

Enteroaggregative Escherichia coli (EAEC) is an E. coli pathotype associated with diarrhea and growth faltering. EAEC virulence gene expression is controlled by the autoactivated AraC family transcriptional regulator, AggR. AggR activates transcription of a large number of virulence genes, including Aar, which in turn acts as a negative regulator of AggR itself. Aar has also been shown to affect expression of E. coli housekeeping genes, including H-NS, a global regulator that acts at multiple promoters and silences AT-rich genes (such as those in the AggR regulon).
KEYWORDS: ANR, Aar, AggR, enteroaggregative E. coli
ABSTRACT
Enteroaggregative Escherichia coli (EAEC) is an E. coli pathotype associated with diarrhea and growth faltering. EAEC virulence gene expression is controlled by the autoactivated AraC family transcriptional regulator, AggR. AggR activates transcription of a large number of virulence genes, including Aar, which in turn acts as a negative regulator of AggR itself. Aar has also been shown to affect expression of E. coli housekeeping genes, including H-NS, a global regulator that acts at multiple promoters and silences AT-rich genes (such as those in the AggR regulon). Although Aar has been shown to bind both AggR and H-NS in vitro, functional significance of these interactions has not been shown in vivo. In order to dissect this regulatory network, we removed the complex interdependence of aggR and aar by placing the genes under the control of titratable promoters. We measured phenotypic and genotypic changes on downstream genes in EAEC strain 042 and E. coli K-12 strain DH5α, which lacks the AggR regulon. In EAEC, we found that low expression of aar increases aafA fimbrial gene expression via H-NS; however, when aar is more highly expressed, it acts as a negative regulator via AggR. In DH5α, aar affected expression of E. coli genes in some cases via H-NS and in some cases independent of H-NS. Our data support the model that Aar interacts in concert with AggR, H-NS, and possibly other regulators and that these interactions are likely to be functionally significant in vivo.
INTRODUCTION
Enteroaggregative Escherichia coli (EAEC) is a common cause of traveler’s diarrhea in industrial and developing countries and has been linked to growth failure in children (1–4). Host colonization of EAEC is attributed to the presence of virulence genes that are controlled by AggR, a member of the AraC family of bacterial transcriptional regulators (5–7). A small protein named Aar (AggR activated regulator), whose expression is activated by AggR, has been described as a negative regulator of AggR (5, 8). Further characterization of Aar found that it belongs to a large family of proteins termed AraC negative regulators (ANR). The ANR family is found in hundreds of Gram-negative pathogens, and phylogenetically close homologs are able to complement function in ANR mutants (8).
In addition to regulating AggR expression, Aar has also been found to regulate genes encoding proteins outside of the AggR regulon, such as H-NS (9). H-NS is a global regulatory protein, which usually acts as a repressor at a wide variety of promoters and genes that are AT-rich and therefore intrinsically curved (10, 11). H-NS, AggR, and Aar have a complex dynamic. H-NS transcriptionally silences AraC transcriptional regulators; however, AraC transcriptional regulators may act as antirepressors that counteract H-NS silencing in selected environments (12, 13). It has been hypothesized that regulation of AggR and H-NS by Aar is via Aar binding directly to either AggR or H-NS. Aar has been shown to bind both AggR and H-NS via surface plasmon resonance, the bacterial two-hybrid system, pulldown assays, and electrophoretic mobility shift assays (9, 14); however, functional significance of these interactions has not been elucidated.
We have previously postulated that Aar could be acting on AggR through direct formation of Aar/AggR complexes and/or through the formation of Aar/H-NS complexes, which could act to lift H-NS silencing of the regulon (9). The benefit to the bacterium of regulating virulence genes by two different interactions effected by one protein is unclear. In this study, we sought to better understand the mechanism by which Aar downregulates AggR-regulated genes and the functional significance (if any) of the hypothesized Aar/AggR and Aar/H-NS binding events.
RESULTS
Independent expression of aggR and aar affects biofilm formation.
Although we have observed that Aar binds both AggR and H-NS (9, 14), the mutual interdependence of these genes obfuscates the functional implications of these putative protein-protein interactions. Specifically, (i) H-NS has been shown to bind to AT-rich structural genes (10, 11), which include both AggR and Aar; (ii) AggR is the activator of Aar gene expression (5); (iii) Aar has been shown to repress AggR expression (8); and (iv) transcriptomic data suggested that Aar may activate expression of the H-NS-encoding gene (9). Therefore, in order to better dissect the roles and contributions of these interdependent regulators in the control of gene expression in EAEC, we assembled systems in which expression of the genes could be controlled independently. Accordingly, we first constructed a derivative of EAEC strain 042 that harbored mutations in aar and aggR and then introduced plasmids that carried the structural genes of aar and aggR under independently controllable promoters.
Plasmid pPrham-aar (designated here paar) features the aar gene under the control of the rhamnose promoter; the plasmid is built on a pBR322 backbone (pMB1 replicon) and confers resistance to ampicillin. In preliminary experiments, we demonstrated that there were growth differences between LB and LB with rhamnose after 4 h, likely due to rhamnose catabolism, so all experiments were performed at 3 h postinduction unless stated otherwise (see Fig. S1 in the supplemental material). Similarly, plasmid pPlacz-aggR (designated here paggR) features aggR under the lacZ promoter and is built on a pACYC177 backbone (p15A replicon) that confers resistance to kanamycin (km).
In EAEC, AggR production induces the expression of aafA, leading to the formation of a bacterial biofilm (5, 15); thus, biofilm formation is a ready phenotypic screen for aggR expression. Strain 042Δaar ΔaggR did not produce an observable biofilm on a polystyrene substratum after 3 h of incubation at 37°C (data not shown). To assess the effect of paggR expression in strain 042Δaar ΔaggR, we subjected the strain to increasing concentrations of isopropyl-β-d-thiogalactopyranoside (IPTG). As predicted, we observed a dose-dependent increase in biofilm formation (Fig. 1A).
FIG 1.
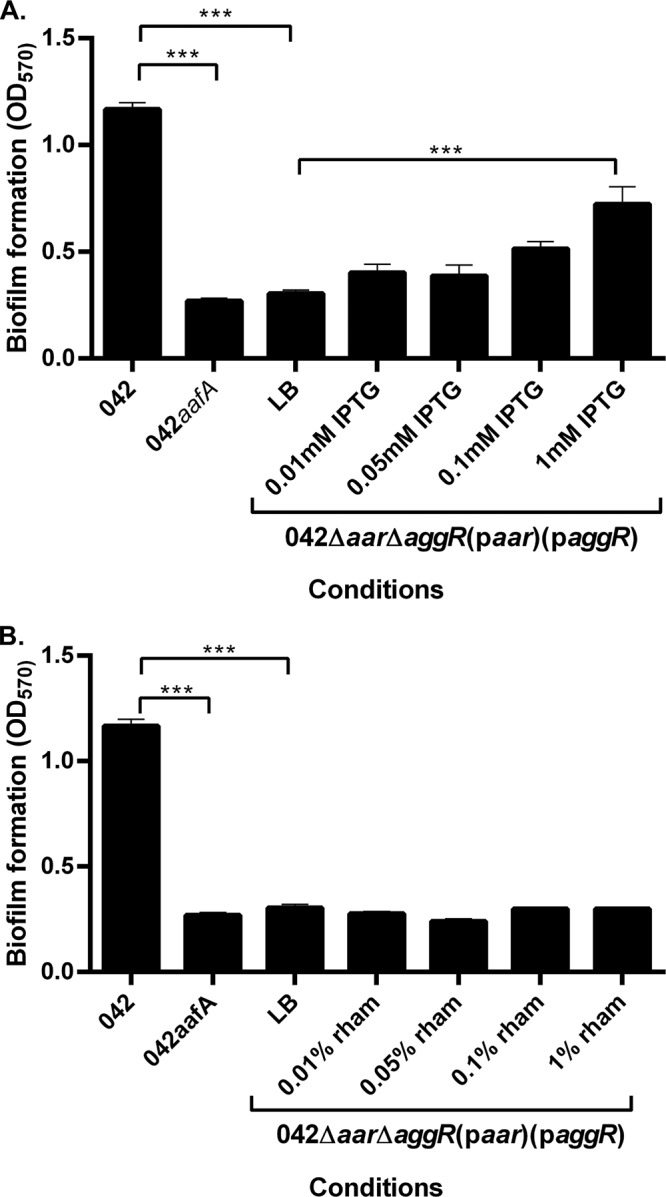
Biofilm formation in the presence and absence of inducer molecules. (A) Biofilm formation was measured using crystal violet staining after 3 h in 042 and 042ΔaafA in DMEM high glucose and in 042Δaar ΔaggR(paar)(paggR) in LB with varying concentrations of IPTG. (B) Biofilm formation was measured using crystal violet staining after 3 h in 042 and 042ΔaafA in DMEM high glucose and in 042Δaar ΔaggR(paar)(paggR) in LB with varying concentrations of rhamnose. Biofilm data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
Expression of aar in 042Δaar ΔaggR via introduction of plasmid paar was affected by increasing concentrations of rhamnose; such a construct did not display expression of aggR. As predicted from our previous observations that aafA and resultant biofilm formation requires AggR (5, 15), we observed no change in biofilm formation in this construct under conditions of increasing rhamnose concentrations (Fig. 1B).
To confirm that changes seen in biofilm formation correlated with changes in the expression of aggR, aar, and aafA, quantitative reverse transcriptase PCR (qRT-PCR) was performed for these gene transcripts. We discovered that inducer concentrations lower than that which produced observable biofilms were found to maximize mRNA transcript production by qRT-PCR; induction curves for qRT-PCR demonstrated that lower concentrations of the inducers were necessary to detect differences at the RNA level (Fig. 2A and B). The combination of aar and aggR expression induced by the lower concentrations of rhamnose and IPTG, respectively, leads to measurable changes in aafA expression (Fig. 2C). Gene expression of aggR, aar, and aafA confirmed that biofilm formation parallels aafA gene expression (Fig. 2A to C).
FIG 2.
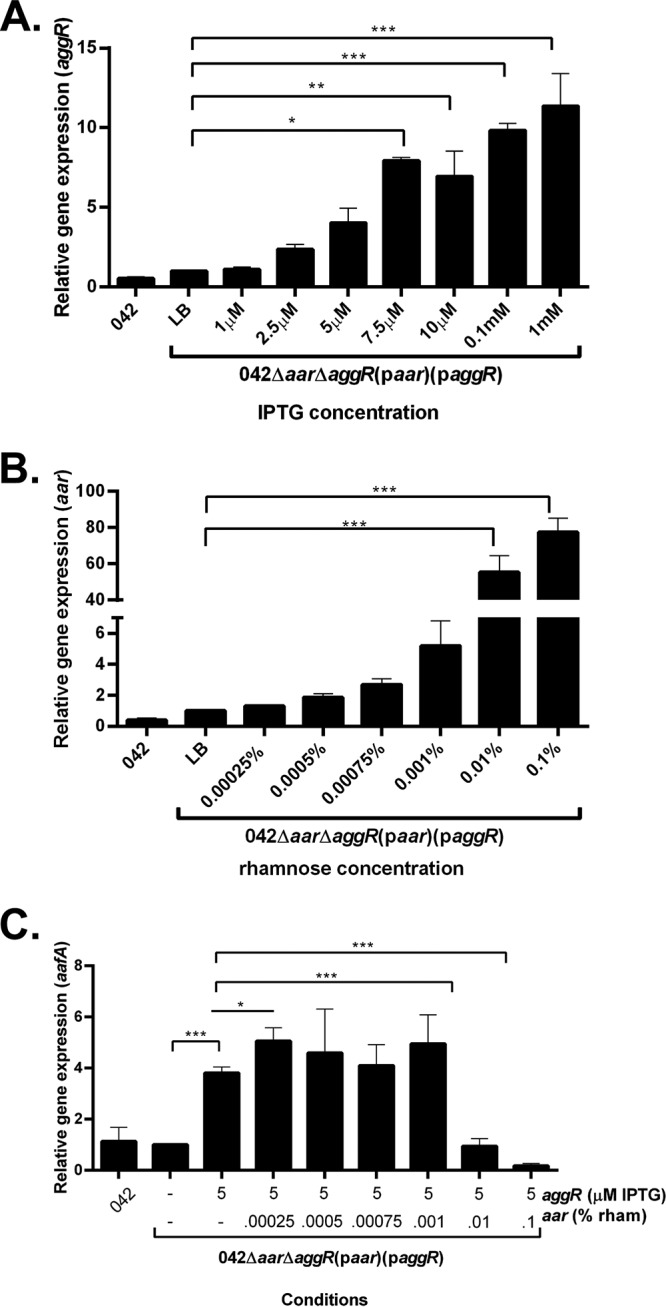
Gene expression after induction of aggR by IPTG or aar by rhamnose. (A) Three-hour aggR expression measured using qRT-PCR on 042 in DMEM high glucose and 042Δaar ΔaggR(paar)(paggR) in LB with varying concentrations of IPTG. (B) Three-hour aar expression measured using qRT-PCR on 042 in DMEM high glucose and 042Δaar ΔaggR(paar)(paggR) in LB with varying concentrations of rhamnose. (C) Three-hour aafA expression measured using qRT-PCR on 042 in DMEM high glucose and 042Δaar ΔaggR(paar)(paggR) in LB with 0.005 mM IPTG and a range of concentrations of rhamnose. qRT-PCR data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005). The difference between aggR (5 μM) and aggR (5 μM) with aar (0.00025%) was found to be significant with a two-tailed paired t test.
Aar has a paradoxical effect on aafA expression.
We have previously observed that Aar serves as a negative regulator of the AggR regulon and that the two proteins bind to each other in vitro (8, 14). As predicted from this model, we observed a dose-dependent decrease in biofilm formation with increasing expression of aar (increasing concentrations of rhamnose) under conditions of constant aggR expression (Fig. 3A, colored bars with same fill pattern). qRT-PCR measurements of aafA transcription support that this decrease in biofilm formation was associated with a decrease in aafA expression (Fig. 3B, colored bars with same fill pattern) under constant aggR expression and increasing aar expression (see Fig. S2A and B, colored bars with same fill pattern, in the supplemental material).
FIG 3.

Biofilm formation and gene expression of aafA in 042Δaar ΔaggR titrated with aar and aggR. (A) Biofilm growth at 3 h postinduction with increasing concentration of IPTG and rhamnose. aggR expression was induced with 0.01 mM IPTG (horizontal fill pattern), 0.1 mM IPTG (diagonal fill pattern), or 1 mM IPTG (vertical fill pattern). aar expression was induced with 0.01% rhamnose (rham) (blue), 0.05% rham (red), or 0.1% rham (green). (B) qRT-PCR analysis of aafA using titratable aar and aggR. aggR expression was induced with either 5 μM IPTG (horizontal fill pattern) or 7.5 μM IPTG (diagonal fill pattern). aar expression was induced with 0.00025% rham (blue), 0.01% rham (red), or 0.1% rham (green). Biofilm data and qRT-PCR data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
Unexpectedly, given our model, we observed a paradoxical effect: in the presence of aggR, low levels of aar expression lead first to increased biofilm (Fig. 3A, dark gray bars to blue bars) followed by the expected dose-dependent decrease in biofilm formation at higher aar concentrations. This effect was not due to the effects of the inducers themselves (see Fig. S3A and B in the supplemental material).
The paradoxical effect of Aar requires H-NS.
Transcriptome sequencing (RNA-seq) data suggested that Aar has an effect on expression of the histone-like protein H-NS, and binding assays suggested that the two proteins physically interact (9). Like many members of the AraC family of transcriptional activators, AggR and the genes that it regulates (i.e., aafA) are repressed by H-NS (12, 13). Members of the AraC family of transcriptional regulators are thought to counteract H-NS-induced silencing in select environments (12, 13). The paradoxical effect of Aar expression on AggR-dependent aafA expression suggested the action of another regulator, and we hypothesized that this regulator was H-NS.
To test this hypothesis, we constructed 042Δaar ΔaggR Δhns, transformed the strain with paar and paggR, and measured biofilm production with varying IPTG and rhamnose concentrations. 042Δaar ΔaggR Δhns had a growth defect compared to that of 042Δaar ΔaggR; growth of 042Δaar ΔaggR Δhns at 5 h produced an optical density at 600 nm (OD600) similar to that observed in 042Δaar ΔaggR at 3 h (see Fig. S4 in the supplemental material). Rhamnose catabolism had no effect on the growth phase of 042Δaar ΔaggR Δhns at 5 h (Fig. S4). Similar to 042Δaar ΔaggR, a low-level increase in aar led to a decrease in biofilm growth (Fig. 4A, colored bars with same fill pattern). However, in the absence of hns, the increase of biofilm formation at low levels of aar was no longer observed (Fig. 4A, dark gray bars to blue bars). The effect of low levels of Aar on biofilm production was rescued when hns was restored (Fig. 4B). qRT-PCR analysis confirmed that in 042Δaar ΔaggR Δhns, under conditions of constant aggR expression (see Fig. S5A in the supplemental material) but increasing aar expression (Fig. S5B), the direct dose-dependent expression of aafA by aggR is maintained as is the negative effect by aar (Fig. 4C); however, the paradoxical effect of aar on aafA is lost in the absence of H-NS (Fig. 4C). As seen in the biofilm assay (Fig. 4B), qRT-PCR supported that the restoration of hns rescued the paradoxical effect (Fig. 4D) under conditions of constant aggR expression (Fig. S5C) but increasing aar expression (Fig. S5D). The paradoxical effect was only seen in the presence of AggR production; i.e., aar expression by itself did not affect aafA expression regardless of the presence of hns. Taken together, these data suggest a tripartite model of AggR/Aar/H-NS interaction, consistent with our in vitro observation that Aar binds to both AggR and H-NS.
FIG 4.

Biofilm formation and expression of aafA in 042Δaar ΔaggR Δhns and hns repair titrated with aar and aggR. (A) Biofilm growth in 042Δaar ΔaggR Δhns at 5 h postinduction with increasing concentration of IPTG and rhamnose. (B) Biofilm growth in hns repair at 3 h postinduction with increasing concentration of IPTG and rhamnose. aggR expression was induced with 0.01 mM IPTG (horizontal fill pattern), 0.1 mM IPTG (diagonal fill pattern), or 1 mM IPTG (vertical fill pattern). aar expression was induced with 0.01% rham (blue), 0.05% rham (red), or 0.1% rham (green). (C) qRT-PCR analysis of aafA using titratable aar and aggR in 042Δaar ΔaggR Δhns after 5 h. (D) qRT-PCR analysis of aafA using titratable aar and aggR in the hns repaired 042Δaar ΔaggR after 3 h. aggR expression was induced with either 5 μM IPTG (horizontal fill pattern) or 7.5 μM IPTG (diagonal fill pattern). aar expression was induced with 0.00025% rham (blue), 0.01% rham (red), or 0.1% rham (green). Biofilm data and qRT-PCR data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
aggR diminishes the aar-induced upregulation of non-AggR-regulated genes in E. coli K-12 via aar.
The results of our AggR and Aar controllable gene expression studies reveal dose-dependent effects of the two regulators consistent with the previously published model of AggR/Aar protein-protein binding (14), i.e., that aafA expression may depend on the concentration of AggR unbound to Aar. If the mechanism is in fact due to protein-protein interaction, then binding of AggR to Aar might also reduce the activity of the latter protein. We sought to utilize a simplified system with which to probe hypothetical interference of Aar activity by AggR. Such an effect would add another regulatory dimension to the tripartite protein-protein interaction system.
We have previously observed in strain 042 that Aar activates housekeeping genes that are AggR independent (9); if true, we would hypothesize that expression of such genes would similarly be affected in a K-12 background. Interrogating this effect in K-12 would eliminate the effect of AggR on other genes of the AggR regulon, which does not exist in K-12.
We transformed E. coli DH5α separately with paar and paggR or their corresponding empty vector controls (pBR322 and pACYC177, respectively). To interrogate a possible inhibitory effect of AggR on Aar, we chose to study two chromosomal E. coli core genes previously shown to be affected by aar in strain 042 (orf1228 and orf2223) (9). As predicted, we observed that in K-12 strain DH5α, when aar was expressed in paar, the expression levels of orf1228 and orf2223 were increased (Fig. 5A). The expression of aggR alone in DH5α had no effect on the expression of either target gene in the absence of aar (Fig. 5A).
FIG 5.

The effect of aar and aggR on gene expression in DH5α transformed with paar and/or paggR/paggR-D. (A) DH5α was transformed with paar and paggR expressing full-length aggR or their corresponding empty vectors pBR322 and pACYC177, respectively. Transcriptional levels of E. coli chromosomal genes orf1228 and orf2223 were analyzed by qRT-PCR. (B) DH5α was transformed with paar and paggR-D expressing the AggR dimerization domain or their corresponding empty vectors. Transcriptional levels of E. coli chromosomal genes orf1228 and orf2223 were analyzed by qRT-PCR. RT-PCR data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
The expression of aggR simultaneously with aar caused only a small reduction to the observed increase in gene expression (Fig. 5A). Given that AggR is a DNA-binding protein, we repeated this experiment using an aggR construct comprising only the AggR dimerization domain (amino acids 69 to 181), therefore, lacking the DNA binding helix-turn-helix C-terminal region (pPlacZ-aggR-D is referred to as paggR-D) (14); we previously reported that the AggR dimerization domain binds Aar in the bacterial two-hybrid system (14). As seen with the full-length aggR, the expression of the aggR-D in DH5α had no effect on the expression of orf1228 and orf2223 (Fig. 5B); when aggR-D and aar were both expressed in DH5α, the expression levels of the two queried genes were both significantly decreased compared to the level of expression when aar was expressed alone (Fig. 5B), suggesting that the expression of aggR-D in this system affects the expression of orf1228 and orf2223 via aar, consistent with protein-protein interaction of the two proteins. The expression levels of aar and aggR were similar whether expressed alone or together (see Fig. S6A in the supplemental material); the same was true for aar and aggR-D (Fig. S6B).
aar upregulates gene expression in E. coli K-12 via hns.
Several of the Aar-controlled genes affected in 042 that are AggR independent are thought to be under H-NS control based on previous literature (9, 16–18). We hypothesize that the effect of Aar on these genes is via the proposed model of Aar/H-NS protein-protein binding.
We used DH5α and DH5αΔhns transformed with paar to probe the expression of ompX, a gene known to be under the regulation of H-NS (17). The expression of ompX was increased in the absence of hns, confirming that ompX expression in DH5α is regulated by hns (Fig. 6A). In DH5α, we found that the expression of ompX was significantly increased when aar was induced (Fig. 6A). However, when aar was induced in DH5αΔhns, there was no change in the expression of ompX (Fig. 6A), suggesting that hns is required for the effect of aar on ompX.
FIG 6.

The effect of aar on gene expression in the presence or absence of hns in DH5α. (A) DH5α and DH5αΔhns were transformed with paar or its corresponding empty vector pBR322. Transcriptional levels of ompX were analyzed by qRT-PCR. (B) DH5αΔhns was transformed with paar and phns or their corresponding empty vectors pBR322 and pKNT25, respectively. Transcriptional levels of orf1228 were analyzed by qRT-PCR. RT-PCR data are representative of at least three independent experiments. Asterisks indicate significant differences by ANOVA (*, P < 0.05; **, P < 0.005; ***, P < 0.0005).
hns diminishes the aar-induced upregulation of non-H-NS-regulated genes in E. coli K-12 via aar.
We reasoned that if Aar bound H-NS in the bacterium, the interactions of the two genes may be mutually interfering, and the activity of Aar in the bacterium may be diminished by expression of hns, similarly to what was demonstrated with AggR (Fig. 5A and B). We, therefore, sought to use the DH5α system to probe hypothetical interference of Aar activity by H-NS.
We transformed E. coli DH5αΔhns separately with paar and our previously published pKNTHNS (designated here phns) (9) or their corresponding empty vector controls pBR322 and pKNT25, respectively. We determined that orf1228 was not affected by H-NS expression (see Fig. S7 in the supplemental material); therefore, we chose to probe the expression of this gene to interrogate a possible inhibitory effect of H-NS on Aar activity. We performed the experiment in DH5αΔhns to remove any effects that the native hns could have on the system. When aar was expressed in DH5αΔhns, the expression of orf1228 was increased (Fig. 6B). The expression of hns alone had no effect on the expression of orf1228 in the absence of aar (Fig. 6B). As suspected, when hns and aar are expressed together, the effect of aar on orf1228 is significantly reduced (Fig. 6B). This suggests that the expression of hns is affecting the expression of orf1228 via aar, which is consistent with the proposed model of a direct interaction of the two proteins.
DISCUSSION
Although binding of Aar to AggR and H-NS has been previously demonstrated in artificial systems, we have not yet provided evidence that either of these binding phenomena have a functional role in the bacterium. In this work, we constructed a series of experimental systems to probe potential interrelationships among Aar, AggR, and H-NS, regulators that are expected to have mutual interdependence. By using a system in which we remove the transcriptional interdependence of aar and aggR, our data suggest functional roles for Aar binding to both AggR and H-NS in EAEC 042. In addition, employing E. coli DH5α, we observe evidence for Aar function in the absence of AggR, both via H-NS and potentially other regulators of the core E. coli genome.
As predicted, increasing aar expression led to a dose-dependent decrease in aggR-regulated aafA expression and biofilm formation. This relationship was inversely reciprocal. Expression of aafA was increased by increasing aggR expression but decreased by aar; aafA expression seemed to correlate best with excess aggR abundance over the level of aar expression. Previously published data failed to reveal evidence that Aar binds to DNA (9, 14), and because we removed the transcriptional dependence of aggR and aar on one another, our data best support the model that Aar binds and sequesters AggR and that aafA expression levels would be determined by free AggR protein concentrations.
In titration experiments, we were surprised to observe a phenotypic effect on biofilm formation with low expression of aar that was contrary to our hypothesis regarding how Aar would affect the expression of AggR-regulated genes. At the lowest levels of aar expression, we observed a paradoxical increase in aafA expression, independent of aggR expression. This effect was abrogated in an hns mutant. These data suggest a potential role for dual binding of both AggR and H-NS by Aar. It is tempting to speculate that Aar has a higher affinity for H-NS than for AggR, given that aar has a positive effect on aafA through hns first and then a negative effect via aggR. Such a nuanced effect could permit early expression of aafA in vivo before the time required for cycles of aggR transcription and translation and subsequent binding to the aafA promoter. Given that there is evidence for EAEC infection of both the duodenal (19) and the colonic mucosae (20), this dual regulation could provide distinct pathogenic timing.
It has previously been shown that Aar acts upon H-NS-regulated promoters differentially (9). Due to the low expression of aar that is necessary to observe changes in aafA through hns, our data suggest that Aar may remove H-NS from the aafA gene (possibly the structural gene itself), thereby permitting AggR to upregulate expression. This affinity for removing H-NS may extend to other AggR-regulated promoters. It is possible that Aar could add specificity to the removal of H-NS at AggR-regulated genes over other H-NS regulated genes, thus allowing for a timed derepression of those specific genes. The underlying mechanism of how Aar is leading to differential expression of various genes is unclear.
H-NS is a global regulator of E. coli gene expression (10, 11), and the putative binding of Aar to H-NS suggested that Aar may have global effects on EAEC gene expression beyond the AggR regulon. For this to prevail in vivo, one would expect effects of Aar on gene expression in E. coli K-12, which is devoid of the AggR regulon; we not only observed such effects in a K-12 system, but our data suggest still more global complexity accompanying aar expression. Our observations in a K-12 system rule out the need for a pathogen-specific intermediary protein.
Although we posit that Aar acts via protein-protein interaction, demonstration of protein-protein binding is not definitive evidence that this phenomenon occurs in vivo. The use of multiple assays suggested AggR/Aar binding: surface plasmon resonance, bacterial two-hybrid system, and electrophoretic mobility shift assay (EMSA) (14). The in vivo data presented here confirm interrelationships among these regulatory proteins in ways that would be difficult to ascribe to alternate mechanisms. Importantly, the expression of aggR alone had no effect on orf1228 and orf2223; therefore, the decrease in expression of these two genes in the presence of both aar and aggR suggests that AggR may be binding free Aar and preventing Aar from activating the genes. By demonstrating an aar effect on gene expression through aggR (in EAEC 042) and an aggR effect on gene expression through aar (in DH5α), our data support the previously published model that the effect of aar and aggR is through protein-protein binding.
Targeting the E. coli gene ompX, previously reported to be under H-NS control (17), we confirmed that expression of aar induced expression of ompX in an hns-dependent manner. Surprisingly, however, our data suggest that the Aar effect on orf1228 expression persists even in an hns mutant, suggesting that Aar may act in concert with still another regulator beyond AggR and H-NS. E. coli possesses additional histone-like proteins, which may be responsible for the effect of aar on orf1228 expression, and these are the targets of ongoing research in our laboratory. As we predicted, expression of the AggR dimerization domain (which does not occur naturally in K-12 and does not bind DNA but has been shown to bind Aar in vitro) demonstrated an Aar inhibitory effect in a K-12 background. These data strongly support the hypothetical model wherein AggR and Aar bind directly, thereby inhibiting activity of both proteins. The effects we observed show interdependence of AggR, Aar, and H-NS; however, the data do not prove that the mechanism is direct protein-protein interactions.
Based on our data and previous studies (8, 9, 14), we propose a model to illustrate a dual function of Aar in EAEC virulence gene expression (Fig. 7). In abiotic environments, H-NS binds AT-rich genes and silences their expression (i.e., aggR and aggR-regulated genes). When the bacteria reach the host, temperature change and inducer molecules induce aggR expression. AggR upregulates aar expression, and at early stages of induction when aar expression is low but detectable, Aar binds and relieves H-NS silencing from AggR-regulated genes. This results in immediate upregulation of previously silenced genes by AggR. As the expression of aar increases, Aar begins to bind AggR in addition to H-NS, preventing AggR dimerization and therefore reducing activation of aggR-regulated genes.
FIG 7.

Proposed mechanism of AggR-Aar-Hns interaction in vivo. When the concentration of Aar (red circles) is low, Aar removes H-NS (gray ovals) repression at AT-rich genes. This allows AggR (green ovals) to abundantly upregulate gene expression. When the concentration of Aar is high, Aar removes H-NS repression but also binds to AggR. AggR is still able to upregulate gene expression but not as abundantly.
The data presented in this paper support the model that Aar is binding to both AggR and H-NS and that both interactions have functional significance. In EAEC strain 042, Aar has a dual function in virulence gene expression. First, when present at low concentrations, Aar removes the inhibitory effect of H-NS on fimbrial gene expression; and then when the concentration of Aar increases, Aar acts as a negative regulator, turning off AggR-activated virulence genes. Our data suggest that not only is Aar an antiactivator, but it can also act as an antirepressor. The role of Aar on genes of the core E. coli genome is more difficult to decipher but could play a role in the switch from the nonpathogenic to the pathogenic lifestyle. Further research will address the concerted action of this complex regulatory circuitry.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions.
Bacterial strains and plasmids used in this study can be found in Table S1 in the supplemental material. Bacteria were grown in Luria Broth (LB) and Dulbecco’s modified Eagle’s medium with 0.4% glucose (DMEM high glucose) (Gibco, Grand Island, NY) as previously described (5). When indicated, medium was supplemented with carbenicillin (100 μg/ml) and/or kanamycin (50 μg/ml). For phenotypic titration studies, 0.01 mM or 1 mM IPTG and 0.01%, 0.05%, or 0.1% rhamnose were added as indicated below. For transcriptional studies, 5 μM or 7.5 μM IPTG and 0.00025%, 0.01%, or 0.1% rhamnose were added as indicated below. Inducer concentrations were selected after a range of concentrations was screened to determine which had detectable effects on the expression of aggR or aar.
Mutagenesis of aggR and hns in 042Δaar was accomplished by using lambda red technology (21). The loci (41,080 to 41,877 and 1,376,831 to 1,377,244; GenBank accession number FN554767.1) in 042Δaar were replaced with the kanamycin (km) resistance marker as previously reported (21). The 042Δaar ΔaggR and 042Δaar ΔaggR Δhns strains were identified by PCR using specific primers for aggR, hns, and a km resistance marker (see Table S2 in the supplemental material). Deletion strains were cured of the km resistance using pCP20 as previously reported (21). Repair of hns in 042Δaar ΔaggR Δhns was done by using the lambda red recombination protocol to recombine a PCR product of hns with large flanking regions from 042 with the 042Δaar ΔaggR Δhns km-resistant strain and testing for recombination via km sensitivity. Primers for recombination of hns and screening for the repair are shown in Table S2.
Mutagenesis of hns in DH5α was accomplished by using lambda red technology, and mutants were PCR screened and cured of km resistance as stated above.
Titratable expression of aggR and aar.
For the independent expression of aggR and aar, plasmids pPlacZ-aggR, pPrham-aar, and pPlacZ-aggR-D were generated in this study (Table S1). Briefly, 1,086-bp fragments containing the lacZ promoter region, the entire aggR gene, a hemagglutination (HA) tag, a termination sequence, and flanked by restriction enzyme sites were synthesized by Genewiz Inc. by fragmentGENE synthesis. The synthesized fragment was inserted into BamHI and PstI sites in pACYC177; the resulting plasmid was designated pPlacZ-aggR. pPlacz-aggR-D was similar to pPlacZ-aggR but only contains the dimerization site of aggR, comprising amino acid residues 69 to 181. pPrham-aar was generated similarly but harbored a 548-bp fragment containing the rhamnose promoter region, the entire aar gene, a 6×histidine tag, a termination sequence, and was flanked by restriction enzyme sites. The synthesized fragment was inserted into the BamHI and HindIII sites in pBR322.
Biofilm production.
The biofilm assay previously described by Sheikh et al. (15) was modified. Briefly, bacterial strains were grown in LB overnight at 37°C shaking. Overnight cultures of wild-type (WT) 042 and 042ΔaafA were diluted 1:20 in DMEM high glucose, and titration constructs were diluted 1:20 in LB with or without IPTG and rhamnose and inoculated into a 24-well polystyrene plate (Sigma-Aldrich). Bacteria were incubated for 3 h at 37°C. After incubation, plates were washed two times with phosphate-buffered saline (PBS) and fixed with 75% ethanol. The fixed biofilms were dried and stained with 0.5% crystal violet (Sigma). Biofilms were washed 4 times with PBS after staining and solubilized in 95% ethanol. The absorbance was determined at 570 nm. Biofilms for 042Δaar ΔaggR Δhns were incubated for 5 h at 37°C due to impaired growth.
RNA extraction and qRT-PCR.
For quantitative reverse transcriptase PCR (qRT-PCR), EAEC strain 042, 042Δaar ΔaggR, and 042Δaar ΔaggR Δhns titration strains were grown aerobically in LB overnight at 37°C with shaking and then diluted 1:100 in DMEM high glucose or LB supplemented with IPTG and rhamnose concentrations as indicated and grown at 37°C. RNA from three biological replicates of each condition was extracted after 3 h or 5 h for the 042Δaar ΔaggR Δhns titration strain. RNA was extracted using RNAprotect bacteria reagent (Qiagen) followed by an RNeasy minikit (Qiagen). Primers used were previously published for EAEC (8, 9). qRT-PCR was performed using a one-step reaction in an ABI 7500 Fast sequence detection system (Applied Biosystems). All data were normalized to the levels of rpoA and analyzed using the comparative cycle threshold (CT) method (22). The relative quantification method was used to determine the expression levels of target genes. Statistical significance was determined by analysis of variance (ANOVA) with post hoc Tukey, and a P value of ≤0.05 was considered significant.
For qRT-PCR on aar and aggR in E. coli K-12 strain DH5α was transformed with titratable aar, aggR, and their corresponding empty vector plasmids and was grown aerobically in LB overnight at 37°C with shaking. A 1:100 dilution was made in LB with 0.1 mM IPTG and 0.1% rhamnose and grown shaking at 37°C for 3 h. RNA was extracted and qRT-PCR performed as above. Primers used were previously published for EAEC (8, 9).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (P01-AI125181 and R01-AI033096) to J.P.N. A.S.M. was supported in part by the National Institutes of Health through the University of Virginia Biodefense training grant (5T32-AI055432).
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Mohamed JA, Huang DB, Jiang Z-D, DuPont HL, Nataro JP, Belkind-Gerson J, Okhuysen PC. 2007. Association of putative enteroaggregative Escherichia coli virulence genes and biofilm production in isolates from travelers to developing countries. J Clin Microbiol 45:121–126. doi: 10.1128/JCM.01128-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Huang DB, Nataro JP, DuPont HL, Kamat PP, Mhatre AD, Okhuysen PC, Chiang T. 2006. Enteroaggregative Escherichia coli is a cause of acute diarrheal illness: a meta-analysis. Clin Infect Dis 43:556–563. doi: 10.1086/505869. [DOI] [PubMed] [Google Scholar]
- 3.Acosta GJ, Vigo NI, Durand D, Riveros M, Arango S, Zambruni M, Ochoa TJ. 2016. Diarrheagenic Escherichia coli: prevalence and pathotype distribution in children from Peruvian rural communities. Am J Trop Med Hyg 95:574–579. doi: 10.4269/ajtmh.16-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen MB, Nataro JP, Bernstein DI, Hawkins J, Roberts N, Staat MA. 2005. Prevalence of diarrheagenic Escherichia coli in acute childhood enteritis: a prospective controlled study. J Pediatr 146:54–61. doi: 10.1016/j.jpeds.2004.08.059. [DOI] [PubMed] [Google Scholar]
- 5.Morin N, Santiago AE, Ernst RK, Guillot SJ, Nataro JP. 2013. Characterization of the AggR regulon in enteroaggregative Escherichia coli. Infect Immun 81:122–132. doi: 10.1128/IAI.00676-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nataro JP, Yikang D, Yingkang D, Walker K. 1994. AggR, a transcriptional activator of aggregative adherence fimbria I expression in enteroaggregative Escherichia coli. J Bacteriol 176:4691–4699. doi: 10.1128/JB.176.15.4691-4699.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morin N, Tirling C, Ivison SM, Kaur AP, Nataro JP, Steiner TS. 2010. Autoactivation of the AggR regulator of enteroaggregative Escherichia coli in vitro and in vivo. FEMS Immunol Med Microbiol 58:344–355. doi: 10.1111/j.1574-695X.2009.00645.x. [DOI] [PubMed] [Google Scholar]
- 8.Santiago AE, Ruiz-Perez F, Jo NY, Vijayakumar V, Gong MQ, Nataro JP. 2014. A large family of antivirulence regulators modulates the effects of transcriptional activators in Gram-negative pathogenic bacteria. PLoS Pathog 10:e1004153. doi: 10.1371/journal.ppat.1004153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Santiago AE, Yan MB, Hazen TH, Sauder B, Meza-Segura M, Rasko DA, Kendall MM, Ruiz-Perez F, Nataro JP. 2017. The AraC negative regulator family modulates the activity of histone-like proteins in pathogenic bacteria. PLoS Pathog 13:e1006545. doi: 10.1371/journal.ppat.1006545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Porter ME, Mitchell P, Roe AJ, Free A, Smith DGE, Gally DL. 2004. Direct and indirect transcriptional activation of virulence genes by an AraC-like protein, PerA from enteropathogenic Escherichia coli. Mol Microbiol 54:1117–1133. doi: 10.1111/j.1365-2958.2004.04333.x. [DOI] [PubMed] [Google Scholar]
- 11.Dorman CJ. 2004. H-NS: a universal regulator for a dynamic genome. Nat Rev Microbiol 2:391–400. doi: 10.1038/nrmicro883. [DOI] [PubMed] [Google Scholar]
- 12.Yang J, Tauschek M, Robins-Browne RM. 2011. Control of bacterial virulence by AraC-like regulators that respond to chemical signals. Trends Microbiol 19:128–135. doi: 10.1016/j.tim.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 13.Egan SM. 2002. Growing repertoire of AraC/XylS activators. J Bacteriol 184:5529–5532. doi: 10.1128/JB.184.20.5529-5532.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santiago AE, Yan MB, Tran M, Wright N, Luzader DH, Kendall MM, Ruiz-Perez F, Nataro JP. 2016. A large family of anti-activators accompanying XylS/AraC family regulatory proteins. Mol Microbiol 101:314–332. doi: 10.1111/mmi.13392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheikh J, Hicks S, Dall'Agnol M, Phillips AD, Nataro JP. 2001. Roles for Fis and YafK in biofilm formation by enteroaggregative Escherichia coli. Mol Microbiol 41:983–997. doi: 10.1046/j.1365-2958.2001.02512.x. [DOI] [PubMed] [Google Scholar]
- 16.Krin E, Danchin A, Soutourina O. 2010. Decrypting the H-NS-dependent regulatory cascade of acid stress resistance in Escherichia coli. BMC Microbiol 10:273. doi: 10.1186/1471-2180-10-273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hommais F, Krin E, Laurent-Winter C, Soutourina O, Malpertuy A, Le Caer JP, Danchin A, Bertin P. 2001. Large-scale monitoring of pleiotropic regulation of gene expression by the prokaryotic nucleoid-associated protein, H-NS. Mol Microbiol 40:20–36. doi: 10.1046/j.1365-2958.2001.02358.x. [DOI] [PubMed] [Google Scholar]
- 18.Gutierrez C, Gordia S, Bonnassie S. 1995. Characterization of the osmotically inducible gene osmE of Escherichia coli K-12. Mol Microbiol 16:553–563. doi: 10.1111/j.1365-2958.1995.tb02418.x. [DOI] [PubMed] [Google Scholar]
- 19.Nataro JP, Deng Y, Cookson S, Cravioto A, Savarino SJ, Guers LD, Levine MM, Tacket CO. 1995. Heterogeneity of enteroaggregative Escherichia coli virulence demonstrated in volunteers. J Infect Dis 171:465–468. doi: 10.1093/infdis/171.2.465. [DOI] [PubMed] [Google Scholar]
- 20.Henderson IR, Hicks S, Navarro-Garcia F, Elias WP, Philips AD, Nataro JP. 1999. Involvement of the enteroaggregative Escherichia coli plasmid-encoded toxin in causing human intestinal damage. Infect Immun 67:5338–5344. doi: 10.1128/IAI.67.10.5338-5344.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.PerkinElmer Corp. 1997. Applied Biosystems Prism 7700 sequence detection system: user bulletin no. 2. PerkinElmer Corp, Norwalk, CT. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.