

Summary
Astrocytic glycogen is an important energy reserve in the brain and is believed to supply fuel during energy crisis. However, the pattern of glycogen metabolism in ischemic stroke and its potential therapeutic impact on neurological outcomes are still unknown. Here, we found extensive brain glycogen accumulation after reperfusion in ischemic stroke patients and primates. Glycogenolytic dysfunction in astrocytes is responsible for glycogen accumulation, caused by inactivation of the protein kinase A (PKA)-glycogen phosphorylase kinase (PhK)-glycogen phosphorylase (GP) cascade accompanied by the activation of glycogen synthase kinase-3β (GSK3β). Genetic or pharmacological augmentation of astrocytic GP could promote astrocyte and neuron survival and improve neurological behaviors. In addition, we found that insulin exerted a neuroprotective effect, at least in part by rescuing the PKA-PhK-GP cascade to maintain homeostasis of glycogen metabolism during reperfusion. Together, our findings suggest a promising intervention for undesirable outcomes in ischemic stroke.
Subject Areas: Neuroscience, Molecular Neuroscience, Cellular Neuroscience
Graphical Abstract
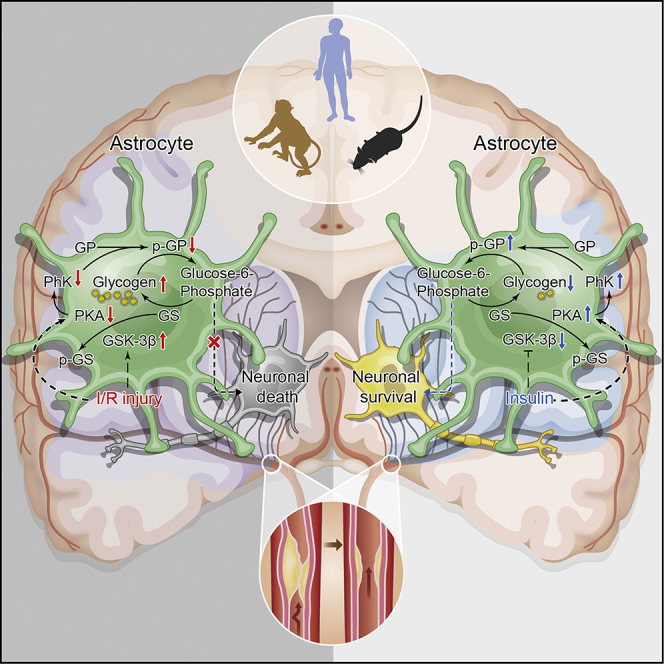
Highlights
-
•
Glycogen accumulates upon cerebral reperfusion in humans, primates, and rodents
-
•
Impaired glycogenolysis underlies excess glycogen during cerebral reperfusion
-
•
Activating glycogenolysis protects against acute and subacute reperfusion insult
-
•
Insulin mediates neuroprotection partly by rescuing glycogenolysis upon reperfusion
Neuroscience; Molecular Neuroscience; Cellular Neuroscience
Introduction
Stroke is the leading cause of mortality and disability in the adult population (GBD 2015 DALYs and HALE Collaborators, 2016, GBD 2015 Mortality and Causes of Death Collaborators, 2016), and approximately 70% of all strokes are ischemic strokes (Hankey, 2017). Restoration of blood flow to the brain with thrombolysis or endovascular thrombectomy after ischemic stroke onset is the most effective therapeutic strategy to reduce the infarct region and salvage the cells in the ischemic penumbra (Ma et al., 2019). However, reperfusion itself contributes to cerebral injury and greatly increases the incidence of cerebral edema and hemorrhagic transformation (Bar and Biller, 2018). Blood deficiency and resupply during ischemia/reperfusion (I/R) disrupts brain energy homeostasis, which inevitably aggravates glutamate excitotoxicity, calcium overload, free radical formation, and inflammation, known as the traditional underlying mechanisms of I/R injury (Krewson et al., 2020, Pundik et al., 2012).
The brain accounts for only 2% of the body weight but consumes 20% of the energy of the whole body (Magistretti and Allaman, 2015). Thus, slight energy deficiency in the brain causes severe dysfunction. Glycogen is the only endogenous energy reserve for the brain during cerebrovascular obstruction and is steadily and dynamically maintained through the balance between glycogenesis and glycogenolysis (Bak et al., 2018). Glycogen synthase kinase-3 (GSK3) and protein kinase A (PKA) comodulate the activity of glycogen synthase (GS), the rate-limiting enzyme in glycogenesis (Zois and Harris, 2016). Glycogen phosphorylase (GP), the key enzyme in glycogenolysis, is controlled by the PKA-glycogen phosphorylase kinase (PhK) cascade (Zois and Harris, 2016). Glycogen is mainly located in astrocytes, but recently, very sensitive assays revealed that it exists in small amounts in neurons with active glycogen metabolism (Duran et al., 2019, Saez et al., 2014). Glycogen plays important roles in astrocyte energetics, including pumping Ca2+ into the endoplasmic reticulum (ER) controlling the extracellular K+ concentration and managing oxidative stress (Dienel, 2019, Dienel and Cruz, 2015). In addition, the stored glycogen in astrocytes can be rapidly triggered to generate metabolic support for neighboring neurons by switching astrocytic glycolysis from blood-borne glucose to glycogen, which spares an equivalent amount of blood-borne glucose for neurons (Dienel and Rothman, 2019). However, little is known about the alterations in glycogen metabolism that occur during recanalization and their impacts on neurological outcomes after ischemic stroke.
Here, we provide evidence that strongly suggests glycogen accumulation at the onset of reperfusion is associated with the development of I/R injury in stroke patients and animal models. In addition, dysfunction of the PKA-PhK-GP cascade is involved in glycogenolytic reprogramming in astrocytes. Genetic and pharmacological augmentation of glycogen breakdown during recirculation could rescue astrocyte and cocultured neuron survival and improve neurological outcomes after ischemic stroke. In addition, we found that insulin exhibits neuroprotective effects, at least in part by rescuing the PKA-PhK-GP cascade to maintain homeostasis of glycogen metabolism during reperfusion.
Results
Glycogen Accumulates in the Human, Primate, and Rodent Brain during Recanalization after Ischemia
Firstly, we conducted studies on brain tissues from 4 stroke patients who received thrombolytic treatment within 6 h after the onset of stroke. The postmortem interval was approximately 30–40 min, and the interval between thrombolysis and death for these patients ranged from 11 h to 14 h (Table S1). A schematic of the penumbra in the ipsilateral hemisphere and the homologous region in the contralateral hemisphere was shown in Figure 1A (top panel). The levels of glycogen significantly increased in the penumbra compared with the contralateral region after recanalization in stroke patients, as indicated by periodic acid-Schiff (PAS) staining and biochemical assays (Figure 1A, bottom and right panel). Glycogen accumulation was also observed in monkey and mouse brains at 12 h after reperfusion (Figure 1A, bottom and right panel), indicating that reperfusion-induced glycogen accumulation is common across species. The oxygen-glucose deprivation/reoxygenation (OGD/R) model was further used to mimic I/R stress in vitro. Consistent with the results obtained in vivo, extensive glycogen accumulation was observed in cultured astrocytes in vitro after OGD/R, as demonstrated by PAS staining and biochemical assays (Figure 1B). In addition, cellular localization was investigated using electron microscopy, and we observed that a large amount of glycogen was mainly distributed in astrocytes but not neurons at 12 h in the mouse I/R model (Figure 1C).
Figure 1.

Cerebral Glycogen Is Substantially Increased in Human, Primate, Rodent, and Cultured Astrocytes at the Onset of Reperfusion
(A) A representative diagram showing the core infarct and penumbral regions in the ipsilateral hemisphere after I/R onset (top). Glycogen accumulated in the ischemic penumbra of the ipsilateral hemisphere compared with the contralateral hemisphere in humans (n = 4, paired samples ttest), monkeys (n = 6, paired samples ttest, at 12 h after reperfusion), and mice (n = 8, paired samples ttest, at 12 h after reperfusion) after reperfusion, as indicated by PAS staining. The glycogen levels in the ischemic penumbra of the ipsilateral hemisphere and the homologous contralateral hemisphere were quantified with a biochemical assay. The arrows indicate glycogen-positive cells. Scale bars represent 50 μm.
(B) Increased glycogen in cultured astrocytes, as revealed by PAS staining and a biochemical assay at 12 h after reoxygenation (n = 8, independent ttest). The arrows indicate glycogen-positive cells. Scale bars represent 100 μm.
(C) Excessively elevated glycogen was localized in astrocytes but not neurons at 12 h after reperfusion in the mouse brain, as revealed using electron microscopy. The arrows indicate glycogen granules. Nu represents the nucleus. Cyto represents the cytoplasm. The blue dashed lines represent nuclear membranes, and the red dashed lines represent cell membranes. Scale bars represent 1 μm.
The data are presented as the mean ± SEM. ∗∗p < 0.01, ∗∗∗p < 0.001. See also Table S1 and Figures S1 and S14.
Next, dynamic changes in glycogen accumulation were investigated separately using electron microscopy and biochemical assays. Glycogen granule levels began to increase 2 h after reperfusion, peaked at 12 h, and accumulated for at least 72 h in the mouse model of middle cerebral artery occlusion/reperfusion (MCAO/R) (Figures 2A–2C). Consistent with these in vivo data, glycogen levels were substantially elevated in cultured astrocytes after OGD/R (Figures 2D–2F). The glycogen levels in cultured astrocytes began to increase 6 h after reoxygenation, were at least two-fold higher than the initial levels at 12 h and remained elevated for at least 72 h in the OGD/R model (Figures 2E and 2F). In addition, we observed that glycolytic capacity was inhibited and ATP production decreased at 12 h after reperfusion in the cultured astrocytes (Figure S1).
Figure 2.

Glycogen Accumulation Lasts for at Least 72 h after I/R in Rodents and Cultured Astrocytes
(A) Representative electron microscopy images of brain glycogen in mice subjected to MCAO/R. The arrows indicate glycogen granules. Scale bars represent 1 μm.
(B and C) Quantified glycogen granules (B, n = 6, one-way ANOVA with the Dunnett T3 multiple comparisons test) and glycogen levels (C, n = 6, factorial analysis) in the ischemic penumbra after reperfusion.
(D) Representative electron microscopy images of glycogen in cultured astrocytes during reoxygenation. The arrows indicate glycogen granules. Scale bars represent 1 μm.
(E and F) Quantified glycogen granules (E, n = 6, one-way ANOVA with the Dunnett T3 multiple comparisons test) and glycogen levels (F, n = 5, one-way ANOVA with the Dunnett T3 multiple comparisons test) in cultured astrocytes after reoxygenation.
The data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Dysfunction of Astrocytic GP Is Responsible for the Extensive Glycogen Accumulation Caused by Suppression of PKA/PhK
The basal glycogen levels in astrocytes depend on the balance between glycogenesis and glycogenolysis (Brewer and Gentry, 2019). We first detected the expression of key enzymes in glycogenesis and glycogenolysis in cultured astrocytes. In addition to GS, glycogen branching enzyme (GBE1) plays a role in cerebral glycogenesis to some extent (Brewer and Gentry, 2019). We found that the mRNA and protein levels of GS and GBE1 were relatively stable at different time points during OGD/R stress (Figures 3A, 3B, S2A, and S2B). GP has three isoforms in the brain: PYGB (brain isoform of GP), PYGM (muscle isoform of GP), and PYGL (liver isoform of GP) (Brewer and Gentry, 2019), and we found the mRNA proportion of PYGB, PYGM, and PYGL was 91.7%, 6.1%, and 2.2% in cultured astrocytes, respectively (Figure S2C). The changes in the protein and mRNA levels of PYGB, PYGM, and PYGL during OGD/R are presented in Figures 3C–3E and S2D–S2F. Protein level was lowest for PYGB at 12 h, when its mRNA expression was also half of the normal level (Figures 3C and S2D). Both the mRNA and protein levels of glycogen debranchingenzyme (AGL), another key enzyme in glycogenolysis, showed no significant changes (Figures 3F and S2G). Subsequently, we investigated the activity of GS and GP. The activity of GS was not markedly affected after reoxygenation, as determined by biochemical analysis (Figure 3G). However, total GP activity was decreased at 12 h after reoxygenation according to a biochemical assay (Figure 3H).
Figure 3.

Relative mRNA Expressions and In Vitro Activities of Enzymes Involved in Glycogen Metabolism Are Selectively Reduced at 12 h after OGD/R in Cultured Astrocytes
(A–F) The mRNA levels of GS (A), GBE1 (B), PYGB (C), PYGM (D), PYGL (E), and AGL (F) were determined by quantitative reverse transcription polymerase chain reaction (RT-qPCR) at 12 h after reoxygenation in cultured astrocytes (n = 8, independent t test).
(G–L) Quantified GS (G), GP (H), PhK (I), PKA (J), GSK3α (K), and GSK3β (L) activities in astrocytes at 12 h after reoxygenation in cultured astrocytes (n = 8, independent t test).
The data are presented as the mean ± SEM. ∗∗∗p < 0.001. See also Figure S2 and Table S3.
The next question is why GP, but not GS, is dysfunctional during I/R. Previous evidence has suggested that the PKA-PhK-GP pathway controls glycogen degradation and that both PKA and GSK3 can inhibit the activity of GS (Zois and Harris, 2016). Here, we found that the activity of PhK was decreased at 12 h after reoxygenation following OGD (Figure 3I). The activity of PKA was also reduced during reoxygenation (Figure 3J). There was no change in the activity of GSK3α, an isoform of GSK3 (Figure 3K), but the activity of GSK3β was upregulated (Figure 3L).
Given that cultured astrocytes are unlikely to completely represent the in vivo situation, we adopted double-labeled immunofluorescence to specifically quantify the relative expressions of key enzymes in astrocytes in a mouse model of MCAO. First, the fluorescence intensity of S100β, used as an astrocytic marker in immunofluorescence, remained stable at 12 h after reperfusion (Figure S3). Second, we found that the expression of GS, GBE1, PYGM, PYGL, and AGL was relatively stable at 12 h after reperfusion in astrocytes (marked by S100β, Figures 4A, 4B, and 4D–4F). However, a 53.5% reduction in PYGB expression was observed at 12 h after MCAO/R (Figure 4C). Previous studies suggested that phosphorylated GS and N-terminal serine phosphorylated GSK-3β were the inactivated forms and that phosphorylation is the activated form for GP and PKA (Taylor et al., 2012, Wang et al., 2014, Zois and Harris, 2016). We found that the level of phosphorylated GS in astrocytes was not changed after reperfusion (Figure 4G). We also synthesized a new antibody against phosphorylated PYGB (Figure S4) and found that the level of phosphorylated PYGB in astrocytes dropped to 5.1% of that in the sham group at 12 h after MCAO/R (Figure 4H). In addition, we found that the level of phosphorylated PKA was significantly decreased at 12 h after reperfusion (Figure 4I) and that there was a decrease in the expression of inactivated phosphorylated GSK3β at 12 h after recanalization (Figure 4J).
Figure 4.

Protein Levels of Enzymes Involved in Glycogen Metabolism Are Selectively Reduced after Reperfusion in a Mouse Stroke Model
(A–F) Left panels: coronal immunofluorescence images of frontal cortex area 1 in the ischemic penumbra after staining with an antibody against S100β and antibodies against GS (A), GBE1 (B), PYGB (C), PYGM (D), PYGL (E), and AGL (F). Right panels: quantification of relative fluorescence intensity of GS (A), GBE1 (B), PYGB (C), PYGM (D), PYGL (E), and AGL (F) in astrocytes of the ischemic penumbra at 12 h after reperfusion (n = 8, independent t test). Astrocytes were marked with S100β. The relative fluorescence intensity of the target protein was calculated as the percentage of fluorescence intensity in the colocalization area (denoted as S100β and target protein) divided by the fluorescence intensity in the S100β+ area. Scale bars represent 50 μm.
(G–J) Left panels: coronal immunofluorescence images of frontal cortex area 1 in the ischemic penumbra after staining with an antibody against S100β and antibodies against phosphorylated GS (p-GS, G), phosphorylated PYGB (p-PYGB, H), phosphorylated PKA (p-PKA, I) and phosphorylated GSK3β (p-GSK3β, J). Right panels: quantification of relative fluorescence intensity of p-GS (G), p-PYGB (H), p-PKA (I), and p-GSK3β (J) in astrocytes of the ischemic penumbra at 12 h after reperfusion (n = 8, independent ttest). Astrocytes were marked with S100β. Scale bars represent 50 μm.
The data are presented as the mean ± SEM. ∗∗∗p < 0.001. See also Figure S3, S4, and S13 and Table S2.
Augmenting Astrocytic PYGB Increases the Survival of Both Astrocytes and Cocultured Neurons and Improves Neurological Outcomes after I/R
To determine the potential clinical significance of glycogen disorders in cerebrovascular disease, we first used lentiviruses to construct PYGB overexpression (Ve-Pygb) and CRISPR/Cas9-mediated PYGB knockdown (Sg-Pygb) models in vitro (Figure S5). We found that GP mRNA expression and in vitro activity were partially restored at 12 h after OGD/R in PYGB-overexpressing astrocytes, whereas PYGB knockdown reduced GP mRNA level and in vitro activity (Figures 5A and 5B). Elevated glycogen was decreased in the PYGB overexpression model but was further increased with PYGB knockdown after OGD/R (Figure 5C). Next, the cell viability of astrocytes increased with PYGB overexpression and decreased with PYGB knockdown under OGD/R insult (Figure 5D). The proportion of apoptotic astrocytes also decreased with PYGB overexpression (Figure 5E). In addition, we found that a pharmacological PKA agonist (8-Br-cAMP, 10 μM) significantly activated GP and suppressed GS at 12 h after OGD/R (Figures S6A and S6B). Glycogen levels also decreased and the viability of astrocytes increased upon treatment with 8-Br-cAMP (Figures S6C and S6D).
Figure 5.

Enhancement of Astrocytic PYGB Improves the Survival of Cultured Astrocytes and Cocultured Neurons During OGD/R
(A) Quantified PYGB mRNA levels in PYGB-overexpressing (Ve-Pygb) and PYGB-knockdown (Sg-Pygb) cultured astrocytes at 12 h after reoxygenation (n = 8, one-way ANOVA with the Dunnett T3 multiple comparisons test). Sg represents astrocytes infected with scrambled sgRNA lentiviruses. Ve represents astrocytes infected with blank vector lentiviruses.
(B) Quantified GP activity in PYGB overexpressing and knockdown cultured astrocytes at 12 h after reoxygenation (n = 8, one-way ANOVA with the Dunnett T3 multiple comparisons test).
(C) Glycogen concentrations in PYGB overexpressing and knockdown cultured astrocytes during OGD/R stress (n = 3, factorial analysis).
(D) The relative cell viability of cultured astrocytes was determined by a Cell Counting Kit-8 (CCK-8) assay at 24 h after the onset of reoxygenation (n = 9, one-way ANOVA with the LSD multiple comparisons test). The control condition means that the astrocytes received only culture medium changes (containing glucose) at the same timepoints as the OGD/R group but no OGD stress.
(E) The numbers of apoptotic cultured astrocytes at 24 h after OGD/R were analyzed by TUNEL staining (n = 7, independent t test). The top panel represents the total cells stained by DAPI, and the bottom panel represents the apoptotic astrocytes stained by TUNEL. Scale bars represent 100 μm.
(F) Diagram of the astrocyte-neuron coculture system.
(G) Viability of neurons at 24 h after reoxygenation in the coculture system (n = 8, one-way ANOVA with the LSD multiple comparisons test). The control condition means that the cocultured neurons received only culture medium changes (containing glucose) at the same timepoints as the OGD/R group but no OGD stress.
(H) Apoptosis analysis of neurons at 24 h after reoxygenation in the coculture system (n = 7, independent t test). Scale bars represent 100 μm.
(I) LDH release in the coculture medium (n = 8, one-way ANOVA with the Dunnett T3 multiple comparisons test).
The data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figure S5, S6, and S15 and Table S3.
Considering the metabolic coupling between astrocytes and neurons, a coculture system was used that allowed the two cell types to share diffusible metabolic substrates but remain divided by a physical filter (Figure 5F). After genetic enhancement of astrocytic PYGB, the viability of neurons was significantly increased, whereas the number of apoptotic neurons was markedly decreased during reoxygenation (Figures 5G and 5H). In contrast, neuronal viability decreased when astrocytic PYGB was silenced (Figure 5G). The observed decreases in lactate dehydrogenase (LDH) in the coculture medium further revealed that the overall survival of neurons and astrocytes was promoted by the augmentation of astrocytic PYGB during OGD/R (Figure 5I).
To evaluate whether genetic enhancement of astrocyticglycogenolysis alleviates brain injury, a knock-in mouse model with astrocyte-specific PYGB overexpression (KI-Pygb) was constructed, and the KI-Pygb mice showed no differences in appearance but had increases in the expressions of PYGB and phosphorylated PYGB compared with those of their wild-type (WT) littermate controls (Figure S7). Histological and neurobehavioral analyses were performed in the acute and subacute phases (Mullins, 2006) after MCAO/R, according to the timeline shown in Figure 6A. Immunofluorescence analysis revealed that the expression of PYGB and phosphorylated PYGB in astrocytes was partially restored at 12 h after reperfusion in transgenic mice compared with WT mice (Figures 6B and 6C). Accordingly, brain glycogen accumulation was significantly attenuated at 12 h after reperfusion in KI-Pygb mice compared with WT mice (Figure 6D). The infarct volumes were notably decreased in KI-Pygb mice during the acute phase after reperfusion (Figure 6E). In addition, a corner test was performed to assess sensorimotor dysfunction (Zhang et al., 2002), and the number of right turns was clearly decreased in the KI-Pygb mice compared with the WT group after reperfusion (Figure 6F). A grid-walking test was also performed to detect the degree of motor impairment (Barbosa et al., 2016), and PYGB enhancement led to significant increases in total steps and decreases in the foot fault ratio after restoration of circulation (Video S1 and Figure 6G). The infarct volumes were markedly decreased in the KI-Pygb mice during the subacute phase after MCAO/R, as revealed by Nissl staining (Figure 6H).
Figure 6.

Enhancement of Astrocytic PYGB Ameliorates Ischemic Outcomes during MCAO/R
(A) Left panel: schematic of the corner test and grid-walking test patterns. Right panel: timeline of biochemical, neurobehavioral, and neuropathological analyses after MCAO/R treatment.
(B) Left panel: coronal immunofluorescence images of frontal cortex area 1 in the ischemic penumbra after staining with an antibody against S100β and an antibody against PYGB. Right panel: quantification of relative fluorescence intensity of PYGB in astrocyte-specific PYGB knock-in (KI-Pygb) mice and WT mice at 12 h after reperfusion (n = 8, independent t test). Astrocytes were marked with S100β. The relative fluorescence intensity of PYGB was calculated as the percentage of fluorescence intensity in the colocalization area (denoted as S100β and PYGB) divided by the fluorescence intensity in the S100β+ area. Scale bars represent 50 μm.
(C) Left panel: coronal immunofluorescence images of frontal cortex area 1 in the ischemic penumbra after staining with an antibody against S100β and an antibody against phosphorylated PYGB (p-PYGB). Right panel: quantification of relative fluorescence intensity of p-PYGB in KI-Pygb mice and WT mice at 12 h after reperfusion (n = 8, independent t test). Astrocytes were marked with S100β. Scale bars represent 50 μm.
(D) Cerebral glycogen levels in the ischemic penumbra at 12 h after MCAO/R (n = 6, independent t test).
(E) Left panel: representative brain slice images of triphenyltetrazolium chloride (TTC) staining at 24 h after MCAO/R. Right panel: the quantified infarct volumes of TTC staining (WT: n = 8; KI-Pygb: n = 7, independent ttest). Scale bars represent 1 mm.
(F) A corner test was performed to analyze the numbers of right turns in 10 trials before (Pre) and after reperfusion in the mouse model of MCAO (WT: n = 8; KI-Pygb: n = 7, repeated measures analysis).
(G) A grid-walking test was performed to assess the total steps (left panel) and foot fault ratios (right panel) before (Pre) and after reperfusion in the mouse model of MCAO (WT: n = 8; KI-Pygb: n = 7, repeated measures analysis).
(H) Nissl staining was used to assess infarct volumes at day 14 after MCAO/R (WT: n = 10; KI-Pygb: n = 9, independent ttest). Scale bar represents 2 mm.
The data are presented as the mean ± SEM. ∗∗p < 0.01. ∗∗∗p < 0.001. See also Figure S7 and Video S1 and Table S2.
Insulin Mediates Neuroprotection by Rebalancing Glycogen Metabolism via Activation of PKA/PhK and Suppression of GSK-3β
Insulin is a proteohormone that is critical for the maintenance of hepatic glycogen homeostasis (Moore et al., 2012) and mediates cardioprotective effects on I/R in heart (Bertrand et al., 2008). Here, we found that the increased glycogen levels gradually returned to normal levels after treatment with increasing concentrations of insulin (IS) during reoxygenation (Figure 7A). In addition, a significant increase in GP activity, but no effect on GS activity, was found in cultured astrocytes at 12 h after OGD/R with insulin (1 μM) treatment (Figures 7B, 7C, and S8). Unexpectedly, insulin was observed to enhance GS activity rather than GP activity in normal cultured astrocytes (Figure S9). This discrepancy prompted us to uncover the upstream alterations in glycogen metabolism. We found that both PhK and PKA were upregulated at 12 h after reoxygenation after insulin treatment (Figures 7D and 7E). The activity of GSK3β was decreased with insulin treatment, whereas the activity of GSK3α was unchanged (Figures 7F and 7G). Subsequently, similar to the above data in vitro, intracerebroventricular insulin (10 μM) treatment increased the levels of phosphorylated PYGB, PKA, and GSK3β, with no effects on phosphorylated GS in mouse brains at 12 h after blood recirculation (Figures 7H–7K). And the fluorescence intensity of S100β remained stable with intracerebroventricular insulin treatment for 12 h (Figure S10).
Figure 7.

Insulin Activates GP, PhK, and PKA Activities and Suppresses GSK3β Activity in Cultured Astrocytes and Increases Their Relative Fluorescence in Rodents after I/R
(A) Glycogen concentrations in cultured astrocytes treated with different doses of insulin after reoxygenation (n = 4, factorial analysis). Insulin was added to the medium immediately after reoxygenation. VEH represents the vehicle group, and IS represents the insulin group.
(B–G) The activities of GS (B), GP (C), PhK (D), PKA (E), GSK3α (F), and GSK3β (G) were quantified at 12 h after reoxygenation in cultured astrocytes treated with insulin (1 μM) (n = 8, independent t test).
(H–K) Left panels: coronal immunofluorescence images of frontal cortex area 1 in the ischemic penumbra after staining with an antibody against S100β and antibodies against phosphorylated GS (p-GS, H), phosphorylated PYGB (p-PYGB, I), phosphorylated PKA (p-PKA, J), and phosphorylated GSK3β (p-GSK3β, K). Right panels: quantification of relative fluorescence intensity of p-GS (H), p-PYGB (I), p-PKA (J), and p-GSK3β (K) in the ischemic penumbra at 12 h after reperfusion (n = 8, independent t test). Astrocytes were marked with S100β. Insulin (10 μM) was injected into the lateral ventricle immediately after reperfusion to achieve a final concentration of approximately 1 μM in the cerebrospinal fluid. The relative fluorescence intensity of the target protein was calculated as the percentage of fluorescence intensity in the colocalization area (denoted as S100β and target protein) divided by the fluorescence intensity in the S100β+ area. Scale bars represent 50 μm.
The data are presented as the mean ± SEM. ∗∗∗p < 0.001. See also Figures S8–S10 and Table S2.
We next examined the role of astrocyticglycogenolysis in insulin-mediated neuroprotection using Sg-Pygb models in vitro. The insulin-induced decrease in glycogen was attenuated in GP knockdown astrocytes during OGD/R (Figure 8A). Astrocyte survival appeared to be increased with insulin treatment but decreased with concomitant PYGB knockdown lentivirus treatment during reoxygenation, as determined by cell viability and apoptosis rate analyses (Figures 8B and 8C). Cocultured neuron survival was notably improved by insulin treatment during OGD/R, and this improvement could be blocked by coculture with GP knockdown astrocytes (Figures 8D and 8E). LDH release into the coculture medium also decreased with insulin treatment and increased when astrocytic PYGB was inhibited during reoxygenation (Figure 8F).
Figure 8.

Insulin Decreases Astrocyte and Neuron Death and Protects the Brain Damage during I/R Injury by Enhancing Astrocytic GP Activity
(A) Glycogen level in cultured astrocytes at 12 h after OGD/R (n = 8, one-way ANOVA with the LSD multiple comparisons test). Insulin (1 μM) was added to the medium immediately after reoxygenation. VEH represents the vehicle group, and IS represents the insulin group. Sg represents astrocytes infected with scrambled sgRNA lentiviruses. Sg-Pygb represents GP-knockdown astrocytes.
(B) Relative cell viability of the cultured astrocytes at 24 h after reoxygenation (n = 8, one-way ANOVA with the LSD multiple comparisons test). The control condition means that the astrocytes received culture medium changes (containing glucose) without OGD/R stress.
(C) The numbers of apoptotic cultured astrocytes were analyzed by TUNEL staining at 24 h after reoxygenation (n = 7, one-way ANOVA with the LSD multiple comparisons test). Scale bars represent 100 μm.
(D) Viability of neurons treated at 24 h after reoxygenation in the astrocyte-neuron coculture system (n = 8, one-way ANOVA with the LSD multiple comparisons test). The control condition means that the cocultured neurons received culture medium changes (containing glucose) without OGD/R stress.
(E) The numbers of apoptotic neurons in coculture were analyzed by TUNEL staining at 24 h after reoxygenation (n = 7, one-way ANOVA with the LSD multiple comparisons test). Scale bars represent 100 μm.
(F) LDH release in the coculture medium was analyzed (n = 8, one-way ANOVA with the Dunnett T3 multiple comparisons test).
(G) Glycogen levels in the ischemic penumbra at 12 h after MCAO/R in mice (n = 7, one-way ANOVA with the LSD multiple comparisons test). Insulin (10 μM) was injected into the lateral ventricle immediately after reperfusion to achieve a final concentration of approximately 1 μM in the cerebrospinal fluid. Sh represents mice infected with scrambled shRNA AAVs. Sh-Pygb represents PYGB knockdown mice.
(H) The infarct volumes at 24 h after MCAO/R in mice were analyzed by TTC staining (n = 9, one-way ANOVA with the LSD multiple comparisons test). Scale bars represent 1 mm.
(I) A corner test was performed to analyze the numbers of right turns in 10 trials before (Pre) and after reperfusion in the mouse model of MCAO (n = 7, repeated measures analysis).
(J) A grid-walking test was performed to assess the total steps (left panel) and foot fault ratios (right panel) before (Pre) and after reperfusion in the mouse model of MCAO (n = 7, repeated measures analysis).
(K) Nissl staining was used to assess infarct volumes at day 14 after MCAO/R in mice (n = 7, one-way ANOVA with the LSD multiple comparisons test). Scale bar represents 2 mm.
The data are presented as the mean ± SEM. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. See also Figures S11 and S12.
To conditionally suppress astrocytic PYGB expression in vivo, an adeno-associated virus (AAV) containing the astrocyte-specific glial fibrillary acidic protein (GFAP) promoter (Brenner et al., 1994) and an exogenous short hairpin RNA (shRNA) targeting the pgyb gene was constructed (Sh-Pygb). Exogenous shRNA, which was marked by FLAG, was predominantly localized in astrocytes, which were marked by GFAP (Figure S11A). The expression of astrocytic PYGB and phosphorylated PYGB was clearly downregulated in the PYGB knockdown mice (Figures S11B–S11D). The accumulated glycogen in the mouse brain was also diminished with insulin treatment after reperfusion, and the decreases in glycogen were suppressed in mice with pygb-modified astrocytes (Figure 8G). Furthermore, the infarct volumes were decreased after insulin administration, and astrocytic PYGB inhibition blocked this effect at 24 h after MCAO/R (Figure 8H). Consistent with the above acute-phase data, neurobehavioral and neuropathological impairments were revealed to be significantly alleviated with insulin treatment in the corner test (Figure 8I) and grid-walking test (Figure 8J) and by Nissl staining (Figure 8K) during the subacute phase of recirculation; these effects were markedly blocked when astrocyticglycogenolysis was suppressed. In addition, intraventricular injection of insulin did not affect blood glucose after MCAO/R operation (Figure S12).
Discussion
The present study reveals that the accumulation of glycogen is strongly associated with the development of I/R injury in mice subjected to transient cerebral ischemia and cultured astrocytes treated with OGD. Our findings provide the evidence that glycogen accumulation is common across species following transient cerebral ischemia. We found that PYGB, the brain isoform of GP in glycogenolysis, plays a substantial role in glycogen-accumulation-associated neuropathy. The PKA-PhK-GP cascade participates in the reprogramming of glycogenolysis during recanalization after ischemic stroke and reoxygenation after OGD.
In this study, we examined cerebral glycogen levels after stroke in humans and nonhuman primates and found that glycogen was excessively accumulated in the ischemic penumbra of humans and nonhuman primates after cerebral I/R, which is consistent with findings from previous observations in rodents (Folbergrova et al., 1996, Gurer et al., 2009, Hossain et al., 2014, Kajihara et al., 2001). To determine the time window for glycogen accumulation after cerebral I/R, brain glycogen levels were continuously detected until 72 h after reperfusion in a focal ischemia rodent model, and we showed that astrocytic glycogen accumulated at 6 h and peaked at 12 h after cerebral reperfusion. Notably, postmortem tissue handling caused glycogen loss in the contralateral hemisphere of stroke patients in this study, because glycogen levels in Figure 1A were reduced to approximately 13% compared with those reported in a previous study (Kirsch and Leitner, 1967). Postmortem loss of glycogen could also be expected in the monkey and mouse brains and cultured astrocytes due to the lag time between decapitation and freezing or cell lysis (See Transparent Methods).
Considering the pivotal role of glycogen in maintaining cerebral physiological function, elucidation of the underlying mechanism of reperfusion-induced glycogen accumulation is necessary. Limited evidence suggests that GSK3beta activity is upregulated during I/R stress (Ramagiri and Taliyan, 2017). Theoretically, GSK3β inhibits GS activity through phosphorylation, and inactivation of GS could decrease the levels of glycogen during I/R (Pederson, 2019). However, where is the excessive glycogen in astrocytes derived from after recirculation? The PKA-PhK-GP pathway has been revealed to determine glycogen degradation in the liver, and PKA can suppress GS activity as well (Zois and Harris, 2016). These clues prompted us to investigate the alterations of these key enzymes in glycogen metabolism. We found that the astrocytic PKA-PhK-GP cascade was significantly inactivated during reperfusion. The activity of GS remained at normal levels due to the neutralizing effects of PKA suppression and GSK3β activation. Interestingly, inequality of changes in mRNA level and protein level related to enzyme activity was found in this study. For instance, Figure 4C showed a 53.5% reduction in PYGB expression, whereas the level of phosphorylated PYGB in astrocytes dropped to 5.1% at 12 h after MCAO/R in Figure 4H. Also, in Figure 7C, the in vitro activity of GP increased after insulin-treated, whereas its protein level was still decreased at 12 h after OGD/R (Figure S8C). In addition, we observed that the expression of PYGB was preferentially affected in mice suffered from MCAO/R and in cultured astrocytes subjected to OGD/R compared with that of PYGM and PYGL. We are not sure whether only PYGB activity was also decreased during reperfusion because the activities of PYGM and PYGL were not detected in this study. No evidence suggests that the PKA-PhK pathway regulates only PYGB not PYGM or PYGL activity. Therefore, we speculate that PYGB is vulnerable to I/R mainly because it accounts for a large proportion of GP mRNA isoforms in cultured astrocytes (91.7%).
Our findings seem to be inconsistent with the existing idea that glycogen storage caused by pharmacological inhibition of glycogenolysis can prevent energy crisis and alleviate brain damage. Constant perfusion of the glycogen breakdown inhibitor, ingliforib, to increase glycogen levels 30 min before myocardial ischemia shows a cardioprotective effect, because glycogen is rapidly degraded to glucose-1-phosphate to provide additional energy (Tracey et al., 2004). Some GP inhibitors, such as CP-316819 and maslinic acid, have also been used to elevate the levels of brain glycogen before brain ischemia (Guan et al., 2011, Xu and Sun, 2010). Release of prestored glycogen has been viewed as a promising therapeutic strategy to rapidly supply energy under ischemia and alleviate brain damage (Xu and Sun, 2010). However, pharmacological treatments targeting glycogenolysis should be carefully used. Our results showed that reprogramming of GP and impairment of glycogen breakdown, instead of glycogen deficit, occurred during the reperfusion phase. Previous studies also revealed that the elevation of glycogen levels during recirculation is prolonged in proportion to the duration of ischemia (Long et al., 1972, Mrsulja et al., 1975, Mrsulja et al., 1979). We strongly suspect that sustained pharmacological action of these GP inhibitors during the reperfusion stage will lead to an aggravated ischemic outcome. Therefore, the timing of pharmacological intervention is particularly important, and short-acting GP antagonists and GP agonists may need be administered separately before ischemia and after reperfusion, respectively, according to the patient's condition.
In this study, we revealed that the mobilization of glycogenolysis in astrocytes contributes to the survival of neighboring neurons after reperfusion. This phenomenon is interesting, but the underlying mechanism might be complicated. Previous studies have provided clues about how degraded glycogen attenuates neuronal damage. First, extra glycogen could fuel Na+/K+-ATPase to enhance K+ uptake in astrocytes and improve neuronal excitation recovery (Xu et al., 2013). The activation of Na+/K+-ATPase also accelerates excitatory glutamate uptake by stimulating the excitatory amino acid transporter and attenuates glutamate excitotoxicity to neurons (Xu et al., 2014, Yin et al., 2019). In addition, glycogen might provide energy for loading Ca2+ into the ER and relieve the neuronal damage caused by calcium overload (Müller et al., 2014). Finally, increased glucose-6-phosphate resulting from glycogen feedback inhibits hexokinase, thereby reducing astrocyte consumption of blood-borne glucose and sparing glucose in neurons (Dienel and Rothman, 2019). Notably, we observed a 43% reduction in glycolytic capacity during OGD/R stress in this study. However, this did not confirm that degraded glycogen could not be used as a fuel for astrocytes because the capacity of most enzymes is suggested to greatly exceed the flux through the metabolic step (Lowry and Passonneau, 1964). In a word, we speculate that in the context of ischemia, compromised astrocytes can boost their energy reserves by reprogramming astrocyte-neuron interactions after reperfusion. Further study should focus on these pathways to elucidate the underlying metabolic profiles during glycogen mobilization and to determine their implications for clinical stroke therapy.
During the past several decades, the glucose, insulin, and potassium (GIK) metabolic cocktail has been widely used in the clinic for cardioprotection after myocardial infarction. Growing evidence has revealed that insulin, but not glucose or potassium, plays an important role in the protective effects of GIK against I/R injury (Zhang et al., 2006). The underlying mechanism of insulin-mediated neuroprotection has been reported to primarily depend on maintenance of calcium homeostasis, inhibition of inflammation, and downregulation of free radical release in heart (Bertrand et al., 2008). In addition, insulin can stimulate erythropoietin production (Masuda et al., 1997), regulate gliotransmission (Cai et al., 2018), and modulate glucose metabolism (Fernandez et al., 2017) in astrocytes and stimulate glucose utilization (Ashrafi et al., 2017, Pearson-Leary et al., 2018) in neurons, which may also contribute to recovery after I/R. However, we cannot ignore the fact that insulin is an important regulator of glycogen homeostasis in the body and that its glycogenic target in the context of neuroprotection is far from completely understood. Recent studies revealed that insulin suppresses the activity of GSK3β through activation of the phosphatidylinositol 3-kinase (PI3K)-protein kinase B (PKB) pathway upon I/R stress (Bertrand et al., 2008, Mishra et al., 2018). Notably, the levels of brain glycogen decreased with insulin treatment during reperfusion in this study, which seems to contradict the finding that insulin promotes glycogen synthesis by inhibiting GSK3β. Here, we found that insulin could also activate the PKA-PhK-GP pathway and that the neuroprotective effects of insulin could be attenuated by PYGB knockdown, which may account for the contribution of insulin-mediated recovery during the acute and subacute phases after stroke. Therefore, insulin in the brain is more likely to act as a coordinator to maintain glycogen metabolic homeostasis rather than to simply accelerate glycogen synthesis when the level of glycogen has already substantially increased during reperfusion.
Collectively, our findings provide evidence that glycogen accumulation occurs in humans and monkeys following transient ischemia and that reprogramming of glycogenolysis leads to astrocytic glycogen accumulation and brain damage. Enhancing glycogenolytic metabolism during the acute stage of reperfusion may protect the brain from I/R injury. These results also support the notion that the activated PKA-PhK-GP cascade underlies insulin-mediated neuroprotection. Thus, glycogenolysis is a potential intervention target for ischemic stroke, but the precise application of targeting strategies should be carefully considered according to the timing of ischemia.
Limitations of the Study
One limitation is the possibility that I/R insult has effects on malin or laforin, leading to an abnormal structure of glycogen that then accumulates, as in Lafora disease (Dukhande et al., 2011, Gentry et al., 2005), which is independent of deficits in GP and its upstream signaling pathways. In our study, we did not detect the activities of laforin and malin in I/R injury. However, we observed that cerebral reperfusion did not induce changes in the expression of laforin and malin in astrocytes (Figure S13). In addition, a previous study revealed that accumulated glycogen is not restricted to astrocytes but can be detected in neurons in Lafora disease (Augé et al., 2018). As shown in Figure 1C, no accumulated glycogen granules were found in neurons using electron microscopy in the mouse MCAO/R model. Therefore, it is conceivable that glycogen accumulation was not due to laforin or malin dysfunction during cerebral reperfusion disorders, but this remains to be further tested.
Another limitation is the existence of some methodological weaknesses in this study. First, the lag time of dissection to obtain the penumbra tissue in sample handling during the glycogen quantitative assay caused glycogen loss. Previous studies suggest that at least 50% of glycogen degrades in 30 s after decapitation in adult mice (Lowry et al., 1964), and our glycogen levels in the mouse cortex are approximately half of those reported previously (Oe et al., 2016). In addition, all methods to harvest tissue and cells for glycogen assays had a lag time before freezing or lysis during which glycogenolysis might have occurred, particularly in the control samples where GP activity was not affected. This would cause underestimation of glycogen concentration in the control samples and overestimation of relative postischemic concentration. Therefore, microwave fixation of mouse brain tissue, which preserves glycogen much better than decapitation (Oe et al., 2016), should be adopted for glycogen quantitative assays in future studies. Secondly, double-labeled immunofluorescence has some disadvantages for the quantification of enzyme activity in vivo. The S100β is mainly in astrocytic soma that reports only about 15% of the cell volume (Bushong et al., 2002), and most of the volume that also contains about half of the glycogen (Oe et al., 2016) and presumably related enzymes is actually in the small processes that were not analyzed in the present study. In addition, the expression of phosphorylated enzyme does not equal the actual activity of the enzyme, and the relationship between fluorescence intensity and expression of the target protein is not linear (Barnett et al., 2019, Odell and Cook, 2013), which illustrates that immunofluorescence is imprecise and only a semiquantitative method to evaluate enzyme activities in vivo. More precise methods might be developed to quantify astrocyte-specific enzyme activity in vivo. Thirdly, hyperglycemia is known to worsen stroke outcome (Zhang et al., 2013) and use of 25 mM glucose in the cultures might exaggerate damage caused by OGD in this study. Normal glucose medium (5.5 mM glucose) should be used in the future related studies.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Lize Xiong (mzkxlz@126.com).
Materials Availability
Viruses and mouse lines generated in this study have been deposited in the laboratory of Department of Anesthesiology and Perioperative Medicine, Xijing Hospital, Fourth Military Medical University. Viruses and mouse lines generated in this study will be made available on request, but we may require a payment and a completed Materials Transfer Agreement if there is potential for commercial application.
Data and Code Availability
All relevant data are available from the corresponding author (L.X.) upon reasonable request. This study did not generate code.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by funds from the Major Programs of the National Natural Science Foundation of China (No. 81590954), the International Cooperation and Exchange of the National Natural Science Foundation of China (No.81420108013), the National Natural Science Foundation of China (Nos.81471110 and 81671184), and the Assistance Program of Xijing Hospital (No.XJZT18MJ19). We thank the China Brain Bank, Zhejiang University, for providing the human brain tissues, and we thank the included stroke patients and their families for their trust.
Author Contributions
L.X. and Y.L. conceived, designed, and supervised the study. Y.C., H.G., Z.F., and X.Z. performed most of the experiments. Y.C. established the OGD/R model and completed most of the cell-based experiments. H.G. performed the TTC staining, Nissl staining, and behavioral tests. Z.F. performed the immunoblotting, RT-qPCR, and immunofluorescence staining. X.Z. performed intracerebroventricular injection. W.T. performed the cell-metabolism-associated assays using the Seahorse system. D.W. and X.J. performed the experiments involving rhesus monkeys. T.G. isolated primary astrocytes and neurons. S.W. performed the MCAO surgery on rodents. A.Y. and L.T. analyzed the data. L.X., Y.L., and H.D. drafted and revised the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: May 22, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101136.
Contributor Information
Yan Li, Email: liyanxjtu@xjtu.edu.cn.
Lize Xiong, Email: mzkxlz@126.com.
Supplemental Information
References
- Ashrafi G., Wu Z., Farrell R.J., Ryan T.A. GLUT4 mobilization supports energetic demands of active synapses. Neuron. 2017;93:606–615.e3. doi: 10.1016/j.neuron.2016.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augé E., Pelegrí C., Manich G., Cabezón I., Guinovart J.J., Duran J., Vilaplana J. Astrocytes and neurons produce distinct types of polyglucosan bodies in Lafora disease. Glia. 2018;66:2094–2107. doi: 10.1002/glia.23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bak L.K., Walls A.B., Schousboe A., Waagepetersen H.S. Astrocytic glycogen metabolism in the healthy and diseased brain. J. Biol. Chem. 2018;293:7108–7116. doi: 10.1074/jbc.R117.803239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar B., Biller J. Select hyperacute complications of ischemic stroke: cerebral edema, hemorrhagic transformation, and orolingual angioedema secondary to intravenous alteplase. Expert Rev. Neurother. 2018;18:749–759. doi: 10.1080/14737175.2018.1521723. [DOI] [PubMed] [Google Scholar]
- Barbosa E.H., Vallim J.H., Lachat J.J., de Castro V.L. Assessments of motor abnormalities on the grid-walking and foot-fault tests from undernutrition in wistar rats. J. Mot. Behav. 2016;48:5–12. doi: 10.1080/00222895.2015.1024824. [DOI] [PubMed] [Google Scholar]
- Barnett D., Hall J., Haab B. Automated identification and quantification of signals in multichannel immunofluorescence images: the SignalFinder-IF platform. Am. J. Pathol. 2019;189:1402–1412. doi: 10.1016/j.ajpath.2019.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand L., Horman S., Beauloye C., Vanoverschelde J.L. Insulin signalling in the heart. Cardiovasc. Res. 2008;79:238–248. doi: 10.1093/cvr/cvn093. [DOI] [PubMed] [Google Scholar]
- Brenner M., Kisseberth W.C., Su Y., Besnard F., Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J. Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brewer M.K., Gentry M.S. Brain glycogen structure and its associated proteins: past, present and future. Adv. Neurobiol. 2019;23:17–81. doi: 10.1007/978-3-030-27480-1_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushong E.A., Martone M.E., Jones Y.Z., Ellisman M.H. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J. Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W., Xue C., Sakaguchi M., Konishi M., Shirazian A., Ferris H.A., Li M.E., Yu R., Kleinridders A., Pothos E.N. Insulin regulates astrocyte gliotransmission and modulates behavior. J. Clin. Invest. 2018;128:2914–2926. doi: 10.1172/JCI99366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel G.A. Brain glucose metabolism: integration of energetics with function. Physiol. Rev. 2019;99:949–1045. doi: 10.1152/physrev.00062.2017. [DOI] [PubMed] [Google Scholar]
- Dienel G.A., Cruz N.F. Contributions of glycogen to astrocytic energetics during brain activation. Metab. Brain Dis. 2015;30:281–298. doi: 10.1007/s11011-014-9493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dienel G.A., Rothman D.L. Glycogenolysis in cerebral cortex during sensory stimulation, acute hypoglycemia, and exercise: impact on astrocytic energetics, aerobic glycolysis, and astrocyte-neuron interactions. Adv. Neurobiol. 2019;23:209–267. doi: 10.1007/978-3-030-27480-1_8. [DOI] [PubMed] [Google Scholar]
- Dukhande V.V., Rogers D.M., Romá-Mateo C., Donderis J., Marina A., Taylor A.O., Sanz P., Gentry M.S. Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity. PLoS One. 2011;6:e24040. doi: 10.1371/journal.pone.0024040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran J., Gruart A., Varea O., López-Soldado I., Delgado-García J.M., Guinovart J.J. Lack of neuronal glycogen impairs memory formation and learning-dependent synaptic plasticity in mice. Front. Cell. Neurosci. 2019;13:374. doi: 10.3389/fncel.2019.00374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez A.M., Hernandez-Garzón E., Perez-Domper P., Perez-Alvarez A., Mederos S., Matsui T., Santi A., Trueba-Saiz A., García-Guerra L., Pose-Utrilla J. Insulin regulates astrocytic glucose handling through cooperation with IGF-I. Diabetes. 2017;66:64–74. doi: 10.2337/db16-0861. [DOI] [PubMed] [Google Scholar]
- Folbergrova J., Katsura K.I., Siesjo B.K. Glycogen accumulated in the brain following insults is not degraded during a subsequent period of ischemia. J. Neurol. Sci. 1996;137:7–13. doi: 10.1016/0022-510x(96)82226-x. [DOI] [PubMed] [Google Scholar]
- GBD 2015 DALYs and HALE Collaborators Global, regional, and National disability-adjusted life-years (DALYs) for 315 diseases and injuries and healthy life expectancy (HALE), 1990-2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1603–1658. doi: 10.1016/S0140-6736(16)31460-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GBD 2015 Mortality and Causes of Death Collaborators Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the global burden of disease study 2015. Lancet. 2016;388:1459–1544. doi: 10.1016/S0140-6736(16)31012-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentry M.S., Worby C.A., Dixon J.E. Insights into Lafora disease: malin is an E3 ubiquitin ligase that ubiquitinates and promotes the degradation of laforin. Proc. Natl. Acad. Sci. U S A. 2005;102:8501–8506. doi: 10.1073/pnas.0503285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan T., Qian Y., Tang X., Huang M., Huang L., Li Y., Sun H. Maslinic acid, a natural inhibitor of glycogen phosphorylase, reduces cerebral ischemic injury in hyperglycemic rats by GLT-1 up-regulation. J. Neurosci. Res. 2011;89:1829–1839. doi: 10.1002/jnr.22671. [DOI] [PubMed] [Google Scholar]
- Gurer G., Gursoy-Ozdemir Y., Erdemli E., Can A., Dalkara T. Astrocytes are more resistant to focal cerebral ischemia than neurons and die by a delayed necrosis. Brain Pathol. 2009;19:630–641. doi: 10.1111/j.1750-3639.2008.00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankey G.J. Stroke. Lancet. 2017;389:641–654. doi: 10.1016/S0140-6736(16)30962-X. [DOI] [PubMed] [Google Scholar]
- Hossain M.I., Roulston C.L., Stapleton D.I. Molecular basis of impaired glycogen metabolism during ischemic stroke and hypoxia. PLoS One. 2014;9:e97570. doi: 10.1371/journal.pone.0097570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajihara H., Tsutsumi E., Kinoshita A., Nakano J., Takagi K., Takeo S. Activated astrocytes with glycogen accumulation in ischemic penumbra during the early stage of brain infarction: immunohistochemical and electron microscopic studies. Brain Res. 2001;909:92–101. doi: 10.1016/s0006-8993(01)02640-3. [DOI] [PubMed] [Google Scholar]
- Kirsch W.M., Leitner J.W. Glycolytic metabolites and co-factors in human cerebral cortex and white matter during complete ischemia. Brain Res. 1967;4:358–368. doi: 10.1016/0006-8993(67)90165-5. [DOI] [PubMed] [Google Scholar]
- Krewson E.A., Sanderlin E.J., Marie M.A., Akhtar S.N., Velcicky J., Loetscher P., Yang L.V. The proton-sensing GPR4 receptor regulates paracellular gap formation and permeability of vascular endothelial cells. iScience. 2020;23:100848. doi: 10.1016/j.isci.2020.100848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long D.M., Mossakowski M.J., Klatzo I. Glycogen accumulation in spinal cord motor neurons due to partial ischemia. Acta Neuropathol. 1972;20:335–347. doi: 10.1007/BF00691750. [DOI] [PubMed] [Google Scholar]
- Lowry O.H., Passonneau J.V. The relationships between substrates and enzymes of glycolysis in brain. J. Biol. Chem. 1964;239:31–42. [PubMed] [Google Scholar]
- Lowry O.H., Passonneau J.V., Hasselberger F.X., Schulz D.W. Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem. 1964;239:18–30. [PubMed] [Google Scholar]
- Müller M.S., Fox R., Schousboe A., Waagepetersen H.S., Bak L.K. Astrocyte glycogenolysis is triggered by store-operated calcium entry and provides metabolic energy for cellular calcium homeostasis. Glia. 2014;62:526–534. doi: 10.1002/glia.22623. [DOI] [PubMed] [Google Scholar]
- Ma H., Campbell B.C.V., Parsons M.W., Churilov L., Levi C.R., Hsu C., Kleinig T.J., Wijeratne T., Curtze S., Dewey H.M. Thrombolysis guided by perfusion imaging up to 9 hours after onset of stroke. N. Engl. J. Med. 2019;380:1795–1803. doi: 10.1056/NEJMoa1813046. [DOI] [PubMed] [Google Scholar]
- Magistretti P.J., Allaman I. A cellular perspective on brain energy metabolism and functional imaging. Neuron. 2015;86:883–901. doi: 10.1016/j.neuron.2015.03.035. [DOI] [PubMed] [Google Scholar]
- Masuda S., Chikuma M., Sasaki R. Insulin-like growth factors and insulin stimulate erythropoietin production in primary cultured astrocytes. Brain Res. 1997;746:63–70. doi: 10.1016/s0006-8993(96)01186-9. [DOI] [PubMed] [Google Scholar]
- Mishra N., Lata S., Deshmukh P., Kamat K., Surolia A., Banerjee T. Insulin signaling pathway protects neuronal cell lines by Sirt3 mediated IRS2 activation. Biofactors. 2018;44:224–236. doi: 10.1002/biof.1413. [DOI] [PubMed] [Google Scholar]
- Moore M.C., Coate K.C., Winnick J.J., An Z., Cherrington A.D. Regulation of hepatic glucose uptake and storage in vivo. Adv. Nutr. 2012;3:286–294. doi: 10.3945/an.112.002089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrsulja B.B., Mrsulja B.J., Ito U., Walker J.T., Jr., Spatz M., Klatzo I. Experimental cerebral ischemia in Mongolian gerbils. II. Changes in carbohydrates. Acta Neuropathol. 1975;33:91–103. doi: 10.1007/BF00687536. [DOI] [PubMed] [Google Scholar]
- Mrsulja B.J., Spatz M., Walker J.T., Jr., Klatzo I. Histochemical investigation of the Mongolian gerbil's brain during unilateral ischemia. Acta Neuropathol. 1979;46:123–131. doi: 10.1007/BF00684813. [DOI] [PubMed] [Google Scholar]
- Mullins M.E. Modern emergent stroke imaging: pearls, protocols, and pitfalls. Radiol. Clin. North Am. 2006;44:41–62. doi: 10.1016/j.rcl.2005.08.002. vii–viii. [DOI] [PubMed] [Google Scholar]
- Odell I.D., Cook D. Immunofluorescence techniques. J. Invest. Dermatol. 2013;133:e4. doi: 10.1038/jid.2012.455. [DOI] [PubMed] [Google Scholar]
- Oe Y., Baba O., Ashida H., Nakamura K.C., Hirase H. Glycogen distribution in the microwave-fixed mouse brain reveals heterogeneous astrocytic patterns. Glia. 2016;64:1532–1545. doi: 10.1002/glia.23020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson-Leary J., Jahagirdar V., Sage J., McNay E.C. Insulin modulates hippocampally-mediated spatial working memory via glucose transporter-4. Behav. Brain Res. 2018;338:32–39. doi: 10.1016/j.bbr.2017.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson B.A. Structure and regulation of glycogen synthase in the brain. Adv. Neurobiol. 2019;23:83–123. doi: 10.1007/978-3-030-27480-1_3. [DOI] [PubMed] [Google Scholar]
- Pundik S., Xu K., Sundararajan S. Reperfusion brain injury: focus on cellular bioenergetics. Neurology. 2012;79:S44–S51. doi: 10.1212/WNL.0b013e3182695a14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramagiri S., Taliyan R. Remote limb ischemic post conditioning during early reperfusion alleviates cerebral ischemic reperfusion injury via GSK-3beta/CREB/BDNF pathway. Eur. J. Pharmacol. 2017;803:84–93. doi: 10.1016/j.ejphar.2017.03.028. [DOI] [PubMed] [Google Scholar]
- Saez I., Duran J., Sinadinos C., Beltran A., Yanes O., Tevy M.F., Martínez-Pons C., Milán M., Guinovart J.J. Neurons have an active glycogen metabolism that contributes to tolerance to hypoxia. J. Cereb. Blood Flow.Metab. 2014;34:945–955. doi: 10.1038/jcbfm.2014.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor S.S., Ilouz R., Zhang P., Kornev A.P. Assembly of allosteric macromolecular switches: lessons from PKA. Nat. Rev. Mol. Cell Biol. 2012;13:646–658. doi: 10.1038/nrm3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey W.R., Treadway J.L., Magee W.P., Sutt J.C., McPherson R.K., Levy C.B., Wilder D.E., Yu L.J., Chen Y., Shanker R.M. Cardioprotective effects of ingliforib, a novel glycogen phosphorylase inhibitor. Am. J. Physiol. Heart Circ. Physiol. 2004;286:H1177–H1184. doi: 10.1152/ajpheart.00652.2003. [DOI] [PubMed] [Google Scholar]
- Wang H., Kumar A., Lamont R.J., Scott D.A. GSK3beta and the control of infectious bacterial diseases. Trends Microbiol. 2014;22:208–217. doi: 10.1016/j.tim.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Song D., Bai Q., Zhou L., Cai L., Hertz L., Peng L. Role of glycogenolysis in stimulation of ATP release from cultured mouse astrocytes by transmitters and high K+ concentrations. ASN Neuro. 2014;6:e00132. doi: 10.1042/AN20130040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Song D., Xue Z., Gu L., Hertz L., Peng L. Requirement of glycogenolysis for uptake of increased extracellular K+ in astrocytes: potential implications for K+ homeostasis and glycogen usage in brain. Neurochem. Res. 2013;38:472–485. doi: 10.1007/s11064-012-0938-3. [DOI] [PubMed] [Google Scholar]
- Xu L., Sun H. Pharmacological manipulation of brain glycogenolysis as a therapeutic approach to cerebral ischemia. Mini Rev. Med. Chem. 2010;10:1188–1193. doi: 10.2174/1389557511009011188. [DOI] [PubMed] [Google Scholar]
- Yin A., Guo H., Tao L., Cai G., Wang Y., Yao L., Xiong L., Zhang J., Li Y. NDRG2 protects the brain from excitotoxicity by facilitating interstitial glutamate uptake. Transl. Stroke Res. 2019 doi: 10.1007/s12975–12019–00708–12979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.X., Zang Y.M., Huo J.H., Liang S.J., Zhang H.F., Wang Y.M., Fan Q., Guo W.Y., Wang H.C., Gao F. Physiologically tolerable insulin reduces myocardial injury and improves cardiac functional recovery in myocardial ischemic/reperfused dogs. J. Cardiovasc. Pharmacol. 2006;48:306–313. doi: 10.1097/01.fjc.0000249873.73197.c3. [DOI] [PubMed] [Google Scholar]
- Zhang L., Schallert T., Zhang Z.G., Jiang Q., Arniego P., Li Q., Lu M., Chopp M. A test for detecting long-term sensorimotor dysfunction in the mouse after focal cerebral ischemia. J. Neurosci. Methods. 2002;117:207–214. doi: 10.1016/s0165-0270(02)00114-0. [DOI] [PubMed] [Google Scholar]
- Zhang Z., Yan J., Shi H. Hyperglycemia as a risk factor of ischemic stroke. J. Drug Metab.Toxicol. 2013;4:153. doi: 10.4172/2157-7609.1000153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zois C.E., Harris A.L. Glycogen metabolism has a key role in the cancer microenvironment and provides new targets for cancer therapy. J. Mol. Med. (Berl.) 2016;94:137–154. doi: 10.1007/s00109-015-1377-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant data are available from the corresponding author (L.X.) upon reasonable request. This study did not generate code.