA, Grandis JR, The mutational landscape of head and neck squamous cell carcinoma, Science, 333 (2011) 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, Cherniack AD, Ambrogio L, Cibulskis K, Bertelsen B, Romero-Cordoba S, Trevino V, Vazquez-Santillan K, Guadarrama AS, Wright AA, Rosenberg MW, Duke F, Kaplan B, Wang R, Nickerson E, Walline HM, Lawrence MS, Stewart C, Carter SL, McKenna A, Rodriguez-Sanchez IP, Espinosa-Castilla M, Woie K, Bjorge L, Wik E, Halle MK, Hoivik EA, Krakstad C, Gabino NB, Gomez-Macias GS, Valdez-Chapa LD, Garza-Rodriguez ML, Maytorena G, Vazquez J, Rodea C, Cravioto A, Cortes ML, Greulich H, Crum CP, Neuberg DS, Hidalgo-Miranda A, Escareno CR, Akslen LA, Carey TE, Vintermyr OK, Gabriel SB, Barrera-Saldana HA, Melendez-Zajgla J, Getz G, Salvesen HB, Meyerson M, Landscape of genomic alterations in cervical carcinomas, Nature, 506 (2014) 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Koneva LA, Zhang Y, Virani S, Hall PB, McHugh JB, Chepeha DB, Wolf GT, Carey TE, Rozek LS, Sartor MA, HPV Integration in HNSCC Correlates with Survival Outcomes, Immune Response Signatures, and Candidate Drivers, Mol Cancer Res, 16 (2018) 90–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mondello P, Cuzzocrea S, Mian M, Pim kinases in hematological malignancies: where are we now and where are we going?, J Hematol Oncol, 7 (2014) 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Horiuchi D, Camarda R, Zhou AY, Yau C, Momcilovic O, Balakrishnan S, Corella AN, Eyob H, Kessenbrock K, Lawson DA, Marsh LA, Anderton BN, Rohrberg J, Kunder R, Bazarov AV, Yaswen P, McManus MT, Rugo HS, Werb Z, Goga A, PIM1 kinase inhibition as a targeted therapy against triple-negative breast tumors with elevated MYC expression, Nat Med, 22 (2016) 1321–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ferris RL, Martinez I, Sirianni N, Wang J, Lopez-Albaitero A, Gollin SM, Johnson JT, Khan S, Human papillomavirus-16 associated squamous cell carcinoma of the head and neck (SCCHN): a natural disease model provides insights into viral carcinogenesis, Eur J Cancer, 41 (2005) 807–815. [DOI] [PubMed] [Google Scholar]
- [40].Chiang WF, Yen CY, Lin CN, Liaw GA, Chiu CT, Hsia YJ, Liu SY, Up-regulation of a serine-threonine kinase proto-oncogene Pim-1 in oral squamous cell carcinoma, Int J Oral Maxillofac Surg, 35 (2006) 740–745. [DOI] [PubMed] [Google Scholar]
- [41].Szydlowski M, Prochorec-Sobieszek M, Szumera-Cieckiewicz A, Derezinska E, Hoser G, Wasilewska D, Szymanska-Giemza O, Jablonska E, Bialopiotrowicz E, Sewastianik T, Polak A, Czardybon W, Galezowski M, Windak R, Zaucha JM, Warzocha K, Brzozka K, Juszczynski P, Expression of PIM kinases in Reed-Sternberg cells fosters immune privilege and tumor cell survival in Hodgkin lymphoma, Blood, 130 (2017) 1418–1429. [DOI] [PubMed] [Google Scholar]
- [42].Asano J, Nakano A, Oda A, Amou H, Hiasa M, Takeuchi K, Miki H, Nakamura S, Harada T, Fujii S, Kagawa K, Endo I, Yata K, Sakai A, Ozaki S, Matsumoto T, Abe M, The serine/threonine kinase Pim-2 is a novel anti-apoptotic mediator in myeloma cells, Leukemia, 25 (2011) 1182–1188. [DOI] [PubMed] [Google Scholar]
- [43].Jimenez-Garcia MP, Lucena-Cacace A, Robles-Frias MJ, Narlik-Grassow M, Blanco-Aparicio C, Carnero A, The role of PIM1/PIM2 kinases in tumors of the male reproductive system, Sci Rep, 6 (2016) 38079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Xu J, Zhang T, Wang T, You L, Zhao Y, PIM kinases: an overview in tumors and recent advances in pancreatic cancer, Future Oncol, 10 (2014) 865–876. [DOI] [PubMed] [Google Scholar]
- [45].Braso-Maristany F, Filosto S, Catchpole S, Marlow R, Quist J, Francesch-Domenech E, Plumb DA, Zakka L, Gazinska P, Liccardi G, Meier P, Gris-Oliver A, Cheang MC, Perdrix-Rosell A, Shafat M, Noel E, Patel N, McEachern K, Scaltriti M, Castel P, Noor F, Buus R, Mathew S, Watkins J, Serra V, Marra P, Grigoriadis A, Tutt AN, PIM1 kinase regulates cell death, tumor growth and chemotherapy response in triple-negative breast cancer, Nat Med, 22 (2016) 1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Rebello RJ, Huglo AV, Furic L, PIM activity in tumours: A key node of therapy resistance, Adv Biol Regul, 67 (2018) 163–169. [DOI] [PubMed] [Google Scholar]
- [47].Jinesh GG, Mokkapati S, Zhu K, Morales EE, Pim kinase isoforms: devils defending cancer cells from therapeutic and immune attacks, Apoptosis, 21 (2016) 1203–1213. [DOI] [PubMed] [Google Scholar]
- [48].Chen J, Kobayashi M, Darmanin S, Qiao Y, Gully C, Zhao R, Yeung SC, Lee MH, Pim-1 plays a pivotal role in hypoxia-induced chemoresistance, Oncogene, 28 (2009) 2581–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Puram SV, Rocco JW, Molecular Aspects of Head and Neck Cancer Therapy, Hematol Oncol Clin North Am, 29 (2015) 971–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Zhang Y, Wang Z, Li X, Magnuson NS, Pim kinase-dependent inhibition of c-Myc degradation, Oncogene, 27 (2008) 4809–4819. [DOI] [PubMed] [Google Scholar]
- [51].Le X, Antony R, Razavi P, Treacy DJ, Luo F, Ghandi M, Castel P, Scaltriti M, Baselga J, Garraway LA, Systematic Functional Characterization of Resistance to PI3K Inhibition in Breast Cancer, Cancer Discov, 6 (2016) 1134–1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Song JH, Singh N, Luevano LA, Padi SKR, Okumura K, Olive V, Black SM, Warfel NA, Goodrich DW, Kraft AS, Mechanisms Behind Resistance to PI3K Inhibitor Treatment Induced by the PIM Kinase, Mol Cancer Ther, 17 (2018) 2710–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H, Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming, Cancer Cell, 22 (2012) 66–79. [DOI] [PubMed] [Google Scholar]
- [54].Mologni L, Magistroni V, Casuscelli F, Montemartini M, Gambacorti-Passerini C, The Novel PIM1 Inhibitor NMS-P645 Reverses PIM1-Dependent Effects on TMPRSS2/ERG Positive Prostate Cancer Cells And Shows Anti-Proliferative Activity in Combination with PI3K Inhibition, J Cancer, 8 (2017) 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].An N, Xiong Y, LaRue AC, Kraft AS, Cen B, Activation of Pim Kinases Is Sufficient to Promote Resistance to MET Small-Molecule Inhibitors, Cancer Res, 75 (2015) 5318–5328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Harada M, Benito J, Yamamoto S, Kaur S, Arslan D, Ramirez S, Jacamo R, Platanias L, Matsushita H, Fujimura T, Kazuno S, Kojima K, Tabe Y, Konopleva M, The novel combination of dual mTOR inhibitor AZD2014 and pan-PIM inhibitor AZD1208 inhibits growth in acute myeloid leukemia via HSF pathway suppression, Oncotarget, 6 (2015) 37930–37947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Mazzacurati L, Lambert QT, Pradhan A, Griner LN, Huszar D, Reuther GW, The PIM inhibitor AZD1208 synergizes with ruxolitinib to induce apoptosis of ruxolitinib sensitive and resistant JAK2-V617F-driven cells and inhibit colony formation of primary MPN cells, Oncotarget, 6 (2015) 40141–40157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Mazzacurati L, Collins RJ, Pandey G, Lambert-Showers QT, Amin NE, Zhang L, Stubbs MC, Epling-Burnette PK, Koblish HK, Reuther GW, The pan-PIM inhibitor INCB053914 displays potent synergy in combination with ruxolitinib in models of MPN, Blood Adv, 3 (2019) 3503–3514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Liu Q, Yu S, Zhao W, Qin S, Chu Q, Wu K, EGFR-TKIs resistance via EGFRindependent signaling pathways, Mol Cancer, 17 (2018) 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Niederst MJ, Engelman JA, Bypass mechanisms of resistance to receptor tyrosine kinase inhibition in lung cancer, Sci Signal, 6 (2013) re6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.