

Abstract
Arene dearomatization reactions are an important class of synthetic technologies for the rapid assembly of unique chemical architectures. Herein, we report a catalytic protocol to initiate a carboamination/dearomatization cascade that proceeds through transient sulfonamidyl radical intermediates formed from native sulfonamide N–H bonds leading to 1,4-cyclohexadiene-fused sultams. Importantly, this work demonstrates a facile approach to employ two-dimensional aromatic compounds as modular building blocks to generate richly substituted, three-dimensional compounds. These reactions occur at room temperature under visible light irradiation and are catalyzed by the combination of an iridium(III) photocatalyst and a dialkyl phosphate base. Reaction optimization, substrate scope, mechanistic features, and synthetic applications of this transformation are presented.
Subject terms: Photocatalysis, Synthetic chemistry methodology
Arene dearomatization reactions allow chemists to rapidly build unique chemical architectures from largely available starting materials. Here, the authors show a photocatalytic carboamination/dearomatization cascade process leading to 1,4-cyclohexadiene-fused sultams via N-centered radicals.
Introduction
Arenes obtained from inexpensive petrochemical feedstocks are incorporated on industrial scale into pharmaceuticals, agrochemicals, and organic materials. Selective dearomatization reactions1,2 of these flat, two-dimensional aromatic compounds are modular strategies to access substituted, three-dimensional chemical space3, including fused, complex heterocyclic skeletons4,5. In addition, the potential to form stereogenic centers through substituent addition concomitant with the dearomatization process is particularly appealing. Of the selective dearomatization methods reported (including UV-promoted photochemical cycloadditions6,7, oxidative8, enzymatic9–11, transition metal-mediated12–14, and nucleophilic dearomatizations15), the Birch reduction of arenes to 1,4-cyclohexadienes (1,4-CHD) is most well-known relying on liquid ammonia as solvent and pyrophoric alkali metals at cryogenic temperatures (Fig. 1a)16,17. Modifications of the canonical Birch reduction conditions have expanded the scope and synthetic utility of the reaction18–22. Although both electrochemical23 and visible light-mediated24–28 arene dearomatization reactions have been reported with proven utility, they have not been integrated with other reaction pathways via reactive intermediates. A complementary strategy addressing the inherent drawbacks of the classic Birch reduction is to promote an arene dearomatization via a radical cascade sequence. Radical cascades, processes in which multiple chemical bonds are formed in a single operation, are step and atom-economical means to rapidly build complex organic molecules (Fig. 1b)29–31. Moreover, initiating cascade sequences from strong N–H bonds leading to dearomatized molecular frameworks has the potential to impact a variety of synthetic endeavors.
Fig. 1. Inspiration and design for our carboamination/dearomatization cascade reaction platform.
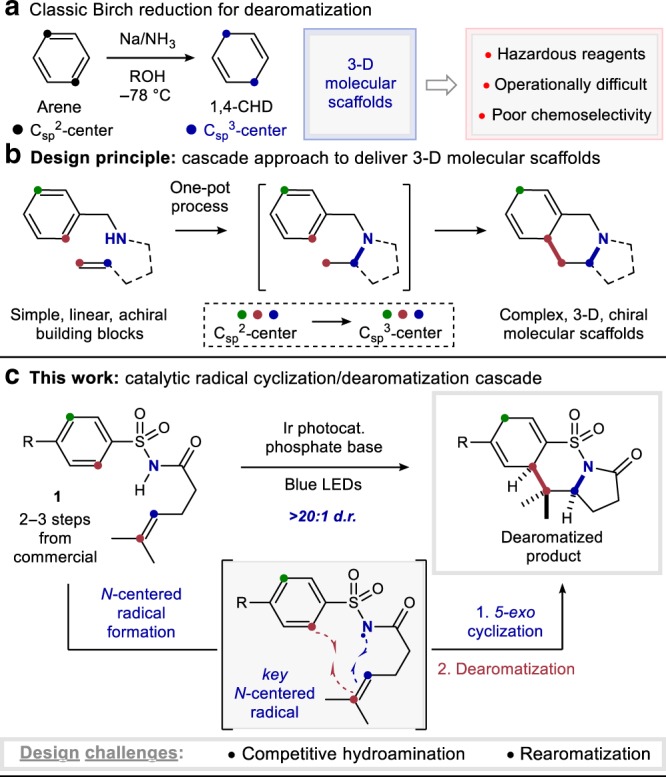
a The classic Birch reduction for arene dearomatization. b The design principle of using a radical cascade approach for arene dearomatization. c This work: catalytic radical carboamination/dearomatization cascade reactions.
Multiple-site concerted proton-electron transfer (MS-CPET) has emerged as a powerful strategy for homolytic activation of strong N–H bonds, often in the presence of weaker ones, avoiding the need for N-pre-functionalization32–34. The Knowles group recently reported the activation of both amide35 and sulfonamide36 N–H bonds for intramolecular and intermolecular hydroamination reactions, respectively, of electron-neutral olefins. The authors propose an excited-state redox catalyst and a weakly coordinating phosphate base, jointly mediate the concerted homolytic activation of the strong N–H bonds under visible light irradiation to afford transient N-centered amidyl and sulfonamidyl radicals capable of adding to olefins with anti-Markovnikov regioselectivity. Separately, Chu and Rovis37 described the generation of amidyl radicals from trifluoroacetamide derivatives for sequential 1,5-HAT and δ-C–H functionalization. By using superstoichiometric K3PO4 as the base, trifluoroacetamides are deprotonated to the amidyl anions then oxidized to the amidyl radical by the excited state of an iridium(III) photocatalyst.
With respect to sulfonamide-based N-radical alkene carboamination platforms, Kinoshita and co-workers38 demonstrated that N-phenylbenzenesulfonamidyl radicals derived from homolysis of a symmetrical sulfonylhydrazide could add to alkenes to give mixtures of products, including sultams. Chemler and co-workers have developed racemic and enantioselective intramolecular Cu(II)-mediated oxidative N-radical cyclizations of alkenyl arylsulfonamides at elevated temperatures (120 °C) to garner benzosultams in good yields39–41. Later, Kanai and co-workers42 reported an intermolecular alkene carboamination reaction of aliphatic alkenes under Cu(I) catalysis and N-fluorobenzenesulfonimide (NFSI) as both a bifunctional reagent, for C–C and C–N bond formation, and an oxidant for the concise synthesis of six-membered sultams. Zhao et al.43 have disclosed a radical cascade cyclization/allylation of β,γ-unsaturated N-aryl hydrazones that gives dihydropyrazoles and tetrahydropyridazines. Very recently, Zhao et al.44 have also described a process for a radical 5-exo cyclization/addition/aromatization cascade of β,γ-unsaturated N-tosyl hydrazones under cooperative photocatalysis and cobalt catalysis enabling the synthesis of dihydropyrazole-fused benzosultams.
We reasoned that the work outlined above, along with our ongoing interests in alkene carboamination reactions with bifunctional arylsulfonamide reagents45, would serve as a basis for developing a catalytic protocol to initiate a carboamination/dearomatization cascade starting from γ,δ-unsaturated N-arylsulfonyl enamides 1 (Fig. 1c). Ideally, this process would proceed through a transient sulfonamidyl radical intermediate originating from formal removal of the N–H hydrogen atom. This strategy obviates the need for N-pre-functionalization, elevated reaction temperatures, and stoichiometric oxidants. The reaction would lead to the selective synthesis of 1,4-cyclohexadiene-fused sultams. Of note, fused sultams have been shown to exhibit a broad range of biological activities46–48.
At the outset, we recognized two major challenges in implementing this design strategy. The first was the potential for competitive hydroamination, wherein the vicinal carbon-centered radical that is formed following N-radical cyclization would undergo premature hydrogen atom transfer or reduction to give the hydroamination product33,35,36,49–54. The second challenge was rearomatization of the cyclohexadienyl radical intermediate. To circumvent these challenges, it was necessary to identify an efficient photocatalytic system capable of selectively reducing the cyclohexadienyl radical to the cyclohexadienyl anion and reestablish the ground state of the photocatalyst. We believed such challenges could be surmounted with judicious choice of reaction additives and parameters. Herein, we describe the combination of the aforementioned reaction classes, whereby visible light-mediated photoredox catalysis is used to promote and control a sulfonamidyl radical cyclization/dearomatization cascade reaction.
Results
Reaction optimization
Initially, we chose arylsulfonamide 1a as the model substrate to test the feasibility of the visible light-induced carboamination/dearomatization cascade reaction and representative results are summarized in Table 1. The expected reaction occured with 1 mol% Ir-photocatalyst A ([Ir(dF(CF3ppy)2)(5,5′-CF3-bpy)]PF6, IrIII*/II = 1.68 V versus SCE in MeCN) and 65 mol% of tetrabutylammonium dibutylphosphate base providing the desired 1,4-cyclohexadiene-fused sultam 2a in 48% 19F-NMR yield and excellent diastereoselectivity (>20:1) following irradiation with blue LEDs in trifluorotoluene (0.2 M) at room temperature (entry 1). Dearomatized product 2a was unambiguously characterized by single-crystal X-ray analysis. Other iridium photocatalysts structurally similar to A (B = [Ir(dF(CF3ppy)2)(dtbbpy)]PF6, IrIII*/II = 1.21 V versus SCE in MeCN; C = [Ir(dF(Meppy)2)(dtbbpy)]PF6, IrIII*/II = 0.97 V versus SCE in MeCN)55,56 were also effective in these reactions (entries 2, 3), however the reaction yields diminished as the oxidation potential of the excited-state species decreased. Importantly, decreasing the reaction concentration (0.05 M) delivered the desired product in 67% yield (entry 4). Also, the reaction is moderately to equally successful in other solvents (entry 5-7) with tert-butanol providing a noticeable increase in yield (entry 8). A 1:1 mixed solvent system of trifluorotoluene/tert-butanol (0.05 M) was finally identified as the optimal solvent combination (entry 9). When the reactions were run using conditions developed by Rovis, relying on step-wise deprotonation/oxidation37, no product was isolated (See Supplementary Tables 1 and 2 for complete optimization details). Control reactions of 1a lacking photocatalyst, base, or light were uniformly unsuccessful, and the starting material was recovered unchanged (entries 10–12).
Table 1.
Reaction optimization and control experiments.
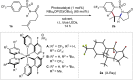 | |||
|---|---|---|---|
| Entry | Photocatalyst | Solvent | Yield (%) |
| 1 | A | PhCF3 (0.2 M) | 48 |
| 2 | B | PhCF3 (0.2 M) | 32 |
| 3 | C | PhCF3 (0.2 M) | 7 |
| 4 | A | PhCF3 (0.05 M) | 67 |
| 5 | A | DMF (0.05 M) | 50 |
| 6 | A | CH2Cl2 (0.05 M) | 47 |
| 7 | A | 1,2-DCE (0.05 M) | 65 |
| 8 | A | t-BuOH (0.05 M) | 73 |
| 9 | A | PhCF3/t-BuOH (0.05 M) | 83 (75)a |
| Variation from best conditions (Entry 9) | ||
|---|---|---|
| 10 | No blue LEDs | 0 |
| 11 | No photocatalyst | 0 |
| 12 | No NBu4OP(O)(OBu)2 | 0 |
Substrate scope
With optimized conditions established, we investigated the generality of this carboamination/dearomatization cascade reaction with a series of diversely substituted arylsulfonamides (Fig. 2). For example, arenes bearing various electron-withdrawing (2a, 2b, 2f–2i), electron-neutral (2c–2e), and electron-donating (2j) groups delivered the desired dearomatized products in moderate to good yields (12–80%) and with excellent diastereoselectivity (>20:1). Unsurprisingly, the latter electron-rich product 2j was prone to rapid oxidative decomposition leading to diminished yields. Dearomatized product 2h was isolated as an inseparable mixture with the aromatized benzosultam product. Next, we investigated modifications to the alkene tether which allowed for the synthesis of an all-carbon spirocycle (2k), fused tetracycle (2l), and cyclic carbamate (2m) in moderate to good yields (21–77%). Interestingly, 3-fluoro and 3,4-difluoro substituted arylsulfonamide substrates generated single dearomatized regioisomers in good yields (2n, 2o) highlighting the regio- and chemoselective nature of the C–C bond forming cyclization step. Furthermore, this methodology proved tolerant of diverse functionality including electron-rich heterocycles (2p), olefins (2q), amino acids (2r), aliphatic carbocycles (2s), and benzyl groups (2t) selectively furnishing the corresponding dearomatized products in good yields. Lastly, a tetrasubstituted olefin substrate (1u) allowed direct access to dearomatized tert-alkylamine57 product 2u, suggesting the initial 5-exo N-radical cyclization is tolerant to steric encumbrance. Terminal olefin 3 failed to convert to the desired dearomatized product (Fig. 2c) likely due to the instability of the resultant vicinal primary radical following C–N bond formation. To probe the importance of the carbonyl moiety and to determine whether it is a necessary functionality for the reaction to proceed, we prepared and subjected o-prenyl aniline derivative 4 and alkyl sulfonamide 5 to the optimized reaction conditions and found they were unreactive, returning the starting material. These experiments highlight the potential importance of the carbonyl moiety for this process to occur by lowering the pKa of the substrate (pKa for 1 ≈ 5)58.
Fig. 2. Reaction scope.

All yields are isolated yields. Relative configurations of products were assigned by analogy to 2a. a General reaction scheme; all reactions were run on 0.2 mmol scale and degassed by sparging with argon for 15 min prior to exposure to optimized conditions. b Arylsulfonamide modifications. c Control experiments with differentially substituted alkene side-chains. *Product isolated as an inseparable mixture of diene and arene products (3:1, diene:arene).
Synthetic applications
The richly functionalized 1,4-cyclohexadiene-fused sultams accessible with this methodology can be readily diversified, leveraging the functional groups tolerated by the dearomative cyclization (Fig. 3). The N-sulfonyl lactam carbonyl is susceptible to nucleophilic addition and subsequent displacement of the sulfonamide leaving group. Thus, 2d is rapidly converted to bicyclic methyl ester 3d at room temperature in excellent yield when treated with excess K2CO3 in methanol. Reaction of 2d with lithium aluminum hydride in THF directly provides the bicyclic alcohol 4d. Of note, 3d and 4d are the products of the formal intermolecular dearomative reaction between a primary sulfonamide N-centered radical and a γ,δ-unsaturated ester or a bis-homoallylic alcohol, respectively. Palladium-catalyzed Suzuki–Miyaura cross-coupling of 2d with N-methylindole-5-boronic acid under unoptimized anhydrous conditions gave a moderate yield of alkenyl indole 5d. Under similar conditions used to prepare 3d, tricyclic sultam 2s containing alkenyl ester functionality converts in good yield to the γ-sulfamoyl enone 3s as a single diastereomer as revelaed by X-ray crystallographic analysis. Due to the resonance stabilizing effects of the enone on the resulting anion, 3s is deprotonated alpha to the sulfonamide by K2CO3 to give a dienolate that is alkylated by benzyl bromide alpha to the carbonyl. Enone 4s is thus isolated following re-conjugation of the remaining alkene with the ketone. N-benzylated products were not observed.
Fig. 3. Synthetic modifications of fused-sultam products.
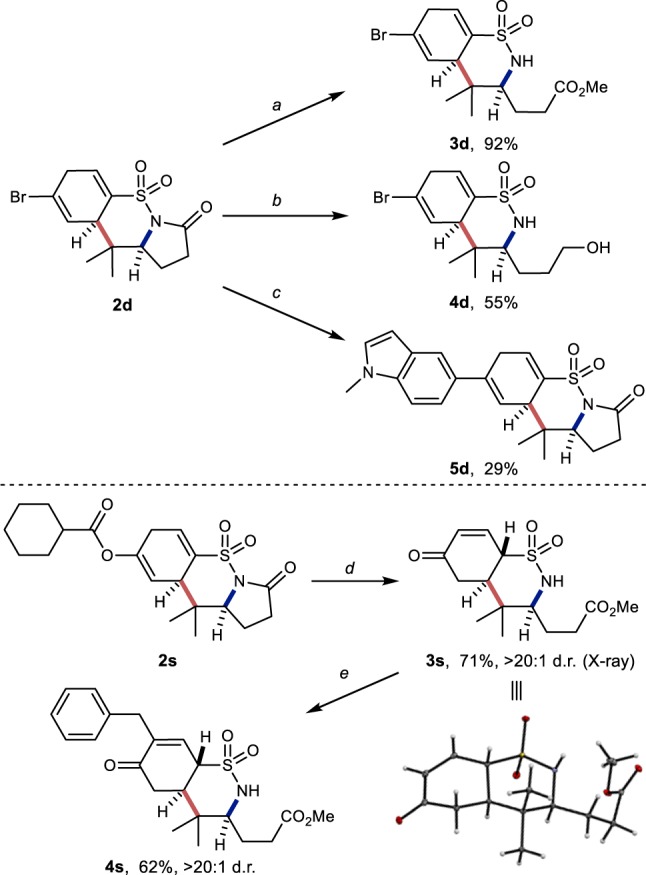
All yields are isolated yields. Conditions: a Sat. aq. K2CO3, MeOH, rt, 15 min. b LiAlH4, THF, −78 °C to room temp., 1.5 h. c 1-(methylindol-5-yl)boronic acid, CsF, PPh3, Pd(OAc)2, THF, reflux, 14 hr. d K2CO3, MeOH, rt, 15 min. e K2CO3, benzyl bromide, DMF, rt, 2 h.
Mechanistic investigations
To gain insight into the reaction mechanism, we performed Stern–Volmer luminescence quenching studies with the model substrate 1a (Supplementary Figs. 73–76). It was revealed that there is no luminescence quenching of the photoexcited state of catalyst A when 1a is used alone, suggesting that direct oxidation, followed by deprotonation, of the substrate is unlikely. This conclusion was further supported by cyclic voltammetry analysis (CH2Cl2 containing 0.1 M NBu4PF6) indicating that direct oxidation of substrate 1a occurs at potentials >1.6 V (versus SCE) (Supplementary Fig. 77). Comparatively, when tetrabutylammonium dibutylphosphate base is used alone in the Stern–Volmer analysis, significant nonlinear photoluminescence quenching of the photoexcited state of catalyst A is observed. This observation aligns with the reports of Knowles59, and others60, suggesting the formation of a less emissive iridium-phosphate complex which may be the active ground-state catalytic species in solution. Importantly, no additional photoluminescence quenching is observed upon addition of substrate 1a as one would expect for a MS-CPET process. Cyclic voltammetry studies of 1a in the presence of varying concentrations of monobasic dibutylphosphate base revealed that current response increased for increasing concentrations of phosphate base but did not show a shifted, less positive potential for the oxidation of substrate 1a (Supplementary Fig. 77). With no evidence for a MS-CPET process, we next investigated whether sequential sulfonamide deprotonation and one-electron oxidation was the operative mechanism for N-centered radical generation. Deprotonation of 1g with sodium hydride followed by salt metathesis gave the sulfonamidyl anion product [NBu4][1g]. When this compound was exposed to the typical reaction conditions in the absence of phosphate, product 2g was not detected and only starting material was observed by NMR analysis of the crude reaction (Fig. 4a). This result suggests that single-electron oxidation of a sulfonamidyl anion is not the dominant route to the nitrogen-centered radical. Qualitatively, these results together are consistent with a phosphate radical being generated under the reaction conditions and serving as a hydrogen-atom abstracting species and is at least partly responsible for the generation of the nitrogen radical61,62. HAT between heteroatoms is known to occur rapidly, even in the presence of weaker C–H bonds63. Next, a deuterium labeling experiment was conducted to probe the origin of the additional proton on the carbocycle in the products. By performing the reaction in the presence of tert-BuOD, the desired product (2a-D) was produced in 51% yield with complete deuterium incorporation (Fig. 4b). This result suggests that the tert-BuOH is serving as a reaction-terminating proton source of a 1,4-cyclohexadienyl anion intermediate in accord with previous mechanistic studies of the classic Birch reduction reaction64,65. Interestingly, the mechanism for sulfonamidyl radical formation presented herein differs from the proposal by Knowles and co-workers36. This observed divergence may be due to differences in acidity between the primary benzenesulfonamides used by Knowles and the N-carbonyl benzenesulfonamides used in this study.
Fig. 4. Experiment to probe mechanism and proposed catalytic cycle.
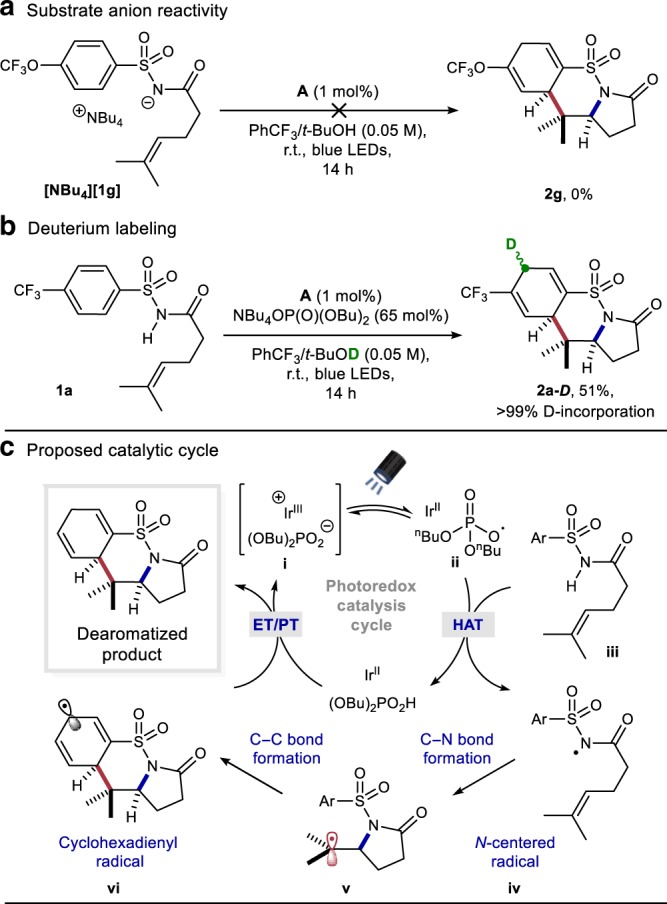
a Deprotonated substrate is unreactive under reaction conditions. b Deuterium labeling experiment. c Proposed catalytic cycle.
Proposed catalytic cycle
A catalytic cycle accounting for these observations, and those made during reaction optimization, is shown in Fig. 4c. We suspect that the Ir-photocatalyst A and phosphate base first form a ground-state iridium-phosphate complex (i). Upon excitation, the Ir-photocatalyst can undergo single-electron transfer from the phosphate salt, generating an oxygen-centered radical (ii). Abstraction of the most activated H-atom of the substrate (iii) generates a transient sulfonamidyl radical intermediate (iv). Next, the N-radical can undergo 5-exo cyclization onto the tethered alkene to furnish a new C–N bond and a vicinal carbon-centered radical (v). This alkyl radical is poised to undergo cyclization with the appended arene to generate a stabilized cyclohexadienyl radical (vi) that can undergo single-electron reduction by the reduced Ir(II) state of the photocatalyst (E1/2[IrIII/IrII]= −1.07 V versus Fc+/Fc)36. Favorable proton transfer between the cyclohexadienyl anion and tert-BuOH (tert-butoxide can then return dibutyl phosphoric acid66 back to the phosphate salt) should deliver the dearomatized product and regenerate both catalysts.
Discussion
In summary, we have reported the discovery and development of a photoredox-mediated N-centered radical strategy to facilitate a carboamination/dearomatization cascade reaction. Simple γ,δ-unsaturated N-arylsulfonyl enamides were converted into complex and stereodefined 1,4-cyclohexadiene-fused sultams in satisfactory yields and excellent diastereoselectivity. This mild and efficient catalytic system demonstrates a broad substrate scope and high functional group tolerance while avoiding premature hydroamination or undesired rearomatization reactivity. Overall, we believe the photochemical strategy outlined here will inspire future synthetic endeavors aimed at employing simple arene building blocks for the rapid synthesis of complex, three-dimensional molecular frameworks in a single operation.
Methods
Representative procedure
To a dry 2-dram vial was added γ,δ-unsaturated N-arylsulfonyl enamide 1 (0.2 mmol), base, (65 mol%), and photocatalyst (1 mol%). The vial contents were then dissolved in a 1:1 mixture of t-BuOH:PhCF3 (0.05 M). The solution was degassed by sparging with argon for 15 min. The reaction was irradiated with two, H150 blue Kessil lamps positioned ~2 cm away and cooled with an overhead fan for 14 h. The reaction was concentrated in vacuo, loaded onto silica, and purified by flash column chromatography to afford the desired 1,4-cyclohexadiene-fused sultam 2. No unexpected or unusually high safety hazards were encountered.
Supplementary information
Acknowledgements
We thank Dr. Jeff W. Kampf for assistance with X-ray crystallographic analysis. We acknowledge the financial support for this research from the NIH-NIGMS (GM127774) and the University of Michigan. This material is based upon work supported by the National Science Foundation Graduate Research Fellowship for RCM (Grant No. DGE 1256260).
Author contributions
R.C.M. and E.A.N. performed the experiments; R.C.M., E.A.N., and C.R.J.S. designed the experiments; R.C.M., E.A.N., and C.R.J.S. prepared the manuscript for publication.
Data availability
Experimental data as well as 1H and 13C NMR spectra for all new compounds prepared in the course of these studies are provided in the Supplementary Information of this manuscript. The solid-state X-ray crystal structures of 2a and 3s are available free of charge from the Cambridge Crystallographic Data Centre under reference numbers CCDC 1952459 and CCDC 1984122, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All other data including synthetic procedures are available in the Supplementary Information file.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information is available for this paper at 10.1038/s41467-020-16369-4.
References
- 1.Wertjes WC, Southgate EH, Sarlah D. Recent advances in chemical dearomatization of nonactivated arenes. Chem. Soc. Rev. 2018;47:7996–8017. doi: 10.1039/c8cs00389k. [DOI] [PubMed] [Google Scholar]
- 2.Zhuo C-X, Zhang W, You S-L. Catalytic asymmetric dearomatization reactions. Angew. Chem. Int. Ed. 2012;51:12662–12686. doi: 10.1002/anie.201204822. [DOI] [PubMed] [Google Scholar]
- 3.Lovering F, Bikker J, Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009;52:6752–6756. doi: 10.1021/jm901241e. [DOI] [PubMed] [Google Scholar]
- 4.Roche, S. P. & Porco, J. A. Jr. Dearomatization strategies in the synthesis of complex natural products. Angew. Chem. Int. Ed.50, 4068–4093. [DOI] [PMC free article] [PubMed]
- 5.Przydacz A, Skrzyńska A, Albrecht Ł. Breaking aromaticity with aminocatalysis: a convenient strategy for asymmetric synthesis. Angew. Chem. Int. Ed. 2019;58:63–73. doi: 10.1002/anie.201808197. [DOI] [PubMed] [Google Scholar]
- 6.Wagner PJ, Sakamoto M. Intramolecular triplet state cyclization of but-3-enoxyacetonaphthones. J. Am. Chem. Soc. 1989;111:9254–9256. [Google Scholar]
- 7.Wagner PJ. Photoinduced ortho [2 + 2] cycloaddition of double bonds to triplet benzenes. Acc. Chem. Res. 2001;34:1–8. doi: 10.1021/ar000113n. [DOI] [PubMed] [Google Scholar]
- 8.Pouységu L, Deffieux D, Quideau S. Hypervalent iodine-mediated phenol dearomatization in natural product synthesis. Tetrahedron. 2010;66:2235–2261. [Google Scholar]
- 9.Hudlicky T, Reed JW. Celebrating 20 years of SYNLETT—special account on the merits of biocatalysis and the impact of arene cis-dihydrodiols on enantioselective synthesis. Synlett. 2009;2009:685–703. [Google Scholar]
- 10.Boyd DR, Bugg TDH. Arene cis-dihydrodiol formation: from biology to application. Org. Biomolecular Chem. 2006;4:181–192. doi: 10.1039/b513226f. [DOI] [PubMed] [Google Scholar]
- 11.Boll M. Dearomatizing benzene ring reductases. J. Mol. Microbiol. Biotechnol. 2005;10:132–142. doi: 10.1159/000091560. [DOI] [PubMed] [Google Scholar]
- 12.Pape AR, Kaliappan KP, Kündig EP. Transition-metal-mediated dearomatization reactions. Chem. Rev. 2000;100:2917–2940. doi: 10.1021/cr9902852. [DOI] [PubMed] [Google Scholar]
- 13.Giustra ZX, Ishibashi JSA, Liu S-Y. Homogeneous metal catalysis for conversion between aromatic and saturated compounds. Coord. Chem. Rev. 2016;314:134–181. [Google Scholar]
- 14.Zheng C, You S-L. Catalytic asymmetric dearomatization by transition-metal catalysis: a method for transformations of aromatic compounds. Chem. 2016;1:830–857. [Google Scholar]
- 15.López Ortiz F, Iglesias MJ, Fernández I, Andújar Sánchez CM, Ruiz Gómez G. Nucleophilic dearomatizing (DNAr) reactions of aromatic C,H-systems. A mature paradigm in organic synthesis. Chem. Rev. 2007;107:1580–1691. doi: 10.1021/cr030207l. [DOI] [PubMed] [Google Scholar]
- 16.Birch AJ. The reduction of organic compounds by metal-ammonia solutions. Q. Rev. Chem. Soc. 1950;4:69–93. [Google Scholar]
- 17.Holy NL. Reactions of the radical anions and dianions of aromatic hydrocarbons. Chem. Rev. 1974;74:243–277. [Google Scholar]
- 18.Dye JL, et al. Alkali metals plus silica gel: powerful reducing agents and convenient hydrogen sources. J. Am. Chem. Soc. 2005;127:9338–9339. doi: 10.1021/ja051786+. [DOI] [PubMed] [Google Scholar]
- 19.Lei P, et al. A practical and chemoselective ammonia-free Birch reduction. Org. Lett. 2018;20:3439–3442. doi: 10.1021/acs.orglett.8b00891. [DOI] [PubMed] [Google Scholar]
- 20.Donohoe TJ, Thomas RE. The partial reduction of electron-deficient pyrroles: procedures describing both Birch (Li/NH3) and ammonia-free (Li/DBB) conditions. Nat. Protoc. 2007;2:1888–1895. doi: 10.1038/nprot.2007.245. [DOI] [PubMed] [Google Scholar]
- 21.Yoo BI, Kim YJ, You Y, Yang JW, Kim SW. Birch reduction of aromatic compounds by inorganic electride [Ca2N]+·e– in an alcoholic solvent: an analogue of solvated electrons. J. Org. Chem. 2018;83:13847–13853. doi: 10.1021/acs.joc.8b02094. [DOI] [PubMed] [Google Scholar]
- 22.Szostak M, Spain M, Procter DJ. Determination of the effective redox potentials of SmI2, SmBr2, SmCl2, and their complexes with water by reduction of aromatic hydrocarbons. Reduction of anthracene and stilbene by samarium(II) iodide–water complex. J. Org. Chem. 2014;79:2522–2537. doi: 10.1021/jo4028243. [DOI] [PubMed] [Google Scholar]
- 23.Peters BK, et al. Scalable and safe synthetic organic electroreduction inspired by Li-ion battery chemistry. Science. 2019;363:838. doi: 10.1126/science.aav5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Southgate EH, Pospech J, Fu J, Holycross DR, Sarlah D. Dearomative dihydroxylation with arenophiles. Nat. Chem. 2016;8:922–928. doi: 10.1038/nchem.2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Southgate EH, Holycross DR, Sarlah D. Total synthesis of lycoricidine and narciclasine by chemical dearomatization of bromobenzene. Angew. Chem. Int. Ed. 2017;56:15049–15052. doi: 10.1002/anie.201709712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hernandez LW, Pospech J, Klöckner U, Bingham TW, Sarlah D. Synthesis of (+)-pancratistatins via catalytic desymmetrization of benzene. J. Am. Chem. Soc. 2017;139:15656–15659. doi: 10.1021/jacs.7b10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Okumura M, Shved AS, Sarlah D. Palladium-catalyzed dearomative syn-1,4-carboamination. J. Am. Chem. Soc. 2017;139:17787–17790. doi: 10.1021/jacs.7b11663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.James MJ, Schwarz JL, Strieth-Kalthoff F, Wibbeling B, Glorius F. Dearomative cascade photocatalysis: divergent synthesis through catalyst selective energy transfer. J. Am. Chem. Soc. 2018;140:8624–8628. doi: 10.1021/jacs.8b03302. [DOI] [PubMed] [Google Scholar]
- 29.Sebren LJ, Devery JJ, Stephenson CRJ. Catalytic radical domino reactions in organic synthesis. ACS Catal. 2014;4:703–716. doi: 10.1021/cs400995r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skubi KL, Blum TR, Yoon TP. Dual catalysis strategies in photochemical synthesis. Chem. Rev. 2016;116:10035–10074. doi: 10.1021/acs.chemrev.6b00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Plesniak MP, Huang H-M, Procter DJ. Radical cascade reactions triggered by single electron transfer. Nat. Rev. Chem. 2017;1:0077. [Google Scholar]
- 32.Yayla HG, Knowles RR. Proton-coupled electron transfer in organic synthesis: novel homolytic bond activations and catalytic asymmetric reactions with free radicals. Synlett. 2014;25:2819–2826. [Google Scholar]
- 33.Miller DC, Tarantino KT, Knowles RR. Proton-coupled electron transfer in organic synthesis: fundamentals, applications, and opportunities. Top. Curr. Chem. 2016;374:30. doi: 10.1007/s41061-016-0030-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gentry EC, Knowles RR. Synthetic applications of proton-coupled electron transfer. Acc. Chem. Res. 2016;49:1546–1556. doi: 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR. Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature. 2016;539:268–271. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Q, Graff DE, Knowles RR. Intermolecular anti-Markovnikov hydroamination of unactivated alkenes with sulfonamides enabled by proton-coupled electron transfer. J. Am. Chem. Soc. 2018;140:741–747. doi: 10.1021/jacs.7b11144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu JCK, Rovis T. Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature. 2016;539:272–275. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miura Y, Ohnishi T, Kinoshita M, Kinoshita T. Addition of a sulfonamidyl radical to unsaturated aromatic and aliphatic hydrocarbons. Bull. Chem. Soc. Jpn. 1990;63:57–63. [Google Scholar]
- 39.Sherman ES, Chemler SR, Tan TB, Gerlits O. Copper(II) acetate promoted oxidative cyclization of arylsulfonyl-o-allylanilines. Org. Lett. 2004;6:1573–1575. doi: 10.1021/ol049702+. [DOI] [PubMed] [Google Scholar]
- 40.Zeng W, Chemler SR. Copper(II)-catalyzed enantioselective intramolecular carboamination of alkenes. J. Am. Chem. Soc. 2007;129:12948–12949. doi: 10.1021/ja0762240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chemler SR. The enantioselective intramolecular aminative functionalization of unactivated alkenes, dienes, allenes and alkynes for the synthesis of chiral nitrogen heterocycles. Org. Biomol. Chem. 2009;7:3009–3019. doi: 10.1039/B907743J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaneko K, Yoshino T, Matsunaga S, Kanai M. Sultam synthesis via Cu-catalyzed intermolecular carboamination of alkenes with N-fluorobenzenesulfonimide. Org. Lett. 2013;15:2502–2505. doi: 10.1021/ol4009848. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Q-Q, Chen J, Yan D-M, Chen J-R, Xiao W-J. Photocatalytic hydrazonyl radical-mediated radical cyclization/allylation cascade: synthesis of dihydropyrazoles and tetrahydropyridazines. Org. Lett. 2017;19:3620–3623. doi: 10.1021/acs.orglett.7b01609. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Q-Q, Hu X-Q, Yang M-N, Chen J-R, Xiao W-J. A visible-light photocatalytic N-radical cascade of hydrazones for the synthesis of dihydropyrazole-fused benzosultams. Chem. Commun. 2016;52:12749–12752. doi: 10.1039/c6cc05897c. [DOI] [PubMed] [Google Scholar]
- 45.Monos TM, McAtee RC, Stephenson CRJ. Arylsulfonylacetamides as bifunctional reagents for alkene aminoarylation. Science. 2018;361:1369–1373. doi: 10.1126/science.aat2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wells GJ, Tao M, Josef KA, Bihovsky R. 1,2-Benzothiazine 1,1-dioxide P2–P3 peptide mimetic aldehyde calpain I inhibitors. J. Medicinal Chem. 2001;44:3488–3503. doi: 10.1021/jm010178b. [DOI] [PubMed] [Google Scholar]
- 47.Xie Y, et al. Convenient preparation of N-8-quinolinyl benzenesultams as novel NF-κB inhibitors. Tetrahedron Lett. 2008;49:2320–2323. [Google Scholar]
- 48.Silver LH. Clinical efficacy and safety of brinzolamide (Azopt™), a new topical carbonic anhydrase inhibitor for primary open-angle glaucoma and ocular hypertension. Am. J. Ophthalmol. 1998;126:400–408. doi: 10.1016/s0002-9394(98)00095-6. [DOI] [PubMed] [Google Scholar]
- 49.Musacchio AJ, et al. Catalytic intermolecular hydroaminations of unactivated olefins with secondary alkyl amines. Science. 2017;355:727–730. doi: 10.1126/science.aal3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miller DC, Choi GJ, Orbe HS, Knowles RR. Catalytic olefin hydroamidation enabled by proton-coupled electron transfer. J. Am. Chem. Soc. 2015;137:13492–13495. doi: 10.1021/jacs.5b09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nguyen TM, Manohar N, Nicewicz DA. Anti-Markovnikov hydroamination of alkenes catalyzed by a two-component organic photoredox system: direct access to phenethylamine derivatives. Angew. Chem. Int. Ed. 2014;53:6198–6201. doi: 10.1002/anie.201402443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morse PD, Nicewicz DA. Divergent regioselectivity in photoredox-catalyzed hydrofunctionalization reactions of unsaturated amides and thioamides. Chem. Sci. 2015;6:270–274. doi: 10.1039/c4sc02331e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Guo H-M, Zhao Q-Q, Chen J-R, Xiao W-J. Visible light-driven photocatalytic generation of sulfonamidyl radicals for alkene hydroamination of unsaturated sulfonamides. Chem. Commun. 2018;54:6780–6783. doi: 10.1039/c7cc09871e. [DOI] [PubMed] [Google Scholar]
- 54.Lardy SW, Schmidt VA. Intermolecular radical mediated anti-markovnikov alkene hydroamination using N-hydroxyphthalimide. J. Am. Chem. Soc. 2018;140:12318–12322. doi: 10.1021/jacs.8b06881. [DOI] [PubMed] [Google Scholar]
- 55.Prier CK, Rankic DA, MacMillan DWC. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013;113:5322–5363. doi: 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Teegardin K, Day JI, Chan J, Weaver J. Advances in photocatalysis: a microreview of visible light mediated ruthenium and iridium catalyzed organic transformations. Org. Process Res. Dev. 2016;20:1156–1163. doi: 10.1021/acs.oprd.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trowbridge A, Reich D, Gaunt MJ. Multicomponent synthesis of tertiary alkylamines by photocatalytic olefin-hydroaminoalkylation. Nature. 2018;561:522–527. doi: 10.1038/s41586-018-0537-9. [DOI] [PubMed] [Google Scholar]
- 58.Lassalas P, et al. Structure property relationships of carboxylic acid isosteres. J. Med. Chem. 2016;59:3183–3203. doi: 10.1021/acs.jmedchem.5b01963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Morton CM, et al. C–H alkylation via multisite-proton-coupled electron transfer of an aliphatic C–H bond. J. Am. Chem. Soc. 2019;141:13253–13260. doi: 10.1021/jacs.9b06834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ardo S, Sun Y, Castellano FN, Meyer GJ. Excited-state electron transfer from ruthenium-polypyridyl compounds to anatase TiO2 nanocrystallites: evidence for a stark effect. J. Phys. Chem. B. 2010;114:14596–14604. doi: 10.1021/jp102349m. [DOI] [PubMed] [Google Scholar]
- 61.Margrey KA, Czaplyski WL, Nicewicz DA, Alexanian EJ. A general strategy for aliphatic C–H functionalization enabled by organic photoredox catalysis. J. Am. Chem. Soc. 2018;140:4213–4217. doi: 10.1021/jacs.8b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wakaki T, et al. C(sp3)–H cyanation promoted by visible-light photoredox/phosphate hybrid catalysis. Chem. A Eur. J. 2018;24:8051–8055. doi: 10.1002/chem.201801746. [DOI] [PubMed] [Google Scholar]
- 63.Mayer JM. Understanding hydrogen atom transfer: from bond strengths to Marcus theory. Acc. Chem. Res. 2011;44:36–46. doi: 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zimmerman HE, Wang PA. Regioselectivity of the Birch reduction. J. Am. Chem. Soc. 1990;112:1280–1281. [Google Scholar]
- 65.Zimmerman HE, Wang PA. The regioselectivity of the Birch reduction. J. Am. Chem. Soc. 1993;115:2205–2216. [Google Scholar]
- 66.Rueping M, Nachtsheim BJ, Ieawsuwan W, Atodiresei I. Modulating the acidity: highly acidic brønsted acids in asymmetric catalysis. Angew. Chem. Int. Ed. 2011;50:6706–6720. doi: 10.1002/anie.201100169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Experimental data as well as 1H and 13C NMR spectra for all new compounds prepared in the course of these studies are provided in the Supplementary Information of this manuscript. The solid-state X-ray crystal structures of 2a and 3s are available free of charge from the Cambridge Crystallographic Data Centre under reference numbers CCDC 1952459 and CCDC 1984122, respectively. These data can be obtained free of charge from The Cambridge Crystallographic Data Center via www.ccdc.cam.ac.uk/data_request/cif. All other data including synthetic procedures are available in the Supplementary Information file.