

Abstract
Loss of biodiversity from lower to upper trophic levels reduces overall productivity and stability of coastal ecosystems in our oceans, but rarely are these changes documented across both time and space. The characterisation of environmental DNA (eDNA) from sediment and seawater using metabarcoding offers a powerful molecular lens to observe marine biota and provides a series of ‘snapshots’ across a broad spectrum of eukaryotic organisms. Using these next-generation tools and downstream analytical innovations including machine learning sequence assignment algorithms and co-occurrence network analyses, we examined how anthropogenic pressures may have impacted marine biodiversity on subtropical coral reefs in Okinawa, Japan. Based on 18 S ribosomal RNA, but not ITS2 sequence data due to inconsistent amplification for this marker, as well as proxies for anthropogenic disturbance, we show that eukaryotic richness at the family level significantly increases with medium and high levels of disturbance. This change in richness coincides with compositional changes, a decrease in connectedness among taxa, an increase in fragmentation of taxon co-occurrence networks, and a shift in indicator taxa. Taken together, these findings demonstrate the ability of eDNA to act as a barometer of disturbance and provide an exemplar of how biotic networks and coral reefs may be impacted by anthropogenic activities.
Subject terms: Biodiversity, Ecological genetics, Molecular ecology, Marine biology, Environmental impact
Introduction
Coastal ecosystems are biologically rich and provide resources for millions of people through fisheries, tourism, shipping, and resource extraction. An important component of coastal ecosystems in both tropical and subtropical environments are zooxanthellate scleractinian corals, a matrix of calcium carbonate skeleton and live animal polyps that provide habitat, food, and refuge, directly leading to high levels of marine biological diversity. In recent years, significant declines in coral habitat have been attributed to deteriorating coastal water quality1, anthropogenically induced climate change2,3, and coastal development4. These stressors also impede the ability of corals to compete for resources with other reef organisms such as macroalgae or sponges1,5, thereby causing compositional shifts in reef communities. For example, on degrading Caribbean reefs, phenotypically plastic coral genera such as Porites, Siderastrea, or Agaricia replaced more sensitive genera such as Acropora and Orbicella6. As another example, in the Red Sea near Eilat, disturbance to mesophotic scleractinian corals was hypothesised to allow the colonisation of other animals such as octocorals, tunicates, sponges, bryozoans, polychaetes, and molluscs on dead portions of their external skeleton7. These shifts in community structure often occur at small spatial scales, or over short periods of time, and these ephemeral changes can be difficult to detect.
Environmental DNA (eDNA) sampling in combination with next-generation sequencing (NGS) metabarcoding can provide an immediate option as a marine monitoring tool in both temperate8–10 and tropical11–13 environments, as well as areas of transition between the two14,15. This approach presents a rapid assessment of unicellular eukaryotes to resident small- and large-bodied animals by amplifying their DNA. In part, eDNA circumvents the need to recruit multiple taxonomic experts and the logistical constraints of exhaustive surveys of a large geographic area. That said, metabarcoding data have been largely restricted to size-fractionated plankton communities in the pelagic zone16,17 or based on material extracted from long-term (>1 year) benthic collection devices18,19. eDNA has therefore rarely been surveyed in a rapid and repeated manner or focused on multiple localised positions along reef and rocky coastlines.
We here chose to focus our surveys on coastal ecosystems in Okinawa (Ryukyu Archipelago, southern Japan), which is known for its high marine biodiversity, including several endemic taxa20. A total of 340 species of hard coral21–23 and at least 1200 species of fish24,25 have been documented from Okinawa, although much less is known about lower order invertebrate taxa26. The coastal areas of many islands in Okinawa are facing increasing anthropogenic pressures due primarily to coastal development and land reclamation projects4,27–29, but also due to the input of marine pollution including red soil runoff, endocrine disrupters, and excessive nutrients30–33. This human impact is mostly prevalent at the main island of Okinawa (land area: 1208 km2; population: ~1.3 million)4,34, with the southern portion of the island characterised by coral reef lagoons that have been reclaimed, high nutrient input from iron-rich terrestrial soil, and high fishing pressure. The Ryukyu Archipelago was also subjected to severe coral bleaching during the 1998 ENSO event22, and more recently from 2016 to 201735–37, with branching and tabular hard coral taxa decreasing the most compared to stress-tolerant genera (e.g. Porites, Dipsastraea, Favites, and Leptastrea). Thus, while some aspects of the coral reefs of Okinawa have been well surveyed, there is a notable paucity of research focused on the wider biological impacts of these anthropogenic drivers26.
In this study, using (i) a ‘universal’ metabarcoding assay targeting 18 S ribosomal RNA (18 S rRNA) of most marine eukaryotes, as well as (ii) a more taxon-focused internal transcribed spacer 2 of ribosomal RNA (ITS2) assay targeting cnidarians and sponges, we investigated whether anthropogenic disturbances affected taxonomic diversity and richness. We additionally tested the connectedness and fragmentation among taxa across sites that experience different levels of anthropogenic pressures, and identified putative keystone taxa (i.e. highly connected nodes within networks). These data contribute to the growing body of eDNA literature and support the utility of metabarcoding in biological monitoring of our ocean ecosystems.
Results
Total biodiversity detected – 18 S rRNA
Using our ‘universal’ assay targeting the 18 S rRNA region, a total of 14,003,698 amplicon reads were generated from 42 sediment samples (2,945,188 sequences) and 164 seawater samples (11,058,510 sequences) to provide a snapshot of marine eukaryotic biodiversity along an anthropogenic pressure gradient at 14 sites in Okinawa, Japan (see Table S2). One sediment sample (now N = 41) and 31 seawater samples (now N = 133) failed to amplify, amplified poorly, or did not pass our sequencing depth threshold set at 20,000 reads. The number of assigned versus unassigned unique sequences per site ranged from 261 to 2114 (mean assigned ± SE, 1218 ± 157) or 389 to 2127 (mean unassigned ± SE, 1054 ± 131) sequences for sediment samples, and 701 to 7151 (mean assigned ± SE, 3658 ± 510) or 664 to 5904 (mean unassigned ± SE, 2646 ± 377) for seawater samples, respectively, when both years of data were combined.
The 18 S rRNA metabarcoding data overall assigned to 39 eukaryotic phyla, 107 classes, 261 orders, and 498 families (Fig. 1, Table S2, Appendix S3). For sediment samples, families within the phyla Arthropoda and Porifera were only present (at > 10% of the total number of families per site) at low pressure sites, Annelida (with one site exception, i.e. Rukan) only appeared at medium and high pressure sites, and Ascomycota and Nematoda only appeared at high pressure sites. Similarly, for seawater samples, with one site exception (i.e. Oura Bay), families within the phylum Arthropoda only appeared (at > 10% of the total number of families per site) at low pressure sites, whereas families within the phyla Mollusca and, with one site exception (i.e. Cape Manza), Annelida only appeared at medium and high pressure sites.
Figure 1.

Sediment and seawater samples collected at 14 sites off the coast of Okinawa, Japan. Circles on the map are shaded according to the level of anthropogenic pressure that they experience (low pressure = light grey, medium pressure = intermediate grey, high pressure = dark grey). Bar graphs indicate the number of taxonomic families assigned at each site based on 18 S rRNA sequences; phyla where the number of families are greater than 10% of the total families for that site are coloured as indicated in the legend. An asterisk above bar graphs indicate sites that were sampled in one year only; sites without an asterisk were sampled twice, over two consecutive years. The figure was created with a combination of QGIS v 3.6 (https://www.qgis.org/en/site/) and Adobe Illustrator v CS6.
Family diversity (i.e. richness) analysis – 18 S rRNA
Based on 18 S rRNA metabarcode sequences, family diversity (i.e. richness) for sediment samples was 44.29 (±SE 4.69) for the low anthropogenic pressure sites, 59.00 (±SE 20.11) for the medium pressure sites, and 62.75 (± SE 6.92) for the high pressure sites (Fig. 1). For seawater samples, family diversity was 98.57 (±SE 12.37) for the low anthropogenic pressure sites, 108.00 (±SE 30.32) for the medium pressure sites, and 124.75 (±SE 21.20) for the high pressure sites (Fig. 1). There were no statistical differences in the family-level taxonomic richness among the three anthropogenic pressure categories for sediment samples (Kruskal-Wallis test: chi-square = 4.8427, df = 2, P = 0.089), but there were differences among seawater samples (Kruskal-Wallis test: chi-square = 18.819, df = 2, P < 0.0001), with richness at low pressure sites significantly lower than that at medium (Pairwise Wilcoxon Rank Sum test: P < 0.001) and high pressure (Pairwise Wilcoxon Rank Sum test: P < 0.001) sites; medium and high pressure sites were no different from each other (Pairwise Wilcoxon Rank Sum test: P = 0.88).
Distinguishing community assemblages - 18 S rRNA
Based on 18 S rRNA metabarcoding sequences and PERMANOVA tests, although there was no significant effect of anthropogenic pressure on the assemblage of families recovered from sediment samples (P = 0.104, df = 2, MS = 5130, Pseudo-F = 1.2181), there was a significant effect for seawater samples (P = 0.0137, df = 2, MS = 15615, Pseudo-F = 1.732) (Fig. 2). Indeed, like family richness, the community assemblages at low pressure sites were different from medium pressure and high pressure sites, whereas medium and high pressure sites were no different from each other.
Figure 2.
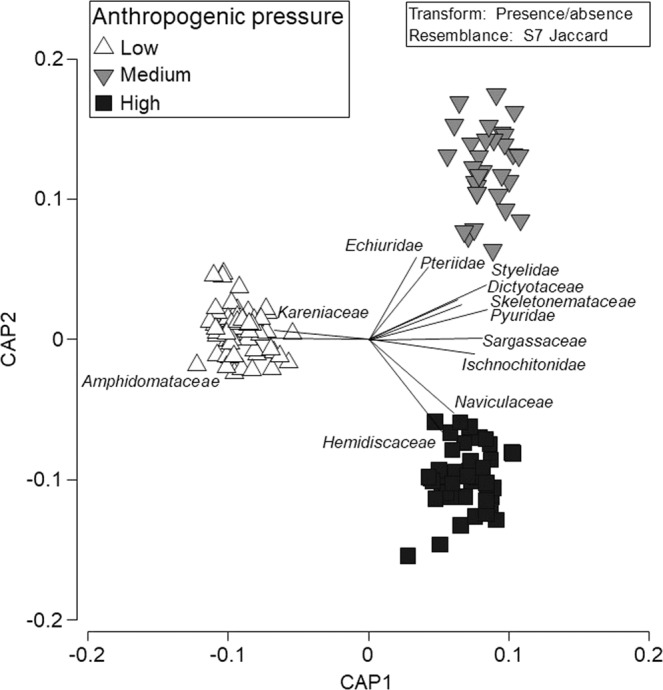
Canonical Analysis of Principle Coordinates (CAP) ordination plot of the presence/absence of eukaryotic families detected based on seawater samples collected at 14 sites in Okinawa, Japan and 18 S rRNA sequences. The relationship of eukaryotic community assemblages identified in each sample using a Jaccard’s coefficient for factor “Impact” is shown. Pearson correlation vectors (r > 0.4) represent the eukaryotic taxa driving the relationship among samples.
Based on follow-up IndVal analyses to resolve taxonomic families of importance in community classifications across anthropogenic pressure categories in sediment (Table 1), we identified chitons (family Chitonidae), free-living flatworms (family Macrostomidae), and sludge or sewage worms (family Naididae) as indicators of high pressure sites, and polychaete worms (family Capitellidae) as an indicator of medium and high pressure sites. For seawater samples, we identified siphonophores (family Agalmatidae), small dinoflagellates (family Ceratiaceae and Kareniaceae), siliceous haptophytes (family Prymnesiaceae), and demosponges (family Petrosiidae) as indicators of low pressure sites. We also identified marine algae (family Fragilariaceae and Rhodellaceae), unicellular flagellates (family Bicosoecidae), and marine slime molds (family Labyrinthula) as indicators of high pressure sites, as well as venus clams (family Veneridae), brown algae (family Dictyotaceae), marine algae (family Sargassaceae), a different group of demosponges (family Callyspongiidae), and haminoeid bubble snails (family Haminoeidae) as indicators of medium and high pressure sites.
Table 1.
Indicator value (IndVal) analyses based on sediment and seawater samples collected at 14 sites in Okinawa, Japan and 18 S rRNA sequences for low, medium, and high anthropogenic pressure sites.
| Low pressure | Medium pressure | High pressure | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Family | Common name | stat/P | Family | Common name | stat/P | Family | Common name | stat/P | |
| Sediment | Capitellidae | Polychaete worms | 0.89/0.050 | Capitellidae | Polychaete worms | 0.89/0.050 | |||
| Chitonidae | Chitons | 0.87/0.022 | |||||||
| Macrostomidae | Free-living flatworms | 0.87/0.016 | |||||||
| Naididae | Sewage worms | 0.87/0.018 | |||||||
| Water | Agalmatidae | Siphonophores | 0.82/0.021 | Veneridae | Venus clams | 1.00/0.003 | Veneridae | Venus clams | 1.00/0.003 |
| Kareniaceae | Dinoflagellates | 0.82/0.046 | Dictyotaceae | Brown algae | 0.93/0.017 | Dictyotaceae | Brown algae | 0.93/0.017 | |
| Prymnesiaceae | Siliceous haptophytes | 0.82/0.037 | Sargassaceae | Marine algae | 0.87/0.011 | Sargassaceae | Marine algae | 0.87/0.011 | |
| Ceratiaceae | Siliceous haptophytes | 0.80/0.049 | Callyspongiidae | Demosponges | 0.79/0.047 | Callyspongiidae | Demosponges | 0.79/0.047 | |
| Petrosiidae | Demosponges | 0.80/0.046 | Haminoeidae | Bubble snails | 0.79/0.029 | Haminoeidae | Bubble snails | 0.79/0.029 | |
| Rhodellaceae | Marine algae | 0.89/0.012 | |||||||
| Bicosoecidae | Unicellular flagellates | 0.87/0.030 | |||||||
| Labyrinthula | Marine slime molds | 0.87/0.025 | |||||||
| Fragilariaceae | Marine algae | 0.84/0.035 | |||||||
P-values were selected at a 0.05 significance level.
Network analyses
Visualisation of the three seawater networks based on sites experiencing low, medium, and high levels of anthropogenic pressure are provided in Fig. 3. The properties of each network and the top ten most connected nodes for each network are summarised in Tables 2 and 3, respectively. The number of nodes captured in each of three networks varied markedly, with the low pressure network having almost half (N = 55) the number of nodes compared to the high pressure network (N = 105). Despite the relatively lower number of nodes, the number of interactions as well as the proportion of positive and negative interactions was similar between the low pressure and high pressure networks. Interestingly, the medium pressure network had relatively few interactions, and in contrast to the other networks, most of these interactions were negative. Despite containing the fewest number of nodes, the low pressure network was the most densely populated with edges (clustering co-efficient = 0.344). Indeed, even with its elevated number of nodes, the high pressure network still contained a lower density of edges (clustering co-efficient = 0.165) than the low pressure network, indicating that fewer nodes can be reached via other nodes38.
Figure 3.

Co-occurrence networks of eukaryotic families detected based on seawater samples collected at 14 sites in Okinawa, Japan, and 18 S rRNA sequences for (a) low, (b) medium, and (c) high pressure sites. Green and red lines represent positive and negative interactions, respectively.
Table 2.
Network properties of the low, medium and high anthropogenic pressure co-occurrence networks based on presence/absence of eukaryotic families detected based on seawater samples collected at 14 sites in Okinawa, Japan and 18 S rRNA sequences.
| Attributes | Low pressure | Medium pressure | High pressure |
|---|---|---|---|
| number of nodes | 55 | 80 | 105 |
| number of interactions | 439 | 174 | 446 |
| number of positive interactions | 334 | 70 | 387 |
| number of negative interactions | 105 | 104 | 59 |
| clustering co-efficient | 0.344 | 0.066 | 0.165 |
| connected components | 1 | 6 | 2 |
Table 3.
The ten most connected nodes from the low, medium and high anthropogenic pressure co-occurrence networks based on presence/absence of eukaryotic families detected based on seawater samples collected at 14 sites in Okinawa, Japan and 18 S rRNA sequences.
| Pressure | Degree | Closeness centrality | Lineage |
|---|---|---|---|
| Low | 30 | 0.6800 | Stramenopiles–Ochrophyta–Bacillariophyceae–Cymatosirales–Cymatosiraceae |
| Low | 30 | 0.8000 | Stramenopiles–Ochrophyta–Bacillariophyceae–Cymatosirales |
| Low | 30 | 0.9091 | Stramenopiles–Ochrophyta–Bacillariophyceae–Cymatosirales–Cymatosiraceae |
| Low | 29 | 0.8333 | Metazoa |
| Low | 26 | 0.6818 | Plantae–Chlorophyta–Chlorodendrophyceae–Chlorodendrales–Chlorodendraceae |
| Low | 26 | 0.6545 | Plantae–Chlorophyta–Chlorodendrophyceae–Chlorodendrales |
| Low | 26 | 0.7500 | Plantae–Chlorophyta–Chlorodendrophyceae–Chlorodendrales–Chlorodendracea |
| Low | 26 | 0.8571 | Plantae–Chlorophyta–Chlorodendrophyceae |
| Low | 25 | 0.6308 | Metazoa–Porifera–Demospongiae–Haplosclerida–Chalinidae |
| Low | 25 | 0.5698 | Metazoa–Porifera–Demospongiae |
| low average (all nodes) | 16 | 0.5832 | |
| Medium | 12 | 0.4714 | Metazoa–Mollusca–Bivalvia–Mytilida–Mytilidae |
| Medium | 12 | 0.4714 | Metazoa–Mollusca–Bivalvia–Mytilida–Mytilidae |
| Medium | 12 | 0.4714 | Metazoa–Mollusca–Bivalvia–Mytilida |
| Medium | 11 | 0.5000 | Metazoa–Chordata–Ascidiacea–Phlebobranchia–Ascidiidae |
| Medium | 11 | 0.5000 | Metazoa–Chordata–Ascidiacea–Phlebobranchia |
| Medium | 11 | 0.5000 | Metazoa–Chordata–Ascidiacea–Phlebobranchia–Ascidiidae |
| Medium | 9 | 0.5500 | Chromista–Myzozoa–Dinophyceae–Peridiniales–Heterocapsaceae |
| Medium | 9 | 0.5500 | Chromista–Myzozoa–Dinophyceae–Peridiniales–Heterocapsaceae |
| Medium | 8 | 0.3548 | Metazoa–Annelida–Polychaeta–Phyllodocida |
| Medium | 8 | 0.3548 | Metazoa–Annelida–Polychaeta–Phyllodocida–Syllidae |
| medium average (all nodes) | 4 | 0.5135 | |
| High | 40 | 0.5361 | Plantae–Chlorophyta–Ulvophyceae–Ulvales–Ulvellaceae |
| High | 39 | 0.5298 | Plantae–Chlorophyta–Ulvophyceae–Ulvales |
| High | 38 | 0.5235 | Plantae–Chlorophyta–Ulvophyceae–Ulvales–Ulvellaceae |
| High | 25 | 0.4611 | Metazoa–Mollusca–Polyplacophora–Chitonida–Ischnochitonidae |
| High | 24 | 0.4564 | Metazoa–Mollusca–Polyplacophora–Chitonida–Ischnochitonidae |
| High | 23 | 0.4279 | Metazoa–Mollusca–Polyplacophora |
| High | 23 | 0.4279 | Metazoa–Mollusca–Polyplacophora–Chitonida |
| High | 21 | 0.4198 | Stramenopiles–Ochrophyta–Bacillariophyceae–Thalassiosirales–Thalassiosiraceae |
| High | 21 | 0.4198 | Stramenopiles–Ochrophyta–Bacillariophyceae–Thalassiosirales–Thalassiosiraceae |
| High | 20 | 0.4428 | Plantae–Chlorophyta–Ulvophyceae–Cladophorales |
| high average (all nodes) | 9 | 0.3676 |
The medium pressure network was weakly connected as indicated by its fragmentation into six components (Fig. 3); an additional small component as a result of fragmentation was also observed in the high pressure network. In contrast, the low pressure network consisted of single component. We additionally observed higher heterogeneities in both the medium and high pressure networks indicating a greater tendency for the creation of hub nodes. In general, the nodes from the low pressure network (mean number of neighbours = 15.96) were far more connected than those from both the medium (mean number of neighbours = 4.35) and high pressure (mean number of neighbours = 8.88) network.
The top ten putatively important taxa as determined by the highest degrees and closeness centrality varied among the networks (Tables 2 and 3). For the low pressure network, the important taxa identified included diatoms, chlorophytes from the order Cymatosirales, and demosponges. On average the low pressure nodes had a higher degree and higher closeness centrality versus those in the more disturbed networks. In the medium pressure network, these important nodes included mytillid bivalves, ascidians (order Phlebobranchia), dinoflagellates (family Heterocapsaceae), and polychaetes (order Phyllodocida). For the high pressure network, these important nodes included Ulvophyceae predominately from the order Ulvales, chitons (family Ischnochitonidae), and a different diatom from the low pressure network (Thalassiosirales, family Thalassiosiraceae).
Anthozoa and Demospongiae richness and community composition analysis – ITS2
A total of 4,220,102 ITS2 amplicon reads were generated from 26 sediment samples (2,261,264 sequences) and 71 seawater samples (1,958,838 sequences) (Table S1). Following subsampling, there were 13,776 unique sediment sequences and 4,213 unique seawater sequences from 200,000 and 44,000 total sediment and seawater reads, respectively. Of the unique sediment reads, 500 could be assigned to the class Anthozoa and 1,311 to Demospongiae; 11,184 sequences were not assigned and the remaining 781 sequences assigned to other eukaryotic taxa. Of the Anthozoa and Demospongiae, 262 could be assigned at the family level with confidence (five cnidarian families: Agariciidae, Merulinidae, Poritidae, Siderastreidae, and Sphenopidae), and these were distributed among only five of the ten sampling sites (Bise, Cape Hedo, Kaichu Doro S1, Makiminato, and Mizugama). Of the 4,213 unique reads subsampled from the seawater replicates, 772 could be assigned to the class Anthozoa and 1,161 to the class Demospongiae; 2,125 sequences were not assigned and the remaining 155 sequences assigned to other eukaryotic taxa. Of the Anthozoa and Demospongiae, 509 were assigned to 19 Anthozoa and Demospongiae families, with at least one ID positively assigned at all 11 sites. The most ubiquitous families were Poritidae (7 sites) and Halichondriidae (6 sites), whereas the families Agariciidae, Alcyoniidae, Aplysinellidae, Astrocoeniidae, Irciniidae, Lobophylliidae, Merulinidae, Mussidae, Parazoanthidae and Spirastrellidae were detected at only one site each (Fig. 4). Family richness for seawater ITS2 replicates was 3.6 (±SE 0.25) and 4.5 (± SE 2.23) for the low and high anthropogenic pressure categories, respectively. The increase in the mean and error for the latter was largely driven by the sampling site Mizugama, for which 15 families were detected. There were no statistical differences in the family-level taxonomic richness among the two anthropogenic pressure categories (Kruskal-Wallis test: chi-square = 0.86072, df = 1, P = 0.35), nor were there any differences in community composition detected between low and high anthropogenic pressure sites (P = 0.104, df = 2, MS = 5130, Pseudo-F = 1.2181).
Figure 4.

ITS2 sequences from seawater samples collected at low (a,b), medium (c,d), and high pressure sites (e,f) that were assigned to either class Anthozoa (left) or class Demospongiae (right) by the INSECT algorithm. Segment sizes and figures in parentheses are the absolute (i.e. not unique) number of sequences assigned at each taxonomic rank (order, family, and genus from inner to outer sections). Missing segments indicate the number of sequences unidentified at each rank.
Discussion
We demonstrated that the biotic composition of marine taxa based on eDNA signatures in seawater was consistently transformed by anthropogenic pressure after controlling for the effects of spatial and temporal partitioning. We find that even though taxonomic richness increased with anthropogenic pressure based on seawater eDNA sampling (but not sediment eDNA sampling), it may not be reflective of compositional changes. Indeed, based on biotic community composition analyses, we found that sites that had been subjected to medium or high levels of anthropogenic pressure did not always lose keystone taxonomic groups, but instead may have replaced them with more opportunistic members within those groups. In fact, many of the indicator families were unicellular chlorophytes and diatoms, possibly due to higher turnover rates and sensitivity to environmental factors39, which stresses the necessity of focusing on a broad taxonomic spectrum to assess disturbance. Diatoms within the division Bacillariophyta are indeed one of the most important groups of microalgae given that they control 40–45% of primary production in our oceans40. Based on co-occurrence network analysis, we also found a decrease in connectedness and increase in fragmentation among the identified eukaryotic taxa, suggesting that disturbance may impact ecological functions and ecosystem resilience.
Family diversity (i.e. richness) analysis – 18 S rRNA
Estimation of taxon richness based on families assigned from 18 S rRNA exhibited differences at small spatial scales among our study sites (a few to tens of kilometres) that were more likely attributed to anthropogenic pressures than abiotic variables, although we did not explicitly quantify the latter. Family-level richness went up with medium and high pressure, which suggests that surveys that rely exclusively on alpha-diversity as a proxy for impact, with higher diversity being deemed “good”, particularly for microbial communities41, may not capture the important compositional changes that can occur. One potential mechanism proposed for this increase in taxon richness or diversity in coastal ecosystems is that disturbances acting via bioerosion or the removal of existing species can provide microhabitat or create the space necessary for the recruitment of a different suite of species7. Indeed, albeit in soft-sediments, Ellis et al. (2017)42 found that the communities therein demonstrated the highest alpha-diversity in areas associated with mild disturbance. The “intermediate disturbance theory” also suggests that richness is the highest at intermediate levels of disturbance43,44. In such scenarios, rapid colonisers and more competitive species can co-occur. Chariton et al. (2015) found eDNA derived taxonomic richness to be consistently higher in anthropogenically influenced estuaries, with the authors suggesting that this was likely due to a confluence of inputs being deposited in these systems14. Recent studies based at offshore gas platforms in the North Adriatic Sea revealed that richness of benthic and planktonic eukaryotes however obtained from water samples did not show a clear pattern along a distance gradient from the putative source of disturbance45. Collectively, the evidence suggests that high eDNA derived taxonomic richness may not necessarily be indicative of a healthy environment. Indeed, variation in alpha-diversity is often non-linear and difficult to directly compare between studies given differences in methodology41,46. We strongly recommend always combining taxon richness metrics with an examination of the biotic composition, in addition to co-occurrence network analyses that can identify breakdowns in the cohesiveness of the community (see below).
Distinguishing community assemblages - 18 S rRNA
It is established that different substrates will give a different picture of the marine environment based on the taxa that are recovered with eDNA metabarcoding9. In our case, the community composition of taxa recovered from sediment versus seawater did not overlap (Fig. S1), and so these were considered in isolation of each other for most of the analyses. In terms of biotic assemblages detected in the sediment, we found that animals from the phyla Arthropoda (e.g. horseshoe crabs, sea spiders, crustaceans) and Porifera (i.e. sponges) largely disappeared with anthropogenic pressure and were replaced by animals from the phyla Annelida (segmented worms) and Nematoda (marine round worms), although these shifts were not significant overall. We also saw an increase in worms of the family Naididae at high pressure sites, which are considered an indicator of low water quality47. Nematodes account for approximately 80% of all individual animals on earth48 and are characterised by high abundances in meiofaunal assemblages49. The fact that nematodes only appeared at high pressure sites in our study may reflect preferential amplification of other taxa present at less impacted sites50 or a shift from k-selected to r-selected taxa that is often associated with pollution51. We additionally identified a specific family of sediment-associated polychaete worm (Capitellidae) indicative of medium pressure and high pressure sites; these organisms have previously been recommended as proxies for environmental disturbances52. The lack of sensitivity of sediment sampling versus seawater sampling is consistent with previous work9,53, and may be due to the legacy of DNA in different substrates. For instance, eDNA does not last as long in the water column as it does in the sediment, which means the latter substrate captures a greater time window. This supports the idea that rapid community turnover may be easier to detect in the water column.
In terms of the biotic assemblages in seawater, we found that animals from the phylum Arthropoda disappeared with anthropogenic pressure and were replaced by animals from the phyla Annelida and Mollusca (Fig. 1). Low pressure sites were dominated by hydrozoans (family Agalmatidae), dinoflagellates (family Kareniaceae and Ceratiaceae), and demosponges (family Petrosiidae), whereas marine slime molds (family Labyrinthulidae), bubble snails (family Haminoeidae), venus clams (Veneridae), and a different group of demosponges (family Callyspongiidae) were putative indicators of medium pressure or high pressure sites. Some demosponges, including species of the Callyspongiidae family, are hypothesised to have negative effects on neighbouring scleractinian corals via the toxic compounds that they produce54. This same group of sponges are also hypothesised to dominate coral reef ecosystems under future climate change scenarios55.
Network analyses
We found that the broad topological features of co-occurrence networks based on 18 S rRNA taxonomic assignments in seawater samples differed considerably among the three levels of anthropogenic pressure. We detected the presence of multiple independent components and a lower clustering coefficient in the higher-pressure categories, which led to fracturing of the networks56,57. This shift was particularly evident in the medium pressure network, which was also dominated by negative (instead of positive) taxon interactions. This shift also indicates a markedly different behaviour where amensalism and/or competition were driving the interactions between nodes, rather than commensalism and mutualism interactions observed in the low and high pressure networks. Although experimental evidence is limited, this is important because it has been suggested that an increase in positive inter-taxa interactions may improve the resilience of an ecosystem to stressors56. Several disturbance studies have shown similar reductions in the degree of links observed within networks, and this downward shift supports the potential utility of endpoints as metrics for detecting changes in co-occurrence networks58–61.
The networks identified a range of hub nodes (i.e. those that were highly connected) in both low and high pressure networks, indicating that they were intrinsically linked via numerous positive interactions. Interestingly, shifts in the taxonomy of the diatom nodes towards a more integrated role of Thalassiosiraceae as a response to anthropogenic pressures was also observed by Chariton et al. (2015) in Australian estuarine sediments14. Furthermore, using a Bayesian network, Graham et al. (2019) predicted a shift towards an increasing dominance of dinoflagellates, and indeed, this group was also observed within key nodes for the medium pressure network62. Surprisingly, many of the nodes identified in the medium and high pressure networks were associated with benthic species (e.g. spionid polychaetes, chitons, and mytillids). One possible explanation for this finding is that there is a distribution of the larval form of these animals in the water column. For example, spionids and mytillids are known to include broadcast spawners with planktotrophic or lecithotrophic larval stages63,64. The alternative is that DNA from these organisms is capable of mixing in the water column and intermittently becomes suspended as plankton.
While there are number of caveats associated with interpretation of the networks, given the spatial and temporal nature of which samples were collected, it does provide an additional line of evidence for understanding how communities respond to disturbances. Both theoretical and empirical studies of the relationship between diversity and ecosystem stability have generally found a positive association between the two, resulting in increased ecosystem resilience to external forces and greater support of ecosystem services65,66. However, as illustrated in this study and in Chariton et al. (2015), metabarcoding diversity may indeed be enriched in disturbed systems, suggesting that richness and diversity may be a more appropriate indicator of condition14.
A surprising result was the extent of fracturing and lower cohesion in the medium pressure network. One possible explanation is that these systems are in a state of flux and susceptible to increased negative interactions based around competition and parasitism57. In contrast, the shift from the low pressure to high pressure networks may be reflective of hysteresis, where the ecological communities are relatively stable in undisturbed or highly disturbed ecosystems, albeit founded on different nodes (i.e. taxa)67. While the fundamental hypothesis that connectivity is intrinsically linked to resilience is still being debated, the greater level of overall connectivity at both the network and key node scale suggest that low pressure systems have a greater level of built in redundancy, enabling composition turnover to occur without a loss of key biotic interactions.
Anthozoa and Demospongiae richness and community composition analysis – ITS2
Our ITS2 metabarcoding assay focused on two important functional groups in coral reef ecosystems, animals from the classes Anthozoa and Demospongiae. Despite no statistical differences in the family-level taxonomic richness or community composition among two anthropogenic pressure categories considered here (low pressure and high pressure only due to small sample sizes), there were some striking qualitative patterns (Fig. 4). For class Anthozoa at the genus level, we noted an increasing trend in the proportion of sequences with anthropogenic pressure for Anthopleura (speciose genus of sea anemones), Palythoa (sand-encrusted colonial anemone), and Zoanthus (non-sand-encrusted colonial anemone). This trend is important because recent phase shifts from scleractinian hard corals to other anthozoan groups such as corallimorpharians68 and zoantharians such as Palythoa69,70 and Zoanthus71 have been reported, although often the underlying causes for their increases are unclear.
For class Demospongiae at the genus level, we noted an increase with anthropogenic pressure in the proportion of sequences for Cliona (speciose genera of sponge), Hymeniacidon (includes mobile sponge species), Ircinia (speciose genera of sponge), and Suberea (horny sponges). We also noted the highest diversity, or at least the highest proportion of successfully identified taxa at Mizugama, with 15 different Anthozoa or Demospongiae families present at this site. In a previous study, Mizugama was shown to be the only site out of eight investigated around Okinawa Island that was dominated by hard corals and Palythoa (family Sphenopidae)69, which is consistent with our observations here. That said, successful amplification for ITS2 was weak overall (Table S1), and a thorough evaluation would require additional sampling or sequencing coverage using this assay.
Caveats
We have confidence in our 18 S rRNA metabarcoding findings and the repeatability of our assays given the grouping of replicates together from the same site, and the approximate grouping of site replicates together sampled in different years (Fig. S2). However, there are still important caveats of the eDNA metabarcoding method and therefore our data set as presented here. First, a significant fraction of our 18 S rRNA sequences post-filtering could not be assigned at the family level (19–63% of unique reads; Table S1). This deficiency reinforces the need for both improved DNA reference databases and a robust taxonomic framework. The INSECT algorithm72 used for ITS2 data, on the other hand, assigns taxon ID to sequences from complex environmental samples using hidden Markov models, and is designed to minimize false discoveries that persist in k-mer based assignment methods73. However, while misclassifications and over-classifications are generally rare, INSECT often under-classifies (i.e. does not assign an ID) if the reference database is underpopulated72. This, once again, stresses the need for the continued development of curated genetic databases representing multiple genes based on taxonomically identified specimens. Such initiatives can be supplemented with algorithms that can simultaneously minimize both type I and type II assignment errors. The present study, and others following similar approaches, do not include abiotic data in the analyses. We recommend collecting environmental data together with genetic material to provide a more robust explanation of changes in marine biological communities. Long-term collection of environmental samples in situ is preferred to remote sensing approaches, when feasible, given the difficulty of “scaling down” with the latter.
Conclusions
Taken together this study adds to the growing body of literature that shows the utility of eDNA in providing a better understanding of marine environments. Even in its current state of development, with large apparent gaps in DNA reference databases, eDNA can act as a powerful method that complements existing survey methods. According to the results, 18 S provided a better understanding of the response by biological communities versus the assay targeting ITS2. This suggests a focus on developing the former versus the latter if a multi-assay approach is not possible based on limited funds. That said, the ITS2 assay has yet to be fully tested given insufficient sequencing coverage in this study. As marine biomonitoring increasingly moves towards a ‘ecosystem-based’ approach to track anthropogenic impacts these metabarcoding data support the ability of eDNA to deliver a more holistic survey of biota and identify indicator taxa. This aligns with new initiatives related to marine monitoring, and may additionally provide a standardized tool, outlined by a recent UN‐sponsored report by the Intergovernmental Science-Policy Platform on Biodiversity and Ecosystem Services (IPBES). Moreover, the continuation of temporal and spatial sampling with sufficient replication for more nuanced co-occurrence network analyses should further enrich the survey of both pristine and degraded marine environments across the globe.
Materials and Methods
Sampling sites and anthropogenic pressure scale
The selected sampling sites in Okinawa were differentially impacted by natural and anthropogenic pressures (Fig. 1). Although environmental data are available for the coastal ecosystems of this region, including sea surface temperature (SST; see Japan Meteorological Agency, https://ds.data.jma.go.jp/tcc/tcc/products/elnino/cobesst/cobe-sst.html), wave height (see Japan Meteorological Agency, https://www.data.jma.go.jp/gmd/kaiyou/db/wave/chart/daily/coastwave.html?year=2019&month=3&day=4&hour=12), additional water parameters (see Okinawa Prefecture, https://www.pref.okinawa.jp/site/kankyo/hozen/mizu_tsuchi/water/public_water.html), and for some areas, live coral cover (see Japan Ministry of Environment http://www.biodic.go.jp/trialSystem/top_en.html and4), there are not comprehensive or detailed enough to allow for the assessment of relative anthropogenic pressures at the geographic resolution we wished to examine (e.g. <2 km). Accordingly, we adopted a point-based assessment system in order to rapidly rank sampling sites based on cumulative anthropogenic impacts. Each of the sites was initially allotted 10 points, and points were then subtracted from this total based on in situ observations and publicly available information. The criteria considered and references justifying these guidelines were:
Distance from shore: -1 point if the sampling site was less than 5 km from the shoreline74;
Distance from heavily populated areas: -1 point if >10,000 people lived within 5 km of the sampling site74;
Freshwater input: -0.5 points if there was a natural river within 2 km of the sampling site; -1 point if there was treated wastewater input within 2 km of the sampling site74;
Fishing pressure: -1 point if there was significant fishing pressure (high number of fishing boat visits, shore fishing, and evidence of lost fishing lines, lures, and weights75);
Coastal development: -1 point if there was recent (<10 years) coastal development, including reclaimed land, “coastal defence” tetrapod placement, road construction, artificial reefs, dredging, or any other major alterations in the immediate vicinity; -0.5 points if there was less recent (>10 years ago) coastal development;28,29
Human recreation area: -1 point if the sampling site was adjacent to a human recreational area, including artificial beaches and frequent scuba diving or snorkeling locations;76,77
Hermatypic coral cover: -1 point if coral cover was between 0% to 25% at ~8 metres depth, -0.5 points if between 25% to 50%78;
Other notable pressures: -1 point if there was evidence of eutrophication, loss of water flow, pollution, or the site was dominated by macroalgae79.
To increase replication, sites were grouped by final scores into low anthropogenic pressure (scores >7), medium anthropogenic pressure (scores >4 but <7), and high anthropogenic pressure (scores <4) treatment groups.
Sample collection and DNA extraction
Sampling was conducted in July 2016 and October 2017 at several sites around Okinawa (Fig. 1). For each sampling site, eight 1 L seawater replicates were collected from 30 cm below the surface using sterile Nalgene bottles. All water samples were immediately stored on ice and filtered at the University of the Ryukyus or in the field within eight hours. 750 ml of each water sample was filtered onto 47 mm polyethersulfone filters with a 0.2 m pore size (Pall Life Sciences, New York, USA) using a Sentino peristaltic pump (Pall Life Sciences). The filtration apparatus was cleaned by soaking in 10% bleach between samples for at least 15 minutes. Approximately 10 g of marine sediment (up to four replicates per sampling site) was also collected with sterile 15 ml falcon tubes between 1 and 10 m depth snorkeling or on scuba. The falcon tubes remained closed until the moment of sampling and were closed immediately after sampling. Sediment samples were immediately frozen and stored at -20 °C until DNA extraction in a PCR-free extraction laboratory at Curtin University in Australia.
DNA bound to filter membranes was extracted using Qiagen DNeasy Blood and Tissue kits (Qiagen; Venlo, Netherlands) that was partially automated on a Qiacube (Qiagen) to minimise human handling and cross-contamination. Total nucleic acids were extracted from sediment samples following homogenization of 0.5 g to 0.8 g of organic material per replicate using bead tubes mixed on a Minilys homogenisation machine (Bertin Technologies, France); homogenised replicates were transferred into sterile 2 ml microfuge tubes. Due to the co-purification of inhibitors in sediment samples, a MoBio Powersoil extraction kit (MoBio Laboratories, CA) was used following the manufacturers protocol. A 2× volume of the homogenate was used at the digestion stage and the DNA was eluted in 100 µl of sterile elution buffer. DNA extraction controls (blanks) were carried out for every set of extractions and the appropriate level of input DNA for metabarcoding was determined by qPCR (see below).
Fusion-tag qPCR
A universal primer set targeting 18 S rRNA (V1-3 hypervariable region; 18S_uni_1F: 5′ - GCCAGTAGTCATATGCTTGTCT - 3′; 18S_uni_400R: 5′ - GCCTGCTGCCTTCCTT - 3′) with an amplicon length of ~340-420 bp was used to maximise the eukaryotic fraction of marine diversity detected80. These data were fed into the taxonomy-dependent family-level richness and assemblage analyses outlined below. We used a second assay targeting ITS2 of Cnidaria and Porifera (scl58SF: 5′ - GARTCTTTGAACGCAAATGGC - 3′; scl28SR: 5′ - GCTTATTAATATGCTTAAATTCAGCG - 3′) to increase the taxonomic resolution of Anthozoa and Demospongiae detected within each environmental sample81. The former class includes anemones, stony corals, soft corals, and gorgonians, whereas the latter class includes 81% of all sponge species. Both groups play an important role in the functioning of coral reef ecosystems, such as recycling dissolved organic matter82.
Quantitative PCR (qPCR) experiments were set up in a separate ultra-clean laboratory at Curtin University designed for ancient DNA work using a QIAgility robotics platform13 (Qiagen Inc., CA). In brief, fusion-tag qPCR was performed in duplicate to mitigate reaction stochasticity on a StepOnePlus Real-Time PCR System (Applied Biosystems, CA, USA) under the following conditions for 18 S: initial denaturation at 95 °C for 5 min, followed by 45 cycles for 30 s at 95 °C, 30 s at 52 °C, and 45 s at 72 °C, with a final extension for 10 min at 72 °C. PCR was performed under the following conditions for ITS2: initial denaturation at 95 °C for 5 min, followed by 45 cycles of 30 s at 95 °C, 30 s at 55 °C, and 45 s at 72 °C, with a final extension for 10 min at 72 °C. Duplicate reactions were combined into a larger library of amplicons by pooling at equimolar ratios based on qPCR CT values and the endpoint of amplification curves. These larger libraries were size-selected using a Pippin-Prep (Sage Science, Beverly, USA) to remove any amplicons outside of the target range. Size-selected libraries were then purified using the QIAquick PCR Purification Kit (Qiagen Inc.) and quantified against DNA standards of known molarity on a LabChip GX Touch (PerkinElmer Health Sciences, MA). Next-generation sequencing was conducted on an Illumina MiSeq platform (Illumina, San Diego, CA) housed in the Trace and Environmental DNA (TrEnD) Laboratory at Curtin University, Western Australia.
Bioinformatic filtering
All sequence data were quality filtered (QF) prior to taxonomic assignment using taxonomy-dependent workflows. Metabarcoding reads recovered by paired-end sequencing were first stitched together using the Illumina MiSeq analysis software (MiSeq Reporter v 2.5) under the default settings. Sequences were then assigned to samples based on their unique index combinations and trimmed in Geneious Pro v 4.8.483. In order to eliminate low quality sequences, only those with 100% identity matches to Illumina adaptors, index barcodes, and template specific oligonucleotides were kept for downstream analyses. Sequences were further processed in USEARCH v 9.273, which was used to trim ambiguous bases, remove sequences with average error rates >1% and those that were <200 bp in length, dereplicate each sample down to unique sequences, abundance filter the unique sequences (minimum of two identical reads)13, and remove chimeras. To enable comparison among replicates, the 18 S sequences in each replicate were sub-sampled to 20,000 reads prior to dereplication. Due to lower amplification success of the ITS2 marker, replicates within sites were merged and subsampled to 20,000 and 4,000 reads prior to dereplication for sediment and seawater samples, respectively.
Taxonomic assignment
Unique 18 S sequences that passed QF were queried against the National Center for Biotechnology Information (NCBI) nucleotide database using BLASTn and a high-performance computer (Magnus) located at the Pawsey Supercomputing Centre in Western Australia. BLASTn results were imported into MEtaGenome ANalyzer (MEGAN) v 5.11.384, and taxonomic identities assigned at the family-level based on the lowest common ancestor (LCA) algorithm (minimum bit score = 600; top percent of reads = 5%; max expected = 0.01). Rarefaction analyses were performed in MEGAN v 5.11.3, and all taxonomic nomenclature was based on the World Register of Marine Species85, MycoBank, UniProt, and Algaebase. ITS2 sequences that passed QF were identified to family-level (or lower) where possible using the ‘insect’ R package v 1.3.072 with the cnidarian_ITS2_marine classifier v 5. This classifier was trained on 8154 cnidarian, sponge, and other ITS2 reference sequences downloaded from NCBI GenBank on 20 Sep 2018 (10.5281/zenodo.3247241). Sensitivity settings were as follows: threshold = 0.8, decay = FALSE, ping = 0.99, mincount = 2, offset = 0. Sequences that were not identified as Anthozoa or Demospongiae were subsequently removed from the analyses.
Network construction and analyses
To examine the connections between 18 S rRNA family-level taxon assignments identified in seawater samples collected from sites under varying levels of anthropogenic pressure, irrespective of year, co-occurrence networks were constructed using CoNet v 1.1 (http://systemsbiology.vub.ac.be/conet)86. Networks were derived by grouping all replicate samples into their appropriate anthropogenic pressure category (low, N = 60; medium, N = 31; high, N = 42). Families that were identified in less than 25% of the samples were discarded prior to computation and pairwise scores were determined using five similarity measures (Bray Curtis dissimilarity, Kullback‐Leibler dissimilarity, Pearson correlation, Spearman correlation, mutual information similarity). For each measure, 1000 renormalized permutation and bootstrap scores were generated86. Measure-specific P-values were merged using Brown’s method87, with correction for multiple-testing performed using the Benjamini–Hochberg procedure. Only network edges that agreed for all measures and resulted in the same interaction type (i.e. positive or negative) were retained. Co-occurrences were not produced using the sediment samples (low pressure, N = 17; medium pressure, N = 10; high pressure, N = 14) or the ITS2 data due to the relatively low number of replicates and limited representation of families across multiple samples. Indeed, the number of samples used to produce co-occurrence networks has a pronounced effect on the reliability of the network’s features88, with Reshef et al. (2011)89 recommending that at least 20 replicates is required to produce a network.
Visualisation of the networks was performed using Cytoscape v 3.7 (http://apps.cytoscape.org/apps/conet)90. The topographical features of the networks were examined using Network Analyzer v 2.791, which included the number of nodes, the number of total positive and negative interactions, the clustering co-efficient (i.e. measure of the degree to which nodes form interactive modules)38, the number of modules, and the average number of neighbours91. Putative hub nodes (i.e. nodes that play a key role in network organisation) were determined by examining the top ten most connected nodes within each network92. In addition, the closeness centrality of the hub nodes was estimated based on the reciprocal of the sum of distances to all other nodes. Collectively, this information provided an indirect test of ecological redundancy (e.g. the “insurance hypothesis”)93, whereby if multiple taxa in an ecosystem perform similar tasks one taxon can replace the role of another.
Statistical analyses
Taxonomic composition of marine eukaryotes at the family-level for 18 S was analysed in presence/absence format (Jaccard similarity matrix) to assess the effect of anthropogenic pressure (low, medium, high) and year (2016, 2017) on biological assemblages using PRIMER v 794. Initial PERMANOVA tests showed that the community assemblage recovered with each method was significantly different (P < 0.0001, df = 1, MS = 43340, Pseudo-F = 12.003), resulting in a lack of overlap for families detected with each substrate (Fig. S1). Data from both methods (sediment versus seawater) were therefore analysed in isolation unless otherwise noted. Initial PERMANOVA tests showed that the community assemblage recovered from each year was not significantly different (P = 0.138, df = 1, MS = 11889, Pseudo-F = 1.9556), and so these data were combined for downstream analyses. PERMANOVA (9999 permutations) was therefore used to quantify the differences between anthropogenic pressure categories, and constrained Canonical Analysis of Principal coordinates (CAP) was used to visualise the hypothesis that assemblages detected at differently impacted locations were not the same. Indicator value (IndVal) analyses95 were also conducted using the Indicspecies package (v 1.7.8) in R96,97 to determine the eukaryotic families of importance in community classifications when compared among anthropogenic pressure categories. Family richness among pressure categories was compared using Kruskal-Wallis (KW) tests in R v 3.5.397. Post-hoc pairwise rank sum Wilcoxon tests were used to compare between each pressure category.
The seawater family-level assignments of Anthozoa or Demospongiae ITS2 sequences were analysed in presence/absence format to assess the effect of anthropogenic pressure on community composition and richness using the vegdist (method = “jaccard”) and adonis (permutations = 9999) functions in the vegan R package (v 2.5–6)97,98, and the Kruskal-Wallis test in R v 3.5.397. The effects of anthropogenic pressure on community assemblages were tested at two levels for the seawater ITS2 analysis (low pressure versus high pressure, with a score of 5 acting as the threshold) instead of three levels (low, medium, high) due to the relatively low number of sites that passed the subsampling threshold for sequencing depth (e.g. seawater samples from only ten sites yielded at least 4,000 ITS2 sequences). Moreover, taxonomic assignment of the sediment ITS2 samples failed to identify sufficient Anthozoa and Demospongiae sequences to assess the impact of anthropogenic pressure; only five cnidarian families were identified in total (Agariciidae, Merulinidae, Poritidae, Siderastreidae, and Sphenopidae), and only five sites yielded at least one Anthozoa or Demospongia family-level identification (see Results).
Supplementary information
Acknowledgements
This study was funded by ARC Linkage Projects (LP160100839 and LP160101508) to J.D.D., M.S., and M.B., a Joint Usage and Collaborative Research Grant from the Tropical Biosphere Research Center (TBRC) at the University of the Ryukyus to J.D.D. and J.D.R., a scholarship from the Campus World Program via the Università Politecnica delle Marche to G.D.M. and P.B., as well as Curtin University Early Career Research Fellowship (ECRF) to J.D.D. and Environment and Agriculture Visiting Scholarship to J.D.R. The authors would like to acknowledge Matthew Power, Megan Coghlan, and Adam Koziol for DNA sequencing assistance. This work was also supported by resources provided by the Pawsey Supercomputing Centre with funding from the Australian Government and the Government of Western Australia. In Okinawa, we thank Yoshihiro Katsushima for field work assistance, as well as members of the MISE Laboratory at the University of the Ryukyus.
Author contributions
J.D.D., J.D.R., and M.B. contributed to study design; J.D.D., J.D.R., G.D.M., P.B. contributed to sample acquisition and laboratory processing; J.D.D., J.D.R., M.S., G.D.M., P.B., M.D.B., S.P.W. and A.A.C. contributed to data analysis and figure production; J.D.D., J.D.R., M.S., G.D.M., P.B., M.D.B., S.P.W., A.A.C. and M.B. contributed to manuscript preparation.
Data availability
All data needed to evaluate the study are available from Dryad Digital Repository 10.5061/dryad.m37pvmczf.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-64858-9.
References
- 1.Vega Thurber RL, et al. Chronic nutrient enrichment increases prevalence and severity of coral disease and bleaching. Glob. Chang. Biol. 2014;20:544–554. doi: 10.1111/gcb.12450. [DOI] [PubMed] [Google Scholar]
- 2.Hughes TP, et al. Coral reefs in the Anthropocene. Nature. 2017;546:82. doi: 10.1038/nature22901. [DOI] [PubMed] [Google Scholar]
- 3.Hughes TP, et al. Global warming transforms coral reef assemblages. Nature. 2018;556:492. doi: 10.1038/s41586-018-0041-2. [DOI] [PubMed] [Google Scholar]
- 4.Heery EC, et al. Urban coral reefs: Degradation and resilience of hard coral assemblages in coastal cities of East and Southeast Asia. Mar. Pollut. Bull. 2018;135:654–681. doi: 10.1016/j.marpolbul.2018.07.041. [DOI] [PubMed] [Google Scholar]
- 5.Zaneveld JR, et al. Overfishing and nutrient pollution interact with temperature to disrupt coral reefs down to microbial scales. Nat. Commun. 2016;7:11833. doi: 10.1038/ncomms11833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pandolfi JM, Jackson JB. Ecological persistence interrupted in Caribbean coral reefs. Ecol. Lett. 2006;9:818–826. doi: 10.1111/j.1461-0248.2006.00933.x. [DOI] [PubMed] [Google Scholar]
- 7.Pinheiro, H. T., Eyal, G. G., Shepherd, B. B. & Rocha, L. A. Ecological insights from environmental disturbances in mesophotic coral ecosystems. Ecosphere10 (2019).
- 8.Kelly RP, et al. Genetic and manual survey methods yield different and complementary views of an ecosystem. Front. Mar. Sci. 2017;3:283. doi: 10.3389/fmars.2016.00283. [DOI] [Google Scholar]
- 9.Koziol AA, et al. Environmental DNA metabarcoding studies are critically affected by substrate selection. Mol. Ecol. Res. 2019;19:366–376. doi: 10.1111/1755-0998.12971. [DOI] [PubMed] [Google Scholar]
- 10.Stat MM, et al. Combined use of eDNA metabarcoding and video surveillance for the assessment of fish biodiversity. Conserv. Biol. 2019;33:196–205. doi: 10.1111/cobi.13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stat MM, et al. Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Sci. Rep. 2017;7:12240. doi: 10.1038/s41598-017-12501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boussarie GG, et al. Environmental DNA illuminates the dark diversity of sharks. Sci. Adv. 2018;4:eaap9661. doi: 10.1126/sciadv.aap9661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DiBattista JD, et al. Digging for DNA at depth: rapid universal metabarcoding surveys (RUMS) as a tool to detect coral reef biodiversity across a depth gradient. PeerJ. 2019;7:e6379. doi: 10.7717/peerj.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chariton AA, et al. Metabarcoding of benthic eukaryote communities predicts the ecological condition of estuaries. Environmental Pollution. 2015;203:165–174. doi: 10.1016/j.envpol.2015.03.047. [DOI] [PubMed] [Google Scholar]
- 15.Berry TE, et al. Marine environmental DNA biomonitoring reveals seasonal patterns in biodiversity and identifies ecosystem responses to anomalous climatic events. PLoS Genet. 2019;15:e1007943. doi: 10.1371/journal.pgen.1007943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.De Vargas CC, et al. Eukaryotic plankton diversity in the sunlit ocean. Science. 2015;348:1261605. doi: 10.1126/science.1261605. [DOI] [PubMed] [Google Scholar]
- 17.Deagle BE, et al. Genetic monitoring of open ocean biodiversity: An evaluation of DNA metabarcoding for processing continuous plankton recorder samples. Mol. Ecol. Res. 2018;18:391–406. doi: 10.1111/1755-0998.12740. [DOI] [PubMed] [Google Scholar]
- 18.Pearman JK, Anlauf HH, Irigoien XX, Carvalho SS. Please mind the gap –Visual census and cryptic biodiversity assessment at central Red Sea coral reefs. Mar. Env. Res. 2016;118:20–30. doi: 10.1016/j.marenvres.2016.04.011. [DOI] [PubMed] [Google Scholar]
- 19.Ransome EE, et al. The importance of standardization for biodiversity comparisons: A case study using autonomous reef monitoring structures (ARMS) and metabarcoding to measure cryptic diversity on Mo’orea coral reefs, French Polynesia. PLoS One. 2017;12:e0175066. doi: 10.1371/journal.pone.0175066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts CM, et al. Marine biodiversity hotspots and conservation priorities for tropical reefs. Science. 2002;295:1280–1284. doi: 10.1126/science.1067728. [DOI] [PubMed] [Google Scholar]
- 21.Nishihira, M. M. & Veron, J. E. N. Hermatypic corals of Japan. Kaiyusha, Tokyo, 439 (1995).
- 22.Tsuchiya, M. M., Nadaoka, K. K., Kayanne, H. H. & Yamano, H. H. Coral Reefs of Japan, Coral Reefs of Japan (2004).
- 23.Hongo CC, Yamano HH. Species-specific responses of corals to bleaching events on anthropogenically turbid reefs on Okinawa Island, Japan, over a 15-year period (1995–2009) PloS One. 2013;8:e60952. doi: 10.1371/journal.pone.0060952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Motomura, H., Hagiwara, K., Senou, H. & Nakae, M. (eds.) Identification guide to fishes of the Amami Islands, Japan. Kagoshima University Museum, Kagoshima, Yokosuka City Museum, Yokosuka, Kanagawa Prefectural Museum of Natural History, Odawara, and National Museum of Nature and Science, Tsukuba. (2018).
- 25.Mochida II, Motomura HH. An annotated checklist of marine and freshwater fishes of Tokunoshima Island in the Amami Islands, Kagoshima, southern Japan, with 214 new records. Bulletin of the Kagoshima University Museum. 2018;10:1–80. [Google Scholar]
- 26.Reimer JD, et al. Marine biodiversity research in the Ryukyu Islands, Japan: Current status and trends. PeerJ. 2019;7:e6532. doi: 10.7717/peerj.6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano, Y. Y. Direct impacts of coastal development. In Ministry of the Environment & Japanese Coral Reef Society (eds.), Coral Reefs of Japan (Chapter 2, pp. 60–63.), Tokyo, Japan: Ministry of the Environment (2004).
- 28.Reimer JD, et al. Effects of causeway construction on environment and biota of subtropical tidal flats in Okinawa, Japan. Mar. Pollut. Bull. 2015;94:153–167. doi: 10.1016/j.marpolbul.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 29.Masucci, G. D. & Reimer, J. D. Expanding walls and shrinking beaches: Loss of natural coastline in Okinawa Island, Japan. PeerJ in press (2019). [DOI] [PMC free article] [PubMed]
- 30.Ramos AA, Inoue YY, Ohde SS. Metal contents in Porites corals: Anthropogenic input of river run-off into a coral reef from an urbanized area, Okinawa. Mar. Pollut. Bull. 2004;48:281–94. doi: 10.1016/j.marpolbul.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 31.Imo ST, et al. Distribution and possible impacts of toxic organic pollutants on coral reef ecosystems around Okinawa Island, Japan. Pac. Sci. 2008;62:317–326. doi: 10.2984/1534-6188(2008)62[317:DAPIOT]2.0.CO;2. [DOI] [Google Scholar]
- 32.Shilla DJ, Mimura II, Takagi KK, Tsuchiya MM. Preliminary survey of the nutrient discharge characteristics of Okinawa Rivers, and their potential effects on inshore coral reefs. Galaxea, JCRS. 2013;15:172–181. doi: 10.3755/galaxea.15.172. [DOI] [Google Scholar]
- 33.Yamano HH, et al. An integrated approach to tropical and subtropical island conservation. J. Ecol. Env. 2015;38:271–279. doi: 10.5141/ecoenv.2015.028. [DOI] [Google Scholar]
- 34.Okinawa Prefecture. 2019. February 2019 census report. https://www.pref.okinawa.jp/toukeika/estimates/2019/pop201903.pdf [08 April (2019).
- 35.Kayanne HH, Suzuki RR, Liu GG. Bleaching in the Ryukyu Islands in 2016 and associated Degree Heating Week threshold. Coral Reef Studies. 2017;19:17–18. doi: 10.3755/galaxea.19.1_17. [DOI] [Google Scholar]
- 36.Ministry of the Environment. Iriomote-Ishigaki National Park Survey: results of coral bleaching phenomenon of Sekisei lagoon. http://kyushu.env.go.jp/naha/pre_2017/post_28.html. (in Japanese) (2017).
- 37.Masucci, G. D., Biondi, P. P., Negro, E. E. & Reimer, J. D. After the long summer: Death and survival of coral communities in the shallow waters of Kume island, from the Ryukyu archipelago. Reg. Stud. Mar. Sci. 100578 (2019).
- 38.Proulx SR, Promislow DE, Phillips PC. Network thinking in ecology and evolution. Trends Ecol. Evol. 2005;20:345–353. doi: 10.1016/j.tree.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 39.Martín GG, de los Reyes Fernández MM. Diatoms as indicators of water quality and ecological status: Sampling, analysis and some ecological remarks. Ecol. Water Qual. 2012;9:183–204. [Google Scholar]
- 40.Mann DG. The species concept in diatoms. Phycologia. 1999;38:437–495. doi: 10.2216/i0031-8884-38-6-437.1. [DOI] [Google Scholar]
- 41.Shade AA. Diversity is the question, not the answer. ISME Journal. 2017;11:1–6. doi: 10.1038/ismej.2016.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ellis JJ, et al. Cross shelf benthic biodiversity patterns in the Southern Red Sea. Sci. Rep. 2017;7:1–14. doi: 10.1038/s41598-016-0028-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Connell JH. Diversity in tropical rain forests and coral reefs. Science. 1978;199:1302–1310. doi: 10.1126/science.199.4335.1302. [DOI] [PubMed] [Google Scholar]
- 44.Townsend CR, Scarsbrook MR, Dolédec SS. The intermediate disturbance hypothesis, refugia, and biodiversity in streams. Limnol. Oceanogr. 1997;42:938–949. doi: 10.4319/lo.1997.42.5.0938. [DOI] [Google Scholar]
- 45.Cordier TT, et al. Multi-marker eDNA metabarcoding survey to assess the environmental impact of three offshore gas platforms in the North Adriatic Sea (Italy) Mar. Environ. Res. 2019;146:24–34. doi: 10.1016/j.marenvres.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 46.Blowes SA, et al. The geography of biodiversity change in marine and terrestrial assemblages. Science. 2019;366:339–345. doi: 10.1126/science.aaw1620. [DOI] [PubMed] [Google Scholar]
- 47.Learner MA. The distribution and ecology of the Naididae (Oligochaeta) which inhabit the filter-beds of sewage-works in Britain. Water Res. 1979;13:1291–1299. doi: 10.1016/0043-1354(79)90174-X. [DOI] [Google Scholar]
- 48.Fonseca VG, et al. Second-generation environmental sequencing unmasks marine metazoan biodiversity. Nat. Comm. 2010;1:98. doi: 10.1038/ncomms1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lambshead, P. J. D. in Nematode Morphology, Physiology and Ecology Vol. 1 (eds Chen, Z., Chen, S. & Dickson, D.) 438–492, Tsinghua Univ Press (2004).
- 50.Nichols RV, et al. Minimizing polymerase biases in metabarcoding. Mol. Ecol. Res. 2018;18:927–939. doi: 10.1111/1755-0998.12895. [DOI] [PubMed] [Google Scholar]
- 51.Warwick RM. A new method for detecting pollution effects on marine microbenthic communities. Mar. Biol. 1986;92:557–562. doi: 10.1007/BF00392515. [DOI] [Google Scholar]
- 52.Dean, H. K. (2001) Capitellidae (Annelida: Polychaeta) from the Pacific Coast of Costa Rica. Revista de Biología Tropical 69–84. [PubMed]
- 53.Holman LE, et al. Detection of introduced and resident marine species using environmental DNA metabarcoding of sediment and water. Sci. Rep. 2019;9:1–10. doi: 10.1038/s41598-019-47899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hoeksema BW, De Voogd NJ. On the run: free-living mushroom corals avoiding interaction with sponges. Coral Reefs. 2012;31:455–459. doi: 10.1007/s00338-011-0856-x. [DOI] [Google Scholar]
- 55.Bell JJ, Davy SK, Jones TT, Taylor MW, Webster NS. Could some coral reefs become sponge reefs as our climate changes? Glob. Chang. Biol. 2013;19:2613–2624. doi: 10.1111/gcb.12212. [DOI] [PubMed] [Google Scholar]
- 56.Tylianakis JM, Laliberté EE, Nielsen AA, Bascompte JJ. Conservation of species interaction networks. Biol. Cons. 2010;143:2270–2279. doi: 10.1016/j.biocon.2009.12.004. [DOI] [Google Scholar]
- 57.Karimi BB, et al. Microbial diversity and ecological networks as indicators of environmental quality. Environ. Chem. Lett. 2017;15:265–281. doi: 10.1007/s10311-017-0614-6. [DOI] [Google Scholar]
- 58.Zhou JJ, Deng YY, Luo FF, He ZZ, Yang YY. Phylogenetic molecular ecological network of soil microbial communities in response to elevated CO2. mBio. 2011;2:1–9. doi: 10.1128/mBio.00122-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lupatini MM, et al. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Environ. Sci. 2014;2:1–11. doi: 10.3389/fenvs.2014.00010. [DOI] [Google Scholar]
- 60.Karimi BB, Meyer CC, Gilbert DD, Bernard NN. Air pollution below WHO levels decreases by 40% the links of terrestrial microbial networks. Environ. Chem. Lett. 2016;14:467–475. doi: 10.1007/s10311-016-0589-8. [DOI] [Google Scholar]
- 61.Zappelini CC, et al. Diversity and complexity of microbial communities from a chlor-alkali tailings dump. Soil Biol. Biochem. 2015;90:101–110. doi: 10.1016/j.soilbio.2015.08.008. [DOI] [Google Scholar]
- 62.Graham SE, Chariton AA, Landis WG. Using Bayesian networks to predict risk to estuary water quality and patterns of benthic environmental DNA in Queensland. Integr. Environ. Assess. Manag. 2019;15:93–111. doi: 10.1002/ieam.4091. [DOI] [PubMed] [Google Scholar]
- 63.Blake JA, Arnofsky PL. Reproduction and larval development of the spioniform Polychaeta with application to systematics and phylogeny. Hydrobiologia. 1999;402:57–106. doi: 10.1023/A:1003784324125. [DOI] [Google Scholar]
- 64.Springer SA, Crespi BJ. Adaptive gamete-recognition divergence in a hydridizing Mytilus population. Evolution. 2007;61:772–783. doi: 10.1111/j.1558-5646.2007.00073.x. [DOI] [PubMed] [Google Scholar]
- 65.McCann KS. The diversity–stability debate. Nature. 2000;405:228. doi: 10.1038/35012234. [DOI] [PubMed] [Google Scholar]
- 66.Balvanera PP, et al. Quantifying the evidence for biodiversity effects on ecosystem functioning and services. Ecol. Lett. 2006;9:1146–1156. doi: 10.1111/j.1461-0248.2006.00963.x. [DOI] [PubMed] [Google Scholar]
- 67.Scheffer MM, et al. Early-warning signals for critical transitions. Nature. 2009;461:53. doi: 10.1038/nature08227. [DOI] [PubMed] [Google Scholar]
- 68.Work TM, Aeby GS, Maragos JE. Phase shift from a coral to a corallimorph-dominated reef associated with a shipwreck on Palmyra Atoll. PLoS One. 2008;3:e2989. doi: 10.1371/journal.pone.0002989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang SY, Bourgeois CC, Ashworth CD, Reimer JD. Palythoa zoanthid ‘barrens’ in Okinawa: examination of possible environmental causes. Zool. Stud. 2013;52:39. doi: 10.1186/1810-522X-52-39. [DOI] [Google Scholar]
- 70.Cruz IC, Meira VH, de Kikuchi RK, Creed JC. The role of competition in the phase shift to dominance of the zoanthid Palythoa cf. variabilis on coral reefs. Mar. Env. Res. 2016;115:28–35. doi: 10.1016/j.marenvres.2016.01.008. [DOI] [PubMed] [Google Scholar]
- 71.Wee HB, et al. Zoantharian abundance in coral reef benthic communities at Terengganu Islands, Malaysia. Reg. Stud. Mar. Sci. 2017;12:58–63. [Google Scholar]
- 72.Wilkinson, S. P., Stat, M. M., Bunce, M. M. & Davy, S. K. Taxonomic identification of environmental DNA with informatic sequence classification trees. Preprint at 10.7287/peerj.preprints.26812v1 (2018).
- 73.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 74.West KK, van Woesik RR. Spatial and temporal variance of river discharge on Okinawa (Japan): inferring the temporal impact on adjacent coral reefs. Mar. Pollut. Bull. 2001;42:864–872. doi: 10.1016/S0025-326X(01)00040-6. [DOI] [PubMed] [Google Scholar]
- 75.Roberts CM. Effects of fishing on the ecosystem structure of coral reefs. Conserv. Biol. 1995;9:988–995. doi: 10.1046/j.1523-1739.1995.9051332.x-i1. [DOI] [PubMed] [Google Scholar]
- 76.Barker NH, Roberts CM. Scuba diver behaviour and the management of diving impacts on coral reefs. Biol. Conserv. 2004;120:481–489. doi: 10.1016/j.biocon.2004.03.021. [DOI] [Google Scholar]
- 77.Mottaghi AA, Shimomura MM, Wee HB, Reimer JD. Investigating the effects of disturbed beaches on crustacean biota in Okinawa, Japan. Reg. Stud. Mar. Sci. 2017;10:75–80. [Google Scholar]
- 78.Gomez ED, Alcala AC. Status of Philippine coral reef. The project, Int. Symp. Biogeogr. Evol. S. Hem. Auckland New Zealand. 1978;2:663–669. [Google Scholar]
- 79.Van Woesik RR, Ripple KK, Miller SL. Macroalgae reduces survival of nursery‐reared Acropora corals in the Florida reef tract. Restor. Ecol. 2018;26:563–569. doi: 10.1111/rec.12590. [DOI] [Google Scholar]
- 80.Pochon XX, Bott NJ, Smith KF, Wood SA. Evaluating detection limits of next-generation sequencing for the surveillance and monitoring of international marine pests. PloS One. 2013;8:e73935. doi: 10.1371/journal.pone.0073935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brian JI, Davy SS, Wilkinson SP. Elevated Symbiodiniaceae richness at Atauro Island (Timor-Leste): a highly biodiverse reef system. Coral Reefs. 2019;38:123–136. doi: 10.1007/s00338-018-01762-9. [DOI] [Google Scholar]
- 82.Rix LL, et al. Coral mucus fuels the sponge loop in warm-and cold-water coral reef ecosystems. Sci. Rep. 2016;6:18715. doi: 10.1038/srep18715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Drummond, A. J. et al. Geneious v 4.8.4, Available from http://www.geneious.com (2009).
- 84.Huson DH, Weber NN. Microbial community analysis using MEGAN. Methods Enzymol. 2013;531:465–485. doi: 10.1016/B978-0-12-407863-5.00021-6. [DOI] [PubMed] [Google Scholar]
- 85. WoRMS Editorial Board, World Register of Marine Species. Available from http://www.marinespecies.org at VLIZ. Accessed 2019-08-10. 10.14284/170 (2017).
- 86.Faust, K. K. & Raes, J. J. CoNet app: inference of biological association networks using Cytoscape. F1000Research 5 (2016). [DOI] [PMC free article] [PubMed]
- 87.Brown MB. 400: a method for combining non-independent, one-sided tests of significance. Biometrics. 1975;31:987–992. doi: 10.2307/2529826. [DOI] [Google Scholar]
- 88.Tylianakis JM, Morris RJ. Ecological networks across environmental gradients. Annu. Rev. Ecol. Evol. S. 2017;48:25–48. doi: 10.1146/annurev-ecolsys-110316-022821. [DOI] [Google Scholar]
- 89.Reshef DN, et al. Detecting novel associations in large data sets. Science. 2011;334:1518–1524. doi: 10.1126/science.1205438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Saito RR, et al. A travel guide to Cytoscape plugins. Nature Methods. 2012;9:1069. doi: 10.1038/nmeth.2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Assenov YY, Ramírez FF, Schelhorn SE, Lengauer TT, Albrecht MM. Computing topological parameters of biological networks. Bioinformatics. 2008;24:282–284. doi: 10.1093/bioinformatics/btm554. [DOI] [PubMed] [Google Scholar]
- 92.Dong JJ, Horvath SS. Understanding network concepts in modules. BMC Syst. Biol. 2007;1:24. doi: 10.1186/1752-0509-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yachi SS, Loreau MM. Biodiversity and ecosystem productivity in a fluctuating environment: the insurance hypothesis. PNAS. 1999;96:1463–1468. doi: 10.1073/pnas.96.4.1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clarke, K. K. & Gorley, R. R. PRIMER V7: User Manual/tutorial. PRIMER-E Ltd, Plymouth, UK, pp. 296 (2015).
- 95.Dufrêne MM, Legendre PP. Species assemblages and indicator species: The need for a flexible asymmetrical approach. Ecol. Monogr. 1997;67:345–366. [Google Scholar]
- 96.De Cáceres MM, Legendre PP. Associations between species and groups of sites: indices and statistical inference. Ecology. 2009;90:3566–3574. doi: 10.1890/08-1823.1. [DOI] [PubMed] [Google Scholar]
- 97.Development Core Team. R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing (2015).
- 98.Oksanen JJ, et al. vegan: Community Ecology Package. R package version. 2019;2:5–4. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the study are available from Dryad Digital Repository 10.5061/dryad.m37pvmczf.