

The zoonotic bacterial pathogen Coxiella burnetii is the causative agent of Q fever, a febrile illness which can cause a serious chronic infection. C. burnetii is a unique intracellular bacterium which replicates within host lysosome-derived vacuoles. The ability of C. burnetii to replicate within this normally hostile compartment is dependent on the activity of the Dot/Icm type 4B secretion system. In a previous study, a transposon mutagenesis screen suggested that the disruption of the gene encoding the novel protein CBU2072 rendered C. burnetii incapable of intracellular replication.
KEYWORDS: Coxiella burnetii, bacterial pathogenesis, host-pathogen interactions, virulence factor, type IV secretion system, virulence factors
ABSTRACT
The zoonotic bacterial pathogen Coxiella burnetii is the causative agent of Q fever, a febrile illness which can cause a serious chronic infection. C. burnetii is a unique intracellular bacterium which replicates within host lysosome-derived vacuoles. The ability of C. burnetii to replicate within this normally hostile compartment is dependent on the activity of the Dot/Icm type 4B secretion system. In a previous study, a transposon mutagenesis screen suggested that the disruption of the gene encoding the novel protein CBU2072 rendered C. burnetii incapable of intracellular replication. This protein, subsequently named EirA (essential for intracellular replication A), is indispensable for intracellular replication and virulence, as demonstrated by infection of human cell lines and in vivo infection of Galleria mellonella. The putative N-terminal signal peptide is essential for protein function but is not required for localization of EirA to the bacterial inner membrane compartment and axenic culture supernatant. In the absence of EirA, C. burnetii remains viable but nonreplicative within the host phagolysosome, as coinfection with C. burnetii expressing native EirA rescues the replicative defect in the mutant strain. In addition, while the bacterial ultrastructure appears to be intact, there is an altered metabolic profile shift in the absence of EirA, suggesting that EirA may impact overall metabolism. Most strikingly, in the absence of EirA, Dot/Icm effector translocation was inhibited even when EirA-deficient C. burnetii replicated in the wild type (WT)-supported Coxiella containing vacuoles. EirA may therefore have a novel role in the control of Dot/Icm activity and represent an important new therapeutic target.
INTRODUCTION
Coxiella burnetii is a Gram-negative intracellular bacterium which causes the disease Q fever (1). In humans, around half of infected individuals present with acute, febrile illness. In some cases, the infection may progress to chronic disease, which is lethal without treatment (1). A typical infection with C. burnetii occurs via inhalation of contaminated aerosols into the lungs, where bacteria are phagocytosed by alveolar macrophages (2, 3). C. burnetii passively undergoes endocytic maturation, and fusion of the lysosome with the C. burnetii-containing phagosome is essential for intracellular replication (4).
Fusion of bacterium-harboring phagosomes/endosomes with the lysosome, coupled with the acidification of the phagosome, triggers the conversion of the pathogen from the nonreplicative and environmentally stable small-cell variant (SCV) form to the replicative large-cell variant (LCV) (4, 5). Fusion with the host lysosome also triggers the activation of the C. burnetii Dot/Icm type 4B secretion system (T4BSS) (6, 7). This multiprotein apparatus is essential for the pathogen to replicate within the host cell, as genetic disruptions of T4BSS genes lead to nonreplicative but viable bacteria retained within lysosome-associated membrane protein-1 (LAMP-1)-positive vacuoles (8, 9). The T4BSS translocates approximately 130 effector proteins into the host cell, which modulate host cell responses and directly regulate the establishment of a unique replicative niche called the Coxiella-containing vacuole (CCV) (10).
The extent to which C. burnetii remodels the lysosomal function and composition remains poorly defined. For many years, it was thought the intravacuolar pH of this compartment was similar to that in mature lysosomes, at approximately 4.5 (11). However, recent studies have suggested that the intraluminal pH of the CCV is approximately 5.2, and that maintenance of this higher pH occurs in a T4BSS-dependent manner (12–14). In addition, T4BSS effector proteins may contribute to the increased fusogenic properties of the CCV, which can fuse with other CCVs, autophagosomes, and vesicles from the endocytic pathway (15–18). This process leads to the maturation of the CCV, which is akin to an autolysosome (17, 18). Replication proceeds up to 6 to 7 days within the host cell, at which point the bacteria are able to exit the host cell via a currently uncharacterized mechanism (5).
The exact mechanisms allowing C. burnetii to withstand lysosomal degradation and replicate within this unique niche are still being investigated. A recent transposon mutagenesis screen revealed that disruption of the gene encoding the C. burnetii protein CBU2072 leads to a severe intracellular growth defect (17). This mutant strain, similar to Dot/Icm mutants, remains in LAMP-1-positive vacuoles but is unable to establish a CCV or replicate (17). Interestingly, this mutant presents no growth defect in axenic culture, suggesting that this defect is specific to the intracellular niche (17). CBU2072 is not a T4BSS effector protein, based on initial screens using β-lactamase (BlaM)-fused CBU2072 in a reporter translocation assay (17). However, previous research using mass spectrometry has demonstrated that EirA is present in acidified citrate cysteine medium-2 (ACCM-2) culture medium during axenic cultivation of C. burnetii (19). This was validated using an inducible, plasmid-expressed C-terminal 3×FLAG-tagged EirA (19).
This study investigates the role of this protein, referred to here as essential for intracellular replication A (EirA, CBU2072), during C. burnetii infection. A putative N-terminal signal sequence was demonstrated to be essential for function, as the loss of this region resulted in attenuations to intracellular replication in both human and G. mellonella. The lack of this N-terminal signal peptide did not impede EirA secretion into the culture medium. EirA was also detected within the bacterial cytoplasm and inner membrane. Without EirA, the C. burnetii T4BSS does not translocate effector proteins, suggesting a role for EirA in T4BSS activity. These findings demonstrate that EirA is a novel protein required for C. burnetii replication within the host cell and as such may be an important target of novel therapeutic approaches.
RESULTS
EirA is essential for intracellular replication of C. burnetii and contains a key functional region.
A transposon mutant disrupting cbu2072 122 bp from the start codon is unable to replicate within HeLa cells (17). PSI-BLAST analysis of the 165-amino-acid protein encoded by cbu2072 revealed 33% homology between amino acids 24 to 96 of CBU2072 to soluble transhydrogenases encoded by Escherichia coli (20). Transhydrogenases are metabolic enzymes involved in hydride transfer between NAD+ and NADP molecules (21–23). In addition, HHpred analysis of EirA suggests this protein has 20 to 40% structural homology to hydrolases and lipoproteins in Gram-positive and Gram-negative bacteria, as well as outer membrane proteins found in Gram-negative bacteria, between amino acids 1 to 58 (24). Despite the presence of these small regions of low homology, the vast majority of the protein sequence for CBU2072 showed no close homology to any other proteins.
To determine whether this intracellular growth defect phenotype could be attributed to transposon disruption of cbu2072, this strain was complemented with a constitutively plasmid-expressed version of CBU2072 containing an N-terminal 3×FLAG tag (2072::Tn pFLAG-2072 mutant). Constitutive expression of 3×FLAG-CBU2072 restored intracellular replication of the 2072::Tn mutant in both HeLa epithelial cells and THP-1 macrophage-like cells to levels comparable with C. burnetii Nine Mile phase II, referred to here as the wild type (WT) (Fig. 1). At 3 days postinfection of HeLa cells, the C. burnetii genome equivalent (GE) fold changes, compared to day 0, were 168 ± 103 for the WT, 2 ± 4 for the 2072::Tn mutant (P = 3.30 × 10−2 compared to the WT), and 134 ± 119 for the 2072::Tn pFLAG-2072 complemented mutant (Fig. 1A). Visual confirmation of this phenotype demonstrated that, in both HeLa and THP-1 cells, the 2072::Tn mutant was only ever observed as individual bacteria tightly enveloped in LAMP-1-positive membranes (Fig. 1C). This defect in replication was only visible during intracellular replication, with no significant differences between the C. burnetii WT, 2072::Tn mutant, and 2072::Tn pFLAG-2072 mutant replication during axenic culture (see Fig. S1 in the supplemental material), consistent with previous findings (25). Confirmation of this striking phenotype led to CBU2072 being assigned the name EirA (essential for intracellular replication A).
FIG 1.

CBU2072 is essential for intracellular replication of C. burnetii. (A and B) Intracellular replication of C. burnetii WT (red), 2072::Tn mutant (blue), 2072::Tn pFLAG-2072 mutant (green), and 2072::Tn pFLAG-207224–165 mutant (purple) in HeLa CCL2 cells (n = 4) (A) and THP1 cells (n = 5) (B). THP1 cell data depict fold changes at 3 days postinfection. Error bars represent standard deviation. *, P < 0.05. P values were determined using an unpaired Student t test. (C) Representative confocal immunofluorescence (IF) images at 3 days postinfection for HeLa CCL2 and THP1 cells. Cells were stained with anti-LAMP1 (green), anti-C. burnetii (red), and DAPI (blue). Scale bar = 10 μm. Arrows indicate individual intracellular C. burnetii cells.
TMHMM2.0 and TMPred are both bioinformatic analysis tools used to predict transmembrane domains (26, 27). These programs predict that this small protein (18.3 kDa) contains an N-terminal signal peptide sequence or transmembrane domain from amino acids 1 to 29 or 1 to 25, respectively. To determine the role of this N-terminal signal peptide for EirA, a strain expressing truncated protein was generated (eirA::Tn pFLAG-EirA24–165 mutant). This strain was unable to restore the intracellular growth defect to WT levels (Fig. 1), suggesting that the N-terminal region is important for the biological function of EirA. At 3 days postinfection of HeLa cells, the fold change in C. burnetii GE compared to day 0 was 2 ± 0.2 (P = 3.17 × 10−2 compared to the 168 ± 103 fold change observed for the WT) (Fig. 1A), with similar differences observed in THP-1 cells (Fig. 1B).
Recent studies have utilized Galleria mellonella larvae to study C. burnetii virulence in vivo. In this animal model, infection with phase II strains is lethal over an 11-day period (18, 28, 29). To assess whether EirA is required for virulence in this model, G. mellonella larvae were infected with 106 GE of C. burnetii WT or eirA::Tn, eirA::Tn pFLAG-EirA, and eirA::Tn pFLAG-EirA24–165 mutant strains. Survival of the infected larvae was monitored every 24 h for 11 days. All WT and eirA::Tn pFLAG-EirA mutant-infected larvae died within 11 days, while all eirA::Tn mutant- and eirA::Tn pFLAG-EirA24–165 mutant-infected larvae survived, alongside the phosphate-buffered saline (PBS) control (Fig. 2A). Infected G. mellonella hemocytes, stained with anti-C. burnetii and 4′,6-diamidino-2-phenylindole (DAPI), showed CCV formation in the WT and eirA::Tn pFLAG-EirA mutant-infected cells, but eirA::Tn mutant- and eirA::Tn pFLAG-EirA24–165 mutant-infected cells presented only individual intracellular C. burnetii (Fig. 2B), similar to previous observations in HeLa and THP-1 cell models (Fig. 1C). Overall, these observations support an essential role for C. burnetii EirA in intracellular replication and highlight the essentiality of the N-terminal signal peptide sequence for function.
FIG 2.

EirA is required for virulence in G. mellonella. Survival of G. mellonella following infection with C. burnetii WT (red), eirA::Tn mutant (blue), eirA::Tn pFLAG-EirA mutant (green), and eirA::Tn pFLAG-EirA24–165 mutant (purple) at 106 GE (A). A PBS control (orange outline) was also included. Results are shown as a representative of two independent biological replicates, each with 12 larvae per condition. (B) Representative confocal IF images at 3 days postinfection of G. mellonella hemocytes. Cells were stained with anti-C. burnetii (red) and DAPI (blue). Scale bar = 10 μm. Arrows indicate individual intracellular C. burnetii.
EirA mutant growth is restored within replication-permissive vacuoles.
We next investigated whether EirA is required for the growth of individual bacteria, or whether the protein regulates intracellular replication indirectly by modulating the properties of the CCV. To address this, HeLa cells were coinfected with C. burnetii WT and the eirA::Tn mutant at a multiplicity of infection (MOI) of 5 for each strain, and replication of the bacteria was monitored every 24 h for 7 days. The transposon disrupting eirA also encodes the fluorescent protein mCherry, allowing mutant and WT bacteria to be differentiated during coinfection. By monitoring mCherry fluorescence, the C. burnetii eirA::Tn mutant displayed intracellular replication in the presence of the WT, with large CCVs containing many mCherry-positive C. burnetii (eirA::Tn mutant) strains observed by 3 days postinfection (Fig. 3). This demonstrates that WT C. burnetii expressing EirA is able to create an intracellular environment conducive to replication by EirA-deficient C. burnetii, thereby supporting a role for EirA in the establishment of the intracellular replicative niche rather than being required by individual C. burnetii for replication.
FIG 3.
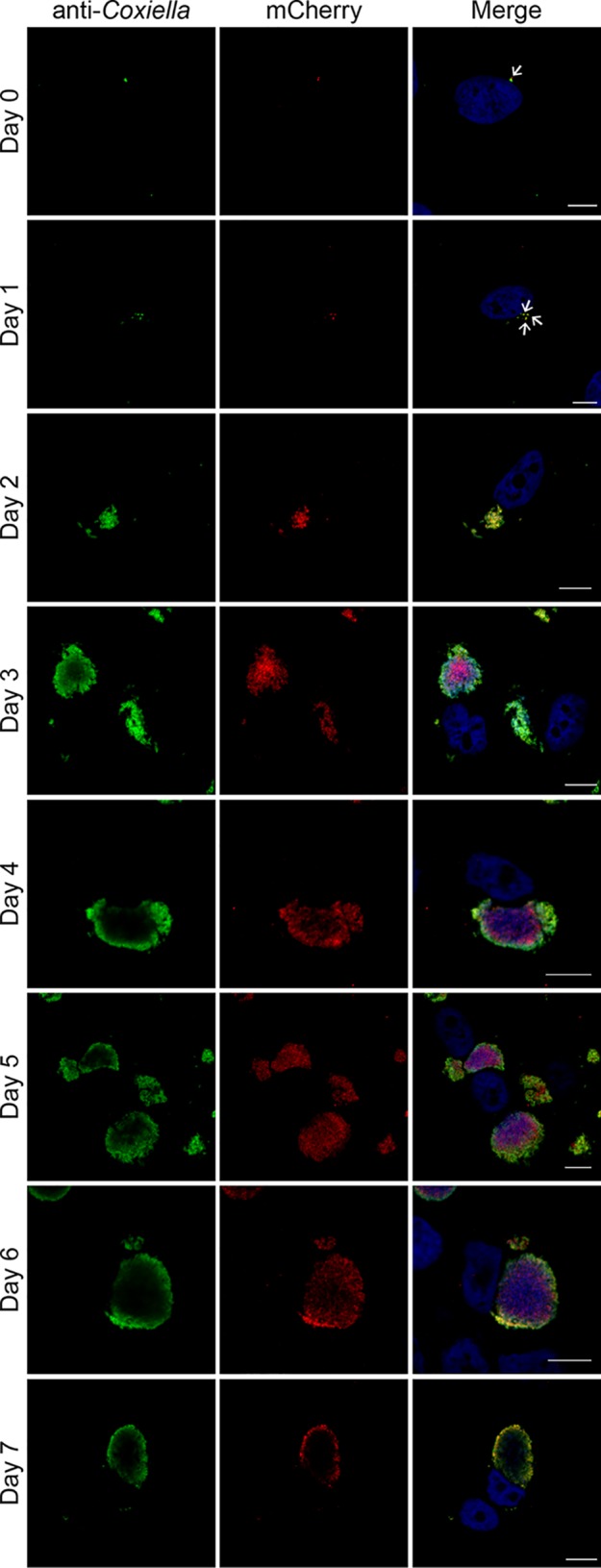
C. burnetii WT cells are able to trans-complement the eirA::Tn mutant during intracellular replication. HeLa CCL2 cells coinfected with C. burnetii WT and the eirA::Tn mutant were fixed and stained every 24 h across a 7-day infection period. Cells were stained with anti-C. burnetii (green) and DAPI (blue), while the eirA::Tn mutant was identified through transposon expression of mCherry (red). Images are representative of three independent biological replicates. Scale bar = 10 μm. Arrows indicate individual intracellular C. burnetii cells.
EirA is not required for the intracellular viability of C. burnetii.
The C. burnetii eirA::Tn mutant persists within individual, tight-fitting LAMP-1-positive vacuoles in both HeLa and THP-1 cells (Fig. 1C). To determine the viability of these persistent cells, HeLa cells were infected with the C. burnetii eirA::Tn mutant at an MOI of 100, and the infection was left to progress for 5 days, allowing for host cell clearance of dead bacteria. At 5 days postinfection, cells were superinfected with either the WT or C. burnetii eirA::Tn mutant at an MOI of 100, and the infection was left to progress for a further 3 days, allowing any viable C. burnetii eirA::Tn mutant cells to form CCVs in the presence of WT. At this stage, cells were fixed and stained for mCherry and C. burnetii to determine whether the eirA::Tn mutant was able to replicate (Fig. 4A). Quantification of fluorescence signals shows significantly higher levels of mCherry, thereby reflecting the expansion of the eirA::Tn mutant within the host cell following superinfection with WT (Fig. 4B). This was further supported by the presence of mCherry-positive CCVs, which were clearly visible in the presence of the WT (Fig. 4C). The C. burnetii eirA::Tn mutant superinfected with the mutant showed no CCV formation, with only isolated bacteria present intracellularly (Fig. 4C, arrows). This demonstrates that the persistent C. burnetii eirA::Tn mutant in individual vacuoles is still viable within epithelial host cells.
FIG 4.

EirA is not required for intracellular viability of C. burnetii. (A) Schematic representation of the experimental procedure, in which HeLa cells were infected with C. burnetii eirA::Tn mutant and incubated for 5 days before being superinfected with either the C. burnetii WT or eirA::Tn mutant. (B) After 3 days, cells were fixed and stained, and total 568-nm fluorescence levels relative to host cell number were measured using Fiji (49) in order to quantify mCherry expressing eirA::Tn replication; n = 6. Error bars represent the standard deviation, and the P value was determined using an unpaired Student t test. (C) Confocal immunofluorescence microscopy images of representative cells stained for all C. burnetii (green), mCherry expressed by the eirA::Tn mutant (red) and DAPI (blue). Scale bar = 10 μm. Arrows indicate individual intracellular C. burnetii cells.
EirA is present at the inner membrane and in the culture supernatant of axenically cultivated C. burnetii.
Previous research has shown that EirA is found in ACCM-2 during axenic cultivation of C. burnetii (19). Therefore, in order to confirm this finding, C. burnetii eirA::Tn, eirA::Tn pEirA-FLAG, eirA::Tn pFLAG-EirA, and eirA::Tn pFLAG-EirA24–165 mutants were harvested at 6 days postinoculation in ACCM-2, and the presence of EirA in both the bacterial whole-cell and axenic culture media was analyzed by immunoblotting (Fig. S2).
Despite plasmid-encoded expression being driven by the same promoter, differences in protein abundance were observed between each strain (Fig. S2). EirA-3×FLAG is more abundant than 3×FLAG-EirA (Fig. S2C). This may indicate a cleavage event at the N terminus of EirA. This cleavage may occur due to the N-terminal signal peptide sequence, which is not present in 3×FLAG-EirA24–165. The highly abundant EirA-3×FLAG was detected in both the whole-cell lysate (WCL) and the culture media (Fig. S2C). However, there was no observed shift in molecular mass resulting from any cleavage events in EirA-3×FLAG present in the supernatant. A band corresponding to a protein of a lower molecular weight was, however, present in the WCL (Fig. S2B). This was in contrast to the previous observation of a doublet in ACCM-2 (19). In addition, EirA-3×FLAG was present at relatively low levels in ACCM-2, in comparison to the nonfunctional 3×FLAG-EirA24–165, which was found at a higher proportion in the culture medium (Fig. S2B and D). This suggests that functional, full-length EirA may be more associated with the bacterial cell rather than with the extracellular environment.
To address the potential loss of 3×FLAG tag from EirA, antibody against EirA was raised using recombinant maltose-binding protein (MBP)-EirA24–165 (Walter and Eliza Hall Institute [WEHI] antibody facility) (Fig. S3). As anticipated, plasmid-driven production of EirA produced more EirA than what is produced under native conditions, as can be seen in a comparison of expression levels between the eirA::Tn pFLAG-EirA mutant and the WT (Fig. S3). A comparison of EirA expression using EirA and FLAG antibody demonstrates that while 3×FLAG-EirA expression seems higher than that of 3×FLAG-EirA24–165 using anti-EirA antibodies, the opposite is true when using anti-FLAG antibodies. This further supports the possibility that the 3×FLAG tag is specifically cleaved in the eirA::Tn pFLAG-EirA mutant strain.
To further investigate the localization of EirA within the bacterial cell, axenically grown C. burnetii WT and eirA::Tn, eirA::Tn pFLAG-EirA, and eirA::Tn pFLAG-EirA24–165 mutant strains were fractionated, and EirA subcellular localization was monitored in each strain (Fig. 5). Each isolated fraction was also probed with antibodies against the T4BSS outer membrane (OM) core complex protein IcmK, periplasmic protein IcmX, inner membrane (IM) component IcmD, and cytoplasmic (Cyto) ATPase DotB (30) to validate the fractionation process.
FIG 5.
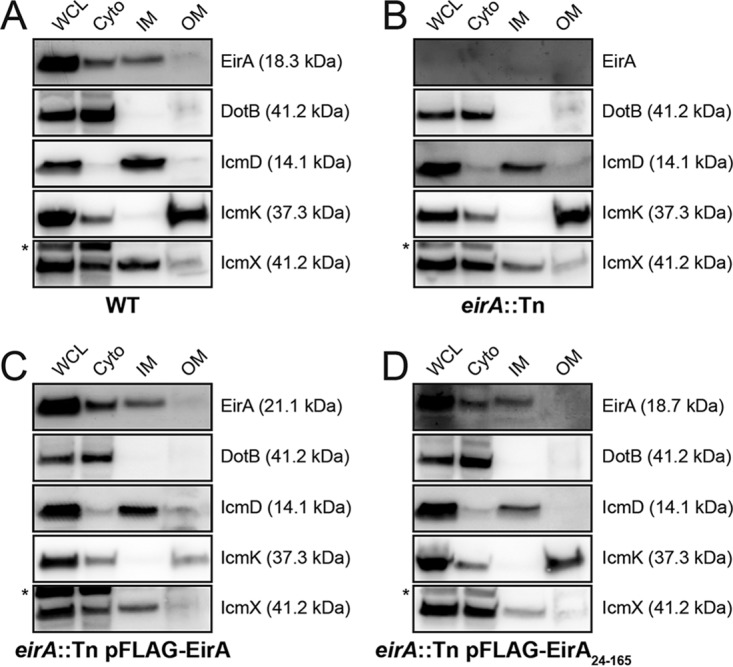
EirA localize to the bacterial cytoplasm and TX-100-soluble membrane fraction. (A to D) Subcellular fractionations of C. burnetii WT (A), eirA::Tn mutant (B), eirA::Tn pFLAG-EirA mutant (C), and eirA::Tn pFLAG-EirA24–165 mutant (D) were performed to observe the subcellular localization of EirA within the bacterial cell. DotB (cyto/cytoplasm), IcmK (TX-100-insoluble/OM/outer membrane), IcmD (TX-100-soluble/IM/inner membrane), and IcmX (periplasm) were used to denote specific subcellular localizations as well as whole-cell lysate (WCL). Blots are representative of three independent biological replicates. *, nonspecific bands.
All forms of EirA, including truncated 3×FLAG-EirA24–165, were detected in both the bacterial cytoplasm and the TX-100-soluble membrane fraction (referred to throughout as the inner membrane fraction). EirA in the inner membrane fraction consistently had a slightly higher apparent molecular weight than that of the cytoplasmic band (Fig. 5), suggesting that the inner membrane-localized EirA may be modified.
EirA influences C. burnetii metabolism.
EirA contains low homology to soluble transhydrogenases of E. coli between amino acids 24 to 96 (20–23). In order to determine whether EirA has a metabolic function, polar metabolites were extracted from axenically grown C. burnetii WT and eirA::Tn mutant strains and analyzed by gas chromatography-mass spectrometry (GC-MS) and liquid chromatography-MS (LC-MS) (Fig. 6 and Tables 1, 2, and S1). The loss of EirA was associated with a significant increase in intracellular levels of NADH and decrease in levels of NADP, consistent with a potential role for this protein in regulating the equilibrium between these essential cofactors. Given that these cofactors are utilized in both anabolic and catabolic processes, the loss of EirA was also associated with global changes in many other metabolites, including most amino acids, and cell wall precursors (meso-2,6-diaminopimelate and ll-2,6-diaminopimelate) (Fig. 6) (31). The decreased intermediates in the tricarboxylic acid (TCA) cycle (malate and succinate) could reflect decreased oxidative phosphorylation and/or increased anaplerotic synthesis of nonessential amino acids such as aspartate and glutamate, which were both increased in the mutant (Fig. 6).
FIG 6.

Absence of EirA leads to an accumulation of some amino acids and significantly lower glycolytic and TCA cycle activities. Shown are combined metabolites detected by GC-MS and LC-MS which were significantly different in abundance between the C. burnetii eirA::Tn mutant and WT strains. Red denotes metabolites which were significantly higher in abundance in the C. burnetii eirA::Tn mutant, and blue denotes metabolites which were significantly lower in abundance in the C. burnetii eirA::Tn mutant. P < 0.05, BH-adjusted unpaired t test. Pale blue denotes metabolites which were not significant. Pale green denotes enzymes which are missing based on genome annotation data in KEGG. Purple denotes putative and defined metabolite transporters. Dotted arrows indicate pathways which have been abbreviated. Metabolite abbreviations are listed in Table S1 in the supplemental material.
TABLE 1.
Metabolites identified as significantly different in abundance between C. burnetii eirA::Tn mutant and WT strains using GC-MS
| Metabolite | Fold changea | BH-adjusted P valueb |
|---|---|---|
| 3P-d-glycerate | −0.68 | 1.20 × 10−2 |
| Succinate | −0.60 | 2.24 × 10−2 |
| Malate | −0.57 | 3.02 × 10−2 |
| Adenine | 4.44 | 5.93 × 10−5 |
| l-Ala | 0.54 | 2.37 × 10−2 |
| l-Asp | 0.88 | 2.95 × 10−3 |
| l-Glu | 4.66 | 1.73 × 10−4 |
| l-Ile | −0.58 | 7.73 × 10−3 |
| l-Phe | −0.58 | 2.13 × 10−2 |
| l-Pro | −1.33 | 1.52 × 10−5 |
| l-Ornithine | −0.82 | 2.95 × 10−3 |
| ll-2,6-Diaminopimelate | 0.90 | 1.20 × 10−2 |
| Glycerol | 4.66 | 3.57 × 10−2 |
| sn-Glycerol-1P | −0.94 | 6.97 × 10−4 |
Positive fold change values denote metabolites with significantly higher abundance, while negative fold change values denote metabolites with significantly lower abundance in C. burnetii eirA::Tn mutant than in the WT.
BH, Benjamini-Hochberg.
TABLE 2.
Metabolites identified as significantly different in abundance between C. burnetii eirA::Tn mutant and WT strains using LC-MS
| Metabolite | Fold changea | BH-adjusted P valueb |
|---|---|---|
| l-Asp | 0.62 | 3.56 × 10−3 |
| l-Glu | 0.70 | 2.55 × 10−5 |
| l-Leu | 0.38 | 2.42 × 10−2 |
| l-Ser | 0.59 | 1.60 × 10−3 |
| l-Met | 0.43 | 1.04 × 10−3 |
| l-Lys | 1.04 | 9.28 × 10−3 |
| l-Tyr | 0.24 | 1.19 × 10−2 |
| 3′-Methyladenine | 0.57 | 1.55 × 10−2 |
| meso-2,6-diaminopimelate | 0.86 | 3.43 × 10−3 |
| GDP-d-mannose | −0.32 | 1.55 × 10−2 |
| UDP-GlcNAc | −0.37 | 1.41 × 10−2 |
| NADH | 0.33 | 3.67 × 10−2 |
| NADP | −0.50 | 1.22 × 10−2 |
Positive fold change values denote metabolites with significantly higher abundance, while negative fold change value denote metabolites with significantly lower abundance in C. burnetii eirA::Tn mutant than in the WT.
BH, Benjamini-Hochberg.
Interestingly, overexpression of 3×FLAG-EirA in the eirA::Tn mutant strain generated further global changes in intracellular metabolite levels, indicating a possible stress response. Fold change data comparing the eirA::Tn pFLAG-EirA mutant strain to the WT and eirA::Tn mutant are presented in Tables S2 and S3. Overall, these data strongly suggest that EirA regulates bacterial cell metabolism, possibly by regulating intracellular levels of NADH and NADP.
EirA is not required for cell membrane integrity.
The accumulation of meso-2,6-diaminopimelate and ll-2,6-diaminopimelate in the C. burnetii eirA::Tn mutant indicated that EirA may indirectly or directly modulate metabolic pathways involved in cell wall biosynthesis. Such a role would also be consistent with partial localization of EirA to the inner membrane of C. burnetii. We therefore investigated whether the absence of EirA made C. burnetii more susceptible to stressors of cell membrane integrity. Axenically grown C. burnetii WT and eirA::Tn, eirA::Tn pFLAG-EirA mutant, and eirA::Tn pFLAG-EirA24–165 mutant strains were treated with either ampicillin, a cell wall stressor, or polymyxin B, an outer membrane stressor, at various concentrations at 4 days postinoculation. Both before antibiotic treatment and at 24 h post-antibiotic exposure, samples were harvested for genome equivalent quantification, and serial dilutions of C. burnetii were plated for CFU quantification. No significant differences in antibiotic susceptibility were observed between the strains across all concentrations of antibiotics (Fig. 7A and B). This suggests that EirA may not have a functional role in cell membrane integrity. In support of this, transmission electron microscopy (TEM) images of the C. burnetii WT, eirA::Tn mutant, and eirA::Tn pFLAG-EirA mutant strains demonstrates no gross differences in bacterial morphology or cell membrane structure (Fig. 7C). Despite observations of significant differences in metabolite composition, these experiments could not demonstrate any EirA-dependent alterations to cell membrane integrity or bacterial morphology.
FIG 7.

Absence of EirA does not impact susceptibility to cell membrane stressors or effect overall gross morphology in C. burnetii. C. burnetii WT and eirA::Tn, eirA::Tn pFLAG-EirA, and eirA::Tn pFLAG-EirA24–165 mutant strains were exposed to various concentrations of either ampicillin or polymyxin B at 4 days postinoculation, and the impact on bacterial replication was quantified at 24 h posttreatment by genome equivalents (GE) (A) and CFU (B) per milliliter. Fold change depicts the bacterial numbers at 24 h posttreatment relative to the numbers pretreatment. No significant differences in GE and CFU were observed based on an unpaired Student t test. (C) C. burnetii WT, eirA::Tn mutant, and eirA::Tn pFLAG-EirA mutant strains were grown for 6 days in ACCM-2 before being imaged using transmission electron microscopy (TEM). Images are representative of two technical replicates.
Translocation of effector proteins via the T4BSS is blocked in the absence of EirA.
EirA is not a substrate of the T4BSS, as a β-lactamase (BlaM)-tagged EirA fusion protein was not translocated into the host cell cytosol during infection (17). Considering the inner membrane localization of EirA and the mutant phenotype reminiscent of disruption or deletion mutants of genes encoding T4BSS apparatus components (8, 9), we hypothesized that EirA may contribute to T4BSS function. In order to determine whether EirA is involved in T4BSS effector translocation, a BlaM translocation assay was performed using the known C. burnetii effector protein MceA, a robust reporter system which has been extensively used to monitor T4BSS activity (7, 8, 17, 32, 33). BlaM fusion protein translocation was determined by monitoring the cleavage of the fluorescent BlaM substrate CCF2-AM and calculating the 450-nm/520-nm fluorescence emission ratio 2 h postaddition of CCF2-AM. HeLa cells were infected with either WT, WT pBlaM-MceA, eirA::Tn mutant, or eirA::Tn pBlaM-MceA mutant strains or a combination of these strains (Fig. 8A and B). At both 48 and 72 h postinfection, translocation of BlaM-MceA was observed in the WT pBlaM-MceA strain, as expected (Fig. 8A and B). However, even when replication was restored through coinfection with the WT, the eirA::Tn pBlaM-MceA mutant-infected cells did not show any translocation of BlaM-MceA.
FIG 8.

EirA is important for Dot/Icm type IV secretion system effector translocation. Plasmids encoding transcriptionally fused β-lactamase (BlaM) and T4BSS effector MceA were introduced into the C. burnetii WT and eirA::Tn mutant. These strains, along with the C. burnetii WT and eirA::Tn mutant strains, were used to infect HeLa CCL2 cells at an MOI of either 100 (red circles), 150 (blue squares), or 300 (green triangles), or at a coinfection ratio of 1:1, 1:2, or 1:5 WT to eirA::Tn mutant, respectively, where two different strains were used (A and B). At either 48 h (A) or 72 h (B) postinfection, the fluorescent β-lactamase substrate CCF2-AM was added to cells, and cleavage of the substrate was determined by calculating the ratio of fluorescence at 450 nm to 520 nm, relative to uninfected cells; n = 3. (C) Translocation assays for C. burnetii expressing BlaM-0425 were performed at 72 h postinfection with an MOI of 300. Coinfections of the WT and eirA::Tn mutant derivatives were conducted with a ratio of 1:5. (D) HeLa CCL2 cells were infected with C. burnetii strains expressing 3×FLAG-MceA and then fixed and stained at 3 days postinfection. Cells were stained with anti-FLAG (green), MitoTracker (red), and DAPI (blue). Images are representative of three independent biological replicates. Scale bar = 10 μm. Asterisks (*) indicate CCVs.
A recent study demonstrated that the translocation of a subset of T4BSS effectors is regulated by the chaperone IcmS (34). This research showed that MceA was dependent on IcmS for translocation via the T4BSS. Therefore, in order to determine whether the EirA-dependent defect in T4BSS translocation is linked to IcmS function, the coinfection translocation assay was performed using the WT and eirA::Tn mutant expressing BlaM fused to an IcmS-independent effector protein, CBU0425 (BlaM-0425) (34) (Fig. 8C). Significant translocation of the BlaM-0425 fusion protein by the WT was consistently observed; however, no translocation of BlaM-0425 was detected by eirA::Tn pBlaM-0425 mutant, even in the presence of the WT (Fig. 8C). This suggests that the defect in T4BSS translocation seen in the absence of EirA is not linked to IcmS function.
The dependence of EirA for T4BSS effector translocation was further confirmed using immunofluorescence microscopy. C. burnetii expressing 3×FLAG-MceA shows distinct anti-FLAG signal at host mitochondria during infection (32), while this was not observed with the eirA::Tn mutant expressing 3×FLAG-MceA, even when coinfection with C. burnetii WT facilitated CCV formation (Fig. 8D). Both the BlaM reporter assay and immunofluorescence microscopy approaches employed here demonstrate that EirA is required for C. burnetii T4BSS effector translocation during infection.
Given that the absence of EirA prevents T4BSS effector translocation, it is possible that EirA may have a functional role regulating the assembly of the T4BSS apparatus or expression levels. However, the subcellular fractionations performed here demonstrate no change in DotB, IcmD, IcmK, and IcmX expression and localization in the absence of functional EirA (Fig. 5).
DISCUSSION
The mechanisms used by the intracellular pathogenic bacteria C. burnetii to thrive and replicate within the normally hostile host phagolysosome are not fully understood. Previous studies have highlighted the important role of C. burnetii lipopolysaccharide (LPS) as an important virulence factor, with avirulent phase II strains lacking mature LPS unable to infect humans or mice (35, 36). However, these strains are still able to establish and thrive within CCVs in tissue culture models of infection. The development of axenic culture conditions and tools for genetic manipulation has made it possible to identify other important proteinaceous C. burnetii factors required for intracellular replication (10). Landmark mutational analyses have highlighted that the T4BSS is another major virulence determinant of C. burnetii, along with a subset of the effector proteins translocated by this system. Genetic disruption of T4BSS components results in the total abolition of intracellular replication, which highlights the importance of this apparatus to C. burnetii (8, 9, 37). In this study, we have identified and characterized a novel protein, EirA, essential for intracellular replication of C. burnetii. It is important to note, however, that these studies have been performed in C. burnetii phase II. Phase I and II strains replicate with similar kinetics in macrophages and share the same endosomal/lysosomal markers on the CCV (38). However, phase II strains cannot cause disease in animals such as mice or humans due to the truncated LPS (39–41). Future work could determine whether the EirA phenotype is consistent in phase I variants of C. burnetii and therefore represents a true virulence factor of C. burnetii.
The precise mechanism through which EirA facilitates C. burnetii virulence remains to be elucidated. Previous work using mass spectrometry detected EirA in the culture medium during axenic growth of C. burnetii (19). Given that EirA does not appear to be a T4BSS effector protein (17), this suggests that EirA is secreted out of the bacterial cell in a T4BSS-independent manner. The probable mechanism for this secretion is via outer membrane vesicles, which were shown to be formed during both axenic and intracellular cultivation (19). However, the data presented in this study demonstrate that functional EirA is equally associated with the bacterial cell, with localization detected in the C. burnetii cytoplasm and inner membrane. Further investigations are warranted to determine the localization at which EirA impacts C. burnetii biology, which may help elucidate protein function.
This research has shown that the N-terminal 23 amino acids, encoding a putative signal peptide sequence, are required for EirA function within host cells. It is interesting to note that while truncated EirA cannot restore WT-level phenotypes, fractionation experiments demonstrate similar localization to the bacterial cytoplasm and inner membrane. The lack of the N terminus may impact the structure of EirA at the inner membrane or its capacity to interact with other bacterial factors. The subcellular fractionation of C. burnetii also indicates that membrane-associated EirA is modified, independent of the N terminus, appearing to have a slightly higher molecular weight than in the cytoplasmic fraction.
The absence of EirA is not lethal for C. burnetii during infection, as the bacteria are not cleared by the host cell and can be rescued through coinfection or superinfection, 5 days post-initial infection, with T4BSS competent C. burnetii strains. As mentioned earlier, this phenotype is identical to that in mutants of the T4BSS apparatus, such as the C. burnetii icmL::Tn (8) and icmD::Tn (9) mutants. The icmD::Tn mutant in particular was also shown to maintain viability in host cells, as replication could be restored 24 h postinfection using an inducible complementation construct (9). Our similar findings again highlight the innate ability of C. burnetii to withstand the host phagolysosomal environment, independent of T4BSS activity and replication.
As EirA partially localizes to the inner membrane, and the loss of this protein results in changes to the intracellular levels of cell wall intermediates, we investigated whether EirA has a direct role in cell wall biosynthesis. However, the absence of EirA did not appear to affect cell membrane integrity in C. burnetii, as assessed by their sensitivity to antibiotics and TEM ultrastructure. However, the resolution which was used to examine the impact of EirA on the bacterial cell membrane may have been insufficient to observe more subtle contributions of EirA to the cell membrane. Observing bacterial membrane integrity or T4BSS assembly at higher resolutions may be of interest, for instance, through the use of cryogenic electron microscopy.
Both the β-lactamase translocation reporter assay and the immunofluorescence microscopy performed in this study show that absence of EirA blocks the translocation of both IcmS-dependent and IcmS-independent Dot/Icm effector proteins. Even when replication was restored by coinfection with WT, the EirA mutant showed no T4BSS activity. This, taken together with the observation that the EirA mutant has an identical rescuable replication defect phenotype to T4BSS mutants, combined with the membrane localization of EirA, led to the hypothesis that EirA contributes to C. burnetii T4BSS function. Given that eirA is not encoded within the T4BSS gene loci, and there are no EirA homologues encoded by Legionella species, which depend on a functionally analogous T4BSS (42), it is unlikely that EirA forms part of the T4BSS apparatus. EirA does not affect DotB, IcmD, IcmK, and IcmX expression levels nor their subcellular localization. However, it remains possible that EirA contributes to the recruitment of other specific Dot/Icm components and/or assembly at the bacterial poles.
Recent studies in Legionella pneumophila have shown that this pathogen uses second messenger signaling to influence T4BSS effector translocation (43). L. pneumophila appears to regulate effector translocation using c-di-GMP signaling via a diguanylate cyclase enzyme. Mutants lacking this enzyme show delayed translocation of the L. pneumophila effector protein LepA, and RNA sequencing data suggest that this enzyme has a posttranscriptional regulatory role for T4BSS effector translocation (43). C. burnetii lacks diguanylate cyclases but may have evolved similar posttranslational regulatory mechanisms to temporally regulate effector translocation during infection. EirA contains a region with homology to soluble transhydrogenases, which interact with NAD(P) cofactors. Here, we show that a loss or overexpression of EirA results in reciprocal changes in the intracellular levels of key cofactors NADH and NADP, as well as global changes in other metabolites. This suggests that EirA may regulate multiple metabolic pathways, one or more of which may in turn regulate T4BSS assembly and function. Future work could determine possible the enzymatic activity of EirA and any protein interacting partners of EirA, which may help elucidate the mechanism through which EirA impacts T4BSS activity.
This study has characterized a novel and unique C. burnetii inner membrane protein, EirA, which is essential for intracellular replication and virulence. Further investigations are required to determine the exact mechanisms by which EirA facilitates virulence. However, the data presented here indicate that EirA may represent a novel factor contributing to T4BSS function. Elucidating the mechanisms through which EirA exerts virulence capacity is important for understanding the virulence of C. burnetii. Given the unique aspects of this protein, determining the function of EirA and subsequently developing the capacity to specifically inhibit EirA function may serve as an important and powerful therapeutic strategy.
MATERIALS AND METHODS
Bacterial strains and mammalian cell lines.
Escherichia coli XL1-Blue (recA1 endA1 gyrA96 thi1 hsdR17 supE44 relA1 lac [F′proAB lacIqZΔM15 Tn10 [TetR]]) and DH5α [F− ø80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) phoA] (Clontech) strains were grown in Luria-Bertani medium supplemented with 50 μg/ml kanamycin (Sigma-Aldrich), 10 μg/ml chloramphenicol (Boehringer Mannheim), or 100 μg/ml ampicillin (A. G. Scientific) when required.
C. burnetii phase II Nine Mile strain RSA439, referred to as the wild type (WT) throughout, and an eirA::Tn mutant, first described in reference 17, were routinely grown in acidified citrate cysteine medium-2 (ACCM-2), as previously described (9). ACCM-2 was supplemented with 350 μg/ml kanamycin or 3 μg/ml chloramphenicol when required.
HeLa human carcinoma cells (CCL2; ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with GlutaMAX (Gibco) supplemented with 10% fetal calf serum (FCS; Gibco) or 5% FCS during infection. THP-1 human monocytic cells (ATCC) were cultured in RPMI 1640 with GlutaMAX (Gibco) supplemented with 10% FCS. Both cell lines were maintained at 37°C with 5% CO2.
Cloning and plasmids.
Genomic DNA (gDNA) of C. burnetii was isolated using the Zymo gDNA extraction kit, as per the manufacturer’s protocol. Oligonucleotides used to amplify ompA, eirA or cbu2072 fragments, mceA or cbu0077, and cbu0425 were obtained from Sigma-Aldrich. All oligonucleotide sequences are listed in Table 3. Restriction endonucleases were obtained from NEB and used according to the manufacturer’s protocol. Plasmid DNA was isolated using the QIAprep spin miniprep kit (Qiagen). pJB-Kan constructs were transformed into relevant strains, as previously described (17), and are listed with all other plasmids in Table 4.
TABLE 3.
Oligonucleotides used in this study
| Oligonucleotide | Sequence (5′ to 3′) | Comments and/or referencea |
|---|---|---|
| eirA_F1 | AAAGGATCCATGCGTTATCCGAAATTCAGT | Used to amplify eirA to clone into 3′ end of 3×FLAG tag in pJB-Kan:3×FLAG-MCS |
| eirA_F2 | ATAAGCTTCTGCAGATGCGTTATCCGAAATTCAGTT | Used to amplify eirA to clone into 5′ end of 3×FLAG tag in pJB-Kan:3×FLAG |
| eirA24–165_F | AAAGGATCCGCCCCTGCAACG | Used to amplify eirA24–165 to clone into 3′ end of 3×FLAG tag in pJB-Kan:3×FLAG-MCS |
| eirA_R1 | AAGCGGCCGCTTAAAATCCAATTTCTTGTTG | Used to amplify eirA and eirA24–165 to clone into 3′ end of 3×FLAG tag in pJB-Kan:3×FLAG-MCS |
| eirA_R2 | ATGTCGACCTACTTGTCATCGTCATCCTT | Used to amplify eirA to clone into 5′ end of 3×FLAG tag in pJB-Kan:3×FLAG |
| mceA_F | AAGGTCGACATGAGACAACTCGTTTCAATTAAA | 32 |
| mceA_R | AAGGTCGACTTACATAATAGAACACCCACGA | 32 |
| cbu0425_F | AAGGATCCATGGAAGGCGTAATCTATTCA | Used to amplify cbu0425 to clone into pJB-Kan:BlaM-MCS (8) |
| cbu0425_R | AACTGCAGTTATGTGGATGCGCTGGGC | Used to amplify cbu0425 to clone into pJB-Kan:BlaM-MCS (8) |
| ompA_F | CAGAGCCGGGAGTCAAGCT | 44 |
| ompA_R | CTGAGTAGGAGATTTGAATCGC | 44 |
MCS, multiple-cloning site.
TABLE 4.
Plasmids used in this study
| Plasmid | Propertiesa | Reference or source |
|---|---|---|
| pJB-Kan:BlaM | C. burnetii expression vector encoding BlaM from Bacillus subtilis under constitutive expression by the cbu1169 promoter (P1169), Kanr | Heinzen Laboratory |
| pJB-Kan:BlaM-MCS | C. burnetii expression vector encoding BlaM from B. subtilis and MCS from pcDNA4/TO under constitutive expression by the P1169 promoter, Kanr | This study |
| pJB-Kan:BlaM-0425 | Encodes 5′-BlaM tag and cbu0425, Kanr | This study |
| pJB-Kan:BlaM-MceA | Encodes 5′-BlaM tag and mceA or cbu0077, Kanr | Newton Laboratory |
| pJB-Kan:3×FLAG | C. burnetii expression vector encoding 5′-3×FLAG tag under constitutive expression by the C. burnetii P1169 promoter, Kanr | 48 |
| pJB-Kan:3×FLAG-MCS | C. burnetii expression vector encoding 5′-3×FLAG tag and MCS from pcDNA4/TO under constitutive expression by the C. burnetii P1169 promoter, Kanr | This study |
| pJB-Kan:3×FLAG-MceA | Encodes 5′-BlaM tag and mceA or cbu0077, Kanr | Newton Laboratory |
| pJB-Kan:3×FLAG-EirA | pJB-Kan:3×FLAG encoding 5′-3×FLAG tag and eirA, Kanr | This study |
| pJB-Kan:EirA-3×FLAG | pJB-Kan:3×FLAG encoding 3′-3×FLAG-tagged eirA, Kanr | This study |
| pJB-Kan:3×FLAG-EirA24–165 | pJB-Kan:3×FLAG encoding 5′-3×FLAG tag and truncated eirA from bp 70–498 (amino acid residues 24–165), Kanr | This study |
| pMAL-c2X:EirA24–165 | pMAL-c2X encoding 5′-MBP tag and truncated eirA from bp 70–498 (amino acid residues 24–165), Ampr | This study |
Kanr, kanamycin resistance; Ampr, ampicillin resistance.
Quantification of C. burnetii genome equivalents.
Genome equivalents (GE) of C. burnetii were quantified by quantitative PCR (qPCR), using oligonucleotides specific for ompA, as previously described (44), or by using the Quant-iT PicoGreen double-stranded DNA (dsDNA) assay kit (Thermo Fisher Scientific), according to the manufacturer’s protocol. The qPCR was performed using either an MX3005P qPCR system (Agilent Technologies) or QuantStudio 7 Flex real-time PCR system (Thermo Fisher Scientific). Data export was performed using their respective analysis software programs. Microsoft Excel was used for further data analysis and generation of standard curves. PicoGreen assay results were read using the CLARIOStar plate reader (BMG Labtech). Data were processed using the MARS analysis software (BMG Labtech) and analyzed using Microsoft Excel.
Metabolite quenching and extraction for profiling studies.
C. burnetii WT, eirA::Tn mutant, and eirA::Tn pFLAG-EirA mutant strains, incubated in ACCM-2 for 6 days, were continuously agitated and rapidly quenched to 0°C in a dry ice-ethanol bath to halt metabolic activity. Cultures were centrifuged (3,220 × g, 15 min, 0°C) to pellet the bacteria. The bacterial pellet was washed once in 10 ml ice-cold phosphate-buffered saline (PBS), centrifuged (3,220 × g, 15 min, –2°C), and then washed again with 1 ml PBS. The bacteria were centrifuged (17,100 × g, 15 min, –2°C), and metabolites were extracted using chloroform-methanol-water (CHCl3-CH3OH-H2O, 1:3:1 [vol/vol/vol]) containing 1 nmol [13C6]sorbitol and 10 nmol 13C5-15N-labeled valine as internal standards. After the initial addition of 300 μl methanol to 99 μl water with 0.5 μl of each of the internal standards (CH3OH-H2O, 3:1 [vol/vol]), bacterial cells were sheared by repeat freeze-thaw following immersion of tubes in liquid nitrogen for 30 s and then a dry ice-ethanol bath for 30 s, for a total of 10 times each. One hundred microliters of chloroform was then added to bring the solution to the aforementioned 1:3:1 (vol/vol/vol) ratio. Following vortex mixing, samples were incubated on ice for 10 min. Cell debris was removed by centrifugation (17,100 × g, 15 min, –2°C), and the supernatant was adjusted to 1:3:3 CHCl3-CH3OH-H2O (vol/vol) by the addition of 200 μl water. The mixture was vortex mixed and centrifuged (17,100 × g, 15 min, –2°C) to induce phase separation. The upper aqueous phase, containing polar metabolites, was transferred to a fresh precooled 1.5-ml microcentrifuge tube and stored at –80°C until gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS) analysis.
Metabolite derivatization and GC-MS analysis.
Aqueous-phase samples were transferred into glass vial inserts and dried in a rotational vacuum concentrator (RVC-2-33; John Morris Scientific), with an additional 30 μl of 100% methanol added to ensure that the samples were completely dry. Free aldehyde groups were derivatized in 20 μl methoxyamine chloride (30 mg/ml in pyridine) with continuous mixing for 2 h at 37°C. Twenty microliters of N,O-bis(trimethylsilyl)trifluoroacetamide (BSTFA) containing 1% trimethylchlorosilane (TMCS) was then added. Samples were incubated for 1 h at 37°C, with continuous shaking, using a Gerstel MPS2 autosampler robot. For GC-MS analysis, 1 μl of derivatized sample was injected into an Agilent 7890A gas chromatograph (split/splitless inlet, 250°C) containing a VF-5ms column (30 m/250 μm/0.25 μm/10 m Eziguard precolumn) coupled to an Agilent 5975C mass selective detector. Helium gas provided at a constant flow rate of 1 ml/min was used as the carrier gas. The GC temperature was ramped from 35°C, initially held for 2 min, to 325°C at 25°C/min, and it was then held for 5 min at 325°C. The 5975C mass selective detector was used in scan mode, and mass spectra data were collected at a rate of 9.19 scans/s over an m/z range of 50 to 600 atomic mass units.
Metabolite analysis on LC-MS.
Polar metabolites were also analyzed by LC-MS using an Agilent Technologies 1200 series LC system. Samples were stored in an autosampler at 4°C. Ten microliters of sample was injected on a SeQuant ZIC-HILIC column (150 mm by 2.1 mm, 5 μm) maintained at 40°C with solvent A [20 mM (NH4)2CO3 (pH 9.0)] and solvent B (100% acetonitrile) at a flow rate of 250 μl/min. The gradients used were time (t) 0 min, 90% B; t 0.5 min, 90% B; t 12 min, 40% B; t 14 min, 40% B; t 15 min, 5% B, t 18 min, 5% B; and t 19 min, 90% B. Analysis was performed on an Agilent Technologies 6520 series quadrupole time of flight (TOF) mass spectrometer. The LC flow was directed to an electrospray ionization (ESI) source, where metabolite ionization was performed with an N2 drying gas pressure of 30 lb/in2 with a gas flow rate of 7 liters/min, 325°C capillary gas temperature, capillary voltage of 3,500 V, and fragmentor skimmer cap voltages of 125 V and 65 V, respectively. Data were collected in centroid mode with a scan range of m/z 50 to 1,700 at an acquisition rate of 1.2 spectra/s in negative MS mode.
Prior to analysis, mass calibration was performed for the negative mode to 0.5 ppm accuracy of the m/z value. Internal mass calibration was performed using the Agilent ESI-TOF reference mass solution containing purine (m/z 119.036320) and hexakis(1H,1H,3H-tetrafluoropropoxy)phosphazene (m/z 981.99509), which was continuously infused into the ESI source at a flow rate of 200 μl/min.
Identification of metabolites.
For GC-MS, metabolites from C. burnetii were identified from chromatograms by using the Agilent mass-selective detector (MSD) Productivity ChemStation for GC and GC-MS. Fragmented ion patterns were used to identify each metabolite using NIST, Fiehn, and Wiley libraries, and the retention times were matched to in-house libraries to confirm identifications (45). Data matrices were generated using PyMS (46).
For LC-MS, the profiles obtained were filtered to remove noise peaks above the specific abundance threshold. Metabolites were then identified using in-house standards from the Metabolomics Australia library by comparing retention times and molecular masses. Metabolites not present in the in-house standards were putatively identified using the MAVEN database, with molecular masses requiring a >70% mass score match to be assigned a putative identification.
Statistical analysis and comparison of metabolite profiles between C. burnetii WT, eirA::Tn mutant, and eirA::Tn pFLAG-EirA mutant strains.
Data matrices generated from the PyMS analyses were missing value imputed, log transformed, and then median normalized, as outlined previously (46, 47). R analysis was used to perform univariate analysis to generate a list of metabolites which were significantly different (P < 0.05, Benjamini-Hochberg [BH] adjusted P < 0.05) between strains. Metabolites which were both significantly and nonsignificantly different between the two conditions were identified as described above.
Mapping identified metabolites onto metabolic pathways.
Metabolites identified in these studies were mapped onto predicted metabolic pathways using the C. burnetii RSA 493 strain from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database as a guide. Any enzymes missing from the KEGG database annotations were searched for in C. burnetii RSA 439 using BLASTp (20), using L. pneumophila, E. coli, and other bacterial and human sequences as references.
Treatment of axenically grown C. burnetii with antibiotics.
C. burnetii strains grown for 6 days in ACCM-2 were quantified for GE as described above. Strains were passaged at 106 GE/ml and grown for a further 4 days, at which point cultures were supplemented with defined concentrations of ampicillin or polymyxin B (Sigma-Aldrich). One hundred microliters from each sample was taken and quantified using qPCR or PicoGreen, as described above, or plated in serial dilutions on ACCM-2 agar plates (48), modified to 0.5% agarose, which were left for 12 days before viability counts were performed. Cultures were left to incubate at 37°C, 5% CO2, and 2.5% O2 for 24 h, where 100 μl from each condition was taken and analyzed as per the previous day. Fold change values for GE and CFU per milliliter were calculated as 24 h posttreatment relative to pretreatment with antibiotics.
C. burnetii infections of tissue culture cell lines.
Growth curves in HeLa CCL2 and THP-1 cells were performed as previously described (32). Briefly, HeLa CCL2 cells were seeded at 5 × 104 cells/well into 24-well flat-bottom tissue culture plates (Corning) and incubated for 24 h. THP-1 cells were seeded at 5 × 105 cells/well and were treated and differentiated with 10 nM phorbol 12-myristate 13-acetate (PMA; Adipogen Life Sciences) for 3 days. Samples for immunofluorescence (IF) were seeded onto 12-mm sterile glass coverslips. HeLa CCL2 cells were infected at a multiplicity of infection (MOI) of 100, and THP-1 cells were infected at an MOI of 25. To determine the MOI, C. burnetii cultures were quantified as described above. Upon infection, cells were incubated for 4 h at 37°C and 5% CO2 before being washed once in PBS and supplemented with fresh medium to remove extracellular bacteria. Cells were lysed with nuclease-free water (Qiagen) for C. burnetii quantification at defined time points. For 1, 3, and 5 day postinfection samples, media from each duplicate well were pooled and collected alongside the lysed cells. Following lysis, samples were centrifuged at 17,000 × g for 15 min before gDNA was extracted using the Zymo gDNA extraction kit (Zymo Research) and quantified using the ompA-specific qPCR. For immunofluorescence, samples were processed at relevant time points, as outlined below.
For 7 day coinfection and phenotype rescue experiments, HeLa CCL2 cells were seeded at 5 × 104 cells/well into 24-well flat-bottom tissue culture plates (Corning) containing 12-mm glass coverslips and incubated for 24 h until infection. For 7 day coinfection experiments, the C. burnetii WT and eirA::Tn mutant strains were used to infect at an MOI of 5. For phenotype rescue experiments, cells were first infected with the eirA::Tn mutant at an MOI of 100. After 5 days of incubation, cells were superinfected with either the WT or eirA::Tn mutant at an MOI of 100 for each strain. Following 3 days of incubation, cells were fixed and stained as outlined below. Cells from both experiments were incubated in DMEM plus 5% FCS for 24 h postinfection before extracellular bacteria were removed by washing once in PBS and replacing with fresh DMEM plus 5% FCS.
Immunofluorescence microscopy.
All primary antibodies were diluted in PBS with 2% bovine serum albumin (BSA; Roche) and 0.05% saponin (Sigma-Aldrich), unless otherwise stated. The staining protocols were performed as previously described (32), unless otherwise stated. For growth curves, infected HeLa CCL2 and THP-1 cells were fixed for 20 min at RT with 4% (wt/vol) paraformaldehyde (in PBS) at 3 days postinfection and stained using 1:500 anti-LAMP-1 (Developmental Studies Hybridoma Bank) and 1:1,000 rabbit anti-C. burnetii antibodies (Roy Laboratory, Yale University) as primary antibodies. Anti-mouse Alexa Fluor 488 and anti-rabbit Alexa Fluor 568 antibodies (Thermo Fisher Scientific) were used at a dilution of 1:2,000. For 7 day coinfections and phenotype rescue experiments, cells were fixed and stained at the given time points described above, except that mouse anti-C. burnetii (Walter and Eliza Hall Institute antibody facility) diluted 1:1,000 and anti-mCherry (Novus Biologicals) diluted 1:500 were used as primary antibodies. For 3×FLAG-MceA staining, cells were fixed and stained at the given time points described above, except that MitoTracker red CMXRos (Thermo Fisher Scientific) staining was performed prior to fixing, according to the manufacturer’s protocol, and anti-FLAG (Sigma-Aldrich) primary antibodies were diluted 1:250. All samples were imaged using a Zeiss LSM700 instrument with the Zen software. All images were analyzed using Fiji (49).
For phenotype rescue experiments, the total 568-nm fluorescence from a ×20 objective image was quantified using Fiji (49). This numerical value was then made relative to the total number of host cell nuclei in the image.
Coinfection β-lactamase translocation assay.
HeLa CCL2 cells were seeded at 5 × 103 cells/well in a 96-well flat clear-bottom black assay plate (Corning) and incubated at 37°C and 5% CO2 for 24 h. Relevant C. burnetii strains were quantified using qPCR, as described above, and added to cells at an MOI of either 100, 150, or 300. For coinfection conditions with two different strains, infections were performed with the C. burnetii WT and eirA::Tn mutant strains at a 1:1 (total MOI, 100), 1:3 (total MOI, 150), or 1:5 (total MOI, 300) ratio. At 46 h or 70 h postinfection, CCF2-AM (Thermo Fisher Scientific) was added to the cells, according to the manufacturer’s protocol. After 2 h of incubation in the dark, fluorescence was measured using a CLARIOStar plate reader (BMG Labtech). Data were processed using the MARS analysis software (BMG Labtech) and analyzed using Microsoft Excel.
Analysis of EirA present in axenic culture media.
One hundred milliliters of day 6 C. burnetii ACCM-2 cultures was centrifuged at 3,220 × g for 15 min at 4°C. Supernatants were passed through a 0.2-μm filter to remove any bacterial cells before being precipitated in 20% trichloroacetic acid (Sigma-Aldrich) overnight at 4°C on ice. Samples were centrifuged at 20,000 × g for 20 min at 4°C before the resulting protein pellets were washed once in 80% ice-cold acetone (Chem Supply) and centrifuged at 17,000 × g for 20 min at 4°C. Protein pellets were dried at 60°C until all residual acetone was removed before being resuspended in 50 μl of 2× SDS-PAGE sample buffer (125 mM Tris, 4% SDS [Amresco], 20% glycerol, 0.2% bromophenol blue [Bio-Rad], 5% 2-mercaptoethanol [Sigma-Aldrich] [pH 6.8]). C. burnetii cells harvested from the initial centrifugation step to separate the ACCM-2 supernatant were washed once in ice-cold PBS and then resuspended in 1 ml ice-cold PBS. This resuspension was diluted 1:10 to a total volume of 50 μl in 2× SDS-PAGE sample buffer. All samples were processed for immunoblot analysis.
Fractionation of C. burnetii.
One hundred milliliters of day 6 C. burnetii ACCM-2 cultures was centrifuged at 3,220 × g for 15 min at 4°C and washed once in ice-cold PBS before being resuspended in 3 ml ice-cold lysis buffer (50 mM Tris-HCl [pH 7.6]; Chem Supply), 1 mM EDTA (Chem Supply), cOmplete EDTA-free protease inhibitor cocktail tablet (Sigma-Aldrich), and 10% glycerol (British Drug Houses [BDH]). Samples were sonicated on ice using a 600-W S-4000 sonicator (Misonix) at 50 W for 10 s, followed by 20 s of rest, until the total sonication time reached 3 min. Samples were then centrifuged at 3,220 × g for 15 min at 4°C to remove intact bacterial cells. Fifty microliters of the supernatant was reserved as the whole-cell lysate sample. Supernatants containing lysed bacteria were centrifuged for 1 h at 100,000 × g and 4°C to separate the membranes from the cytoplasmic fraction. Fifty microliters of the resulting supernatant was collected as a portion of the cytoplasmic fraction. The pellet was resuspended in lysis buffer using a 26-G needle (Terumo) and centrifuged as described before to remove residual cytoplasmic proteins. Pelleted proteins containing the membrane fractions were resuspended in 500 μl ice-cold membrane solubilization buffer (50 mM Tris-HCl [pH 7.6], 200 mM MgCl2 [Chem Supply], 1% Triton X-100 [Sigma-Aldrich]) using a 26-G needle and then left to incubate for 2 h on a rotating wheel at 4°C. Samples were made up to an equivalent total volume as the supernatant representing the cytoplasmic fraction, using solubilization buffer, before being centrifuged as described above to separate the outer and inner membranes. The TX-100-soluble fraction was considered to contain inner membrane contents, and the TX-100-insoluble fraction was considered to represent the outer membrane. Fifty microliters of the TX-100-soluble fraction was reserved for further analysis. The TX-100-insoluble pellets were resuspended in 2× SDS-PAGE sample buffer in volumes equivalent to those of other fractions, and 50 μl was reserved for further analysis. All reserved samples were diluted 1:1 in 2× SDS-PAGE sample buffer and boiled at 60°C for 30 min before immunoblot analysis.
Protein purification.
pMAL-c2X:EirA24–165 was transformed into E. coli Rosetta 2 by electroporation. Briefly, 100 ng of plasmid DNA was electroporated into E. coli Rosetta 2 at 200 Ω, 1.8 kV, and 25 μF in 2-mm electroporation cuvettes. Two hundred milliliters of log-phase E. coli Rosetta 2 pMAL-c2X:EirA24–165 was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside, and cells were incubated overnight at 18°C. Cultures were centrifuged at 10,000 × g for 15 min before pellets were resuspended in 25 ml column buffer (20 mM Tris-HCl [pH 7.5], 200 mM NaCl [Chem Supply], 1 mM EDTA, 20 μM phenylmethylsulfonyl fluoride [Sigma-Aldrich], 1 mM dithiothreitol [Astral Scientific]). Cells were lysed using the Dounce homogenizer and EmulsiFlex-C3 high-pressure homogenizer (Avestin), according to the manufacturer’s protocol. Purification was performed according to the manufacturer’s protocol (New England BioLabs), except that samples were diluted 1:2 in column buffer prior to resin binding. All elutions were pooled for dialysis, and samples were incubated overnight at 4°C in 2 liters of Tris-buffered saline (TBS; 20 mM Tris 150 mM NaCl [pH 8.0]) with gentle mixing, using Cellu·Sep regenerated cellulose tubular membrane (Membrane Filtration Products). Protein expression and purification were confirmed using NuPAGE Bis-Tris gels (Life Technologies) with morpholineethanesulfonic acid (MES) running buffer (Life Technologies), where a BSA standard was run alongside to determine concentrations. All elutions were then diluted to 2 mg/ml for long-term storage at –80°C. Antibodies against MBP-EirA24–165 (referred to as anti-EirA) were generated at the WEHI antibody facility.
Immunoblot analysis.
Whole-cell lysates of C. burnetii WT, eirA::Tn mutant, and related strains grown to day 6 postinoculation, as well as samples from fractionation studies, were prepared for Western blot analysis, as previously described (50). Proteins were transferred onto nitrocellulose membranes using the iBlot2 system (Life Technologies). Membranes were blocked in TBS with 0.1% Tween 20 (TBST; Thermo Fisher Scientific) and 5% skim milk for 1 h at room temperature (RT) before being incubated in primary antibodies for 1 h at RT. Anti-FLAG antibodies (Sigma-Aldrich) and anti-EirA antibodies (rabbit; WEHI antibody facility) were diluted 1:2,000 in TBST plus 5% skim milk, while anti-DotB, anti-IcmD, anti-IcmK, and anti-IcmX antibodies (rabbit, kindly donated by Robert A. Heinzen and Edward I. Shaw) were diluted 1:2,000 in TBST plus 5% BSA. Membranes were washed three times in TBST before anti-mouse-horseradish peroxidase (anti-mouse-HRP) or anti-rabbit-HRP secondary antibodies (PerkinElmer) diluted 1:3,000 in TBST plus 5% skim milk were added. The membranes were then incubated for 1 h at RT before being washed as described before. Proteins were detected using the Clarity Western ECL reagent (Bio-Rad) and visualized using the Amersham Imager 600 (GE Healthcare).
Transmission electron microscopy of C. burnetii.
One hundred milliliters of ACCM-2 cultures of C. burnetii was centrifuged for 15 min at 3,220 × g at RT before being washed once in PBS before centrifuging as described before. Pellets were resuspended in 2.5% glutaraldehyde (ProSciTech) and fixed for 2 h at RT. Bacteria were then rinsed twice in PBS without resuspension to remove any residual fixative. Samples were postfixed in 1% osmium tetroxide (Electron Microscopy Sciences), dehydrated in a graded series of acetone (Merck), and infiltrated and embedded with EPON resin (ProSciTech). Ninety-nanometer-thick sections were cut and stained with uranyl acetate (BDH) and lead citrate (BDH) before imaging on a CM120 transmission electron microscope (Phillips) at 120 kV.
Infection of Galleria mellonella.
G. mellonella larvae were grown in-house and kept at 30°C in the dark until use. Infections were conducted as described previously (37). Briefly, 106 GE of C. burnetii WT and eirA::Tn mutant strains were injected in the right proleg of G. mellonella larvae. Survival of the larvae incubated at 37°C was monitored every 24 h across 11 days. PBS controls were included alongside, and each condition consisted of 12 larvae.
To harvest hemocytes for immunofluorescence microscopy, three larvae per condition were anaesthetized on ice for 15 min before a small incision was made near the tail end of the larvae with a scalpel. Hemolymph from the incisions from the three larvae was pooled and immediately resuspended in 500 μl PBS. These resuspended cells were seeded onto 12-mm coverslips in a 24-well flat-bottom plate, which was then centrifuged for 10 min at 500 × g to allow cell adherence. Cells were then fixed and stained as outlined above.
Supplementary Material
ACKNOWLEDGMENTS
We kindly thank Robert A. Heinzen (Rocky Mountain Laboratories, NIAID, NIH, USA) and Edward I. Shaw (Oklahoma State University, USA) for the generation and donation of T4BSS antibodies. Confocal imaging was performed at the Biological Optical Microscopy Platform at the University of Melbourne (https://microscopy.unimelb.edu.au/).
This research was financially supported by NHMRC grants 1120344 and ARC DP180101298 awarded to H.J.N. A.H. is supported by the H2020-MSCA-Global Fellowship under grant number 657766. M.J.M. is an NHMRC principal research fellow. M.K. is supported by an Australian Research Training Program (RTP) scholarship.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Schneeberger PM, Wintenberger C, van der Hoek W, Stahl JP. 2014. Q fever in the Netherlands – 2007–2010: what we learned from the largest outbreak ever. Med Mal Infect 44:339–353. doi: 10.1016/j.medmal.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 2.Graham JG, MacDonald LJ, Hussain SK, Sharma UM, Kurten RC, Voth DE. 2013. Virulent Coxiella burnetii pathotypes productively infect primary human alveolar macrophages. Cell Microbiol 15:1012–1025. doi: 10.1111/cmi.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Raoult D, Marrie T, Mege J. 2005. Natural history and pathophysiology of Q fever. Lancet Infect Dis 5:219–226. doi: 10.1016/S1473-3099(05)70052-9. [DOI] [PubMed] [Google Scholar]
- 4.Hackstadt T, Williams JC. 1981. Biochemical stratagem for obligate parasitism of eukaryotic cells by Coxiella burnetii. Proc Natl Acad Sci U S A 78:3240–3244. doi: 10.1073/pnas.78.5.3240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coleman SA, Fischer ER, Howe D, Mead DJ, Heinzen RA. 2004. Temporal analysis of Coxiella burnetii morphological differentiation. J Bacteriol 186:7344–7352. doi: 10.1128/JB.186.21.7344-7352.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson CL, Martinez E, Beare PA, Jeffrey B, Heinzen RA, Bonazzi M. 2016. Right on Q: genetics begin to unravel Coxiella burnetii host cell interactions. Future Microbiol 11:919–939. doi: 10.2217/fmb-2016-0044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Newton HJ, McDonough JA, Roy CR. 2013. Effector protein translocation by the Coxiella burnetii Dot/Icm type IV secretion system requires endocytic maturation of the pathogen-occupied vacuole. PLoS One 8:e54566. doi: 10.1371/journal.pone.0054566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carey KL, Newton HJ, Luhrmann A, Roy CR. 2011. The Coxiella burnetii Dot/Icm system delivers a unique repertoire of type IV effectors into host cells and is required for intracellular replication. PLoS Pathog 7:e1002056. doi: 10.1371/journal.ppat.1002056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beare PA, Gilk SD, Larson CL, Hill J, Stead CM, Omsland A, Cockrell DC, Howe D, Voth DE, Heinzen RA. 2011. Dot/Icm type IVB secretion system requirements for Coxiella burnetii growth in human macrophages. mBio 2:e00175-11. doi: 10.1128/mBio.00175-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lührmann A, Newton HJ, Bonazzi M. 2017. Beginning to understand the role of the type IV secretion system effector proteins in Coxiella burnetii pathogenesis. Curr Top Microbiol Immunol 413:243–268. doi: 10.1007/978-3-319-75241-9_10. [DOI] [PubMed] [Google Scholar]
- 11.Akporiaye ET, Rowatt JD, Aragon AA, Baca OG. 1983. Lysosomal response of a murine macrophage-like cell line persistently infected with Coxiella burnetii. Infect Immun 40:1155–1162. doi: 10.1128/IAI.40.3.1155-1162.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulye M, Samanta D, Winfree S, Heinzen RA, Gilk SD. 2017. Elevated cholesterol in the Coxiella burnetii intracellular niche is bacteriolytic. mBio 8:e02313-16. doi: 10.1128/mBio.02313-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mansilla Pareja ME, Bongiovanni A, Lafont F, Colombo MI. 2017. Alterations of the Coxiella burnetii replicative vacuole membrane integrity and interplay with the autophagy pathway. Front Cell Infect Microbiol 7:112. doi: 10.3389/fcimb.2017.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Samanta D, Clemente TM, Schuler BE, Gilk SD. 2019. Coxiella burnetii type 4B secretion system-dependent manipulation of endolysosomal maturation is required for bacterial growth. PLoS Pathog 15:e1007855. doi: 10.1371/journal.ppat.1007855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Howe D, Melnicakova J, Barak I, Heinzen RA. 2003. Fusogenicity of the Coxiella burnetii parasitophorous vacuole. Ann N Y Acad Sci 990:556–562. doi: 10.1111/j.1749-6632.2003.tb07426.x. [DOI] [PubMed] [Google Scholar]
- 16.Veras PS, de Chastellier C, Moreau MF, Villiers V, Thibon M, Mattei D, Rabinovitch M. 1994. Fusion between large phagocytic vesicles: targeting of yeast and other particulates to phagolysosomes that shelter the bacterium Coxiella burnetii or the protozoan Leishmania amazonensis in Chinese hamster ovary cells. J Cell Sci 107:3065–3076. [DOI] [PubMed] [Google Scholar]
- 17.Newton HJ, Kohler LJ, McDonough JA, Temoche-Diaz M, Crabill E, Hartland EL, Roy CR. 2014. A screen of Coxiella burnetii mutants reveals important roles for Dot/Icm effectors and host autophagy in vacuole biogenesis. PLoS Pathog 10:e1004286. doi: 10.1371/journal.ppat.1004286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kohler LJ, Reed SCO, Sarraf SA, Arteaga DD, Newton HJ, Roy CR. 2016. Effector protein Cig2 Decreases host tolerance of infection by directing constitutive fusion of autophagosomes with the Coxiella-containing vacuole. mBio 7:e01127-16. doi: 10.1128/mBio.01327-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stead CM, Omsland A, Beare PA, Sandoz KM, Heinzen RA. 2013. Sec-mediated secretion by Coxiella burnetii. BMC Microbiol 13:222. doi: 10.1186/1471-2180-13-222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boonstra B, French CE, Wainwright I, Bruce NC. 1999. The udhA gene of Escherichia coli encodes a soluble pyridine nucleotide transhydrogenase. J Bacteriol 181:1030–1034. doi: 10.1128/JB.181.3.1030-1034.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cao Z, Song P, Xu Q, Su R, Zhu G. 2011. Overexpression and biochemical characterization of soluble pyridine nucleotide transhydrogenase from Escherichia coli. FEMS Microbiol Lett 320:9–14. doi: 10.1111/j.1574-6968.2011.02287.x. [DOI] [PubMed] [Google Scholar]
- 23.Sauer U, Canonaco F, Heri S, Perrenoud A, Fischer E. 2004. The soluble and membrane-bound transhydrogenases UdhA and PntAB have divergent functions in NADPH metabolism of Escherichia coli. J Biol Chem 279:6613–6619. doi: 10.1074/jbc.M311657200. [DOI] [PubMed] [Google Scholar]
- 24.Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Newton P, Latomanski EA, Newton HJ. 2016. Applying fluorescence resonance energy transfer (FRET) to examine effector translocation efficiency by Coxiella burnetii during siRNA silencing. J Vis Exp :e54210. doi: 10.3791/54210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. 2001. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 27.Hofmann K. 1993. TMbase–a database of membrane spanning protein segments. Biol Chem Hoppe Seyler 374:166. [Google Scholar]
- 28.Kuba M, Neha N, De Souza DP, Dayalan S, Newson JPM, Tull D, McConville MJ, Sansom FM, Newton HJ. 2019. Coxiella burnetii utilizes both glutamate and glucose during infection with glucose uptake mediated by multiple transporters. Biochem J 476:2851–2867. doi: 10.1042/BCJ20190504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez E, Allombert J, Cantet F, Lakhani A, Yandrapalli N, Neyret A, Norville IH, Favard C, Muriaux D, Bonazzi M. 2016. Coxiella burnetii effector CvpB modulates phosphoinositide metabolism for optimal vacuole development. Proc Natl Acad Sci U S A 113:E3260–E3269. doi: 10.1073/pnas.1522811113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vincent CD, Friedman JR, Jeong KC, Buford EC, Miller JL, Vogel JP. 2006. Identification of the core transmembrane complex of the Legionella Dot/Icm type IV secretion system. Mol Microbiol 62:1278–1291. doi: 10.1111/j.1365-2958.2006.05446.x. [DOI] [PubMed] [Google Scholar]
- 31.Barreteau H, Kovač A, Boniface A, Sova M, Gobec S, Blanot D. 2008. Cytoplasmic steps of peptidoglycan biosynthesis. FEMS Microbiol Rev 32:168–207. doi: 10.1111/j.1574-6976.2008.00104.x. [DOI] [PubMed] [Google Scholar]
- 32.Fielden LF, Moffatt JH, Kang Y, Baker MJ, Khoo CA, Roy CR, Stojanovski D, Newton HJ. 2017. A farnesylated Coxiella burnetii effector forms a multimeric complex at the mitochondrial outer membrane during infection. Infect Immun 85:e01046-16. doi: 10.1128/IAI.01046-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McDonough JA, Newton HJ, Klum S, Swiss R, Agaisse H, Roy CR. 2013. Host pathways important for Coxiella burnetii infection revealed by genome-wide RNA interference screening. mBio 4:e00606-12. doi: 10.1128/mBio.00606-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Larson CL, Beare PA, Heinzen RA. 2019. Dependency of Coxiella burnetii type 4B secretion on the chaperone IcmS. J Bacteriol 201:e00431-19. doi: 10.1128/JB.00431-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beare PA, Jeffrey BM, Long CM, Martens CM, Heinzen RA. 2018. Genetic mechanisms of Coxiella burnetii lipopolysaccharide phase variation. PLoS Pathog 14:e1006922. doi: 10.1371/journal.ppat.1006922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoover TA, Culp DW, Vodkin MH, Williams JC, Thompson HA. 2002. Chromosomal DNA deletions explain phenotypic characteristics of two antigenic variants, phase II and RSA 514 (crazy), of the Coxiella burnetii nine mile strain. Infect Immun 70:6726–6733. doi: 10.1128/iai.70.12.6726-2733.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Norville IH, Hartley MG, Martinez E, Cantet F, Bonazzi M, Atkins TP. 2014. Galleria mellonella as an alternative model of Coxiella burnetii infection. Microbiology 160:1175–1181. doi: 10.1099/mic.0.077230-0. [DOI] [PubMed] [Google Scholar]
- 38.Howe D, Shannon JG, Winfree S, Dorward DW, Heinzen RA. 2010. Coxiella burnetii phase I and II variants replicate with similar kinetics in degradative phagolysosome-like compartments of human macrophages. Infect Immun 78:3465–3474. doi: 10.1128/IAI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoker MG, Fiset P. 1956. Phase variation of the Nine Mile and other strains of Rickettsia burnetii. Can J Microbiol 2:310–321. doi: 10.1139/m56-036. [DOI] [PubMed] [Google Scholar]
- 40.Moos A, Hackstadt T. 1987. Comparative virulence of intra- and interstrain lipopolysaccharide variants of Coxiella burnetii in the guinea pig model. Infect Immun 55:1144–1150. doi: 10.1128/IAI.55.5.1144-1150.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, Gilk SD, Williams KP, Sobral BW, Kupko JJ III, Porcella SF, Samuel JE, Heinzen RA. 2009. Comparative genomics reveal extensive transposon-mediated genomic plasticity and diversity among potential effector proteins within the genus Coxiella. Infect Immun 77:642–656. doi: 10.1128/IAI.01141-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Isaac DT, Isberg R. 2014. Master manipulators: an update on Legionella pneumophila Icm/Dot translocated substrates and their host targets. Future Microbiol 9:343–359. doi: 10.2217/fmb.13.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Allombert J, Jaboulay C, Michard C, Andréa C, Charpentier X, Vianney A, Doublet P. 2019. Orchestrated delivery of Legionella effectors by the Icm/Dot secretion system. bioRxiv. doi: 10.1101/754762. [DOI] [PubMed]
- 44.Jaton K, Peter O, Raoult D, Tissot JD, Greub G. 2013. Development of a high throughput PCR to detect Coxiella burnetii and its application in a diagnostic laboratory over a 7-year period. New Microbes New Infect 1:6–12. doi: 10.1002/2052-2975.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saunders EC, Ng WW, Kloehn J, Chambers JM, Ng M, McConville MJ. 2014. Induction of a stringent metabolic response in intracellular stages of Leishmania mexicana leads to increased dependence on mitochondrial metabolism. PLoS Pathog 10:e1003888. doi: 10.1371/journal.ppat.1003888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.O'Callaghan S, De Souza DP, Isaac A, Wang Q, Hodkinson L, Olshansky M, Erwin T, Appelbe B, Tull DL, Roessner U, Bacic A, McConville MJ, Likić VA. 2012. PyMS: a Python toolkit for processing of gas chromatography-mass spectrometry (GC-MS) data. Application and comparative study of selected tools. BMC Bioinformatics 13:115. doi: 10.1186/1471-2105-13-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Masukagami Y, De Souza DP, Dayalan S, Bowen C, O’Callaghan S, Kouremenos K, Nijagal B, Tull D, Tivendale KA, Markham PF, McConville MJ, Browning GF, Sansom FM. 2017. Comparative metabolomics of Mycoplasma bovis and Mycoplasma gallisepticum reveals fundamental differences in active metabolic pathways and suggests novel gene annotations. mSystems 2:e00055-17. doi: 10.1128/mSystems.00055-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011. Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated by an improved axenic growth medium. Appl Environ Microbiol 77:3720–3725. doi: 10.1128/AEM.02826-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Latomanski EA, Newton HJ. 2018. Interaction between autophagic vesicles and the Coxiella-containing vacuole requires CLTC (clathrin heavy chain). Autophagy 14:1710–1725. doi: 10.1080/15548627.2018.1483806. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.