

The opportunistic pathogen Pseudomonas aeruginosa is a leading cause of morbidity and mortality worldwide. To survive in both the environment and the host, P. aeruginosa must cope with redox stress. In P. aeruginosa, a primary mechanism for protection from redox stress is the antioxidant glutathione (GSH). GSH is a low-molecular-weight thiol-containing tripeptide (l-γ-glutamyl-l-cysteinyl-glycine) that can function as a reversible reducing agent.
KEYWORDS: Pseudomonas aeruginosa, glutathione, lung infection, chronic wound infection, burn wound infection, antibiotics
ABSTRACT
The opportunistic pathogen Pseudomonas aeruginosa is a leading cause of morbidity and mortality worldwide. To survive in both the environment and the host, P. aeruginosa must cope with redox stress. In P. aeruginosa, a primary mechanism for protection from redox stress is the antioxidant glutathione (GSH). GSH is a low-molecular-weight thiol-containing tripeptide (l-γ-glutamyl-l-cysteinyl-glycine) that can function as a reversible reducing agent. GSH plays an important role in P. aeruginosa physiology and is known to modulate several cellular and social processes that are likely important during infection. However, the role of GSH biosynthesis during mammalian infection is not well understood. In this study, we created a P. aeruginosa mutant defective in GSH biosynthesis to examine how loss of GSH biosynthesis affects P. aeruginosa virulence. We found that GSH is critical for normal growth in vitro and provides protection against hydrogen peroxide, bleach, and ciprofloxacin. We also studied the role of P. aeruginosa GSH biosynthesis in four mouse infection models, including the surgical wound, abscess, burn wound, and acute pneumonia models. We discovered that the GSH biosynthesis mutant was slightly less virulent in the acute pneumonia infection model but was equally virulent in the three other models. This work provides new and complementary data regarding the role of GSH in P. aeruginosa during mammalian infection.
INTRODUCTION
Infections caused by the bacterium Pseudomonas aeruginosa are a major public health threat (1). This Gram-negative bacterium is most commonly isolated from areas associated with human activity, including contaminated soil, water, and hospital fomites (1–3). As an opportunistic pathogen, it primarily infects people with compromised immune systems and underlying comorbidities, particularly cystic fibrosis and diabetes (1). It can infect multiple body sites, including the skin and soft tissues, lungs, eyes, bloodstream, and urinary tract (1). P. aeruginosa is naturally tolerant to many conventional disinfectants and antibiotics, and there is increasing global incidence of P. aeruginosa strains with multidrug resistance traits (4–6). There is currently no vaccine against P. aeruginosa, and therapeutic regimens used to treat this bacterium are often ineffective. For these reasons, there is an urgent need to better understand P. aeruginosa infection biology.
To survive both in the environment and in the host, P. aeruginosa must cope with redox stress. Glutathione (GSH) is a major cellular regulator of redox homeostasis (7, 8). It is a low-molecular-weight thiol-containing tripeptide (l-γ-glutamyl-l-cysteinyl-glycine) that can function as a reversible reducing agent either directly by interacting with reactive oxygen (ROS) and nitrogen (RNS) species or indirectly by serving as an enzymatic cofactor (8, 9). GSH is produced by most Gram-negative bacteria and by a few Gram-positive bacteria, and it is ubiquitous and essential in eukaryotic cells (7, 8, 10). Bacteria that do not biosynthesize GSH often produce functionally similar molecules, such as mycothiol or bacillithiol (11–13). In addition to protection from oxidative and nitrosative stress, GSH is involved in many important cellular functions, such as detoxification of electrophilic compounds, lipid peroxides, toxic metabolites and xenobiotics; regulation of cellular osmolality and pH (7, 14); and metal homeostasis (7, 8).
In P. aeruginosa, genetic defects in GSH biosynthesis result in a myriad of altered phenotypes both in vitro and in vivo that suggest an important role during infection. GSH biosynthesis has been shown to affect quorum sensing (15), type III and VI secretion systems (T3SS and T6SS) (16), production of the secondary metabolites pyocyanin and pyoverdine (17, 18), motility (16–18), biofilm formation (16–18), sensitivity to oxidative and nitrosative stress (16–18), and sensitivity to antibiotics (19, 20). GSH-deficient P. aeruginosa mutants have attenuated virulence in Drosophila (17) and Caenorhabditis elegans (21) infection models, as well as a mouse model of acute pneumonia (16). Glutathione-deficient mutants in other bacterial species are also attenuated for virulence in mouse models of infection, including Salmonella enterica in an intraperitoneal model (22), Listeria monocytogenes in an intravenous model (9), and Burkholderia pseudomallei in an acute pneumonia model (23).
A large number of in vitro and in vivo models are used to study P. aeruginosa virulence, and it is clear that the traits required for P. aeruginosa fitness in these models can vary (24–26). This may be due to differences in nutrient availability, innate immune defenses, or other environmental factors, such as oxygen tension or pH. However, the role of GSH in P. aeruginosa pathogenesis in mammals, particularly at different sites of infection, is still unclear. In this study, we first examined how GSH deficiency affects P. aeruginosa’s growth rate and susceptibility to antimicrobials in vitro. We then explored how GSH deficiency affects the fitness of P. aeruginosa in four mouse models of infection, including surgical wound, abscess, burn wound, and acute pneumonia models.
RESULTS AND DISCUSSION
P. aeruginosa requires GSH for normal growth in vitro.
In P. aeruginosa, GSH biosynthesis is catalyzed by a two-step reaction. In the rate-limiting first step, γ-glutamylcysteine synthetase, the product of the gshA gene, produces γ-glutamylcysteine. In the second step, GSH is produced from γ-glutamatecysteine by glutathione synthetase, the product of the gshB gene (7, 8). To study the importance of GSH biosynthesis in P. aeruginosa biology, we constructed a gshA deletion (ΔgshA) mutant in the P. aeruginosa strain PAO1 background. We chose to inactivate gshA and not gshB not only because it disrupts the first step in glutathione biosynthesis but also because gshB mutants still produce γ-glutamylcysteine, which can partially compensate for the lack of GSH (27). This is likely the reason why P. aeruginosa gshA mutants generally have more severe growth rate defects and less antimicrobial tolerance than gshB mutants (17, 28).
Previous studies have shown that P. aeruginosa ΔgshA has a growth defect in minimal media, but growth rates are restored to wild-type (WT) levels in LB broth (18). However, the reason for the differences in growth rate for gshA mutants in the two media was unclear. To address this question, we measured the growth rates of P. aeruginosa WT and ΔgshA in several media. We tested MOPS (morpholinepropanesulfonic acid) minimal medium supplemented with 20 mM glucose (MOPS-glucose), MOPS-glucose containing 1 mM GSH, MOPS-glucose containing yeast extract (5 g/liter), MOPS-glucose containing tryptone (10 g/liter), chemically defined medium (CDM) (29), and LB broth. We selected 1 mM GSH because this concentration is within the range of typical intracellular GSH concentrations (18, 30–32). We chose 5 g/liter yeast extract and 10 g/liter of tryptone because these are the concentrations present in LB. Finally, CDM is a rich medium that contains all amino acids and nucleotides and many metabolites essential for growth, but it does not contain GSH (29).
Consistent with previous studies (18), we observed a growth rate defect for P. aeruginosa ΔgshA compared to the WT in MOPS-glucose, but not in LB (Fig. 1). While it was previously hypothesized that the reason for this difference in growth rate was stress caused by nutritional restriction in minimal medium (18), we discovered that P. aeruginosa ΔgshA also exhibited a growth rate defect in CDM, a rich defined medium, as well as MOPS-glucose with tryptone. These data indicate that nutritional stress is unlikely to be the cause of the growth rate defect for P. aeruginosa ΔgshA in MOPS-glucose. Importantly, the growth rate of the mutant was restored to WT levels with the addition of GSH or yeast extract. GSH is abundant in yeast cells (33); thus, the growth rate of P. aeruginosa ΔgshA is likely restored to WT levels in LB due to the presence of GSH in yeast extract.
FIG 1.
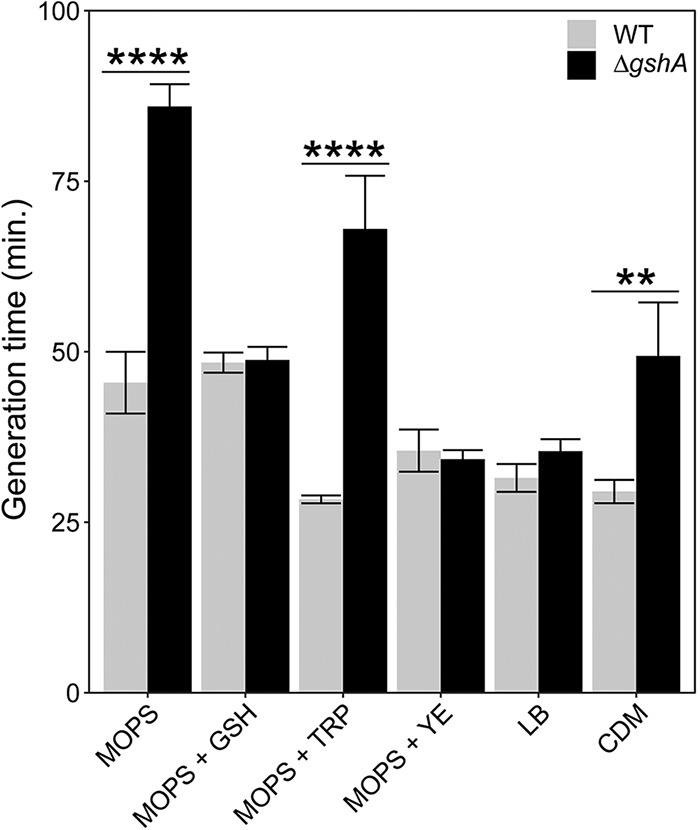
Growth rates of P. aeruginosa WT and P. aeruginosa ΔgshA in different media. n = 3 to 5. Significance was calculated by two-way analysis of variance (ANOVA) followed by Bonferroni’s multiple-comparison test with 95% confidence interval. **, P < 0.01; ****, P < 0.0001. Error bars represent 1 standard error. GSH, glutathione; TRP, tryptone; YE, yeast extract; CDM, chemically defined medium.
These results underscore the importance of using a medium that does not contain GSH when GSH biosynthesis is being studied, since the presence of GSH in a growth medium such as LB can confound interpretation of experimental results. For example, compared to the WT, GSH-deficient P. aeruginosa mutants reportedly produce less biofilm when grown in M9 minimal medium (18) than in LB (17), likely because of the high levels of GSH in the yeast extract component of LB (33). Furthermore, unbuffered GSH added to medium at high concentrations, such as 10 mM, can significantly lower the pH of the medium and induce an acid shock response in bacteria (10). Finally, even bacteria grown in the same medium but in different culture vessels or culture volumes display greatly different oxidative stress responses (34). Taken together, these results show that the experimental growth environment should be carefully considered and that differences in growth conditions between laboratories likely explain conflicting findings.
P. aeruginosa requires GSH for protection against some disinfectants and antibiotics.
GSH has been implicated in tolerance to several antimicrobials. To kill bacteria during infection, neutrophils and macrophages produce ROS such as hydrogen peroxide (H2O2) and hypochlorous acid (HOCl) (35, 36). GSH is one of the cell’s primary mechanisms for protection against ROS (7, 8), and GSH-deficient P. aeruginosa mutants are sensitive to ROS-generating agents, including methyl viologen and paraquat (17, 18). Several recent studies have also argued that different classes of bactericidal antibiotics, but not bacteriostatic antibiotics, can generate ROS and kill bacteria through secondary mechanisms (37, 38). In some Gram-negative bacteria, GSH also helps maintain cellular osmolality by regulating intracellular potassium (7, 14), and mutants that are deficient in either GSH biosynthesis or potassium transport are hypersusceptible to antimicrobial peptides (AMPs) (39–42).
To explore the role of GSH biosynthesis in protection of P. aeruginosa from different antimicrobials, we performed disc diffusion assays with a panel of disinfectants and antibiotics (Table 1). We grew P. aeruginosa WT and P. aeruginosa ΔgshA on MOPS-glucose–tryptone agar plates with or without 1 mM GSH, representing permissive and nonpermissive conditions for the ΔgshA mutant, respectively. We tested two disinfectants, hydrogen peroxide and bleach (NaOCl), and six antibiotics, polymyxin B, colistin, ciprofloxacin, carbenicillin, tetracycline, and chloramphenicol. Polymyxin B and colistin are bactericidal AMPs (43), ciprofloxacin and carbenicillin are bactericidal antibiotics, and tetracycline and chloramphenicol are bacteriostatic antibiotics.
TABLE 1.
P. aeruginosa zones of inhibition following exposure to 8 antimicrobials
| Antimicrobial | GSH | Zone of inhibition (mm)a
|
P valueb | |
|---|---|---|---|---|
| WT | ΔgshA | |||
| Hydrogen peroxide | − | 38.0 ± 0.9 | 49.6 ± 1.9 | <0.0001 |
| + | 38.3 ± 1.1 | 39.3 ± 1.2 | >0.9999 | |
| Bleach | − | 37.0 ± 2.1 | 47.4 ± 1.3 | 0.0002 |
| + | 21.3 ± 1.8 | 22.3 ± 2.2 | >0.9999 | |
| Polymyxin B | − | 13.8 ± 0.2 | 16.0 ± 0.4 | >0.9999 |
| + | 13.8 ± 0.4 | 15.2 ± 0.6 | >0.9999 | |
| Colistin | − | 15.0 ± 0.3 | 18.1 ± 0.6 | 0.4750 |
| + | 14.9 ± 0.3 | 16.0 ± 0.6 | >0.9999 | |
| Ciprofloxacin | − | 28.4 ± 0.9 | 35.8 ± 1.6 | <0.0001 |
| + | 27.9 ± 0.8 | 36.6 ± 1.6 | <0.0001 | |
| Carbenicillin | − | 19.9 ± 1.5 | 14.3 ± 0.6 | 0.0789 |
| + | 17.6 ± 1.1 | 14.4 ± 1.3 | >0.9999 | |
| Tetracycline | − | 10.9 ± 0.8 | 8.8 ± 2.7 | >0.9999 |
| + | 10.6 ± 0.7 | 12.3 ± 0.7 | >0.9999 | |
| Chloramphenicol | − | 14.3 ± 1.6 | 14.3 ± 1.2 | >0.9999 |
| + | 13.3 ± 1.0 | 13.1 ± 0.8 | >0.9999 | |
Values are average zone of inhibition ± standard error of the mean for P. aeruginosa WT and the ΔgshA mutant with (+) or without (–) 1 mM glutathione (GSH) added to the medium. n = 3 to 8 for each condition.
P values of <0.05, determined using two-way ANOVA followed by Bonferroni’s multiple-comparison test with 95% confidence interval, are in bold.
As shown in Table 1, P. aeruginosa ΔgshA had a larger zone of inhibition (ZOI) than the WT when exposed to hydrogen peroxide or bleach, and the ZOI was restored to WT levels with the addition of GSH. P. aeruginosa ΔgshA was also more susceptible to the bactericidal antibiotic ciprofloxacin; however, this enhanced susceptibility was not chemically complemented by addition of GSH. P. aeruginosa ΔgshA was also slightly more resistant to the bactericidal antibiotic carbenicillin (P = 0.079). Finally, we observed no difference in the ZOIs of P. aeruginosa ΔgshA and the WT when they were treated with the AMPs polymyxin B and colistin or the bacteriostatic antibiotics chloramphenicol and tetracycline.
Taken together, these results show that P. aeruginosa GSH is critical for protection from ROS. In our previous transposon sequencing (Tn-seq) experiments that assessed the fitness of thousands of P. aeruginosa mutants simultaneously upon exposure to antimicrobials, a fitness defect for P. aeruginosa gshA mutants was observed upon exposure to bleach but not to H2O2 (28). This lack of a fitness defect upon exposure to H2O2 was likely due to rapid detoxification of H2O2 by P. aeruginosa catalases, which prevented identification of a number of functions important for P. aeruginosa H2O2 tolerance in that study, including catalase (28). The importance of GSH for tolerance to ROS raises the possibility that GSH biosynthesis inhibitors may be a reasonable therapeutic strategy for enhancing bacterial clearance by host-generated ROS. However, this approach would likely require development of GSH biosynthesis inhibitors that are bacterium specific, as common nonspecific GSH inhibitors, such as buthionine sulfoximine (BSO) (30) and diethyl maleate (DEM) (44), are also toxic to eukaryotic host cells.
The slight increase in tolerance to carbenicillin (Table 1) is likely due to the lower growth rate of P. aeruginosa ΔgshA in MOPS-glucose with tryptone (Fig. 1), as it is known that slower-growing cells are more resistant to the beta-lactam class of antibiotics (45, 46). This phenotype was mitigated by the addition of GSH, although this was due to an increase in tolerance of WT P. aeruginosa to carbenicillin in the presence of GSH. Thus, the mechanisms controlling carbenicillin tolerance and GSH are more complex than that observed for ROS. The mechanism of ciprofloxacin hypersusceptibility also appears complex, as tolerance of P. aeruginosa ΔgshA was not restored by addition of GSH. Finally, P. aeruginosa ΔgshA showed no change in tolerance to AMPs, as has been observed in other bacteria (39), although this does agree with recent Tn-seq data showing that inactivation of gshA had no effect on P. aeruginosa fitness in the presence of polymyxin B (28).
GSH biosynthesis provides a fitness benefit to P. aeruginosa in a mouse model of acute pneumonia but is dispensable in surgical wound, abscess, and acute burn wound infections.
P. aeruginosa is a versatile pathogen that can infect multiple body sites, among the most common of which are soft tissues and lungs. P. aeruginosa is one of the organisms most commonly isolated from human chronic wound infections, burn wound infections, and hospital-associated acute pneumonia. The presence of P. aeruginosa in these infections is also associated with increased infection severity and mortality (43, 47–50). Previous studies (15–18, 21), as well as our in vitro experiments, suggest an important role for GSH biosynthesis for P. aeruginosa during infection. Here, we comprehensively assessed the fitness of P. aeruginosa WT and ΔgshA in four mouse models of infection: the surgical wound, abscess, acute burn wound, and acute pneumonia models.
We first tested two nonlethal soft tissue infection models. The surgical wound mouse model involves surgically removing a full-thickness area of skin from the shaved backs of the mice, applying a semipermeable bandage over the wound, and administering the bacterial inoculum to the wound topically underneath the bandage (24, 51–53). The abscess model involves a subcutaneous injection of bacteria into the shaved inner thigh of the mice (54, 55). For both models, we infected mice with ∼106 CFU of P. aeruginosa WT or P. aeruginosa ΔgshA and enumerated bacterial loads after 4 and 3 days postinfection for the surgical wound and abscess models, respectively. Results from these experiments revealed no significant difference between bacterial loads obtained with the ΔgshA mutant and the WT in the surgical wound model (Fig. 2A) or in the abscess model (Fig. 2B).
FIG 2.

Surgical wound and abscess mouse infection models. Mice were infected with approximately 1 × 106 CFU of P. aeruginosa WT or ΔgshA in the surgical wound (A) and abscess (B) mouse models of infection. Mice were sacrificed at 4 and 3 days postinfection, respectively. Mouse infections were performed with three mice in each group and repeated independently twice (n = 6).
We next tested two lethal mouse infection models, the acute burn wound and acute pneumonia models. The mouse burn wound model (24, 56) involves exposing the shaved backs of mice to 90°C water for 10 s, creating a uniform third-degree burn. The acute pneumonia model (57, 58) involves a noninvasive intratracheal administration of bacteria directly into the lung. We infected mice with ∼103 CFU and 2.5 × 107 CFU P. aeruginosa WT and P. aeruginosa ΔgshA for the burn model and the pneumonia model, respectively. Survival was monitored over a period of 7 days. As shown in Fig. 3A, there was no significant difference in survival between mice infected with P. aeruginosa WT and those with P. aeruginosa ΔgshA in the burn wound model. However, in the pneumonia model (Fig. 3B) we observed a modest but statistically significant increase in survival for mice infected with P. aeruginosa ΔgshA compared to those infected with the WT. The pneumonia model data are consistent with a previous study showing decreased bacterial loads in the lung for P. aeruginosa ΔgshA in a mouse acute pneumonia model (16). The increased importance of P. aeruginosa GSH biosynthesis in the lung and not soft tissue is likely explained by the fact that GSH-deficient P. aeruginosa mutants have reduced expression of the type III secretion system (16), which is important for lung pathogenesis (43).
FIG 3.
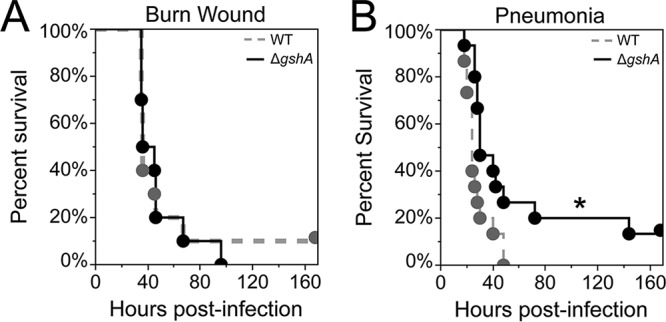
Burn wound and acute pneumonia infection models. (A) For the mouse burn wound infection model, mice were infected with approximately 1 × 103 CFU of P. aeruginosa WT or ΔgshA. Experiments were performed with five mice in each group and repeated independently twice (n = 10). (B) For the mouse acute pneumonia infection, mice were infected with approximately 2.5 × 107 CFU (2× LD50) of P. aeruginosa. Mouse infections were performed with five mice in each group and repeated independently three times (n = 15). All mice were monitored for 7 days or until moribund and humanely euthanized. Results are presented as Kaplan-Meier survival curves; differences in survival were calculated by the log-rank test. *, P < 0.05.
While P. aeruginosa ΔgshA does show a statistically significant decrease in lethality in the pneumonia model, the effect is quite modest compared to that of other mutants that are considered attenuated in this model, such as rhlR (58) and aroA (59) mutants. These results, along with the lack of an infection phenotype for P. aeruginosa ΔgshA in the burn, surgical wound, and abscess mouse models, reveal that GSH biosynthesis plays little if any role in virulence in commonly used mouse models of P. aeruginosa infection. This result is somewhat surprising given the role of GSH in the pathogenesis of other bacteria, the high susceptibility of P. aeruginosa ΔgshA to ROS (Table 1), and the heightened inflammatory responses, oxidative stress, and tissue GSH depletion that are characteristic of the mouse models that we used (60–65). One simple explanation for our results is that P. aeruginosa can scavenge sufficient bioavailable GSH from the host in these infection models, despite the lowered tissue GSH levels associated with infection, thus mitigating the ΔgshA phenotype. This explanation is supported by our previous Tn-seq studies showing that P. aeruginosa gshA mutants were more fit in the burn and surgical wound mouse models than in a minimal MOPS-succinate medium without GSH (24). These studies also indicate that the gshA mutant is not trans-complemented by other P. aeruginosa mutants in the infection; thus, GSH does not appear to be a public good that is shared among community members (24). It should be noted that we followed commonly used methods in our experiments, including the method of inoculation and inoculating dose. Thus, while we think the difference in results is unlikely due to the diversity of models and inoculating doses used, it is possible that altering these parameters could change the outcome of our experiments and reveal a role for GSH biosynthesis in P. aeruginosa pathogenesis.
Due to its potent antioxidant properties, there is increasing interest in using GSH as a treatment strategy for a variety of human pathologies. Uncontrolled oxidative stress is a key pathophysiological feature of chronic wounds (66), burn wounds (63), and pneumonia (65, 67). Experimental rodent and human clinical studies have shown that administration of GSH can result in significantly improved healing, reduced tissue damage, and lower mortality in chronic wounds (68, 69), burn wounds (62, 70, 71), and pneumonia (72–74). However, it is unclear how administration of GSH in human subjects will affect the physiology of P. aeruginosa during infection. While our in vitro data suggest that inhibiting GSH biosynthesis may render P. aeruginosa more susceptible to ROS, addition of GSH can restore tolerance to the gshA mutant and in some cases alter the tolerance of WT P. aeruginosa (Table 1). Thus, GSH administration may in fact promote unwanted P. aeruginosa-mediated infection phenotypes. Further studies are necessary to elucidate the safety and efficacy of GSH administration during P. aeruginosa infection. In conclusion, our results provide new and complementary data regarding the role of GSH in P. aeruginosa virulence.
MATERIALS AND METHODS
Bacterial strains and growth media.
P. aeruginosa strain PAO1 was obtained from Colin Manoil (University of Washington). The PAO1 ΔgshA deletion mutant was constructed as previously described (25). Briefly, ∼700-bp fragments flanking the gshA gene were amplified by PCR with Phusion Hot Start II DNA polymerase (Thermo Scientific, Waltham, MA) to replace the coding sequence with the sequence 5′-GCGGCCGCC-3′ flanked by the native start codon. The PCR primers were 5′-TTCTGCAGGTCGACTCTAGACGAGAAGGTCGAAGGCCAGC-3′ and 5′-CCGGCTTGGCTCGGGATTCCTTGTGATCGCGT-3′ for the upstream region and 5′-GAATCCCGAGCCAAGCCGGCGCGCC-3′ and 5′-GAATTCGAGCTCGAGCCCGGGCTTGCTGGACGCATCCGGC-3′ for the downstream region. These two amplicons and the suicide vector pEXG2 (75) were assembled using Gibson assembly as described previously (76), transformed initially into Escherichia coli DH5α λpir, and then transformed into E. coli SM10 λpir for conjugation into P. aeruginosa strain PAO1. Successful transconjugants were selected for with gentamicin followed by selection on sucrose to obtain the chromosomal deletion. This mutation was then verified by PCR. For routine growth, P. aeruginosa strains were grown on LB agar plates overnight. Unless otherwise stated, overnight P. aeruginosa cultures from single colonies on the LB agar plates were grown in morpholinepropanesulfonic acid (MOPS)-buffered minimal medium with 20 mM glucose (MOPS-glucose), which does not contain GSH.
Growth curves.
P. aeruginosa WT PAO1 and the ΔgshA mutant were grown overnight in MOPS-glucose, MOPS-glucose plus 1 mM GSH (l-glutathione reduced; Sigma), MOPS-glucose plus 5 g/liter yeast extract (Sigma), MOPS-glucose plus 10 g/liter Bacto tryptone (Fisher), LB broth (Fisher), or a rich chemically defined medium (CDM) (29). Subcultures were diluted in the same medium to an optical density at 600 nm (OD600) of 0.05 and grown in culture tubes at 37°C with shaking. OD600 measurements were recorded by diluting subculture samples to between 0.09 and 0.5. The generation times were calculated using OD600 measurements taken during exponential growth.
Disc diffusion assays.
Overnight P. aeruginosa cultures were grown in MOPS-glucose, adjusted to an OD of 0.4, and swabbed onto MOPS-glucose–tryptone agar plates (MOPS-glucose supplemented with 10 g/liter Bacto tryptone; Fisher) with or without added 1 mM GSH. Sterile quarter-inch filter discs were placed on the center of each plate, and 20 μl of each compound tested was pipetted onto the disc. Stock concentrations of each antibiotic (polymyxin B [Sigma], colistin [Sigma], ciprofloxacin [Sigma], carbenicillin [Alfa Aesar], tetracycline [Sigma], and chloramphenicol [IBI Scientific]) were dissolved at concentrations of 1 mg/ml. For the disinfectants, 3% hydrogen peroxide or 0.85% sodium hypochlorite was used. An additional 20 μl of hydrogen peroxide was added to the hydrogen peroxide plates after 7 h of growth. Plates were incubated at 37°C overnight. Two measurements of the diameter of the zone of inhibition were recorded for each plate and averaged.
Mouse infections.
All mouse procedures were carried out in strict accordance with established guidelines at each respective university following recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (77) as well as local, state, and federal laws. For all mouse experiments, P. aeruginosa strains were struck on LB agar plates, grown overnight in LB broth, and then subcultured in LB broth to logarithmic-phase growth. Bacterial inoculums were diluted to the appropriate OD600 in phosphate-buffered saline (PBS).
For the mouse acute thermal injury infection model (also referred to as the mouse burn wound model) (24, 56), all animals were treated humanely and in accordance with protocol number 96020 approved by the Institutional Animal Care and Use Committee (IACUC) at Texas Tech University Health Sciences Center in Lubbock, TX. Adult 6- to 8-week-old female Swiss Webster mice (Charles River Laboratories, Inc.) weighing between 20 and 25 g were anesthetized by intraperitoneal injection of 100 mg/kg sodium pentobarbital (Nembutal; Diamondback Drugs). Their backs were then shaved, and the remaining hair was removed with a depilatory agent (Nair). Prior to injury, 0.5 mg/kg buprenorphine (ZooPharm) was administered subcutaneously for pain management. A third-degree, full-thickness thermal injury affecting 15% of the total body surface area was induced by submerging an exposed area of the dorsal surface in a 90°C water bath for 10 s. Mice were then given a subcutaneous injection of 500 μl physiological saline for fluid support. Approximately 103 CFU (in 100 μl) of either P. aeruginosa PAO1 WT or P. aeruginosa PAO1 ΔgshA was injected subcutaneously directly underneath the freshly injured tissue. The animals were then monitored for 7 days after injury and infection for signs of sepsis (lethargy, weight loss, tremors, etc.). In the event that mice became moribund, they were euthanized by intraperitoneal injection of 200 μl (390 mg/ml) Fatal-Plus (Vortech Pharmaceuticals, Ltd.) and included in the experimental results. Surviving mice were similarly euthanized after the 7-day experimental endpoint.
For the mouse surgical incision infection model (also referred to as the mouse chronic wound model) (24, 51), female 6- to 8-week-old adult Swiss Webster mice weighing between 20 and 25 g were anesthetized by intraperitoneal injection of 100 mg/kg sodium pentobarbital. Their backs were then shaved, and the remaining hair was removed with a depilatory agent. Prior to injury, animals were given 0.5 mg/kg buprenorphine subcutaneously for pain management. A dorsal, 1.5- by 1.5-cm excisional skin wound to the level of the panniculus muscle was administered. Wounds were then covered with transparent, semipermeable polyurethane dressings (Opsite dressings). Approximately 1 × 106 CFU (in 100 μl) of bacterial cells was injected under the dressing on top of the wound to establish the infection. The adhesive dressing prevents contractile healing and ensures that these wounds heal by deposition of granulation tissue, much like human wounds. After 4 days postinfection the animals were sacrificed by intraperitoneal injection of 200 μl (390 mg/ml) Fatal-Plus, and wounds were harvested. Wounds were homogenized in PBS for 30 s in BeadBug tubes with 2.8-mm steel beads (Sigma-Aldrich) using a Mini-Beadbeater-16 (BioSpec Products). Bacterial loads in the wounds were enumerated by serial dilution and plating on Pseudomonas isolation agar (Sigma).
For the mouse acute pneumonia model (57, 58), all animals were treated humanely and in accordance with protocol number DAR-201700441 approved by the IACUC at Emory University in Atlanta, GA. Adult 6- to 8-week-old female BALB/c mice (Jackson Laboratories, Bar Harbor, ME) were anesthetized by an intraperitoneal injection of 200 μl of a mixture of (80 mg/kg) ketamine and (10 mg/kg) xylazine (Med-Vet International). Mice were infected by noninvasive intratracheal instillation of 50 μl of ∼2.5 × 107 CFU (twice the 50% lethal dose [LD50]) of P. aeruginosa PAO1 WT or PAO1 ΔgshA. The animals were then monitored for 7 days postinfection for survival. In the event that mice became moribund, they were humanely euthanized by CO2 asphyxiation and included in the experimental results. Surviving mice were similarly euthanized after the 7-day experimental endpoint.
For the mouse abscess model (54, 55), all animals were treated humanely and in accordance with protocol number 00136 approved by the IACUC of The University of Texas at Austin (Austin, TX) and protocol number A17086 approved by the IACUC of Georgia Institute of Technology (Atlanta, GA). Adult 6- to 8-week-old female Swiss Webster mice (Charles River Laboratories, Inc.) were anesthetized with inhaled isoflurane (1 to 3% for maintenance; up to 5% for induction) in oxygen from a precision vaporizer. While the mice were anesthetized, the inner thigh was shaved and any remaining hair removed with a depilatory agent (Nair). A 75% ethanol solution was used to clean the area prior to infection. Abscesses were initiated by injecting 100 μl of approximately 1 × 106 CFU of P. aeruginosa PAO1 WT or PAO1 ΔgshA subcutaneously into the inner thigh. Mice were monitored for signs of sepsis, and in the event that mice became moribund, they were humanely euthanized by CO2 asphyxiation. Three days postinfection, mice were euthanized by CO2 asphyxiation, and abscesses were harvested. Abscesses were homogenized in PBS for 30 s in BeadBug tubes with 2.8-mm steel beads (Sigma-Aldrich) using a Mini-Beadbeater-16 (BioSpec Products). Bacterial loads were enumerated by serial dilution and plating on Pseudomonas isolation agar (Sigma).
ACKNOWLEDGMENTS
We acknowledge the Whiteley lab for discussion of the manuscript.
This work was supported by National Institutes of Health grant R01GM116547 (to M.W. and K.P.R.) and Cystic Fibrosis Foundation grant WHITEL16G0 (to M.W.). M.W. is a Burroughs Wellcome Investigator in the Pathogenesis of Infectious Disease.
REFERENCES
- 1.Moradali MF, Ghods S, Rehm BH. 2017. Pseudomonas aeruginosa lifestyle: a paradigm for adaptation, survival, and persistence. Front Cell Infect Microbiol 7:39. doi: 10.3389/fcimb.2017.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crone S, Vives-Flórez M, Kvich L, Saunders AM, Malone M, Nicolaisen MH, Martínez-García E, Rojas-Acosta C, Catalina Gomez-Puerto M, Calum H, Whiteley M, Kolter R, Bjarnsholt T. 10 November 2019. The environmental occurrence of Pseudomonas aeruginosa. APMIS doi: 10.1111/apm.13010. [DOI] [PubMed] [Google Scholar]
- 3.Ringen LM, Drake CH. 1952. A study of the incidence of Pseudomonas aeruginosa from various natural sources. J Bacteriol 64:841–845. doi: 10.1128/JB.64.6.841-845.1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jovcic B, Lepsanovic Z, Suljagic V, Rackov G, Begovic J, Topisirovic L, Kojic M. 2011. Emergence of NDM-1 metallo-β-lactamase in Pseudomonas aeruginosa clinical isolates from Serbia. Antimicrob Agents Chemother 55:3929–3931. doi: 10.1128/AAC.00226-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mataseje L, Peirano G, Church DL, Conly J, Mulvey M, Pitout J. 2016. Colistin-nonsusceptible Pseudomonas aeruginosa sequence type 654 with blaNDM-1 arrives in North America. Antimicrob Agents Chemother 60:1794–1800. doi: 10.1128/AAC.02591-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Potron A, Poirel L, Nordmann P. 2015. Emerging broad-spectrum resistance in Pseudomonas aeruginosa and Acinetobacter baumannii: mechanisms and epidemiology. Int J Antimicrob Agents 45:568–585. doi: 10.1016/j.ijantimicag.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 7.Smirnova G, Oktyabrsky O. 2005. Glutathione in bacteria. Biochemistry (Mosc) 70:1199–1211. doi: 10.1007/s10541-005-0248-3. [DOI] [PubMed] [Google Scholar]
- 8.Lushchak VI. 2012. Glutathione homeostasis and functions: potential targets for medical interventions. J Amino Acids 2012:736837. doi: 10.1155/2012/736837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reniere ML, Whiteley AT, Hamilton KL, John SM, Lauer P, Brennan RG, Portnoy DA. 2015. Glutathione activates virulence gene expression of an intracellular pathogen. Nature 517:170–173. doi: 10.1038/nature14029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ku JW, Gan Y-H. 2019. Modulation of bacterial virulence and fitness by host glutathione. Curr Opin Microbiol 47:8–13. doi: 10.1016/j.mib.2018.10.004. [DOI] [PubMed] [Google Scholar]
- 11.Newton GL, Rawat M, La Clair JJ, Jothivasan VK, Budiarto T, Hamilton CJ, Claiborne A, Helmann JD, Fahey RC. 2009. Bacillithiol is an antioxidant thiol produced in bacilli. Nat Chem Biol 5:625–627. doi: 10.1038/nchembio.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ung KS, Av-Gay Y. 2006. Mycothiol-dependent mycobacterial response to oxidative stress. FEBS Lett 580:2712–2716. doi: 10.1016/j.febslet.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 13.Fahey RC. 2013. Glutathione analogs in prokaryotes. Biochim Biophys Acta 1830:3182–3198. doi: 10.1016/j.bbagen.2012.10.006. [DOI] [PubMed] [Google Scholar]
- 14.Meury J, Kepes A. 1982. Glutathione and the gated potassium channels of Escherichia coli. EMBO J 1:339–343. doi: 10.1002/j.1460-2075.1982.tb01171.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhou H, Wang M, Smalley NE, Kostylev M, Schaefer AL, Greenberg EP, Dandekar AA, Xu F. 2019. Modulation of Pseudomonas aeruginosa quorum sensing by glutathione. J Bacteriol 201:e00685-18. doi: 10.1128/JB.00685-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Zhang C, Du X, Zhou Y, Kong W, Lau GW, Chen G, Kohli GS, Yang L, Wang T, Liang H. 2019. Glutathione activates type III secretion system through Vfr in Pseudomonas aeruginosa. Front Cell Infect Microbiol 9:164. doi: 10.3389/fcimb.2019.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wongsaroj L, Saninjuk K, Romsang A, Duang-Nkern J, Trinachartvanit W, Vattanaviboon P, Mongkolsuk S. 2018. Pseudomonas aeruginosa glutathione biosynthesis genes play multiple roles in stress protection, bacterial virulence and biofilm formation. PLoS One 13:e0205815. doi: 10.1371/journal.pone.0205815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Van Laar TA, Esani S, Birges TJ, Hazen B, Thomas JM, Rawat M. 2018. Pseudomonas aeruginosa gshA mutant is defective in biofilm formation, swarming, and pyocyanin production. mSphere 3:e00155-18. doi: 10.1128/mSphere.00155-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Duan K. 2009. Glutathione exhibits antibacterial activity and increases tetracycline efficacy against Pseudomonas aeruginosa. Sci China C Life Sci 52:501–505. doi: 10.1007/s11427-009-0074-8. [DOI] [PubMed] [Google Scholar]
- 20.Goswami M, Jawali N. 2010. N-Acetylcysteine-mediated modulation of bacterial antibiotic susceptibility. Antimicrob Agents Chemother 54:3529–3530. doi: 10.1128/AAC.00710-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feinbaum RL, Urbach JM, Liberati NT, Djonovic S, Adonizio A, Carvunis A-R, Ausubel FM. 2012. Genome-wide identification of Pseudomonas aeruginosa virulence-related genes using a Caenorhabditis elegans infection model. PLoS Pathog 8:e1002813. doi: 10.1371/journal.ppat.1002813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Song M, Husain M, Jones-Carson J, Liu L, Henard CA, Vázquez-Torres A. 2013. Low-molecular-weight thiol-dependent antioxidant and antinitrosative defences in Salmonella pathogenesis. Mol Microbiol 87:609–622. doi: 10.1111/mmi.12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gamage AM, Liao C, Cheah IK, Chen Y, Lim DRX, Ku JWK, Chee RSL, Gengenbacher M, Seebeck FP, Halliwell B, Gan Y-H. 2018. The proteobacterial species Burkholderia pseudomallei produces ergothioneine, which enhances virulence in mammalian infection. FASEB J 32:6395–6409. doi: 10.1096/fj.201800716. [DOI] [PubMed] [Google Scholar]
- 24.Turner KH, Everett J, Trivedi U, Rumbaugh KP, Whiteley M. 2014. Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection. PLoS Genet 10:e1004518. doi: 10.1371/journal.pgen.1004518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turner KH, Wessel AK, Palmer GC, Murray JL, Whiteley M. 2015. Essential genome of Pseudomonas aeruginosa in cystic fibrosis sputum. Proc Natl Acad Sci U S A 112:4110–4115. doi: 10.1073/pnas.1419677112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ibberson CB, Stacy A, Fleming D, Dees JL, Rumbaugh K, Gilmore MS, Whiteley M. 2017. Co-infecting microorganisms dramatically alter pathogen gene essentiality during polymicrobial infection. Nat Microbiol 2:17079. doi: 10.1038/nmicrobiol.2017.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fuchs JA, Warner HR. 1975. Isolation of an Escherichia coli mutant deficient in glutathione synthesis. J Bacteriol 124:140–148. doi: 10.1128/JB.124.1.140-148.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murray JL, Kwon T, Marcotte EM, Whiteley M. 2015. Intrinsic antimicrobial resistance determinants in the superbug Pseudomonas aeruginosa. mBio 6:e01603-15. doi: 10.1128/mBio.01603-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Socransky S, Dzink J, Smith C. 1985. Chemically defined medium for oral microorganisms. J Clin Microbiol 22:303–305. doi: 10.1128/JCM.22.2.303-305.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuppusamy P, Li H, Ilangovan G, Cardounel AJ, Zweier JL, Yamada K, Krishna MC, Mitchell JB. 2002. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res 62:307–312. [PubMed] [Google Scholar]
- 31.Turnbull AL, Surette MG. 2010. Cysteine biosynthesis, oxidative stress and antibiotic resistance in Salmonella typhimurium. Res Microbiol 161:643–650. doi: 10.1016/j.resmic.2010.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Shukla A, Rasik AM, Dhawan BN. 1999. Asiaticoside-induced elevation of antioxidant levels in healing wounds. Phytother Res 13:50–54. doi:. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Wei G, Chen J. 2004. Glutathione: a review on biotechnological production. Appl Microbiol Biotechnol 66:233–242. doi: 10.1007/s00253-004-1751-y. [DOI] [PubMed] [Google Scholar]
- 34.Kram KE, Finkel SE. 2014. Culture volume and vessel affect long-term survival, mutation frequency, and oxidative stress of Escherichia coli. Appl Environ Microbiol 80:1732–1738. doi: 10.1128/AEM.03150-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Babior BM. 2000. Phagocytes and oxidative stress. Am J Med 109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 36.Wang G, Nauseef WM. 2015. Salt, chloride, bleach, and innate host defense. J Leukoc Biol 98:163–172. doi: 10.1189/jlb.4RU0315-109R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Han M-L, Zhu Y, Creek DJ, Lin Y-W, Gutu AD, Hertzog P, Purcell T, Shen H-H, Moskowitz SM, Velkov T, Li J. 2019. Comparative metabolomics and transcriptomics reveal multiple pathways associated with polymyxin killing in Pseudomonas aeruginosa. mSystems 4:e00149-18. doi: 10.1128/mSystems.00149-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ. 2014. Antibiotics induce redox-related physiological alterations as part of their lethality. Proc Natl Acad Sci U S A 111:E2100–E2109. doi: 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tzeng Y-L, Ambrose KD, Zughaier S, Zhou X, Miller YK, Shafer WM, Stephens DS. 2005. Cationic antimicrobial peptide resistance in Neisseria meningitidis. J Bacteriol 187:5387–5396. doi: 10.1128/JB.187.15.5387-5396.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Parra-Lopez C, Lin R, Aspedon A, Groisman E. 1994. A Salmonella protein that is required for resistance to antimicrobial peptides and transport of potassium. EMBO J 13:3964–3972. doi: 10.1002/j.1460-2075.1994.tb06712.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bengoechea JA, Skurnik M. 2000. Temperature-regulated efflux pump/potassium antiporter system mediates resistance to cationic antimicrobial peptides in Yersinia. Mol Microbiol 37:67–80. doi: 10.1046/j.1365-2958.2000.01956.x. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y-C, Chuang Y-C, Chang C-C, Jeang C-L, Chang M-C. 2004. A K+ uptake protein, TrkA, is required for serum, protamine, and polymyxin B resistance in Vibrio vulnificus. Infect Immun 72:629–636. doi: 10.1128/iai.72.2.629-636.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sadikot RT, Blackwell TS, Christman JW, Prince AS. 2005. Pathogen–host interactions in Pseudomonas aeruginosa pneumonia. Am J Respir Crit Care Med 171:1209–1223. doi: 10.1164/rccm.200408-1044SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Wei Y. 2009. Impact of glutathione on the gene expression of exoY and exoS in Pseudomonas aeruginosa. Wei Sheng Wu Xue Bao 49:603–608. [PubMed] [Google Scholar]
- 45.Solapure S, Dinesh N, Shandil R, Ramachandran V, Sharma S, Bhattacharjee D, Ganguly S, Reddy J, Ahuja V, Panduga V, Parab M, Vishwas KG, Kumar N, Balganesh M, Balasubramanian V. 2013. In vitro and in vivo efficacy of β-lactams against replicating and slowly growing/nonreplicating Mycobacterium tuberculosis. Antimicrob Agents Chemother 57:2506–2510. doi: 10.1128/AAC.00023-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tuomanen E, Cozens R, Tosch W, Zak O, Tomasz A. 1986. The rate of killing of Escherichia coli by β-lactam antibiotics is strictly proportional to the rate of bacterial growth. J Gen Microbiol 132:1297–1304. doi: 10.1099/00221287-132-5-1297. [DOI] [PubMed] [Google Scholar]
- 47.Gjødsbøl K, Christensen JJ, Karlsmark T, Jørgensen B, Klein BM, Krogfelt KA. 2006. Multiple bacterial species reside in chronic wounds: a longitudinal study. Int Wound J 3:225–231. doi: 10.1111/j.1742-481X.2006.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dowd SE, Sun Y, Secor PR, Rhoads DD, Wolcott BM, James GA, Wolcott RD. 2008. Survey of bacterial diversity in chronic wounds using pyrosequencing, DGGE, and full ribosome shotgun sequencing. BMC Microbiol 8:43. doi: 10.1186/1471-2180-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Parker CM, Kutsogiannis J, Muscedere J, Cook D, Dodek P, Day AG, Heyland DK, Canadian Critical Care Trials Group. 2008. Ventilator-associated pneumonia caused by multidrug-resistant organisms or Pseudomonas aeruginosa: prevalence, incidence, risk factors, and outcomes. J Crit Care 23:18–26. doi: 10.1016/j.jcrc.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 50.Sittig K, Deitch EA. 1988. Effect of bacteremia on mortality after thermal injury. Arch Surg 123:1367–1370. doi: 10.1001/archsurg.1988.01400350081012. [DOI] [PubMed] [Google Scholar]
- 51.Dalton T, Dowd SE, Wolcott RD, Sun Y, Watters C, Griswold JA, Rumbaugh KP. 2011. An in vivo polymicrobial biofilm wound infection model to study interspecies interactions. PLoS One 6:e27317. doi: 10.1371/journal.pone.0027317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolcott RD, Rumbaugh KP, James G, Schultz G, Phillips P, Yang Q, Watters C, Stewart PS, Dowd SE. 2010. Biofilm maturity studies indicate sharp debridement opens a time-dependent therapeutic window. J Wound Care 19:320–328. doi: 10.12968/jowc.2010.19.8.77709. [DOI] [PubMed] [Google Scholar]
- 53.Holder IA, Brown RL, Greenhalgh DG. 1997. Mouse models to study wound closure and topical treatment of infected wounds in healing-impaired and normal healing hosts. Wound Repair Regen 5:198–204. doi: 10.1046/j.1524-475X.1997.50213.x. [DOI] [PubMed] [Google Scholar]
- 54.Lewin GR, Stacy A, Michie KL, Lamont RJ, Whiteley M. 2019. Large-scale identification of pathogen essential genes during coinfection with sympatric and allopatric microbes. Proc Natl Acad Sci U S A 116:19685–19694. doi: 10.1073/pnas.1907619116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stacy A, Fleming D, Lamont RJ, Rumbaugh KP, Whiteley M. 2016. A commensal bacterium promotes virulence of an opportunistic pathogen via cross-respiration. mBio 7:e00782-16. doi: 10.1128/mBio.00782-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haynes A III, Ruda F, Oliver J, Hamood AN, Griswold JA, Park PW, Rumbaugh KP. 2005. Syndecan 1 shedding contributes to Pseudomonas aeruginosa sepsis. Infect Immun 73:7914–7921. doi: 10.1128/IAI.73.12.7914-7921.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Revelli DA, Boylan JA, Gherardini FC. 2012. A non-invasive intratracheal inoculation method for the study of pulmonary melioidosis. Front Cell Infect Microbiol 2:164. doi: 10.3389/fcimb.2012.00164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mukherjee S, Moustafa D, Smith CD, Goldberg JB, Bassler BL. 2017. The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer. PLoS Pathog 13:e1006504. doi: 10.1371/journal.ppat.1006504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Priebe GP, Brinig MM, Hatano K, Grout M, Coleman FT, Pier GB, Goldberg JB. 2002. Construction and characterization of a live, attenuated aroA deletion mutant of Pseudomonas aeruginosa as a candidate intranasal vaccine. Infect Immun 70:1507–1517. doi: 10.1128/iai.70.3.1507-1517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lazarus GS, Cooper DM, Knighton DR, Margolis DJ, Pecoraro RE, Rodeheaver G, Robson MC. 1994. Definitions and guidelines for assessment of wounds and evaluation of healing. Arch Dermatol 130:489–493. doi: 10.1001/archderm.1994.01690040093015. [DOI] [PubMed] [Google Scholar]
- 61.Sen CK, Gordillo GM, Roy S, Kirsner R, Lambert L, Hunt TK, Gottrup F, Gurtner GC, Longaker MT. 2009. Human skin wounds: a major and snowballing threat to public health and the economy. Wound Repair Regen 17:763–771. doi: 10.1111/j.1524-475X.2009.00543.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deniz M, Borman H, Seyhan T, Haberal M. 2013. An effective antioxidant drug on prevention of the necrosis of zone of stasis: N-acetylcysteine. Burns 39:320–325. doi: 10.1016/j.burns.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 63.Parihar A, Parihar MS, Milner S, Bhat S. 2008. Oxidative stress and anti-oxidative mobilization in burn injury. Burns 34:6–17. doi: 10.1016/j.burns.2007.04.009. [DOI] [PubMed] [Google Scholar]
- 64.Bertin-Maghit M, Goudable J, Dalmas E, Steghens J, Bouchard C, Gueugniaud P, Petit P, Delafosse B. 2000. Time course of oxidative stress after major burns. Intensive Care Med 26:800–803. doi: 10.1007/s001340051250. [DOI] [PubMed] [Google Scholar]
- 65.Cemek M, Çaksen H, Bayiroğlu F, Cemek F, Dede S. 2006. Oxidative stress and enzymic–non-enzymic antioxidant responses in children with acute pneumonia. Cell Biochem Funct 24:269–273. doi: 10.1002/cbf.1220. [DOI] [PubMed] [Google Scholar]
- 66.Mustoe TA, O’Shaughnessy K, Kloeters O. 2006. Chronic wound pathogenesis and current treatment strategies: a unifying hypothesis. Plast Reconstr Surg 117:35S–41S. doi: 10.1097/01.prs.0000225431.63010.1b. [DOI] [PubMed] [Google Scholar]
- 67.Chow C-W, Herrera Abreu MT, Suzuki T, Downey GP. 2003. Oxidative stress and acute lung injury. Am J Respir Cell Mol Biol 29:427–431. doi: 10.1165/rcmb.F278. [DOI] [PubMed] [Google Scholar]
- 68.Mudge BP, Harris C, Gilmont RR, Adamson BS, Rees RS. 2002. Role of glutathione redox dysfunction in diabetic wounds. Wound Repair Regen 10:52–58. doi: 10.1046/j.1524-475X.2002.10803.x. [DOI] [PubMed] [Google Scholar]
- 69.Aktunc E, Ozacmak V, Ozacmak H, Barut F, Buyukates M, Kandemir O, Demircan N. 2010. N-acetyl cysteine promotes angiogenesis and clearance of free oxygen radicals, thus improving wound healing in an alloxan-induced diabetic mouse model of incisional wound. Clin Exp Dermatol 35:902–909. doi: 10.1111/j.1365-2230.2010.03823.x. [DOI] [PubMed] [Google Scholar]
- 70.Al-Jawad F, Sahib A, Al-Kaisy A. 2008. Role of antioxidants in the treatment of burn lesions. Ann Burns Fire Disasters 21:186–191. [PMC free article] [PubMed] [Google Scholar]
- 71.Ocal K, Avlan D, Cinel I, Unlu A, Ozturk C, Yaylak F, Dirlik M, Camdeviren H, Aydin S. 2004. The effect of N-acetylcysteine on oxidative stress in intestine and bacterial translocation after thermal injury. Burns 30:778–784. doi: 10.1016/j.burns.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 72.Rahman I, MacNee W. 2000. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radic Biol Med 28:1405–1420. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- 73.Lai K, Ng W, Chan P, Wong K, Cheng F. 2010. High-dose N-acetylcysteine therapy for novel H1N1 influenza pneumonia. Ann Intern Med 152:687–688. doi: 10.7326/0003-4819-152-10-201005180-00017. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Q, Ju Y, Ma Y, Wang T. 2018. N-acetylcysteine improves oxidative stress and inflammatory response in patients with community acquired pneumonia: a randomized controlled trial. Medicine 97:e13087. doi: 10.1097/MD.0000000000013087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rietsch A, Vallet-Gely I, Dove SL, Mekalanos JJ. 2005. ExsE, a secreted regulator of type III secretion genes in Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 102:8006–8011. doi: 10.1073/pnas.0503005102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA III, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 77.National Research Council. 2009. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]