

Abstract
Vγ9Vδ2 T cell-based anticancer immunotherapy has shown some promise in early-phase clinical trials but there is still large room for improvement. Using the extracellular domain of the human NKG2D, a stimulatory receptor expressed by Vγ9Vδ2 T cells, we constructed NKG2D ligand-specific chimeric antigen receptors (CARs). We adopted a non-viral CAR approach via mRNA electroporation to modify Vγ9Vδ2 T cells and demonstrated that, upon interaction with the NKG2D ligand-positive cancer cells, the CARs substantially enhanced the cytotoxic activity of the modified cells toward multiple cultured solid tumor cell lines, including those resistant to Zometa treatment. Repeated doses of the CAR-expressing cells resulted in tumor regression in mice with established tumors, extending median survival time by up to 132% as compared to the PBS control group. The findings suggest clinical potential for RNA CAR-modified Vγ9Vδ2 T cells to treat a wide variety of NKG2D ligand-expressing cancers.
Keywords: gamma delta T cells, chimeric antigen receptor, NKG2D receptor, cancer therapy
Graphical Abstract
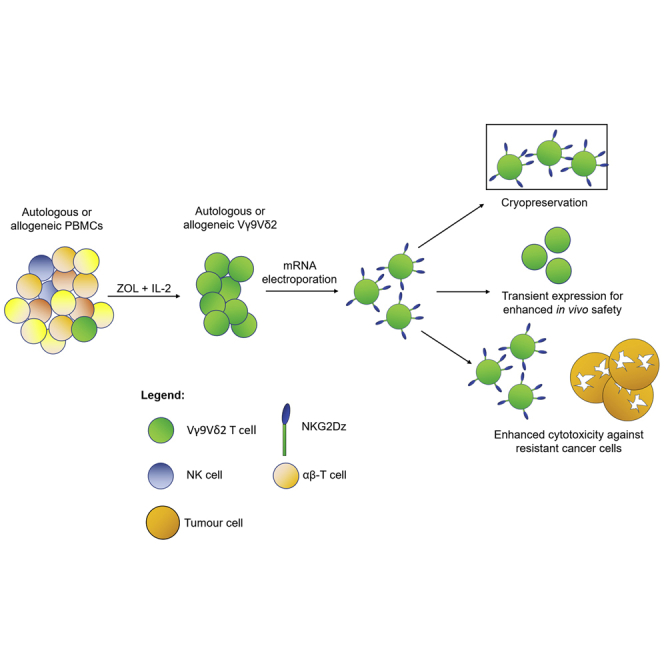
NKG2Dz-expressing Vγ9Vδ2 CAR-T cells were generated through mRNA electroporation and demonstrated enhanced anti-tumor activity against NKG2D ligand-positive cancer cells, including those resistant to Zometa treatment. While a transient mRNA expression allows management of on-target off-tumor effects, repeated infusions of mRNA CAR-T cells greatly enhanced survival of tumor-bearing mice.
Introduction
Peripheral blood gamma delta (γδ) T cells that express γδ heterodimer of T cell receptor (TCR) chains are a minor population (1%–5% in healthy adults), the majority of which express the variable gene segments Vγ9 and Vδ2 (Vγ9Vδ2 T cells).1 Through their TCR, γδ T cells can recognize and interact in a non-MHC restricted fashion with tumor-associated antigens, including phosphoantigens that are produced during metabolic dysregulation in tumor cells.1, 2, 3, 4, 5 Once activated, γδ T cells exert potent effector functions to kill target cells by secreting cytotoxic molecules like granzyme and perforin, as well as involving the death receptor-ligand systems.
γδ TCR recognition of tumor-associated antigens is strengthened by the expression of co-stimulatory receptors, among which is the natural killer group 2 member D (NKG2D) receptor.2 The NKG2D receptor, expressed by human natural killer (NK) cells, CD8+ T cells, γδ T cells, and NKT cells, can interact with eight NKG2D ligands (NKG2DLs) belonging to two families: two MHC class I chain-related proteins MICA and MICB and six human cytomegalovirus (HCMV) UL16-binding proteins (ULBP1-6). The interaction between NKG2D and its ligands can efficiently induce NK cell activation. In γδ T cells and CD8+ T cells, the interaction has been described to provide a potent co-stimulatory signal for antigen-mediated activation.6 Furthermore, similar to the NKG2D-mediated activation of NK cells, antigen-independent, NKG2D-mediated activation has been demonstrated in Vγ9Vδ2 T cells.7 NKG2DLs are not usually expressed on healthy tissues, and can, however, be upregulated upon DNA damage, infection, and cell transformation, thus being commonly detected on carcinomas.8, 9, 10 Because of their tumor-associated overexpression feature, NKG2DLs have been a favorable therapeutic target for anticancer strategies.8, 9, 10
Critically for cancer therapy, γδ T cells are capable of infiltrating a range of human malignancies and viewed as an important component of tumor-infiltrating lymphocytes (TILs).11,12 With a meta-analysis of expression signatures from ~18,000 human tumors, the presence of intratumoral γδ T cells has been ranked as the single most prognostically favorable immune-cell infiltrate identified across a panel of 39 tumor types.13 Vγ9Vδ2 T cells have been tested in several early-phase clinical trials for adoptive cancer immunotherapy by taking advantage of the efficient ex vivo expansion of Vγ9Vδ2 T cells with isopentenyl pyrophosphate (IPP), its synthetic analog of bromohydrin pyrophosphate (BrHPP), or Zoledronate or zoledronic acid (Zometa).11,14 Zometa is a FDA-approved, commercially available bisphosphonate drug that has been used to treat patients with postmenopausal osteoporosis. While T cells with αβ TCRs play a central role in inducing graft-versus host-disease (GvHD), Vγ9Vδ2 T cells are less prone to alloreactivity and, therefore, should be less likely to cause GvHD.15 Indeed, adoptive transfer of allogeneic γδ T cells did not lead to acute or chronic GvHD and was accompanied by anti-tumor activity in humans.16 The observations of these clinical trials suggest that the regimen is very well tolerated and can yield positive clinical outcomes, but failures to achieve primary clinical end-points are still common in the trials.11,14
To improve the efficacy of adoptive T cell therapy, chimeric antigen receptors (CARs), composed of an antigen recognition domain and an intracellular signaling domain of CD3zeta chain, have been developed to modify immune effector cells by gene transfer. CARs can redirect the specificity of immune cells to surface antigens, including NKG2DLs, expressed on tumor cells.17, 18, 19, 20 We hypothesized that after introducing a CAR specific to NKG2DLs into expanded Vγ9Vδ2 T cells, the binding of the CAR to the ligands expressed on tumor cells could activate the cells directly through CD3zeta, thus enhancing the antitumor immunity of Vγ9Vδ2 T cells. To test the hypothesis, we have constructed several CARs that use the extracellular domain (ED) of the human NKG2D receptor to target NKG2DLs. In order to minimize the potential risk of on-target/off-tumor toxicity against normal tissues, we adopted an RNA CAR approach to transiently enhance the specificity of Vγ9Vδ2 T cells toward NKG2DLs and their tumor cell killing activity.
Results
Vγ9Vδ2 T Cells Electroporated with NKG2Dz RNA CAR Display an Improved In Vitro Killing Activity against Multiple Human Solid Tumor Cell Lines
Four different NKG2DL-targeting CAR constructs were prepared initially, which share the same fragments of the human NKG2D ED, a CD8α hinge and transmembrane region, and the intracellular signaling domain CD3zeta. These CAR constructs differ in co-stimulatory domains, varying from no co-stimulatory domain (1st generation CAR), one co-stimulatory domain (2nd generation CAR), to two co-stimulatory domains (3rd generation CAR). The control vector mGFP CAR was generated by replacing the NKG2D-ED fragment with the GFP sequence. To introduce CAR-encoding mRNA into Vγ9Vδ2 T cells, we used a K562 artificial antigen-presenting cell (aAPC)-based method previously established in the lab for the expansion of Vγ9Vδ2 T cells and electroporated the expanded cells.21 RNA electroporation was optimized using the mGFP CAR, with the transfection efficiency reaching 96%, the cell viability being approximately 65%, and the transgene expression lasting for at least 7 days in Vγ9Vδ2 T cells (Figure S1). We compared the cell viability and the tumor cell killing activities of the 4 constructs after electroporation of their mRNA molecules into Vγ9Vδ2 T cells and selected the first generation NKG2D CAR (NKG2Dz, Figure 1A) that showed the highest activity among the 4 tested RNA CARs (Figure S2) for detailed investigations in the current study.
Figure 1.

Tumor Cell Lysis Induced by Vγ9Vδ2 T Cells Modified with NKG2D CAR
(A) Schematics of the plasmid constructs used for CAR mRNA production: NKG2D CAR containing the NKG2D extracellular domain (ED) and CD3ζ, and a control CAR replacing NKG2D ED with the membrane binding GFP (mGFP CAR). The DNA templates of the CARs were PCR amplified using a CMV forward primer and reverse primer with 150 Ts. The PCR amplicons were then used for RNA transcription to generate mRNA molecules encoding the CARs for the electroporation of Vγ9Vδ2 T cells (γδ T). (B) Flow cytometric analysis to demonstrate the NKG2D expression on Vγ9Vδ2 T cells. Black lines represent wild-type γδ T cells stained with an isotype control antibody. Red lines represent wild-type γδ T cells stained with an anti-NKG2D antibody to show the expression of endogenous NKG2D receptor. Blue lines represent γδ T cells electroporated with a CAR construct and stained with the anti-NKG2D antibody. Cell samples were collected 24 h post-electroporation for staining. The results of one representative experiment out of three independent experiments with three different donors are shown. (C) Time-lapse analysis of NKG2D CAR expression after electroporation. Vγ9Vδ2 T cell samples were collected at the indicated time points post-electroporation for staining for flow cytometric analysis. Black lines represent wild-type Vγ9Vδ2 T cells stained with an isotype control antibody. Red lines represent wild-type γδ T cells stained with an anti-NKG2D antibody to show the expression of endogenous NKG2D receptor. Blue lines represent γδ T cells electroporated with mGFP CAR and stained with the anti-NKG2D antibody. Yellow lines represent cells electroporated with NKG2D CAR and stained with the anti-NKG2D antibody. The results of one representative experiment out of three independent experiments with three different donors are shown. (D) Delfia EuTDA cytotoxicity assay (3 h EuTDA culturing) used to assess tumor cell lysis efficiency. Human HepG2 hepatocellular carcinoma cell line, MCF7 breast adenocarcinoma cell line, CAOV3 ovarian cancer cell line, Detroit562 pharyngeal carcinoma cell lines that express NKG2D ligands (NKG2DL+), and A549 pulmonary adenocarcinoma cell line that does not express NKG2D ligands (NKKG2DL−) were tested. The results of one representative experiment out of at least two independent experiments with different donors are shown. The differences between NKG2D CAR γδ T and mGFP CAR γδ T are statistically significant (p < 0.001) for all tested tumor cell lines except A549.
While electroporation with the mGFP CAR did not increase NKG2D expression, the electroporation of Vγ9Vδ2 T cells with NKG2Dz RNA CAR significantly increased NKG2D expression (Figures 1B). Time-lapse analysis demonstrated that the increased NKG2Dz CAR expression maintained for at least 5 days (Figure 1C).
The cytotoxicity of NKG2Dz CAR-equipped Vγ9Vδ2 T cells was investigated against various types of human tumor cell lines, including NKG2DL-positive human HepG2 hepatocellular carcinoma cell line, MCF7 breast adenocarcinoma cell line, CAOV3 ovarian cancer cell line, and Detroit562 pharyngeal carcinoma cell line and NKG2DL-negative A549 human lung adenocarcinoma cell line (Figure S3). HepG2, MCF7, CAOV3, and Detroit562 tumor cells were highly susceptible to NKG2Dz CAR-equipped Vγ9Vδ2 T cells, whereas A549 cells were resistant, only showing approximately 20% cell lysis at an effector to target (E:T) ratio of 20:1 (Figure 1D).
Cryopreservation of CAR-modified immune cells is an important component of adoptive cell therapy and is often required for safety testing prior to clinical application. We investigated whether cryopreservation and storage at −150°C affected the viability and potency of Vγ9Vδ2 T cells electroporated with NKG2Dz RNA CAR. The survival of Vγ9Vδ2 T cells after electroporation and 1 month of cryopreservation was 49.7% ± 14.6%. We allowed NKG2Dz-modified Vγ9Vδ2 T cells to rest for 24 h after thawing and before performing the GFP or NKG2D CAR expression analysis and cytotoxicity assays. Although GFP and NKG2D expression levels on the thawed cells were slightly lower compared to ones on freshly manipulated cells (Figure 2A), the cytolytic activities of fresh and thawed NKG2Dz-modified Vγ9Vδ2 T cells were similar when HCT116-Luc human colorectal cancer and SKOV3-Luc human ovarian cancer cells, the two cell lines used for the following animal experiments were tested, both mediating 100% killing of the tumor cells at E:T ratio of 20:1 (Figure 2B). When cytokine secretion was examined, incubation of thawed NKG2Dz-modified Vγ9Vδ2 T cells with SKOV3 cells resulted in robust production of interferon-γ (IFN-γ), tumor necrosis factor alpha (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF) (Figure 2C). Thus, cryopreservation did not affect the potency of NKG2Dz-modified Vγ9Vδ2 T cells and the cryopreserved CAR-Vγ9Vδ2 T cells were used in the following animal experiments.
Figure 2.

Effects of Cryopreservation on the Cytotoxicity of Vγ9Vδ2 T Cells Modified with NKG2D CAR
(A) Flow cytometric analysis of GFP and NKG2D expression on Vγ9Vδ2 T cells (γδ T) and Vγ9Vδ2 T cells modified with NKG2D CAR (NKG2Dz) before freezing (24 h post-electroporation) and 24 h after thawing. The results of one representative experiment out of three independent experiments with three different donors are shown. (B) Delfia EuTDA cytotoxicity assay (2 h EuTDA culturing) to assess tumor cell lysis efficiency. Fresh: NKG2D CAR mRNA electroporated cells were analyzed before freezing (24 h post-electroporation). Thawed: the electroporated cells were cryopreserved, stored in a liquid nitrogen tank for 1 month, and analyzed 24 h after thawing. The results of one representative experiment out of three with different donors are shown. The differences between NKG2Dz-γδ T and mGFP-γδ T are statistically significant (p < 0.001) in all tested groups. (C) Thawed Vγ9Vδ2 T cells modified with NKG2D CAR produce robust levels of cytokines in response to SKOV3-Luc tumor cells when compared with mock-γδ T (∗∗∗) or mGFP CAR-modified γδ T (+++). Data represent the mean ± SD of Vγ9Vδ2 T cells from three different PBMC samples. ∗∗∗p < 0.001 and +++p < 0.001, statistical significance between NKG2Dz-γδ T and mock-γδ T and between NKG2Dz-γδ T and mGFP-γδ T, respectively.
Multiple Injections of Vγ9Vδ2 T Cells Electroporated with NKG2Dz RNA CAR Delay Disease Progression in Tumor-Bearing Mice
To obtain a proof-of-concept of the in vivo tumor killing effect of the NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells, we established a colorectal cancer mouse model by intraperitoneal (i.p.) injection of 1 × 107 HCT116-Luc aggressive human colorectal cancer cells into NSG mice. 7 days post-tumor cell injection, tumor-bearing mice were treated with i.p. injection of the NKG2Dz or control mGFP CAR modified Vγ9Vδ2 T cells (1 × 107 cells per injection) or PBS, twice a week for 3 weeks (Figure 3A). Tumor growth was monitored with non-invasive whole-body bioluminescent imaging (BLI) of HCT16-Luc cells from day 7 to day 42 (Figures 3B and 3C). The BLI intensities, indicative of tumor burden and distribution, demonstrated that HCT116 tumors progressed aggressively in the control groups treated with PBS or Vγ9Vδ2 T cells only. The repeated injections of NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells markedly suppressed tumor progression, resulting in the tumor growth inhibition in one out of the 5 treated mice and slowing down tumor growth in the rest 4 mice as compared to the controls (Figure 3C). Tumor regrowth was noticed after termination of the treatment. Kaplan-Meier analysis of survival data is shown in Figure 3D. Due to rapid disease progression, mice receiving PBS treatment had to be euthanized by day 41 and their median survival time was 39.5 days. Treatments with Vγ9Vδ2 T cells only increased median survival to 43 days. The repeated injections of the NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells resulted in median survival of 57 days, prolonging median survival time by 44% as compared to that in the PBS group (p < 0.0001).
Figure 3.

Effects of NKG2D CAR-Expressing Vγ9Vδ2 T Cells in a Human CRC Xenograft Model
(A) Experimental outline for the animal study to examine in vivo killing capacity against intraperitoneal (i.p.) xenografts of human HCT116-Luc colorectal cancer (CRC) cells. NSG mice were i.p. injected with 1 × 107 HCT116-luc cells on day 0. The mice were randomized into different groups (n = 6 per group) before beginning CAR-modified Vγ9Vδ2 T cell (γδ T) treatment on day 7. The treatment was conducted twice a week for 3 weeks, 1 × 107 cells per i.p. injection, using PBS and γδ T cells only as controls. (B) Bioluminescent images prior to treatment (day 7) and following the treatments on days 21 and 35. (C) Tumor burden over time by bioluminescent imaging after the treatment. Each mouse is represented by one line. (D) Kaplan-Meier analysis of survival. Statistical analysis of survival between groups was performed using the log-rank test. ∗p < 0.05; ∗∗∗p < 0.001.
We established another xenograft mouse model of human ovarian carcinoma by i.p. injection of 1 × 107 SKOV3-Luc human ovarian cancer cells into NSG mice and tested whether an increase in number of injections would improve the efficacy of multiple injections of NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells. Four groups of animals were used in this experiment: a PBS control group and three treatment groups in which animals received 6, 10, or 14 i.p. injections of CAR-T cells. CAR-T cell injection started on day 7 post-tumor inoculation, twice a week for 3, 5, or 7 weeks (Figure 4A). After SKOV3-Luc inoculation, tumors progressed rapidly in the PBS group (Figures 4B and 4C), resulting in animal death/euthanasia due to being moribund within 43 days (median survival: 41 days, Figure 4D). Tumor burdens after the treatment of NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells were obviously reduced relative to the initial tumor burdens in all three treatment groups (Figures 4B and 4C). With the increase in number of injections, greater reduction in tumor burden was observed. The effects were sustained during the treatment periods, although tumors were not completely eradicated even after 14 injections and regrew within 1 week after termination of the treatments (Figure 4C). The tumor burden reduction correlated with an increased survival of the treated mice (Figure 4D). The median survival times after 6, 10, and 14 injections of the CAR-T cells were 69, 90, and 95 days, respectively, extending median survival by 68%, 120%, and 132%, respectively, as compared to the PBS control (p < 0.0001).
Figure 4.

Effects of Multiple i.p. Injections of Vγ9Vδ2 T Cells Electroporated with NKG2D CAR (NKG2Dz) on Disease Progression in Tumor-Bearing Mice
(A) Experimental outline of the animal study. NSG mice (n = 6 per group) were i.p. injected with SKOV3-Luc human ovarian cancer cells (1 × 107 per mouse). The treatment started 7 days after tumor cell inoculation, twice a week for 3, 5, and 7 weeks, 1 × 107 CAR-modified Vγ9Vδ2 T cells (γδ T) per injection. PBS injection, twice a week for 3 weeks, was included as a control. (B) Bioluminescent images prior to treatment (day 7) and following the treatments on days 28 and 42. (C) Tumor burden over time by bioluminescent imaging after multiple i.p. injections of CAR-modified Vγ9Vδ2 T cells. Each mouse is represented by one line. (D) Kaplan-Meier analysis of survival. Statistical analysis of survival between groups was performed using the log-rank test. ∗p < 0.05; ∗∗p < 0.01.
In Vivo Treatment with CAR-Modified Vγ9Vδ2 T Cells Is More Effective in Delaying Tumor Progression than That with the Vγ9Vδ2 T Cells Plus Zometa
Since Zometa treatment can increase intracellular levels of IPP, thus sensitizing tumor cells to attack by Vγ9Vδ2 T cells, Zometa pre-treatment has been used for adoptive Vγ9Vδ2 T cell therapy. We examined the effects of Zometa pre-treatment on Vγ9Vδ2 T cell- and NKG2Dz RNA CAR-mediated in vitro tumor cell killing at an E:T ratio of 10:1 (Figure 5A). Without Zometa pre-treatment of tumor cells, Vγ9Vδ2 T cells or mGFP-modified Vγ9Vδ2 T cells did not kill the 3 tested tumor cell lines, including human SW480 colon carcinoma cells, HepG2 hepatocarcinoma cells, and SKOV3 ovarian cancer cells. In contrast, NKG2Dz-modified Vγ9Vδ2 T cells were able to kill these tumor cells that were not pre-treated with Zometa, with the efficiency varying from 40% to 80%. The sensitivity of SW480 and SKOV3 cells, but not HepG2 cells, toward Vγ9Vδ2 T cells was significantly increased by Zometa pre-treatment. However, probably because NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells were already highly effective in killing tumor cells even without Zometa pre-treatment, Zometa pre-treatment did not significantly increase the in vitro tumor cell sensitivity toward these CAR-modified Vγ9Vδ2 T cells.
Figure 5.

Zometa Treatment and Effects of Vγ9Vδ2 T Cells Expressing NKG2D CAR (NKG2Dz)
(A) In vitro cytolytic effects on Zometa-treated tumor cells. Target tumor cells tested were HepG2 human hepatocarcinoma cells, SW480 human colon carcinoma cells, and SKOV3 human ovarian cancer cells. Tumor cells were either pre-treated with 1 μM Zometa (+Zometa) or without Zometa treatment. Effector cells tested include Vγ9Vδ2 T cells (γδ T), γδ T electroporated with mGFP CAR, and γδ T electroporated with NKG2Dz RNA CAR. Delfia EuTDA cytotoxicity assay (3 h EuTDA culturing) was used to assess tumor cell lysis efficiency at an effector to target ratio of 10:1. The results of one representative experiment out of three are shown. Data shown are mean ± SD of triplicates. (B) Experimental outline of the animal study. NSG mice (n = 6 per group) were i.p. injected with the SKOV3-Luc human ovarian cancer cells (1 × 107 per mouse). The treatment started 7 days after tumor cell inoculation, twice a week for 3 weeks. Zometa (1 mg/kg) was i.p. injected 1 day before cell injection, followed by 1 × 107 Vγ9Vδ2 T cells per i.p. injection 1 day later. The mice were followed with serial weekly imaging to assess the tumor burden. (C–F) In vivo effects. (C) Tumor burden images by bioluminescent imaging (BLI) on days 7, 28, and 42. (D) Quantitative analysis of SKOV3-luc cell bioluminescence signals obtained in the in vivo experiment. Tumor burden over time by BLI is shown. Each mouse is represented by one line. (E) Kaplan-Meier analysis of survival of the in vivo experiment. Statistical analysis of survival between groups was performed using the log-rank test. (F) Blood human Vγ9Vδ2 T cells. Mouse blood samples were collected 7 days after the last CAR-T cell injection for flow cytometric analysis. Examples of anti-human TCR Pan γδ antibody and anti-human TCR Vδ2 antibody double staining from each group with mean percentages (SD) of human Vγ9Vδ2 T cells are shown. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; NS, not significant.
We then compared in vivo tumor killing activity of NKG2Dz CAR-modified Vγ9Vδ2 T cells in mice bearing SKOV3-Luc xenografts with or without Zometa treatment (Figure 5B). 7 days after tumor inoculation, three groups of mice started to receive treatments of PBS, Vγ9Vδ2 cells transfected with the control mGFP RNA CAR plus Zometa (γδ T-Z), or NKG2Dz RNA CAR-modified Vγ9Vδ2 T cells plus Zometa (NKG2Dz-γδ T-Z). Cells (1 × 107 per dose) were i.p. injected twice a week for 3 weeks. Zometa (100 μg/kg) was i.p. injected 1 day before cell injection. Figure 5C shows images of mice in each group at 3 different time points post-tumor cell injection. BLI demonstrated the obvious inhibitory effect of the NKG2Dz-γδ T-Z treatment on tumor growth as compared with the PBS group and the γδ T-Z group (Figure 5D). On day 28, while established tumors continued to grow in PBS-treated mice or remained stable in mice treated with γδ T-Z, the tumor burdens in mice receiving NKG2Dz-γδ T-Z were significantly reduced relative to the initial levels (p < 0.001). Noticeably, significant reduction of the disease by the NKG2Dz-γδ T-Z treatment maintained for >2 weeks in 4 out of 6 treated mice after termination of the treatment (Figure 5D). Consequently, the NKG2Dz-γδ T-Z treatment significantly prolonged the survival of the tumor-bearing mice over the two controls (Figure 5E), and the median survival time increased from 42 days in the PBS control group and 51.5 days in the Vγ9Vδ2 T-Z group to 72.5 days (p < 0.0001). Thus, the NKG2D-γδ T-Z treatment prolonged median survival by 73% and 41% as compared to the PBS group and the γδ T-Z group, respectively. We also compared mean photon flux values of tumor burden on day 28 from 10 groups of mice used in the three animal experiments shown in Figures 3, 4, and 5 and the values from mice treated with PBS were used as an internal control for cross-experiment comparison (Table S1). The comparison indicates that among the 4 treatment regimens examined in our work, Vγ9Vδ2 T cell alone, mGFP CAR Vγ9Vδ2 T+Zometa, NKG2D CAR Vγ9Vδ2 T, and NKG2D CAR Vγ9Vδ2 T+Zometa, the last was the most effective one.
We further analyzed mouse peripheral blood samples collected from the three groups of mice shown in Figure 5 7 days after the last treatment and analyzed percentages of human Vγ9Vδ2 T cells. As expected, no human Vγ9Vδ2 T cells were detected in mice treated with PBS. An increased percentage of human Vγ9Vδ2 T cells in the mouse peripheral blood was detectable after the NKG2Dz-γδ T-Z treatment, as compared to that of the Vγ9Vδ2 T-Z group (Figure 5F), indicating that improved in vivo persistence or cell proliferation of NKG2Dz CAR-modified Vγ9Vδ2 T cells could contribute to the higher in vivo tumor killing activity of the CAR-modified cells. Overall, while Vγ9Vδ2 T cells plus Zometa treatment were able to kill some solid tumor cells, the tumor-killing efficacy was further enhanced significantly by the modification of the effector cells with CAR gene transfer.
Discussion
Several previous preclinical studies have tested NKG2DL-targeting CAR constructs with the full-length NKG2D receptor fused with CD3zeta, in which the NKG2D adaptor protein DAP10 is engaged to provide co-stimulation and enhance the surface expression of the NKG2D CAR.22, 23, 24, 25 Other designs of NKG2D CARs have used the NKG2D ED only, which is fused to a hinge and transmembrane domain followed by CD28 or 4-1BB signaling domain in tandem with CD3zeta.26, 27, 28, 29 These NKG2D CAR studies were pursued with either αβ T cells or NK cells.22, 23, 24,26, 27, 28, 29, 30, 31, 32, 33, 34, 35 Positive outcomes from the preclinical investigations led to the initiation of a phase I clinical trial in 2015 to evaluate the safety and feasibility of a single dose of autologous αβ T cells expressing a first-generation NKG2D CAR to treat patients with acute myeloid leukemia or multiple myeloma (ClinicalTrials.gov Identifier: NCT02203825). After confirming the initial safety and tolerability of NKG2D CAR-expressing T cells in NCT0220382, a multinational open-label phase I study with a multiple dosing approach (NCT03018405, THINK trial) was launched in January 2017 to assess the safety and clinical activity of αβ T cells modified with the same NKG2D CAR in solid tumors and hematological tumors.
Although it has been well recognized that γδ T cell function requires NKG2D,6 the impact of these cells as immune effector cells in NKG2D CAR-mediating anti-tumor activity has not been evaluated before. In the current study, we tested NKG2DL-targeting CAR by fusing the NKG2D ED with the CD3zeta signaling domain only, without the use of a costimulatory domain such as CD28 or 4-1BB, in light of our pilot study demonstrating the superior cytolytic activity of the 1st generation of NKG2D RNA CAR against cancer cells as compared to the 2nd and 3rd generation RNA CARs (Figure S2). A previous toxicology study also showed that the NKG2D CAR with the full-length receptor fused to cytoplasmic CD3zeta alone was minimally toxic, in contrast to the one co-expressed with DAP10 and the NKG2D-CD28-CD3zeta CAR.36
Almost all the previous NKG2D CAR studies were pursued with immune effector cells transduced with integrating viral vectors. This viral transduction approach provides the advantage for long-term CAR expression on infused cells across multiple cell divisions, thus achieving functional persistence of CAR-expressing cells. However, transduction of effector cells with a viral vector also comes with certain practical constraints, such as being laborious and expensive for large-scale, cGMP-compliant virus manufacturing. Theoretically, the use of integrating viral vectors is associated with the concern of insertional mutagenesis. When applied clinically, permanent DNA CAR expression mediated by integrating viral vectors cannot be shut off quickly on the occasion of the occurrence of severe toxicity associated with cytokine storm or on-target/off-tumor toxicity. VanSeggelen et al.36 have reported a strain- and preconditioning-dependent lethal toxicity in mice treated with αβ T cells transduced with retroviral vectors encoding NKG2D CARs, especially when a 2nd generation NKG2D CAR (NKG2D28ζ) and a NKG2D CAR co-expressed with DAP10 (NKG2Dζ10) were used.37 Similarly, retrovirally transduced, CD19-specific CAR T cells, when tested in fully syngeneic mouse models, induced toxicity associated with 2nd generation CD28-containing CAR constructs as compared to the 1st generation CAR constructs.34 The αβ T cells modified with the 2nd generation CAR were able to eradicate established tumor and persisted for extended periods, but induced B cell aplasia and chronic toxicity possibly because of the chronic effects of the stable CAR expression on emerging normal CD19-positive B cells from the bone marrow. These observations suggest that the pursuit of virus-transduced CAR-T cell therapies should be undertaken with caution and additional safety measures to stop CAR gene expression may be necessary.
CARs can also be expressed by transfection through mRNA electroporation.38,39 Using short-lived CAR-expressing cells modified with this non-integrating technology, the duration and potency of CAR effects can be controlled by different dosing and infusion schemes. Understandably, transient CAR expression necessitates multiple infusions of a relatively higher dose of CAR-modified immune effector cells to achieve antitumor effects. It, however, provides convenience in controlling CAR toxicity. Through discontinuing the infusion of the modified effector cells, an excessive response caused by the toxicity related to recognition of normal tissues and/or cytokine storms can be stopped. Moreover, transfection of mRNA encoding a CAR is more economical in testing new CARs, particularly when performing an initial clinical trial of a novel CAR. There were two previous studies that assessed in vitro effects of NKG2D CAR mRNA transfected αβ T cells and NK cells, respectively,24,26 but the in vivo impact of these RNA CARs in an animal model has not been evaluated previously. To further obviate the safety concern for on target/off tumor toxicity, we adopted a local i.p. injection procedure, instead of commonly used systemic intravenous (i.v.) injection of CAR-T cells, in our animal studies.
Expression of the NKG2Dz RNA CAR in the current study significantly augmented the cytolytic activity of Vγ9Vδ2 T cells against all cultured tumor cell lines tested, consistent with the finding that NKG2D ligands are widely upregulated on a variety of cancer types. As the main effort of the study, we investigated whether Vγ9Vδ2 T cells with NKG2Dz RNA CAR display significant anti-tumor effects in two mouse models with established tumors, where SKOV3-Luc ovarian cancer and HCT116-Luc colorectal cancer cells were injected i.p., respectively. Both animal models displayed fast progressive tumor growth, leading to extensive peritoneal carcinomatosis with hemorrhagic ascites and metastases to serous membranes, liver, spleen, and in some animals, lung (data not shown). Without cell therapy, mice died or had to be euthanized approximately 41 days post-tumor inoculation in the HCT116-Luc model and 52 days in the SKOV3-Luc model. Probably because of the difference in tumor aggressiveness between the two tumor models, we observed obvious tumor regression during CAR-T cell treatment in the SKOV3-Luc model, while the HCT116-Luc model appeared to be therapeutically challenging: the treatment was not sufficient to induce tumor regression but resulted in slowed tumor growth only (Figure 3). We further demonstrated that an increase in dosing frequency (Figure 4) or multiple injections in combination with Zometa treatment (Figure 5) provide additional survival benefits to tumor-bearing mice. Collectively, the present study provides convincing evidence that repeated doses of Vγ9Vδ2 T cells modified with an NKG2D RNA CAR can result in notable therapeutic outcomes in animal tumor models.
The relatively low frequency of Vγ9Vδ2 T cells in the peripheral blood is one of the practical challenges in generating CAR-engrafted Vγ9Vδ2 T cells and necessitates the need of using a powerful method for the enrichment and expansion of CAR Vγ9Vδ2 T cells. We have successfully developed a K562-aAPC based method for massive expansion of Vγ9Vδ2 T in gas-permeable G-Rex vessels in a good manufacturing practice (GMP)-compliant manner.21 An extremely high expansion rate has been achieved, enabling the generation of large clinically relevant quantities (2 to 15 billion) of functional Vγ9Vδ2 T cells from 2 mL of peripheral blood. With this powerful technology, costly and risky apheresis procedures can probably be eliminated for large-scale production of Vγ9Vδ2 T cells and the generation of multiple aliquots of cryopreserved cells for repeated infusions becomes possible and easy. Moreover, as demonstrated in the current study, the generated Vγ9Vδ2 T cells can be used as CAR-engrafted effector cells.
Vγ9Vδ2 T cells possess distinct features over αβ T cells for adoptive immunotherapy.40, 41, 42 While αβ TCRs recognize specific peptide fragments presented on MHC, γδ TCRs recognize cancer-associated, non-peptide phosphoantigens in an MHC-independent manner. Thus, Vγ9Vδ2 T cells theoretically offer attractive possibilities to develop “off-the-shelf” cell therapeutics for allogeneic use, giving them a key advantage over αβ T cells. Due to their unique cytokine release profile, low secretion of interleukin-2 (IL-2), IFN-γ, and TNF as compared to αβ T cells, the use of Vγ9Vδ2 T cells considerably has a lower risk of mediating unwanted side effects, including cytokine release syndrome. A recent study has shown that CAR-modified Vγ9Vδ2 T cells display a cytolytic activity similar to that of αβ T cells modified with the same CAR but a lower level of cytokine secretion,43 suggesting the possible superiority of CAR-Vγ9Vδ2 T cell therapy in risk mitigation for cytokine release syndrome. A crucial biological feature of Vγ9Vδ2 T cells, as potent innate immune effector cells, is the intrinsic antitumor activity against a large array of tumor types, which is mediated by their endogenous γδ TCRs and activating receptors and usually not determined by mutation-derived neoantigens. This may help in alleviating the rate of antigen escape relapse, a clinical dilemma that has been described in CD19-directed CAR T cell therapy. The APC-like phenotype and function of Vγ9Vδ2 T cells have been confirmed in clinical trials.44 CAR-grafted Vγ9Vδ2 T cells would retain the APC ability to take up tumor antigens and cross present the processed peptides to conventional CD4+ and CD8+ αβ T cells.45 Therefore, it is important to recognize that Vγ9Vδ2 T cells may mediate systemic immunity and provide indirect antitumor effects in cancer patients. The limited in vivo persistence of adoptively transferred Vγ9Vδ2 T cells appears to be a problem. This, however, potentially enable reducing the risks of chronic toxicity associated with prolonged presence of effector cells.
In conclusion, the current study demonstrates that electroporation of NKG2Dz RNA CAR can harness the tumor killing power of Vγ9Vδ2 T cells. Given that an RNA CAR approach can potentially minimize the risk of on-target/off-tumor toxicity, adoptive transfer of the transiently modified cells represents a novel alternative for CAR-T cell therapy against NKG2DL-positive tumors.
Materials and Methods
Cells and Cell Culture Conditions
Buffy coats of healthy donors were collected from National University Hospital Singapore, Department of Laboratory Medicine Blood Transfusion Service, as approved by the institutional review board of National University of Singapore (NUS-IRB Reference Code B-14-133E). Vγ9Vδ2 T cells were prepared as described in our previous publications.21,46 Human PBMCs were isolated from fresh buffy coats and activated by 5 μM Zometa (Sigma-Aldrich, St. Louis, MO, USA) plus 300 IU/mL IL-2 in 1 mL AIM-V (Life Technologies, Carlsbad, CA, USA) supplemented with 5% human AB serum (Valley Biomedical). After 7 days of Zometa treatment, cells were mixed with γ-irradiated K562 Clone A aAPCs at an immune cell:K562 ratio of 1:100 and the anti-CD3 antibody OKT3 (60 ng/mL, eBioscience, San Diego, CA, USA) for further expansion for 10 days. Our previous publication demonstrated that including K562 aAPC/OKT3 can significantly promote numerical expansion of Vγ9Vδ2 T cells (approximately 500,000-fold expansion), assist the maintenance of the TEM cell population, and promote the downregulation of immune checkpoint receptor on cell surface.21 Using this method, Vγ9Vδ2 T, with an initial frequency of approximately 2% in PBMCs, could be enriched to 89% in 17 days (NK cells: 1.0% and αβ T cells: 7.4% by day 17).
Eight different human solid tumor cell lines from ATCC (Manassas, VA, USA) were used in the current study. They are human HCT116-Luc and SW480 colorectal cancer cell lines, SKOV3-Luc and CAOV3 ovarian cancer cell lines, HepG2 hepatocellular carcinoma cell line, MCF7 breast adenocarcinoma cell line, Detroit562 pharyngeal carcinoma cell line, and A549 pulmonary adenocarcinoma cell line. SKOV3-Luc cells were cultured in McCoy’s 5A (Lonza Biotech, Basel, Switzerland) supplemented with 10% FBS. The rest tumor cell lines were cultured in in DMEM (Lonza) supplemented with 10% FBS (Hyclone, Logan, UT, USA).
Construction of Chimeric NKG2D CAR Vectors and Generation of CAR T Cells
To construct NKG2D RNA CAR vectors, we amplified the extracellular domain of human NKG2D by PCR from a PBMC cDNA library. The 1st, 2nd, and 3rd generation NKG2D CAR vectors were generated by fusing NKG2D-ED to the CD8α hinge and transmembrane region and CD3ζ signaling moiety, with or without the intracellular costimulatory domain of CD27, CD28, and CD28-41BB. The mGFP control vector was generated by replacing the NKG2D ED part of the NKG2Dz vector with the GFP encoding sequence. PCR was performed to generate mRNA molecules encoding the NKG2D CARs, which were then electroporated into Vγ9Vδ2 T cells.
Flow Cytometric Analysis and Cytotoxicity Assay
Flow cytometric analysis was performed with Accuri C6 cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Purity of Vγ9Vδ2 T cells was analyzed by co-staining of the Vδ2 (clone B6; BioLegend, USA) and Vγ9 (clone B3; BioLegend, USA) receptors. The cytolytic activity of CAR-modified T cells was examined with a non-radioactive method (DELFIA EuTDA Cytotoxicity Reagents kit, PerkinElmer, MA, USA).
Animal Experiments
Non-obese diabetic/severe combined immunodeficiency/IL-2Rγcnull (NSG) mice (6 to 8 weeks old, female) were used in the current study. All handling and care of animals was performed according to the guidelines for the Care and Use of Animals for Scientific Purposes issued by the National Advisory Committee for Laboratory Animal Research, Singapore. The animal study protocol was reviewed and approved by Institutional Animal Care and Use Committee (IACUC), the Biological Resource Centre, the Agency for Science, Technology and Research (A∗STAR), Singapore (permit number: BRC IACUC 110612).
NSG mice were inoculated via i.p. injection of 1 × 107 HCT116-luc or SKOV3-Luc cells to generate tumor models. To investigate in vivo anti-tumor effects of CAR-T cells, we i.p. injected 1 × 107 of human Vγ9Vδ2 T cells electroporated with NKG2Dz RNA CAR into tumor-bearing mice twice a week for 3 weeks. Mice treated with PBS or cells with a control mGFP CAR were used as controls. For the acute toxicology study, healthy NSG mice were given one dose of either PBS, 1 × 107 or 6 × 107 Vγ9Vδ2 T cells electroporated with NKG2Dz RNA CAR containing the mouse NKG2D ED.
Statistical Analysis
Data are presented as mean ± standard deviation (SD). All statistics were performed GraphPad Prism 7 (San Diego, CA, USA). p values <0.05 were considered significant. For details, see Supplemental Materials and Methods.
Author Contributions
Conceived and designed the experiments: S.W., J.Z., and H.C.T. Performed the experiments: W.X.A., Y.Y.N., L.X., C.C., Z.L., Z.C., J.C.K.T., and W.K.T. Analyzed the data: W.X.A., L.X., C.C., and Y.Y.N. Wrote the manuscript: S.W., W.X.A., and Y.Y.N.
Conflicts of Interest
The authors S.W., J.Z., and W.X.A. have filed a patent application for the NKG2D CAR-modified Vγ9Vδ2 T cells that has been licensed to CytoMed Therapeutics, Singapore. J.Z. and W.K.T. are currently employees of the company.
Acknowledgments
This work was supported by the Singapore Ministry of Health’s National Medical Research Council (NMRC/CIRG/1406/2014 and NMRC/OFLCG/003/2018) and Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore). C.C. received salary support from Tessa Therapeutics.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omto.2020.04.013.
Supplemental Information
References
- 1.Kabelitz D. Human γδ T cells: From a neglected lymphocyte population to cellular immunotherapy: A personal reflection of 30years of γδ T cell research. Clin. Immunol. 2016;172:90–97. doi: 10.1016/j.clim.2016.07.012. [DOI] [PubMed] [Google Scholar]
- 2.Legut M., Cole D.K., Sewell A.K. The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 2015;12:656–668. doi: 10.1038/cmi.2015.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Silva-Santos B., Serre K., Norell H. γδ T cells in cancer. Nat. Rev. Immunol. 2015;15:683–691. doi: 10.1038/nri3904. [DOI] [PubMed] [Google Scholar]
- 4.Tyler C.J., Doherty D.G., Moser B., Eberl M. Human Vγ9/Vδ2 T cells: Innate adaptors of the immune system. Cell. Immunol. 2015;296:10–21. doi: 10.1016/j.cellimm.2015.01.008. [DOI] [PubMed] [Google Scholar]
- 5.Chitadze G., Oberg H.H., Wesch D., Kabelitz D. The Ambiguous Role of γδ T Lymphocytes in Antitumor Immunity. Trends Immunol. 2017;38:668–678. doi: 10.1016/j.it.2017.06.004. [DOI] [PubMed] [Google Scholar]
- 6.Zhang J., Basher F., Wu J.D. NKG2D Ligands in Tumor Immunity: Two Sides of a Coin. Front. Immunol. 2015;6:97. doi: 10.3389/fimmu.2015.00097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rincon-Orozco B., Kunzmann V., Wrobel P., Kabelitz D., Steinle A., Herrmann T. Activation of V gamma 9V delta 2 T cells by NKG2D. J. Immunol. 2005;175:2144–2151. doi: 10.4049/jimmunol.175.4.2144. [DOI] [PubMed] [Google Scholar]
- 8.Spear P., Wu M.R., Sentman M.L., Sentman C.L. NKG2D ligands as therapeutic targets. Cancer Immun. 2013;13:8. [PMC free article] [PubMed] [Google Scholar]
- 9.López-Soto A., Huergo-Zapico L., Acebes-Huerta A., Villa-Alvarez M., Gonzalez S. NKG2D signaling in cancer immunosurveillance. Int. J. Cancer. 2015;136:1741–1750. doi: 10.1002/ijc.28775. [DOI] [PubMed] [Google Scholar]
- 10.Demoulin B., Cook W.J., Murad J., Graber D.J., Sentman M.L., Lonez C., Gilham D.E., Sentman C.L., Agaugue S. Exploiting natural killer group 2D receptors for CAR T-cell therapy. Future Oncol. 2017;13:1593–1605. doi: 10.2217/fon-2017-0102. [DOI] [PubMed] [Google Scholar]
- 11.Fournié J.J., Sicard H., Poupot M., Bezombes C., Blanc A., Romagné F., Ysebaert L., Laurent G. What lessons can be learned from γδ T cell-based cancer immunotherapy trials? Cell. Mol. Immunol. 2013;10:35–41. doi: 10.1038/cmi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lo Presti E., Dieli F., Meraviglia S. Tumor-Infiltrating γδ T Lymphocytes: Pathogenic Role, Clinical Significance, and Differential Programing in the Tumor Microenvironment. Front. Immunol. 2014;5:607. doi: 10.3389/fimmu.2014.00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gentles A.J., Newman A.M., Liu C.L., Bratman S.V., Feng W., Kim D., Nair V.S., Xu Y., Khuong A., Hoang C.D. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kobayashi H., Tanaka Y. γδ T Cell Immunotherapy-A Review. Pharmaceuticals (Basel) 2015;8:40–61. doi: 10.3390/ph8010040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Torikai H., Cooper L.J. Translational Implications for Off-the-shelf Immune Cells Expressing Chimeric Antigen Receptors. Mol. Ther. 2016;24:1178–1186. doi: 10.1038/mt.2016.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilhelm M., Smetak M., Schaefer-Eckart K., Kimmel B., Birkmann J., Einsele H., Kunzmann V. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J. Transl. Med. 2014;12:45. doi: 10.1186/1479-5876-12-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sadelain M., Rivière I., Riddell S. Therapeutic T cell engineering. Nature. 2017;545:423–431. doi: 10.1038/nature22395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gilham D.E., Maher J. ‘Atypical’ CAR T cells: NKG2D and Erb-B as examples of natural receptor/ligands to target recalcitrant solid tumors. Immunotherapy. 2017;9:723–733. doi: 10.2217/imt-2017-0045. [DOI] [PubMed] [Google Scholar]
- 19.Pang D.J., Neves J.F., Sumaria N., Pennington D.J. Understanding the complexity of γδ T-cell subsets in mouse and human. Immunology. 2012;136:283–290. doi: 10.1111/j.1365-2567.2012.03582.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao L., Chen C., Li Z., Zhu S., Tay J.C., Zhang X., Zha S., Zeng J., Tan W.K., Liu X. Large-scale expansion of Vγ9Vδ2 T cells with engineered K562 feeder cells in G-Rex vessels and their use as chimeric antigen receptor-modified effector cells. Cytotherapy. 2018;20:420–435. doi: 10.1016/j.jcyt.2017.12.014. [DOI] [PubMed] [Google Scholar]
- 22.Zhang T., Lemoi B.A., Sentman C.L. Chimeric NK-receptor-bearing T cells mediate antitumor immunotherapy. Blood. 2005;106:1544–1551. doi: 10.1182/blood-2004-11-4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang T., Barber A., Sentman C.L. Generation of antitumor responses by genetic modification of primary human T cells with a chimeric NKG2D receptor. Cancer Res. 2006;66:5927–5933. doi: 10.1158/0008-5472.CAN-06-0130. [DOI] [PubMed] [Google Scholar]
- 24.Chang Y.H., Connolly J., Shimasaki N., Mimura K., Kono K., Campana D. A chimeric receptor with NKG2D specificity enhances natural killer cell activation and killing of tumor cells. Cancer Res. 2013;73:1777–1786. doi: 10.1158/0008-5472.CAN-12-3558. [DOI] [PubMed] [Google Scholar]
- 25.Sentman C.L., Meehan K.R. NKG2D CARs as cell therapy for cancer. Cancer J. 2014;20:156–159. doi: 10.1097/PPO.0000000000000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lehner M., Götz G., Proff J., Schaft N., Dörrie J., Full F., Ensser A., Muller Y.A., Cerwenka A., Abken H. Redirecting T cells to Ewing’s sarcoma family of tumors by a chimeric NKG2D receptor expressed by lentiviral transduction or mRNA transfection. PLoS ONE. 2012;7:e31210. doi: 10.1371/journal.pone.0031210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song D.G., Ye Q., Santoro S., Fang C., Best A., Powell D.J., Jr. Chimeric NKG2D CAR-expressing T cell-mediated attack of human ovarian cancer is enhanced by histone deacetylase inhibition. Hum. Gene Ther. 2013;24:295–305. doi: 10.1089/hum.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernández L., Metais J.Y., Escudero A., Vela M., Valentín J., Vallcorba I., Leivas A., Torres J., Valeri A., Patiño-García A. Memory T Cells Expressing an NKG2D-CAR Efficiently Target Osteosarcoma Cells. Clin. Cancer Res. 2017;23:5824–5835. doi: 10.1158/1078-0432.CCR-17-0075. [DOI] [PubMed] [Google Scholar]
- 29.Han Y., Xie W., Song D.G., Powell D.J., Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells. J. Hematol. Oncol. 2018;11:92. doi: 10.1186/s13045-018-0635-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barber A., Rynda A., Sentman C.L. Chimeric NKG2D expressing T cells eliminate immunosuppression and activate immunity within the ovarian tumor microenvironment. J. Immunol. 2009;183:6939–6947. doi: 10.4049/jimmunol.0902000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barber A., Sentman C.L. Chimeric NKG2D T cells require both T cell- and host-derived cytokine secretion and perforin expression to increase tumor antigen presentation and systemic immunity. J. Immunol. 2009;183:2365–2372. doi: 10.4049/jimmunol.0900721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spear P., Barber A., Rynda-Apple A., Sentman C.L. Chimeric antigen receptor T cells shape myeloid cell function within the tumor microenvironment through IFN-γ and GM-CSF. J. Immunol. 2012;188:6389–6398. doi: 10.4049/jimmunol.1103019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spear P., Barber A., Rynda-Apple A., Sentman C.L. NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunol. Cell Biol. 2013;91:435–440. doi: 10.1038/icb.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Spear P., Barber A., Sentman C.L. Collaboration of chimeric antigen receptor (CAR)-expressing T cells and host T cells for optimal elimination of established ovarian tumors. OncoImmunology. 2013;2:e23564. doi: 10.4161/onci.23564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xiao L., Cen D., Gan H., Sun Y., Huang N., Xiong H., Jin Q., Su L., Liu X., Wang K. Adoptive Transfer of NKG2D CAR mRNA-Engineered Natural Killer Cells in Colorectal Cancer Patients. Mol. Ther. 2019;27:1114–1125. doi: 10.1016/j.ymthe.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.VanSeggelen H., Hammill J.A., Dvorkin-Gheva A., Tantalo D.G., Kwiecien J.M., Denisova G.F., Rabinovich B., Wan Y., Bramson J.L. T Cells Engineered With Chimeric Antigen Receptors Targeting NKG2D Ligands Display Lethal Toxicity in Mice. Mol. Ther. 2015;23:1600–1610. doi: 10.1038/mt.2015.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lynn R.C., Powell D.J., Jr. Strain-dependent Lethal Toxicity in NKG2D Ligand-targeted CAR T-cell Therapy. Mol. Ther. 2015;23:1559–1561. doi: 10.1038/mt.2015.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheadle E.J., Sheard V., Rothwell D.G., Bridgeman J.S., Ashton G., Hanson V., Mansoor A.W., Hawkins R.E., Gilham D.E. Differential role of Th1 and Th2 cytokines in autotoxicity driven by CD19-specific second-generation chimeric antigen receptor T cells in a mouse model. J. Immunol. 2014;192:3654–3665. doi: 10.4049/jimmunol.1302148. [DOI] [PubMed] [Google Scholar]
- 39.Zhao Y., Moon E., Carpenito C., Paulos C.M., Liu X., Brennan A.L., Chew A., Carroll R.G., Scholler J., Levine B.L. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer Res. 2010;70:9053–9061. doi: 10.1158/0008-5472.CAN-10-2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harrer D.C., Dörrie J., Schaft N. Chimeric Antigen Receptors in Different Cell Types: New Vehicles Join the Race. Hum. Gene Ther. 2018;29:547–558. doi: 10.1089/hum.2017.236. [DOI] [PubMed] [Google Scholar]
- 41.Rotolo R., Leuci V., Donini C., Cykowska A., Gammaitoni L., Medico G., Valabrega G., Aglietta M., Sangiolo D. CAR-Based Strategies beyond T Lymphocytes: Integrative Opportunities for Cancer Adoptive Immunotherapy. Int. J. Mol. Sci. 2019;20:E2839. doi: 10.3390/ijms20112839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebestyen Z., Prinz I., Déchanet-Merville J., Silva-Santos B., Kuball J. Translating gammadelta (γδ) T cells and their receptors into cancer cell therapies. Nat. Rev. Drug Discov. 2020;19:169–184. doi: 10.1038/s41573-019-0038-z. [DOI] [PubMed] [Google Scholar]
- 43.Harrer D.C., Simon B., Fujii S.I., Shimizu K., Uslu U., Schuler G., Gerer K.F., Hoyer S., Dörrie J., Schaft N. RNA-transfection of γ/δ T cells with a chimeric antigen receptor or an α/β T-cell receptor: a safer alternative to genetically engineered α/β T cells for the immunotherapy of melanoma. BMC Cancer. 2017;17:551. doi: 10.1186/s12885-017-3539-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sawaisorn P., Tangchaikeeree T., Chan-On W., Leepiyasakulchai C., Udomsangpetch R., Hongeng S., Jangpatarapongsa K. Antigen-Presenting Cell Characteristics of Human γδ T Lymphocytes in Chronic Myeloid Leukemia. Immunol. Invest. 2019;48:11–26. doi: 10.1080/08820139.2018.1529039. [DOI] [PubMed] [Google Scholar]
- 45.Capsomidis A., Benthall G., Van Acker H.H., Fisher J., Kramer A.M., Abeln Z., Majani Y., Gileadi T., Wallace R., Gustafsson K. Chimeric Antigen Receptor-Engineered Human Gamma Delta T Cells: Enhanced Cytotoxicity with Retention of Cross Presentation. Mol. Ther. 2018;26:354–365. doi: 10.1016/j.ymthe.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Du S.H., Li Z., Chen C., Tan W.K., Chi Z., Kwang T.W., Xu X.H., Wang S. Co-Expansion of Cytokine-Induced Killer Cells and Vγ9Vδ2 T Cells for CAR T-Cell Therapy. PLoS ONE. 2016;11:e0161820. doi: 10.1371/journal.pone.0161820. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.