

Abstract
Gallinamide A, originally isolated with modest antimalarial activity, was subsequently re-isolated and characterized as a potent, selective, and irreversible inhibitor of the human cysteine protease cathepsin L. Molecular docking identified potential modifications to improve binding, which were synthesized as a suite of analogs., Resultingly, this current study produced the most potent gallinamide analog yet tested against cathepsin L (10, Ki = 0.0937 ± 0.01 nM and kinact /Ki = 8,730,000). From a protein structure and substrate preference perspective, cruzain, an essential Trypanosoma cruzi cysteine protease, is a highly homologous. Our investigations revealed that gallimamide and its analogs potently inhibit cruzain and are exquisitely toxic towards T. cruzi in the intracellular amastigote stage. The most active compound, 5, had an IC50 = 5.1 ± 1.4 nM, but was relatively inactive to both the epimastigote (insect stage) and the host cell, and thus represents a new candidate for treatment of Chagas disease.
Keywords: Molecular-modeling, organic synthesis, structure activity relationship, gallinamide A, cathepsin L, Trypanosoma cruzi
Graphic Abstract
Introduction
Natural products (NPs) have long served an important role in drug discovery through identifying new chemical scaffolds that interact with novel targets within cellular systems. Indeed, NPs of different molecular weight classes have led to approximately 70% of all clinically useful agents, either directly, by derivation, or inspiration.1 Marine life has only recently been prospected in earnest as a resource for new drug leads, but already thirteen agents have been derived or inspired from marine NPs for use in the clinic, and another 31 are in various stages of clinical development.2,3 Nevertheless, some therapeutic areas have lagged behind others in terms of development of new pharmacotherapies; neurological disorders and parasitic disease represent two such conditions.4, 5 Interestingly, these two disease areas are mechanistically connected in that cysteine proteases have been identified as excellent potential drug targets in both.6
Gallinamide A (1) was first isolated from a Panamanian collection of marine cyanobacteria in 2009 for activity against the malaria-causing parasite Plasmodium falciparum. In this work the full planar structure was solved but the stereochemistry of the N,N-dimethylisoleucine residue was not resolved.7 It was subsequently and independently isolated by Taori and coworkers from a Floridian cyanobacterial collection and given the name ‘symplostatin 4’.8 At that time of the second isolation it was posited on the basis of NMR data that the stereochemistry of the N,N-dimethylisoleucine was different between gallinamide A and symplostatin 4.8 Conroy and coworkers confirmed the absolute stereochemistry of symplostatin 4 by total synthesis, and also suggested the stereochemistry of the N,N-dimethylisoleucine tail was different in gallinamide A.9 These authors subsequently corrected this surmise and showed by careful synthesis of all four diastereomers at this terminal amino acid that gallinamide A also contained a 2S,3S-N,N-dimethyl isoleucine residue, and thus the two compounds were identical;10 in this publication we shall refer to this compound exclusively as ‘gallinamide A’.
Subsequent to our initial discovery and structural description of 1,7 our interest in this area was re-kindled by a screening program that detected it as an extremely potent and highly selective inhibitor of human cathepsin L.11 Preincubation-dilution and activity-based probe experiments revealed an irreversible mode of cathepsin L inhibition, and molecular docking plus molecular dynamics simulations identified a likely pose for 1 binding to the active site. This, as well as another modeling study by Omotunyi and coworkers, found that the cyclic methylmethoxypyrrolinone (MMP) residue was important for binding by positioning the pharmacophore enone proximate to the thiolate nucleophile.12
Human cathepsin L is a clan CA family C1 protease that participates in producing peptide neurotransmitters (neuropeptides)13 for synaptic function which is compromised in neurological diseases,14 and functions in lysosomes which participate in neurodegenerative diseases.15 Interestingly, it shows high sequence identity and structural similarity to many proteases found in parasites responsible for neglected tropical diseases.16 When compared by sequence identity, cathepsin L is most similar to human cathepsin V (78% by amino acid sequence), and clades closely with human cathepsins K and S. However, when compared by substrate selectivity, a more appropriate metric for assessing functional similarity, cathepsins L and V separate from the other human enzymes and clade with cruzain (from T. cruzi) and cathepsin L (from Leishmania mexicana).17 Thus, it can be hypothesized that inhibitors tailored to human cathepsin L will have significant crossover with these parasitic enzymes, and provide useful scaffolds for the development of anti-Chagas disease and perhaps antileishmaniasis agents.
Thus, for a variety of reasons, including its antiparasitic activity, inhibition of cathepsin L, and unique linear lipopeptide structure, there has been considerable interest in 1 and its analogs. For example, 1 was synthesized by Stolze and coworkers, who showed that it inhibited one or more of the Plasmodium food vacuole falcipain cysteine proteases, predominately FP2, FP2’ and FP3 enzymes involved in heme degradation and essential for the survival of the parasite.18 They also demonstrated a likely irreversible mechanism of action based on an enzyme kinetics approach, and that the principal target in mammalian cells was cathepsin L. Stolze also investigated via chemical synthesis some of the structural features in gallinamide A necessary for antimalarial activity, concluding that the cyclic nature of the MMP head group was crucial for bioactivity. Further, they produced rhodamine fluorophore labeled derivatives, and showed that these labeled food vacuoles and the key FP2, FP2’ and FP3 protease enzymes in a manner consistent with their being the relevant biological target. Through selective hydrogenation of the double bonds, the enamide olefin was shown to be necessary for high inhibition potency, supporting our previous hypothesis of a Michael-addition like reaction with the active site cysteine thiol,11 as well as experiments described below by Conway.19
The Conway group subsequently reported the synthesis of a number of analogs of 1 and their relative potency as inhibitors of the FP2 and FP3 proteases, as well as activity in the drug sensitive 3D7 and drug resistant Dd2 strains of Plasmodium falciparum.19 The pharmacophore for this protease inhibition was again identified as the enone of the unusual 4(S)-amino-2(E)-pentenoic acid unit through production of saturated analogs in this residue and separately in the pyrrolinone ring and demonstration of reduced potency to FP2 and FP3, as well as to P. falciparum. This latter work again revealed the critical nature of the MMP group for production of high potency agents in this class, and by addition of a phenyl or indole ring to the methyl-pyrrolinone ring, they were able to produce derivatives with low nM potencies to both of the P. falciparum strains.
Our previous in-silico docking and molecular dynamics simulations with 1 produced a stable and reaction-ready pose for the natural product in the active site of the cathepsin L protease.11 This pose, as well as insights from the literature pertaining to selective cathepsin inhibitors and their substrate specificity, was used to design a new suite of analogs for improved binding in cathepsin L as well as the falcipains. Drawing from the literature, 1 was synthesized using a combination of steps from both Conroy et al. and Stolze et al.,9,10,18,19 modifying steps as necessary due to constraints on reagent availability or to improve convenience or yield. Informed by our modeling, new analogs of 1 were designed and synthesized by replacement of the amino acid building blocks. As the analogs covered in this work were mostly similar to the natural product (e.g. phenylalanine versus alanine), minimal modification of the reaction or purification conditions was necessary.
This effort afforded a perspicacious suite of compounds (1-16) that were available to test against cathepsin L. The modeled protein structure of cathepsin L suggested a structure and substrate scope similar to cruzain, another cysteine protease necessary for the survival of Trypanosoma cruzi, the causative agent of Chagas disease. Current treatments for Chagas disease, benzidazole and nifurtimox, are associated with significant adverse effects, require long treatments up to 60 days, and have controversial efficacy in the chronic phase of the disease.20 New treatments that might target the amastigote stage of the chronic infection are much needed, so this panel of compounds was also tested against cruzain and in a cell based in vitro T. cruzi assay. The gallinamide A scaffold thus represents a new model for potential drug development to this Neglected Tropical Disease (NTD).
Notable among the analogs synthesized, a modification was made to introduce a terminal alkyne to serve as a handle for potential click chemistry, as well as another analog that exhibited reversible binding characteristics to the enzyme. Two analogs were measured as sub-nanomolar inhibitors of human cathepsin L (analogs 10, 11), with one that displays single digit nanomolar antiparasitic activity against T. cruzi in a cell-based assay (analog 5) without any toxicity towards host cells up to 10 micromolar. Cathepsin L inhibition is not fatal, as demonstrated by cathepsin L gene knockout mice which are viable and live through development to adulthood and reproduce progeny.21,22 Studies of cruzain, the most abundant cysteine protease of T. cruzi23 and a validated drug target,24,25 along with the findings presented in this manuscript, illustrate the productive strategy of investigating related human and parasitic cysteine proteases for the design and synthesis of potent inhibitors as candidate pharmaceutical agents.
Results
Molecular modeling and rational design of analogs
The co-crystal structure of human cathepsin L with a bound covalent nitrile inhibitor (2xu3.pdb)26 was imported to the integrated drug discovery and computational chemistry software package Molecular Operating Environment (Computational Chemistry Group). The nitrile ligand was removed, and the protein was prepared for docking using MOE’s Structure Preparation module to fix structural errors, protonated with the Protonate 3D module set to pH 5.5 (maximal activity for cathepsin L), and then energy minimized using the Amber14:EHT force field equations.27 Compound 1 was imported into MOE and similarly protonated, energy minimized using the same criteria, and then docked to the prepared cathepsin L enzyme using an induced fit protocol to allow a flexible ligand and receptor. The docked poses were filtered based on proximity of the reactive carbon of the enamide to the cysteine thiol, and the top pose (S=−10.70 score for binding energy using GBVI/WSA dG) was compared to the pose reported previously.11 The orientation of the core enamide in both cases was identical, and the carbonyl of this functional group was well supported by hydrogen bonding in the oxyanion hole of the active site cleft, an important feature for stabilization of the intermediate inhibitory species (Figure 1).
Figure 1: Gallinamide A docked pose in cathepsin L and ligand interactions.
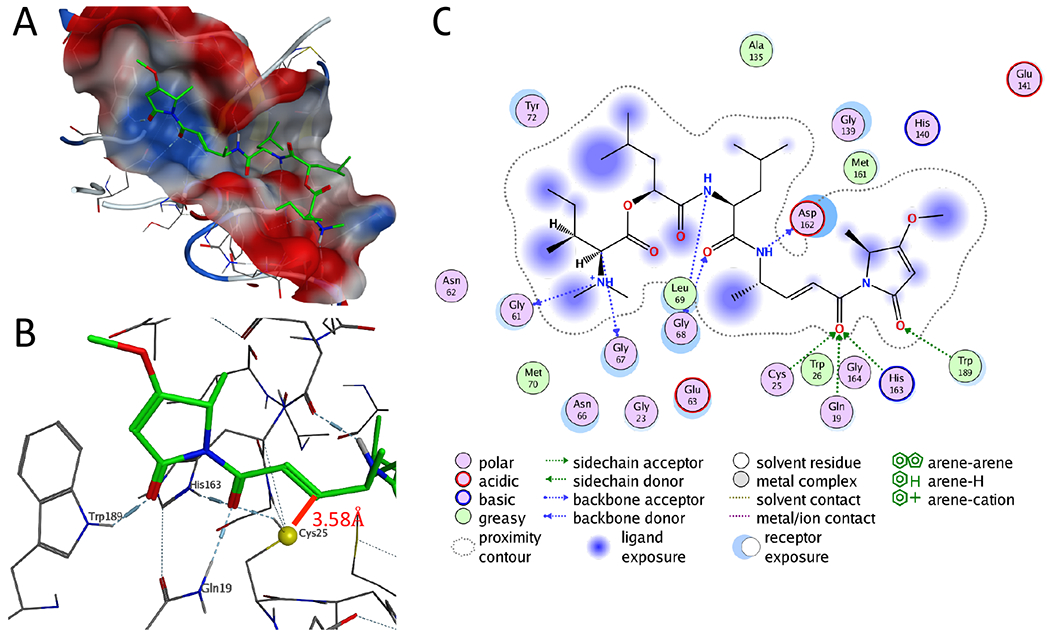
A. Docked pose for gallinamide A in the active site cleft of human cathepsin L. The pose was generated through the induced-fit docking protocol, followed by energy minimization. The enzyme surface map is colored by electrostatic potential, with red indicating a negative and blue indicating positive surface potential B. Detailed representation of the hydrogen bond network (blue dotted lines) and proximity of the enamide pharmacophore to the active site cysteine (yellow sphere). The red line indicates the distance between the sulfur atom and reactive carbon atom of the enone. C. 2D ligand interactions. Importantly, the carbonyl of the reactive enamide core is stabilized by hydrogen bonds in the enzyme oxyanion hole.
Using this as a starting binding pose, in silico modifications were made to the structure of 1, energy minimized and then re-scored to assess predicted changes to binding affinity. The R’ and R” residues (Figure 2), both l-alanine in the natural product (1), were each changed to l-phenylalanine based on comparisons with common cathepsin substrates and examination of the binding sites by MOE. Inclusion of l-phenylalanine at the R’’ (3) residue showed modest improvement to the binding score (S = −10.95), as well as at the R’ position (6) (S = −11.29). Inclusion of l-phenyalanine at both positions (8) further improved binding, resulting in an S score of −11.47. While changing the R’’ position residue to d-phenylalanine resulted in only mildly worse binding compared to the opposite configuration (S = −10.46 versus S = −10.95 for 3), inclusion of a bulky d-amino acid in the R’ position appeared to cause a steric clash that prevented the reactive carbon of the enamide from being accessed by the cysteine-25 thiol. This modification reduced the binding score to −7.58 and the distance between the reactive carbon and the sulfur increased from 3.64 Å in 6 to 5.11 Å in this analog.
Figure 2: In silico structural modifications to gallinamide A and their resulting docking scores.
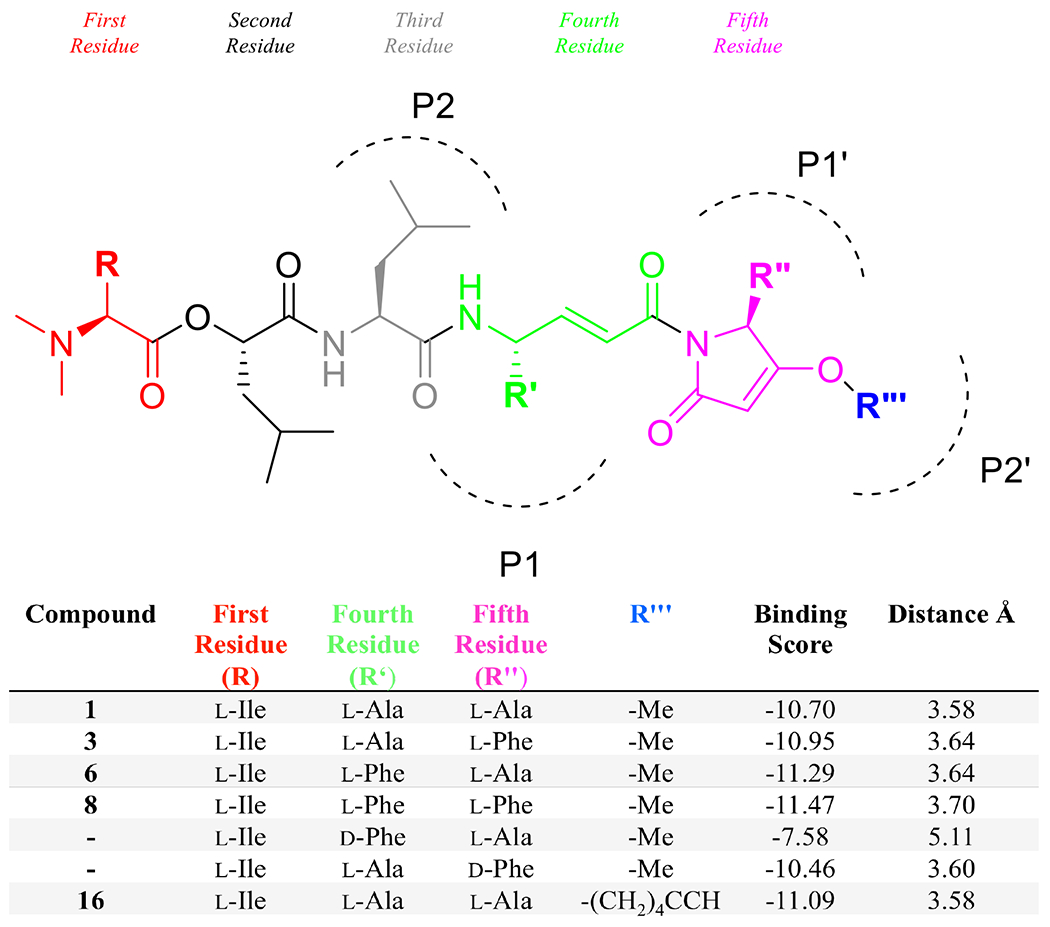
The residues within the gallinamide analogs are labeled according to Schechter-Berger nomenclature, with the fourth and fifth residues residing at P1 and P1’, respectively. Changes to the substrate residues (at R, R’, R” and R”’) are shown with a corresponding binding score, calculated with GBVI/WSA dG. Binding scores with a lower number (more negative) are predicted to bind with higher affinity.
Interestingly, a lipophilic surface was identified beyond the binding site of the methylmethoxypyrrolinone (MMP) group, which could be accessed by extensions to the methoxy group on the pyrrolinone ring, while simultaneously including a click handle for later structural modifications. An analog with an O-hexynyl moiety at this position (16) was modeled and the binding energy was slightly improved to S = −11.09 (Figure 2). Not modeled was the analog where the fourth residue alkene in the enamide was hydrogenated to the alkane (17); mechanistically, this was expected to be a reversible inhibitor.11,18,19 Based on these in silico data, summarized in Figure 2, a prioritized set of analogs incorporating various combinations of these modifications was identified for total synthesis (compounds 2-16). These compounds were designed to evaluate whether incorporation of phenylalanine residues would improve binding in vitro against cathepsin L, or if the incorporation of d-amino acids would interfere with binding. Leucine was included as a replacement for the phenylalanine residue to test whether the aromatic system, or only the hydrophobic residue, was important to binding. Finally, analogs were designed wherein several of these modifications were combined to evaluate for potential synergistic interactions.
Total synthesis of gallinamide A and analogs
Guided by previous syntheses of 1,9,10,18,19 a retrosynthetic analysis sectioned the molecule into three components: an enamide core, the cyclized head group, and the lipophilic tail comprised of aliphatic amino acids. With the exception of l-leucic acid, all of the starting materials were variously protected standard amino acids. To accomplish the total synthesis of 1, reactions were appropriated from the published routes with additional modifications introduced that were found to improve or facilitate the reactions, or necessary due to other constraints (Figures 3, 4).
Figure 3:
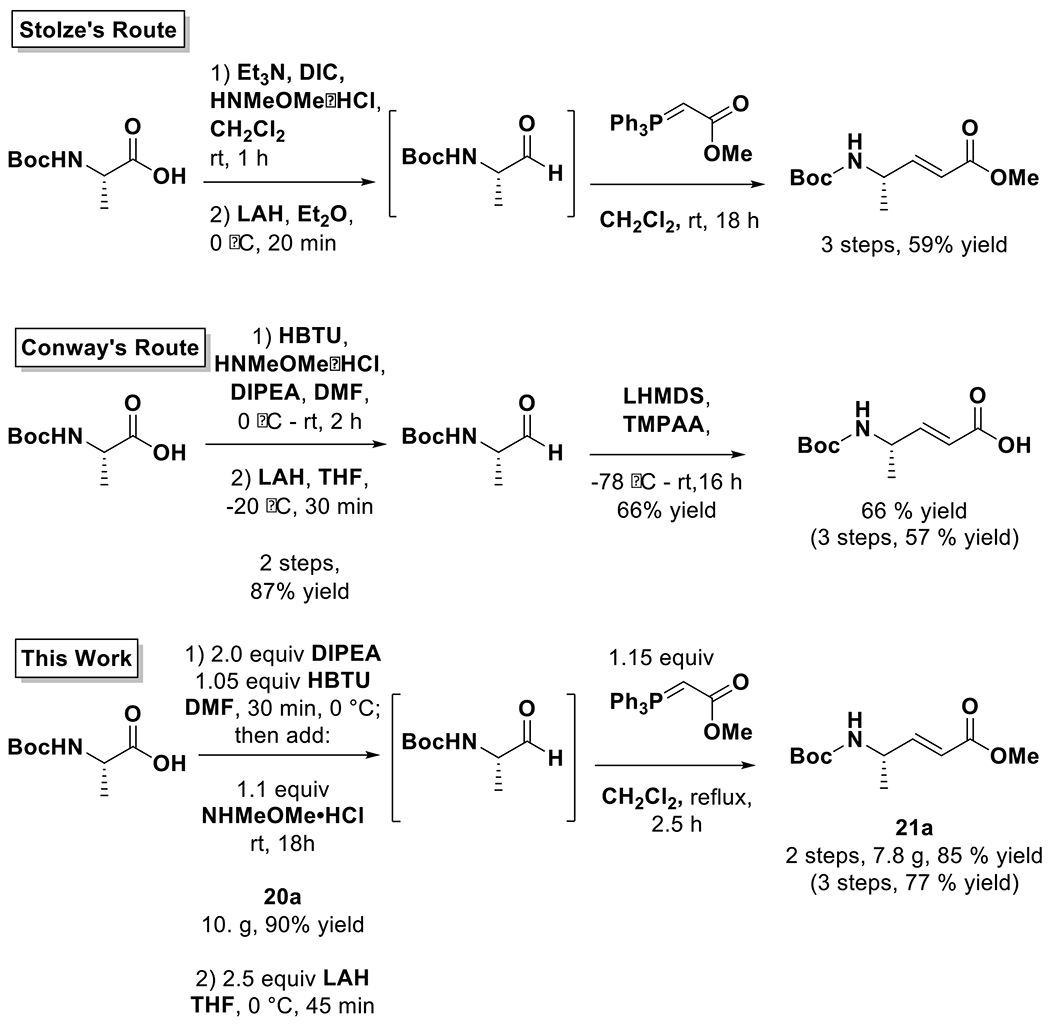
Comparison of the synthetic routes to the enamide core of gallinamide A (1).
Figure 4: Total synthesis of gallinamide A.
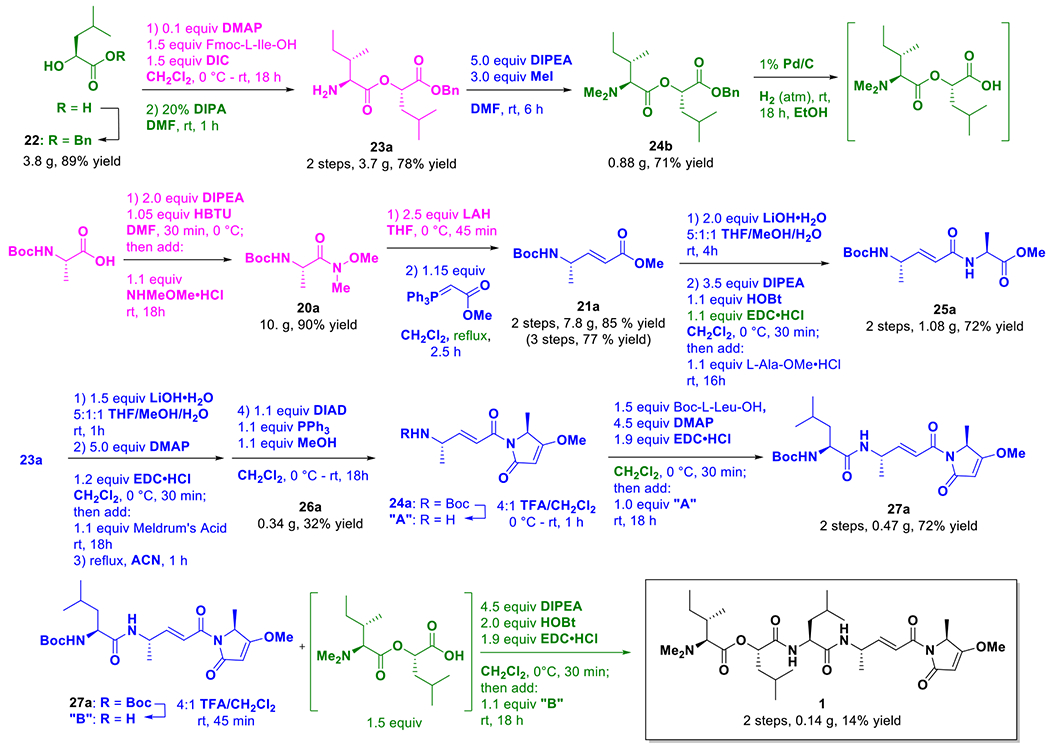
The total synthesis was completed using reactions drawn from the Conroy (shown in magenta) and Stolze (shown in blue) synthetic routes, as well as new reactions to improve efficiency and yield (shown in green).
An example of this hybrid scheme towards the synthesis is provided by the first steps involving the formation of the enamide core from Boc-L-Alanine-OH (Figure 3). The first reaction formed the Weinreb amide (20a), which was then reduced to an aldehyde and reacted with an anylidene to form the conjugated system of the enamide as a methyl ester (21a). Conway’s scheme was emulated for forming 20a as the purification via crystallization produced better results;10 however, Stolze’s route of reacting the aldehyde without purification with the ester anylidene to form 21a proved more effective than purifying the aldehyde and subsequently reacting it with free acid anylidene.18 It was also found that refluxing the Wittig reaction, rather than running it at room temperature, improved the yield and shortened the reaction time.
The synthetic route for the head group was largely drawn from Stolze’s route, using an 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride salt (EDC) based coupling reaction to append an alanine and form the enamide core (25a), which was subsequently deprotected, extended with Meldrum’s acid, and cyclized.18 The resulting enol was trapped via a Mitsunobu reaction to complete the head group (26a). In the final steps to form the tail section and couple it to the enamide core, the route largely followed Stolze’s procedure,18 but with modifications. For example, piperidine was used in Conroy’s work to deprotect the Fmoc group10 whereas thionyl chloride was employed in Stolze’s route as a coupling reagent;18 both are controlled substances in California, and thus these reaction steps were modified for the convenience of working with unregulated chemicals.
To append the tail section, Boc-L-Leu-OH was first attached via EDC coupling to the free amine derived from trifluoracetic acid (TFA) deprotection of 26a to afford the leucine extended species (27a). Compound 27a was then coupled to a dipeptide ester prepared similarly to Stolze’s and Conroy’s routes, but using different protecting groups. l-Leucic acid was protected as the benzyl ester (22) rather than a tert-butyl or methyl ester because synthesis of the tert-butyl ester was difficult to replicate whereas cleavage of the methyl ester with lithium hydroxide risked cleavage of the desired ester product. The benzyl ester of l-leucic acid was connected to Fmoc-protected l-isoleucine (or a different amino acid) via carbondiimide coupling using N,N-diisopropylcarbodiimide (DIC) to afford the desired ester. The product was filtered to remove DIC derived urea, but was not otherwise purified as this was not observed to improve the yield before Fmoc deprotection. Fmoc deprotection with diisopropyl amine gave a free amine (23a). The amine was then dimethylated with methyl iodide (24a). Finally the benzyl group was removed via hydrogenation to provide the desired ester intermediate. The free acid of this two-residue ester was used immediately in an EDC coupling with the free amine derived from the TFA deprotection of 27a, to afford the final product 1.
By replacing the various starting amino acids, a variety of analogs could be synthesized. The modifications focused on four components of 1: the first residue, the amino acid bearing the demethylated amino terminus; the fourth residue, the amino acid extended into the enamide core; the fifth residue, the amino acid in the cyclic head group; and the alcohol used to trap the enol via a Mitsunobu reaction in the head group (R”‘).
To modify the R substituent, Fmoc-l-isoleucine was replaced as the first residue with Fmoc-l-valine or Fmoc-l-phenylalanine. The enamide core residue R’ could easily be modified by replacing the starting Boc-l-alanine by the equivalent carbamates of d- and l-phenylalanine; however, the Weinreb amides derived from phenylalanine (20b and 20c) could not be crystallized from N,N-dimethyl formamide (DMF), and thus were purified by flash chromatography. In the same fashion, the head group extension to form the initial enamide core could be modified by using an amino acid methyl ester other than l-alanine methyl ester. Analogs were synthesized with the equivalent esters of l-leucine and d- or l-phenylalanine, which combined with different enamide starting materials, led to the production of nine different core-head group pairings (25b-i, see Methods and Supporting Information). The final modification was the use of an alcohol other than methanol to trap the enol in the Mitsunobu reaction; 5-hexyn-1-ol was used to create a new analog (26j, see Methods and Supporting Information) at this position (R”‘) as it also provided the opportunity to attach a probe via click chemistry in future work.
An interesting aspect of the spectroscopic data for all synthetic compounds possessing the pyrrolidone ring (e.g. all compounds in series 26 and 27 as well as 1) was the appearance of 1:1 twinned signals for several of the 1H and 13C NMR resonances assigned to the acyclic enone as well as the pyrrolidone ring structure (Figure S65). This observation of twinning was also made by Stolze who created the pyrrolidone system using this same sequence of synthetic reactions (personnel communication).18 We reasoned that this was either due to racemization of C-4 of the 4-(S)-amino-2-(E)-pentenoic acid (Apa) unit or the presence of two stable conformers about the amide bond connecting the Apa unit to the pyrrolidone ring (Figure S67). To distinguish between these two possibilities, we subjected synthetic gallinamide A (1) to ozonolysis, hydrolysis and Marfey’s analysis. This revealed that the alanyl residue released from the Apa unit was present as a 6:1 S:R mixture, and was thus inconsistent with the 1:1 twinning observed by 13C NMR (Figure S66). This was additionally probed by variable temperature 1H NMR in which the −OCH3 group appeared as a pair of signals at 22 °C; heating of the sample to 55 °C in CDCl3 resulted in their partial coalescence to a broadened single peak. Calculation of the energy barrier to rotation about this tertiary amide also supports this interpretation (between 12.2 and 13.5 kcal/mol using the B3LYP method with the 6-31+G(d) basis set). As the 13C NMR of natural product gallinamide A (1) did not display twinned resonances in this section of the molecule, it suggests that the final pyrrolidone biosynthetic cyclization reaction occurs with this secondary amide in the E or Z conformation, and upon formation of the tertiary amide, it becomes locked as a single conformer at room temperature.
Kinetic analysis of cathepsin L inhibition
Irreversible inhibitors display a time-dependent shift in the observed IC50 with increased incubation of inhibitor and enzyme concentrations, making end-point dose-response experiments inappropriate for assessing inhibitor potency.12 Thus, a kinetic approach was used to determine the rate and potency of inhibition, in which inhibitor and enzyme were mixed with 10 μM of the fluorogenic substrate cbz-FR-AMC, and product formation monitored for 90 minutes. Progress curves were produced with the concentration of inhibitor ranging from 0.0169 nM to 1000 nM and fit to a non-linear least-squares regression based on the 2-step irreversible reaction scheme (Figure 5A and 5B). The kinetic constants kaI, kaS, and kbS, were fixed at 200, 20, and 1, respectively. The constants kcat , kbI and kinact were explicitly estimated by the model, and used to calculate Ki, the association constant for initial reversible inhibitor binding. Furthermore, the kinact /Ki value was calculated to more accurately assess the complete inhibitory potential of each compound. The regressed Ki = 4.67 ± 0.40 nM for gallinamide A (1) was in agreement with our previously reported IC50 value of 5.0 nM, and the kinact/Ki = 901,000 confirms that it is an extremely potent inhibitor.7,11 The regression failed to fit the progress curve data to reaction schemes involving reversible binding or 1-step reaction mechanisms.
Figure 5: Kinetic analysis of gallinamide A inhibition of cathepsin L.
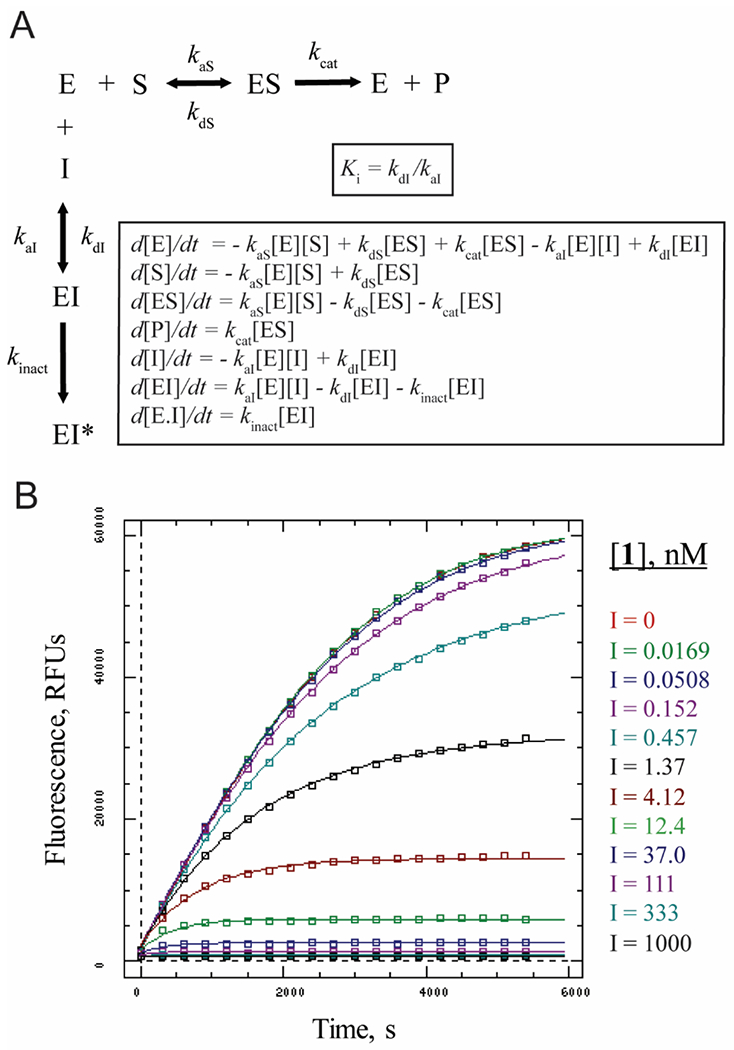
A. Reaction mechanism and differential equations used to explicitly estimate kinetic constants from product formation curves. B. Representative data for gallinamide A (1) inhibition of cathepsin L. Boxes are experimentally measured progress curves of cathespin L activity with various concentrations of inhibitor, and lines were fit by non-linear regression. Note that I = 0 concentration overlaps with the lowest doses at 0.0169 and 0.0508 nM.
Alteration to the first residue, the amino acid bearing the dimethyl amine, had modest but consistent effects on the cathepsin L inhibitory potency of the analogs (Figure 8). Substitution of the isoleucine at this position with a valine residue resulted in decreased potency with all other structural features held constant (e.g. compare: 1 and 2, 10 and 11), whereas replacement with a phenylalanine increased potency moderately (e.g. compare: 3 and 4, 8 and 9). Interestingly, and in contrast to the docking results, inclusion of an l-phenylalanine at the fourth residue was moderately detrimental to potency (e.g. compare: 1 and 6). Inversion of the fifth residue stereocenter increased potency in every case, as demonstrated by the improvement in kinact /Ki from compound 8 to 12 from 719,000 ± 72,000 to 1,030,000 ± 77,000. Comparing these two compounds to the analogs 14 and 15 where there is inversion of the stereocenter at the fourth residue reveals reduced inhibitory potency as demonstrated by kinact /Ki values of 6,900 ± 587 and 23,900 ± 1840, respectively. This result was consistent with the modeling and likely results from a steric clash caused by the fourth residue’s side chain, which forces the reactive carbon of the enamide core to be positioned further from the active site cysteine.
Figure 8:
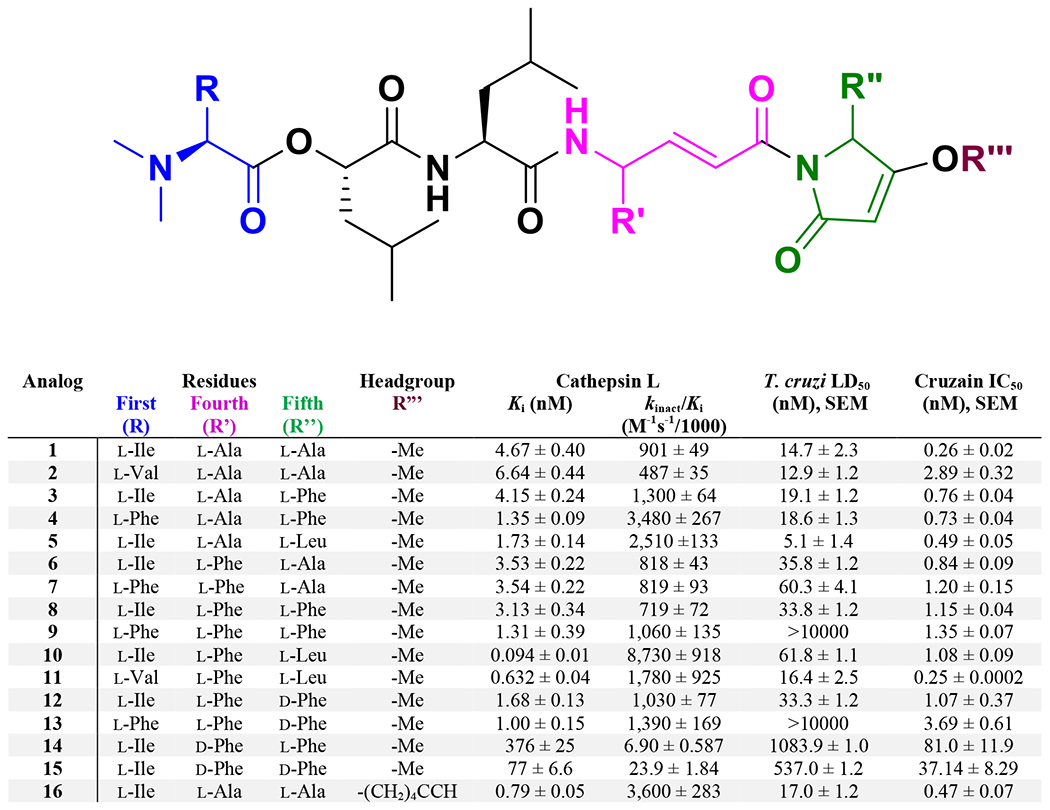
Structure-activity relationships of synthetic gallinamides, listing structural modifications to the gallinamide A (1) structure and corresponding inhibitory activities to cathepsin L and T. cruzi CA-I/72. kinact/Ki values for cathepsin L have been divided by 1000 to allow easier viewing of the data. Ki and IC50 values are given in nM. The LD50’s to the murine host cells were all over 10 μM. N/A = Not assessed.
Remarkably, inclusion of a leucine side chain at the fifth residue significantly improved potency, with the most active compound (10) possessing a Ki = 0.094 ± 0.01 nM and kinact /Ki = 8,730,000 ± 918,000 (Figure 8). Additionally, the inclusion of an alkynyl tail at the R”‘ position as in 16, predicted to interact with the lipophilic pocket beyond the prime-side binding pockets, resulted in a sub-nanomolar Ki and a kinact /Ki = 3,600,000 ± 283,000. These results suggested that the most important alterations in terms of improving inhibitory potency were those that modified the head-group at the amino acid side chain and R”‘ positions, where the structure of the side chain, its stereochemistry, and the inclusion of the O-hexynyl tail had the most significant impacts. Future SAR studies could combine several of these structural modifications to explore their collective impact on inhibition of cathepsin L activity and toxicity to T. cruzi.
Interestingly, the kinact was remarkably similar for most of the compounds produced in this study, rarely deviating more than 0.001 s−1. This indicates that the driving force for inhibitor potency among these gallinamide analogs is the rate of formation of the reversible E-I complex, as opposed to the rate of covalent modification. Leveraging this information, future analogs could be designed with this characteristic in mind. A summary of all kinetic parameters calculated for each analog can be found in Table S63.
Saturation of the enamide olefin produces a reversible gallinamide analog
It has been proposed that gallinamide A is an irreversible inhibitor of the cysteine cathepsins based on 1,4-nucleophilic addition to the enamide core.11,18,19 Thus, we hypothesized that hydrogenation of the enamide olefin would produce an analog with reversible binding characteristics. Analog 17 was synthesized from gallinamide A (1) by palladium catalyzed hydrogenation, selectively reducing the enamide double bond. Reversibility was assessed using a preincubation and dilution experiment adapted from Copeland et al., in which concentrated enzyme and inhibitor are incubated before a rapid dilution with buffer containing substrate (Figure 6A).28 Reversible inhibitors are released from the active site upon dilution and the resulting activity is proportional to the final concentration at 0.1 x IC50. By contrast, an irreversible inhibitor is not released upon dilution, and thus no enzymatic function will be recovered because the activity remains proportional to the initial concentration of 10 x IC50.
Figure 6: Saturation of the enamide olefin produces a reversible gallinamide analog (17).
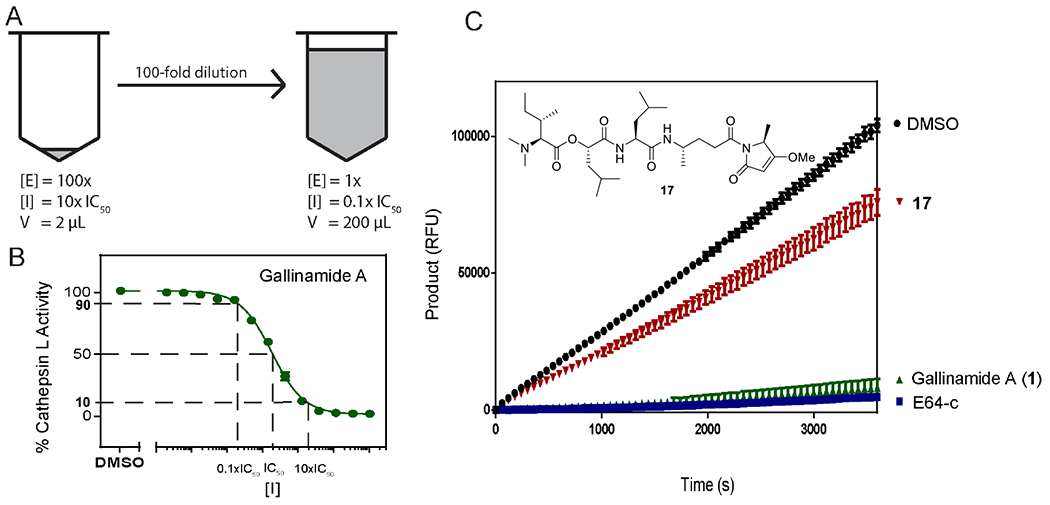
A. A concentrated solution of the enzyme and inhibitor was incubated in assay buffer before a rapid dilution with assay buffer that contained substrate at the standard assay concentration. As noted in section B, this dilution reduces the gallinamide concentration from approximately 90% enzyme inhibition to a predicted 10% of enzyme inhibition. C. Following this dilution, product formation is monitored over 2 hours. Compound 17, the hydrogenated gallinamide analog (red), reduced the enzymatic reaction rate by 25% compared to vehicle control (black), while the intact gallinamide A (green) reduced product formation by 95%, and this activity was not restored over a 2 hour period post-dilution. This latter result matched that of the known irreversible inhibitor E-64c (blue).
Upon dilution, cathepsin L incubated with natural 1 displayed approximately 5% catalytic activity, and displayed no signs of recovery after 1 hour of kinetic readings. Conversely, incubation of 17 with cathepsin L followed by dilution gave approximately 75% of initial activity, and remained linear over 1 hour of monitoring. E-64c, a known irreversible inhibitor of cathepsin L, was used as a control, and matched the response of natural product 1 (Figure 6C). These results clearly demonstrated the conversion of the irreversible natural product inhibitor (1), into a reversible analog (17), confirming that the enamide core is the pharmacophore of the natural product.
Gallinamide A and analog activities against Chagas’ disease (Trypanosoma cruzi)
Based on the structural and functional similarities between cathepsin L and cruzain, gallinamide A (1) and its synthetic analogs were assayed against the amastigote stage of T. cruzi. Murine myoblast host cells were infected with T. cruzi trypomastigotes before addition of test compounds, and after a 3-day incubation the plates were fixed and stained for analysis (Figure 7A).
Figure 7: Gallinamide A (1) potently eliminates T. cruzi from murine host cells.
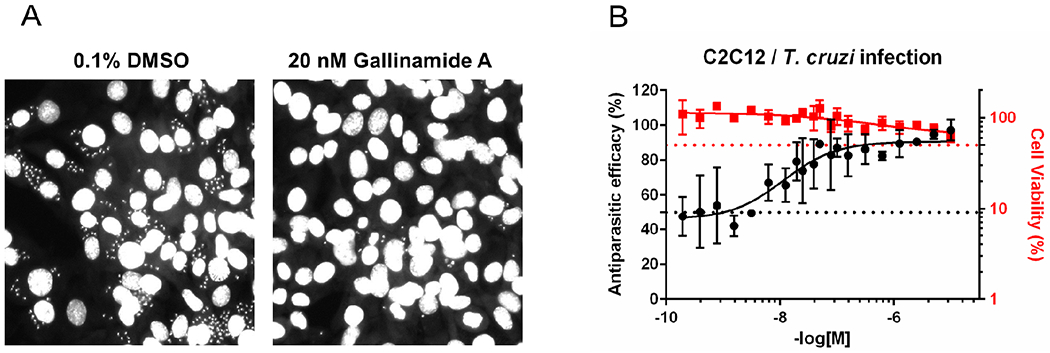
A. Representative images of gallinamide A inhibition of T. cruzi infection. The nuclear stain allows visualization of the host and parasite nuclei in the presence of DMSO vehicle or 20 nM gallinamide A; the latter treatment eliminates the majority of parasites in infected murine cells. B. The dose response curve to intracellular parasite with IC50 = 14.7 nM ± 2.3.
Gallinamide A (1) showed remarkable activity in a cell-based intracellular T. cruzi amastigote assay with an LD50 = 14.7 nM ± 2.3 (Figure 7B) and confirmed inhibitory activity against recombinant cruzain with a sub-nanomolar IC50 (0.26 nM ± 0.02). This is significantly better compared to the positive control, benznidazole,29 which had an LD50 of 1.5 μM in the same cell-based assay. Furthermore, no cytotoxicity to the host murine cells was observed at concentrations up to 10 μM. Compound 5 was the most potent analog with an LD50 of just 5.1 ± 1.4 nM, again without toxicity to host cells up to 10 μM. While cytotoxicity to the host murine cells does not necessarily relate to reduced toxicity in a whole animal system, the presence of highly homologous enzymes in murine cells (e.g. murine cathepsin L) indicates that inhibition of these enzymes does not necessarily lead to cell death. This lack of mammalian cell toxicity is likely due to the redundancy of proteolytic enzymes in these latter cells which can compensate for the inhibition of specific proteases in the lysosome or elsewhere.30 Cruzain, however, is indispensable to the survival of the parasite, and thus its inhibition rapidly leads to cell death of T. cruzi.31
Discussion and Conclusions
An integrated approach of molecular modeling and total chemical synthesis identified several structural features in 1 that could be optimized to improve binding to human cathepsin L (Figure 8). The structure-activity relationships seen to cathepsin L were partly mirrored in the Chagas’ disease antiparasitic activity data (Figure 8). This can be rationalized by recognition of the close similarity in substrate binding and specificity between cathepsin L and cruzain, the cysteine protease target of 1 in Chagas.16
While changing the first residue from l-Ile to l-Val did not improve potency against cathepsin L, replacing this with a l-Phe residue sometimes improved and sometimes decreased activity when in combination with other residue replacements. The fourth residue (R’) shows a distinct preference for l-amino acids, with d-Phe at this position leading to a substantial (up to >100 fold) reduction in activity (e.g. comparing 8 with 14). Substitution of the l-Ala derived residue at this position with a larger aromatic group, such as l-Phe, had a relatively neutral impact on potency (e.g. comparing 1 with 6 and 7). Our finding that the activity to cathepsin L increases when the fifth residue is a larger aliphatic or aromatic residue (e.g. comparing 6 and 10) is consistent with Conway’s finding that a Trp residue at this location enhances potency.19 This is also consistent with our molecular modeling results of cathepsin L with bound gallinamide.11 Finally, and again consistent with our modeling results, placement of a lipophilic group at R”‘ enhances binding affinity and potency of the resulting agent (e.g. comparing 1 and 16). Ultimately, it appears that interaction between these various residues can lead to a maximally potent ligand; in our case l-Ile at the first residue, l-Phe at the fourth residue and l-Leu at the fifth provides the highest affinity binding agent to cathepsin L (10, Ki = 0.094 ± 0.01 nM). Interestingly, this highest affinity analog also showed an enhanced binding score by molecular modeling (−11.31). Against T. cruzi, the highest potency gallinamide derivative was achieved with the first and fourth residues comprised of l-Ile and l-Ala with the fifth residue as l-Leu (5, LD50 = 5.1 ± 1.4 nM). It will be of interest to continue to evaluate the combination of optimized substituents at R, R’ and R” as in compound 10 with the enhancing properties of an aliphatic group at R”‘ as in compound 16.
Chemical synthesis of an analog of 1 lacking the mid-section enone (17) allowed biochemical experiments that provided conclusive proof that this electrophilic group is the pharmacophore of the molecule. As predicted by our previous modeling studies,11 the active site cysteine thiol serves as the nucleophile and adds via 1,4-conjugate addition to this enamide. As discussed above, previous work by Stolze using a kinetic approach,18 and Conway using compound potency as a metric,19 also identified this same conjugated system as the electrophilic pharmacophore of 1.
Because of the close structural and functional relationship of human cathepsin L and the T. cruzi cruzain, we also evaluated 1 and its analogs to the intracellular, disease relevant, form of the parasite. Previous studies with 1, including the original isolation work in 2005,7 also evaluated the compound for activity against T. cruzi. However, those studies reported very low or no activity to this parasite,7 and the focus was shifted toward its anti-malarial properties.9,18 In these previous studies, however, the assay utilized the epimastigotes (insect) stage of T. cruzi. This is likely the cause for the observed lack of activity, as epimastigotes do not rely on cruzain for catabolism of host proteins at this life stage,31 highlighting the importance of screening for antiparasitic agents using diseaserelevant stages of the parasite.
Preliminary docking studies with 1 in cruzain resulted in a similar binding score of S = −9.568 as found for cathepsin L, with a strong hydrogen bonding network in the oxyanion hole (Figure S69). The parasite enzyme contains a deep S2 binding pocket lined by a prominent glutamate residue, a feature that could be taken advantage of through inclusion of arginine in place of the third residue, which corresponds to the P2 residue of natural substrates.32 Modifications could also be made to increase the compound’s stability and solubility in vivo. For example, the ester linkage is not indicated to be important for drug binding in the enzyme (docking scored this analog very similar to the parent ester compound at −10.74), and could be replaced with a standard amide bond to increase plasma stability. In fact, this modification was previously made by Conway, and resulted in an analog of increased stability, although of two-fold reduced potency to P. falciparum.19
Summarizing, the gallinamide A skeleton, comprised of amino acid and polyketide sections, represents an inspiring scaffold with potent binding and inhibition properties to cysteine proteases, including human cathepsin L and T. cruzi cruzain. Moreover, the gallinamide skeleton is distinct from other anti-Chagas disease drugs and lead molecules.33 As a result, this natural product and its analogs have significant potential in diverse therapeutic areas, such as regulation of pain through modulation of neuropeptide processing and antiparasitic activity through inhibition of an enzyme involved in cell invasion23, parasite differentiation24 and metabolism.25 Continuing efforts are focused at further enhancement of the selectivity of this drug class as well as improvement of fundamental pharmaceutical properties and stability.
Experimental Section
General Experimental.
Unless specifically noted otherwise, synthetic reactions were carried out in flame or oven-dried glassware under Argon, with the exception of reactions run in water as co-solvent. All reactions were stirred using a magnetic stir bar. Deuterated chloroform for NMR, with 0.03% TMS as reference, was purchased from Cambridge Isotope Laboratories. NMR spectra were collected on a Varian 500 MHz spectrometer or a Varian 500 MHz spectrometer equipped with an inverse cryo-probe. HR-ESI-TOF-MS data were collected by the Molecular Mass Spectrometry Facility at UCSD. Purity of the final products 1-17 was assessed as being above ≥95% by evaluation of their 1H NMR and 13C NMR spectra.
Docking protocol.
Gallinamide A was positioned into the cathepsin L active site using the docking module of MOE. No hydrogen bond constraints were applied. Initial placements were scored with London dG, induced fit refinement was used with the default parameters, and final poses were scored with GBVI/WSA dG during the final step. Poses were assessed individually and filtered based on distance between the reactive carbon of the enamide Michael acceptor system and the cysteine thiol.
General Chemical Synthesis
Final Coupling Reaction (1-16).
As a general procedure, a vial was charged with the benzyl ester protected dimer then this material was dissolved in EtOH, the palladium (as a 1% loading on carbon) was added in one portion, and the atmosphere was replaced with hydrogen. The reaction was then stirred overnight and the reaction mixture was passed through a ca. 1 cm diatomaceous earth plug, prepared in a pipette with glass wool, into a 25 mL pear flask using three 1 mL DCM rinses. This material was then dried under N2 (g) flow. In a separate vial the carbamate of the leucine extended product was dissolved in DCM and cooled to 0 °C by ice bath and the TFA was added dropwise. After 0.5-1 hours at 0 °C the reaction mixture was dried under N2 (g) flow, then azeotroped three times with three drops of toluene.
The free acid dimer was dissolved DCM and to this solution was added the DIPEA, dropwise, and the HOBt, in one portion. The reaction mixture was cooled to 0 °C by ice bath and the EDC was added in one portion. After 30-45 min the deprotected lecuine extended product was taken up in DCM and added to the reaction mixture with two DCM rinses. The reaction mixture was then allowed to warm to room temperature overnight. To work up the reaction, the mixture was diluted into DCM and washed with 5% citric acid (aq); the aqueous layer was then back extracted three times with hexanes and the combined organics were washed with brine and dried over MgSO4. The filtrate of this step was then concentrated to dryness by rotary evaporator and purified by a RP-SPE using elutions of 25%/60%/80% CH3CN/H2O and CH3CN. The fractions containing product as detected by LCMS, usually the 60% and 80% fraction were combined and further purified by RP-HPLC using an CH3CN/H2O elution, then dried with by rotary evaporation and lyophilization. Purity of the final products 1-17 was assessed as being above ≥95% by evaluation of their 1H NMR and 13C NMR spectra.
Gallinamide A (1).
Collected as 124.1 mg of a colorless oil, 47% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (ddd, J = 15.4, 9.2, 1.8, 1H), 6.99 (ddd, J = 15.6, 7.9, 4.9, 1H), 6.53 (dd, J = 8.5, 5.3, 1H), 6.45 (dd, J = 14.8, 8.1, 1H), 5.17 (m, 1H), 5.05 (d, J = 3.1, 1H), 4.72 (m, 1H), 4.62 (m, 1H), 4.47 (tt, J = 8.7, 5.2 1H), 3.87 (s, 3H), 2.94 (d, J = 10.2, 1H), 2.31 (s, 6H), 1.85 (m, 1H), 1.75 (m, 2H), 1.64 (m, 4H), 1.49 (dd, J = 6.5, 2.5, 3H), 1.29 (dd, J = 6.9, 3H), 1.17 (m, 1H), 0.94 (m, 12H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.7*, 171.0*, 170.6*, 170.4*, 169.7*, 164.1*, 148.3*, 122.2*, 93.0*, 72.4(0), 72.3(8), 58.7*, 55.7*, 51.4*, 46.1*, 41.6, 41.1*, 40.8, 33.4, 25.0, 24.7, 24.4, 23.2, 23.0, 22.0*, 21.4, 19.9*, 17.1*, 15.8, 10.5. *Showed twinning. νmax 3292, 3083, 2960, 2875, 2787, 1727, 1632, 1546, 1458, 1332, 1289, 1247, 1177, 1051, 978, 809, 701 cm−1. −64.9 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 593.3909, 0.0 ppm (calculated for C31H53N4O7+, 593.3909).
(S)-1-(((S)-1-(((S,E)-5-((S)-3-Methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-valinate (2).
Collected as 15.0 mg of a colorless oil, 6% yield. 1H NMR (500 MHz, CDCl3) δ 7.40 (ddd, J = 15.6, 8.5, 1.7, 1H), 6.97 (ddd, J = 15.5, 7.4, 4.9, 1H), 6.39 (t, J = 7.3, 1H), 6.14 (m, 1H), 5.16 (dt, J = 9.4, 4.4, 1H), 5.03 (s, 1H), 4.72 (m, 1H), 4.61 (qd, J = 6.5, 2.7, 1H), 4.43 (tt, J = 8.9, 5.2, 1H), 3.86 (s, 3H), 2.83 (d, J = 10.4, 1H), 2.33 (s, 6H), 2.02 (m, 1H), 1.76 (m, 3H), 1.57 (m, 3H), 1.49 (dd, J = 6.6, 2.1, 1H), 1.30 (d, J = 6.2, 3H), 0.99 (d, J = 6.7, 3H), 0.95 (m, 12H), 0.90 (d, J = 6.5, 3H). 13C NMR (125 MHz, CDCl3) δ 180.7, 171.1*, 170.6*, 170.4*, 169.6, 164.2*, 148.1*, 122.3*, 93.0, 74.3, 72.5*, 58.7, 55.7*, 51.5, 46.1*, 41.5, 41.0, 40.9*, 27.7, 24.8, 24.5, 23.2, 23.0, 22.0*, 21.4, 19.9*, 19.7, 19.4, 17.1*. *Showed twinning. νmax 3293, 3081, 2958, 2875, 2789, 1727, 1658, 1632, 1546, 1458, 1332, 1290, 1243, 1179, 1053, 978, 809, 701 cm−1. −33.5 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 579.3751, −0.2 ppm (calculated for C30H51N4O7+, 579.3752).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (3).
Collected as 85.9 mg of a colorless oil, 51% yield. 1H NMR (500 MHz, CDCl3) δ 7.34 (dd, J = 15.5, 10.2, 1H), 7.20 (m, overlap with chloroform signal, 3H), 7.06 (dd, J = 15.5, 4.9, 1H), 6.94 (m, 2H), 6.51 (m, 1H), 6.37 (m, 1H), 5.17 (m, 1H), 4.89 (m, 1H), 4.83 (d, J = 5.6, 1H), 4.75 (m, 1H), 4.45 (m, 1H), 3.82 (s, 3H), 3.54 (m, 1H), 3.12 (d, J = 13.8, 1H), 2.94 (d, J = 10.2, 1H), 2.32 (s, 6H), 1.84 (m, 1H), 1.74 (m, 3H), 1.64 (m, 3H), 1.56 (m, 1H), 1.31 (dd, J = 7.0, 3.2, 3H), 1.15, (m, 1H), 0.93 (m, 12H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 178.0*, 171.1*, 170.7*, 170.4*, 169.8*, 164.6*, 148.8*, 134.3*, 129.7, 128.3*, 127.1*, 122.3*, 95.0, 72.6, 72.5, 59.8*, 58.5, 51.6, 46.1*, 41.7, 41.2*, 41.0, 34.7*, 33.5, 25.2, 24.8, 24.5, 23.2, 23.1*, 22.2*, 21.5, 20.0*, 15.9, 10.6. *Showed twinning. νmax 3293, 3076, 2959, 2874, 2787, 1728, 1635, 1545, 1455, 1347, 1248, 1166, 1052, 972, 732 cm−1. −59.0 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 669.4222, 0.1 ppm (calculated for C37H57N4O7+, 669.4223).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-phenylalaninate (4).
Collected as 50.5 mg of a colorless foam, 29% yield. 1H NMR (500 MHz, CDCl3) δ 1H 7.35 (ddd, J = 15.5, 8.4, 1.7, 1H), 7.29 (m, 2H), 7.21 (m, 6H), 7.05 (dd, J = 15.5, 4.9, 2.5, 1H), 6.94 (m, 2H), 6.30 (dd, J = 20.1, 8.2, 1H), 6.21 (dd, J = 15.4, 8.1, 1H), 5.09 (dt, J = 8.3, 4.2, 1H), 4.89 (m, 1H), 4.81 (d, J = 3.1, 1H), 4.76 (m, 1H), 4.36 (m, 1H), 3.81 (s, 3H), 3.56 (m, 2H), 3.12 (m, 1H), 3.01 (m, 2H), 2.42 (d, J = 1.7, 6H), 1.67 (m, 4H), 1.57 (m, 1H), 1.45 (m, 1H), 1.33 (dd, J = 7.0, 3.1, 3H), 0.91 (m, 12H). 13C NMR (125 MHz, CDCl3) δ 177.8, 170.9*, 170.5*, 170.4*, 169.7, 164.5*, 148.7*, 137.7*, 134.3*, 129.6, 128.9, 128.6, 128.2*, 127.0*, 126.8*, 122.2*, 94.9*, 72.9*, 69.2*, 59.7*, 58.4*, 51.4, 46.1*, 41.9, 40.8, 40.4*, 35.7, 34.6*, 24.8, 24.4, 23.1, 23.0*, 21.9*, 21.5, 20.0*. *Showed twinning. νmax 3292, 3067, 2956, 2873, 2789, 1727, 1635, 1548, 1455, 1349, 1245, 1165, 1069, 1023, 972, 807, 735, 701 cm−1. −34.4 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 703.4063, −0.3 ppm (calculated for C40H55N4O7+, 703.4065).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (5).
Collected as 71.4 mg of a colorless oil, 52% yield. 1H NMR (500 MHz, CDCl3) δ 7.40 (ddd, J = 15.5, 9.6, 1.8, 1H), 6.98 (m, 1H), 6.51 (m, 1H), 6.36 (dd, J = 23.7, 8.0, 1H), 5.18 (m, 1H), 5.06 (d, J = 5.4, 1H), 4.73 (m, 1H), 4.68 (m, 1H), 4.46 (m, 1H), 3.86 (s, 3H), 2.94 (d, J = 10.1, 1H), 2.32 (s, 6H), 1.83 (m, 3H), 1.75 (m, 4H), 1.65 (m, 3H), 1.57 (m, 1H), 1.29 (d, J = 7.0, 3H), 1.17 (m, 1H), 0.93 (m, 18H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.6*, 171.0, 170.6*, 170.4*, 170.1*, 164.3*, 148.2*, 122.4*, 93.6, 72.5, 72.4, 58.6*, 51.5, 46.1*, 41.6, 41.1*, 40.9, 39.1*, 33.4, 25.1, 24.8, 24.4, 24.2, 23.7*, 23.2, 23.0, 22.7, 22.1*, 21.4, 19.9, 15.8, 10.5. *Showed twinning. νmax 3292, 3086, 2959, 2874, 2788, 1728, 1630, 1546, 1457, 1336, 1224, 1173, 1052, 984, 809, 685 cm−1. −58.2 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 635.4376, −0.3 ppm (calculated for C34H59N4O7+, 635.4378).
(S)-1-(((S)-1-(((S,E)-5-((S)-3-Methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (6).
Collected as 54.6 mg of a glassy colorless oil, 43% yield. 1H NMR (500 MHz, CDCl3) δ 7.40 (ddd, J = 15.6, 12.2, 1.8, 1H), 7.27 (m, overlap with chloroform signal, 2H), 7.21 (m, 1H), 7.18 (m, 2H), 7.02 (m, 1H), 6.47 (dd, J = 8.2, 3.7, 1H), 6.37 (dd, J = 20.7, 8.4, 1H), 5.16 (m, 1H), 5.02 (d, J = 4.0, 1H), 4.94 (m, 1H), 4.59 (p, J = 6.4, 1H), 4.40 (m, 1H), 3.85 (s, 3H), 2.92 (m, 3H), 2.30, (s, 6H), 1.84 (m, 1H), 1.74 (m, 3H), 1.64 (m, 3H), 1.53 (m, 1H), 1.47 (dd, J = 8.4, 6.5, 3H), 1.15 (m, 1H), 0.92 (m, 15H), 0.85 (d, J = 6.6, 3H). 13C NMR (125 MHz, CDCl3) δ 180.6*, 170.9*, 170.8, 170.3*, 169.6*, 163.9*, 146.5*, 136.5*, 129.3*, 128.6, 126.9, 123.0*, 93.0, 72.6, 72.3, 58.7*, 55.7*, 51.6*, 51.4*, 41.6, 41.1, 40.9*, 40.5*, 33.5, 25.2, 24.7*, 24.4, 23.2, 23.0, 22.1*, 21.5, 17.1*, 15.7, 10.5. *Showed twinning. −35.2 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 669.4221, −0.3 ppm (calculated for C37H54N4O7+, 669.4222).
(S)-1-(((S)-1-(((S,E)-5-((S)-3-Methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-phenylalaninate (7).
Collected as 81.5 mg of colorless oil, 52% yield. 1H NMR (500 MHz, CDCl3) δ 7.42 (ddd, J = 15.5, 10.4, 1.7, 1H), 7.25 (m, overlap with chloroform signal, 6H) 7.17 (m, 4H), 7.02 (td, J = 15.7, 15.6, 5.2, 1H), 6.49 (m, 1H), 6.19 (dd, J = 17.6, 8.0, 1H), 5.10 (m, 1H), 5.02 (d, J = 2.9, 1H), 4.94 (m, 1H), 4.59 (m, 1H), 4.31 (m, 1H), 3.85 (s, 3H), 3.49 (m, 1H), 3.01 (m, 2H), 2.96 (m, 1H), 2.89, (m, 1H), 2.39 (s, 6H), 1.65 (m, 4H), 1.53 (m, 1H), 1.47 (t, J = 6.6, 3H), 1.41 (m, 1H), 0.89 (m, 12H). 13C NMR (125 MHz, CDCl3) δ 180.6*, 170.7(2)*, 170.6(8), 170.4*, 169.5*, 164.0*, 146.7*, 137.6*, 136.6*, 129.3*, 128.9*, 128.6*, 128.5, 126.9, 126.8, 123.0*, 93.0, 72.6*, 69.5*, 58.7*, 55.7, 515.*, 51.4, 41.9*, 40.7, 40.4*, 40.0*, 35.9*, 24.7*, 24.4, 23.1, 23.0, 21.9*, 21.6*, 17.1*. *Showed twinning. νmax 3295, 3064, 3032, 2955, 2873, 2787, 1725, 1662, 1628, 1544, 1455, 1334, 1289, 1245, 1175, 1063, 971, 739, 700 cm−1. −44.3 (CH2O2). HR-ESI-TOF-MS [M + Na]+ m/z 725.3886, 0.1 ppm (calculated for C40H54N4NaO7+, 725.3885).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (8).
Collected as 34.7 mg of a glassy colorless oil, 29% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 2H), 7.23 (m, 1H), 7.19 (m, 6H), 7.09 (ddd, J = 15.5, 7.6, 5.1, 1H), 6.90 (m, 2H), 6.48 (dd, J = 8.2, 4.1, 1H), 6.35 (dd, J = 70.5, 8.5, 1H), 5.16 (m, 1H), 4.98 (m, 1H), 4.88 (m, 1H), 4.80 (d, J = 3.7, 1H), 4.40, (tt, J = 8.6, 5.6, 1H), 3.81 (d, J = 1.3, 1H), 3.53 (ddd, J = 17.2, 13.9, 5.1, 1H), 3.10 (ddd, J = 14.0, 7.4, 3.0, 1H), 2.93 (m, 3H), 2.30 (d, J = 2.05, 6H), 1.84 (m, 1H), 1.76 (m, 2H), 1.64 (m, 4H), 1.52 (m, 1H), 1.15 (m, 1H), 0.95 (m, 6H), 0.90 (m, 9H), 0.86 (dd, J =6.7, 1.6, 3H). 13C NMR (125 MHz, CDCl3) δ 177.8*, 170.9*, 170.7*, 170.2*, 169.5*, 164.2*, 146.8*, 136.4*, 134.2*, 129.6, 129.4*, 128.6, 128.2*, 127.0*, 126.9*, 122.9*, 94.9, 72.6, 72.3*, 59.7*, 58.4*, 51.6*, 51.3*, 41.6, 41.0*, 40.9, 40.5*, 34.5*, 33.5, 25.2, 24.7, 24.4, 23.2, 22.9*, 22.1*, 21.5*, 15.8, 10.5. *Showed twinning. νmax 3301, 3066, 3030, 2958, 2874, 2785, 1726, 1662, 1630, 1543, 1455, 1342, 1249, 1168, 1125, 1050, 970, 735, 700 cm−1. −48.2 (CH2O2). HR-ESI-TOF-MS [M + Na]+ m/z 767.4354, −0.2 ppm (calculated for C43H60N4NaO7+, 767.4354).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-phenylalaninate (9).
Collected as 39.4 mg of a colorless oil, 28% yield. 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 3H), 7.20 (m, overlap with chloroform signal, 11H), 7.08 (ddd, J = 15.5, 8.2, 5.2, 1H), 6.91 (m, 2H), 6.41 (dd, J = 29.3, 8.3, 1H), 6.18 (t, J = 8.2, 1H), 5.09 (dd, J = 8.6, 3.9, 1H), 4.96 (m, 1H), 4.88 (m, 1H), 4.79 (s, 1H), 4.29 (q, J = 7.4, 1H), 3.81 (s, 3H), 3.51 (m, 2H), 3.10 (m, 1H), 3.00 (m, 3H), 2.93 (m, 1H), 2.39 (d, J = 2.1, 6H), 1.65 (m, 4H), 1.52 (m, 1H), 1.40 (m, 1H), 0.89 (m, 12H). 13C NMR (125 MHz, CDCl3) δ 177.8, 170.7*, 170.6, 170.4*, 169.5*, 164.3*, 147.0*, 137.6*, 136.5*, 134.2*, 129.6*, 129.3*, 128.9, 128.6*, 128.2*, 127.0*, 126.9, 126.8*, 122.9*, 94.9, 72.6*, 69.5*, 59.7*, 58.3*, 51.5*, 51.4*, 41.9, 40.7, 40.5*, 40.0*, 35.9*, 34.5*, 24.7*, 24.4, 23.1, 22.9*, 22.0*, 21.6*. *Showed twinning. νmax 3298, 3031, 2954, 2872, 2789, 1726, 1662, 1630, 1544, 1453, 1342, 1246, 1164, 1066, 1023, 969, 808, 737, 700 cm−1. −33.2 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 801.4196, −0.2 ppm (calculated for C46H58N4NaO7+, 801.4198).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (10).
Collected as 27.9 mg of a colorless oil, 22% yield. 1H NMR (500 MHz, CDCl3) δ 7.37 (ddd, J = 19.0, 15.5, 1.8, 1H), 7.27 (m, overlaps with the chloroform signal), 7.21 (m, 1H), 7.17 (m, 2H), 7.00 (ddd, J = 15.5, 9.8, 5.1, 1H), 6.46 (dd, J = 8.2, 5.0, 1H), 6.29 (dd, J = 39.0, 8.5, 1H), 5.16 (ddd, J = 9.5, 6.9, 4.0, 1H), 5.03 (d, J = 3.5, 1H), 4.95 (m, 1H), 4.66 (dt, J = 6.4, 4.2, 1H), 4.39 (dtd, J = 14.3, 8.6, 5.6, 1H), 3.85 (s, 3H), 2.91 (m, 3H), 2.30 (d, J = 2.2, 6H), 1.78 (m, 6H), 1.62 (m, 3H), 1.53 (m, 1H), 1.15 (m, 1H), 0.93 (m, 18H), 0.86 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.6, 170.9*, 170.8*, 170.3*, 170.0*, 164.0*, 146.4*, 136.4*, 129.3*, 128.6*, 126.9, 123.1*, 93.6*, 72.6, 72.3*, 58.6*, 58.5*, 51.6*, 51.3*, 41.6, 41.0 40.9*, 40.5*, 39.0*, 33.5, 25.2*, 24.7*, 24.4, 24.2*, 23.7*, 23.2, 22.9*, 22.7*, 22.1*, 21.5, 15.7*, 10.5. νmax 3299, 3069, 2958, 2872, 2786, 1725, 1660, 1627, 1543, 1458, 1335, 1250, 1173, 1051, 982, 740, 699 cm−1. −42.4 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 711.4691, 0.0 ppm (calculated for C40H63N4O7+, 711.4691).
(S)-1-(((S)-1-(((S,E)-5-((S)-2-Isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-valinate (11).
Collected as 19.5 mg of a colorless oil, 16% yield. 1H NMR (500 MHz, CDCl3) δ 7.37 (ddd, J = 17.6, 15.5, 1.8, 1H), 7.26 (m, overlap with chloroform signal, 5H), 7.21 (m, 1H), 7.17 (m, 2H), 6.99 (ddd, J = 15.6, 9.1, 5.1, 1H), 6.42 (dd, J = 8.1, 5.8, 1H), 6.23 (dd, J = 30.8, 8.4, 1H), 5.16 (m, 1H), 5.03 (d, J = 2.8, 1H), 4.94 (m, 1H), 4.66 (m, 1H), 4.38 (m, 1H), 3.85 (s, 3H), 2.92 (m, 2H), 2.81, (dd, J = 10.2, 2.2, 1H), 2.31 (d, J = 1.9, 6H), 2.01 (m, 1H), 1.77 (m, 6H), 1.62 (m, 2H), 1.52 (m, 1H), 0.91 (m, 24H). 13C NMR (125 MHz, CDCl3) δ 180.6, 171.0*, 170.8*, 170.2*, 170.0*, 164.0*, 146.4*, 136.4*, 129.3*, 128.6*, 126.9, 123.1*, 93.6, 74.3, 72.3*, 58.6*, 58.5*, 51.6*, 51.3*, 41.5, 41.0*, 40.9, 40.5*, 39.0*, 27.7, 24.7*, 24.4, 24.2*, 23.7*, 23.2, 22.9*, 22.7*, 22.1*, 21.5, 19.6*, 19.4. *Showed twinning. νmax 3291, 3075, 2957, 2873, 2787, 1724, 1661, 1627, 1545, 1458, 1336, 1228, 1171, 1049, 982, 809, 740, 699 cm−1. −40.7 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 719.4353, −0.1 ppm (calculated for C39H60N4NaO7+, 719.4354).
(S)-1-(((S)-1-(((S,E)-5-((R)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (12).
Collected as 47.5 mg of a colorless oil, 32% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 3H), 7.23 (m, 1H), 7.19 (m, 5H), 7.08 (ddd, J = 15.6, 7.8, 5.1, 1H), 6.90 (m, 2H), 6.45 (m, 1H), 6.27 (m, 1H), 5.15 (m, 1H), 4.97 (m, 1H), 4.87 (td, J = 5.0, 2.9, 1H), 4.80 (d, J = 2.8, 1H), 4.38 (tdd, J = 8.8, 5.7, 3.6, 1H), 3.81 (d, J = 1.2, 3H), 3.53 (ddd, J = 15.7, 14.0, 5.1, 1H), 3.10 (ddd, J = 13.9, 6.9, 2.9, 1H), 2.93 (m, 3H), 2.30 (d, J = 1.9, 6H), 1.84 (m, 1H), 1.76 (m, 2H), 1.64 (m, 4H), 1.51 (m, 1H), 1.15 (m, 1H), 0.95 (m, 6H), 0.90 (m, 9H), 0.86 (m, 3H). 13C NMR (125 MHz, CDCl3) δ 177.8, 170.9, 170.7, 170.2*, 169.5*, 164.2*, 146.8*, 136.4*, 134.2*, 129.6, 129.4*, 128.6, 128.2*, 127.0*, 126.9*, 122.9*, 94.9, 72.6, 72.3*, 59.7*, 58.4*, 51.6*, 51.3*, 41.6, 41.0*, 40.9, 40.6, 40.4, 34.5*, 33.5, 25.2, 24.7, 24.4, 23.2, 22.9*, 22.1*, 21.5*, 15.7, 10.5. *Showed twinning. −11.9 (CH2O2). HR-ESI-TOF-MS [M + H]+ m/z 745.4538, −0.3 ppm (calculated for C43H61N4O7+, 745.4540).
(S)-1-(((S)-1-(((S,E)-5-((R)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-phenylalinate (13).
Collected as 51.7 mg of a colorless oil, 37% yield. 1H NMR (500 MHz, CDCl3) δ 7.32 (m,2H), 7.19 (m, overlap with chloroform signal, 12H), 7.09 (m, 1H), 6.91 (m, 2H), 6.46 (dd, J = 41.0, 8.4, 1H), 6.21 (dd, J = 11.3, 8.0, 1H), 5.09 (m, 1H), 4.96 (m, 1H), 4.87 (m, 1H), 4.79 (s, 1H), 4.30 (m, 1H), 3.80 (s, 3H), 3.50 (m, 2H), 3.10 (m, 1H), 3.00 (m, 3H), 2.93 (m, 1H), 2.39 (s, 6H), 1.64 (m, 4H), 1.52 (m, 1H), 1.40 (m, 1H), 0.89 (m, 12H). 13C NMR (125 MHz, CDCl3) δ 177.7, 170.6(1)*, 170.5(7)*, 170.3*, 169.5*, 164.2*, 147.0*, 137.6*, 136.5*, 134.2*, 129.5*, 129.3*, 128.8, 128.5(1), 128.4(6), 128.1*, 126.9*, 129.8(1), 126.7(8)*, 122.9*, 94.9, 72.6*, 69.5*, 59.6*, 58.3*, 51.4*, 51.3*, 41.8, 40.7, 40.4*, 40.0*, 35.8*, 34.5*, 24.6*, 24.3, 23.1, 22.8*, 21.9*, 21.5*. *Showed twinning. νmax 3298, 3031, 2954, 2872, 2789, 1726, 1662, 1630, 1543, 1454, 1343, 1247, 1164, 1068, 1023, 970, 737, 700 cm−1. −32.7 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 801.4196, −0.2 ppm (calculated for C46H58N4NaO7+, 801.4198).
(S)-1-(((S)-1-(((R,E)-5-((S)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (14).
Collected as 24.3 mg of a colorless oil, 13% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 3H), 7.24 (m, 1H), 7.20 (m, 5H), (m, 1H), 6.92 (m, 2H), 6.46 (dd, J = 13.9, 8.1, 1H), 6.14 (dd, J = 25.5, 8.5, 1H), 5.16 (m, 1H), 4.97 (m, 1H), 4.87 (m, 1H), 4.79 (m, 1H), 4.38 (m, 1H), 3.81 (d, J = 2.3, 3H), 3.52 (td, J = 13.5, 5.2, 1H), 3.12 (m, 1H), 3.02 (m, 1H), 2.91 (m, 2H), 2.29 (s, 6H), 1.82, (m, 1H), 1.72 (m, 2H), 1.59 (m, 3H), 1.42 (m, 2H), 1.13 (m, 1H), 0.86 (m, 18H). 13C NMR (125 MHz, CDCl3) δ 177.9*, 171.1, 170.8*, 170.4*, 169.6*, 164.2*, 146.8*, 136.4*, 134.3*, 129.5, 129.4*, 128.6, 128.2, 127.0, 126.9*, 122.9*, 94.9*, 72.6*, 72.5*, 59.7*, 58.4, 51.5, 51.3*, 41.6*, 40.8*, 40.7*, 40.3*, 34.6*, 33.4*, 25.2*, 24.6*, 24.4*, 23.2*, 22.9*, 21.9*, 21.5*, 15.7*, 10.5*. *Showed twinning. νmax 3292, 3067, 2957, 2872, 2787, 1727, 1633, 1542, 1454, 1345, 1250, 1165, 1050, 971, 806, 735, 700, 516 cm−1. +8.4 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 745.4535, −0.7 ppm (calculated for C43H61N4O7+, 745.4540).
(S)-1-(((S)-1-(((R,E)-5-((R)-2-Benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (15).
Collected as 63.9 mg of a colorless oil, 36% yield. 1H NMR (500 MHz, CDCl3)) δ 7.30 (m, 3H), 7.25 (m, 1H), 7.20 (m, 5H), 7.8 (ddd, J = 15.5, 14.0, 5.1, 1H), 6.91 (m, 2H), 6.49 (dd, J = 13.1, 8.2, 1H), 6.22 (dd, J = 28.5, 8.5, 1H), 5.16 (m, 1H), 4.97 (m, 1H), 4.86 (m, 1H), 4.79 (d, J = 6.2, 1H), 4.38 (m, 1H), 3.81 (d, J = 2.6, 3H), 3.53 (td, J = 13.7, 5.2, 1H), 3.11 (ddd, J = 13.9, 7.7, 3.0, 1H), 3.03 (ddd, J = 27.4, 13.9, 6.3, 1H), 2.91 (m, 2H), 2.28 (d, J = 2.2, 6H), 1.82 (m, 1H), 1.74 (m, 2H), 1.59 (m, 3H), 1.41 (m, 2H), 1.13 (m, 1H), 0.86 (m, 18H). 13C NMR (125 MHz, CDCl3) δ 177.8*, 171.0, 170.7*, 170.3*, 169.5*, 164.1*, 146.8*, 136.4*, 134.2*, 129.4, 129.3*, 128.5, 128.1, 126.9, 126.8*, 122.8*, 94.8*, 72.5*, 72.4*, 59.6*, 58.3, 51.4, 51.2*, 41.5*, 40.8*, 40.6*, 40.2*, 34.5*, 33.3*, 25.1*, 24.5*, 24.3*, 23.1*, 22.8*, 21.9*, 21.4*, 15.6*, 10.4*. *Showed twinning. νmax 3291, 3032, 2958, 2873, 2787, 1727, 1633, 1542, 1455, 1345, 1251, 1165, 1050, 971, 805, 735, 700 cm−1. −23.7 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 745.4538, −0.3 ppm (calculated for C43H61N4O7+, 745.4540).
(S)-1-(((S)-1-(((S,E)-5-((S)-3-(Hex-5-yn-1-yloxy)-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (16).
Collected as 81.5 mg of a yellow oil, 35% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (ddd, J = 15.6, 8.9, 1.8, 1H), 6.99 (ddd, J = 15.6, 8.4, 5.0, 1H), 6.53 (m, 1H), 6.44 (dd, J = 14.3, 8.2, 1H), 5.17 (m, 1H), 5.02 (d, J = 3.8, 1H), 4.72 (m, 1H), 4.61 (m, 1H), 4.47 (tt, J = 8.7, 5.5, 1H), 4.06 (m, 1H), 4.01 (m, 1H), 2.94 (d, J = 10.2, 1H), 2.31 (s, 6H), 2.28 (td, J = 6.9, 2.6, 2H), 2.0 (t, J = 2.6, 1H), 1.93 (m, 2H), 1.85 (m, 1H), 1.76 (m, 3H), 1.66 (m, 5H), 1.59 (m, 1H), 1.49 (dd, J = 6.7, 2.8, 3H), 1.29 (d, J = 6.9, 3H), 1.17 (m, 1H), 0.94 (m, 12H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 179.7*, 171.0, 170.6*, 170.4*, 169.8*, 164.1*, 148.2*, 122.3*, 93.2, 83.4, 72.5, 72.3, 71.4, 69.1, 55.8*, 51.5, 46.1*, 41.6, 41.1*, 40.9, 33.4, 27.4, 25.1, 24.8, 24.6, 24.4, 23.2, 23.0, 22.1*, 21.5, 19.9*, 18.0, 17.2*, 15.8, 10.5. *Showed twinning. νmax 3295, 3086, 2958, 2875, 2788, 1726, 1659, 1625, 1546, 1459, 1331, 1288, 1235, 1176, 1046, 978, 703, 637 cm−1. −50.7 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 659.4376, −0.3 ppm (calculated for C36H59N4O7+, 659.4378).
Hydrogenation of gallinamide A (1) to form compound 17.
In a vial 15.2 mg of gallinamide A was dissolved in 1.00 mL ethanol and ca. 7.6 mg of palladium on carbon (1% loading) was added in one portion, and the atmosphere was replaced with hydrogen. After stirring overnight the reaction was quenched with 1.0 mL of dimethyl sulfoxide DMSO and stirred for 6 hours at which point it was passed through a ca. 1 cm diatomaceous earth plug, prepared in a pipette with glass wool, with 10 mL of DCM and the filtrate was washed with 10 mL of brine. The aqueous layer was extracted three times with equal volume DCM and the combined organics were dried over MgSO4. This material was concentrated to dryness by rotovap and purified by RP-SPE using two elutions 10% CH3CN/H2O and CH3CN. The CH3CN fraction was further purified by RP-HPLC using a CH3CN/H2O elution to provide pure compound 17.
(S)-1-(((S)-1-(((S)-5-((S)-3-Methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)amino)-4-methyl-1-oxopentan-2-yl dimethyl-l-isoleucinate (17).
1H NMR (500 MHz, CDCl3) δ 6.49 (dd, J = 8.3, 4.3, 1H), 6.36 (dd, J = 11.8, 7.7, 1H), 5.20 (m, 1H), 5.04 (d, J = 2.6, 1H), 4.56 (q, J = 6.6, 1H), 4.40 (m, 1H), 3.92 (m, 1H), 3.87 (d, J = 1.4, 3H), 2.94 (d, J = 10.1, 1H), 2.88 (m, 2H), 2.32 (s, 6H), 1.81 (m, 5H), 1.66 (m, 4H), 1.53 (m, 1H), 1.47 (t, J = 6.2, 3H), 1.18 (m, 1H), 1.13 (d, J= 6.5, 3H), 0.93 (m, 12H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.7*, 172.3*, 170.9*, 170.8*, 170.2, 169.6, 92.8, 72.6, 72.5*, 58.7*, 55.6*, 51.6*, 45.5*, 41.6, 41.5*, 41.1*, 34.0*, 33.5*, 30.9*, 25.2, 24.8*, 24.5, 23.2, 23.0*, 22.1*, 21.5*, 20.8*, 17.1*, 15.8*, 10.5. *Showed twinning. νmax 3292, 3083, 2960, 2874, 2788, 1728, 1642, 1548, 1457, 1377, 1329, 1294, 1247, 1156, 1052, 984, 810, 693 cm−1. −46.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 617.3888, 0.5 ppm (calculated for C31H55N4NaO7+, 617.3885).
Weinreb Amide Synthesis (20a-c).
As a general procedure, the Boc-protected amino acid was dissolved in DMF, the reaction mixture was cooled to 0 °C by ice bath and the N,N-diisopropyl ethyl amine (DIPEA) was added dropwise. The N,N,N,N-tetramethyl-O-(1H-benzotriazol-1-yl)uronium hexafluorophosphate (HBTU) was then added in one portion and the reaction mixture was allowed to warm to room temperature overnight. The alanine product was collected according to the literature procedure by iterative crystallization from cold CH3CN,9 but the phenylalanine cases did not precipitate out of DMF. In the phenylalanine cases the reactions were worked up by diluting the reaction mixtures into diethyl ether. The dilute mixture was then washed twice with 5% citric acid (aq), then brine, and finally dried over MgSO4, filtered, and the filtrate was concentrated to dryness in vacuo on a rotary evaporator. The crude residue was then dissolved in hexanes and filtered through diatomaceous earth, concentrated again using a rotary evaporator and purified by silica gel flash chromatography with an ethyl acetate/hexanes gradient elution.
Tert-butyl (S)-(1-(methoxy(methyl)amino)-1-oxopropan-2-yl)carbamade (20a).
Collected as white crystals, 90% yield. 1H NMR (500 MHz, CDCl3) δ 5.25 (bs, 1H), 4.68 (bs, 1H), 3.77 (s, 3H), 3.21 (s, 3H), 1.44 (s, 9H), 1.31 (d, J = 7.0 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 173.7, 155.3, 79.5, 61.7, 46.6, 32.2, 28.4, 18.7. IR (neat) νmax3295, 2976, 1705, 1660, 1545, 1451, 1363, 1298, 1183, 980, 585 cm−1. λmax (log ε) 205 (6.5) nm, maximum at the end of range. +0.1 (CH2Cl2). HR-ESI-TOS-MS [M + Na]+ m/z 241.1154, −2.0 ppm (calculated for C9H18N2NaO4+, 241.1159).
Tert-butyl (S)-(1-(methoxy(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamade (20b).
Collected as a viscous faint-yellow oil, 84% yield. 1H NMR (500 MHz, CDCl3) δ 7.28 (m, overlap with chloroform signal, 2H), 7.22 (m, 1H), 7.17 (d, J = 7.4, 2H), 5.16 (bd, J = 9.1, 1H), 4.95 (bd, J = 8.2, 1H), 3.66 (s, 3H), 3.17 (s, 3H), 3.05 (dd, J = 13.8, 6.1, 1H), 2.87 (dd, J= 13.8, 7.2, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 172.3, 155.1, 136.6, 129.4, 128.3, 126.7, 79.6, 61.6, 51.5, 38.9, 32.1, 28.3. IR (neat) νmax 3321, 2976, 2935, 1711, 1662, 1498, 1453, 1390, 1367, 1251, 1171 cm−1. λmax (log ε) 205 (6.8) nm, maximum at the end of range. +21.8 (CH2Cl2). HR-ESI-TOS-MS [M + Na]+ m/z 331.1627, −0.3 ppm (calculated for C16H24N2NaO4+, 331.1628).
Tert-butyl (R)-(1-(methoxy(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamade (20c).
Collected as 1.08 g of a faint-yellow oil, 97% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, overlaps with the chloroform signal, 2H), 7.22 (m, 1H), 7.17 (m, 2H), 5.16 (d, J = 8.6, 1H), 4.95 (q, J = 7.1, 1H), 3.66 (s, 3H), 3.17 (s, 3H), 3.06 (dd, J = 13.8, 6.1, 1H), 2.88 (dd, J = 13.7, 7.1, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 172.2, 155.1, 136.5, 129.4, 128.3, 126.7, 79.6, 61.6, 51.5, 38.8, 32.0, 28.3. IR (neat) νmax 3426, 3323, 2976, 2935, 1710, 1661, 1501, 1452, 1378, 1329, 1251, 1170, 989, 750, 701 cm−1. λmax (log ε) 205 (7.0) nm, maximum at the end of range. −21.0 (CH2Cl2). HR-ESI-TOS-MS [M + Na]+ m/z 331.1628, 0.0 ppm (calculated for C16H24N2NaO4+, 331.1628).
Enone Synthesis (21a-c).
As a general procedure, 18a-c was dissolved in tetrahydrofuran (THF), cooled to 0 °C in an ice bath, and then lithium aluminum hydride was added in five equal portions. After 45 min the reaction was quenched with minimal ethyl acetate and diluted into dichloromethane (DCM); this mixture was washed with 10% KH2PO4 (aq), using a small amount of brine to assist phase separation. The aqueous phase was then back extracted iteratively with DCM, and then the combined organic phases were washed with brine and dried over MgSO4. The sample was filtered and the filtrate was collected as a yellow oil and dissolved in DCM. While stirring, the anylidene was added to the reaction mixture in one portion and the reaction was heated to reflux using an oil bath for 2.5 h. The reaction mixture was then concentrated to dryness on a rotary evaporator and purified by silica gel flash chromatography using ethyl acetate/hexanes in a gradient elution.
Methyl (S,E)-4-((tert-butoxycarbonyl)amino)pent-2-enoate (21a).
Collected as 7.81 g of an off white low melting solid, 85% yield. 1H NMR (500 MHz, CDCl3) δ 6.89 (dd, J = 15.4, 4.7 Hz, 1H), 5.91 (dd, J = 15.7, 1.1 1H), 4.51 (bs, 1H), 4.40 (bs, 1H), 3.74 (s, 3H), 1.45 (s, 9H), 1.27 (d, J = 6.9 Hz, 3H). 13C NMR (125 MHz, CDCl3) δ 166.8, 154.9, 149.7, 119.7, 79.8, 51.6, 47.0, 28.4, 20.3. IR (neat) νmax 3354, 2978, 1712, 1519, 1448, 1369, 1275, 1171, 1046 cm−1. λmax (log ε) 205 (6.4) nm, maximum at the end of range. −18.5 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 252.1204, −0.8 ppm (calculated for C11H19NNaO4+, 252.1206).
Methyl (S,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoate (21b).
Collected as 15.16 g of a colorless oil, 78% yield. 1H NMR (500 MHz, CDCl3) δ 7.31 (t, J = 7.4 Hz, 2H), 7.24 (m, overlaps with chloroform signal), 7.17 (d, J = 7.5, 2H), 6.92 (dd, J = 15.7, 5.1, 1H), 5.86 (d, J = 15.5, 1H), 4.62 (bs, 1H), 4.53 (bs, 1H), 3.72 (s, 3H), 2.89, (d, J = 6.7, 2H), 1.40 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.6, 154.9, 147.9, 136.3, 129.4, 128.6, 126.9, 120.7, 79.9, 52.3, 51.7, 40.8, 28.3. IR (neat) νmax 3352, 2978, 1712, 1516, 1439, 1367, 1282, 1168, 1023, 702 cm−1. λmax (log ε) 205 (7.7) nm, maximum at the end of range. −1.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 328.1516, −0.9 ppm (calculated for C17H23NNaO4+, 328.1519).
Methyl (R,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoate (21c).
Collected as 0.86 g of a colorless oil, 80% yield. 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 2H), 7.23 (m, 1H), 7.17 (m, 2H), 6.91 (dd, J = 5.1, 15.7, 1H), 5.86 (dd, J = 1.8, 15.7, 1H), 4.62 (bs, 1H), 4.50 (bs, 1H), 3.73 (s, 3H), 2.89 (d, J = 6.8, 2H), 1.40 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 166.6, 154.9, 147.9, 136.3, 129.4, 128.6, 126.9, 120.7, 79.9, 52.3, 51.6, 40.8, 28.3. IR (neat) νmax 3359, 2977, 2941, 1715, 1515, 1445, 1365, 1280, 1255, 1168, 1023 cm−1. λmax (log ε) 205 (7.3) nm, maximum at the end of range. −4.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 328.1517, −0.6 ppm (calculated for C17H23NNaO4+, 328.1519).
Benzyl Esterification (22).
Leucic acid was dissolved in dimethyl sulfoxide, and while stirring cesium carbonate was added in one portion followed by dropwise addition of the benzyl bromide. After stirring for 6 h at room temperature the reaction mixture was quenched with deionized water and extracted iteratively with diethyl ether. The combined organic phases were washed with brine and then dried over MgSO4. This material was filtered, the filtrate concentrated to an oil in vacuo, and the crude oil was purified by iterative flash chromatography with a gradient elution of EtOAc in hexanes to afford a colorless oil. Running the reaction with potassium carbonate for 18 h afforded the product with a slightly reduced yield (85%).
Benzyl (S)-2-hydroxy-4-methylpentanoate (22).
Collected as 3.77 g of a colorless oil, 89% yield.1H NMR (500 MHz, CDCl3) δ 7.36 (m, 5H), 5.21 (d, J = 2.0, 2H), 4.24 (m, 1H), 2.62 (d, J = 6.1, 1H), 1.88 (m, 1H), 1.57 (m, 2H), 0.94 (t, J = 6.3, 6H). 13C NMR (125 MHz, CDCl3) δ 175.7, 135.2, 128.7, 128.5, 128.3, 69.1, 67.3, 43.4, 24.4, 23.2, 21.5. νmax 3458, 2956, 2874, 1736, 1460, 1375, 1266, 1206, 1140, 1085, 1004, 744, 697, 591, 418 cm−1. λmax (log ε) 205 (6.8) nm, maximum at end of range. −18.4 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 245.1149, 0.4 ppm (calculated for C12H18NaO3+, 245.1148).
Tail Bi-residue Ester Synthesis (23a-c).
As a general procedure, the benzyl ester protected l-leucic acid was dissolved in DCM, and then the pyridine and the Fmoc-protected amino acid were added to the solution in one portion each. The reaction was cooled to 0 °C and DIC was added dropwise. The reaction mixture was allowed to warm to room temperature overnight, after which the reaction was worked up by diluting into DCM, washing with 10% NaHCO3 (aq), 5% citric acid (aq), and brine before being dried over MgSO4. The suspension was filtered, and the filtrate was then concentrated to dryness in vacuo and the DIC-derived urea salt was removed by filtration through a glass frit with iterative EtOAc washes. The filtrate was concentrated to dryness in vacuo and dissolved in DMF. To this solution was added DIPA and the reaction was stirred for 2.5-2.75 h. The reaction mixture was diluted with diethyl ether, washed with 5% citric acid (aq), and brine (if by TLC the citric acid wash showed product it was back extracted with ether). The organic phase was dried over MgSO4, filtered, and the filtrate of this step was then concentrated to dryness by rotary evaporation. The crude residue was purified by silica gel flash chromatography with a gradient elution of diethyl ether in hexanes.
Benzyl (S)-2-((l-isoleucyl)oxy)-4-methylpentanoate (23a).
Collected as a faint-yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.34 (m, 5H), 5.16 (s, 2H), 5.11 (dd, J = 9.6, 4.0, 1H), 3.40 (d, J = 4.9, 1H), 1.80 (m, 2H), 1.74 (m, 1H), 1.67 (m, 1H), 1.51 (m, 3H), 1.21 (m, 1H), 0.92 (m, 12H). 13C NMR (125 MHz, CDCl3) δ 175.2, 170.4, 135.3, 128.6, 128.4, 128.3, 71.2, 67.0, 59.1, 39.8, 39.0, 24.6, 24.4, 23.0, 21.6, 15.5, 11.6. νmax 3384, 3035, 2962, 2877, 1745, 1460, 1380, 1273, 1171, 1074, 1012, 746, 699 cm−1. λmax (log ε) 205 (6.1) nm, maximum at end of range. −11.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 358.1982, −2.0 ppm (calculated for C19H29NNaO4+, 358.1989).
Benzyl (S)-2-((l-valyl)oxy)-4-methylpentanoate (23b).
Collected as 0.94 g of a faint-yellow oil, 65% yield. 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5H), 5.16 (s, 2H), 5.12 (dd, J = 9.7, 4.0, 1H), 3.35 (d, J = 4.7, 1H), 2.11 (m, 1H), 1.81 (m, 1H), 1.73 (m, 1H), 1.66 (m, 1H), 1.41 (dd, J = 6.7, 3.0, 1H), 0.99 (d, J = 6.9, 3H), 0.94 (d, J = 6.6, 3H), 0.91 (d, J = 6.7, 3H), 0.90 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 175.2, 170.4, 128.6, 128.4, 128.3, 71.2, 67.0, 59.8, 39.7, 31.9, 24.6, 23.0, 21.5, 19.4, 16.7. νmax 3388, 2962, 2878, 1745, 1462, 1381, 1275, 1169, 1073, 1010, 747, 699 cm−1. λmax (log ε) 205 (6.7) nm, maximum at end of range. −15.6 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 322.2014, 0.3 ppm (calculated for C18H28NO4+, 322.2013).
Benzyl (S)-2-((l-phenylalanyl)oxy)-4-methylpentanoate (23c).
Collected as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5H), 7.30 (m, 2H), 7.23 (m 3H), 5.17 (m, 3H), 3.79 (dd, J = 8.6, 4.7, 1H), 3.20 (dd, J = 13.8, 4.7, 1H), 2.76 (dd, J = 13.8, 1H), 1.82 (m, 1H), 1.69 (m, 2H), 0.93 (d, J = 6.4, 3H), 0.91 (d, J = 6.3, 3H). 13C NMR (125 MHz, CDCl3) δ 174.8, 170.5, 137.4, 135.3, 129.5, 128.8, 128.7, 128.6, 128.5, 126.9, 71.5, 67.3, 55.7, 40.9, 39.8, 24.7, 23.2, 21.7. νmax 3032, 2958, 1746, 1457, 1380, 1273, 1178, 1075, 1009, 747, 699 cm−1. λmax (log ε) 205 (7.1) nm, maximum at end of range. −22.2 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 370.2014, 0.3 ppm (calculated for C22H28NO4+, 370.2013).
Tail Dimer Methylation (24a-c).
As a general procedure, the free amine was dissolved in DMF and to this solution DIPEA and then iodomethane were sequentially added dropwise. After 6-6.25 hours the reaction mixture was diluted into hexanes and washed with 5% citric acid (aq); the aqueous layer was back extracted three times with hexanes and the combined organics were washed with brine and dried over MgSO4. The suspension was filtered, and the filtrate of this step was concentrated to dryness by rotary evaporation, with caution as the product was observed to be mildly volatile. The crude residue was then purified by silica gel flash chromatography with a gradient elution of diethyl ether in hexanes.
Benzyl (S)-2-((dimethyl-l-isoleucyl)oxy)-4-methylpentanoate (24a).
Collected as 0.88 g of an amber oil, 71%. 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5H), 5.17 (s, 2H), 5.09 (dd, J = 10.3, 3.8, 1H), 2.93 (d, J = 10.5, 1H), 2.30 (s, 6H), 1.83 (m, 2H), 1.77 (m, 1H), 1.67 (m, 1H), 1.61 (m, 1H), 1.14 (m, 1H), 0.95 (d, J = 6.6, 3H), 0.91 (d, J = 6.5, 3H), 0.88 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 171.4, 170.8, 135.4, 128.7, 128.5(4), 128.5(1), 72.4, 70.8, 67.1, 41.5, 39.9, 33.4, 25.1, 24.7, 23.3, 21.3, 15.7, 10.5. νmax 2959, 2875, 2788, 1739, 1461, 1374, 1272, 1150, 1071, 746, 699 cm−1. λmax (log ε) 205 (6.7) nm, maximum at end of range. −44.1 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 350.2328, 0.6 ppm (calculated for C20H32NO4+, 350.2326).
Benzyl (S)-2-((dimethyl-l-valyl)oxy)-4-methylpentanoate (24b).
Collected as 0.73 g of a colorless oil, 63% yield. 1H NMR (500 MHz, CDCl3) δ 7.35 (m, 5H), 5.17 (s, 2H), 5.9, (dd, J = 10.3, 3.8, 1H), 2.82 (d, J = 10.6, 1H), 2.31 (s, 6H), 2.00 (m, 1H), 1.84, (m, 1H), 1.76 (m, 1H), 1.61 (ddd, J = 13.2, 9.2, 3.8, 1H), 0.98 (d, J = 6.6, 3H), 0.95 (d, J = 6.7, 3H), 0.91 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 171.1, 170.6, 135.3, 128.6, 128.3(9), 128.3(6), 74.0, 70.6, 67.0, 41.2, 39.8, 27.6, 24.5, 23.1, 21.1, 19.5, 19.3. νmax 2961, 2875, 2787, 1738, 1460, 1376, 1270, 1135, 1067, 745, 698 cm−1. λmax (log ε) 205 (6.9) nm, maximum at end of range. −37.9 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 364.2485, 0.8 ppm (calculated for C21H34NO4+, 364.2482).
Benzyl (S)-2-((dimethyl-l-phenylalanyl)oxy)-4-methylpentanoate (24c).
Collected as 0.25 g of a yellow oil, 25% yield. 1H NMR (500 MHz, CDCl3) δ 7.34 (m, 6H), 7.24 (m, 1H), 7.20 (m, 3H), 5.13 (m, 2H), 5.04 (dd, J = 9.9, 4.0, 1H), 3.58 (m, 1H), 3.06 (dd, J = 8.2, 14.1, 1H), 2.96 (m, 1H), 2.40 (s, 6H), 1.79 (m, 1H), 1.61 (m, 2H), 0.92 (d, J = 6.5, 3H), 0.88 (d, J = 6.5, 3H). 13C NMR (125 MHz, CDCl3) δ 171.2, 170.5, 138.3, 135.5, 129.1, 128.7, 128.5(3), 128.4(7), 128.4, 126.5, 71.2, 68.9, 67.1, 41.7, 39.9, 35.9, 24.6, 23.2, 21.4. νmax 3032, 2957, 2873, 2789, 1742, 1498, 1455, 1372, 1271, 1155, 1074, 1020, 745, 699 cm−1. λmax (log ε) 205 (7.5) nm, maximum at end of range. −13.6 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 398.2329, 0.8 ppm (calculated for C24H32NO4+, 398.2326).
Enamide Synthesis (25a-i).
As a general procedure, 19 was dissolved into a solution comprised of 5:1:1 THF/MeOH/H2O and lithium hydroxide was added in one portion. The reaction mixture was then vigorously stirred for 3.25 – 4.0 hours during which the color of the solution changed from colorless to a yellow-orange color. The reaction mixture was then diluted with DCM and acidified by washing with 5% citric acid (aq). The aqueous layer was twice back extracted with DCM, and then the combined organic layers were washed with brine and then dried over MgSO4. The crude extract was filtered, and the filtrate of this step was then concentrated to dryness using a rotary evaporator and then diluted into DCM. Next, DIPEA was added dropwise followed by addition of hydroxybenzotriazole (HOBt) as a single portion. The solution was cooled to 0°C in an ice bath before EDC was added in one portion. After 30 min at 0 °C the amino acid methyl ester, as a hydrochloride salt, was added in one portion. The reaction was then allowed to warm to room temperature overnight. The reaction mixture was then worked up by diluting into DCM and washed with 5% citric acid (aq), back extracted three times with DCM, and then the organics were washed with brine and dried over MgSO4. Again, the extract was filtered and the filtrate was dried on a rotary evaporator and purified by iterative silica gel flash chromatography with a 100:1 DCM/methanol elution.
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)pent-2-enoyl)-l-alaninate (25a).
Collected as 1.08 g of an off-white amorphous solid, 72% yield. 1H NMR (500 MHz, CDCl3) δ 6.75 (dd, J = 5.7, 15.2 Hz, 1H), 6.14 (d, J = 6.6, 1H), 5.91 (d, J = 15.3, 1H), 4.66 (p, J = 7.2, 1H), 4.53 (m, 1H), 4.37 (m, 1H), 3.77 (s, 3H), 1.45 (m, 11H), 1.26 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 173.5, 164.8, 154.9, 145.7, 122.2, 79.7, 52.5, 48.1, 47.1, 28.4, 20.6, 18.6. IR (neat) νmax 3347, 2978, 2937, 1722, 1628, 1516, 1454, 1330, 1292, 1247, 1175, 1051, 971 cm−1. λmax (log ε) 208 (6.9) nm. −25.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 323.1575, −0.6 ppm (calculated for C14H24N2NaO5+, 323.1577).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)pent-2-enoyl)-l-phenylalaninate (25b).
Collected as 1.48 g of an amorphous off-white solid, 78% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, overlaps with chloroform signal, 3H), 7.09 (d, J = 6.8, 2H), 6.74 (dd, J = 15.3, 5.4, 1H), 6.00 (d, J = 7.4, 1H), 5.86 (d, J = 15.3, 1H), 4.95 (dt, J = 7.6, 5.7, 1H), 4.52 (m, 1H), 4.38 (m, 1H), 3.73 (s, 3H), 3.16 (t, J = 6.2, 2H), 1.44 (s, 9H), 1.25 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 171.9, 164.9, 154.9, 146.1, 135.8, 129.3, 128.6, 127.2, 121.9, 79.7, 53.2, 52.4, 47.0, 37.9, 28.4, 20.6. IR (neat) νmax 3345, 2977, 2937, 1722, 1630, 1629, 1515, 1452, 1329, 1247, 1170 cm−1. λmax (log ε) 208 (7.1) nm. +30.8 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 399.1888, −0.5 ppm (calculated for C20H28N2NaO5+, 399.1890).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)pent-2-enoyl)-l-leucinate (25c).
Collected as 1.30 g of a light amber oil, 76% yield. 1H NMR (500 MHz, CDCl3) δ 6.75 (dd, J = 15.2, 5.7, 1H), 6.01 (d, J = 8.4, 1H), 5.92 (d, J = 15.2, 1H), 4.71 (td, J = 8.6, 4.4, 1H), 4.58 (m, 1H), 4.39 (m, 1H), 3.75 (s, 3H), 1.67 (m, 2H), 1.57 (m, 1H), 1.45 (s, 9H), 1.26 (d, J = 6.9, 3H), 0.95 (t, J = 6.5, 6H). 13C NMR (125 MHz, CDCl3) δ 173.6, 165.1, 154.9, 145.8, 122.1, 79.7, 52.9, 50.7, 47.0, 41.8, 28.4, 24.8, 22.8, 21.9, 20.5. IR (neat) νmax_3308, 3058, 2966, 2878, 1741, 1683, 1641, 1529, 1450, 1368, 1250, 1169, 1055, 981 cm−1. λmax (log ε) 205 (6.8) nm, maximum at the end of range. −25.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 365.2043, −1.1 ppm (calculated for C17H30N2NaO5+, 365.2047).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-l-alaninate (25d).
Collected as 1.09 g of an amorphous off-white solid, 60% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (t, J = 7.4, 2H), 7.23 (t, J = 7.4, 1H), 7.17 (d, J = 7.5, 2H), 6.80 (m, 1H), 6.05 (d, J = 7.4, 1H), 5.85 (d, J = 15.4, 1H), 4.65 (p, J = 7.4, 1H), 4.59 (bs, 1H), 4.52 (bs, 1H), 3.76 (s, 3H), 2.90 (m, 2H), 1.42 (d, J = 7.2, 3H), 1.40 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 173.4, 164.5, 155.0, 144.0, 136.5, 129.4, 128.5, 126.8, 123.1, 79.8, 52.6, 52.3, 48.1, 41.0, 28.3, 18.6. IR (neat) νmax 3334, 3024, 2983, 2936, 1739, 1676, 1635, 1528, 1449, 1371, 1318, 1267, 1214, 1167, 1050, 1024, 974, 699, 654 cm−1. λmax (log ε) 205 (6.6) nm, maximum at the end of range. −3.4 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 399.1889, −0.2 ppm (calculated for C20H28NNaO5+, 399.1890).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-l-phenyl-alaninate (25e).
Collected as 1.24 g of an amorphous off-white solid, 56% yield. 1H NMR (500 MHz, CDCl3) δ 7.27 (m, 4H), 7.23 (m, 2H), 7.16 (m, 2H), 7.07 (m, 2H), 6.80 (dd, J = 15.2, 5.3, 1H), 5.97 (m, 1H), 5.79 (dd, J = 15.3, 1.6, 1H), 4.93 (dt, J = 7.7, 5.7, 1H), 4.59 (bs, 1H), 4.56 (m, 1H), 3.72 (s, 3H), 3.14 (qd, J = 13.9, 5.7, 1H), 2.88 (m, 1H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.9, 164.6, 154.9, 144.2, 136.5, 135.8, 129.5, 129.3, 128.6, 128.5, 127.1, 126.8, 122.9, 79.8, 53.2, 52.4, 52.3, 41.0, 37.8, 28.3, IR (neat) νmax3338, 3028, 2977, 1741, 1681, 1638, 1529, 1446, 1365, 1326, 1257, 1212, 1170, 699 cm−1. λmax (log ε) 205 (7.1) nm, maximum at the end of range. +48.2 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 475.2207, 0.8 ppm (calculated for C26H32N2NaO5+, 475.2203).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-l-leucinate (25f).
Collected as 1.52 g of an amorphous off-white solid, 74% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 2H), 7.23 (m, 1H), 7.18 (m, 2H), 6.80 (m, 1H), 5.85 (m, 2H), 4.70 (td, J = 8.41, 4.6, 1H), 4.60 (m, 1H), 4,52 (m, 1H), 3.74 (s, 3H), 2.89 (m, 2H), 1.65 (m, 2H), 1.54 (m, 1H), 1.40 (s, 9H), 0.94 (t, J = 7.0, 6H). 13C NMR (125 MHz, CDCl3) δ 173.4, 164.7, 154.9, 144.1, 136.4, 129.5, 128.5, 126.8, 123.0, 79.8, 52.4, 52.3, 50.7, 41.8, 41.0, 28.3, 24.8, 22.8, 22.0. IR (neat) νmax 3343, 3025, 2961, 2874, 1744, 1679, 1638, 1529, 1446, 1368, 1325, 1258, 1206, 1169, 1021, 979, 698, 651 cm−1. λmax (log ε) 205 (6.7) nm, maximum at the end of range. −6.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 441.2367, 1.6 ppm (calculated for C23H34N2NaO5+, 441.2360).
Methyl ((S,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-d-phenyl-alaninate (25g).
Collected as 1.19 g of an amorphous light-yellow solid, 54% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 4H), 7.25 (m, 2H), 7.17 (d, J = 7.2, 2H), 7.05 (m, 2H), 6.81 (dd, J = 15.2, 5.1, 1H), 5.92 (d, J = 7.7, 1H) 5.77 (dd, J = 15.3, 1.7, 1H), 4.95 (dt, J = 7.8, 5.5, 1H), 4.60 (m, 1H), 4.52 (m, 1H), 3.73 (s, 3H), 3.15 (m, 2H), 2.88 (d, J = 6.7, 2H), 1.39, (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.8, 164.4, 155.0, 144.2, 136.4, 135.6, 129.5, 129.2, 128.6, 128.5, 127.1, 126.8, 122.8, 79.8, 53.1, 52.4, 52.3, 41.1, 37.8, 28.3. IR (neat) νmax 3331, 3030, 2978, 1742, 1679, 1638, 1526, 1446, 1366, 1252, 1212, 1170, 1022, 742, 699 cm−1. λmax (log ε) 205 (7.2) nm, maximum at the end of range. −54.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 475.2209, 1.3 ppm (calculated for C26H32N2NaO5+, 475.2203).
Methyl ((R,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-l-phenyl-alaninate (25h).
Collected as 0.35 g of an off-white amorphous solid, 55% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 4H), 7.24 (m, 2H), 7.17 (d, J = 7.3, 2H), 7.05 (d, J = 6.6, 2H), 6.81 (dd, J = 15.2, 5.2, 1H), 5.91 (d, J = 7.8, 1H), 5.77 (d, J = 15.4, 1H), 4.95 (dt, J = 7.8, 5.5, 1H), 4.60 (m, 1H), 4.52 (m, 1H), 3.73 (s, 3H), 3.15 (t, J = 4.8, 2H), 2.88 (d, J = 6.7, 2H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.8, 164.4, 155.0, 144.2, 136.4, 135.6, 129.5, 129.3, 128.6, 128.5, 127.1, 126.8, 122.8, 79.8, 53.1, 52.4, 52.3, 41.1, 37.8, 28.3. IR (neat) νmax 3335, 3026, 2980, 2947, 1748, 1676, 1638, 1528, 1445, 1367, 1323, 1258, 1213, 1169, 699 cm−1. λmax (log ε) 205 (7.1) nm, maximum at the end of range. +59.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 475.2207, 0.8 ppm (calculated for C26H32N2NaO5+, 475.2203).
Methyl ((R,E)-4-((tert-butoxycarbonyl)amino)-5-phenylpent-2-enoyl)-d-phenyl-alaninate (25i).
Collected as 0.44 g of an off-white amorphous solid, 68% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 4H), 7.24 (m, 2H), 7.16 (d, J = 7.59, 2H), 7.06 (d, J = 7.51, 2H), 6.81 (dd, J = 15.1, 5.1, 1H), 5.90 (d, J = 7.2, 1H), 5.78 (d, J = 15.2, 1H), 4.94 (q, J = 5.9, 1H), 4.61 (m, 1H), 4.50 (m, 1H), 3.73 (s, 3H), 3.15 (m, 2H), 2.89 (m, 2H), 1.39 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 171.8, 164.5, 154.9, 144.2, 136.4, 135.7, 129.5, 129.2, 128.6, 128.5, 127.1, 126.8, 122.8, 79.8, 53.2, 52.4, 52.2, 41.0, 37.7, 28.3. IR (neat) νmax 3336, 3029, 2976, 1741, 1680, 1638, 1528, 1446, 1365, 1324, 1256, 1212, 1170, 698 cm−1. λmax (log ε) 206 (7.3) nm. −55.5 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 475.2209, 1.3 ppm (calculated for C26H32N2NaO5+, 475.2203).
Head group Cyclization and Mitsunobu Reaction (26a-j).
As a general procedure, the starting ester was dissolved into the 5:1:1 THF/MeOH/H2O solution and then lithium hydroxide was added in one portion, after which the reaction mixture was stirred vigorously for 1-2.5 h. The reaction mixture was then diluted into DCM and acidified by washing with 5% citric acid (aq); the aqueous layer was back extracted iteratively with DCM, and then the combined organic layers were washed with brine and dried over MgSO4. The extract was filtered and concentrated to dryness in vacuo and diluted into DCM. Pyridine was then added in one portion to this sample, the reaction mixture was cooled to 0 °C on an ice bath, and EDC was added. After 30 min at 0 °C Meldrum’s acid was added in one portion, and the reaction mixture was allowed to warm to room temperature overnight. The reaction mixture was then worked up by diluting into DCM, washing twice with 5% citric acid (aq), and once with brine before being dried over MgSO4. After filtration, the sample was concentrated to dryness and dissolved in CH3CN; this solution was brought to reflux in an oil bath for 1 h. The reaction mixture was then concentrated to dryness in vacuo and dissolved in DCM. To this mixture was added PPh3 in one portion followed by the alcohol via dropwise addition. The reaction mixture was then cooled to 0 °C on an ice bath and diisopropyl azodicarboxylate (DIAD) was added dropwise, after which the reaction mixture was allowed to warm to room temperature overnight. For workup, the reaction mixture was concentrated to dryness in vacuo and then purified by silica gel flash chromatography with a gradient elution of EtOAc in hexanes. The material collected from this column was further subjugated to reverse-phase solid phase extraction (RP-SPE) with a 10 g C-18 column using 20%/40%/(60% or 80%) CH3CN/H2O and then 100% CH3CN. The fraction eluting with 60% or 89% consistently contained the desired analog product. This was collected and dried by rotary evaporation.
Tert-butyl ((S,E)-5-((S)-3-methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)carbamate (26a).
Collected 0.34 g of a colorless oil, 32% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (m, 1H), 7.03 (m, 1H), 5.04 (s, 1H), 4.62 (m, 1H), 4.48 (bs, 1H), 3.87 (s, 3H), 1.49 (dd, J = 6.5, 2.6, 3H), 1.46 (s, 9H), 1.30 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 180.7*, 169.8, 164.4*, 155.0*, 149.5*, 121.6*, 93.0, 79.6, 58.7, 55.7, 47.3*, 28.4, 20.4, 17.1*. *Showed twinning. νmax 3345, 3107, 2978, 2937, 1719, 1628, 1517, 1454, 1332, 1290, 1246, 1175, 1053, 976, 812, 701 cm−1. λmax (log ε) 243 (6.8) nm. −20.5 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 347.1575, −0.6 ppm (calculated for C16H24N2NaO5+, 347.1577).
Tert-butyl ((S,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)carbamate (26b).
Collected as 1.52 g of a light-yellow foam, 37% yield. 1H NMR (500 MHz, CDCl2) δ 7.34 (m, 1H), 7.20 (m, 3H), 7.08 (m, 1H), 6.94 (m, 2H), 4.91 (m, 1H), 4.82 (s, 1H), 4.61 (s, 1H), 4.49 (s, 1H), 3.82 (s, 3H), 3.56 (m, 1H), 3.12 (dd, J = 14.0, 3.0, 1H), 1.45 (s, 9H), 1.32 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 177.8, 169.8, 164.7*, 155.0*, 149.8*, 134.3*, 129.6, 128.2, 127.0*, 121.5, 95.0*, 79.0, 59.7*, 58.4*, 47.3, 34.6*, 28.4*, 20.5*. *Showed twinning. νmax 3343, 2977, 2935, 1719, 1629, 1512, 1452, 1337, 1246, 1171 cm−1. λmax (log ε) 205 (6.9) nm, maximum at end of range. −7.3 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 423.1895, 1.2 ppm (calculated for C22H28N2NaO5+, 423.1890).
Tert-butyl ((S,E)-5-((S)-2-isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)carbamate (26c).
Collected as 0.10 g of a yellow oil, 11% yield. 1H NMR (500 MHz, CDCl3) δ 7.39 (ddd, J = 15.5, 10.7, 1.7, 1H), 7.00 (m, 1H), 5.05 (s, 1H), 4.69 (dt, J = 6.4, 3.0, 1H), 4.65 (m, 1H), 4.47 (s, 1H), 3.86 (s, 3H), 1.83 (m, 2H), 1.75 (m, 1H), 1.45 (d, J = 1.2, 9H), 1.30 (d, J = 6.9, 3H), 0.92 (d, J = 6.4, 3H), 0.88 (d, J = 6.4, 3H). 13C NMR (125 MHz, CDCl3) δ 180.5*, 170.1, 164.4*, 154.9*, 149.3*, 121.7*, 93.5*, 79.4*, 58.5*, 58.4, 47.2, 39.0, 28.3, 24.1*, 23.6*, 22.6*, 20.3*. *Showed twinning. νmax 3349, 2969, 2877, 1719, 1627, 1515, 1456, 1331, 1246, 1174, 1054, 983 cm−1. λmax (log ε) 234 (6.6) nm. −12.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 389.2046, −0.3 ppm (calculated for C19H20N2NaO5+, 389.2047).
Tert-butyl ((S,E)-5-((S)-3-methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26d).
Collected 0.50 g as a light-yellow foam, 43% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (ddd, J = 15.5, 9.1, 1.6, 1H), 7.29 (m, 2H), 7.20 (m, 3H), 7.06 (m, 1H), 5.03 (s, 1H), 4.68 (m, 1H), 4.61 (p, J = 6.3 1H), 4.55 (m, 1H), 3.86 (s, 3H), 2.93 (m, 2H), 1.48 (t, J = 6.4, 3H), 1.40 (s, 9H). 13C NMR (125 MHz, CDCl3) δ 180.7, 169.7, 164.1*, 155.0, 147.8*, 136.7*, 129.4*, 128.5, 126.7, 122.5*, 93.0, 79.7, 58.7, 55.7, 52.6, 41.0, 28.3, 17.1*. *Showed twinning. νmax 3346, 2978, 2935, 1717, 1627, 1509, 1453, 1334, 1290, 1248, 1172, 1098, 1040, 973, 701 cm−1. λmax (log ε) 210 (7.8) nm. −2.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 423.1890, 0.0 ppm (calculated for C22H28N2NaO5+, 423.1890).
Tert-butyl ((S,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26e).
Collected as 0.27 g of a yellow foam, 31% yield. 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 3H), 7.20 (m, 5H), 6.92 (m, 2H), 4.90 (m, 1H), 4.80 (s, 1H), 4.61 (s, 1H), 3.82 (s, 3H), 3.52 (m, 1H), 3.12 (m, 1H), 2.91 (m, 2H), 1.40 (d, J = 2.1, 9H). 13C NMR (125 MHz, CDCl3) δ 177.7*, 169.6, 164.3, 154.8*, 148.0*, 136.6*, 134.1*, 129.5, 129.4, 128.4, 128.1, 126.9, 126.6*, 94.9*, 79.6*, 59.6(1), 59.5(9), 58.3, 53.1*, 52.3, 41.0, 40.8, 37.7*, 34.4, 28.2. νmax 3343, 3028, 2977, 2936, 1717, 1630, 1505, 1449, 1358, 1248, 1170, 1021, 970, 738, 701 cm−1. λmax (log ε) 205 (7.4) nm, maximum at end of range. 1.0 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 499.2205, 0.4 ppm (calculated for C28H32N2NaO5+, 499.2203).
Tert-butyl ((S,E)-5-((S)-2-isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26f).
Collected as 0.35 g of a yellow oil, 32% yield. 1H NMR (500 MHz, CDCl3) δ 7.38 (m, 1H), 7.29 (t, J = 7.4, 2H), 7.22 (m, 1H), 7.19 (m, 2H), 7.05 (m, 1H), 5.04 (s, 1H), 4.69 (dt, J = 6.9, 3.5, 1H), 4.62 (m, 1H), 4.57 (m, 1H), 3.85 (d, J = 1.2, 3H), 2.94 (m, 2H) 1.83 (m, 2H), 1.73 (m, 1H), 1.40 (s, 9H), 0.92 (dd, J = 6.5, 2.6, 3H), 0.87 (dd, J = 6.5, 3.2, 3H). 13C NMR (125 MHz, CDCl3) δ 180.6*, 170.2*, 164.2, 155.0*, 147.7, 136.7*, 129.5*, 128.5*, 126.7, 122.5*, 93.6*, 79.7, 58.6*, 58.5(4), 58.5(0), 52.6*, 41.0*, 38.9*, 28.3, 24.2*, 23.7*, 22.7*. *Showed twinning. νmax_3346, 2960, 2872, 1720, 1626, 1505, 1454, 1334, 1250, 1173, 981 cm−1. λmax (log ε) 210 (6.6) nm. 6.2 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 465.2364, 0.9 ppm (calculated for C25H34N2NaO5+, 465.2360).
Tert-butyl ((S,E)-5-((R)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26g).
Collected as 0.31 g of a light-yellow foam, 32% yield. 1H NMR (500 MHz, CDCl3) δ 7.31 (m, 3H), 7.21 (m, 5H), 6.92 (m, 2H), 4.90 (td, J = 4.7, 3.1, 1H), 4.80 (s, 1H), 4.71 (s, 1H), 4.62 (s, 1H), 3.82 (s, 3H), 3.54 (m, 1H), 3.11 (m, 1H), 2.94 (m, 2H), 1.40 (d, J = 2.2, 9H). 13C NMR (125 MHz, CDCl3) δ 177.8*, 169.7, 164.4*, 155.0*, 148.1*, 136.7*, 134.2*, 129.6, 129.5, 128.5*, 128.2, 126.7*, 122.3*, 95.0*, 79.7, 59.7*, 58.4, 52.6*, 41.0*, 34.5, 28.3. *Showed twinning. νmax 3338, 3026, 2977, 2934, 1719, 1629, 1502, 1451, 1347, 1248, 1171, 1121, 1021, 969, 738, 701 cm−1. λmax (log ε) 205 (7.2) nm, maximum at end of range. 2.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 499.2205, 0.4 ppm (calculated for C28H32N2NaO5+, 499.2203).
Tert-butyl ((R,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26h).
Collected as 41 mg of an amber oil, 26% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 5H), 7.19 (m, 5H), 6.92 (m, 2H), 4.94 (m, 1H), 4.89 (m, 1H), 4.80 (s, 1H), 4.62 (m, 1H) 3.82 (s, 3H), 3.55 (m, 1H), 3.12 (m, 1H), 2.93 (m, 2H), 1.40 (d, J = 2.2, 9H). 13C NMR (125 MHz, CDCl3) δ 175.9, 167.8, 162.5, 153.0, 146.2*, 134.8*, 132.3*, 127.7*, 127.6, 126.6, 126.3, 125.1, 124.9*, 124.9*, 93.1*, 77.8, 57.8*, 56.5, 51.3*, 39.3*, 32.6, 26.4. *Showed twinning. νmax 3336, 3028, 2977, 2936, 1718, 1630, 1505, 1450, 1356, 1248, 1171, 1117, 1021, 970, 737, 701 cm−1. λmax (log ε) 205 (6.7) nm, maximum at end of range. −6.5 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 477.2387, 0.6 ppm (calculated for C28H23N2O5+, 477.2384).
Tert-butyl ((R,E)-5-((R)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)carbamate (26i).
Collected as 53 mg of an amber oil, 27% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 5H), 7.20 (m, 5H), 6.92 (m, 2H), 4.94 (m, 1H), 4.90 (m, 1H), 4.80 (s, 1H), 4.61 (m, 1H), 3.82 (s, 3H), 3.55 (m, 1H), 3.12 (m, 1H), 2.91 (m, 2H), 1.40 (d, J = 2.2, 9H). 13C NMR (125 MHz, CDCl3) δ 177.8, 169.7, 164.4, 155.0, 1481*, 136.5*, 134.2*, 129.6*, 129.5, 128.5*, 128.2, 127.0, 126.7*, 122.6*, 95.0*, 79.8*, 59.7*, 58.4, 53.2*, 41.4*, 34.5, 28.3. *Showed twinning. νmax 3338, 3029, 2977, 2936, 1718, 1630, 1505, 1450, 1356, 1248, 1170, 1021, 970, 738, 701 cm−1. λmax (log ε) 205 (7.1) nm, maximum at end of range. −7.8 (CH2Cl2). HR-ESI-TOF-MS [M + H]+ m/z 477.2384, 0 ppm (calculated for C28H33N2O5+, 477.2384).
Tert-butyl ((S,E)-5-((S)-3-(hex-5-yn-1-yloxy)-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)carbamate (26j).
Collected as 0.55 g of a light-yellow oil, 37% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (ddd, J = 15.5, 7.1, 1.7, 1H), 7.03 (m, 1H), 5.01 (s, 1H), 4.62 (m, 2H), 4.47 (bs, 1H), 4.05 (m, 1H), 4.00 (m, 1H), 2.28 (td, J = 6.9, 2.7, 2H), 1.99 (t, J = 2.7, 1H), 1.93 (m, 2H), 1.67 (m, 2H), 1.50 (dd, J = 6.6, 2.2, 3H), 1.46 (s, 9H), 1.30 (d, J = 6.9, 3H). 13C NMR (125 MHz, CDCl3) δ 179.7*, 169.9, 164.3*, 155.0*, 149.5*, 121.6*, 93.1, 83.4, 79.6, 71.4, 69.1, 55.8, 47.4*, 28.4, 27.4, 24.6, 20.4, 18.0, 17.2*. *Showed twinning. νmax 3304, 3107, 2976, 2939, 1717, 1624, 1515, 1455, 1330, 1289, 1242, 1174, 1047, 976, 935, 811, 704, 638 cm−1. λmax (log ε) 244 (7.1) nm. −19.8 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 413.2051, 1.0 ppm (calculated for C21H30N2NaO5+, 413.2047).
Leucine Extension (27a-j).
As a general procedure, the carbamate starting material was dissolved in DCM and cooled to 0 °C before TFA was added dropwise. After 1 hour the reaction mixture was concentrated to dryness in vacuo and then further dried by three times forming an azeotrope with three drops of toluene and rotary evaporation. In a separate flask, the Boc-l-Leu-OH was dissolved in DCM; to this mixture was added DIPEA, dropwise, and then HOBt, in one portion. The leucine reaction mixture was cooled to 0 °C and EDC was added in one portion. After 30 min at 0 °C, the deprotected amine was added as a solution in DCM with iterative washes. The reaction mixture was allowed to warm to room temperature overnight, and then worked up by dilution into DCM, washing twice with 5% citric acid (aq), and once with brine before drying over MgSO4. After filtration the product was purified by silica gel flash chromatography with elution using EtOAc/hexanes. The material collected from this column was further subjected to RP-SPE with a 10 g C-18 column using elutions of 25%/40%/75 or 80% CH3CN/H2O and then 100% CH3CN. The desired compound eluted in the 75 or 80% CH3CN/H2O fraction and was collected and dried in vacuo.
Tert-butyl ((S)-1-(((S,E)-5-((S)-3-methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27a).
Collected as 0.47 g of an off-white foam, 72% yield. 1H NMR (500 MHz, CDCl3) δ 7.41 (ddd, J = 15.6, 9.5, 1.7, 1H), 6.99 (m, 1H), 6.17 (m, 1H), 5.03 (s, 1H), 4.89 (m, 1H), 4.77 (m, 1H), 4.62 (qd, J = 6.6, 3.1, 1H), 4.09 (m, 1H), 3.87 (s, 3H), 1.69 (m, 2H), 1.49 (m, 4H), 1.44 (s, 9H), 1.32 (d, J = 7.0, 3H), 0.95 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.7, 171.8*, 169.7*, 164.2*, 155.8, 148.3*, 122.4*, 93.0, 80.0, 58.7, 55.7*, 53.0, 46.0*, 41.1*, 28.3, 24.7*, 23.0, 22.1*, 20.0*, 17.1*. *Showed twinning. νmax 3313, 2969, 2876, 1720, 1667, 1629, 1529, 1456, 1359, 1331, 1288, 1245, 1173, 1039, 976 cm−1. λmax (log ε) 242 (7.0) nm. −35.4 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 460.2425, 1.5 ppm (calculated for C22H35N3NaO6+, 460.2418).
Tert-butyl ((S)-1-(((S,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27b).
Collected as 0.40 g of an off-white foam, 73% yield. 1H NMR (500 MHz, CDCl3) δ 7.34 (t, J = 14.5, 1H), 7.19 (m, 3H), 7.07 (dd, J = 15.6, 4.9, 1H), 6.94 (m, 2H), 6.42 (m, 1H), 5.01, (q, J = 9.3, 1H), 4.90 (m, 1H), 4.81 (m, 2H), 4.12 (m, 1H), 3.83 (s, 3H), 3.55 (m, 1H), 3.45 (m, 1H), 3.12 (m, 1H), 1.67 (m, 2H), 1.50 (m, 1H), 1.43 (s, 9H), 1.33 (d, J = 6.5, 1H), 0.93 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 177.8*, 172.0, 169.7, 164.6*, 155.9*, 148.9*, 134.2*, 129.5, 128.2*, 127.0*, 122.0*, 95.0*, 80.0, 59.7*, 58.4*, 53.0, 45.9, 41.2, 34.5*, 31.6, 28.3, 24.7, 22.9, 22.0, 19.9*. *Showed twinning. νmax 3308, 3063, 3032, 2968, 2876, 1722, 1668, 1632, 1530, 1453, 1357, 1247, 1172, 1119, 1026, 970, 807, 735, 703, 642 cm−1. λmax (log ε) 205 (6.7) nm, maximum at end of range. −20.0 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 536.2738, 1.3 ppm (calculated for C28H39N3NaO6+, 536.2731).
Tert-butyl ((S)-1-(((S,E)-5-((S)-2-isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27c).
Collected as 0.39 go of yellow semi-solids, 42% yield. 1H NMR (500 MHz, CDCl3) δ 7.40 (ddd, J = 15.6, 11.7, 1.8, 1H), 6.98 (ddd, J = 15.6, 7.0, 4.9, 1H), 6.21 (m, 1H), 5.05 (s, 1H), 4.92 (m, 1H), 4.79 (m, 1H), 4.69 (dt, J = 6.9, 3.5, 1H), 4.10 (m, 1H), 3.86 (s, 3H), 1.83 (m, 2H), 1.68 (m, 2H), 1.50 (m, 1H), 1.44 (s, 9H), 1.32 (d, J = 7.0, 3H), 1.27 (m, 1H), 0.95 (m, 6H), 0.92 (d, J = 6.4, 3H), 0.88 (dd, J = 6.5, 1.7, 3H). 13C NMR (125 MHz, CDCl3) δ 180.6*, 171.8*, 170.1*, 164.3*, 155.8, 148.4*, 122.3*, 93.6*, 80.0, 58.6, 58.4*, 52.9, 45.9*, 41.2*, 39.0*, 28.3, 24.7, 24.2*, 23.7*, 23.0, 22.7, 22.0, 20.0*. *Showed twinning. νmax 3309, 3071, 2961, 2875, 1722, 1668, 1628, 1532, 1456, 1359, 1330, 1245, 1173, 1045, 983 cm−1. λmax (log ε) 233 (6.7) nm. −23.3 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 502.2895, 1.4 ppm (calculated for C25H41N3NaO6+, 502.2888).
Tert-butyl ((S)-1-(((S,E)-5-((S)-3-methoxy-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27d).
Collected as 0.33 g of a light-yellow foam, 52% yield. 1H NMR (500 MHz, CDCl3) δ 7.40 (t, J = 16.3, 1H), 7.29 (m, 2H), 7.21 (m, 3H), 7.02 (m, 1H), 6.32 (m,1H), 5.03 (s, 1H), 4.98 (m, 1H), 4.85 (m, 1H), 4.60 (p, J = 6.5, 1H), 4.09 (m, 1H), 3.86 (s, 3H), 2.95 (m, 2H), 1.63 (m, 2H), 1.47 (m, 4H), 1.44 (s, 9H), 0.91 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 180.6, 172.0*, 169.6*, 163.9*, 155.7, 146.6*, 136.5*, 129.4, 128.5, 126.8, 123.0*, 93.0*, 79.9, 58.7, 55.7*, 53.0, 51.1, 41.1*, 40.5, 28.3, 24.7, 22.9, 22.0, 17.1*. *Showed twinning. νmax 3306, 2960, 2874, 1719, 1667, 1628, 1524, 1454, 1333, 1288, 1246, 1174, 701 cm−1. λmax (log ε) 205 (6.7) nm, maximum at end of range. −20.1 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 536.2735, 0.7 ppm (calculated for C28H39N3NaO6+, 536.2735).
Tert-butyl ((S)-1-(((S,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27e).
Collected as 0.33 g of a light-yellow oil, 78% yield, 87% pure with DCM (68% adjusted yield). 1H NMR (500 MHz, CDCl3) δ 7.32 (m, 3H), 7.20 (m, 6H), 7.09 (m, 1H), 6.92 (m, 2H), 6.27 (m, 1H), 5.02 (m, 1H), 4.89 (m, 1H), 4.80 (m, 2H), 4.08 (m, 1H), 3.82 (d, J = 1.9, 3H), 3.54 (m, 1H), 3.11 (m, 1H), 2.96 (m, 2H), 1.62 (m, 2H), 1.43 (m, 10H), 0.90 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 177.7*, 171.9*, 169.5*, 164.2*, 155.6, 146.8*, 136.4*, 134.1*, 129.5*, 129.4*, 128.5*, 128.1*, 126.9*, 126.8*, 122.8*, 94.9*, 79.9, 59.5*, 58.3*, 53.0, 51.0*, 41.0*, 40.4*, 34.3*, 28.2, 24.6, 22.8, 22.0. *Showed twinning. νmax 3308, 3062, 3030, 2957, 2871, 1719, 1667, 1630, 1523, 1451, 1354, 1247, 1170, 1118, 1025, 969, 736, 700 cm−1. λmax (log ε) 205 (7.2) nm, maximum at end of range. −10.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 612.3047, 0.5 ppm (calculated for C34H43N3NaO6+, 612.3044).
Tert-butyl ((S)-1-(((S,E)-5-((S)-2-isobutyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27f).
Collected as 0.37 g of a yellow oil, 43% yield. 1H NMR (500 MHz, CDCl3) ) δ 7.38 (m, 1H), 7.28 (m, 2H), 7.23 (m, 1H), 7.20 (m, 2H), 7.00 (ddd, J = 15.6, 12.1, 5.2, 1H), 6.18 (m, 1H), 5.03 (s, 1H), 4.98 (m, 1H), 4.79 (m, 1H), 4.67 (dt, J = 6.8, 3.5, 1H), 3.85 (s, 3H), 2.95 (m, 2H), 1.82 (m, 3H), 1.72 (m, 1H), 1.64 (m, 2H), 1.44 (d, J = 1.3, 9H), 0.91 (m, 9H), 0.87 (t, J = 6.8, 3H). 13C NMR (125 MHz, CDCl3) δ 180.5*, 171.9*, 170.0*, 164.0*, 155.6, 146.4*, 136.5*, 129.4*, 128.5*, 126.8, 123.2*, 93.6*, 80.0*, 58.6, 58.5*, 53.1, 51.1*, 40.6*, 39.0(0), 38.9(5), 28.3, 24.7*, 24.2*, 23.7(1), 23.6(7), 22.9, 22.7. *Showed twinning. νmax 3305, 3064, 2959, 2874, 1719, 1668, 1627, 1526, 1454, 1333, 1247, 1173, 1043, 980, 812, 742, 699 cm−1. λmax (log ε) 205 (7.2) nm, maximum at end of range. −19.9 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 578.3203, 0.3 ppm (calculated for C31H45N3NaO6+, 578.3201).
Tert-butyl ((S)-1-(((S,E)-5-((R)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27g).
Collected as 0.21 g of a light-brown foam, 58% yield. 1H NMR (500 MHz, CDCl3) δ 7.30 (m, 2H), 7.20 (m, 7H), 7.08 (ddd, J = 15.6, 7.8, 5.2, 1H), 6.91 (m, 2H), 6.15 (m, 1H), 5.01 (m, 1H), 4.89 (dt, J = 5.6, 3.0, 1H), 4.80 (m, 2H), 4.06 (m, 1H), 3.82 (d, J = 1.7, 3H), 3.54 (td, J = 13.8, 5.2, 1H), 3.10 (dt, J = 13.9, 3.5, 1H), 2.97 (m, 2H), 1.62 (m, 2H), 1.43 (d, J = 2.9, 9H), 1.41 (m, 1H), 0.90 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 177.8*, 171.9, 169.5, 164.3*, 155.7, 146.9 *, 146.9*, 136.5*, 134.2*, 129.6*, 129.5*, 128.6*, 128.2*, 127.0*, 126.8*, 123.0*, 95.0*, 80.0, 59.7*, 58.4, 53.1, 51.2*, 41.1, 40.6*, 34.5*, 28.3, 24.7, 22.9, 22.0. *Showed twinning. νmax 3307, 2955, 1720, 1667, 1629, 1523, 1451, 1343, 1247, 1171, 968, 701 cm−1. λmax (log ε) 205 (7.1) nm, maximum at end of range. −9.6 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 612.3046, 0.3 ppm (calculated for C34H43N3NaO6+, 612.3044).
Tert-butyl ((S)-1-(((R,E)-5-((S)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27h).
Collected as 0.16 g of an amber oil, 74% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 3H), 7.19 (m, 6H), 7.09 (m, 1H), 6.93 (m, 2H), 6.27 (m, 1H), 5.01 (m, 1H), 4.88 (m, 1H), 4.79 (s, 1H), 3.82 (s, 3H), 3.54 (m, 1H), 3.10 (m, 2H), 2.95 (m, 1H), 1.61 (m, 1H), 1.52 (m, 1H), 1.42 (m, 10H), 0.86 (t, J = 5.6, 6H). 13C NMR (125 MHz, CDCl3) δ 177.9*, 172.0, 169.6, 164.5*, 156.1, 147.2*, 136.5*, 134.2*, 129.6, 129.4*, 128.5, 128.2*, 127.0*, 126.9*, 122.8*, 94.9*, 80.2, 59.7*, 58.4, 53.0, 51.0*, 41.6, 40.4*, 34.6, 28.3, 24.7, 22.9, 21.8. *Showed twinning. νmax 3305, 3063, 3031, 2958, 1719, 1666, 1633, 1530, 1450, 1360, 1247, 1170, 1119, 1026, 970, 738, 700 cm−1. λmax (log ε) 205 (7.2) nm, maximum at end of range. −6.7 (CH2Cl2). HR-ESI-TOF-MS [M + H] m/z 590.3224, −0.2 ppm (calculated for C34H44N3O6+, 590.3225).
Tert-butyl ((S)-1-(((R,E)-5-((R)-2-benzyl-3-methoxy-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxo-1-phenylpent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27i).
Collected as 0.14 g of an amber oil, 63% yield. 1H NMR (500 MHz, CDCl3) δ 7.29 (m, 3H), 7.19 (m, 6H), 7.09 (m, 1H), 6.93 (m, 2H), 6.28 (m, 1H), 5.01 (m, 1H), 4.88 (dt, J = 5.5, 2.9, 1H), 4.79 (s, 1H), 3.82 (s, 3H), 3.54 (ddd, J = 15.7, 14.0, 5.3, 1H), 3.10 (m, 2H), 2.95 (m, 1H), 1.61 (m, 1H), 1.53 (m, 1H), 1.42 (m, 10H), 0.86 (t, J = 5.6, 6H). 13C NMR (125 MHz, CDCl3) δ 177.9*, 172.0, 169.6, 164.2, 155.9, 147.2*, 136.4*, 134.2*, 129.6, 129.4*, 128.6*, 128.2*, 127.0*, 126.9*, 122.8*, 94.9*, 80.1, 59.7*, 58.4, 53.0, 51.0*, 41.1, 40.3*, 34.6, 28.3*, 24.7, 22.9, 21.8. *Showed twinning. νmax 3311, 3062, 3031, 2958, 2872, 1719, 1668, 1631, 1509, 1451, 1357, 1248, 1170, 1118, 1025, 970, 807, 736, 701 cm−1. λmax (log ε) 205 (7.0) nm, maximum at end of range. −7.2 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 612.3045, 0.3 ppm (calculated for C34H43N3NaO6+, 612.3044).
Tert-butyl ((S)-1-(((S,E)-5-((S)-3-(hex-5-yn-1-yloxy)-2-methyl-5-oxo-2,5-dihydro-1H-pyrrol-1-yl)-5-oxopent-3-en-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (27j).
Collected as 0.34 g of a colorless foam, 69% yield. 1H NMR (500 MHz, CDCl3) δ 7.42 (ddd, J = 15.6, 9.7, 1.9, 1H), 6.99 (m, 1H), 6.24 (bs, 1H), 5.01 (s, 1H), 4.92 (m, 1H), 4.77 (m, 1H), 4.62 (m, 1H), 4.11 (m, 1H), 4.03 (m, 2H), 2.28 (td, J = 6.9, 2.6, 2H), 1.99 (t, J = 2.5, 1H), 1.94 (m, 2H), 1.68 (m, 4H), 1.49 (m, 4H), 1.44 (s, 9H), 1.32 (d, J = 7.0, 3H), 0.95 (m, 6H). 13C NMR (125 MHz, CDCl3) δ 179.7, 171.8*, 169.8*, 164.2*, 155.8, 148.4*, 122.3*, 93.1, 83.4, 80.0, 71.4, 69.1, 55.8*, 52.9, 46.0*, 41.2*, 28.3, 27.4, 24.7*, 24.6, 23.0, 22.0, 20.0*, 18.0 17.2*. *Showed twinning. νmax 3303, 3096, 2961, 2877, 1720, 1668, 1625, 1532, 1457, 1356, 1329, 1288, 1241, 1174, 1038, 978, 940, 862, 810, 703, 637, 506 cm−1. λmax (log ε) 245 (7.0) nm. −30.2 (CH2Cl2). HR-ESI-TOF-MS [M + Na]+ m/z 526.2894, 1.1 ppm (calculated for C27H41N3NaO6+, 526.2888).
Receptor and Ligand Preparation.
A cathepsin L structure (PDB ID: 2XU3) was downloaded from the Protein Data Bank.34 All ligands and H2O molecules were removed, and hydrogens were subsequently added to the protein structure using the Protonate 3D module of MOE at pH 5.5. The protein was then processed using the Structure Preparation module and energy minimized using the Amber12:EHT forcefield equations.25 Gallinamide A was built using Cambridge ChemDraw 2D builder and processed using Protonate 3D at pH 5.5 to mimic assay conditions.
Cathepsin Kinetic Assay.
Kinetic assays were run in 96-well plate format, with final volumes of 50 μL and DMSO was kept below 6%. Inhibitors were prepared in a 3-fold, 11-point serial dilution, with final concentrations ranging from 0.0017 nM to 1.0 μM. Final enzyme concentrations were 0.5 ng/mL for cathepsin L and 2.5 ng/mL for cathepsin V. Z-FR-AMC was used as a substrate, at a concentration of 10 μM (CatL) or 15 μM (CatV). Substrate and inhibitor was added to the wells, and enzyme was rapidly dispensed to initiate the reaction. Product formation was monitored by Spectramax M2 spectrophotometer for 2 h.
Data Fitting.
The concentrations of each reaction species ([E], [S], [P], [I], [ES], [EI], [EI*]) over time were calculated using a set of ordinary differential equations based on each reaction step in the model shown in Figure 4.3. [P] was determined based on AMC standard curve fluorescence, and progress curves were fit to a six-parameter (kaS, kdS, kcat, kaI , kdI , kinact) kinetic inhibition model using DynaFit software.35
Reversibility.
A preincubation-dilution experiment was adapted from Copeland et al.28 Cathepsin L at 100-fold its final assay concentration was incubated with gallinamide A or the hydrogenated analog at 10-fold its respective IC50 value for 30 min in a volume of 2 μL in a 96-well plate. This mixture was diluted 100-fold with assay buffer containing 10 μM Z-FR-AMC substrate to a final volume of 200 μL, resulting in a standard concentration of enzyme and 0.1-times the IC50 value of inhibitor. A rapidly reversible inhibitor will dissociate from the enzyme to restore approximately 90% of enzymatic activity following the dilution event, while an irreversible inhibitor will maintain approximately 10% of enzymatic activity. Fluorescence intensities of the 200-μL wells were monitored continuously for AMC hydrolysis on a Specramax M2 plate reader in kinetic mode for 2 h.
Recombinant cruzain expression and enzymatic inhibition assay.
Recombinant cruzain (GenBank M84342.1) was sub-cloned into piczαA using gblock technology and transformed into Pichia pastoris. The transformed yeast culture was grown at 28 °C in YPD (1% yeast extract, 2% peptone, 2% glucose) until reaching log phase of growth followed by centrifugation at 5,000 rpm. The cell pellet was resuspended in BM (100 mM potassium phosphate pH 6.0, 1.34% yeast nitrogen base, 4X10-5% biotin) and recombinant cruzain was expressed at 28 °C for 24 h with 1% MeOH as a soluble protein. The cleared supernatant was filtered (0.45 μm), concentrated and exchanged into 50 mM Tris pH 8.0. The recombinant cruzain was purified using a monoQ column in the AKTA FPLC (GE). The fluorogenic agent (aminomethylcoumarin - AMC) from the substrate Z-Phe-Arg-AMC was used to measure enzyme activity. All cruzain assays were performed using 0.1 molar sodium acetate (pH 5.5) in the presence of 0.01 % Triton X-100 and 10 μM z-Phe-Arg-AMC. Incubation of the compounds with recombinant enzyme was carried out for 30 min at room temperature.
T. cruzi Inhibition Assay.
Antiparasitic activity and compound cytotoxicity were determined as in Ekins et al. and Jones et al.36,37 Briefly, C2C12 mouse myoblasts (ATUC®CRL-1772) were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen) supplemented with 5% fetal bovine serum (FBS, GE Healthcare) and 1% penicillin-streptomycin (Invitrogen) at 37°C with 5% CO2. T. cruzi parasites, strain CA-I/72 were maintained by weekly co-infection with C2C12 cells. For all activity assays, C2C12 cells were infected at a 15:1 parasite to cell ratio with day 6-7 T. cruzi tissue culture-derived trypomastigotes and treated with compounds in 10-point dilution for 72 h. Plates were then fixed with 4% formaldehyde for at least 1 hour and stained with 0.5 μg/ml of 4’,6-diamidino-2-phenylindole (DAPI) for at least 4 h. Plates were imaged and analyzed with the automated ImageXpress MicroXL microscope (Molecular Devices). Infection levels (parasites per host cell) were normalized to positive control (uninfected wells) and negative control (DMSO vehicle). IC50 (antiparasitic activity) and CC50 (host cell cytotoxicity) values were determined using GraphPad Prism software.
Supplementary Material
Acknowledgments
We thank B. Duggan for helpful suggestions on NMR acquisition, and E. Balskus for access to the Harvard Odyssey Cluster for energy barrier calculations. We gratefully acknowledge NIH grants TW006634, AI127505 and NS094597 for support of this project, and the country of Panama for permission to make the original cyanobacterial collections. L.-I. McCall was supported by a postdoctoral fellowship from the Canadian Institutes of Health Research (338511, http://www.cihr-irsc.gc.ca/).
Abbreviations
- BOC
tert-butyloxycarbonyl
- DIC
N,N-diisopropylcarbodiimide
- EDC
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride salt
- Fmoc
Fluorenylmethyloxycarbonyl chloride
- MMP
methylmethoxypyrrolinone
- MOE
molecular operating environment
- NP
natural product
- TFA
trifluoroacetic acid
Footnotes
Associated Content
Supporting Information. NMR spectra of all intermediates and final products, variable temperature NMR experiments, Marfey’s analysis, Gaussian energy modeling, kinetic analysis, modeling of gallinamide A with cruzain, HPLC chromatograms of final products, and SMILES strings for final compounds 1-17. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
PDB Codes
For PDB ID: 2XU3, authors will release the atomic coordinates and experimental data upon article publication.
References
- 1.Newman DJ; Cragg GM Natural Products as Sources of New Drugs from 1981 to 2014. J. Nat. Prod 2016, 79, 629–661 [DOI] [PubMed] [Google Scholar]
- 2.Gerwick WH; Moore BS Lessons from the Past and Charting the Future of Marine Natural Products Drug Discovery and Chemical Biology. Chem. Biol 2012, 19, 85–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.http://marinepharmacology.midwestern.edu (accessed Jun 13, 2019).
- 4.Panagalow MN; Schechter LE; Hurko O Drug Development for CNS Disorders: Strategies for Balancing Risk and Reducing Attrition. Nat. Rev. Drug Discov 2007, 6, 521–532. [DOI] [PubMed] [Google Scholar]
- 5.Rohwer A; Marhoefer RJ; Caffrey CR; Selzer PM Drug Discovery Approached Toward Anti-Parasitic Agents. Drug Discovery in Infectious Diseases 2011, 2(Apicomplexan Parasites), 3–20. [Google Scholar]
- 6.Jerica S; Janko K Microbial and Fungal Protease Inhibitors—Current and Potential Application. Appl. Microbiol. Biotechnol 2012, 93, 1351–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Linington RG; Clark BR; Trimble EE; Almanza A; Ureña L-D; Kyle DE; Gerwick WH Antimalarial Peptides from Marine Cyanobacteria: Isolation and Structural Elucidation of Gallinamide A. J. Nat. Prod 2009, 72, 14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Taori K; Liu Y; Paul VJ; Luesch H Combinatorial Strategies by Marine Cyanobacteria: Symplostatin 4, an Antimitotic Natural Dolastatin 10/15 Hybrid that Synergizes with the Coproduced HDAC Inhibitor Largazole. ChemBioChem. 2009, 10, 1634–1639. [DOI] [PubMed] [Google Scholar]
- 9.Conroy T; Guo JT; Hunt NH; Payne RJ Total Synthesis and Antimalarial Activity of Symplostatin 4. Org. Lett 2010, 12, 5576–5579. [DOI] [PubMed] [Google Scholar]
- 10.Conroy T; Guo JT; Linington RG; Hunt NH; Payne RJ Total Synthesis, Stereochemical Assignment, and Antimalarial Activity of Gallinamide A. Chem.: Eur. J 2011, 17, 13544–13552. [DOI] [PubMed] [Google Scholar]
- 11.Miller B; Friedman AJ; Choi H; Hogan J; McCammon JA; Hook V; Gerwick WH The Marine Cyanobacterial Metabolite Gallinamide A Is a Potent and Selective Inhibitor of Human Cathepsin L. J. Nat. Prod 2014, 77, 92–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omotuyi OI Methyl-methoxylpyrrolinone and Flavinium Nucleus Binding Signatures on Falcipain-2 Active Site J. Mol. Model 2014, 20, 2386. [DOI] [PubMed] [Google Scholar]
- 13.Funkelstein L; Beinfeld M; Minokadeh A; Sadina J; Hook V Unique Biological Function of Cathepsin L in Secretory Vesicles for Biosynthesis of Neuropeptides. Neuropeptides 2010, 44, 457–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lepeta K; Lourenco MV; Schweitzer BC; Martino Adami PV; Banerjee P; Catuara-Solarz S; de La Fuente Revenga M; Guillem AM; Haidar M; Ijomone OM; Nadorp B; Qi L; Perera ND; Refsgaard LK; Reid KM; Sabbar M; Sahoo A; Schaefer N; Sheean RK; Suska A; Verma R; Vicidomini C; Wright D; Zhang XD; Seidenbecher C Synaptopathies: Synaptic Dysfunction in Neurological Disorders. J. Neurochem 2016, 138, 785–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stoka V; Turk V; Turk B Lysosomal Cathepsins and their Regulation in Aging and Neurodegeneration. Ageing Res. Rev 2016, 32, 22–37. [DOI] [PubMed] [Google Scholar]
- 16.Turk B Targeting Proteases: Successes, Failures and Future Prospects. Nat. Rev. Drug Discov 2006, 5, 785–799. [DOI] [PubMed] [Google Scholar]
- 17.Choe Y; Leonetti F; Greenbaum DC; Lecaille F; Bogyo M; Brömme D; Ellman JA; Craik CS Substrate Profiling of Cysteine Proteases Using a Combinatorial Peptide Library Identifies Functionally Unique Specificities. J. Biol. Chem 2006, 281, 12824–12832. [DOI] [PubMed] [Google Scholar]
- 18.Stolze SC; Deu E; Kaschani F; Li, Florea BI; Richau KH; Colby T; van der Hoorn RAL; Overkleeft HS; Bogyo M; Kaiser M The Antimalarial Natural Product Symplostatin 4 is a Nanomolar Inhibitor of the Food Vacuole Falcipains. Chem. Biol 2012, 19, 1546–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conroy T; Guo JT; Elias N; Cergol KM; Gut J; Legac J; Khatoon L; Liu Y; McGowan S; Rosenthal PJ; N. H; Payne RJ Synthesis of Gallinamide A Analogues as Potent Falcipain Inhibitors and Antimalarials. J. Med. Chem 2014, 57, 10557–10563. [DOI] [PubMed] [Google Scholar]
- 20.Lazarin-Bidoia D; Desoti VC; Martins SC; Ribeiro FM; Ud Din Z; Rodrigues-Filho E; Ueda-Nakamura T; Nakamura CV; Silva SO Dibenzylideneacetones are Potent Trypanocidal Compounds that Affect the Trypanosoma cruzi Redox System. Antimicrob. Agents Chemother 2016, 60, 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yasothornsrikul S; Greenbaum D; Medzihradszky KF; Toneff T; Bundey R; Miller R; Schilling B; Petermann I; Dehnert J; Logvinova A; Goldsmith P; Neveu JM; Lane WS; Gibson B; Reinheckel T; Peters C; Bogyo M; Hook V Cathepsin L in Secretory Vesicles Functions as a Prohormone-Processing Enzyme for Production of the Enkephalin Peptide Neurotransmitter. Proc. Natl. Acad. Sci. USA 2003, 100, 9590–9595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funkelstein L; Toneff T; Mosier C; Hwang SR; Beuschlein F; Lichtenauer UD; Reinheckel T; Peters C; Hook V Major Role of Cathepsin L for Producing the Peptide Hormones ACTH, Beta-Endorphin, and Alpha-MSH, Illustrated by Protease Gene Knockout and Expression. J. Biol. Chem 2008, 283, 35652–35659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cazzulo JJ; Stoka V; Turk V Cruzipain, the Major Cysteine Proteinase from the Protozoan Parasite Trypanosoma cruzi. Biol Chem. 1997, 378, 1–10. [DOI] [PubMed] [Google Scholar]
- 24.Barr SC; Warner KL; Kornreic BG; Piscitelli J; Wolfe A; Benet L; McKerrow JH A Cysteine Protease Inhibitor Protects Dogs from Cardiac Damage During Infection by Trypanosoma cruzi. Antimicrob Agents Chemother. 2005, 49, 5160–5161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Engel JC; Doyle PS; Hsieh I; McKerrow JH Cysteine Protease Inhibitors Cure an Experimental Trypanosoma cruzi Infection. J Exp Med. 1998, 188, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hardegger LA; Kuhn B; Spinnler B; Anslem L; Ecabert R; Stihle M; Gsell B; Thoma R; Diez J; Benz J; Plancher J-M; Hartmann G; Banner DW; Happ W; Diederich F Systematic Investigation of Halogen Bonding in Protein–Ligand Interactions. Angew. Chem. Int. Ed 2011, 50, 314–318. [DOI] [PubMed] [Google Scholar]
- 27.Case DA; Darden TA; Cheatham TE III; Simmerling CL; Wang J; Duke RE; Luo R; Walker RC; Zhang W; Merz KM; Roberts B; Hayik S; Roitberg A; Seabra G; Swails J; Götz AW; Kolossváry I; Wong KF; Paesani F; Vanicek J; Wolf RM; Liu J; Wu X; Brozell SR; Steinbrecher T; Gohlke H; Cai Q; Ye X; Wang J; Hsieh M-J; Cui G; Roe DR; Mathews DH; Seetin MG; Salomon-Ferrer R; Sagui C; Babin V; Luchko T; Gusarov S; Kovalenko A; Kollman PA AMBER 12. 2012. University of California, San Francisco: http://ambermd.org/doc12/Amber12.pdf (accessed May 7, 2019).. [Google Scholar]
- 28.Copeland RA Evaluation of Enzyme Inhibitors in Drug Discovery. A Guide for Medicinal Chemists and Pharmacologists. Methods Biochem. Anal. 2005, 46, 1–265. [PubMed] [Google Scholar]
- 29.Perez-Molina JA; Perez-Ayala A; Moreno S; Fernandez-Gonzalez MC; Zamora J; Lopez-Velez R Use of Benznidazole to Treat Chronic Chagas’ Disease: a Systematic Review with a Meta-Analysis. J. Antimicrob. Chemother 2009, 64, 1139–1147. [DOI] [PubMed] [Google Scholar]
- 30.Harrold JM; Ramanathan M; Mager DE Network-Based Approaches in Drug Discovery and Early Development. Clin. Pharmacol. Ther 2013, 94, 651–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ferreira LG; Andricopulo AD Targeting Cysteine Proteases in Trypanosomatid Disease Drug Discovery. Pharmacol. Ther 2017, 180, 49–61. [DOI] [PubMed] [Google Scholar]
- 32.Gillmore SA, Craik CS, Fletterick RJ Structural Determinants of Specificity in the Cysteine Protease Cruzain. Protein Science 1997, 6, 1603–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alonso-Padilla J; Rodriguez A High Throughput Screening for Anti-Trypanosoma Cruzi Drug Discovery. PLoS Neglected Trop. Dis 2014, 8, e3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berman HM; Westbrook J; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kuzmic P Program DYNAFIT for the Analysis of Enzyme Kinetic Data: Application to HIV Proteinase. Anal. Biochem 1996, 237, 260–273. [DOI] [PubMed] [Google Scholar]
- 36.Ekins S; Siqueira-Neto JL; McCall L-I; Sarker M; Yadav M; Ponder EL; Kallel EA; Kellar D; Chen S; Arkin M; Bunin BA; McKerrow JH; Talcott C Machine Learning Models and Pathway Genome Data Base for Trypanosoma cruzi Drug Discovery. PLoS Negl. Trop. Dis 2015, 9, e0003878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones BD; Tochowicz A; Tang Y; Cameron MD; McCall L-I; Hirata K; Siqueira-Neto JL; Reed SL; McKerrow JH; Roush WR Synthesis and Evaluation of Oxyguanidine Analogues of the Cysteine Protease Inhibitor WRR-483 Against Cruzain. ACS Med. Chem. Lett 2016, 7, 77–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.