

Chagas disease is a major public health issue, affecting ∼10 million people worldwide. Transmitted by a protozoan named Trypanosoma cruzi, this infection triggers a chronic inflammatory process that can lead to cardiomyopathy (Chagas disease). Resolvin D1 (RvD1) is a novel proresolution lipid mediator whose effects on inflammatory diseases dampens pathological inflammatory responses and can restore tissue homeostasis. Current therapies are not effective in altering the outcome of T. cruzi infection, and as RvD1 has been evaluated as a therapeutic agent in various inflammatory diseases, we examined if exogenous RvD1 could modulate the pathogenesis of Chagas disease in a murine model.
KEYWORDS: Trypanosoma cruzi, RvD1, inflammation, cardiac tissue, fibrosis, resolvin, cardiomyopathy, host response, immunopathogenesis, therapy
ABSTRACT
Chagas disease is a major public health issue, affecting ∼10 million people worldwide. Transmitted by a protozoan named Trypanosoma cruzi, this infection triggers a chronic inflammatory process that can lead to cardiomyopathy (Chagas disease). Resolvin D1 (RvD1) is a novel proresolution lipid mediator whose effects on inflammatory diseases dampens pathological inflammatory responses and can restore tissue homeostasis. Current therapies are not effective in altering the outcome of T. cruzi infection, and as RvD1 has been evaluated as a therapeutic agent in various inflammatory diseases, we examined if exogenous RvD1 could modulate the pathogenesis of Chagas disease in a murine model. CD-1 mice infected with the T. cruzi Brazil strain were treated with RvD1. Mice were administered 3 μg/kg of body weight RvD1 intraperitoneally on days 5, 10, and 15 to examine the effect of RvD1 on acute disease or administered the same dose on days 60, 65, and 70 to examine its effects on chronic infection. RvD1 therapy increased the survival rate and controlled parasite replication in mice with acute infection and reduced the levels of interferon gamma and transforming growth factor β (TGF-β) in mice with chronic infection. In addition, there was an increase in interleukin-10 levels with RvD1 therapy in both mice with acute infection and mice with chronic infection and a decrease in TGF-β levels and collagen content in cardiac tissue. Together, these data indicate that RvD1 therapy can dampen the inflammatory response, promote the resolution of T. cruzi infection, and prevent cardiac fibrosis.
INTRODUCTION
The interaction of healthy tissues with a stressor can send alarm signals in the form of chemical messengers, initiating a rapid acute inflammatory response to harmful stimuli. Aimed primarily at removing injurious agents, ideally, this first line of defense should be self-limited and followed by a complete resolution of the initial inflammatory response, returning tissues to homeostasis. The nature of the stimulus, the intensity of the stimulus, the location of the injury, and the responsiveness of the host dictate the outcome of this initial response. If inflammation is not properly controlled, the initial response can result in chronic complications (1–3). Resolution of the inflammation is the optimal outcome of an acute response and is initiated by an active class switch in the release of proinflammatory lipid markers, ranging from the production of arachidonic acid (AA) molecules in the omega-6 family, such as prostaglandins and leukotrienes, to the production of proresolving mediators (SPMs). Endogenous SPMs actively participate in the dampening of the host responses and in the resolution of inflammation (4–7). Mainly produced by macrophages and neutrophils in distinct omega-3 polyunsaturated fatty acid (PUFA) pathways, the four distinct families of SPMs, lipoxins, maresins, protectins, and resolvins, have proven to act as initiators of resolution of acute inflammation; and therefore, they limit polymorphonuclear neutrophil (PMN) infiltration, counterregulate the production of cytokines and chemokines, and enhance macrophage-mediated actions (3, 6, 8–10).
Many inflammatory diseases are often the result of recurrent or excessive inflammatory responses (11). Infection with the hemoflagellate Trypanosoma cruzi, i.e., Chagas disease, often leads to chronic cardiomyopathy (12, 13). The pathophysiology of Chagas disease is characterized by an inflammatory imbalance with the associated destruction of cardiomyocytes, myocarditis, fibrosis, and changes in the heart architecture and functionality (13–15). Among the many acute inflammatory mediators implicated in this infection (16–19), AA derivatives play a major role by increasing the expression of endothelial cell adhesion molecules (ECAMs) and allowing immune cell migration to the site of infection. They are, therefore, important potential drivers of T. cruzi acute inflammation and progression to chronic cardiac damage. Interestingly, T. cruzi itself is capable of synthesizing these derivatives by the action of T. cruzi old yellow enzyme (TcOYE) (and perhaps other enzymes); hence, both host- and parasite-derived autacoids probably play a substantial role in cardiac remodeling and the ultimate outcome of infection (13, 14, 20–24).
Colas and colleagues (25) have demonstrated that lysates of T. cruzi trypomastigotes (the Tulahuen and Brazil strains) contained increased levels of SPM proresolving mediators and that T. cruzi (Brazil strain)-infected CD-1 mice exhibit elevated plasma levels of resolvin D1 (RvD1). RvD1 (7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid) (26) is produced in vivo through docosahexaenoic acid (DHA) by the enzymatic activity of 15-lipoxygenase (15-LO) and 5-LO (27, 28). This SPM has been shown to counterregulate the actions of PMNs and the production of chemokines and cytokines, stimulate efferocytosis, block nuclear factor κB (NF-κB) activation, and attenuate pain (27, 29, 30). In bacterial infections, RvD1 administration has reduced bacterial titers, reversed hypothermia, increased survival, and enhanced microbial containment and killing by phagocytic cells (10).
There is a significant therapeutic need for effective agents to prevent the development of and/or to treat chronic Chagas disease. The ability of T. cruzi to synthesize both proinflammatory and proresolution mediators from lipid pathways represents a mechanism by which the parasite alters host inflammatory processes, ultimately resulting in chronic persistent infection. Modulation of these pathways represents a new therapeutic target for affecting the pathogenesis and progression of T. cruzi infection. We therefore sought to determine if the exogenous administration of RvD1 in a mouse model of Chagas disease could alter the microenvironment in the host, resulting in improved outcomes following T. cruzi infection. The data presented in this report confirm this hypothesis and demonstrate the efficacy of RvD1 in promoting the resolution of T. cruzi infection and improving the disease outcome in the host. SPMs represent a promising novel adjunctive therapeutic approach for this chronic infection.
RESULTS
RvD1 therapy decreased parasitemia in T. cruzi-infected CD-1 mice and improved the survival rate.
Parasitemia was monitored following T. cruzi infection in a murine model of Chagas disease (Fig. 1) (15). Parasites were first detected at 7 days postinfection (dpi), and on days 21 and 28, the parasite levels were significantly higher in infected control mice than in infected mice receiving RvD1 treatment (Fig. 2A). Parasitemia was undetectable by microscopy in the RvD1-treated animals from days 28 to 35 postinfection. Parasites were again detected at 42 dpi, which corresponded to the time of observed mortality. No parasites were seen in either group after 45 days.
FIG 1.

Experimental design. Mice were infected with T. cruzi and then treated with RvD1, as noted, or observed without treatment. After 45 days, a group of infected untreated mice was then used for the study of RvD1 treatment during chronic infection.
FIG 2.
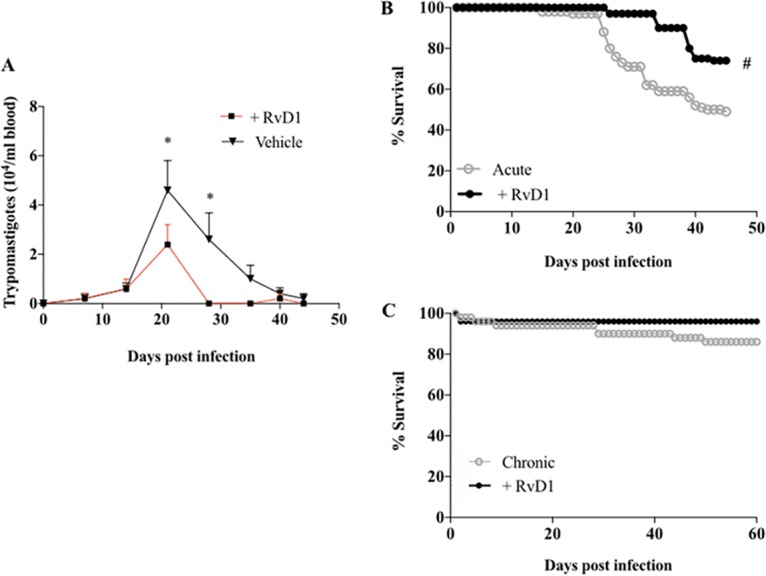
Parasitemia and survival curves. The level of parasitemia during RvD1 treatment during acute infection (A), the survival curve during RvD1 treatment during acute infection (B), and the survival curve during RvD1 treatment during chronic infection (C) for CD-1 mice infected with the T. cruzi Brazil strain are shown. No parasites were seen during RvD1 treatment during chronic infection (curve not shown). The data at each point of the parasitemia curve represent the average parasitemia per day for each group. *, P < 0.05 for differences in the circulating levels of parasites in the T. cruzi-infected control group in comparison to those in infected mice receiving RvD1 (two-way ANOVA); #, P < 0.0001 for differences in the survival rate of infected animals treated or not treated with RvD1 (log-rank [Mantel-Cox] and Gehan-Breslow-Wilcoxon tests).
A survival curve demonstrated an increase in the survival rate for animals receiving RvD1 (3 μg/kg of body weight) during acute infection. Animals receiving RvD1 had a 74% survival rate and infected untreated animals had a 49% survival rate at day 45 postinfection (P < 0.0001) (Fig. 2B).
After 45 days of infection, a subset of the surviving mice was utilized for a study of the effects of RvD1 on chronic infection (see Fig. 1 for the experimental design). The infected untreated animals were randomly separated into two different groups; one group was given RvD1 (infected mice that received RvD1), and the other group was monitored without treatment (the infected control group). Both groups were then monitored for 120 days. The treated animals received their first shot of RvD1 on day 60 postinfection, followed by second and third shots on 65 and 70 dpi, respectively. Parasitemia was undetectable in both groups (data not shown), and animals receiving the RvD1 treatment in the chronic stage of infection displayed a higher survival rate than animals in the T. cruzi-infected control group (Fig. 2C; day 60 survival rates, 83% for the RvD1 group versus 67% for the control group).
RvD1-treated mice had improved cardiac mass measurements compared to that for T. cruzi-infected controls in both stages of infection.
Clinically infected control mice (Fig. 3 and 4) appeared sick (less mobility, poor grooming, lethargy, and “pancake” appearance), whereas mice that received RvD1 remained active and resembled uninfected control animals. Interestingly, the weights of the infected control mice were higher than those of the infected mice that received RvD1, which appeared to be due to fluid retention due to heart dysfunction in the infected animals (Fig. 3B and 4B). We believe that the loss of weight in the RvD1-treated infected mice (Fig. 3B and 4B) was a consequence of infection (probably due to infection-associated cytokines), as the uninfected control mice that received RvD1 did not have any weight loss and did not display any clinical differences from the uninfected untreated control mice.
FIG 3.

Relative heart weight-to-body weight ratio during acute infection. The clinical appearance (A), body weight (B), and relative heart weight (C) of CD-1 mice with acute infection with the T. cruzi Brazil strain (gray bars) and uninfected mice (black bars) treated or not treated with RvD1 are shown. P values were determined by one-way ANOVA with Tukey’s posttest. *, P < 0.05 in comparison to the uninfected control group; &, P < 0.05 in comparison to the uninfected group treated with RvD1.
FIG 4.

Relative heart weight-to-body weight ratio during chronic infection. The clinical appearance (A), body weight (B), and relative heart weight (C) of CD-1 mice with chronic infection with the T. cruzi Brazil strain (gray bars) and uninfected mice (black bars) treated or not treated with RvD1 are shown. P values were determined by one-way ANOVA with Tukey’s posttest. *, P < 0.05 in comparison to the uninfected control group; &, P < 0.05 in comparison to uninfected mice treated with RvD1.
There was an increase in the relative heart mass in infected mice in both acute and chronic infection and a reduction in the relative heart mass in infected animals that received RvD1 (Fig. 3C and 4C). In the infected group that received RvD1 in the setting of chronic infection, the relative heart mass was not significantly higher than that seen in uninfected mice of the same age (Fig. 3C).
RvD1 is able to dampen inflammation by modulating the production of proinflammatory and regulatory cytokines in both stages of T. cruzi infection.
The ability of RvD1 therapy to shift the pattern of the interferon gamma (IFN-γ), transforming growth factor β (TGF-β), and interleukin-10 (IL-10) cytokines during T. cruzi infection was evaluated. No significant difference between these cytokines was seen in the serum of uninfected animals (either treated or untreated). Infected mice demonstrated a significant increase in the levels of all of these cytokines during both acute and chronic infection. IFN-γ (Fig. 5A and D) was used as a representative of a T helper 1 (Th1) cytokine, and after 45 and 120 dpi, it was possible to observe an increase in its level of production in the serum of infected animals. However, the T. cruzi control group had significantly higher levels of IFN-γ than animals treated with RvD1 during chronic infection. The same pattern was observed for TGF-β (Fig. 5B and E). IL-10 levels have been demonstrated in other studies to be elevated in response to RvD1, and higher levels of IL-10 have been implicated as a marker of an improved prognosis in chagasic heart disease. Figure 5C and F demonstrate an increase in IL-10 production in RvD1-treated animals during both acute and chronic infection (P < 0.05).
FIG 5.

The serum levels of the cytokines IFN-γ (A, D), TGF-β (B, E), and IL-10 (C, F) in CD-1 mice infected with the T. cruzi Brazil strain and uninfected mice treated or not treated with RvD1 were measured by enzyme-linked immunosorbent assay. The concentrations of the cytokines in serum were measured at 45 dpi (acute phase) (A to C) and at 120 dpi (chronic phase) (D to F) after infection with the T. cruzi Brazil strain. The scatterplot graph represents the mean ± SD for each group. P values were determined by one-way ANOVA with Tukey’s posttest. &, P < 0.001 when the uninfected group treated with RvD1 was compared with the infected groups; *, P < 0.001 when the uninfected control group was compared with the infected groups.
RvD1 reduces cellular infiltration and fibrosis formation in cardiac tissue.
The effects of RvD1 administration on the inflammatory infiltration of cardiac tissue during both the acute (Fig. 6) and the chronic (Fig. 7A and B) stages of infection were also examined. The production of the circulating cytokines IFN-γ and TGF-β were elevated at 45 and 120 days postinfection in CD-1 mice following infection with the Brazil strain of T. cruzi. In agreement with these findings, an evaluation of cardiac tissue demonstrated more cellular infiltration in mice infected with T. cruzi, and infected mice treated with RvD1 in both the acute and chronic stages had significantly reduced leukocyte infiltration into their hearts (Fig. 6B and 7B). Similarly, the development of fibrosis significantly decreased in infected mice treated with RvD1 (Fig. 7C and D), as revealed by picrosirius red staining. This finding agrees with the results presented in Fig. 5E and 8 of a reduction in the level of TGF-β serum production and expression in these mice.
FIG 6.

Quantification of cellular nuclei in the cardiac tissue of mice treated with RvD1 during acute infection. (A) Representative photomicrographs of histological sections of the heart. U, uninfected mice; U+RvD1, uninfected mice treated with RvD1; I, infected mice; +RvD1, infected mice treated with RvD1. (B) Quantitation of cellular infiltration after treatment during acute infection. Data are shown as the means ± SD. P values were determined by one-way ANOVA with Tukey’s posttest. *, P <0.001 when the uninfected group was compared to the infected groups; &, P < 0.001 when the uninfected group treated with RvD1 was compared with the infected groups. Black bars, uninfected mice and uninfected mice treated with RvD1; gray bars, CD-1 mice infected with the Brazil strain of T. cruzi and infected CD-1 mice treated with RvD1 during acute infection.
FIG 7.

Quantification of cellular nuclei and fibrosis formation in the cardiac tissue of mice treated with RvD1 during chronic infection. (A) Representative photomicrographs of histological sections of the heart. U, uninfected mice; U+RvD1, uninfected mice treated with RvD1; I, infected mice; +RvD1, infected mice treated with RvD1. (B) Quantitation of cellular infiltration after treatment during chronic infection. (C) Sections of hearts stained with picrosirius red after 120 days of infection. (D) Quantitation of the collagen content (the percentage of the red area in stained sections) as a marker of cardiac fibrosis severity. Data are represented as the means ± SD. P values were determined by one-way ANOVA with Tukey’s posttest. *, P < 0.001 when the uninfected group was compared to the infected groups; &, P < 0.001 when the uninfected group treated with RvD1 was compared with the infected groups. Black bars, uninfected mice and uninfected mice treated with RvD1; gray bars, CD-1 mice infected with the Brazil strain of T. cruzi and infected CD-1 mice treated with RvD1 during chronic infection.
FIG 8.
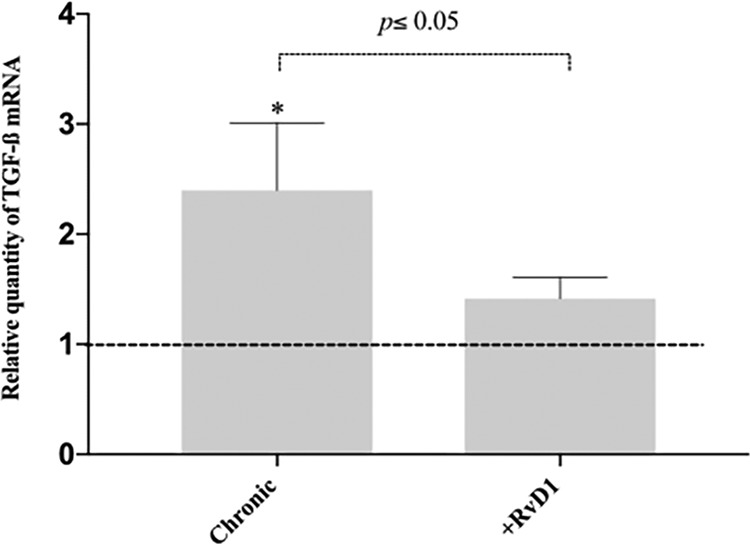
Increased TGF-β expression in the heart. TGF-β mRNA levels in the hearts of infected CD-1 mice (2−ΔCT threshold cycle [CT] values are plotted) treated or not treated with RvD1 were determined after 120 days of infection. The dashed line represents the value for the group of uninfected controls. Data represent the mean ± SD for 3 independent mice per group. *, P < 0.05 between the infected and uninfected groups (two-tailed Student's t test).
RvD1 treatment reduces TGF-β expression, resulting in less fibrosis during chronic infection.
In order to investigate the participation of TGF-β in fibrosis formation, we analyzed its expression in heart fragments of animals in the chronic stage of infection by quantitative PCR (qPCR). Cardiac tissue is the main target of T. cruzi, and the upregulation of TGF-β has been pointed out to be a marker of the fibrotic process in the pathogenesis of Chagas heart disease. We observed that TGF-β mRNA levels were significantly higher in the hearts of infected and untreated animals than in those of infected animals that received RvD1 therapy (Fig. 8). These data correlate with the reduction of TGF-β serum levels and the collagen content stained in cardiac tissue in RvD1-treated animals.
RvD1 therapy improved the LV mass during chronic infection.
Echocardiographic data from CD-1 mice at 120 days (uninfected mice, infected mice, and infected mice treated with RvD1 in the chronic stage of infection) are shown in Fig. 9 and Table 1. The heart rate ranged from 400 to 600 beats per minute in uninfected controls and in the group of infected animals that received RvD1, and although no significant differences were seen between the groups, a lower heart rate was noted in the infected control mice (Table 1). A significant increase in left ventricular (LV) mass was observed in the infected control animals (173.4 ± 36.4 g) compared to that in infected animals receiving RvD1 (118.7 ± 29.12 g). The LV ejection fraction was not statistically significantly altered between the experimental groups (uninfected mice, 57.56% ± 6.89%; infected mice, 56.52% ± 8.3%; infected animals receiving RvD1, 69.89% ± 8.1%), although the overall trend points to improved function with RvD1 therapy. M-mode baseline echocardiographic measurements for left ventricular diastolic inner dimensions (LVIDd) were <5 mm in all 3 groups (Fig. 9). As a result of infection, there was an increase in LVIDd in the infected control animals (4.7 ± 0.2 mm) compared to that in uninfected animals and infected animals treated with RvD1 (4.3 ± 0.4 and 4.1 ± 0.6 mm, respectively). No significant changes in relative wall thickness (RWT) or in fractional shortening were seen between the groups, but there was a tendency toward a reduction in infected control animals compared to that in the animals in the other two groups.
FIG 9.

Effects of RvD1 on cardiac structure and function in T. cruzi-infected CD-1 mice at 90 dpi. Images were determined in the two-dimensional (2D) mode for the parasternal long-axis view (top) and by representative M-mode transthoracic echocardiography of left ventricular function (bottom) in uninfected CD-1 mice (A), infected CD-1 mice (B), and infected CD-1 mice treated with RvD1 during chronic infection with T. cruzi (C). LV, left ventricle; AV, atrial ventricle; LA, left aorta; IVS, interventricular septum; LVPW, left ventricle posterior wall.
TABLE 1.
Echocardiographic analyses of diastolic and systolic diameters from left ventricle of T. cruzi-infected CD-1 mice at 90 dpia
| Parameter | Value for the following group: |
||
|---|---|---|---|
| U | I | +RvD1 | |
| Body wt (g) | 45.94 ± 1.28 | 42.84 ± 2.63 | 36.44 ± 2.22#,* |
| Heart rate (no. of beats/min) | 528.8 ± 52.25 | 465 ± 82.1 | 490.4 ± 87.27 |
| Ejection fraction (%) | 57.56 ± 6.81 | 56.52 ± 8.3 | 69.89 ± 11.12 |
| Fractional shortening (%) | 30.65 ± 6.67 | 29.71 ± 5.3 | 35.97 ± 4.786 |
| LV mass (g) | 121.7 ± 31.54 | 173.4 ± 36.4 | 118.7 ± 29.12# |
| LVIDd (mm) | 4.3 ± 0.37 | 4.7 ± 0.20 | 4.1 ± 0.60 |
| LVIDs (mm) | 3.0 ± 0.3 | 3.2 ± 0.4 | 2.8 ± 0.4 |
| RWT (mm) | 0.40 ± 0.06 | 0.31 ± 0.05 | 0.38 ± 0.12 |
U, uninfected; I, infected; +RvD1, infected and treated with RvD1 during chronic infection; LVIDs, left ventricular systolic inner dimension. *, significant differences (P < 0.05) in comparison to the uninfected control; #, significant differences (P < 0.05) in comparison to the infected controls. P values were determined by one-way ANOVA with Tukey’s posttest.
DISCUSSION
In T. cruzi infection, the absence of an appropriate balance in the acute inflammatory response generated can dictate the outcome of disease. About one-third of infected humans progress to severe forms of chronic Chagas cardiomyopathy (15). The resolution of inflammation is an understudied field of research and is orchestrated by endogenous proresolution molecules with a crucial role in the acute and chronic settings of inflammatory diseases, activating specific mechanisms to promote homeostasis. Among the four distinct families of proresolving mediators that have been studied, the majority of studies have focused on D-series resolvins, a new genus of autacoids that can counterregulate this excessive inflammation (10, 31). RvD1 has potent activity at pico- to nanogram concentrations in the modification of cytokine production, leukocyte recruitment, efferocytosis, the switching of macrophages to a nonphlogistic phenotype, and the promotion of healing to restore organ function (27–29, 31). RvD1 is not immune suppressive, and the immune response to pathogens is not inhibited by this compound. Thus, RvD1 has functions that suggest that exogenous treatment with this compound may help modulate the immune response in T. cruzi infection.
Since it has been demonstrated that lysates from T. cruzi trypomastigotes have detectable RvD1 and that CD-1 mice have increased plasma levels of RvD1 during infection (25), it was not clear if exogenous RvD1 would be beneficial during either acute or chronic T. cruzi infection. The current study demonstrates that a short course of treatment with exogenous RvD1 produced both histological and clinical benefits in experimental murine T. cruzi infection. This highlights a new therapeutic approach to modulate the pathogenesis of this infection by affecting the kinetics of the resolution of the immune response.
Previous studies have shown that the progression of chronic Chagas disease probably involves an imbalance in T helper 1 (Th1) and T helper 2 (Th2) responses, leading to the excessive production of proinflammatory cytokines (15). After infection with the T. cruzi Brazil strain, CD-1 mice displayed increased systemic production of the cytokines IFN-γ, TGF-β, and IL-10 that was modulated by RvD1 treatment. The upregulation of TGF-β is a marker of the fibrotic process in the pathogenesis of Chagas heart disease, and elevated TGF-β levels are associated with a worse clinical outcome for patients with the indeterminate form of this disease as well as for experimental models of this infection (32). In the chronic stage, RvD1 therapy decreased TGF-β expression in the heart and also in the serum and was associated with a decrease in collagen content in the heart. This provides evidence for a protective role for exogenous RvD1 in preventing the development of cardiac fibrosis.
The efficacy of RvD1 in regulating the production of various inflammatory cytokines, such as IFN-γ, has been described in previous investigations (33–35). IFN-γ is a major Th1-like cytokine that is responsible for the activation of other immune cells and that is a promoter of chemokine release and nitric oxide production from M1 macrophages; together, these functions contribute to parasite control (36). T. cruzi infection triggers the upregulation of IFN-γ, which allows the host to control intracellular parasite replication (37–40). Despite being essential for parasite control in acute infection, higher levels of IFN-γ in chronic infection correlate with the development of damage and cardiomyopathy in Chagas disease (41–43). The aspirin-triggered epimer of RvD1 (AT-RvD1) has been effective in decreasing IFN-γ levels in peripheral blood mononuclear cells (PBMC), which suggests that RvD1 can have a beneficial role in reducing cardiac damage (44). In the current study, RvD1 treatment was associated with a decrease in IFN-γ levels, which could be a factor associated with the better outcome in these RvD1-treated animals. RvD1-treated animals had an improved survival rate in comparison to that for the infected controls during both acute and chronic infection.
We hypothesize that the reduced levels of IFN-γ led to less leukocyte infiltration. Since IFN-γ appears to have a positive interaction with the production of chemokines, such as CCL2 and CCL5, which have previously been implicated in augmented cellular infiltration and retention into the inflammatory foci in T. cruzi infection, it could later steer the host toward cardiac dysfunction and higher mortality rates (39, 45, 46). IL-10 production in response to RvD1 therapy has been shown in previous studies, and this could be correlated with the lower levels of production of IFN-γ for these animals, since IL-10 is capable of inhibiting IFN-γ-mediated macrophage activation (47–49).
In acute infection, an increase in IL-10 levels has been associated with host susceptibility to infection. A study performed with IL-10−/− mice infected with the T. cruzi Tulahuen strain demonstrated that control wild-type mice had a lower parasite burden and higher levels of serum tumor necrosis factor (TNF), IL-12, and IFN-γ than infected IL-10+/+ mice (50). The observed IL-10 increase was not associated with an increase in parasitemia, which was a possibility, as IL-10 is associated with a decrease in host immune responses. Studies have shown that in patients with chronic Chagas disease, cardiac pathology is associated with an inflammatory response and elevated levels of TNF and IFN-γ as predominant markers in the damaged tissue. In contrast, higher levels of IL-10 are correlated with a decrease in cardiac pathology (e.g., cardiomyopathy) and persistence in the indeterminate phase of infection (47–49). As IL-10 is consistently associated with the generation of a protective tissue phenotype, the higher levels of IL-10 in animals treated with RvD1 suggests that treatment with RvD1 shifts the immune response to a favorable profile in these animals.
Animals treated with RvD1 exhibited a significant decrease in body mass compared to that of infected untreated animals in both the acute- and the chronic-stage treatment models. This may be a consequence of cardiac dysfunction and increased edema in infected untreated animals. In addition, as RvD1 also actively regulates uncontrolled inflammation in adipose tissue from obese individuals (51), this finding may also reflect changes in adipocyte biology due to infection that are being modulated by the exogenous RvD1. This effect could be important, as it has been shown that adipocytes serve as a reservoir for T. cruzi, playing an important role in the pathogenesis of Chagas disease.
In conclusion, T. cruzi infection persists as an important cause of mortality in Latin American countries, and this infection is now found globally due to immigration from regions of endemicity. In both humans and experimental animal models of T. cruzi infection, the immune response to this pathogen is critical, as the damage that occurs from the inflammatory response eventually leads to cardiac pathology and cardiomyopathy. RvD1 is a novel proresolution lipid mediator whose effects on inflammation dampen pathological inflammatory responses and can restore tissue homeostasis. Modulation of RvD1 is a new approach to modulate the pathogenesis of T. cruzi infection, resulting in improved outcomes. The current treatment for Chagas disease is nitroheterocyclic drugs, such as benznidazole, and future studies of the addition of RvD1 to benznidazole are indicated to evaluate if this combination can provide improved outcomes in the management of T. cruzi infection.
MATERIALS AND METHODS
Mice and infection.
Ten- to 12-week-old male CD-1 mice (The Jackson Laboratory, Bar Harbor, ME) were intraperitoneally infected with 4 × 104 trypomastigote forms of the T. cruzi Brazil strain, obtained by culture passage of an infected cell culture of human foreskin fibroblasts (HFF) that were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Life Technologies) supplemented with 10% fetal bovine serum (FBS), penicillin, and streptomycin (Life Technologies). All procedures described in the current study were performed with the approval of the Institutional Animal Care and Use Committee of the Albert Einstein College of Medicine (approval number 20180902). All efforts were made to minimize suffering during surgical procedures.
Resolvin D1.
RvD1 (7S,8R,17S-trihydroxy-4Z,9E,11E,13Z,15E,19Z-docosahexaenoic acid) was purchased from Cayman Chemical (catalog no. 10012554; Cayman Chemical, Ann Arbor, MI). It was diluted in phosphate buffer to a final concentration of 3 μg/kg of RvD1.
Treatment scheme and experimental groups.
A total of 140 CD-1 male mice (The Jackson Laboratory, Bar Harbor, ME) were used for this protocol. Mice were treated and assessed in both the acute and the chronic phases of T. cruzi infection (Fig. 1), and the time periods for the acute (45 dpi) or chronic (60 to 120 dpi) stage were selected based on previous reports that evaluated immunological/parasitological parameters in T. cruzi-infected mice (15). During the acute phase, the study mice were divided into four different groups: uninfected control mice (n = 15), uninfected mice treated with RvD1 (n = 5), infected control mice (n = 90), and infected mice treated with RvD1 (n = 30). These mice were treated intraperitoneally with 3 shots of 3 μg/kg of RvD1. The first shot was given on day 5 postinfection, followed by the second and third shots on days 10 and 15 postinfection, respectively. On day 45 postinfection, blood was collected from the orbital venous sinus of some of the surviving animals to obtain serum for immunological analyses, and these mice were then euthanized. During this procedure, the heart was carefully removed and weighed, a portion of the cardiac tissue was fixed in 10% formalin, and the other portion was frozen at −80°C for molecular analyses. The remaining animals from both the uninfected and the infected control groups were used for the chronic-stage experiment and were arranged in the following groups: uninfected control mice (n = 5), uninfected mice treated with RvD1 (n = 5), infected control mice (n = 18), and infected mice treated with RvD1 (n = 18). These animals had their first treatment of RvD1 on day 60, followed by treatment of days 65 and 70 postinfection, and were sacrificed at day 120 postinfection.
Parasitological parameters.
The levels of parasitemia were assessed using 10 μl of blood obtained from the tail vein of infected mice, collected every 7 days. Parasites were evaluated by counting in a hemocytometer chamber as previously described (52) until the 44th day of infection for animals with acute infection. Animals in the acute treatment arm of the study were sacrificed on the 45th day, and the surviving uninfected and infected control animals were kept for the chronic treatment arm studies. Parasitemia for animals in the chronic treatment arm was performed twice, at days 56 and 70 postinfection. The survival rate was recorded daily until the end of the experiment.
Relative heart weight-to-body weight ratio.
The body weight of each mouse was measured before euthanasia. During necropsy, ex vivo hearts were carefully excised and gently dried on absorbent paper, followed by perfusion in phosphate saline buffer to remove any remaining blood. Subsequently, the organ removed from each mouse was immediately weighed on an analytical balance, and the relative heart weight was calculated using the mouse heart weight/body weight and used to evaluate the cardiac mass measurement at the time of euthanasia in all four groups studied in both the acute and the chronic phases (45 and 120 dpi, respectively) of T. cruzi infection (n = 6).
Immunoassays.
To analyze the interference of RvD1 therapy with the inflammatory response associated with T. cruzi experimental infection, an enzyme-linked immunosorbent assay (ELISA) was performed to detect the circulating levels of the inflammatory mediators transforming growth factor β (TGF-β), interferon gamma (IFN-γ), and interleukin-10 (IL-10), according to the manufacturer’s instructions (R&D Systems, Minneapolis, MN, USA), in serum samples collected from the orbital venous sinus on days 45 and 120 postinfection. The levels in all samples were simultaneously measured in triplicate, and the limit of detection was as specified by the manufacturer (32 to 3,000 pg/ml).
Histopathological analysis.
After removal, the heart fragments were immediately fixed in 10% (vol/vol) buffered formalin solution. Briefly, tissues were dehydrated, cleared, and embedded in paraffin, and the blocks were cut into 4-mm-thick sections and stained with hematoxylin and eosin (H&E) to quantify leukocyte infiltration in both the acute and the chronic infection groups. The stained sections were imaged using a 3DHistec Pannoramic 250 Flash II slide scanner; the pictures shown in this work were acquired at a ×20 magnification and also at a ×40 magnification for a total of 74,931 μm2, which is an area equivalent to 50 fields of the analyzed myocardium. The inflammatory process was assessed through the number of cellular nuclei found in the infected heart tissue and compared to the background of the cardiac cellular nuclei found in the uninfected mice, quantified using a Leica Qwin (v3) image analyzer. Heart sections were also stained with fibrillar collagen-specific picrosirius red to visualize the collagen content and identify sites of fibrosis in the heart tissue at 120 days postinfection. The fibrosis index was obtained by quantification of the proportion of tissue staining positive for collagen; an increase in collagen content compared to that in the uninfected controls was considered indicative of myocardial fibrosis. Images were obtained using the 3DHistec Pannoramic 250 Flash II slide scanner, and polarized micrographs were obtained via a Zeiss Axioskop II microscope (a Zeiss Stemi SV11 microscope with a Nikon Macro lens). The collagen defined by the micrographs was measured by the use of ImageJ (v1.45s) software, which was developed by Wayne Rasband and which is available at http://rsb.info.nih.gov/ij/index.html (National Institutes of Health, Bethesda, MD).
RNA extraction and quantitative real-time PCR.
The coding region for TGF-β1 was analyzed by quantitative PCR (qPCR). Total RNA was isolated from heart tissue using a Qiagen RNeasy minikit (catalog no. 74134; Qiagen, Germantown, MD) and quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc.). RNA was converted into cDNA by reverse transcription (RT) using a OneTaq RT-PCR kit (catalog no. E5310S; New England Biolabs). TGF-β1 was amplified using the following primers: TGF-β1-forward (5′-CTTGTACGGCAGTGGCTGAAC-3′) and TGF-β1-reverse (5′-GGGTGGCCATGAGGAGCAGG-3′) (52). Relative mRNA levels were normalized against the level of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA using the primers GAPDH-forward (AACGACCCCTTCATTGA) and GAPDH-reverse (TTCACGACATACTCAGCA) (53). All data are expressed as the mean ± standard deviation (SD) for three independent mice (each in triplicate).
Echocardiography.
To evaluate whether chronic T. cruzi infection would affect left ventricular (LV) function, we performed echocardiography on the surviving mice at 90 days postinfection (dpi) to assess function and morphology. Acquisitions and analyses were performed by a certified and experienced physician blind to the data using a 30-MHz RMV-707 B scanhead interfaced with a Vevo 770 high-resolution imaging system (VisualSonics, Toronto, ON, Canada). The total number of mice available for study was five in all three analyzed groups (uninfected control mice, infected control mice, infected mice treated with RvD1). Mice were placed in the supine position, and light anesthesia was achieved with 1.5% isoflurane inhalation (54). After anesthesia, the chest wall was carefully shaved, an ultrasound transmission gel was applied to the precordium, and transthoracic transmission was performed. The heart was first imaged in the two-dimensional (2D) mode in the parasternal long-axis view. The parameters left ventricular inner dimension (left ventricular end diastolic diameter [LVID], i.e., left ventricular end diastolic diameter), interventricular septum thickness (IVS), and left ventricular posterior wall thickness (LVPW) were measured from three to six consecutive cardiac cycles using M-mode imaging. The left ventricle internal diameter in diastole was obtained at the point of the greatest cavity dimension, and systole measurements were obtained at the point of minimal cavity dimension. Also, the relative wall thickness (2× left ventricular diastolic wall thickness/end diastolic diameter) was calculated as an additional parameter. All measurements were done according to the leading method of the American Society of Echocardiography guidelines (55).
Statistical analyses.
All analyses were performed using GraphPad Prism (v8.3) software (GraphPad Software, San Diego, CA, USA). Data are expressed as the mean ± standard deviation (SD), and the patterns of normality were confirmed using the Shapiro-Wilk test. Parasitemia was evaluated using two-way analysis of variance (ANOVA) with Sidak’s posttest for multiple comparisons. Statistical differences in the survival curve were detected using the log-rank (Mantel-Cox) test and the Gehan-Breslow-Wilcoxon test. For analyses comparing the means for two populations, we used a two-tailed Student's t test and a one-way ANOVA for multiple comparisons with Tukey’s posttest. P values of <0.05 were considered statistically significant.
ACKNOWLEDGMENTS
We dedicate this study to our departed colleague Herbert B. Tanowitz, whose insights into the pathogenesis of T. cruzi infection and interest in the role of resolvin and other inflammatory lipid mediators formed the basis for the current study.
We thank Jun Shu for the excellent technical assistance.
The H&E (SIG 1S10OD019961-01) and picrosirius red (SIG 1S10OD023591-01) pictures were obtained in the Advanced Imaging Facility at the Albert Einstein College of Medicine, which is supported by NCI Cancer Center support grant P30CA013330, with assistance from Hillary Guzik. This work was supported by NIH grant AI1421110 (to H.H.) and the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior—Brasil (CAPES; finance code 001). A.L.H. was supported by a grant from the Department of Pathology (Albert Einstein College of Medicine).
REFERENCES
- 1.Nathan C. 2002. Points of control in inflammation. Nature 420:846–852. doi: 10.1038/nature01320. [DOI] [PubMed] [Google Scholar]
- 2.Bannenberg G, Serhan CN. 2010. Specialized pro-resolving lipid mediators in the inflammatory response: an update. Biochim Biophys Acta 1801:1260–1273. doi: 10.1016/j.bbalip.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buckley CD, Gilroy DW, Serhan CN, Stockinger B, Tak PP. 2013. The resolution of inflammation. Nat Rev Immunol 13:59–66. doi: 10.1038/nri3362. [DOI] [PubMed] [Google Scholar]
- 4.Cotran RS, Kumar V, Robbins SL. 1994. Robbins' pathologic basis of disease, 5th ed W. B. Saunders, Philadelphia, PA. [Google Scholar]
- 5.Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN. 2001. Lipid mediator class switching during acute inflammation: signals in resolution. Nat Immunol 2:612–619. doi: 10.1038/89759. [DOI] [PubMed] [Google Scholar]
- 6.Serhan CN, Savill J. 2005. Resolution of inflammation: the beginning programs the end. Nat Immunol 6:1191–1197. doi: 10.1038/ni1276. [DOI] [PubMed] [Google Scholar]
- 7.Serhan CN. 2009. Systems approach to inflammation resolution: identification of novel anti-inflammatory, and pro-resolving mediators. J Thromb Haemost 7:44–48. doi: 10.1111/j.1538-7836.2009.03396.x. [DOI] [PubMed] [Google Scholar]
- 8.Serhan CN. 2004. A search for endogenous mechanisms of anti-inflammation uncovers novel chemical mediators: missing links to resolution. Histochem Cell Biol 122:305–321. doi: 10.1007/s00418-004-0695-8. [DOI] [PubMed] [Google Scholar]
- 9.Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O'Neill LAJ, Perretti M, Rossi AG, Wallace JL. 2007. Resolution of inflammation: state of the art, definitions and terms. FASEB J 21:325–332. doi: 10.1096/fj.06-7227rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serhan CN, Chiang N, Dalli J, Levy BD. 2014. Lipid mediators in the resolution of inflammation. Cold Spring Harb Perspect Biol 7:a016311. doi: 10.1101/cshperspect.a016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Medzhitov R. 2007. Recognition of microorganisms and activation of the immune response. Nature 449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 12.Marin-Neto JA, Cunha-Neto E, Maciel BC, Simões MV. 2007. Pathogenesis of chronic Chagas heart disease. Circulation 115:1109–1123. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- 13.Machado FS, Tanowitz HB, Ribeiro AL. 2013. Pathogenesis of Chagas cardiomyopathy: role of inflammation and oxidative stress. J Am Heart Assoc 2:e000539. doi: 10.1161/JAHA.113.000539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brener Z, Gazzinelli RT. 1997. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas’ disease. Int Arch Allergy Immunol 114:103–110. doi: 10.1159/000237653. [DOI] [PubMed] [Google Scholar]
- 15.Talvani A, Teixeira MM. 2011. Inflammation and Chagas disease: some mechanisms and relevance. Adv Parasitol 76:171–194. doi: 10.1016/B978-0-12-385895-5.00008-6. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Calderon TM, Berman JW, Braunstein VL, Weiss LM, Wittner M, Tanowitz HB. 1999. Infection of endothelial cells with Trypanosoma cruzi activates NF-kappaB and induces vascular adhesion molecule expression. Infect Immun 67:5434–5440. doi: 10.1128/IAI.67.10.5434-5440.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang H, Chan J, Wittner M, Jelicks LA, Morris SA, Factor SM, Weiss LM, Braunstein VL, Bacchi CJ, Yarlett N, Chandra M, Shirani J, Tanowitz HB. 1999. Expression of cardiac cytokines and inducible form of nitric oxide synthase (NOS2) in Trypanosoma cruzi-infected mice. J Mol Cell Cardiol 1:75–88. doi: 10.1006/jmcc.1998.0848. [DOI] [PubMed] [Google Scholar]
- 18.Tanowitz HB, Huang H, Jelicks LA, Chandra M, Loredo ML, Weiss LM, Factor SM, Shtutin V, Mukherjee S, Kitsis RN, Christ GJ, Wittner M, Shirani J, Kisanuki YY, Yanagisawa M. 2005. Role of endothelin 1 in the pathogenesis of chronic chagasic heart disease. Infect Immun 73:2496–2503. doi: 10.1128/IAI.73.4.2496-2503.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petkova SB, Tanowitz HB, Magazine HI, Factor SM, Chan J, Pestell RG, Bouzahzah B, Douglas SA, Shtutin V, Morris SA, Tsang E, Weiss LM, Christ GJ, Wittner M, Huang H. 2000. Myocardial expression of endothelin-1 in murine Trypanosoma cruzi infection. Cardiovasc Pathol 9:257–265. doi: 10.1016/s1054-8807(00)00045-4. [DOI] [PubMed] [Google Scholar]
- 20.Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. 2001. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol 22:199–204. doi: 10.1016/s1471-4906(01)01863-4. [DOI] [PubMed] [Google Scholar]
- 21.Golias C, Tsoutsi E, Matziridis A, Makridis P, Batistatou A, Charalabopoulos K. 2007. Leukocyte and endothelial cell adhesion molecules in inflammation focusing on inflammatory heart disease. In Vivo 21:757–769. [PubMed] [Google Scholar]
- 22.Ashton AW, Mukherjee S, Nagajyothi FN, Huang H, Braunstein VL, Desruisseaux MS, Factor SM, Lopez L, Berman JW, Wittner M, Scherer PE, Capra V, Coffman TM, Serhan CN, Gotlinger K, Wu KK, Weiss LM, Tanowitz HB. 2007. Thromboxane A2 is a key regulator of pathogenesis during Trypanosoma cruzi infection. J Exp Med 204:929–940. doi: 10.1084/jem.20062432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pavanelli WR, Gutierrez FR, Mariano FS, Prado CM, Ferreira BR, Teixeira MM, Canetti C, Rossi MA, Cunha FQ, Silva JS. 2010. 5-Lipoxygenase is a key determinant of acute myocardial inflammation and mortality during Trypanosoma cruzi infection. Microbes Infect 12:587–597. doi: 10.1016/j.micinf.2010.03.016. [DOI] [PubMed] [Google Scholar]
- 24.Lopez-Munoz RA, Molina-Berrios A, Campos-Estrada C, Abarca-Sanhueza P, Urrutia-Llancaqueo L, Peña-Espinoza M, Maya JD. 2018. Inflammatory and pro-resolving lipids in trypanosomatid infections: a key to understanding parasite control. Front Microbiol 9:1961. doi: 10.3389/fmicb.2018.01961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Colas RA, Ashton AW, Mukherjee S, Dalli J, Akide-Ndunge OB, Huang H, Desruisseaux MS, Guan F, Jelicks LA, Matos Dos Santos F, Nagajyothi J, Zingman MA, Reyes J, Weiss LM, Serhan CN, Tanowitz HB. 2018. Trypanosoma cruzi produces the specialized proresolving mediators resolvin D1, resolvin D5, and resolvin E2. Infect Immun 86:e00688-17. doi: 10.1128/IAI.00688-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun Y-P, Oh SF, Uddin J, Yang R, Gotlinger K, Campbell E, Colgan SP, Petasis NA, Serhan CN. 2007. Resolvin D1 and its aspirin-triggered 17R epimer. Stereochemical assignments, anti-inflammatory properties, and enzymatic inactivation. J Biol Chem 282:9323–9334. doi: 10.1074/jbc.M609212200. [DOI] [PubMed] [Google Scholar]
- 27.Serhan CN, Hong S, Gronert K, Colgan SP, Devchand PR, Mirick G, Moussignac RL. 2002. Resolvins: a family of bioactive products of omega-3 fatty acid transformation circuits initiated by aspirin treatment that counter pro inflammation signals. J Exp Med 196:1025–1037. doi: 10.1084/jem.20020760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong S, Gronert K, Devchand PR, Moussignac RL, Serhan CN. 2003. Novel docosatrienes and 17S-resolvins generated from docosahexaenoic acid in murine brain, human blood, and glial cells. Autacoids in anti-inflammation. J Biol Chem 278:14677–14687. doi: 10.1074/jbc.M300218200. [DOI] [PubMed] [Google Scholar]
- 29.Xu ZZ, Zhang L, Liu T, Park JY, Berta T, Yang R, Serhan CN, Ji RR. 2010. Resolvins RvE1 and RvD1 attenuate inflammatory pain via central and peripheral actions. Nat Med 16:592–597. doi: 10.1038/nm.2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malta-Santos H, Andrade BB, Costa DL, Costa JM, Bozza T, Bandeira-Melo C, Barral A, França-Costa J, Borges VM. 2017. Resolvin D1 drives establishment of Leishmania amazonensis infection. Sci Rep 7:46363. doi: 10.1038/srep46363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spite M, Claria J, Serhan CN. 2014. Resolvins, specialized proresolving lipid mediators, and their potential roles in metabolic diseases. Cell Metab 19:21–36. doi: 10.1016/j.cmet.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Araujo-Jorge TC, Waghabi MC, Hasslocher-Moreno AM, Xavier S, Higuchi MDL, Keramidas M, Bailly S, Feige JJ. 2002. Implication of transforming growth factor-beta1 in Chagas disease myocardiopathy. J Infect Dis 186:1823–1828. doi: 10.1086/345882. [DOI] [PubMed] [Google Scholar]
- 33.Benabdoun HA, Kulbay M, Rondon E-P, Vallières F, Shi Q, Fernandes J, Fahmi H, Benderdour M. 2019. In vitro and in vivo assessment of the proresolutive and antiresorptive actions of resolvin D1: relevance to arthritis. Arthritis Res Ther 21:72. doi: 10.1186/s13075-019-1852-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiurchiu V, Leuti A, Dalli J, Jacobsson A, Battistini L, Maccarrone M, Serhan CN. 2016. Proresolving lipid mediators resolvin D1, resolvin D2, and maresin 1 are critical in modulating T cell responses. Sci Transl Med 8:353ra111. doi: 10.1126/scitranslmed.aaf7483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang X, Wang T, Gui P, Yao C, Sun W, Wang L, Wang H, Xie W, Yao S, Lin Y, Wu Q. 2013. Resolvin D1 reverts lipopolysaccharide-induced TJ proteins disruption and the increase of cellular permeability by regulating I κBα signaling in human vascular endothelial cells. Oxid Med Cell Longev 2013:185715. doi: 10.1155/2013/185715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abrahamsohn IA, Coffman RL. 1996. Trypanosoma cruzi: IL-10, TNF, IFN-γ, and IL-12 regulate innate and acquired immunity to infection. Exp Parasitol 84:231–244. doi: 10.1006/expr.1996.0109. [DOI] [PubMed] [Google Scholar]
- 37.Aliberti JCS, Cardoso MA, Martins GA, Gazzinelli RT, Vieira LQ, Silva JS. 1996. Interleukin-12 mediates resistance to Trypanosoma cruzi in mice and is produced by murine macrophages in response to live trypomastigotes. Infect Immun 64:1961–1967. doi: 10.1128/IAI.64.6.1961-1967.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ropert C, Gazzinelli RT. 2000. Signaling of immune system cells by glycosylphosphatidylinositol (GPI) anchor and related structures derived from parasitic protozoa. Curr Opin Microbiol 3:395–403. doi: 10.1016/s1369-5274(00)00111-9. [DOI] [PubMed] [Google Scholar]
- 39.Talvani A, Ribeiro CS, Aliberti JCS, Michailowsky V, Santos PVA, Murta SMF, Romanha AJ, Almeida IC, Farber J, Lannes-Vieira J, Silva JS, Gazzinelli RT. 2000. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-γ as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect 2:851–866. doi: 10.1016/S1286-4579(00)00388-9. [DOI] [PubMed] [Google Scholar]
- 40.Romanha AJ, Alves RO, Murta SM, Silva JS, Ropert C, Gazzinelli RT. 2002. Experimental chemotherapy against Trypanosoma cruzi infection: essential role of endogenous interferon-gamma in mediating parasitologic cure. J Infect Dis 186:823–828. doi: 10.1086/342415. [DOI] [PubMed] [Google Scholar]
- 41.Pinazo MJ, Thomas MC, Bustamante J, Almeida IC, Lopez MC, Gascon J. 2015. Biomarkers of therapeutic responses in chronic Chagas disease: state of the art and future perspectives. Mem Inst Oswaldo Cruz 110:422–432. doi: 10.1590/0074-02760140435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poveda C, Fresno M, Girones N, Martins-Filho OA, Ramírez JD, Santi-Rocca J, Marin-Neto JA, Morillo CA, Rosas F, Guhl F. 2014. Cytokine profiling in Chagas disease: towards understanding the association with infecting Trypanosoma cruzi discrete typing units (a BENEFIT TRIAL sub-study). PLoS One 9:e91154. doi: 10.1371/journal.pone.0091154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sousa GR, Gomes JA, Fares RC, Damásio MP, Chaves AT, Ferreira KS, Nunes MC, Medeiros NI, Valente VA, Corrêa-Oliveira R, Rocha MO. 2014. Plasma cytokine expression is associated with cardiac morbidity in Chagas disease. PLoS One 9:e87082. doi: 10.1371/journal.pone.0087082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ogata H, Teixeira MM, de Sousa RC, da Silva MV, Correia D, Rodrigues Junior V, Levy BD, Rogério ADP. 2016. Effects of aspirin-triggered resolvin D1 on peripheral blood mononuclear cells from patients with Chagas’ heart disease. Eur J Pharmacol 777:26–32. doi: 10.1016/j.ejphar.2016.02.058. [DOI] [PubMed] [Google Scholar]
- 45.Aliberti JCS, Souto JT, Marino A, Lannes-Vieira J, Teixeira MM, Farber J, Gazzinelli RT, Silva JS. 2001. Modulation of chemokine production and inflammatory responses in interferon-γ- and tumor necrosis factor-R1-deficient mice during Trypanosoma cruzi infection. Am J Pathol 158:1433–1440. doi: 10.1016/S0002-9440(10)64094-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Teixeira MM, Gazzinelli RT, Silva JS. 2002. Chemokines, inflammation and Trypanosoma cruzi infection. Trends Parasitol 18:262–265. doi: 10.1016/s1471-4922(02)02283-3. [DOI] [PubMed] [Google Scholar]
- 47.Titos E, Rius B, González-Périz A, López-Vicario C, Morán-Salvador E, Martínez-Clemente M, Arroyo V, Clària J. 2011. Resolvin D1 and its precursor docosahexaenoic acid promote resolution of adipose tissue inflammation by eliciting macrophage polarization toward an M2-like phenotype. J Immunol 187:5408–5418. doi: 10.4049/jimmunol.1100225. [DOI] [PubMed] [Google Scholar]
- 48.Silva JS, Morrissey PJ, Grabstein KH, Mohler KM, Anderson D, Reed SG. 1992. Interleukin 10 and interferon gamma regulation of experimental Trypanosoma cruzi infection. J Exp Med 175:169–174. doi: 10.1084/jem.175.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cardillo F, Voltarelli JC, Reed SG, Silva JS. 1996. Regulation of Trypanosoma cruzi infection in mice by gamma interferon and interleukin 10: role of NK cells. Infect Immun 64:128–134. doi: 10.1128/IAI.64.1.128-134.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hunter CA, Ellis-Neyes LA, Slifer T, Kanaly S, Grünig G, Fort M, Rennick D, Araujo FG. 1997. IL-10 is required to prevent immune hyperactivity during infection with Trypanosoma cruzi. J Immunol 158:3311–3316. [PubMed] [Google Scholar]
- 51.Costa GC, da Costa Rocha MO, Moreira PR, Menezes CA, Silva MR, Gollob KJ, Dutra WO. 2009. Functional IL-10 gene polymorphism is associated with Chagas disease cardiomyopathy. J Infect Dis 199:451–454. doi: 10.1086/596061. [DOI] [PubMed] [Google Scholar]
- 52.Trischmann TM, Tanowitz H, Wittner M, Bloom BR. 1978. Trypanosoma cruzi: role of the immune response in the natural resistance of inbred strains of mice. Exp Parasitol 45:160–168. doi: 10.1016/0014-4894(78)90055-3. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira RR, de Souza EM, de Oliveira FL, Ferrão PM, Gomes LHF, Mendonça-Lima L, Meuser-Batista M, Bailly S, Feige JJ, de Araujo-Jorge TC, Waghabi MC. 2016. Proteins involved on TGF-β pathway are up-regulated during the acute phase of experimental Chagas disease. Immunobiology 221:587–594. doi: 10.1016/j.imbio.2016.01.009. [DOI] [PubMed] [Google Scholar]
- 54.Chandra M, Shirani J, Shtutin V, Weiss LM, Factor SM, Petkova SB, Rojkind M, Dominguez-Rosales JA, Jelicks LA, Morris SA, Wittner M, Tanowitz HB. 2002. Cardioprotective effects of verapamil on myocardial structure and function in a murine model of chronic Trypanosoma cruzi infection (Brazil strain): an echocardiographic study. Int J Parasitol 32:207–215. doi: 10.1016/S0020-7519(01)00320-4. [DOI] [PubMed] [Google Scholar]
- 55.Schiller NB, Shah PM, Crawford M, DeMaria A, Devereux R, Feigenbaum H, Gutgesell H, Reichek N, Sahn D, Schnittger I, Silverman NH, Tajik AJ. 1989. Recommendations for quantitation of the left ventricle by two-dimensional echocardiography. American Society of Echocardiography Committee on Standards, Subcommittee on Quantitation of Two-Dimensional Echocardiograms. J Am Soc Echocardiogr 2:358–367. doi: 10.1016/S0894-7317(89)80014-8. [DOI] [PubMed] [Google Scholar]