

Bacterial lipoproteins have diverse roles in multiple aspects of bacterial physiology, antimicrobial resistance, and pathogenesis. Dedicated pathways direct the posttranslational lipidation and localization of lipoproteins, but there is considerable variation in these pathways among the proteobacteria. In this study, we characterized the proteins responsible for lipoprotein synthesis and localization in Helicobacter pylori, a member of the Epsilonproteobacteria that contributes to stomach cancer pathogenesis. We also provide evidence suggesting that lipidation of CagT, a component of the H. pylori Cag T4SS, is required for delivery of the H. pylori CagA oncoprotein into human gastric cells. Overall, these results constitute the first systematic analysis of H. pylori lipoprotein production and localization pathways and reveal how these processes in H. pylori differ from corresponding pathways in model proteobacteria.
KEYWORDS: Toll-like receptor 2, Helicobacter pylori, lipoproteins, posttranslational protein modification, type IV secretion systems
ABSTRACT
Our current understanding of lipoprotein synthesis and localization in Gram-negative bacteria is based primarily on studies of Escherichia coli. Newly synthesized E. coli prolipoproteins undergo posttranslational modifications catalyzed by three essential enzymes (Lgt, LspA, and Lnt). The mature lipoproteins are then sorted to the inner or outer membrane via the Lol system (LolABCDE). Recent studies suggested that this paradigm may not be universally applicable among different classes of proteobacteria. In this study, we conducted a systematic analysis of lipoprotein processing and sorting in Helicobacter pylori, a member of the Epsilonproteobacteria that colonizes the human stomach. We show that H. pylori lgt, lspA, and lnt homologs can complement conditionally lethal E. coli mutant strains in which expression of these genes is conditionally regulated. Mutagenesis studies and analyses of conditionally lethal H. pylori mutant strains indicate that lgt and lspA are essential for H. pylori growth but lnt is dispensable. H. pylori lolA and the single lolC (or lolE) homolog are also essential genes. We then explored the role of lipoproteins in H. pylori Cag type IV secretion system (Cag T4SS) activity. Comparative analysis of the putative VirB7 homolog CagT in wild-type and lnt mutant H. pylori strains indicates that CagT undergoes amino-terminal modifications consistent with lipidation, and we show that CagT lipidation is essential for CagT stability and Cag T4SS function. This work demonstrates that lipoprotein synthesis and localization in H. pylori diverge from the canonical pathways and that lipidation of a T4SS component is necessary for H. pylori Cag T4SS activity.
INTRODUCTION
Bacterial lipoproteins undergo posttranslational addition of acyl chains to their amino-terminal ends, which helps localize and anchor the mature lipoproteins (1). Lipoproteins have roles in multiple processes, including nutrient uptake, signal transduction, adhesion, conjugation, sporulation, antibiotic resistance, protein transport, and extracytoplasmic folding of proteins (2–7). In addition, bacterial lipoproteins present a pathogen-associated molecular pattern (PAMP) recognized by Toll-like receptor 2 (TLR2) when heterodimerized with either TLR1 or TLR6 (8). Recognition of lipoproteins through TLR2 stimulates the production of proinflammatory cytokines and antimicrobial effector molecules.
Lipoprotein synthesis in Gram-negative bacteria requires multiple steps (see Fig. S1 in the supplemental material) (9, 10). Newly synthesized lipoproteins are typically recognized and exported via the Sec pathway. The lipidation machinery then recognizes a short cysteine-containing amino acid sequence known as a lipobox, located near the amino terminus of the protein. The first modification is the addition of a diacylglyceride to the cysteine sulfhydryl of the preprolipoprotein, catalyzed by prolipoprotein diacylglyceryl transferase (Lgt). Amino acids preceding the cysteine are then cleaved by prolipoprotein signal peptidase (LspA, signal peptidase II), resulting in a diacylated apolipoprotein. Finally, the amino-terminal cysteine is N-acylated by apolipoprotein N-acyltransferase (Lnt) to produce the mature triacylated lipoprotein. The three lipoprotein-specific enzymes (Lgt, LspA, and Lnt) are essential for growth of the model organism, Escherichia coli. However, Wolbachia pipientis (a Wolbachia endosymbiont of Brugia malayi) lacks a lnt homolog, and mutational analyses demonstrated that lnt is not essential in Sinorhizobium meliloti (11–13) or Francisella tularensis or Neisseria gonorrhoeae (14) or in Acinetobacter species (15).
Posttranslational modifications in synthesis of lipoproteins in Gram-negative bacteria. The first modification is the addition of a diacylglyceride to the cysteine sulfhydryl of the preprolipoprotein, catalyzed by prolipoprotein diacylglyceryl transferase (Lgt). Amino acids preceding the cysteine are cleaved by prolipoprotein signal peptidase (Lsp), resulting in a diacylated apolipoprotein. Finally, a fatty acid is ligated to the amino terminus of the amino-terminal cysteine by apolipoprotein N-acyltransferase (Lnt) to produce the mature triacylated lipoprotein. Download FIG S1, PDF file, 0.1 MB (138.1KB, pdf) .
Copyright © 2020 McClain et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
In the canonical lipoprotein sorting pathway in Gram-negative bacteria, newly synthesized lipoproteins destined for the outer membrane interact with the LolCDE complex in the inner membrane. Lipoproteins retained in the inner membrane include a “Lol avoidance signal” and do not interact with LolCDE (9). LolCDE transfers the newly synthesized lipoprotein to the periplasmic protein LolA, which then transfers the lipoprotein to the outer membrane lipoprotein LolB for insertion into the outer membrane. Additional proteins may then act to transfer a subset of outer membrane lipoproteins to the external leaflet of the outer membrane (16).
Recent studies indicate that lipoprotein sorting in some species of bacteria does not conform to the canonical Lol pathway (17, 18). For example, the Alpha- and Betaproteobacteria, as well as some Gamma-, Delta-, and Epsilonproteobacteria, harbor a single lolF gene encoding a protein that has features of both LolC and LolE (14). Though lnt is typically essential in bacteria containing LolC and LolE, lnt appears to be nonessential in bacteria containing LolF. In the absence of lnt, lipoproteins are expected to be diacylated. Thus, it has been hypothesized that LolF, in contrast to LolC and LolE, can recognize both diacylated and triacylated lipoproteins for sorting to the outer membrane (14, 15). As additional evidence that the canonical pathway may not be broadly applicable, lolB is not found in the Alpha- or Epsilonproteobacteria (16, 19). Furthermore, although lolA and lolB are essential in wild-type (WT) E. coli, either gene may be mutated in strains deficient in Lpp or OsmB, and global transposon mutagenesis of Caulobacter crescentus suggests that lolA is not essential (20, 21). Together, these results suggest that some bacterial classes utilize a lipoprotein sorting pathway distinct from the LolAB pathway (20).
Helicobacter pylori is a Gram-negative bacterium, classified among the Epsilonproteobacteria, that colonizes the gastric mucosa of humans (22–25). Colonization with H. pylori induces gastric mucosal inflammation and is associated with an increased risk for peptic ulcer disease, gastric adenocarcinoma, and gastric lymphoma (26–29). The H. pylori cag pathogenicity island (cag PAI) encodes a secreted effector protein (CagA) and a type IV secretion system (Cag T4SS) that delivers CagA into human gastric cells (30, 31). Individuals colonized with H. pylori strains harboring the cag PAI have a higher risk of gastric cancer or peptic ulcer disease than individuals colonized with cag PAI-negative strains or H. pylori-negative individuals (32). The H. pylori genome is predicted to encode approximately 20 lipoproteins, but these predictions are largely based on informatics-driven identification of putative lipoboxes (short peptide motifs containing the cysteine that becomes lipidated) (33–35). Experimental analysis of putative H. pylori lipoproteins has been hindered by an inability to label lipoproteins in H. pylori using 3H-palmitate, perhaps because H. pylori lacks proteins involved in long-chain fatty acid transport and catabolism (36–38), and by the failure of globomycin to inhibit signal peptide cleavage by H. pylori LspA (36, 39). Thus, characterization of H. pylori lipoproteins by recombinant expression in E. coli, coupled with 3H-palmitate incorporation or globomycin-mediated inhibition of LspA or both, has been undertaken for only a small number of H. pylori lipoproteins (36, 37, 39, 40). H. pylori lipoproteins are currently being investigated as antigens for potential inclusion in H. pylori vaccines (41–47) and are believed to have important functions in bacterial adhesion to mammalian cells and colonization of the stomach (37, 39, 48), altering cell migration and signaling (36, 49) and stimulating gamma interferon (IFN-γ) production (50) and natural transformation competence (51).
H. pylori homologs of the enzymes involved in posttranslational modification of lipoproteins (Lgt, Lsp, and Lnt) have been proposed, as have homologs of LolA and LolF (33, 52–54). However, it remains unclear whether these enzymes are essential for H. pylori growth. One study reported that lspA is essential whereas lolA is nonessential (54). Conversely, a microarray-based analysis of a transposon mutant library in H. pylori detected transposon insertions in lgt, lspA, lnt, and lolF but no insertions in lolA (55). A systematic experimental analysis of lipoprotein synthesis and localization pathways in H. pylori has not yet been performed.
In the present report, we present a comprehensive analysis of H. pylori lipoprotein synthesis and localization pathways. We report that the genes whose products are predicted to mediate posttranslational modification of H. pylori lipoproteins are able to complement E. coli mutants in which the corresponding E. coli genes were conditionally regulated. We constructed gene knockouts or conditional mutants in H. pylori and show that lgt, lspA, lolA, and lolF are essential for H. pylori growth whereas lnt is dispensable. We also provide experimental evidence that CagT (a component of the H. pylori Cag T4SS and a putative VirB7 homolog) undergoes amino-terminal modifications consistent with lipidation. By analyzing a lnt mutant, we show that there is little if any alteration of Cag T4SS activity when H. pylori lipoproteins (including CagT) were diacylated rather than triacylated, and by analyzing mutants in which the CagT lipobox was disrupted, we show that lipidation of CagT is essential for CagT stability and Cag T4SS activity.
RESULTS
Lipoprotein synthesis: functional complementation of conditionally lethal E. coli mutant strains.
H. pylori genomes are predicted to contain homologs of the canonical genes lgt, lspA, and lnt (genes hp0955, hp0074, and hp0180, respectively, in sequenced strain 26695) required for the posttranslational modification of lipoproteins in E. coli (Fig. 1). The predicted H. pylori proteins share a relatively low level of amino acid sequence identity with characterized homologs found in E. coli or Pseudomonas aeruginosa (about 20 to 40% amino acid identity). However, each of the H. pylori proteins is predicted to adopt a three-dimensional fold similar to that of respective counterparts in other bacteria, and critical residues in each enzyme are conserved (Fig. 1). Conserved residues in H. pylori 26695 Lgt include R155 and E163 (corresponding to E. coli Lgt R143 and E151), predicted to bind phosphatidyl glycerol, the HGGL motif (residues 115 to 118 of H. pylori Lgt) which may bind to the peptide substrate, and an H-bond network consisting of R155, R232, E236, and R239 (R143, R239, E243, and R246 in E. coli Lgt) predicted to catalyze the transfer of diacylglycerol to the preprolipoprotein (56–58). H. pylori 26695 LspA includes the catalytic dyad D114 and D131 (D124 and D143 in Pseudomonas aeruginosa LspA) and 10 of 12 additional residues strictly conserved in LspA proteins from 485 organisms (the remainder of the 12 residues, G108 and A109 in P. aeruginosa LspA, are replaced by A98 and G99 in H. pylori LspA) (59). Finally, conserved residues in H. pylori 26695 Lnt include a catalytic triad comprised of E242, K296, and C349 (E267, K335, and C387 in E. coli Lnt) as well as residues Q207, K210, F211, N371, and W374 (Q233, K236, W237, N412, and W415 in E. coli Lnt) that form a pocket surrounding the site where the phosphate head group of the phospholipid donor is predicted to bind (60–62).
FIG 1.

Conservation among lipoprotein synthetic enzymes Lgt, LspA, and Lnt. Ribbon diagrams representing E. coli Lgt (A) (PDB 5AZC [56]), P. aeruginosa LspA (B) (PDB 5DIR [59]), and E. coli Lnt (C) (PDB 5N6H [61]) and predicted structures of H. pylori Lgt (D), Lsp (E), and Lnt (F) (generated by submitting the H. pylori sequences to Phyre2 [92]) are shown. Superimposed structures of Lgt (G), LspA (H), and Lnt (I) were generated using Chimera (93). The amino acid side chains of important conserved residues are shown as green spheres.
To experimentally evaluate whether these H. pylori proteins function as predicted, we sought to complement E. coli mutant strains in which the expression of genes required for posttranslational modification of lipoproteins is conditionally regulated. In these mutant strains, the expression of lgt, lspA, or lnt is under the control of an arabinose-inducible promoter (58, 63, 64). These strains grew on media supplemented with arabinose but did not grow on media supplemented with glucose (Fig. 2), indicating that the regulated enzymes are essential for growth. We introduced plasmids encoding the predicted H. pylori homologs under the control of the lacUV5 or trc promoter into the E. coli strains in which expression of lgt, lspA, or lnt is controlled by the arabinose-inducible promoter. In contrast to the parental strains, the strains containing these plasmids grew on media supplemented with glucose (Fig. 2). These results indicate that H. pylori lgt, lspA, and lnt homologs can functionally complement conditionally lethal E. coli lgt, lspA, and lnt mutant strains, respectively.
FIG 2.

Functional properties of H. pylori lgt, lsp, and lnt expressed in E. coli. E. coli PAP9403, YX238, and KA349 contain lgt, lspA, and lnt, respectively, under the control of an arabinose-inducible promoter (58, 63, 64). Since expression of lgt, lsp, and lnt is essential for bacterial growth, these strains do not grow on medium containing glucose but do grow on medium containing arabinose. Introduction of plasmids containing the homologous H. pylori genes under the control of the lacUV5 or Trc promoter supported growth on glucose. Vector-only control plasmids (C) did not support growth on glucose. Results are representative of 3 experiments.
Lipoprotein synthesis: construction and analysis of H. pylori mutant strains.
We next sought to determine whether the lgt, lspA, or lnt genes are essential in H. pylori. H. pylori strain 26695 was transformed with DNA constructs designed to disrupt lgt, lspA, or lnt by insertion of a gene conferring antibiotic resistance. Derivatives in which lnt was disrupted were readily isolated (e.g., VM211), indicating that lnt is not essential for H. pylori growth. In contrast, we were unable to isolate strains in which lgt or lspA was disrupted, despite repeated attempts.
As a complementary approach for assessing the essentiality of these genes, we constructed derivatives of H. pylori strain 26695 in which lgt or lspA was placed under the control of a TetR-regulated promoter (65). We then compared the growth of the resulting strains, VM176 and VM183, in the absence or presence of anhydrotetracycline (ATc), which results in repressed or derepressed expression of the gene of interest, respectively. Results indicated that strains containing TetR-regulated copies of lgt or lspA grew in the presence but not in the absence of ATc, indicating that lgt and lspA are essential for H. pylori growth (Fig. 3).
FIG 3.

Growth of H. pylori conditional mutants. H. pylori strains engineered to express lgt (VM176), lspA (VM183), lolA (VM189), or lolF (VM191) under the control of a TetR-regulated promoter were inoculated onto media in the presence or absence of anhydrotetracycline (ATc). Representative results from three independent cultures of each mutant are shown.
Lipoproteins represent pathogen-associated molecular patterns that can be recognized by TLR2 heterodimers on host cells. Triacylated lipoproteins are recognized by TLR1/TLR2 heterodimers, whereas diacylated lipoproteins are recognized by TLR2/TLR6 heterodimers. Upon binding of lipoproteins to TLR2 heterodimers (8), host cells respond by activation of NFκB-responsive promoters, including promoters for genes encoding proinflammatory cytokines such as interleukin-8 (IL-8). To compare the TLR2-activating properties of wild-type (WT) and lnt mutant H. pylori, we used HEK293 cell lines stably transfected to express human TLR1/TLR2 or mouse TLR2/TLR6 heterodimers. Addition of lipoprotein-enriched protein extracts from WT H. pylori to TLR2-expressing cells elicited responses similar to the responses induced by the triacylated control peptide Pam3CSK4 (Fig. 4). In contrast, addition of lipoprotein-enriched protein extracts prepared from lnt mutant VM211 elicited responses similar to the responses induced by the diacylated control peptide Pam2CSK4 (Fig. 4). These results are consistent with the presence of triacylated lipoproteins in WT H. pylori and the presence of diacylated lipoproteins in lnt-deficient H. pylori.
FIG 4.
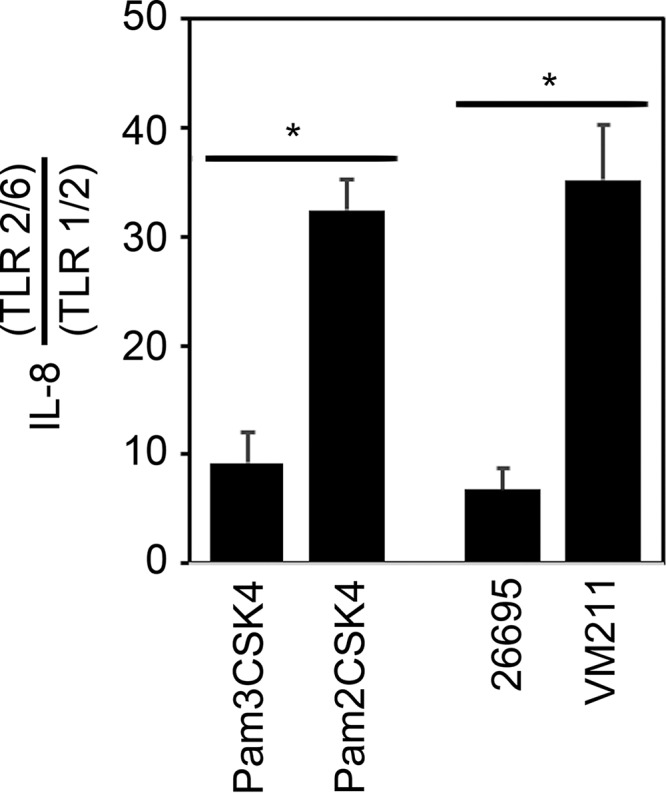
TLR2 activation by H. pylori lipoproteins. Protein extracts enriched in lipoproteins were prepared from H. pylori strain 26695 or the lnt mutant strain VM211 and were added (150 ng per ml) to 293-mTLR1/2 or 293-hTLR2/6 cell lines. As controls, cells also were incubated with triacylated Pam3CSK4 or diacylated Pam2CSK4 (30 ng per ml). Following 24 h of incubation, culture supernatants were recovered and subjected to ELISA to determine IL-8 concentrations. Results are expressed as the ratio of the level of IL-8 produced by 293-hTLR2/6 cells divided by the level of IL-8 produced by 293-hTLR1/2 cells and represent means and standard deviations of three independent experiments, each with triplicate samples. Asterisks denote results that were significantly different from the control results (Student's t test, P < 0.0003).
Lipoprotein localization genes: construction of H. pylori mutant strains.
Of the five genes whose products direct lipoprotein localization in E. coli (lolABCDE), H. pylori appears to lack lolB (16, 19), and a homolog of lolD has not been identified. One of numerous predicted ABC transporter-like ATP-binding proteins likely encodes LolD (53). H. pylori is predicted to encode a homolog of lolA (HP0785) as well as a single homolog of lolC or lolE (which has been termed lolF [HP0787]) to direct localization of newly synthesized lipoproteins (14). We sought to determine whether the lolA or lolF genes are essential in H. pylori. Repeated attempts to isolate strains in which either lolA or lolF was disrupted by insertion of a gene conferring antibiotic resistance were unsuccessful. As an alternative approach for assessing the essentiality of these genes, we constructed derivatives of strain 26695 in which lolA or lolF was placed under the control of a TetR-regulated promoter (65). The resulting strains, VM189 and VM191, were able to grow in the presence of ATc, but growth was arrested when the bacteria were cultured on media lacking ATc (Fig. 3). These results indicate that lolA and lolF are essential in H. pylori.
H. pylori Cag T4SS activity and posttranslational lipidation.
Secretion of the oncoprotein CagA and other effector molecules by H. pylori is mediated through a type IV secretion system (Cag T4SS) (66). Several of the H. pylori proteins required for T4SS activity exhibit sequence relatedness to components of T4SSs in other bacterial species. These include CagT, a putative homolog of the lipoprotein VirB7 (67). CagT (32 kDa) is much larger in size than VirB7 proteins from most bacterial species (for example, Agrobacterium tumefaciens VirB7 is 6 kDa), and comparisons of CagT with putative VirB7 homologs showed a very low level of amino acid sequence relatedness (31). To evaluate whether CagT undergoes N-terminal modifications consistent with lipidation, we introduced a gene encoding CagT with a DDK epitope between amino acids 26 and 27 into an H. pylori strain harboring a deletion of the endogenous cagT gene (Fig. 5A). The resulting strain, BV357, expressed CagT-DDK and retained Cag T4SS function (Fig. 5B and C). We next deleted lnt from the CagT-DDK strain, resulting in strain VM207. Protein extracts were prepared from BV357 and VM207, and the extracts were treated with enterokinase (which cleaves at the C-terminal end of the DDK epitope) (Fig. 5A). Immunoblotting the resulting protein preparations with anti-DDK antibody revealed the presence of DDK-reactive peptides in both BV357 and VM207, each with an apparent molecular mass somewhat higher than expected (predicted molecular weights for the triacylated and diacylated peptides are 2.5 and 2.2 kDa, respectively). Anomalous SDS-PAGE migration of lipoproteins is a common phenomenon (68–71). The DDK-reactive peptide from lnt mutant strain VM207 has a lower molecular mass than the peptide from strain BV357, consistent with a failure to add the third acyl chain in the absence of lnt (Fig. 5). These results are consistent with the presence of an Lnt-dependent N-terminal posttranslational modification of CagT.
FIG 5.

Analyses of CagT-DDK. (A) Amino-terminal amino acid sequence of CagT-DDK and the CagT-DDK peptides following enterokinase treatment. The lipobox is highlighted in red, the DDK epitope is underlined, and the asterisk indicates the site of triacyl lipid modification in WT H. pylori or the site of diacyl lipid modification in lnt mutant H. pylori. Signal peptide cleavage occurs between the “A” and “C” within the lipobox. Enterokinase cleavage occurs after lysine at its cleavage site DDDDK. (B) Expression of CagT was assessed by immunoblotting extracts of strains 26695 (WT), BV199 (ΔcagT), BV321 (restored cagT), and BV357 (cagT-DDK27) using anti-CagT and anti-HspB (as loading control). (C) H. pylori strains were cocultured with AGS cells, and the ability of each strain to induce IL-8 production was determined by ELISA. Asterisks denote results that were significantly different from the BV199 control results (analysis of variance [ANOVA] followed by Dunnett’s post hoc test, P < 0.05). (D) Protein extracts from BV357 and VM207 (each producing CagT-DDK27, the latter in an lnt mutant background) were treated with enterokinase and immunoblotted using anti-DDK monoclonal antibody. Consistent with expectations, the immunoreactive peptides from BV357 and VM207 differed in molecular mass.
We next analyzed Cag T4SS function in wild-type H. pylori 26695 and the lnt mutant strain (VM211). Immunoblotting demonstrated that the steady-state levels of CagT were similar in 26695 and VM211 (Fig. 6A). Cocultured with AGS cells, both 26695 and VM211 were able to induce IL-8 production (a phenotype dependent on Cag T4SS activity), whereas a control strain lacking the cag PAI was not (Fig. 6B). Furthermore, both 26695 and VM211 were able to translocate the effector protein CagA into AGS cells, where the protein then became phosphorylated (Fig. 6C). These results suggest that there is little if any alteration of Cag T4SS activity when H. pylori lipoproteins (including CagT) are diacylated rather than triacylated.
FIG 6.

Activity of diacylated CagT. (A) Whole-cell lysates from H. pylori strains 26695, 26695 ΔPAI, and VM211 (Δlnt) were analyzed by immunoblotting with anti-CagT antisera. A representative blot is shown. (B and C) AGS cells were cultured alone or in the presence of H. pylori strains at an MOI of 100:1 for 7 h. Cell culture supernatants were analyzed for IL-8 by ELISA (B), and cell lysates were analyzed by immunoblotting (C) using an antibody recognizing phospho-Tyr (PY99) to detect phosphorylated CagA (indicated by an arrowhead) or antiserum directed against CagA (to detect total CagA). Multiple additional bands (unrelated to CagA) were detected by the anti-phospho-Tyr antibody in all samples, including AGS cells alone. Results in panel B represent means and standard deviations of three biological replicates, each analyzed in triplicate; results in panel C are representative of three biological replicates. Asterisks denote results that were significantly different from the 26695 ΔPAI control results (ANOVA followed by Dunnett’s post hoc test, P < 0.05).
To determine whether lipidation of CagT is required for Cag T4SS function, we constructed H. pylori strains expressing mutant forms of cagT in which the putative CagT lipobox (a peptide motif within the signal peptide containing an essential cysteine as the site of lipidation) was disrupted (by introducing a C21S mutation, strain BV218) or restored (strain BV260) (Fig. 7). Analyses of steady-state levels of CagT by immunoblotting revealed that disruption of the putative lipobox led to destabilization of CagT whereas introduction of an intact lipobox after the C21S mutation (CagT1) resulted in wild-type levels of CagT (Fig. 7). Consistent with these findings, the C21S mutation abolished the ability of H. pylori to induce IL-8 expression in AGS gastric epithelial cells (Fig. 7).
FIG 7.

Requirement of a lipobox cysteine residue for CagT stability. (A) Amino-terminal amino acid sequences of CagT and mutant forms of CagT analyzed in this study. The CagT lipobox is highlighted in red, and the disrupted lipobox (CagT-C21S) is highlighted in blue. (B) Expression of CagT was evaluated by immunoblotting extracts of strains 26695 (WT), BV199 (ΔcagT), BV321 (in which a WT copy of cagT was introduced into the ureA locus-restored cagT strain), BV218 (cagT-C21S), and BV260 (cagT1) using anti-CagT. Vertical line indicates cropping of an additional unreported lane from the image. (C) H. pylori strains were cocultured with AGS cells, and the ability of each strain to induce IL-8 production was determined by ELISA. Asterisks denote results that were significantly different from the BV199 control results (ANOVA followed by Dunnett’s post hoc test, P < 0.05).
One possible explanation for the results shown in Fig. 7 is that the CagT-C21S mutant protein may lack a functional signal peptide and therefore may not be properly secreted. Analysis of CagT by SignalP-5.0 predicts signal peptide cleavage by LspA between Ala20 and Cys21 (Fig. 8A) (72). In contrast, analysis of CagT-C21S predicts no signal peptide cleavage (data not shown). Therefore, as a complementary approach, we constructed an H. pylori strain, VM253, expressing a mutant protein (CagT2) in which the 20-amino-acid signal peptide of CagT was replaced with the 33-amino-acid signal peptide from the secreted protein VacA (Fig. 8A and B). This mutant CagT also includes a DDK epitope between amino acids 26 and 27. Both wild-type CagT and the CagT2 chimera are expected to undergo signal peptide cleavage immediately preceding a Cys residue (C21 in WT CagT, C34 in CagT2) (Fig. 8A and B). However, the mutant CagT2 is not expected to undergo lipidation due to the absence of a lipobox (Fig. 8B). Immunoblotting of protein extracts from strain VM253 revealed that exchange of the native CagT signal peptide with the signal peptide from VacA led to destabilization of CagT (Fig. 8C). Correspondingly, strain VM253, expressing the mutant protein (CagT2), was defective in inducing IL-8 expression in AGS gastric epithelial cells (Fig. 8D). Therefore, the combined results of Fig. 7 and Fig. 8 indicate that an intact CagT lipobox is required for CagT stability and Cag T4SS activity.
FIG 8.

An intact lipobox is required for CagT stability. (A and B) SignalP 5.0 predicts signal peptide cleavage of wild-type CagT and a mutant CagT protein (CagT2) in which the H. pylori VacA signal peptide was fused to CagT (72). The sites of predicted signal peptide cleavage by LspA (LIPO) or by signal peptidase I (SP) and the corresponding cleavage sites (CS) are shown. (C) Immunoblot showing steady-state levels of CagT 26695 (WT), BV199 (ΔcagT), BV357 (CagT-DDK), and VM253 (cagT2) determined using anti-CagT. Vertical line indicates cropping of an additional unreported lane from the image. (D) H. pylori strains were cocultured with AGS cells, and the ability of each strain to induce IL-8 production was determined by ELISA. Asterisks denote results that were significantly different from the BV199 control results (ANOVA followed by Dunnett’s post hoc test, P < 0.05).
DISCUSSION
Genes HP0955, HP0074, and HP0180 in H. pylori strain 26695 are annotated as homologs of lgt, lspA, and lnt, respectively (33, 52–54). Although there is very limited primary amino acid sequence similarity between the encoded H. pylori proteins and more thoroughly characterized homologs from other species, multiple amino acids predicted to be required for functional activity are conserved (Fig. 1). In the present study, we report that H. pylori HP0955, HP0074, and HP0180 are indeed functional homologs of lgt, lspA, and lnt, based on the ability of each gene to complement conditionally lethal E. coli mutant strains.
Results of previous studies have provided conflicting evidence on whether genes involved in lipoprotein synthesis and sorting in H. pylori are essential (54, 55). The comprehensive analyses conducted in the present study indicated that the lgt and lspA genes encoding lipoprotein synthetic enzymes as well as the genes whose products direct lipoprotein localization, lolA and lolF, are essential in H. pylori. In contrast, lnt is not essential. The latter result is consistent with recent studies suggesting that lnt is not essential in bacterial species containing lolF in place of lolC and lolE (14, 15). The affinity of the LolCDE complex is much higher toward triacylated lipoproteins than diacylated lipoproteins, and this likely explains the essential nature of lnt in species containing lolC and lolE (73). In contrast, the nonessential nature of lnt in species containing lolF suggests that the affinity of LolF toward diacylated lipoproteins (the product of Lgt action) is sufficient to direct them to LolA for sorting.
In the canonical sorting pathway, lipoproteins destined for localization to the outer membrane are transferred from the inner membrane LolCDE complex to the periplasmic LolA protein. LolA then transfers the lipoproteins to LolB anchored in the outer membrane (9). Previous studies have concluded that LolA receives lipoproteins from LolCDE but cannot insert lipoproteins into membranes (74–78), and similar studies have concluded that LolB can insert lipoproteins into membranes but cannot accept lipoproteins from LolCDE (9, 78, 79). There are several possible explanations for the essentiality of lolA in H. pylori and the lack of a recognizable H. pylori lolB (and for the similar apparent absence of lolB in the Alpha- and other Epsilonproteobacteria [16, 19]), including the following: (i) outer membrane lipoproteins are not essential in H. pylori; (ii) H. pylori LolA, unlike E. coli LolA, can insert lipoproteins into membranes; (iii) there is an uncharacterized protein in H. pylori with a function analogous to that of LolB; and/or (iv) H. pylori possesses a LolAB-bypass system, as has recently been suggested in E. coli (20).
H. pylori strains lacking CagT (either through cagT knockout mutation or with repressed cagT gene expression) are deficient in Cag T4SS activity (65, 80, 81), and we now present experimental evidence indicating that CagT undergoes N-terminal modification consistent with lipidation. Lipidation of CagT is necessary for CagT stability and therefore for Cag T4SS activity. Comparisons of Cag T4SS activities in wild-type and mutant H. pylori in which lipidation of CagT is disrupted suggest that CagT functions similarly whether it is triacylated or diacylated. However, disruption of CagT lipidation appears to destabilize CagT, most likely due to defects in secretion or membrane anchoring leading to degradation of the mislocalized protein.
Previous studies demonstrated that CagT is essential for Cag T4SS assembly and function (65, 80, 81). Analyses of the Cag T4SS structure by electron cryotomography (cryo-electron tomography [cryo-ET]) revealed that outer membrane core complexes from cagT mutant strains were highly variable in structure, generally consisting of a central ring but lacking the peripheral densities seen in intact complexes (82). A reduced number of core complexes were visualized by cryo-ET in a cagT mutant strain compared to a WT strain, and a reduced number of core complexes were purified from the cagT mutant strain (82, 83); this might be attributable to reduced stability or impaired localization of the complexes in a cagT mutant strain. Single-particle cryo-electron microscopy analysis of Cag T4SS outer membrane core complexes suggested that the lipidated amino-terminal end of CagT is positioned to interact with the H. pylori outer membrane (Fig. 9) (31). Together, these results suggest that lipidated CagT helps stabilize the core complex and direct it or anchor it to the outer membrane.
FIG 9.

The lipidated amino-terminal end of CagT is positioned at the interface between the Cag T4SS and the H. pylori outer membrane. (A and B) Ribbon representations of CagT, CagX, and CagY (PDB accession no. 6OEE, 6OEG, and 6OEF, respectively [31]) within the Cag T4SS outer membrane core complex are shown in side (A) and top (B) views. The amino-terminal residue resolved in the CagT structure is lysine 26 and is highlighted in red. (C) The amino-terminal amino acid sequence of CagT is shown with an asterisk indicating the site of lipidation, and lysine 26 is highlighted in red.
In summary, this report provides a systemic experimental analysis of lipoprotein synthesis and localization pathways in H. pylori, a member of the Epsilonproteobacteria. The lipoprotein synthetic enzymes Lgt and LspA are essential, implying that one or more lipoproteins are essential for H. pylori. Lnt (which performs the final enzymatic step in the synthesis of triacylated lipoproteins in Gram-negative bacteria) is nonessential in H. pylori. This is consistent with previous reports indicating that Lnt is not required by certain members of the Alpha-, Beta-, and Gammaproteobacteria (11–15). The nonessential nature of Lnt in H. pylori is consistent with current understanding that Lnt is not required by bacteria (such as H. pylori) that produce LolF rather than LolC and LolE (14, 15). Further differentiating lipoprotein sorting in H. pylori from model proteobacteria is the observation that H. pylori, like other members of the Epsilonproteobacteria as well as the Alphaproteobacteria, appears to lack LolB (16, 19). The current study showed that lipidation of CagT is essential for Cag T4SS function, but we did not detect any substantial difference in T4SS activity dependent on whether CagT was diacylated or triacylated. In future studies, it will be important to determine what fitness advantage is provided by the nonessential lnt, to determine how CagT and other lipoproteins are directed to the H. pylori outer membrane in the absence of a lolB homolog, and to define the structural basis by which a lipid moiety in CagT helps direct or anchor the T4SS outer membrane core complex to the outer membrane.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
Strains and plasmids used in this study are listed in Table 1. H. pylori strain 26695 was grown on either Trypticase soy agar (TSA) plates containing 5% sheep blood or in bisulfite-free Brucella broth containing 10% fetal bovine serum (BB-FBS) at 37°C in room air supplemented with 5% CO2. H. pylori mutant strains were selected using metronidazole (15 μg ml−1), kanamycin (12.5 μg ml−1), or chloramphenicol (2.5 μg ml−1). Anhydrotetracycline (ATc) was added as indicated at 100 ng ml−1 (65). Plasmids were maintained using E. coli strain DH5α or strain XL-10 Gold. E. coli strains were grown on Luria-Bertani (LB) agar plates or in LB broth containing ampicillin (100 μg ml−1), chloramphenicol (25 μg ml−1), or kanamycin (25 μg ml−1).
TABLE 1.
Plasmids and strains used in this study
| Plasmid or strain | Description or genotypea | Reference or source |
|---|---|---|
| Plasmids | ||
| pAD1 | Vector for introducing DNA into the H. pylori ureA locus | 94, 95 |
| pBbA5a-RFP | p15A origin, bla, lacI, rfp, lacUV5 promoter | 96 |
| pBbA5c-RFP | p15A origin, cat, lacI, rfp, lacUV5 promoter | 96 |
| pBbE1c-RFP | colE1 origin, cat, lacI, rfp, trc promoter | 96 |
| pBV173 | H. pylori genome region that includes cagT cloned into pGEMT-Easy (Promega) | Current study |
| pBV175 | pBV173 ΔcagT | Current study |
| pBV193 | pGEMT-cagT-C21S | Current study |
| pBV253 | pGEMT-cagT1 | Current study |
| pBV334 | Derivative of pBV173 in which sequences encoding a DDK epitope are inserted into cagT after lysine 26 |
Current study |
| pBV342 | H. pylori cagT cloned into pAD1 | Current study |
| pBV449 | H. pylori lgt cloned into pBbA5a-RFP | Current study |
| pMM690 | H. pylori lspA cloned into pBbE1c-RFP | Current study |
| pMM691 | H. pylori lnt cloned into pBbA5c-RFP | Current study |
| pMM693 | pAD1-cagT2 | Current study |
| Strains | ||
| H. pylori 26695 | 33 | |
| H. pylori 26695 ΔPAI | 26695 Δ(HP520–547) | 89 |
| H. pylori 26695 ΔrdxA | 26695 ΔrdxA | 84 |
| H. pylori 26695 ΔrdxA cagT::cat/rdxA) | 26695 ΔrdxA cagT::cat/rdxA | 80 |
| H. pylori BV199 | 26695 ΔrdxA ΔcagT | Current study |
| H. pylori BV218 | 26695 ΔrdxA cagT-C21S | Current study |
| H. pylori BV260 | 26695 ΔrdxA cagT1 | Current study |
| H. pylori BV321 | 26695 ΔrdxA ΔcagT ureA::(cagT Chlr) | Current study |
| H. pylori BV357 | BV199 ureA::cagT-DDK26 | Current study |
| H. pylori VM124 | 26695 ureA::tetR Chlr | 65 |
| H. pylori VM165 | 26695 ureA::tetR Kanr | Current study |
| H. pylori VM173 | VM165 tetO::PcagU-lgt (630–631, Chlr) | Current study |
| H. pylori VM176 | VM173 Δlgt-rdxA (Mtzr) | Current study |
| H. pylori VM183 | VM124 tetO-PcagU::lspA Kanr | Current study |
| H. pylori VM189 | VM124 PcagU::lolA Kanr | Current study |
| H. pylori VM191 | VM124 PcagU::lolF Kanr | Current study |
| H. pylori VM207 | BV357 Δlnt Kanr | Current study |
| H. pylori VM211 | 26695 Δlnt Kanr | Current study |
| H. pylori VM253 | BV199 ureA::cagT2 (Chlr) | Current study |
| E. coli DH5α | F– Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK– mK+) phoA supE44 λ– thi-1 gyrA96 relA1 |
|
| E. coli PAP9403 | lacIq rrnBT14 ΔlacZWJ16 hsdR514 ΔaraBADAH33 ΔrhaBADLD78 lgt::Kanr pCHAP9231 | 58 |
| E. coli YX238 | MG1655 ΔlspA::Kan pBAD30::lspAEc | 63 |
| E. coli KA349 | ybeX-(Kanr-rrnB TT-araC-PBAD)-lnt lpp::Chlr | 64 |
| E. coli XL-10 Gold |
endA1 glnV44 recA1 thi-1 gyrA96 relA1 lac Hte Δ(mcrA)183 Δ(mcrCB-hsdSMR-mrr) 173 Tetr F'[proAB lacIqZΔM15 Tn10(Tetr Amy Chlr)] |
|
Chlr, chloramphenicol resistance; Kanr, kanamycin resistance; Mtzr, metronidazole resistance; Tetr, tetracycline resistance.
Complementation of E. coli strains with H. pylori lgt, lspA, and lnt.
E. coli strains PAP9403, YX238, and KA349 are conditionally lethal strains in which lgt, lspA, and lnt (respectively) are expressed under the control of arabinose-inducible promoters (58, 63, 64). Expression of the relevant gene in these strains is stimulated by the presence of arabinose and inhibited by the addition of glucose. The lgt, lspA, and lnt genes from H. pylori 26695 were subjected to PCR amplification and cloned into pBbA5a-RFP (pBbA5a-red fluorescent protein), pBbE1c-RFP, and pBbA5c-RFP (Addgene), using EcoRI and BglII restriction sites, to yield pBV449, pMM690, and pMM691, respectively. These plasmids then were transformed into the relevant E. coli mutant strains. E. coli strains were cultured with isopropyl β-d-1-thiogalactopyranoside (IPTG) (Roche) (0.1 mM) and either arabinose (0.2%) or glucose (0.2%).
Construction of recombinant H. pylori strains.
H. pylori strain 26695 was transformed with synthetic constructs (GenScript) designed to delete lgt, lspA, lnt, lolA, or lolF. For lgt, the construct included a deletion of lgt (HP0955) and rdxA (HP0954); deletion of rdxA confers metronidazole resistance (84). For lspA, lnt, lolA, and lolF, the genes of interest were replaced with an antibiotic resistance determinant. We succeeded only in obtaining H. pylori transformants in which lnt was deleted, resulting in strain VM211.
To place lgt under TetR regulation, tetR linked to a kanamycin resistance determinant was inserted into the ureA locus of strain 26695 to yield strain VM165. Then, a synthetic construct (GenScript) in which a chloramphenicol resistance determinant and 3 copies of tetO linked to the promoter of cagUT (65) placed upstream of HP0955 (and changing the ATG start codon to a TTG start codon [85]) was introduced into the intergenic region between HP0630 and HP0631 to yield strain HP173. Finally, the endogenous lgt locus was deleted using the lgt deletion construct described above to yield strain VM176.
To place lspA, lolA, or lolF under TetR regulation, strain VM124 (which contains TetR in the ureA locus) (65) was transformed with synthetic constructs in which a kanamycin resistance determinant and 3 copies of tetO linked to the promoter of cagUT (65) were placed upstream of lspA, lolA, and lolF to yield strains VM183, VM189, and VM191, respectively. The start codons for lolA (ATG) and lspA (GTG) were changed to TTG (85); lolF naturally begins with a TTG start codon.
To delete cagT from H. pylori, a DNA sequence containing cagT along with 500 bp of flanking sequences was amplified from H. pylori and ligated into pGEMT-Easy (Promega) to yield pBV173. Using inverse PCR, cagT was excised to produce pBV175. H. pylori 26695ΔrdxA (cagT::cat/rdxA) (80) was then transformed with pBV175, and metronidazole-resistant transformants were selected to yield H. pylori BV199, which contains an unmarked ΔcagT mutation. The cagT mutation in BV199 was complemented in cis by transformation with pBV342 (ureA::cagT-CAT) to produce strain BV321.
To generate an H. pylori strain expressing DDK-tagged CagT, pBV173 was mutagenized to insert DDK epitope-encoding sequences after the codon for lysine 26 of cagT, yielding pBV334. Plasmid pBV334 was then used to transform H. pylori strain 26695 ΔrdxA cagT::cat/rdxA (80), and metronidazole-resistant transformants were selected to yield H. pylori strain BV357. The lgt locus was deleted from strain BV357 using the lgt deletion construct described above to yield strain VM207.
To generate an H. pylori strain expressing the 33-amino-acid signal peptide from H. pylori VacA fused to CagT at Cys21 and including a DDK epitope inserted after amino acid 26 of CagT, a chimeric sequence including a chloramphenicol resistance determinant was synthesized by GenScript and cloned into plasmid pAD1. The resulting plasmid, pMM692, was used to transform H. pylori strain BV199, and chloramphenicol-resistant colonies were selected. The resulting strain was designated VM253.
Site-directed mutagenesis of cagT was accomplished using a QuikChange-Multi site-directed mutagenesis system (Agilent) and pBV173 as the template. The resulting plasmids (pBV193 [encoding CagT-C21S] and pBV253 [encoding CagT1]) were then used to transform H. pylori 26695 ΔrdxA cagT::cat/rdxA (80), and metronidazole-resistant transformants were selected. The resulting H. pylori strains were designated BV218 and BV260, respectively (Table 1).
Analysis of CagT lipidation in H. pylori.
To evaluate N-terminal modifications of CagT in H. pylori, protein extracts were prepared from strains BV357 and VM207 (described above). H. pylori strains were cultured on TSA plates containing 5% sheep blood for 24 h. Bacteria were resuspended in phosphate-buffered saline (PBS), pelleted, and washed in PBS. Bacterial pellets were resuspended in PBS containing 0.5% SDS. Insoluble material was pelleted at 21,000 × g for 5 min. Proteins in the soluble fraction were recovered using methanol-chloroform and solubilized in 0.1% SDS (86). Protein concentrations were determined by microBCA assay (Pierce), and 50 μg of each extract was incubated with 2 units of enterokinase (EK Max; ThermoFisher) at 37°C for 4 h. Samples then were separated by Tricine–SDS-PAGE using a 16%T/3%C gel containing 6 M urea (87) and transferred to polyvinylidene difluoride (PVDF). The PVDF membrane was developed using mouse monoclonal anti-DDK antibody (clone M2; Sigma) followed by anti-mouse horseradish peroxidase (HRP) (Promega) and chemiluminescence detection (Pierce).
TLR2 activation assay.
Protein extracts enriched in lipoproteins were prepared using Triton X-114 (TX114) (88). H. pylori strains were cultured in broth for 20 h. Cultures were pelleted and washed in PBS, and the bacterial pellets were resuspended in PBS containing 2% TX114 and incubated at 4°C with continuous mixing for 24 h. Insoluble material was pelleted, and the protein extract was warmed and pelleted at 37°C to promote separation of TX114 and aqueous phases. The upper, aqueous phase was discarded, and the lower, detergent phase was extracted two times with the starting volume of PBS. Proteins in the final detergent phase were precipitated with 4× volumes of −20°C acetone. The protein pellets were resuspended in PBS, and the protein concentrations were determined by micro-bicinchoninic acid (BCA) assay (Pierce).
293-mTLR1/2 (m, mouse) and 293-hTLR2/6 (h, human) cells (InvivoGen) were grown in DMEM with glucose (4.5 g liter−1), 2 mM l-glutamine, penicillin (50 U ml−1), streptomycin (50 μg ml−1), 10% FBS, and 10 μg ml−1 at 37°C with 5% CO2. These cells are stably transfected to express mouse TLR1 and TLR2 and to express human TLR2 and TLR6, respectively. Cells were seeded into 96-well tissue culture dishes at 2.5 × 104 cells per well. Serial dilutions of either bacterial TX114 lipoprotein extracts or synthetic control peptides (triacylated lipoprotein Pam3CSK4 or diacylated Pam2CSK4; InvivoGen) were added to each well. Culture supernatant was collected after cells were stimulated for 24 h at 37°C with 5% CO2. TLR2 stimulation was assessed by measuring IL-8 levels in cell culture supernatants by enzyme-linked immunosorbent assay (ELISA) (Genscript).
Assays of H. pylori Cag T4SS activity.
The ability of H. pylori to induce production of IL-8 when cocultured with gastric cells was determined as previously described (84). AGS gastric epithelial cells (ATCC CRL-1739) were grown in RPMI medium containing 25 mM HEPES and 10% FBS and were inoculated with H. pylori (multiplicity of infection [MOI] of 100) from liquid cultures that had been grown overnight. Cocultures were incubated for 4 h at 37°C with 5% CO2. Supernatant was collected, and IL-8 content was determined using an IL-8 ELISA (GenScript), according to the manufacturer’s specifications.
The ability of H. pylori to translocate CagA under conditions of coculture with gastric cells was determined as previously described (89, 90). H. pylori cells were cocultured with AGS cells for 7 h at 37°C with 5% CO2. Monolayers were washed in PBS and lysed in NP-40 lysis buffer containing Complete Mini protease inhibitors (Roche) and Phos-Stop phosphatase inhibitors (Roche). CagA translocation was assessed based on immunoblotting cell lysates with anti-CagA polyclonal antibody (Santa Cruz) and anti-phospho-tyrosine monoclonal antibody (pY-99; Santa Cruz).
Immunoblotting.
Unless otherwise indicated, protein lysates were resolved by SDS-PAGE and proteins were transferred to nitrocellulose membranes. Membranes were blocked using PBS containing 0.1% Tween and 2% nonfat dry milk. Proteins were detected by incubating the membrane with primary antisera (diluted 1:5,000 to 1:10,000), followed by horseradish peroxidase-conjugated secondary antibody. Rabbit antiserum to H. pylori CagT and antiserum to HspB (a GroEL heat shock protein homolog) have been described previously (80, 91). Anti-CagT antiserum was preabsorbed to H. pylori BV199 (26695 ΔrdxA ΔcagT) prior to immunoblotting. Signals were detected by the use of an enhanced chemiluminescence (ECL) methodology.
ACKNOWLEDGMENTS
We thank Nienke Buddelmeijer (Institut Pasteur), Daniel Wall (University of Wyoming), and Timothy Meredith (Pennsylvania State University) for providing E. coli strains PAP9403, YX238, and KA349, respectively.
This work was supported by NIH AI118932, AI039657, CA116087, and GM125264 and the Department of Veterans Affairs (Merit Review grant BX004447).
Footnotes
Citation McClain MS, Voss BJ, Cover TL. 2020. Lipoprotein processing and sorting in Helicobacter pylori. mBio 11:e00911-20. https://doi.org/10.1128/mBio.00911-20.
REFERENCES
- 1.Konovalova A, Silhavy TJ. 2015. Outer membrane lipoprotein biogenesis: Lol is not the end. Philos Trans R Soc Lond B Biol Sci 370:20150030. doi: 10.1098/rstb.2015.0030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sutcliffe IC, Russell RR. 1995. Lipoproteins of gram-positive bacteria. J Bacteriol 177:1123–1128. doi: 10.1128/jb.177.5.1123-1128.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Perego M, Higgins CF, Pearce SR, Gallagher MP, Hoch JA. 1991. The oligopeptide transport system of Bacillus subtilis plays a role in the initiation of sporulation. Mol Microbiol 5:173–185. doi: 10.1111/j.1365-2958.1991.tb01838.x. [DOI] [PubMed] [Google Scholar]
- 4.Mathiopoulos C, Mueller JP, Slack FJ, Murphy CG, Patankar S, Bukusoglu G, Sonenshein AL. 1991. A Bacillus subtilis dipeptide transport system expressed early during sporulation. Mol Microbiol 5:1903–1913. doi: 10.1111/j.1365-2958.1991.tb00814.x. [DOI] [PubMed] [Google Scholar]
- 5.Lampen JO, Nielsen JB. 1984. N-terminal glyceride-cysteine modification of membrane penicillinases in gram-positive bacteria. Methods Enzymol 106:365–368. doi: 10.1016/0076-6879(84)06038-9. [DOI] [PubMed] [Google Scholar]
- 6.Kovacs-Simon A, Titball RW, Michell SL. 2011. Lipoproteins of bacterial pathogens. Infect Immun 79:548–561. doi: 10.1128/IAI.00682-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alloing G, de Philip P, Claverys JP. 1994. Three highly homologous membrane-bound lipoproteins participate in oligopeptide transport by the Ami system of the gram-positive Streptococcus pneumoniae. J Mol Biol 241:44–58. doi: 10.1006/jmbi.1994.1472. [DOI] [PubMed] [Google Scholar]
- 8.Takeda K, Takeuchi O, Akira S. 2002. Recognition of lipopeptides by Toll-like receptors. J Endotoxin Res 8:459–463. doi: 10.1179/096805102125001073. [DOI] [PubMed] [Google Scholar]
- 9.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 10.Buddelmeijer N. 2015. The molecular mechanism of bacterial lipoprotein modification–how, when and why? FEMS Microbiol Rev 39:246–261. doi: 10.1093/femsre/fuu006. [DOI] [PubMed] [Google Scholar]
- 11.Turner JD, Langley RS, Johnston KL, Gentil K, Ford L, Wu B, Graham M, Sharpley F, Slatko B, Pearlman E, Taylor MJ. 2009. Wolbachia lipoprotein stimulates innate and adaptive immunity through Toll-like receptors 2 and 6 to induce disease manifestations of filariasis. J Biol Chem 284:22364–22378. doi: 10.1074/jbc.M901528200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Foster J, Ganatra M, Kamal I, Ware J, Makarova K, Ivanova N, Bhattacharyya A, Kapatral V, Kumar S, Posfai J, Vincze T, Ingram J, Moran L, Lapidus A, Omelchenko M, Kyrpides N, Ghedin E, Wang S, Goltsman E, Joukov V, Ostrovskaya O, Tsukerman K, Mazur M, Comb D, Koonin E, Slatko B. 2005. The Wolbachia genome of Brugia malayi: endosymbiont evolution within a human pathogenic nematode. PLoS Biol 3:e121. doi:10.1371/journal.pbio.0030121 doi: 10.1371/journal.pbio.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tiwari RP, Reeve WG, Dilworth MJ, Glenn AR. 1996. An essential role for actA in acid tolerance of Rhizobium meliloti. Microbiology 142(Pt 3):601–610. doi: 10.1099/13500872-142-3-601. [DOI] [PubMed] [Google Scholar]
- 14.LoVullo ED, Wright LF, Isabella V, Huntley JF, Pavelka MS. 2015. Revisiting the Gram-negative lipoprotein paradigm. J Bacteriol 197:1705–1715. doi: 10.1128/JB.02414-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gwin CM, Prakash N, Christian Belisario J, Haider L, Rosen ML, Martinez LR, Rigel NW. 2018. The apolipoprotein N-acyl transferase Lnt is dispensable for growth in Acinetobacter species. Microbiology 164:1547–1556. doi: 10.1099/mic.0.000726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wilson MM, Bernstein HD. 2016. Surface-exposed lipoproteins: an emerging secretion phenomenon in Gram-negative bacteria. Trends Microbiol 24:198–208. doi: 10.1016/j.tim.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grabowicz M. 2018. Lipoprotein transport: greasing the machines of outer membrane biogenesis: re-examining lipoprotein transport mechanisms among diverse Gram-negative bacteria while exploring new discoveries and questions. Bioessays 40:e1700187. doi: 10.1002/bies.201700187. [DOI] [PubMed] [Google Scholar]
- 18.Hooda Y, Moraes TF. 2018. Translocation of lipoproteins to the surface of Gram negative bacteria. Curr Opin Struct Biol 51:73–79. doi: 10.1016/j.sbi.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 19.Tokuda H, Matsuyama S. 2004. Sorting of lipoproteins to the outer membrane in E. coli. Biochim Biophys Acta 1693:5–13. doi: 10.1016/j.bbamcr.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Grabowicz M, Silhavy TJ. 2017. Redefining the essential trafficking pathway for outer membrane lipoproteins. Proc Natl Acad Sci U S A 114:4769–4774. doi: 10.1073/pnas.1702248114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christen B, Abeliuk E, Collier JM, Kalogeraki VS, Passarelli B, Coller JA, Fero MJ, McAdams HH, Shapiro L. 2011. The essential genome of a bacterium. Mol Syst Biol 7:528. doi: 10.1038/msb.2011.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Atherton JC, Blaser MJ. 2009. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest 119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cover TL, Blaser MJ. 2009. Helicobacter pylori in health and disease. Gastroenterology 136:1863–1873. doi: 10.1053/j.gastro.2009.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Suerbaum S, Michetti P. 2002. Helicobacter pylori infection. N Engl J Med 347:1175–1186. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 25.Gilbreath JJ, Cody WL, Merrell DS, Hendrixson DR. 2011. Change is good: variations in common biological mechanisms in the epsilonproteobacterial genera Campylobacter and Helicobacter. Microbiol Mol Biol Rev 75:84–132. doi: 10.1128/MMBR.00035-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.NIH Consensus Development Panel on Helicobacter pylori in Peptic Ulcer Disease. 1994. NIH Consensus Conference. Helicobacter pylori in peptic ulcer disease. NIH Consensus Development Panel on Helicobacter pylori in Peptic Ulcer Disease. JAMA 272:65–69. doi: 10.1001/jama.1994.03520010077036. [DOI] [PubMed] [Google Scholar]
- 27.IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. 1994. Schistosomes, liver flukes and Helicobacter pylori. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Lyon, 7-14 June 1994. IARC Monogr Eval Carcinog Risks Hum 61:1–241. [PMC free article] [PubMed] [Google Scholar]
- 28.Fox JG, Wang TC. 2007. Inflammation, atrophy, and gastric cancer. J Clin Invest 117:60–69. doi: 10.1172/JCI30111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wroblewski LE, Peek RM Jr, Wilson KT. 2010. Helicobacter pylori and gastric cancer: factors that modulate disease risk. Clin Microbiol Rev 23:713–739. doi: 10.1128/CMR.00011-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Backert S, Tegtmeyer N, Fischer W. 2015. Composition, structure and function of the Helicobacter pylori cag pathogenicity island encoded type IV secretion system. Future Microbiol 10:955–965. doi: 10.2217/fmb.15.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chung JM, Sheedlo MJ, Campbell AM, Sawhney N, Frick-Cheng AE, Lacy DB, Cover TL, Ohi MD. 2019. Structure of the Helicobacter pylori Cag type IV secretion system. Elife 8:e47644. doi: 10.7554/eLife.47644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cover TL. 2016. Helicobacter pylori diversity and gastric cancer risk. mBio 7:e01869-15. doi: 10.1128/mBio.01869-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tomb JF, White O, Kerlavage AR, Clayton RA, Sutton GG, Fleischmann RD, Ketchum KA, Klenk HP, Gill S, Dougherty BA, Nelson K, Quackenbush J, Zhou L, Kirkness EF, Peterson S, Loftus B, Richardson D, Dodson R, Khalak HG, Glodek A, McKenney K, Fitzegerald LM, Lee N, Adams MD, Hickey EK, Berg DE, Gocayne JD, Utterback TR, Peterson JD, Kelley JM, Cotton MD, Weidman JM, Fujii C, Bowman C, Watthey L, Wallin E, Hayes WS, Borodovsky M, Karp PD, Smith HO, Fraser CM, Venter JC. 1997. The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388:539–547. doi: 10.1038/41483. [DOI] [PubMed] [Google Scholar]
- 34.Babu MM, Priya ML, Selvan AT, Madera M, Gough J, Aravind L, Sankaran K. 2006. A database of bacterial lipoproteins (DOLOP) with functional assignments to predicted lipoproteins. J Bacteriol 188:2761–2773. doi: 10.1128/JB.188.8.2761-2773.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. 2003. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci 12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kostrzynska M, O'Toole PW, Taylor DE, Trust TJ. 1994. Molecular characterization of a conserved 20-kilodalton membrane-associated lipoprotein antigen of Helicobacter pylori. J Bacteriol 176:5938–5948. doi: 10.1128/jb.176.19.5938-5948.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Odenbreit S, Till M, Hofreuter D, Faller G, Haas R. 1999. Genetic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol Microbiol 31:1537–1548. doi: 10.1046/j.1365-2958.1999.01300.x. [DOI] [PubMed] [Google Scholar]
- 38.Doig P, de Jonge BL, Alm RA, Brown ED, Uria-Nickelsen M, Noonan B, Mills SD, Tummino P, Carmel G, Guild BC, Moir DT, Vovis GF, Trust TJ. 1999. Helicobacter pylori physiology predicted from genomic comparison of two strains. Microbiol Mol Biol Rev 63:675–707. doi: 10.1128/MMBR.63.3.675-707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O'Toole PW, Janzon L, Doig P, Huang J, Kostrzynska M, Trust TJ. 1995. The putative neuraminyllactose-binding hemagglutinin HpaA of Helicobacter pylori CCUG 17874 is a lipoprotein. J Bacteriol 177:6049–6057. doi: 10.1128/jb.177.21.6049-6057.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim JS, Chang JH, Seo WY, Yu GJ, Chung SI, Yum JS. 2000. Cloning and characterization of a 22 kDa outer-membrane protein (Omp22) from Helicobacter pylori. Mol Cells 10:633–641. doi: 10.1007/s10059-000-0633-0. [DOI] [PubMed] [Google Scholar]
- 41.Flach CF, Svensson N, Blomquist M, Ekman A, Raghavan S, Holmgren J. 2011. A truncated form of HpaA is a promising antigen for use in a vaccine against Helicobacter pylori. Vaccine 29:1235–1241. doi: 10.1016/j.vaccine.2010.11.088. [DOI] [PubMed] [Google Scholar]
- 42.Keenan J, Oliaro J, Domigan N, Potter H, Aitken G, Allardyce R, Roake J. 2000. Immune response to an 18-kilodalton outer membrane antigen identifies lipoprotein 20 as a Helicobacter pylori vaccine candidate. Infect Immun 68:3337–3343. doi: 10.1128/iai.68.6.3337-3343.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xue RY, Guo MF, Guo L, Liu C, Li S, Luo J, Nie L, Ji L, Ma CJ, Chen DQ, Sun S, Jin Z, Zou QM, Li HB. 2019. Synthetic lipopeptide enhances protective immunity against Helicobacter pylori infection. Front Immunol 10:1372. doi: 10.3389/fimmu.2019.01372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo L, Yin R, Xu G, Gong X, Chang Z, Hong D, Liu H, Ding S, Han X, Li Y, Tang F, Liu K. 2017. Immunologic properties and therapeutic efficacy of a multivalent epitope-based vaccine against four Helicobacter pylori adhesins (urease, Lpp20, HpaA, and CagL) in Mongolian gerbils. Helicobacter 22:e12428. doi: 10.1111/hel.12428. [DOI] [PubMed] [Google Scholar]
- 45.Zhang R, Duan G, Shi Q, Chen S, Fan Q, Sun N, Xi Y. 2016. Construction of a recombinant Lactococcus lactis strain expressing a fusion protein of Omp22 and HpaA from Helicobacter pylori for oral vaccine development. Biotechnol Lett 38:1911–1916. doi: 10.1007/s10529-016-2173-5. [DOI] [PubMed] [Google Scholar]
- 46.Zhang R, Peng X, Duan G, Shi Q, Chen S, Wang C, Fan Q, Xi Y. 2016. An engineered Lactococcus lactis strain exerts significant immune responses through efficient expression and delivery of Helicobacter pylori Lpp20 antigen. Biotechnol Lett 38:2169–2175. doi: 10.1007/s10529-016-2209-x. [DOI] [PubMed] [Google Scholar]
- 47.Huang X, Xu B, Duan G, Song C. 2013. The rOmp22-HpaA fusion protein confers protective immunity against Helicobacter pylori in mice. Curr Microbiol 67:487–492. doi: 10.1007/s00284-013-0390-x. [DOI] [PubMed] [Google Scholar]
- 48.de Jonge R, Durrani Z, Rijpkema SG, Kuipers EJ, van Vliet AH, Kusters JG. 2004. Role of the Helicobacter pylori outer-membrane proteins AlpA and AlpB in colonization of the guinea pig stomach. J Med Microbiol 53:375–379. doi: 10.1099/jmm.0.45551-0. [DOI] [PubMed] [Google Scholar]
- 49.Vallese F, Mishra NM, Pagliari M, Berto P, Codolo G, de Bernard M, Zanotti G. 2017. Helicobacter pylori antigenic Lpp20 is a structural homologue of Tipalpha and promotes epithelial-mesenchymal transition. Biochim Biophys Acta Gen Subj 1861:3263–3271. doi: 10.1016/j.bbagen.2017.09.017. [DOI] [PubMed] [Google Scholar]
- 50.Lindgren A, Pavlovic V, Flach CF, Sjoling A, Lundin S. 2011. Interferon-gamma secretion is induced in IL-12 stimulated human NK cells by recognition of Helicobacter pylori or TLR2 ligands. Innate Immun 17:191–203. doi: 10.1177/1753425909357970. [DOI] [PubMed] [Google Scholar]
- 51.Hofreuter D, Odenbreit S, Haas R. 2001. Natural transformation competence in Helicobacter pylori is mediated by the basic components of a type IV secretion system. Mol Microbiol 41:379–391. doi: 10.1046/j.1365-2958.2001.02502.x. [DOI] [PubMed] [Google Scholar]
- 52.Alm RA, Ling LSL, Moir DT, King BL, Brown ED, Doig PC, Smith DR, Noonan B, Guild BC, DeJonge BL, Carmel G, Tummino PJ, Caruso A, Uria-Nickelsen M, Mills DM, Ives C, Gibson R, Merberg D, Mills SD, Jiang Q, Taylor DE, Vovis GF, Trust TJ. 1999. Genomic-sequence comparison of two unrelated isolates of the human gastric pathogen Helicobacter pylori. Nature 397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 53.Liechti G, Goldberg JB. 2012. Outer membrane biogenesis in Escherichia coli, Neisseria meningitidis, and Helicobacter pylori: paradigm deviations in H. pylori. Front Cell Infect Microbiol 2:29. doi: 10.3389/fcimb.2012.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chalker AF, Minehart HW, Hughes NJ, Koretke KK, Lonetto MA, Brinkman KK, Warren PV, Lupas A, Stanhope MJ, Brown JR, Hoffman PS. 2001. Systematic identification of selective essential genes in Helicobacter pylori by genome prioritization and allelic replacement mutagenesis. J Bacteriol 183:1259–1268. doi: 10.1128/JB.183.4.1259-1268.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Salama NR, Shepherd B, Falkow S. 2004. Global transposon mutagenesis and essential gene analysis of Helicobacter pylori. J Bacteriol 186:7926–7935. doi: 10.1128/JB.186.23.7926-7935.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mao G, Zhao Y, Kang X, Li Z, Zhang Y, Wang X, Sun F, Sankaran K, Zhang XC. 2016. Crystal structure of E. coli lipoprotein diacylglyceryl transferase. Nat Commun 7:10198. doi: 10.1038/ncomms10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sankaran K, Gan K, Rash B, Qi HY, Wu HC, Rick PD. 1997. Roles of histidine-103 and tyrosine-235 in the function of the prolipoprotein diacylglyceryl transferase of Escherichia coli. J Bacteriol 179:2944–2948. doi: 10.1128/jb.179.9.2944-2948.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pailler J, Aucher W, Pires M, Buddelmeijer N. 2012. Phosphatidylglycerol::prolipoprotein diacylglyceryl transferase (Lgt) of Escherichia coli has seven transmembrane segments, and its essential residues are embedded in the membrane. J Bacteriol 194:2142–2151. doi: 10.1128/JB.06641-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vogeley L, El Arnaout T, Bailey J, Stansfeld PJ, Boland C, Caffrey M. 2016. Structural basis of lipoprotein signal peptidase II action and inhibition by the antibiotic globomycin. Science 351:876–880. doi: 10.1126/science.aad3747. [DOI] [PubMed] [Google Scholar]
- 60.Noland CL, Kattke MD, Diao J, Gloor SL, Pantua H, Reichelt M, Katakam AK, Yan D, Kang J, Zilberleyb I, Xu M, Kapadia SB, Murray JM. 2017. Structural insights into lipoprotein N-acylation by Escherichia coli apolipoprotein N-acyltransferase. Proc Natl Acad Sci U S A 114:E6044–E6053. doi: 10.1073/pnas.1707813114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiktor M, Weichert D, Howe N, Huang CY, Olieric V, Boland C, Bailey J, Vogeley L, Stansfeld PJ, Buddelmeijer N, Wang M, Caffrey M. 2017. Structural insights into the mechanism of the membrane integral N-acyltransferase step in bacterial lipoprotein synthesis. Nat Commun 8:15952. doi: 10.1038/ncomms15952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lu G, Xu Y, Zhang K, Xiong Y, Li H, Cui L, Wang X, Lou J, Zhai Y, Sun F, Zhang XC. 2017. Crystal structure of E. coli apolipoprotein N-acyl transferase. Nat Commun 8:15948. doi: 10.1038/ncomms15948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xiao Y, Wall D. 2014. Genetic redundancy, proximity, and functionality of lspA, the target of antibiotic TA, in the Myxococcus xanthus producer strain. J Bacteriol 196:1174–1183. doi: 10.1128/JB.01361-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Armbruster KM, Meredith TC. 2017. Identification of the Lyso-form N-acyl intramolecular transferase in low-GC Firmicutes. J Bacteriol 199:e00099-17. doi: 10.1128/JB.00099-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McClain MS, Duncan SS, Gaddy JA, Cover TL. 2013. Control of gene expression in Helicobacter pylori using the Tet repressor. J Microbiol Methods 95:336–341. doi: 10.1016/j.mimet.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cover TL, Lacy DB, Ohi MD. 26 March 2020, posting date The Helicobacter pylori Cag type IV secretion system. Trends Microbiol doi: 10.1016/j.tim.2020.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fischer W. 2011. Assembly and molecular mode of action of the Helicobacter pylori Cag type IV secretion apparatus. FEBS J 278:1203–1212. doi: 10.1111/j.1742-4658.2011.08036.x. [DOI] [PubMed] [Google Scholar]
- 68.Theisen M, Rioux CR, Potter AA. 1992. Molecular cloning, nucleotide sequence, and characterization of a 40,000-molecular-weight lipoprotein of Haemophilus somnus. Infect Immun 60:826–831. doi: 10.1128/IAI.60.3.826-831.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen R, Henning U. 1987. Nucleotide sequence of the gene for the peptidoglycan-associated lipoprotein of Escherichia coli K12. Eur J Biochem 163:73–77. doi: 10.1111/j.1432-1033.1987.tb10738.x. [DOI] [PubMed] [Google Scholar]
- 70.Jung JU, Gutierrez C, Villarejo MR. 1989. Sequence of an osmotically inducible lipoprotein gene. J Bacteriol 171:511–520. doi: 10.1128/jb.171.1.511-520.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takase I, Ishino F, Wachi M, Kamata H, Doi M, Asoh S, Matsuzawa H, Ohta T, Matsuhashi M. 1987. Genes encoding two lipoproteins in the leuS-dacA region of the Escherichia coli chromosome. J Bacteriol 169:5692–5699. doi: 10.1128/jb.169.12.5692-5699.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S, von Heijne G, Nielsen H. 2019. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37:420–423. doi: 10.1038/s41587-019-0036-z. [DOI] [PubMed] [Google Scholar]
- 73.Narita S, Tokuda H. 2011. Overexpression of LolCDE allows deletion of the Escherichia coli gene encoding apolipoprotein N-acyltransferase. J Bacteriol 193:4832–4840. doi: 10.1128/JB.05013-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yakushi T, Masuda K, Narita S, Matsuyama S, Tokuda H. 2000. A new ABC transporter mediating the detachment of lipid-modified proteins from membranes. Nat Cell Biol 2:212–218. doi: 10.1038/35008635. [DOI] [PubMed] [Google Scholar]
- 75.Matsuyama S, Tajima T, Tokuda H. 1995. A novel periplasmic carrier protein involved in the sorting and transport of Escherichia coli lipoproteins destined for the outer membrane. EMBO J 14:3365–3372. doi: 10.1002/j.1460-2075.1995.tb07342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kanamaru K, Taniguchi N, Miyamoto S, Narita S, Tokuda H. 2007. Complete reconstitution of an ATP-binding cassette transporter LolCDE complex from separately isolated subunits. FEBS J 274:3034–3043. doi: 10.1111/j.1742-4658.2007.05832.x. [DOI] [PubMed] [Google Scholar]
- 77.Okuda S, Tokuda H. 2009. Model of mouth-to-mouth transfer of bacterial lipoproteins through inner membrane LolC, periplasmic LolA, and outer membrane LolB. Proc Natl Acad Sci U S A 106:5877–5882. doi: 10.1073/pnas.0900896106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Taniguchi N, Matsuyama S, Tokuda H. 2005. Mechanisms underlying energy-independent transfer of lipoproteins from LolA to LolB, which have similar unclosed {beta}-barrel structures. J Biol Chem 280:34481–34488. doi: 10.1074/jbc.M507388200. [DOI] [PubMed] [Google Scholar]
- 79.Matsuyama S, Yokota N, Tokuda H. 1997. A novel outer membrane lipoprotein, LolB (HemM), involved in the LolA (p20)-dependent localization of lipoproteins to the outer membrane of Escherichia coli. EMBO J 16:6947–6955. doi: 10.1093/emboj/16.23.6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson EM, Gaddy JA, Voss BJ, Hennig EE, Cover TL. 2014. Genes required for assembly of pili associated with the Helicobacter pylori cag type IV secretion system. Infect Immun 82:3457–3470. doi: 10.1128/IAI.01640-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R. 2001. Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42:1337–1348. doi: 10.1046/j.1365-2958.2001.02714.x. [DOI] [PubMed] [Google Scholar]
- 82.Hu B, Khara P, Song L, Lin AS, Frick-Cheng AE, Harvey ML, Cover TL, Christie PJ. 2019. In situ molecular architecture of the Helicobacter pylori Cag type IV secretion system. mBio 10:e00849-19. doi: 10.1128/mBio.00849-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Frick-Cheng AE, Pyburn TM, Voss BJ, McDonald WH, Ohi MD, Cover TL. 2016. Molecular and structural analysis of the Helicobacter pylori cag type IV secretion system core complex. mBio 7:e02001-15. doi: 10.1128/mBio.02001-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Shaffer CL, Gaddy JA, Loh JT, Johnson EM, Hill S, Hennig EE, McClain MS, McDonald WH, Cover TL. 2011. Helicobacter pylori exploits a unique repertoire of type IV secretion system components for pilus assembly at the bacteria-host cell interface. PLoS Pathog 7:e1002237. doi: 10.1371/journal.ppat.1002237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Debowski AW, Sehnal M, Liao T, Stubbs KA, Marshall BJ, Benghezal M. 2015. Expansion of the tetracycline-dependent regulation toolbox for Helicobacter pylori. Appl Environ Microbiol 81:7969–7980. doi: 10.1128/AEM.02191-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wessel D, Flügge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 138:141–143. doi: 10.1016/0003-2697(84)90782-6. [DOI] [PubMed] [Google Scholar]
- 87.Schägger H. 2006. Tricine-SDS-PAGE. Nat Protoc 1:16–22. doi: 10.1038/nprot.2006.4. [DOI] [PubMed] [Google Scholar]
- 88.Bordier C. 1981. Phase separation of integral membrane proteins in Triton X-114 solution. J Biol Chem 256:1604–1607. [PubMed] [Google Scholar]
- 89.Busler VJ, Torres VJ, McClain MS, Tirado O, Friedman DB, Cover TL. 2006. Protein-protein interactions among Helicobacter pylori Cag proteins. J Bacteriol 188:4787–4800. doi: 10.1128/JB.00066-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Loh JT, Torres VJ, Cover TL. 2007. Regulation of Helicobacter pylori cagA expression in response to salt. Cancer Res 67:4709–4715. doi: 10.1158/0008-5472.CAN-06-4746. [DOI] [PubMed] [Google Scholar]
- 91.Ivie SE, McClain MS, Algood HMS, Lacy DB, Cover TL. 2010. Analysis of a -helical region in the p55 domain of Helicobacter pylori vacuolating toxin. BMC Microbiol 10:60. doi: 10.1186/1471–2180-10–60. doi: 10.1186/1471-2180-10-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. 2015. The Phyre2 Web portal for protein modeling, prediction and analysis. Nat Protoc 10:845–858. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera - a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 94.Ando T, Israel DA, Kusugami K, Blaser MJ. 1999. HP0333, a member of the dprA family, is involved in natural transformation in Helicobacter pylori. J Bacteriol 181:5572–5580. doi: 10.1128/JB.181.18.5572-5580.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Forsyth MH, Cover TL. 2000. Intercellular communication in Helicobacter pylori: luxS is essential for the production of an extracellular signaling molecule. Infect Immun 68:3193–3199. doi: 10.1128/IAI.68.6.3193-3199.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee TS, Krupa RA, Zhang F, Hajimorad M, Holtz WJ, Prasad N, Lee SK, Keasling JD. 2011. BglBrick vectors and datasheets: a synthetic biology platform for gene expression. J Biol Eng 5:12. doi: 10.1186/1754-1611-5-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Posttranslational modifications in synthesis of lipoproteins in Gram-negative bacteria. The first modification is the addition of a diacylglyceride to the cysteine sulfhydryl of the preprolipoprotein, catalyzed by prolipoprotein diacylglyceryl transferase (Lgt). Amino acids preceding the cysteine are cleaved by prolipoprotein signal peptidase (Lsp), resulting in a diacylated apolipoprotein. Finally, a fatty acid is ligated to the amino terminus of the amino-terminal cysteine by apolipoprotein N-acyltransferase (Lnt) to produce the mature triacylated lipoprotein. Download FIG S1, PDF file, 0.1 MB (138.1KB, pdf) .
Copyright © 2020 McClain et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.