

Abstract
Pompe disease is an autosomal recessive lysosomal storage disorder characterized by progressive muscle weakness. The disease is caused by mutations in the acid α-glucosidase (GAA) gene. Despite the currently available enzyme replacement therapy (ERT), roughly half of the infants with Pompe disease die before the age of 3 years. Limitations of ERT are immune responses to the recombinant enzyme, incomplete correction of the disease phenotype, lifelong administration, and inability of the enzyme to cross the blood-brain barrier. We previously reported normalization of glycogen in heart tissue and partial correction of the skeletal muscle phenotype by ex vivo hematopoietic stem cell gene therapy. In the present study, using a codon-optimized GAA (GAAco), the enzyme levels resulted in close to normalization of glycogen in heart, muscles, and brain, and in complete normalization of motor function. A large proportion of microglia in the brain was shown to be GAA positive. All astrocytes contained the enzyme, which is in line with mannose-6-phosphate receptor expression and the key role in glycogen storage and glucose metabolism. The lentiviral vector insertion site analysis confirmed no preference for integration near proto-oncogenes. This correction of murine Pompe disease warrants further development toward a cure of the human condition.
Keywords: murine Pompe disease, acid α-glucosidase, lentiviral vector, hematopoietic stem cell transplantation, skeletal muscle, central nervous system
Graphical Abstract
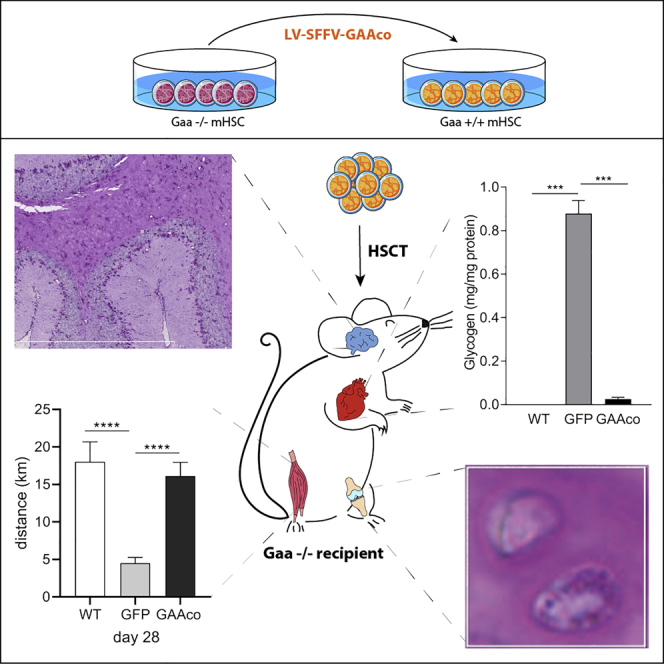
This publication reports that stem cell gene therapy using a codon-optimized gene encoding acid α-glucosidase (GAA) cures the mouse model of Pompe disease, a lysosomal storage disorder.
Introduction
Pompe disease is an autosomal recessive lysosomal storage disorder caused by the deficiency of the lysosomal hydrolase acid α-glucosidase (GAA). GAA deficiency hampers the breakdown of glycogen to glucose and leads to systemic accumulation of glycogen inside the lysosomes.1, 2, 3, 4 The disease manifests as a broad clinical spectrum, ranging from the most severe and rapidly progressive classic infantile form to less progressive forms presenting in late adulthood.5 Classic infantile Pompe disease presents shortly after birth and is characterized by profound and progressive skeletal muscle weakness, respiratory distress, cardiomyopathy, and death within the first year of life when untreated.6,7 Patients with classic infantile Pompe disease have an overall better survival and improved quality of life when treated with enzyme replacement therapy (ERT; registered in 2006 as Myozyme, also marketed as Lumizyme), but this therapy has its limitations. Close to 50% of infants still die at a very young age despite ERT, and residual disease remains in the other patients due to insufficient targeting of the skeletal muscles and a neutralizing immune response, particularly in patients that do not produce any endogenous acid GAA.8, 9, 10, 11, 12, 13 Another limitation of ERT is the inability to cross the blood-brain barrier. White matter abnormalities and signs of cognitive decline have been reported in long-term survivors of classic infantile Pompe disease treated with ERT. In mice, signs of CNS involvement and neural deficits have been reported to attribute to respiratory dysfunction.14, 15, 16
Gene therapy approaches and adjuvant treatments are currently under investigation in preclinical or clinical studies, including the use of adeno-associated viral vectors,17, 18, 19 suppression of autophagy,20 suppression of glycogen biosynthesis,21 the use of chaperones,22,23 and improved ERTs,24,25 but a single intervention that provides long-lasting therapeutic effect with limited risks to develop immune responses against the transgene product is still an unmet medical need.
Transplantation of lentiviral vector gene-modified hematopoietic stem cells (HSCs) has great potential as a delivery tool providing a continuous supply of therapeutic enzyme. Successful clinical trials are currently in progress for X-linked adrenoleukodystrophy (X-ALD) and metachromatic leukodystrophy (MLD).26,27 As for Pompe disease, we have previously reported a proof of principle that gene-modified HSC transplantation ameliorates the murine Pompe phenotype and results in addition in robust immune tolerance to the transgene product, which is attributable to the intrinsic tolerogenic nature of HSC transplantation.28 Although sustained elevated GAA levels were achieved, these levels were not sufficiently high to effectuate complete depletion of the accumulated glycogen in skeletal muscle and brain, and to achieve full correction of locomotor function. In the present preclinical evaluation of gene-modified HSC transplantation we explored whether a new vector with codon optimization29 of the GAA sequence would result in improved reversal of the Pompe disease phenotype.
Results
GAA Enzyme Activities in Blood and Immune Cells
At present, the use of codon-optimized cDNA for improving gene expression is a generally applied and accepted methodology in the development of gene therapeutic approaches. In this study, conducted in GAA-deficient knockout (KO) mice, we have explored a GAAco construct using a multiplicity of infection (MOI) of 20 as a potential vector for lentiviral gene therapy in Pompe disease. GAAco shares 83% nucleotide sequence identity with the native cDNA, with 422 of the 940 codons modified. Untreated KO animals start to manifest symptoms around 6 months of age but their lifespan is not dramatically shortened.30 After long-term follow up, i.e., 10–12 months after transplantation, using a study design identical to that published for the GAA construct to allow for a direct comparison,28 enzyme activity per vector copy in gene-modified spleen cells was increased more than 20-fold by the GAAco vector as compared to the GAA vector (326.42 ± 307.81 versus 14.4 ± 7.7 nmol/h/mg, respectively; Figure 1). Since the percentage of descendants from the transduced stem cells (i.e., chimerism) was purposely kept at 24.1% ± 5.0% in both groups of mice, the vector copy numbers, as determined by quantitative PCR, were corrected for chimerism and were similar in both groups of mice (GAAco, 7.0 ± 2.6; GAA, 7.1 ± 5.3). Since enzyme activity per vector copy is independent of cell type and chimerism, the level of chimerism resulted in an overall 4.4-fold increase in leukocytes, thymus, and spleen cells.
Figure 1.
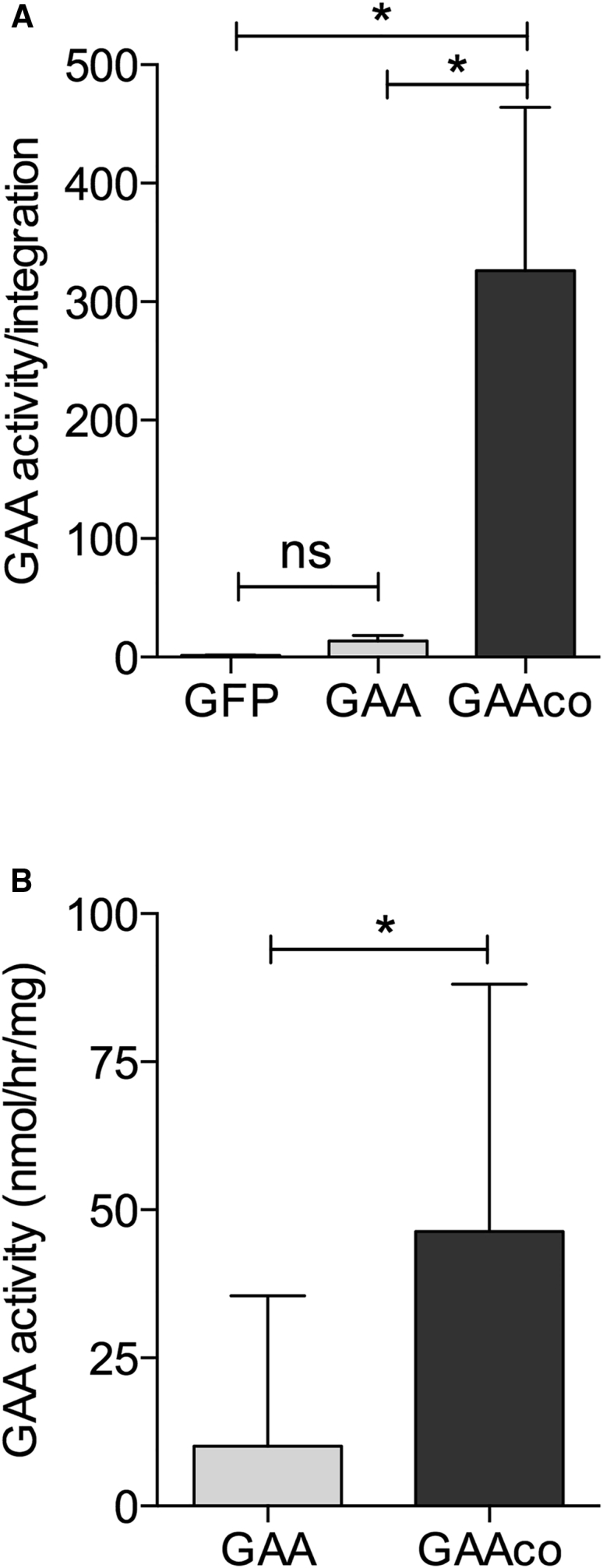
Recoding the GAA Sequence Increases GAA Enzyme Activity per Integration, Resulting in Increased GAA Activity in Leukocytes
(A) Gaa−/− mice transplanted with 5 × 105 Lin− cells transduced with either the lentiviral vector expressing the native GAA cDNA (GAA), a codon-optimized GAA cDNA (GAAco), or GFP (control vector; KO), the transgenes driven by the SF promoter and spleen cells collected 10–12 months after transplantation from n = 3 GAA, n = 5 GAAco, and n = 3 KO mice. GAA activity per vector copy number ± SD. (B) GAA activity in leukocytes of individual mice 2 months after transplantation (GAA, n = 10; GAAco, n = 10). Significance was calculated by a Mann-Whitney U test (∗p < 0.05).
Glycogen Clearance in Heart Tissue
Gaa−/− mice present left ventricle hypertrophy and impaired cardiac function, similar to classic infantile Pompe patients. We previously reported that transplantation of KO mice with GAA-transduced HSCs reversed heart remodeling and led to a substantial improvement of cardiac function.28 In the present study we assessed the GAA activity in heart muscle, the cardiac dimensions, and the heart rate at 10 months post-transplantation. Treatment with the lentiviral GAAco vector led to an increase of GAA activity in heart 5-fold higher than in wild-type (WT) mice (Figure 2A), resulting in near complete normalization of glycogen (Figure 2B). The cardiac hypertrophy present in the untreated Gaa−/− mice normalized in the animals that received HSC-based gene therapy using the GAAco cDNA construct (Figure 2C). Normalization of the left ventricular (LV) wall thickness and the LV lumen diameter (data not shown) resulted in a trend to improvement of the heart rhythm (data not shown) and rate (Figure 2D).
Figure 2.

Delivery of High Levels of GAA to Cardiac Tissue Clears Glycogen Storage and Normalizes Cardiac Dimension and Rhythm
(A–C) High enzyme levels were detectable in heart tissue (A), resulting in almost complete reduction of glycogen deposition (B) and normalization of the left ventricular mass in gene therapy-treated mice (C). WT, wild-type animal (n = 3 in biochemical assays, n = 6 in functional tests); GFP, Gaa−/− animals treated with GFP control vector (n = 3–4 in biochemical assays, n = 4–10 in functional tests); GAAco, Gaa−/− animals treated with GAAco vector (n = 5). Data are means of 4–6 mice ± SD. Significance was calculated by a Mann-Whitney U test (∗p < 0.05).
Glycogen Clearance in Skeletal Muscle and Normalized Motor Function
Gaa−/− mice at 10 months of age have severely impaired skeletal muscle function. Animals transplanted with GAAco-transduced HSCs at age 6–8 weeks had high GAA activity in skeletal muscles when sacrificed 12 months post-transplantation. The muscles studied included tongue, masseter, quadriceps femoris, gastrocnemius, soleus, extensor digitorum longus, and tibialis anterior and displayed near complete reduction of glycogen (Figure 3A). Depending on the particular muscle type, the GAA activity in these muscles was 3- to 40-fold higher than in WT animals, but due to the wide spread between the treated animals there was no significant difference except for the tongue. However, a clear reduction of glycogen can be observed. To investigate the effect on muscle function, the mice were subjected to rotarod and voluntary running wheel tests. Voluntary running wheel activity monitored for 28 days and starting at 10 months post-transplantation displayed no difference between the WT mice and the mice transplanted with GAAco-transduced HSCs, whereas GFP-transplanted mice ran significantly shorter distances per day (Figure 3B). Dissecting the number of rotations during the 28 days in bins of 2 min demonstrated that the Gaa−/− mice often made fewer rotations than did WT and gene therapy-treated mice (Figure 3C). The distribution of rotations per 2-min bin was very much the same for the GAAco-transplanted KO mice and the WT mice, not only in the lower but also in the higher number of rotations per bin. Figure S1 shows actograms of three representative mice from each group, again showing that the GAAco-transplanted and WT mice outperform the GFP-transplanted mice with regard to daily running wheel activity. Furthermore, the average speed in the running wheels during the 28 days was not different between gene therapy-treated Gaa−/− and WT mice, but untreated Gaa−/− mice reached an average speed around 2-fold lower (Figure 3D). Similarly, GAAco-transplanted KO mice managed to stay almost equally long on the accelerating rotarod as did WT mice (Figure 3E), whereas untreated mice fell off rapidly.
Figure 3.

GAA Expression Clears Glycogen Storage and Corrects Skeletal Muscle Function
(A) Near complete reduction of glycogen content in the skeletal muscles (masseter) (MAS), tongue (TON), soleus (SOL), quadriceps femoris (QF), gastrocnemius (GAS), extensor digitorum longus (EDL), and tibialis anterior (TA). Number of mice per group: WT, n = 3–5; GAAco, n = 4–5; GFP, n = 3–5. Rotarod and running wheel performance was determined at 10–12 months post-transplantation and compared to age-matched WT and GFP controls. (B) Distance achieved in a voluntary running wheel. Number of mice per group: WT, n = 3; GAAco, n = 4; GFP, n = 9. (C) The number of rotations made in the running wheel was measured in 2-min bins, categorized in 17 levels of performance, and the frequency per category was scored during a period of 28 days. (D) Performance of mice on a rotarod. Latency indicates the time spent on the rotarod before falling off. Number of mice per group: WT, n = 6; GAAco, n = 5; GFP, n = 4. (E) Average speed in the running wheel. Data are means ± SD and significance was calculated with a Mann-Whitney U test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001).
Glycogen Clearance in Brain Tissue
A few years after ERT became available for patients with Pompe disease, CNS-related symptoms were revealed. It is well accepted that hematopoietic stem cell transplantation (HSCT) can alleviate neurological symptoms of lysosomal enzyme deficiencies by contributing to the pool of enzyme-expressing microglia that resides in the brain.26,31,32 Transplantation of Gaa−/− mice with the GAAco construct-transduced HSCs led to a significant increase of GAA activity to a level of approximately half that in brain of WT mice (Figure 4A), resulting in reduction of glycogen to near normal levels (Figure 4B), with glycogen staining largely restricted to astrocytes (Figure 4C).
Figure 4.

Enzyme Activity and Glycogen Levels in Brain Tissue
(A and B) GAA activity (A) and glycogen content (B) in whole-brain lysates, 12 months after transplantation. Data are expressed as mean ± SD. Number of mice per group: WT, n = 3; GAAco, n = 5; GFP, n = 3. (C) PAS staining of cerebellum, demonstrating selective storage of glycogen in astrocytes and residual glycogen in astrocytes of gene therapy-treated mice. Significance was calculated with a Mann-Whitney U test (∗p < 0.05).
To display the localization of GAA in detail, we performed immunofluorescence staining of brain sections from mice 3.5 months post-transplantation, which demonstrated GAA in roughly half of the microglia and in virtually all astrocytes (Figure 5).
Figure 5.

GAA in Microglia and Astrocytes
(A) Microglia (F4/80, left panel); anti-GAA and Hoechst staining show that around half of microglia are GAA positive (arrowheads) and show the presence of GAA-negative resident microglia (arrows); original magnification, ×20. (B) Virtually all astrocytes appeared to contain GAA protein; two representative examples are shown here. The merged panel of the GFAP and GAA staining demonstrates the localization of GAA; original magnification, ×100. Figure S2 shows the reverse magnifications for microglia and astrocytes, respectively.
Glycogen Clearance in Visceral Organs and Articular Cartilage
Since glycogen deposition occurs in virtually all tissues, visceral organs were measured for enzyme activity and glycogen content. High enzyme activity was also detected in these organs (i.e., liver and kidney), and with full glycogen clearance (Figure 6).
Figure 6.

Reduction of Glycogen Storage in Visceral Organs
Glycogen values determined in visceral organs at 10–12 months after transplantation of gene-modified cells; data are expressed as mean ± SD. Age-matched Gaa−/− (KO) and wild-type (WT) animals were used as controls; n = 4–10 mice. Statistical analysis was calculated with a Mann-Whitney U test (∗p < 0.05).
Cells residing in articular cartilage may be difficult to reach by GAA due to its impenetrable density and low oxygenation from virtually no blood supply. Chondrocytes residing in tibio-femoral joint cartilage of Pompe mice were subjected to periodic acid-Schiff (PAS) staining (Figure 7). Quantification, with the PAS staining intensity of WT chondrocytes set at 0%, demonstrated that ~93% of the chondrocytes were glycogen positive in KO mice, which was reduced to ~45% positive cells in GAAco-treated animals. In addition to increased PAS staining intensity, we noted chondrocyte hypertrophy in the articular cartilage of KO mice, whereas the cartilage of GAAco-treated mice appeared morphologically normal compared to age-matched WT mice.
Figure 7.

Clearance of Glycogen in Articular Cartilage after Gene Therapy Treatment
PAS staining (dark purple granules) of articular cartilage shows glycogen storage in approximately 93% of the chondrocytes of 10-months-old GAA-deficient (GFP) mice. The GAAco (treated mice (GAAco, bottom panel) show a reduction in the number of PAS-positive chondrocytes to approximately 45%, as well as in the intensity of the PAS staining. Wild-type mice (WT, top panel) have no glycogen storage detetctable by PAS staining. Original magnification, ×40; scale bars represent 100 μm.
Vector Integration Site Analysis and Safety Profiling
Bone marrow DNA from five KO mice transplanted with GFP-transduced HSCs and 36 KO mice transplanted with GAAco-transduced HSCs were used as a template for linear amplification-mediated PCR (LAM-PCR) reactions prior to high-throughput sequencing. A total of 757 unique integration sites were mapped to the murine genome: 424 in the DNA from mice transplanted with the GFP vector, and 333 in the DNA from mice transplanted with the GAAco vector. Hits were widely distributed across the genome (Figure 8A), and integrations were annotated to the gene with the nearest transcription start site. Using this approach, the genes nearest to each integration site were sorted into 10 bins based on their expression levels in lineage-negative (Lin−) Sca-1+ c-Kit+ (LSK) bone marrow cells, with genes in the higher expression bins being favored for integration (Figure 8B).
Figure 8.

Vector Integration Analysis of Bone Marrow of Gene Therapy-Treated Mice
(A) Distribution of the retrieved lentiviral integrations on each chromosome of GAAco-treated mice, 10–12 months after transplantation. (B) The expression of the nearest gene to the vector integration site was assigned a bin from low to high expression. Both integrations of GFP- and GAAco-transduced bone marrow are presented. The vector integrations show the expected lentiviral vector distribution with more frequent integrations in the higher expression bins.
Integration site genes were screened against a list of 528 oncogenes compiled by the Retroviral Tagged Cancer Gene Database (RTCGD) database.33 Fifteen oncogenes were found in the annotation of the integration sites in GAAco-treated mice (4.2% of the sample pool), with two oncogenes appearing twice, i.e., Cr2 and Eps15l1. This percentage is similar to the frequency of integrations near oncogenes found in in vivo studies of WT murine Lin− cells transduced with GFP (58 of 1,402, amounting to 4.1%, with oncogene Zfp521 appearing twice), and close to the frequency expected by random chance (528 oncogenes out of 21,886 RefSeq genes in Ensembl, amounting to 2.5% oncogene frequency in the murine genome). Fifteen genes were annotated three or more times in the GAAco-treated group and 25 genes in the GFP-treated group, and these can be considered common integration sites (CISs) (Tables S1 and S2). Most of the common integration sites were intronic, and none of the CIS genes was marked as oncogenes or present in the two highest LSK expression bins.
Discussion
ERT for Pompe disease is not curative and is only partially effective, requires weekly or biweekly enzyme infusions, may result in neutralizing immune responses to the recombinant enzyme, and is very expensive. The most severely affected cross-reactive immunologic material (CRIM)-negative Pompe patients still have a strongly reduced overall survival, a need for respiratory support, and develop high antibody titers against the recombinant protein. Hence, a single intervention with curative intent represents an unmet medical need.
Attempts at gene therapy in Pompe disease include adeno-associated virus (AAV) through in vivo injections, or using lentiviral vectors, and ex vivo lentiviral transduction of HSCs.17,19,28,34, 35, 36, 37 Although AAV therapy may correct disease in Pompe mice, and potentially provide partial reduction of glycogen in brain when secretable GAA fusion proteins are used, to correct Pompe patients would require a large AAV vector quantity, which poses a risk of immune responses against the transgene product and/or AAV capsid.19 In a previous report we showed that gene-modified HSCs provided immune tolerance to GAA, and that this approach ameliorates the Pompe phenotype.28 Supranormal enzyme levels were obtained in hematopoietic cells, and most target tissues reached enzyme activity similar to that of WT mice. However, skeletal muscle function was not fully normalized, and an effect on glycogen levels in brain tissue was absent. The present study is based on the hypothesis that codon optimization of the GAA sequence might result in enzyme levels sufficient for full correction of the Pompe phenotype. The self-inactivating (SIN) third-generation lentiviral vector used in this study contained the strong spleen focus-forming virus (SFFV) viral promoter driving GAAco expression. Recoding GAA resulted on average in a 20-fold increase of the acid GAA activity per viral copy number (VCN) in spleen, which also resulted in increased enzyme activities in target tissues, near complete reduction of glycogen to WT levels, and consequently normalized heart parameters and function in the gene therapy-treated mice as well as locomotor function in voluntary running wheels and rotarod tests. The increase of GAA activity in skeletal muscles resulted in a near complete reduction of glycogen shown in Figure 3A, which ultimately led to a normalization of skeletal muscle function (Figures 3B–3E).
Despite prominent glycogen storage in brain tissue, Pompe disease, in contrast to most other lysosomal enzyme deficiencies, does not result in prominent neurological symptoms. However, some signs of CNS deficits became apparent in classic infantile patients treated with ERT.14,38 In addition, neural deficits in Pompe mice may contribute to respiratory insufficiency39, 40, 41 and even skeletal muscle dysfunction.42 GAA is thought not to pass the blood-brain barrier, so ERT is not expected to have any effect on glycogen levels in the brain, but gene therapy approaches may, which require further investigation. We previously established in the Krabbe mouse model that monocytes pass the blood-brain barrier to contribute to microglia and thereby deliver the needed enzyme.43 Based on this rationale, gene-modified HSCs are used successfully in lentiviral clinical trials for X-ALD and MLD to counteract the neuronal damage, including cognitive decline.26,44 Using the GAA lentiviral vector, GAA levels were very low in brain without much effect on glycogen, but using the vector described in the present study, glycogen levels were reduced close to WT levels, in accordance with a GAA level approximately half that of normal mice. As for the mechanism, approximately half of the microglia appeared to contain GAA and are therefore descendants of transduced stem cells, whereas the resident microglia or the untransduced donor microglia did not display GAA. This finding is consistent with a population of resident microglia, which originate from early yolk sac precursors prior to birth and persist throughout life, and microglia derived from monocytes infiltrating the brain in response to tissue damage or inflammation.45, 46, 47
Somewhat surprisingly, virtually all astrocytes appeared to contain GAA. Astrocytes have emerged as major players in energy delivery, production, utilization, and storage of the brain and almost exclusively store glycogen, as was also observed in the present study and most likely explains the limited neurological symptoms in the Pompe disorder in contrast to most other lysosomal enzyme deficiencies. Astrocytes play a major role in development, maintenance, and modulation of the blood-brain barrier, control energy supply, in particular through the glucose metabolism and specific neuronal capacities such as long-term memory, and express the mannose 6-phosphate receptors by which GAA is internalized,48, 49, 50 which explains why virtually all astrocytes appeared GAA positive after treatment by the gene-modified HSC approach. Such a reduction of the total glycogen content of the brain should contribute to an improved overall outcome following gene therapy in classic infantile patients. Interestingly, the efficacy of our approach was also apparent in a major reduction in intracellular glycogen accumulation in articular cartilage despite the fact that chondrocytes are particularly difficult to target due to the composition and density of the surrounding matrix.
The present results were obtained using the strong gammaretroviral SFFV promoter and a relatively high VCN per cell (an average of 7 VCN per cell) and are in line with the high doses of ERT required to alleviate the Pompe phenotype. In a follow-up study we have been able to reduce the VCN required to 1–2 per cell by glycosylation-independent lysosomal targeting24,51 (unpublished data).
The initial stem cell gene therapy trials for X-linked severe combined immune deficiency (SCID) using gammaretroviral gene transfer vectors resulted in successful restoration of T cell immunity, but, unfortunately, in six patients autonomous T cell clones developed into leukemia, among which one patient did not survive. However, it has meanwhile been recognized that the treated immune deficiency disorders at least co-predispose to leukemia development, given the absence of leukemia in the adenosine deaminase-deficient (ADA)-SCID trial,52,53 a 30% incidence in the X-linked SCID trials,54, 55, 56 and a 90% incidence in the gammaretroviral Wiskott-Aldrich trial.57 Of note, no such adverse effects occurredd in any preclinical evaluation or clinical trial of enzyme deficiencies, including ADA-SCID, which, although listed as a primary immune deficiency, is in essence a systemic enzyme deficiency. Safety issues of the gammaretroviral vectors have been improved by third-generation lentiviral vectors made SIN by deletion of enhancer regions from the long terminal repeat sequences58, 59, 60 to reduce the risk of influencing nearby genes,61 resulting in highly favorable safety profiles, as is also encountered in the analysis of vector integration sites in Pompe mice, which showed the expected lentiviral vector integration pattern. A more thorough comparison of lentiviral vector integration site distributions by vectors containing viral and eukaryotic promoters, various therapeutic transgenes and used in different species (unpublished data) showed no significantly increased risk of the strong SFFV promoter in integration frequency near oncogenes compared to eukaryotic promoters. Also, genotoxic events were absent during prolonged monitoring of mice transplanted with stem cells transduced with the GFP or GAAco vectors.
The present report demonstrates that transplantation of mice at 6–8 weeks of age with HSCs transduced using a highly efficient overnight gene transfer method with a third-generation SIN lentiviral vector containing a codon-optimized transgene driven by the strong SFFV promoter results in high and sustained GAA activity in the tissues that are most affected in Pompe disease. Consequently, biochemical and functional parameters of heart and skeletal muscle tissues normalized, and, in addition, glycogen accumulation in the brain appeared to be reduced to near normal levels. Serious adverse effects were not observed during prolonged monitoring of the treated animals, resulting in a favorable safety profile confirmed by the integration analysis. The therapeutic findings warrant further development including similar, better, and/or safer constructs directed at clinical implementation of the approach as a single intervention therapy with curative intent. Patients with classic infantile and juvenile Pompe disease are presently seen as the first target group because of their relative resistance to the negative side effects of bone marrow transplantation and their upcoming severe pathology when treated with ERT, whereas late-onset Pompe disease requires further consideration and a specific risk/benefit analysis.
Materials and Methods
Animals
Gaa−/− mice (FVB/N background) were generated as previously described.62 Age-matched control female FVB/N mice (WT) were obtained from Charles River Nederland (Maastricht, the Netherlands) and used as controls. All procedures for this study were approved by the Animal Ethics Committee of the Erasmus University Medical Center, Rotterdam in accordance with legislation in the Netherlands.
Construction of Vector Plasmids
The human GAA native construct driven under the SFFV promoter has been described previously (pRRL.PPT.SFFV.GAA.bPRE4∗.SIN [GAA]).28 The OptimumGene algorithm and synthesis were performed by GenScript (Piscataway, NJ, USA) to optimize the human GAA open reading frame for improved expression, including consensus Kozak sequence and an additional TGA stop codon (GAAco). GFP in the pRRL.PPT.SFFV.GFP.bPRE4∗.SIN vector (referred to as GFP) was replaced by GAAco, creating the pRRL.PPT.SFFV.GAAco.bPRE4∗.SIN vector (referred to as GAAco).
Transduction and Transplantation of Murine HSCs
Bone marrow was harvested from the femora and tibiae of 8- to 12-week-old male Gaa−/− mice. Lin− cells were purified using a mouse HSC isolation kit (BD Biosciences) and transduced with the GAAco vector at an MOI of 20 (mice are marked as GAAco) and an MOI of 2 for the GFP vector, the latter to serve for mock therapy (these mice are marked as GFP). Cells were transduced overnight in serum-free enriched Dulbecco’s medium, supplemented with murine stem cell factor (100 ng/mL), human FMS-like tyrosine kinase 3 murine ligand (50 ng/mL), and murine thrombopoietin (10 ng/mL), as previously described.63 Transduced cells (5 × 105 cells) were transplanted via the tail vein of 6- to 8-week-old Gaa−/− female recipients after sublethal irradiation (6 Gy).
Muscle Performance Tests
To determine skeletal muscle function, mice were tested at 12 months after transplantation on an accelerating rotarod (from 4 to 40 rpm in 5 min; Panlab/Harvard Apparatus Rota Rod, Holliston, MA, USA). Mice were given two training sessions to adjust to the apparatus, followed by three runs with an interval of 5 min, and the average latency to fall from the rotarod was determined.
The distance run voluntarily in a running wheel was used to measure activity performance at 10 months after transplantation for 28 days. Mice were individually housed in type III polyester cages, equipped with a steel running wheel (diameter 11.8 cm) connected to a sensor system to detect revolutions of the wheel.64 Running wheel activity (number of revolutions per 2 min) was continuously recorded using the ERS program (University of Groningen, Groningen, the Netherlands). The daily distance that the mice ran was calculated by multiplying the number of revolutions with the circumference of the wheels. The average running speed was calculated for the time that the mice spent in the running wheel.
Tissue Preparation and Histology
At 12 months post-transplantation, the mice were fasted overnight and sacrificed by transcardial perfusion with saline after sedation with ketamine/Sedator mix (Eurovet Animal Health Limited, Bladel, the Netherlands). Tissues were collected and homogenized by TissueLyser II (QIAGEN) twice for 2.5 min at 30 Hz. Histological assessment was performed as described earlier.31 PAS (for glycogen) staining was performed on these slides as previously described with the exception of cartilage. The tibialis bones were collected and undecalcified and embedded in methyl methacrylate (MMA). Positive PAS chondrocytes were counted and displayed as percentages of total chondrocytes.
Immunofluorescence Staining
For localization of GAA in the brain, transplanted mice were overnight fasted and sacrificed by transcardial perfusion with saline followed by 4% formaldehyde (FA) in phosphate-buffered saline (PBS) after administration of a ketamine/Sedator mix, 3.5 months after transplantation (n = 3). The entire brain was dissected out and overnight fixed in 4% FA before being frozen in OCT Tissue Tek (Sakura Finetek Europe). Sections were permeabilized in acetone and methanol (1:4 [v/v]) prior to blocking in 10% fetal calf serum in PBS at room temperature, and subsequently stained for GAA (anti-GAA 1:100), anti-glial fibrillary acidic protein (GFAP; Sigma-Aldrich; 1:500), and F4/80 (AbD Serotec; 1:200) overnight at 4°C. The following secondary antibodies were used: Alexa Fluor 488/555/647 (Invitrogen; 1:500). Hoechst (Sigma-Aldrich; 1:15,000) was used for nuclear staining. Tissue sections were examined by fluorescence microscopy (Leica DMRXA) connected to a Leica DFC 350FX camera.
GAA and Glycogen Assays
GAA activity and glycogen content were determined as described previously64,65 and corrected for protein content (bicinchoninic acid [BCA] kit, Pierce). The glycogen content shown in Figures 3 and 7 were obtained using undiluted samples, which may maximally result in a factor 2 lower compared to the results from titrated samples, however irrelevant for the conclusion of correction of glycogen in the tissues involved.
Quantitative PCR of LV Integrations
Spleen DNA was extracted with the NucleoSpin tissue kit (Bioké). To determine the vector copies per cell, quantitative PCR was performed on DNA extracted from spleen cells with HIV-ΔU3 forward primer (5′-CTGGAAGGGCTAATTCACTC-3′) and HIV-PSI reverse primer (5′-GGTTTCCCTTTCGCTTTCAG-3′) on the ABI Prism 7900 HT sequence detection system (Applied Biosystems). To determine the chimerism, Y chromosome-specific Sry locus primers were used: forward, 5′-CATCGGAGGGCTAAAGTGTCAC-3′, reverse 5′-TGGCATGTGGGTTCCTGTCC-3′. Samples were normalized to mouse Gapdh (forward primer, 5′-CATCACTGCCACCCAGAAGAC-3′, reverse primer, 5′-TGACCTTGCCCACAGCCTTG-3′). Samples were analyzed with SDS 2.2.2 software (Applied Biosystems).
LAM-PCR
High-resolution insertion-site analysis by LAM-PCR66 was performed on bone marrow DNA of Gaa−/− mice transduced with SF.GFP (n = 5) or SF.GAAco (n = 36) LV vectors. Restriction enzyme Tsp509I was used with the lentiviral (HIV) primer set, and PCR products were run on high-resolution polyacrylamide (Spreadex) gel. Sequencing reads were analyzed using MAVRIC,67 and to obtain the nearest RefSeq gene, the following settings were used: no repeat masking; minimum sequence length, 20 bp; minimum BLAST e-value, 0.01; and maximum distance from integration site to look for genes, 100 kb. Sequences were aligned to the murine genome via BLAST (v.37) and nearby genes identified using Ensembl version 59.
Echocardiography
Ten months after transplantation, mice were weighed, anesthetized with isoflurane, and intubated as previously described.68 Animals were placed on a heating pad to maintain body temperature at 37°C while being artificially ventilated with a mixture of O2 and N2 (1:2 [v/v]) to which isoflurane (2.5% [v/v]) was added at a rate of 90 strokes/min using a rodent ventilator (SAR-830/P, CWE, Ardmore, PA, USA) at an inspiratory pressure of 18 cm H2O and a positive end expiratory pressure of 4 cm H2O. To dehair the chest, Veet hair removal cream (Reckitt Benckiser, Parsippany, NJ, USA) was used. Transthoracic echocardiograms of the left ventricle were obtained with an Aloka SSD 4000 echo device (Aloka, Tokyo, Japan) using a 13-MHz probe. Images of the short and long axes were obtained in two-dimensional and M-mode settings, used to measure the LV lumen diameter and wall thickness at end diastole and end systole by SigmaScan Pro 5 image analysis software.
Statistical Analysis
Statistical analysis was performed with GraphPad Prism (GraphPad, La Jolla, CA, USA). Significance of differences was determined by the Mann-Whitney U test. A significant difference was assumed at p <0.05. Error bars represent SD, as indicated in the legends.
Author Contributions
M.S. contributed to the design of experimental work, executed most of the technical work, designed figures, interpreted data, and contributed to writing the manuscript; H.d.B, T.P.V., O.R., H.J., and E.D.v.D. performed technical work and interpreted data; M.W.H. and E.H.J. executed technical work, interpreted data, and contributed to the manuscript; A.J.J.R. and N.P.v.T. contributed to experimental design, interpretation of data, and the manuscript; and G.W. designed the project and contributed to the experimental design, interpretation of data, and writing the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
The authors gratefully acknowledge Ton de Jong and Nicole Kops for assistance with histochemistry. This work was supported by the European Commission’s 5th, 6th, and 7th Framework Programs (contracts QLK3-CT-2001-00 427-INHERINET and LSHB-CT-2004-005242-CONSERT and grant agreement no. 222878-PERSIST); the Netherlands Organization for Health Research (ZonMw) (program grant 434-00-010); and by the Netherlands Organisation for Scientific Research (NWO) (project 021.002.129). E.H.J. received support from the the Netherlands Organisation for Scientific Research (NWO) ZonMw-Veni award (40-00806-98-06082) and the European Research Advisory Board (ERAB) project grant EA 07 14.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2020.04.023.
Supplemental Information
References
- 1.van der Ploeg A.T., Reuser A.J. Pompe’s disease. Lancet. 2008;372:1342–1353. doi: 10.1016/S0140-6736(08)61555-X. [DOI] [PubMed] [Google Scholar]
- 2.Hagemans M.L., Winkel L.P., Van Doorn P.A., Hop W.J., Loonen M.C., Reuser A.J., Van der Ploeg A.T. Clinical manifestation and natural course of late-onset Pompe’s disease in 54 Dutch patients. Brain. 2005;128:671–677. doi: 10.1093/brain/awh384. [DOI] [PubMed] [Google Scholar]
- 3.Hirschhorn R. Glycogen storage disease type II: acid alpha-glucosidase (acid maltase) deficiency. In: Eighth Edition, Scriver C., Beaudet A.L., Sly W.S., Valle D., editors. The Metabolic and Molecular Bases of Inherited Disease. Volume III. McGraw-Hill; 2001. pp. 3389–3420. [Google Scholar]
- 4.Winkel L.P., Hagemans M.L., van Doorn P.A., Loonen M.C., Hop W.J., Reuser A.J., van der Ploeg A.T. The natural course of non-classic Pompe’s disease; a review of 225 published cases. J. Neurol. 2005;252:875–884. doi: 10.1007/s00415-005-0922-9. [DOI] [PubMed] [Google Scholar]
- 5.Güngör D., Reuser A.J. How to describe the clinical spectrum in Pompe disease? Am. J. Med. Genet. A. 2013;161A:399–400. doi: 10.1002/ajmg.a.35662. [DOI] [PubMed] [Google Scholar]
- 6.Noordzij J.G., Verkaik N.S., van der Burg M., van Veelen L.R., de Bruin-Versteeg S., Wiegant W., Vossen J.M., Weemaes C.M., de Groot R., Zdzienicka M.Z. Radiosensitive SCID patients with Artemis gene mutations show a complete B-cell differentiation arrest at the pre-B-cell receptor checkpoint in bone marrow. Blood. 2003;101:1446–1452. doi: 10.1182/blood-2002-01-0187. [DOI] [PubMed] [Google Scholar]
- 7.van den Hout H.M., Hop W., van Diggelen O.P., Smeitink J.A., Smit G.P., Poll-The B.T., Bakker H.D., Loonen M.C., de Klerk J.B., Reuser A.J., van der Ploeg A.T. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics. 2003;112:332–340. doi: 10.1542/peds.112.2.332. [DOI] [PubMed] [Google Scholar]
- 8.van Capelle C.I., Winkel L.P., Hagemans M.L., Shapira S.K., Arts W.F., van Doorn P.A., Hop W.C., Reuser A.J., van der Ploeg A.T. Eight years experience with enzyme replacement therapy in two children and one adult with Pompe disease. Neuromuscul. Disord. 2008;18:447–452. doi: 10.1016/j.nmd.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Strothotte S., Strigl-Pill N., Grunert B., Kornblum C., Eger K., Wessig C., Deschauer M., Breunig F., Glocker F.X., Vielhaber S. Enzyme replacement therapy with alglucosidase alfa in 44 patients with late-onset glycogen storage disease type 2: 12-month results of an observational clinical trial. J. Neurol. 2010;257:91–97. doi: 10.1007/s00415-009-5275-3. [DOI] [PubMed] [Google Scholar]
- 10.Kishnani P.S., Corzo D., Leslie N.D., Gruskin D., Van der Ploeg A., Clancy J.P., Parini R., Morin G., Beck M., Bauer M.S. Early treatment with alglucosidase alpha prolongs long-term survival of infants with Pompe disease. Pediatr. Res. 2009;66:329–335. doi: 10.1203/PDR.0b013e3181b24e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van Capelle C.I., van der Beek N.A., Hagemans M.L., Arts W.F., Hop W.C., Lee P., Jaeken J., Frohn-Mulder I.M., Merkus P.J., Corzo D. Effect of enzyme therapy in juvenile patients with Pompe disease: a three-year open-label study. Neuromuscul. Disord. 2010;20:775–782. doi: 10.1016/j.nmd.2010.07.277. [DOI] [PubMed] [Google Scholar]
- 12.van der Ploeg A.T., Clemens P.R., Corzo D., Escolar D.M., Florence J., Groeneveld G.J., Herson S., Kishnani P.S., Laforet P., Lake S.L. A randomized study of alglucosidase alfa in late-onset Pompe’s disease. N. Engl. J. Med. 2010;362:1396–1406. doi: 10.1056/NEJMoa0909859. [DOI] [PubMed] [Google Scholar]
- 13.van der Ploeg A.T. Where do we stand in enzyme replacement therapy in Pompe’s disease? Neuromuscul. Disord. 2010;20:773–774. doi: 10.1016/j.nmd.2010.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Ebbink B.J., Poelman E., Plug I., Lequin M.H., van Doorn P.A., Aarsen F.K., van der Ploeg A.T., van den Hout J.M. Cognitive decline in classic infantile Pompe disease: an underacknowledged challenge. Neurology. 2016;86:1260–1261. doi: 10.1212/WNL.0000000000002523. [DOI] [PubMed] [Google Scholar]
- 15.Gambetti P., DiMauro S., Baker L. Nervous system in Pompe’s disease. Ultrastructure and biochemistry. J. Neuropathol. Exp. Neurol. 1971;30:412–430. doi: 10.1097/00005072-197107000-00008. [DOI] [PubMed] [Google Scholar]
- 16.Fuller D.D., ElMallah M.K., Smith B.K., Corti M., Lawson L.A., Falk D.J., Byrne B.J. The respiratory neuromuscular system in Pompe disease. Respir. Physiol. Neurobiol. 2013;189:241–249. doi: 10.1016/j.resp.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun B., Zhang H., Bird A., Li S., Young S.P., Koeberl D.D. Impaired clearance of accumulated lysosomal glycogen in advanced Pompe disease despite high-level vector-mediated transgene expression. J. Gene Med. 2009;11:913–920. doi: 10.1002/jgm.1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mah C., Pacak C.A., Cresawn K.O., Deruisseau L.R., Germain S., Lewis M.A., Cloutier D.A., Fuller D.D., Byrne B.J. Physiological correction of Pompe disease by systemic delivery of adeno-associated virus serotype 1 vectors. Mol. Ther. 2007;15:501–507. doi: 10.1038/sj.mt.6300100. [DOI] [PubMed] [Google Scholar]
- 19.Puzzo F., Colella P., Biferi M.G., Bali D., Paulk N.K., Vidal P., Collaud F., Simon-Sola M., Charles S., Hardet R. Rescue of Pompe disease in mice by AAV-mediated liver delivery of secretable acid α-glucosidase. Sci. Transl. Med. 2017;9:eaam6375. doi: 10.1126/scitranslmed.aam6375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raben N., Schreiner C., Baum R., Takikita S., Xu S., Xie T., Myerowitz R., Komatsu M., Van der Meulen J.H., Nagaraju K. Suppression of autophagy permits successful enzyme replacement therapy in a lysosomal storage disorder—murine Pompe disease. Autophagy. 2010;6:1078–1089. doi: 10.4161/auto.6.8.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ashe K.M., Taylor K.M., Chu Q., Meyers E., Ellis A., Jingozyan V., Klinger K., Finn P.F., Cooper C.G., Chuang W.L. Inhibition of glycogen biosynthesis via mTORC1 suppression as an adjunct therapy for Pompe disease. Mol. Genet. Metab. 2010;100:309–315. doi: 10.1016/j.ymgme.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Porto C., Cardone M., Fontana F., Rossi B., Tuzzi M.R., Tarallo A., Barone M.V., Andria G., Parenti G. The pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme replacement therapy in Pompe disease fibroblasts. Mol. Ther. 2009;17:964–971. doi: 10.1038/mt.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Khanna R., Flanagan J.J., Feng J., Soska R., Frascella M., Pellegrino L.J., Lun Y., Guillen D., Lockhart D.J., Valenzano K.J. The pharmacological chaperone AT2220 increases recombinant human acid α-glucosidase uptake and glycogen reduction in a mouse model of Pompe disease. PLoS ONE. 2012;7:e40776. doi: 10.1371/journal.pone.0040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maga J.A., Zhou J., Kambampati R., Peng S., Wang X., Bohnsack R.N., Thomm A., Golata S., Tom P., Dahms N.M. Glycosylation-independent lysosomal targeting of acid α-glucosidase enhances muscle glycogen clearance in pompe mice. J. Biol. Chem. 2013;288:1428–1438. doi: 10.1074/jbc.M112.438663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tiels P., Baranova E., Piens K., De Visscher C., Pynaert G., Nerinckx W., Stout J., Fudalej F., Hulpiau P., Tännler S. A bacterial glycosidase enables mannose-6-phosphate modification and improved cellular uptake of yeast-produced recombinant human lysosomal enzymes. Nat. Biotechnol. 2012;30:1225–1231. doi: 10.1038/nbt.2427. [DOI] [PubMed] [Google Scholar]
- 26.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 27.Biffi A. Hematopoietic stem cell gene therapy for storage disease: current and new indications. Mol. Ther. 2017;25:1155–1162. doi: 10.1016/j.ymthe.2017.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Til N.P., Stok M., Aerts Kaya F.S., de Waard M.C., Farahbakhshian E., Visser T.P., Kroos M.A., Jacobs E.H., Willart M.A., van der Wegen P. Lentiviral gene therapy of murine hematopoietic stem cells ameliorates the Pompe disease phenotype. Blood. 2010;115:5329–5337. doi: 10.1182/blood-2009-11-252874. [DOI] [PubMed] [Google Scholar]
- 29.Ward N.J., Buckley S.M., Waddington S.N., Vandendriessche T., Chuah M.K., Nathwani A.C., McIntosh J., Tuddenham E.G., Kinnon C., Thrasher A.J., McVey J.H. Codon optimization of human factor VIII cDNAs leads to high-level expression. Blood. 2011;117:798–807. doi: 10.1182/blood-2010-05-282707. [DOI] [PubMed] [Google Scholar]
- 30.Bijvoet A.G., Van Hirtum H., Vermey M., Van Leenen D., Van Der Ploeg A.T., Mooi W.J., Reuser A.J. Pathological features of glycogen storage disease type II highlighted in the knockout mouse model. J. Pathol. 1999;189:416–424. doi: 10.1002/(SICI)1096-9896(199911)189:3<416::AID-PATH445>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 31.Brazelton T.R., Rossi F.M., Keshet G.I., Blau H.M. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- 32.Mezey E., Chandross K.J. Bone marrow: a possible alternative source of cells in the adult nervous system. Eur. J. Pharmacol. 2000;405:297–302. doi: 10.1016/s0014-2999(00)00561-6. [DOI] [PubMed] [Google Scholar]
- 33.Akagi K., Suzuki T., Stephens R.M., Jenkins N.A., Copeland N.G. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32:D523–D527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun B., Zhang H., Benjamin D.K., Jr., Brown T., Bird A., Young S.P., McVie-Wylie A., Chen Y.T., Koeberl D.D. Enhanced efficacy of an AAV vector encoding chimeric, highly secreted acid α-glucosidase in glycogen storage disease type II. Mol. Ther. 2006;14:822–830. doi: 10.1016/j.ymthe.2006.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sun B., Bird A., Young S.P., Kishnani P.S., Chen Y.T., Koeberl D.D. Enhanced response to enzyme replacement therapy in Pompe disease after the induction of immune tolerance. Am. J. Hum. Genet. 2007;81:1042–1049. doi: 10.1086/522236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Douillard-Guilloux G., Richard E., Batista L., Caillaud C. Partial phenotypic correction and immune tolerance induction to enzyme replacement therapy after hematopoietic stem cell gene transfer of α-glucosidase in Pompe disease. J. Gene Med. 2009;11:279–287. doi: 10.1002/jgm.1305. [DOI] [PubMed] [Google Scholar]
- 37.Kyosen S.O., Iizuka S., Kobayashi H., Kimura T., Fukuda T., Shen J., Shimada Y., Ida H., Eto Y., Ohashi T. Neonatal gene transfer using lentiviral vector for murine Pompe disease: long-term expression and glycogen reduction. Gene Ther. 2010;17:521–530. doi: 10.1038/gt.2009.160. [DOI] [PubMed] [Google Scholar]
- 38.Ebbink B.J., Aarsen F.K., van Gelder C.M., van den Hout J.M., Weisglas-Kuperus N., Jaeken J., Lequin M.H., Arts W.F., van der Ploeg A.T. Cognitive outcome of patients with classic infantile Pompe disease receiving enzyme therapy. Neurology. 2012;78:1512–1518. doi: 10.1212/WNL.0b013e3182553c11. [DOI] [PubMed] [Google Scholar]
- 39.DeRuisseau L.R., Fuller D.D., Qiu K., DeRuisseau K.C., Donnelly W.H., Jr., Mah C., Reier P.J., Byrne B.J. Neural deficits contribute to respiratory insufficiency in Pompe disease. Proc. Natl. Acad. Sci. USA. 2009;106:9419–9424. doi: 10.1073/pnas.0902534106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner S.M., Hoyt A.K., ElMallah M.K., Falk D.J., Byrne B.J., Fuller D.D. Neuropathology in respiratory-related motoneurons in young Pompe (Gaa−/−) mice. Respir. Physiol. Neurobiol. 2016;227:48–55. doi: 10.1016/j.resp.2016.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee K.Z., Qiu K., Sandhu M.S., Elmallah M.K., Falk D.J., Lane M.A., Reier P.J., Byrne B.J., Fuller D.D. Hypoglossal neuropathology and respiratory activity in Pompe mice. Front. Physiol. 2011;2:31. doi: 10.3389/fphys.2011.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee N.C., Hwu W.L., Muramatsu S.I., Falk D.J., Byrne B.J., Cheng C.H., Shih N.C., Chang K.L., Tsai L.K., Chien Y.H. A neuron-specific gene therapy relieves motor deficits in Pompe disease mice. Mol. Neurobiol. 2018;55:5299–5309. doi: 10.1007/s12035-017-0763-4. [DOI] [PubMed] [Google Scholar]
- 43.Hoogerbrugge P.M., Suzuki K., Suzuki K., Poorthuis B.J., Kobayashi T., Wagemaker G., van Bekkum D.W. Donor-derived cells in the central nervous system of twitcher mice after bone marrow transplantation. Science. 1988;239:1035–1038. doi: 10.1126/science.3278379. [DOI] [PubMed] [Google Scholar]
- 44.Cartier N., Hacein-Bey-Abina S., Bartholomae C.C., Veres G., Schmidt M., Kutschera I., Vidaud M., Abel U., Dal-Cortivo L., Caccavelli L. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science. 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- 45.Jung S., Schwartz M. Non-identical twins—microglia and monocyte-derived macrophages in acute injury and autoimmune inflammation. Front. Immunol. 2012;3:89. doi: 10.3389/fimmu.2012.00089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shemer A., Jung S. Differential roles of resident microglia and infiltrating monocytes in murine CNS autoimmunity. Semin. Immunopathol. 2015;37:613–623. doi: 10.1007/s00281-015-0519-z. [DOI] [PubMed] [Google Scholar]
- 47.Goldmann T., Wieghofer P., Jordão M.J., Prutek F., Hagemeyer N., Frenzel K., Amann L., Staszewski O., Kierdorf K., Krueger M. Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 2016;17:797–805. doi: 10.1038/ni.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magistretti P.J., Sorg O., Yu N., Martin J.L., Pellerin L. Neurotransmitters regulate energy metabolism in astrocytes: implications for the metabolic trafficking between neural cells. Dev. Neurosci. 1993;15:306–312. doi: 10.1159/000111349. [DOI] [PubMed] [Google Scholar]
- 49.Bélanger M., Allaman I., Magistretti P.J. Brain energy metabolism: focus on astrocyte-neuron metabolic cooperation. Cell Metab. 2011;14:724–738. doi: 10.1016/j.cmet.2011.08.016. [DOI] [PubMed] [Google Scholar]
- 50.Brown A.M., Baltan Tekkök S., Ransom B.R. Energy transfer from astrocytes to axons: the role of CNS glycogen. Neurochem. Int. 2004;45:529–536. doi: 10.1016/j.neuint.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 51.Liang Q., van Helsdingen Y., van der Velden G., Stok M., Jacobs E., Duncker D., Reuser A., van der Ploeg A., Vulto V., van Til N.P., Wagemaker G. Lentiviral stem cell gene therapy for Pompe disease. J. Neuromuscul. Dis. 2014;2(Suppl 1):S64. [PubMed] [Google Scholar]
- 52.Aiuti A., Cassani B., Andolfi G., Mirolo M., Biasco L., Recchia A., Urbinati F., Valacca C., Scaramuzza S., Aker M. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J. Clin. Invest. 2007;117:2233–2240. doi: 10.1172/JCI31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aiuti A., Biasco L., Scaramuzza S., Ferrua F., Cicalese M.P., Baricordi C., Dionisio F., Calabria A., Giannelli S., Castiello M.C. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hacein-Bey-Abina S., Pai S.Y., Gaspar H.B., Armant M., Berry C.C., Blanche S., Bleesing J., Blondeau J., de Boer H., Buckland K.F. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N. Engl. J. Med. 2014;371:1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hacein-Bey-Abina S., Von Kalle C., Schmidt M., McCormack M.P., Wulffraat N., Leboulch P., Lim A., Osborne C.S., Pawliuk R., Morillon E. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 56.Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Göhring G., Steinemann D. Gene therapy for Wiskott-Aldrich syndrome—long-term efficacy and genotoxicity. Sci. Transl. Med. 2014;6:227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 58.Naldini L., Blömer U., Gallay P., Ory D., Mulligan R., Gage F.H., Verma I.M., Trono D. In vivo gene delivery and stable transduction of nondividing cells by a lentiviral vector. Science. 1996;272:263–267. doi: 10.1126/science.272.5259.263. [DOI] [PubMed] [Google Scholar]
- 59.Zhang F., Thornhill S.I., Howe S.J., Ulaganathan M., Schambach A., Sinclair J., Kinnon C., Gaspar H.B., Antoniou M., Thrasher A.J. Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood. 2007;110:1448–1457. doi: 10.1182/blood-2006-12-060814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schambach A., Bohne J., Baum C., Hermann F.G., Egerer L., von Laer D., Giroglou T. Woodchuck hepatitis virus post-transcriptional regulatory element deleted from X protein and promoter sequences enhances retroviral vector titer and expression. Gene Ther. 2006;13:641–645. doi: 10.1038/sj.gt.3302698. [DOI] [PubMed] [Google Scholar]
- 61.Cesana D., Ranzani M., Volpin M., Bartholomae C., Duros C., Artus A., Merella S., Benedicenti F., Sergi Sergi L., Sanvito F. Uncovering and dissecting the genotoxicity of self-inactivating lentiviral vectors in vivo. Mol. Ther. 2014;22:774–785. doi: 10.1038/mt.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bijvoet A.G., van de Kamp E.H., Kroos M.A., Ding J.H., Yang B.Z., Visser P., Bakker C.E., Verbeet M.P., Oostra B.A., Reuser A.J., van der Ploeg A.T. Generalized glycogen storage and cardiomegaly in a knockout mouse model of Pompe disease. Hum. Mol. Genet. 1998;7:53–62. doi: 10.1093/hmg/7.1.53. [DOI] [PubMed] [Google Scholar]
- 63.van Til N.P., Wagemaker G. Lentiviral gene transduction of mouse and human hematopoietic stem cells. Methods Mol. Biol. 2014;1185:311–319. doi: 10.1007/978-1-4939-1133-2_21. [DOI] [PubMed] [Google Scholar]
- 64.Van Leenen D., Bijvoet A.G., Visser P., Heuvelsland G.F., Verkerk A., Van Der Horst G.T., Reuser A.J. A low-cost computerized system to monitor running performance and circadian rhythms of twenty mice simultaneously. Contemp. Top. Lab. Anim. Sci. 1999;38:29–32. [PubMed] [Google Scholar]
- 65.Okumiya T., Keulemans J.L., Kroos M.A., Van der Beek N.M., Boer M.A., Takeuchi H., Van Diggelen O.P., Reuser A.J. A new diagnostic assay for glycogen storage disease type II in mixed leukocytes. Mol. Genet. Metab. 2006;88:22–28. doi: 10.1016/j.ymgme.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 66.Schmidt M., Schwarzwaelder K., Bartholomae C., Zaoui K., Ball C., Pilz I., Braun S., Glimm H., von Kalle C. High-resolution insertion-site analysis by linear amplification-mediated PCR (LAM-PCR) Nat. Methods. 2007;4:1051–1057. doi: 10.1038/nmeth1103. [DOI] [PubMed] [Google Scholar]
- 67.van Til N.P., de Boer H., Mashamba N., Wabik A., Huston M., Visser T.P., Fontana E., Poliani P.L., Cassani B., Zhang F. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol. Ther. 2012;20:1968–1980. doi: 10.1038/mt.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.de Waard M.C., van der Velden J., Bito V., Ozdemir S., Biesmans L., Boontje N.M., Dekkers D.H., Schoonderwoerd K., Schuurbiers H.C., de Crom R. Early exercise training normalizes myofilament function and attenuates left ventricular pump dysfunction in mice with a large myocardial infarction. Circ. Res. 2007;100:1079–1088. doi: 10.1161/01.RES.0000262655.16373.37. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.