

Chronic Helicobacter pylori colonization in animal models often leads to downregulation of the type IV secretion system (T4SS), typically by recombination in cagY, which is an essential T4SS gene. However, 17 other cag pathogenicity island (cagPAI) genes, as well as some non-cagPAI genes, are also essential for T4SS function. To get a more complete picture of how H. pylori regulates the T4SS during animal colonization, we examined cagY in 534 mouse-passaged isolates that lost T4SS function, defined as a normalized interleukin-8 (IL-8) value of <0.
KEYWORDS: Helicobacter, mice, type IV secretion system, pylori
ABSTRACT
Chronic Helicobacter pylori colonization in animal models often leads to downregulation of the type IV secretion system (T4SS), typically by recombination in cagY, which is an essential T4SS gene. However, 17 other cag pathogenicity island (cagPAI) genes, as well as some non-cagPAI genes, are also essential for T4SS function. To get a more complete picture of how H. pylori regulates the T4SS during animal colonization, we examined cagY in 534 mouse-passaged isolates that lost T4SS function, defined as a normalized interleukin-8 (IL-8) value of <0.3 relative to the input H. pylori strain PMSS1. In order to analyze the genetic changes in the strains with unchanged cagY, we sequenced the entire pathogenicity island of 60 such isolates using single-molecule, real-time (SMRT) sequencing technology (PacBio, Menlo Park, CA), and we compared the results to the PMSS1 wild type (WT). Of the 534 strains, 271 (51%) showed evidence of recombination in cagY, but we also found indels or nonsynonymous changes in 13 other essential cagPAI genes implicated in H. pylori T4SS function, most commonly cag5, cag10, and cagA. While cagY recombination is the most common mechanism by which H. pylori downregulates T4SS function during murine infection, loss of function is also associated with changes in other essential cagPAI genes.
INTRODUCTION
Helicobacter pylori is a Gram-negative, microaerophilic bacterium that commonly colonizes the human gastric epithelium where it sometimes causes peptic ulcers or gastric adenocarcinoma, the third most common cause of cancer death, after lung cancer and colorectal cancer (1). The pathogenesis of H. pylori infection is most strongly associated with the cag pathogenicity island (cagPAI), an ∼40-kb DNA segment that encodes a type IV secretion system (T4SS) that translocates the CagA oncoprotein into host gastric epithelial cells (2), where it is phosphorylated on tyrosine residues by Src and Abl family kinases (3). Translocation of CagA was initially reported to require T4SS contact with β1 integrin receptors (4, 5), although more recent work suggests the importance of carcinoembryonic antigen-related cell adhesion molecule (CEACAM) receptors binding to the outer membrane protein HopQ (6–8). Under some experimental conditions, both receptors may be important (9). The injection of CagA into the host cell causes complex changes in host cell signaling, which leads to alterations in host cell physiology, including the disruption of tight junctions, cytoskeletal rearrangements, and the production of interleukin-8 (IL-8), a proinflammatory cytokine (10, 11).
T4SS function is typically assessed by identification of phosphorylated CagA by immunoblot, induction of interleukin-8 (IL-8) by enzyme-linked immunosorbent assay (ELISA), or by light microscopic detection of changes in epithelial cell morphology termed the “hummingbird” phenotype. Gene deletion studies have demonstrated that 18 genes on the cagPAI are necessary for a functional T4SS, although occasionally differences are found that are strain or assay dependent (12–14). One of these essential genes is cagY, which encodes a protein that, together with CagX, CagM, CagT, and Cag3, forms the T4SS core complex thought to be located between the inner and outer bacterial cell membrane (15–17). CagY is an ortholog of VirB10, which is an essential component of all T4SSs described to date. In the canonical T4SS of Agrobacterium tumefaciens, VirB10 senses ATP binding or hydrolysis and undergoes a conformational change that is required for the transfer of DNA across the outer bacterial membrane (18). However, cagY encodes an ∼220-kDa protein that is much larger than other virB10 orthologs, and it contains an extraordinary number of DNA repeats in what has been termed the middle repeat region (MRR) or repeat region 1 (19, 20). The DNA repeats permit many in-frame deletions or insertions that can cause either gain or loss of function in the T4SS (21). The passage of H. pylori through mice is known to cause a loss of T4SS function (22, 23). We recently demonstrated that the loss of T4SS function in the murine model results from CD4 T cell- and interferon gamma (IFNγ)-dependent immune pressure that selects for MRR variants of cagY (24), which are expressed but are not functional, perhaps because they fail to bind β1 integrins (25). Recombination in cagY can also cause gain of function in the T4SS, so it serves as a sort of molecular rheostat that appears to modulate the host inflammatory response so as to maintain a favorable environment for persistent infection.
Although we previously found that the loss of T4SS function in mice is usually associated with changes in cagY (21), there are 17 other cagPAI genes that are essential for T4SS function. T4SS function is also dependent upon proteins not encoded on the cagPAI, including HopQ (6, 7), which binds CEACAMs and HtrA, which cleaves the junctional proteins claudin-8, occludin, and E-cadherin (26). Recently, we found that the loss of T4SS function during murine colonization, without a change in cagY, is more common than in our previously published data, suggesting that one or more genes on the cagPAI, or perhaps non-cagPAI genes, may be responsible. Here, we characterized many mouse output strains that lost T4SS function during mouse colonization. The results show that while recombination in cagY accounts for about half of all instances in which T4SS function is lost, a variety of other PAI genes are also subject to change, most commonly cag5, cag10, and cagA.
RESULTS AND DISCUSSION
Loss of T4SS function during mouse colonization is caused by recombination in cagY and by indels in other essential PAI genes.
Here, we examined PMSS1 isolates that lost T4SS function during mouse colonization but without a change in the size or PCR-restriction fragment length polymorphism (RFLP) pattern of cagY, suggesting that the loss of T4SS function resulted from changes in other genes on the PAI. To study this further, we performed cagY PCR-RFLP on 534 mouse-passaged PMSS1 isolates that lost T4SS function, defined as normalized IL-8 values of <0.3 relative to wild type (WT) PMSS1. To analyze the genetic changes in the strains with unchanged cagY, we sequenced the entire pathogenicity island of 60 such isolates using single-molecule, real-time (SMRT) sequencing technology (PacBio, Menlo Park, CA) and compared the results to PMSS1 WT. To avoid the analysis of clones, each sequenced strain was recovered from a unique mouse. All differences between PMSS1 and the output strains were verified by Sanger sequencing, except single-nucleotide indels within a portion of the cagY MRR (bp 2650 to 3370), which could not be sequenced by primer extension. In these cases, Western blots were performed to look for a size difference in the expressed CagY protein, which, if an indel were present, would result in a frameshift and premature stop. If sequence analysis detected an insertion or deletion in an essential gene, this was assumed to be the mechanism for the loss of T4SS function. As cagA is not contiguous with the PAI in PMSS1, it was sequenced separately by the Sanger technique.
Of the 534 strains, 282 (53%) showed evidence of recombination in cagY, typically detected by PCR-RFLP, but occasionally by only a change in size on agarose gel electrophoresis (Fig. 1A) or Western blot (Fig. 1B). While cagY was the gene most commonly affected by mouse passage, we also found changes in 13 other PAI genes implicated in H. pylori T4SS function, most commonly cag5, cag10, and cagA (Fig. 1C). No strain had more than one cagPAI sequence change, and no DNA sequence changes were detected in PAI genes not previously implicated in T4SS function. Specific changes observed are listed in Table 1. While it is possible that the changes observed after in vivo infection might also occur during in vitro passage, this is unlikely. We previously showed that the loss of T4SS function was rare after in vitro passage of H. pylori J166 (21). Although the studies here used strain PMSS1 and not J166, we have also shown that PMSS1 does not lose T4SS function even in vivo, if inoculated into Rag−/− mice or mice without IFN gamma signaling (24), suggesting that immune pressure is essential for the loss of T4SS function.
FIG 1.

PAI genes responsible for the loss of T4SS function in mouse-passaged H. pylori. When cagY PCR-RFLP showed no changes, amplicons were run on low-percent agarose gels to detect size differences (A). Examples of changed cagY size are shown in lanes 3 and 4. The PacBio sequence of cagY was unable to be verified with the Sanger technique due to its repetitive nature. Therefore, Western blots were used to detect changes in the size of CagY resulting from indels causing truncation (B). Lane 1, input WT PMSS1; lane 2, unchanged output strain; lanes 3 to 5, changed output strains. Changes were found most commonly in cagY, but all genes essential for cagPAI function were changed at least once except for cag3, cag4, cag18, and cag20 (C). No changes were found in genes that are nonessential for CagA delivery. Bars indicate frequency of changes during mouse passage for each gene that is essential for cagPAI function.
TABLE 1.
cagPAI gene changes in mouse output strains
| Gene | Nucleotide position of genea | Change typeb | Changed base | Nucleotide position of change | Frequency of change |
|---|---|---|---|---|---|
| cag5 | 759807 | D | A | 157 | 5 |
| cag5 | 759859 | D | C | 209 | 1 |
| cag5 | 759880 | I | T | 230 | 3 |
| cag5 | 760128 | D | T | 478 | 1 |
| cag5 | 760250 | I | A | 600 | 1 |
| cag5 | 761413 | I | C | 1763 | 3 |
| virB11 | 759147 | D | T | 498 | 1 |
| cagY | 752059 | S | T→Cc | 2 | 1 |
| cagY | 752688 | D | A | 631 | 4 |
| cagY | 753394 | I | T | 1337 | 1 |
| cagY | 753612 | D | A | 1555 | 1 |
| cagY | 753686 | I | A | 1629 | 1 |
| cagY | 754887 | D | A | 2830 | 2 |
| cagY | 755292 | D | G | 3235 | 1 |
| cag8 | 750905 | D | A | 431 | 2 |
| cag8 | 751095–751097 | D | AGA | 621–623 | 1 |
| cag8 | 751872 | S | G→Ad | 1398 | 1 |
| cag9 | 749358 | D | T | 543 | 1 |
| cag9 | 749511 | I | T | 696 | 2 |
| cag10 | 748053–748150 | D | Multiple | 1–98 | 1 |
| cag10 | 748061 | I | A | 9 | 3 |
| cag10 | 748165 | I | T | 113 | 1 |
| cag10 | 748539 | D | A | 487 | 1 |
| cag11 | 747396 | S | G→Td | 277 | 1 |
| cag12 | 746825 | I | TA | 156 | 1 |
| cag16 | 743066 | D | A | 270 | 1 |
| cag19 | 739940 | D | A | 607 | 1 |
| cag19 | 740045 | I | C | 712 | 1 |
| cag23 | 734139–734312 | D | Multiple | 182–355 | 2 |
| cag23 | 735493 | I | A | 1542 | 1 |
| cag24 | 733355 | I | T | 36 | 1 |
| cag25 | 733028 | D | G | 55 | 1 |
| cagA | 776684–796971 | D | Multiple | Entire gene | 3 |
| cagA | 777411 | I | G | 136 | 1 |
| cagA | 779419–779553 | D | Multiple | 2144–2278 | 2 |
GenBank accession number CP018823.
I, insertion (downstream of noted position); D, deletion; S, substitution.
Start codon deleted.
Stop codon created.
Selected changes in the PAI were investigated further to determine whether they were, in fact, responsible for the observed loss of T4SS function, including in-frame deletions (cag8 and cagA), single base substitutions (cag25), and an insertion leading to truncation in cag24, whose role in IL-8 induction is apparently strain specific (12, 27). Contraselection was used to replace the gene of interest in PMSS1 with the mutated gene from the mouse-passaged strain or from WT PMSS1 as a control. For cag8, a single glutamic acid deletion (amino acid 210) was shown to be sufficient to cause a loss of T4SS function (Fig. 2A). Cag8 (CagX) is a VirB9 ortholog that, along with CagY, CagM, CagT, and Cag3, forms the outer membrane complex of the H. pylori T4SS nanomachine (28). The deletion of a glutamic acid may change its three-dimensional structure by disruption of a salt bridge, causing destabilization of this core complex. Although the crystal structure of Cag8 has been reported (29), the effect of deleting glutamic acid could not be confirmed because only the soluble C-terminal portion of the protein (amino acids 396 to 498) was solved.
FIG 2.

Verification of the functional significance of selected changes in essential PAI genes after mouse passage. The cagPAI gene of interest was deleted from wild-type PMSS1 (Δ) and replaced with the mutated gene of interest [Δ(Out)] or the WT gene [Δ(WT)] as a positive control. IL-8 was measured and normalized to that of wild-type PMSS1. Western blots show tyrosine phosphorylated translocated CagA (α-PY99) and total CagA (α-CagA). Specific mutations are shown by the schematic at the top of each panel. Changes in cag8 (deletion of glutamic acid at amino acid 210), cag24 (stop codon after lysine at amino acid 31), and cagA (deletion of amino acids 711 to 755) eliminated T4SS function (A, B, C), but a change from alanine to threonine in cag25 did not. Error bars represent SD.
The truncation of cag24 (cagD) after 31 amino acids resulted in a 90% reduction in IL-8 induction and complete elimination of CagA translocation in the PMSS1 background (Fig. 2B). When deleted from 26695, it only moderately reduces IL-8 induction and cagA translocation (12). However, in strain G27, cag24 is essential for the translocation of CagA into gastric epithelial cells and for maximal induction of IL-8 (27). Cag24 localizes to the inner bacterial membrane but also is secreted into the cell supernatant (27). The N-terminus of Cag24 in strain PMSS1 is predicted (SignalP 5.0) to contain a signal peptide with cleavage between residues 41 and 42, which likely explains the lack of T4SS function in the N-terminal 31-amino acid truncated protein.
Although CagA was originally thought to be unnecessary for induction of IL-8 (12), this is strain dependent (14) and is required for PMSS1. A 45-amino acid deletion from cagA (711 to 755) was found to be sufficient for reduction of IL-8 induction to a level similar to a cagA knockout (Fig. 2C). This is, perhaps, surprising since in wild-type cagA, this region does not contain the EPIYA motifs, which are tyrosine phosphorylated in host cells, resulting in deregulation of cell signaling. However, loss of this region was sufficient to eliminate binding to a polyclonal cagA antibody. A single amino acid change in Cag25 (alanine to threonine at amino acid 34) was not sufficient to reduce IL-8 induction or CagA translocation (Fig. 2D).
Seven isolates were found for which no change in the PAI was identified. We studied these isolates further by Sanger sequencing of hopQ and oipA, known to be necessary for a fully functioning T4SS (30, 31). None of these 7 isolates showed disruption of both hopQ alleles (one functioning allele is sufficient for T4SS function) or oipA, leaving the cause for reduced IL-8 unknown. Sanger sequencing of select PAI genes detected a single base insertion within a polynucleotide tract in cag5 in 2 of these isolates, which had not been seen during the original sequencing, an issue often associated with the PacBio technique. Therefore, we do not feel confident concluding that the remaining 5 isolates have an off-island cause for the loss of T4SS function due to the inability of PacBio to accurately detect the length of homopolymeric tracts, of which there are many in the PAI.
cagA copy number frequently changes during mouse infection but is not correlated with the level of IL-8 induction.
PMSS1 has been previously reported to have multiple copies of cagA (32, 33) arranged adjacent to one another in the same orientation. The copy number appears to be highly unstable due to the presence of direct repeats upstream and downstream of the cagA copies, allowing for frequent recombination. Increased copy number has been shown to lead to increased expression of the CagA protein. To investigate whether cagA copy number changed during mouse passage and whether it could be correlated with the loss of T4SS function, we performed real-time PCR on the 60 sequenced output strains. Of the 60 isolates tested, 52 changed their cagA copy number (Fig. 3A) by at least two standard deviations outside the mean of the PMSS1 controls run on each assay. Three isolates were found to have deleted cagA completely, and they had no other cagPAI changes. However, when plotted versus normalized IL-8, no correlation was seen with the number of cagA copies (Fig. 3B). This is inconsistent with previous results showing a positive correlation between cagA copy number and IL-8 (32) but could be due to the fact that all of the isolates were selected for their relatively low level of IL-8 induction.
FIG 3.

cagA copy number and IL-8 induction in mouse-passaged H. pylori. cagA copy number was determined by real-time PCR. Those mouse-passaged isolates with cagA copy number outside two standard deviations of the mean of all wild-type controls run on each assay were defined as changed. Most isolates showed either an increase or decrease in the copy number of cagA (A), and cagA was deleted in three cases, but copy number was not correlated with level of IL-8 induction (B).
PAI genes with longer polynucleotide tracts are more prone to slipped strand mispairing, truncated proteins, and loss of cag island function.
Of the 60 strains examined, 55 showed PAI sequence changes leading to a loss of T4SS function. Of these changes, 42 were single-bp indels, 10 were multiple-bp indels, and 3 were single nucleotide polymorphisms (SNPs). Of the single-bp indels, 30 (71%) occurred in homopolymeric polynucleotide tracts, suggesting that genes with longer polynucleotide tracts had an increased frequency of changes. To examine this, we determined the longest polynucleotide tract in each cagPAI gene and compared the results between those with (n = 14) and without changes (n = 13). Although the difference was small and its functional significance is unclear, genes with changes had longer polynucleotide tracts (Fig. 4). Overall, more than 50% of the changes in cagPAI genes occurred when the length of the polynucleotide tract was 8 nucleotides or greater.
FIG 4.
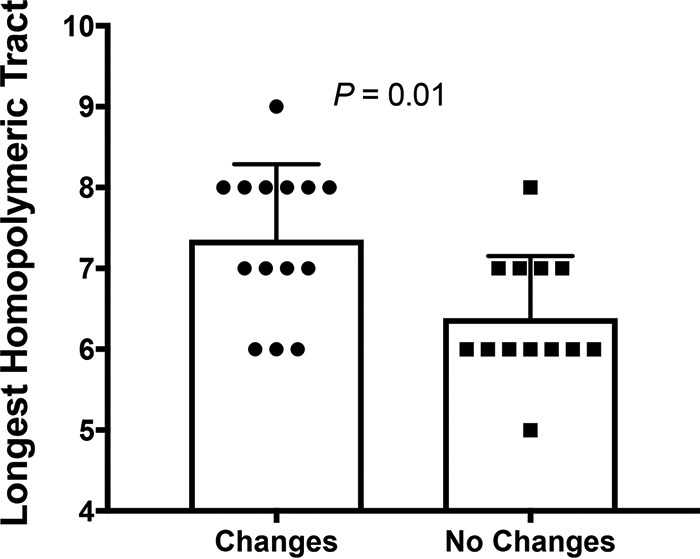
Genes with changes had longer homopolymeric tracts. The longest polynucleotide tract was determined for each island gene. The mean of genes with changes was found to differ significantly from genes without (Mann-Whitney U test; error bars represent SD).
Perspectives.
From the bacterial perspective, H. pylori-induced inflammation is a double-edged sword; it promotes nutrient acquisition on the one hand, but it comes at the cost of reduction in bacterial load and reduced chance of transmission. Thus, H. pylori is probably dependent on the host response but must also closely regulate it. While H. pylori employs many redundant strategies for this regulation (34), genomic change in the cagPAI, particularly but not exclusively recombination in cagY, is a common mechanism.
MATERIALS AND METHODS
Bacterial strains.
We performed cagY PCR-RFLP on 534 mouse-passaged PMSS1 isolates, recovered from 117 mice, that lost T4SS function, defined as normalized IL-8 values of <0.3 relative to WT PMSS1 (35). These strains were obtained from previous experiments as described (21, 24). Briefly, 10- to 12-week-old mice were challenged by oral gavage with 2.5 × 109 CFU of H. pylori PMSS1 suspended in 0.25 ml of brucella broth. Mice were euthanized at 8 weeks postinfection and stomachs were cut longitudinally. Half of each stomarch was homogenized with a glass pestle and serially diluted and plated onto brucella agar (BBL/Becton, Dickinson, Sparks, MD) containing 5% heat-inactivated newborn calf serum (Invitrogen, Carlsbad, CA) and ABPNV antibiotics (amphotericin B, 20 μg/ml; bacitracin, 200 μg/ml; polymyxin B, 3.3 μg/ml; nalidixic acid, 10.7 μg/ml; vancomycin, 100 μg/ml). Following initial culture, brucella agar plates containing 5% serum and TVPA antibiotics were used for passage (trimethoprim, 5 μg/ml; vancomycin, 10 μg/ml; polymyxin B, 2.5 units/ml; amphotericin B, 2.5 μg/ml). Strains were grown at 37°C in microaerobic conditions (5% O2, 7.6% CO2) generated via a fixed O2 concentrator (Anoxomat, Advanced Instruments, Norwood, MA). Cultures were passed minimally and frozen at −80°C in brucella broth with 20% glycerol. Many of these strains had a change in cagY RFLP, which was presumed to be the explanation for the loss of T4SS function. To identify the basis for the loss of T4SS function when cagY was unchanged, we selected 60 such strains, each from a unique mouse, and sequenced the entire cagPAI. All mouse experiments were conducted according to the Animal Welfare Act and National Institutes of Health guidelines, and protocols were approved by the UC Davis Institutional Animal Care and Use Committee.
cagY PCR-RFLP.
PCR-restriction fragment length polymorphism (PCR-RFLP) was used as described previously (21) to detect changes in cagY sequences in strains recovered after mouse colonization. Briefly, cagY was PCR amplified with specific primers (Table 2), purified using the QIAquick PCR purification kit (Qiagen Sciences, Maryland, MD), and diluted in water to a concentration of 105 ng/μl. The samples were then digested overnight at 37°C with DdeI and Sau3AI separately, electrophoresed on a 5.0% agarose gel, and stained with ethidium bromide, using the input PMSS1 as the positive control. If the cagY PCR-RFLP was identical to that of PMSS1, unrestricted cagY fragments were compared in a 0.4% agarose gel to detect size differences that might not have changed the RFLP pattern.
TABLE 2.
Primers used for amplification, sequencing, and cloning
| Primer name by gene and procedure | Sequence (5′–3′) | Usea |
|---|---|---|
| Amplification and sequencing | ||
| cagPAI | ||
| pai_1F | TCT AGC CAG ATC ACA AGC GAA C | A |
| pai_1R | CCT CTA AGG CAT GCT ACT GAA GAA | A |
| pai_2F | TCG CTA AAT TGC TGC TCA AAA | A |
| pai_2R | CAC TAC TTC ATG CCT TTG GAA GAT AA | A |
| pai_3F | CAA ACA TTC TTC ATA GTT GCC ACC | A |
| pai_3R | TCT CCC TAA CTT CTT CCT CTT TGG | A |
| pai_4F | ATC AAT GAA GTG GCA AGA GAA AAA G | A |
| pai_4R | CAG GTT CAG ACA TCT TGC TTG G | A |
| pai_5F | AGA GTA ACA CTC GGT TCA AAG CTG | A |
| pai_5R | TGA TAT ACT CAA GCG ATT GAT TTC AA | A |
| cagA | ||
| cagA_up | GCT TTA CTT TAT GGT GAG CCA TAA C | A |
| cagA_down | AGC AAG GGG TGG TTT TTG CG | A |
| HP0547:548L23 | CAC GCC CAT GAA CTT TTG ATC CG | S |
| HP0547:438U21 | TAT GGA AAA TAT CAT ACA ACC | S |
| HP0547:930U25 | GAG TCA TAA TGG CAT AGA ACC TGA A | S |
| HP0547:1232U28 | ATA ATG CTA AAT TAG ACA ACT TGA GCG A | S |
| HP0547:1704U21 | AGG ATT GTC CCT ACA AGA AGC | S |
| HP0547:2177U19 | CCC TTA AAG GCT CGG TGA A | S |
| HP0547:2554U21 | ACC CTA GTC GGT AAT GGG TTA | S |
| HP0547:3160U24 | GCT AGT TTG TCA GCG AAA CTA GAC | S |
| oipA | ||
| oipA_up | AAA TGT TGG TTA AGC GGT G | A, S |
| HP0639:592U20 | TTG CAT GCT TAT GGT TAT GG | A, S |
| hopQ1B | ||
| HP0726:823U28 | GGC TCT AGC AAT GTG TGG CAG CAA CAA A | A, S |
| HP0724:849U23 | CGC TAA TGA AAT CGC TCA TTC AA | A, S |
| HP1177:371U22 | GAG GTT ATA CCA AAA GTC CAG G | S |
| HP1177:1749L24 | GGT CTA GCG AGA TTG GTT CTC AAG | S |
| hopQ type II | ||
| hopQ_up | CTT AAA AGC TTG ATT GAG CG | A, S |
| HP1176:24L20 | TAA AAA GGA GTT TGC CAT GG | A, S |
| HP1177:1479L18 | TTG ATG TAG GCG TGG TTG | S |
| cagA CN real-time PCR | ||
| RTcagAF | CCC TTA AAG GCT CGG TGA A | A |
| RTcagAR | TTT TCA AGG TCG CTT TTT GC | A |
| RTureAF | AAA AGC CGT TAG CGT GAA AGT | A |
| RTureAR | CCC GCT CGC AAT GTC TAA G | A |
| cagY PCR-RFLP | ||
| cagY:5157L24 | CCG TTC ATG TTC CAT ACA TCT TTG | A |
| cagX:1515U22 | CTA TGG TGA ATT GGA GCG TGT G | A |
| Construction of knockout strains | ||
| cag8::CAT_rpsL | ||
| HP0528:62U24_XhoI | AAC CTC GAG CTA GCG TAA TAG AAG CAG CAG CAC | A, L |
| HP0528:546L26_SacI | AAC GAG CTC GTG GGT TAC TCA TAG CGT TAG TGA GA | A, L |
| HP0528:653U19_HincII | AAC GTC GAC AGG ACA TGC AAG AGC AGG C | A, R |
| HP0528:1545L21_NotI | AAC GCG GCC GCT TAT CTC TGA CAA GAG GGA GC | A, R |
| rpsL_F_SacI | AAC GAG CTC GAT GCT TTA TAA CTA TGG ATT AAA CAC | A, C |
| C2-CatR_HincII | AAC GTC GAC TTA TCA GTG CGA CAA ACT GGG AT | A, C |
| cag24,25::CAT_rpsL | ||
| HP0526up_NotI | AAC GCG GCC GCT GAT CTT GCC TTG GGT TAG TAA CA | A, L |
| HP0546:20L28_SacI | AAC GAG CTC ACT ACA ACT TTC TTG TAG CTG TCA GTG A | A, L |
| HP0545:70U23_HincII | AAC GTC GAC GTT TTC AGT TCG TAT GGG TCA GC | A, R |
| HP0544:523L25_XhoI | AGT CTC GAG GAG AAT AGT TGT TAG TAA GGA TCA C | A, R |
| rpsL_F_SacI | AAC GAG CTC GAT GCT TTA TAA CTA TGG ATT AAA CAC | A, C |
| C2-CatR_HincII | AAC GTC GAC TTA TCA GTG CGA CAA ACT GGG AT | A, C |
| cagA::CAT_rpsL | ||
| cagA_LF_XhoI | AAC CTC GAG CGT TTT TGA AGT GTC GCC TAG GTA TG | A, L |
| cagA_LR_EcoRI | AAC GAA TTC GGA GCG TTT TGA AGC GGA TAA TAT C | A, L |
| cagA_RF_BamHI | AAC GGA TCC TTA CGC CTT TGG AGA TAT GAT GTG TG | A, R |
| HP0549:634U26_NotI | AAC GCG GCC GCG ATG CTA TTG TGG AAT ATT TGC AGC A | A, R |
| rpsL_F_EcoRI | AAC GAA TTC GAT GCT TTA TAA CTA TGG ATT AAA CAC | A, C |
| C2-CatR_BamHI | AAC GGA TCC TTA TCA GTG CGA CAA ACT GGG AT | A, C |
A, amplification; S, sequencing; L, left arm; R, right arm; C, CAT_rpsL.
IL-8 ELISA.
H. pylori induction of IL-8 was measured as described previously (36). Briefly, human AGS gastric adenocarcinoma cells (ATCC, Manassas, VA) were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U/ml penicillin, and 100 μg μg/ml streptomycin in 5% CO2 at 37°C. Approximately 5 × 105 AGS cells were seeded into 6-well plates in 1.8 ml of RPMI/10% fetal bovine serum, and incubated in 5% CO2 at 37°C for 24 h. AGS cells were then cocultured with H. pylori for 20 h at a multiplicity of infection (MOI) of 100:1. Brucella broth with no bacteria served as a baseline control. Culture supernatants were diluted 1:8 and assayed for IL-8 by enzyme-linked immunosorbent assay (human IL-8 ELISA kit; Invitrogen, Camarillo, CA) according to the manufacturer’s protocol. PMSS1 and its isogenic cagY deletion mutant were included on every plate as positive and negative controls, respectively. To account for variability in the assay, IL-8 values were normalized to WT H. pylori PMSS1 and were determined concurrently.
CagA translocation.
Detection of translocated CagA was performed as previously described (21). Briefly, AGS cells were infected with H. pylori at an MOI of 100:1 and incubated overnight. Cells were harvested by scraping and then lysed with NENT (1% NP-40, 5 mM EDTA, 250 mM NaCl, 25 mM Tris, 1 mM sodium orthovanadate, 1 mM phenylmethylsulfonyl fluoride). CagA and phospho-CagA were detected by immunoblot.
Immunoblot.
Immunoblots were performed to detect CagY and to detect CagA translocation as described previously (21). Proteins were separated on 7.5% Mini-Protean TGX gels (Bio-Rad, Hercules, CA) and transferred to a polyvinylidene difluoride (PVDF) membrane. For detection of CagY, primary antibody to CagY and horseradish peroxidase (HRP)-conjugated secondary anti-rabbit antibody were diluted 1:20,000. For detection of translocated CagA, mouse anti-phosphotyrosine IgG (PY-99; Santa Cruz Biotechnology, Santa Cruz, CA) was diluted 1:1,000, and HRP-conjugated secondary anti-mouse antibody was diluted 1:20,000. Bound antibodies were visualized with chemiluminescence provided by ProSignal Dura substrate (Genesee Scientific, San Diego, CA). Following visualization of phospho-CagA, membranes were stripped and reprobed with CagA antibody (Austral Biological, San Ramon, CA) diluted 1:5,000.
DNA sequencing.
PacBio sequencing was used for initial sequencing of pathogenicity islands. Specific primers (Table 2) were designed to amplify five ∼7-kb fragments, which overlapped by approximately 250 bp and covered the entire pathogenicity island. cagA was sequenced separately by the Sanger technique, as it is not directly adjacent to the PAI in PMSS1. Primers were tailed with barcode sequences obtained from Pacific Biosciences (Menlo Park, CA). Fragments were amplified using LongAmp Taq DNA polymerase (New England BioLabs, Ipswich, MA) with barcoded primers and purified with the Nucleofast 96 PCR clean-up kit (Macherey-Nagel, Bethlehem, PA). Fragments from 20 isolates were pooled equimolarly and used to prepare a SMRTbell library. Sequencing was performed in a single SMRT cell v3 with the DNA sequencing kit 4.0 v2 and sequencing polymerase 6 (P6C4) on the PacBio RS II instrumentation. Data were assembled de novo using the SMRT portal (RS long amplicon analysis protocol) via the Amazon Cloud. Assemblies were imported into Geneious 10.2.3 (Biomatters, Ltd., Auckland, New Zealand) where they were aligned to PMSS1.
Sanger sequencing was used to confirm differences found when PacBio sequences were compared with wild-type PMSS1 and also to sequence cagA, hopQ, and oipA using the primers listed in Table 2. DNA fragments were PCR amplified with Herculase II fusion DNA polymerase (Agilent Technologies, Santa Clara, CA). Following purification with the DNA clean and concentrator-25 kit (Zymo Research, Irvine, CA), samples were submitted to the College of Biological Sciences DNA Sequencing Facility at the University of California, Davis, for sequencing with the BigDye Terminator v. 3.1 cycle sequencing kit. An ABI Prism 3730 genetic analyzer was used along with ABI Prism DNA sequencing analysis software v5.2. Sequencher 5.2.4 (Gene Codes, Ann Arbor, MI) was used for postsequencing alignment and analysis.
Contraselection for genetic exchange of PAI genes.
Contraselectible streptomycin sensitivity was used to insert alleles of interest into PMSS1, as previously described (20). Briefly, a CAT_rpsL cassette was inserted between DNA fragments upstream and downstream of the gene of interest and cloned into pBluescript (Stratagene, La Jolla, CA). Resulting plasmids were transformed into streptomycin-resistant PMSS1 to delete target genes. Genomic DNA was isolated from parent strains and transformed into the PMSS1 deletions, with selection on streptomycin. Transformations were carried out essentially as described (37). Overnight cultures of recipient strains were spread in 1-cm circles onto TVPA plates using a loop and incubated in an atmosphere of 2.9% CO2, 7.6% H2, and 5% O2 for 3 to 6 h. Following incubation, approximately 250- to 500-ng plasmid or genomic DNA was added to each circle and mixed with a loop. Cultures were then incubated in the same atmosphere overnight before plating to selective media and incubating at 7.6% CO2, 7.6% H2, and 5% O2. Several clones of each construct were verified by Sanger sequencing of the target gene and PCR-RFLP of the cagY gene. Primers and strains are listed in Table 2 and 3, respectively.
TABLE 3.
Bacterial strains
| Strain | Description | Source or reference |
|---|---|---|
| PMSS1 | Wild type | 35 |
| PMSS1 Out1 | PMSS1 mouse output 8 weeks postinfection | This study |
| PMSS1 Out2 | PMSS1 mouse output 8 weeks postinfection | This study |
| PMSS1 Out3 | PMSS1 mouse output 8 weeks postinfection | This study |
| PMSS1 Out4 | PMSS1 mouse output 8 weeks postinfection | This study |
| PMSS1Δcag8::CAT_rpsL | PMSS1 with bp 578-658 of cag8 replaced by CAT_rpsL | This study |
| PMSS1Δcag8(Out1) | PMSS1Δcag8 transformed with Out1 genomic DNA | This study |
| PMSS1Δcag8(PMSS1) | PMSS1Δcag8 transformed with PMSS1 gDNA | This study |
| PMSS1Δcag24_25::CAT_rpsL | PMSS1 with cag24 1–89 bp and cag25 48–345 bp replaced by CAT_rpsL | This study |
| PMSS1Δcag24_25(Out2) | PMSS1Δcag24_25 transformed with Out2 genomic DNA | This study |
| PMSS1Δcag24_25(Out4) | PMSS1Δcag24_25 transformed with Out4 genomic DNA | This study |
| PMSS1Δcag24_25(PMSS1) | PMSS1Δcag24_25 transformed with PMSS1 gDNA | This study |
| PMSS1ΔcagA::CAT_rpsL | PMSS1 with all copies of cagA replaced by CAT_rpsL | 33 |
| PMSS1ΔcagA(Out3) | PMSS1ΔcagA transformed with Out3 genomic DNA | This study |
| PMSS1ΔcagA(PMSS1) | PMSS1ΔcagA transformed with PMSS1 genomic DNA | This study |
| PMSS1 cagA SF-1 | PMSS1 with a single copy of cagA | 32 |
Quantitation of cagA copy number.
The number of copies of cagA was determined by real-time PCR, as previously described (32). Primers for amplification of a 145-bp amplicon of cagA and a 142-bp amplicon of the reference gene ureA are listed in Table 2. SYBR Premix Ex Taq (TaKaRa, Kusatsu, Japan) was used for amplification with the QuantStudio 6 flex real-time PCR instrument (Applied Biosystems, Foster City, CA). PMSS1 containing a single copy of cagA was used as a calibrator. Wild-type PMSS1 DNA was included in each run as an internal control. QuantStudio real-time PCR software v1.0 was used for data analysis. For two isolates with 135-bp deletions within the cagA gene, which prevented binding of one of the primers used in real-time PCR, Southern blots were used to determine cagA copy number. Genomic DNA was digested with SspI, which cuts outside the cagA repeat region, resulting in a large fragment whose size correlates with the number of cagA copies. Following gel electrophoresis (0.5% agarose, 0.75 V/cm, 18 h), the DNA was transferred to a nylon membrane by capillary transfer. The blot was then hybridized to a biotinylated fragment of cagA (1,217 to 1,514 bp) (primers in Table 2). Labeling and detection were carried out with the North2South chemiluminescent system (Thermo Fisher Scientific, Waltham, MA). Southern blots were also used to verify the absence of cagA in several isolates.
ACKNOWLEDGMENTS
PacBio sequencing was performed at the DNA Technologies and Expression Analysis Core at the UC Davis Genome Center, which is supported by NIH Shared Instrumentation grant 1S10OD010786-01. This work was supported by the National Institutes of Health grant R01 AI108713 to J.V.S.
REFERENCES
- 1.Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. 2018. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Odenbreit S, Püls J, Sedlmaier B, Gerland E, Fischer W, Haas R. 2000. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- 3.Mueller D, Tegtmeyer N, Brandt S, Yamaoka Y, De Poire E, Sgouras D, Wessler S, Torres J, Smolka A, Backert S. 2012. c-Src and c-Abl kinases control hierarchic phosphorylation and function of the CagA effector protein in Western and East Asian Helicobacter pylori strains. J Clin Invest 122:1553–1566. doi: 10.1172/JCI61143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, Rohde M, Pirch T, Jung K, Retta SF, Terradot L, Fischer W, Haas R. 2009. Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog 5:e1000684. doi: 10.1371/journal.ppat.1000684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, Konig W, Backert S. 2007. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- 6.Javaheri A, Kruse T, Moonens K, Mejias-Luque R, Debraekeleer A, Asche CI, Tegtmeyer N, Kalali B, Bach NC, Sieber SA, Hill DJ, Koniger V, Hauck CR, Moskalenko R, Haas R, Busch DH, Klaile E, Slevogt H, Schmidt A, Backert S, Remaut H, Singer BB, Gerhard M. 2017. Helicobacter pylori adhesin HopQ engages in a virulence-enhancing interaction with human CEACAMs. Nat Microbiol 2:16189. doi: 10.1038/nmicrobiol.2016.243. [DOI] [PubMed] [Google Scholar]
- 7.Koniger V, Holsten L, Harrison U, Busch B, Loell E, Zhao Q, Bonsor DA, Roth A, Kengmo-Tchoupa A, Smith SI, Mueller S, Sundberg EJ, Zimmermann W, Fischer W, Hauck CR, Haas R. 2016. Helicobacter pylori exploits human CEACAMs via HopQ for adherence and translocation of CagA. Nat Microbiol 2:16188. doi: 10.1038/nmicrobiol.2016.188. [DOI] [PubMed] [Google Scholar]
- 8.Zhao Q, Busch B, Jimenez-Soto LF, Ishikawa-Ankerhold H, Massberg S, Terradot L, Fischer W, Haas R. 2018. Integrin but not CEACAM receptors are dispensable for Helicobacter pylori CagA translocation. PLoS Pathog 14:e1007359. doi: 10.1371/journal.ppat.1007359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tegtmeyer N, Harrer A, Schmitt V, Singer BB, Backert S. 2019. Expression of CEACAM1 or CEACAM5 in AZ-521 cells restores the type IV secretion deficiency for translocation of CagA by Helicobacter pylori. Cell Microbiol 21:e12965. doi: 10.1111/cmi.12965. [DOI] [PubMed] [Google Scholar]
- 10.Amieva MR, Vogelmann R, Covacci A, Tompkins LS, Nelson WJ, Falkow S. 2003. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 300:1430–1434. doi: 10.1126/science.1081919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Segal ED, Lange C, Covacci A, Tompkins LS, Falkow S. 1997. Induction of host signal transduction pathways by Helicobacter pylori. Proc Natl Acad Sci U S A 94:7595–7599. doi: 10.1073/pnas.94.14.7595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fischer W, Püls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. 2001. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 13.Shaffer CL, Gaddy JA, Loh JT, Johnson EM, Hill S, Hennig EE, McClain MS, McDonald WH, Cover TL. 2011. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog 7:e1002237. doi: 10.1371/journal.ppat.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brandt S, Kwok T, Hartig R, Konig W, Backert S. 2005. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD, Cover TL. 2016. Molecular and structural analysis of the Helicobacter pylori cag type IV secretion system core complex. mBio 7:e02001-15. doi: 10.1128/mBio.02001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kutter S, Buhrdorf R, Haas J, Schneider-Brachert W, Haas R, Fischer W. 2008. Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J Bacteriol 190:2161–2171. doi: 10.1128/JB.01341-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chung JM, Sheedlo MJ, Campbell AM, Sawhney N, Frick-Cheng AE, Lacy DB, Cover TL, Ohi MD. 2019. Structure of the Helicobacter pylori Cag type IV secretion system. Elife 8:e47644. doi: 10.7554/eLife.47644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cascales E, Christie PJ. 2004. Agrobacterium VirB10, an ATP energy sensor required for type IV secretion. Proc Natl Acad Sci U S A 101:17228–17233. doi: 10.1073/pnas.0405843101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aras RA, Fischer W, Perez-Perez GI, Crosatti M, Ando T, Haas R, Blaser MJ. 2003. Plasticity of repetitive DNA sequences within a bacterial (type IV) secretion system component. J Exp Med 198:1349–1360. doi: 10.1084/jem.20030381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Koelblen T, Berge C, Cherrier MV, Brillet K, Jimenez-Soto L, Ballut L, Takagi J, Montserret R, Rousselle P, Fischer W, Haas R, Fronzes R, Terradot L. 2017. Molecular dissection of protein-protein interactions between integrin alpha5beta1 and the Helicobacter pylori Cag type IV secretion system. FEBS J 284:4143–4157. doi: 10.1111/febs.14299. [DOI] [PubMed] [Google Scholar]
- 21.Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM Jr., Cover TL, Solnick JV. 2013. Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9:e1003189. doi: 10.1371/journal.ppat.1003189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crabtree JE, Ferrero RL, Kusters JG. 2002. The mouse colonizing Helicobacter pylori strain SS1 may lack a functional cag pathogenicity island. Helicobacter 7:139–140. doi: 10.1046/j.1083-4389.2002.00071.x. [DOI] [PubMed] [Google Scholar]
- 23.Philpott DJ, Belaid D, Troubadour P, Thiberge JM, Tankovic J, Labigne A, Ferrero RL. 2002. Reduced activation of inflammatory responses in host cells by mouse-adapted Helicobacter pylori isolates. Cell Microbiol 4:285–296. doi: 10.1046/j.1462-5822.2002.00189.x. [DOI] [PubMed] [Google Scholar]
- 24.Barrozo RM, Hansen LM, Lam AM, Skoog EC, Martin ME, Cai LP, Lin Y, Latoscha A, Suerbaum S, Canfield DR, Solnick JV. 2016. CagY is an immune-sensitive regulator of the Helicobacter pylori type IV secretion system. Gastroenterol 151:1164–1175.e3. doi: 10.1053/j.gastro.2016.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Skoog EC, Morikis VA, Martin ME, Foster GA, Cai LP, Hansen LM, Li B, Gaddy JA, Simon SI, Solnick JV. 2018. CagY-dependent regulation of type IV secretion in Helicobacter pylori is associated with alterations in integrin binding. mBio 9:e00717-18. doi: 10.1128/mBio.00717-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tegtmeyer N, Wessler S, Necchi V, Rohde M, Harrer A, Rau TT, Asche CI, Boehm M, Loessner H, Figueiredo C, Naumann M, Palmisano R, Solcia E, Ricci V, Backert S. 2017. Helicobacter pylori employs a unique basolateral type IV secretion mechanism for CagA delivery. Cell Host Microbe 22:552–560.e5. doi: 10.1016/j.chom.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Cendron L, Couturier M, Angelini A, Barison N, Stein M, Zanotti G. 2009. The Helicobacter pylori CagD (HP0545, Cag24) protein is essential for CagA translocation and maximal induction of interleukin-8 secretion. J Mol Biol 386:204–217. doi: 10.1016/j.jmb.2008.12.018. [DOI] [PubMed] [Google Scholar]
- 28.Hu B, Khara P, Song L, Lin AS, Frick-Cheng AE, Harvey ML, Cover TL, Christie PJ. 2019. In situ molecular architecture of the Helicobacter pylori cag type IV secretion system. mBio 10:e00849-19. doi: 10.1128/mBio.00849-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang J, Fan F, Zhao Y, Sun L, Liu Y, Keegan RM, Isupov MN, Wu Y. 2017. Crystal structure of the type IV secretion system component CagX from Helicobacter pylori. Acta Crystallogr F Struct Biol Commun 73:167–173. doi: 10.1107/S2053230X17001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belogolova E, Bauer B, Pompaiah M, Asakura H, Brinkman V, Ertl C, Bartfeld S, Nechitaylo TY, Haas R, Machuy N, Salama N, Churin Y, Meyer TF. 2013. Helicobacter pylori outer membrane protein HopQ identified as a novel T4SS-associated virulence factor. Cell Microbiol 15:1896–1912. doi: 10.1111/cmi.12158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horridge DN, Begley AA, Kim J, Aravindan N, Fan K, Forsyth MH. 2017. Outer inflammatory protein a (OipA) of Helicobacter pylori is regulated by host cell contact and mediates CagA translocation and interleukin-8 response only in the presence of a functional cag pathogenicity island type IV secretion system. Pathog Dis 75:ftx113. doi: 10.1093/femspd/ftx113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jang S, Su H, Blum FC, Bae S, Choi YH, Kim A, Hong YA, Kim J, Kim JH, Gunawardhana N, Jeon YE, Yoo YJ, Merrell DS, Ge L, Cha JH. 2017. Dynamic expansion and contraction of cagA copy number in Helicobacter pylori impact development of gastric disease. mBio 8:e01779-16. doi: 10.1128/mBio.01779-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Draper JL, Hansen LM, Bernick DL, Abedrabbo S, Underwood JG, Kong N, Huang BC, Weis AM, Weimer BC, van Vliet AH, Pourmand N, Solnick JV, Karplus K, Ottemann KM. 2017. Fallacy of the unique genome: sequence diversity within Single Helicobacter pylori strains. mBio 8:e02321-16. doi: 10.1128/mBio.02321-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Javed S, Skoog EC, Solnick JV. 2019. Impact of Helicobacter pylori virulence factors on the host immune response and gastric pathology. Curr Top Microbiol Immunol 421:21–52. doi: 10.1007/978-3-030-15138-6_2. [DOI] [PubMed] [Google Scholar]
- 35.Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, Sparwasser T, Muller A. 2011. Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterology 140:199–209. doi: 10.1053/j.gastro.2010.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Israel DA, Salama N, Arnold CN, Moss SF, Ando T, Wirth HP, Tham KT, Camorlinga M, Blaser MJ, Falkow S, Peek RM Jr.. 2001. Helicobacter pylori strain-specific differences in genetic content, identified by microarray, influence host inflammatory responses. J Clin Invest 107:611–620. doi: 10.1172/JCI11450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moore ME, Lam A, Bhatnagar S, Solnick JV. 2014. Environmental determinants of transformation efficiency in Helicobacter pylori. J Bacteriol 196:337–344. doi: 10.1128/JB.00633-13. [DOI] [PMC free article] [PubMed] [Google Scholar]