

SUMMARY
Schlafen 11 (SLFN11) was recently discovered as a cellular restriction factor against replication stress. Here, we show that SLFN11 increases chromatin accessibility genome wide, prominently at active promoters in response to replication stress induced by the checkpoint kinase 1 (CHK1) inhibitor prexasertib or the topoisomerase I (TOP1) inhibitor camptothecin. Concomitantly, SLFN11 selectively activates cellular stress response pathways by inducing the transcription of the immediate early genes (IEGs), including JUN, FOS, EGR1, NFKB2, and ATF3, together with the cell cycle arrest genes CDKN1A (p21WAF1) and GADD45. Both chromatin remodeling and IEG activation require the putative ATPase and helicase activity of SLFN11, whereas canonical extrinsic IEG activation is SLFN11 independent. SLFN11-dependent IEG activation by camptothecin is also observed across 55 non-isogenic NCI-60 cell lines. We conclude that SLFN11 acts as a global regulator of chromatin structure and an intrinsic IEG activator with the potential to engage the innate immune activation in response to replicative stress.
In Brief
Schlafen 11 (SLFN11), a promising therapeutic biomarker, binds chromatin and sensitizes cancer cells to DNA-targeting agents by blocking DNA replication. Murai et al. show that, in response to replication stress, SLFN11 selectively increases chromatin accessibility at promoters and activates a subset of genes known as the immediate early genes (IEGs).
Graphical Abstract
INTRODUCTION
The discovery of predictive biomarkers for anticancer agents remains an unmet clinical need. In recent years, large cancer cell line databases have been developed to address this shortcoming (Barretina et al., 2012; Garnett et al., 2012). Based on the NCI-60 genomic and pharmacological databases (Paull et al., 1989; Reinhold et al., 2015, 2017a, 2017b; Scherf et al., 2000) and on the subsequent development of the CellMiner bioinformatics tools (Rajapakse et al., 2018; Reinhold et al., 2015, 2017a, 2017b; https://discover.nci.nih.gov/cellminer), our group discovered Schlafen 11 (SLFN11) as a genomic determinant of response to DNA-targeted anticancer drugs, including platinums (cisplatin, carboplatin, and oxaliplatin), topoisomerase I (TOP1) inhibitors (camptothecin, topotecan, and irinotecan and the indenoisoquinolines indotecan LMP400 and indimitecan LMP776), topoisomerase II (TOP2) inhibitors (etoposide, mitoxantrone, and doxorubicin), poly(ADP-ribose) polymerase (PARP) inhibitors (olaparib, rucaparib, niraparib, and talazoparib), and DNA synthesis inhibitors (gemcitabine, cytarabine, and nucleoside analogs) (Murai et al., 2016, 2019; Tang et al., 2018; Zoppoli et al., 2012). The larger database of the Cancer Cell Line Encyclopedia (CCLE) at the Broad Institute also identified SLFN11 as the gene whose mRNA expression is the most highly correlated with sensitivity to topotecan (Barretina et al., 2012). Since the initial discovery in cancer cell lines, the relevance of SLFN11 to drug sensitivities has been consolidated in various tumor models as well as in the clinic (Allison Stewart et al., 2017; Coussy et al., 2020; Deng et al., 2015; Gardner et al., 2017; Goss and Gordon, 2016; Kang et al., 2015; Lok et al., 2017; Murai et al., 2016; Pietanza et al., 2018; Shee et al., 2019; Tian et al., 2014; Zoppoli et al., 2012).
SLFN11 belongs to the Schlafen family encompassing 5 genes in humans and 10 genes in mice (Mavrommatis et al., 2013; Murai et al., 2019). SLFN11 is a nuclear protein with a putative helicase domain and a replication protein A (RPA)-binding domains in its C terminus and a nucleic-acid-binding helicase domain in its N terminus (reviewed in Murai et al., 2019). The molecular mechanisms by which SLFN11 kills cells under replication stress has been elucidated partially (Li et al., 2018; Mezzadra et al., 2019; Mu et al., 2016; Murai et al., 2016, 2018; Zoppoli et al., 2012). SLFN11 is recruited to abnormal replication forks harboring extended RPA-coated single-stranded DNA (Maréchal and Zou, 2013; Mu et al., 2016; Murai et al., 2018), which is generated by the uncoupling of the CDC45/MCM2–7/GINS (CMG) replication helicase complex and the DNA polymerase complex (Murai et al., 2018; Saldivar et al., 2017; Toledo et al., 2013). Binding of SLFN11 to the CMG complex then blocks replication through SLFN11’s putative ATPase activity (Murai et al., 2018). SLFN11 was also recently found to disable the DNA damage response (DDR) by depleting the tRNAs for ataxia telangiectasia and Rad3-related protein (ATR) and ataxia telangiectasia mutated (ATM) (Li et al., 2018). In addition to its role as restriction factor for DNA-replication-targeted anticancer drugs, SLFN11 has been linked with the innate immune response. Like other SLFN genes, SLFN11 is inducible by interferon-γ (IFN-γ) and sensitizes tumor cells to IFN-γ-mediated T cell killing (Mezzadra et al., 2019). SLFN11 has been shown to also act as a restriction factor against HIV-1 replication (Abdel-Mohsen et al., 2013; Kiselinova et al., 2016; Li et al., 2012).
In response to stress and extracellular stimuli, cells activate the immediate early genes (IEGs). Those genes can be transcribed within minutes in response to various external stimuli, such as extracellular-signal-regulated kinase (ERK) and mitogen-activated protein kinase (MAPK) pathways (reviewed in Bahrami and Drabløs, 2016). The number and composition of the IEGs vary depending on the types of stimuli, species, and cell lines (Arner et al., 2015). Around 100 IEGs, including JUN, FOS, ATF3, NFKB2, EGR1, CDKN1A (p21WAF1), and GADD45A/B, are commonly upregulated by various external stimuli, immune response, and cellular stress (Bahrami and Drabløs, 2016). Serum addition after culturing in serum-free medium, growth factors, and cytokines are well known stimulators, and this induction does not need protein synthesis. Hence, it is thought that all required factors for transcription, such as transcription factors, DNA modifications, and chromatin status are under preset conditions for those genes. As for the FOS gene, the regulatory mechanisms of immediate activation have been intensively studied since the 1980s (reviewed in O’Donnell et al., 2012). However, it is not understood whether the regulatory mechanisms for the FOS gene are applicable to other IEGs and related to chromatin accessibility.
In this study, we report two functions of SLFN11 in response to replication stress, namely, global induction of chromatin accessibility measured by assay for transposase-accessible chromatin using sequencing (ATAC-seq), and selective transcriptional activation of the IEGs, which both depend on the putative ATPase and helicase activity of SLFN11.
RESULTS
SLFN11 Induces Genome-Wide Chromatin Accessibility at Promoters
Recent studies revealed that SLFN11 is recruited to RPA-coated single-stranded DNA formed at stressed replication forks and DNA damage sites (Mu et al., 2016, Murai et al., 2018). Both camptothecin (CPT), the canonical TOP1 inhibitor, and prexasertib (LY2606368), a cell cycle checkpoint kinase 1 inhibitor (CHK1i) in early clinical development, induce replication stress. As reported (Murai et al., 2018), both drugs induced SLFN11 foci in the nuclear periphery and the inner nucleus in leukemia CCRF-CEM SLFN11-positive cells within 4 h (Figure 1A). At this 4-h time point, CPT reduced the replicating S-phase population both in SLFN11-positive parental and in the SLFN11-knockout (SLFN11-KO) cells due to S-phase checkpoint activation (Murai et al., 2018; Figure 1B). CHK1i treatment for 6 h also reduced the late S-phase population regardless of SLFN11 (Figure 1B). Yet, the viability after treatment for 3 days was very different between CPT and the CHK1i (Figure 1C). SLFN11-KO cells conferred high resistance to CPT, whereas no viability difference was observed between the parental and the SLFN11-KO cells for prexasertib (Figure 1C). Considering these results, we used a short time treatment (2–6 h) with CPT or prexasertib in the following studies to avoid secondary effects of cell death or cell cycle differences.
Figure 1. SLFN11 Increases Chromatin Accessibility in Response to Replication Stress Induced by TOP1 or CHK1 Inhibitors Irrespective of Subsequent Cell Fate.

(A) Immunofluorescence analyses in CCRF-CEM parental (SLFN11-positive) cells treated as indicated (no treatment control, 100 nM CHK1 inhibitor, or 100 nM CPT) for 4 h. Cells were washed with pre-extraction buffer before fixation, and only chromatin-bound SLFN11 (green) was detected by confocal microscopy.
(B) Immunofluorescence Representative flow cytometry cell cycle and replication in response to CPT (100 nM) or CHK1i (100 nM) for the indicated times in CCRF-CEM parental and isogenic SLFN11-knockout (SLFN11-KO) cells. The percentage of highly replicating cells in the red line boxes are annotated. PI, propidium iodide; BrdU, 5-bromo-2′-deoxyuridine (BrdU was added 30 min before harvest)
(C) Immunofluorescence Viability curves of CCRF-CEM parental and SLFN11-KO cells in response to CPT or CHK1i. Viability curves were determined 72 h after continuous drug treatment. Cellular ATP was used to measure cell viability with untreated cells set as 100%. Results are average of three independent experiments with ± SD.
(D) Immunofluorescence ATAC-seq results of CCRF-CEM parental and SLFN11-KO cells without or with drug treatments (no treatment [0 h], 100 nM CPT for 2 or 4 h). ATAC-seq tag densities compared in a 1,000-bp window around the summit of ATAC peaks under the indicated drug and time conditions are shown. The total number of peaks is indicated at the left bottom. Sample number is one for each condition. The data are representative of three independent experiments.
(E) Immunofluorescence ChIP-seq results using antiacetylated H3K27 antibody in CCRF-CEM parental and SLFN11-KO cells (no treatment [0 h], 100 nM CPT for 2 or 4 h). Tag density of ChIP-seq compared in a 2,000-bp window around the summit of signal peaks under the indicated drug and time conditions are shown. The total number of peaks is annotated at the left bottom. Sample number is one for each condition.
(F–H) ATAC-seq results of CCRF-CEM parental and SLFN11-KO cells without or with drug treatments (no treatment [control], 100 nM CPT for 4 h, and 100 nM CHK1 inhibitor for 2, 4, or 6 h) in parallel. Sample number is one for each condition.
(F) Immunofluorescence IGV sequencing tracks for the JUN locus. Signal in promoters of untreated and treated samples were compared using the Poisson test, which is similarly applied by MACS for peak calling (Zhang et al., 2008). **p ≤ 0.01.
(G) Immunofluorescence Bar graphs representing ATAC peak quantification (fragments per kilobase million [FPKM]) at promoter and intergenic regions (mean ± SEM). The number of ATAC peaks of each region is shown in parentheses. ***p ≤ 0.001.
(H) Immunofluorescence Dot plots representing basal ATAC peak quantification (x axis) and after drug treatment (y axis). Each point represents a single ATAC peak in each of the 2 conditions. The black, red, and blue lines indicate no difference, 2-fold increase, and 2-fold decrease, respectively. CPT: camptothecin; CHK1i, prexasertib.
See also Figure S1.
We previously reported that SLFN11 increases chromatin accessibility (“chromatin remodeling”) in the vicinity of replication origins in response to CPT (Murai et al., 2018). To examine whether the chromatin remodeling is linked to chromatin modification or histone density, we performed, in parallel, ATAC-seq and chromatin immunoprecipitation sequencing (ChIP)-seq for histone H3K27ac, a marker of active and open chromatin (Figures 1D and 1E). As expected, pre-existing ATAC peaks under basal conditions overall corresponded to the peaks of H3K27ac (Figure S1A; Buenrostro et al., 2013). The ATAC-seq peaks were enhanced by CPT treatment in a SLFN11-dependent manner, whereas the signal levels of H3K27ac ChIP-seq remained globally unchanged regardless of SLFN11 and CPT treatment (Figure 1E), indicating that the ATAC-peak enhancement by SLFN11 is not accompanied by detectable core histone depletion.
Next, we examined by ATAC-seq the genome-wide effect of SLFN11 on chromatin in response to CHK1 inhibition by prexasertib compared with CPT by using CCRF-CEM parental and SLFN11-KO cells (Figures 1F–1H). Genome-wide distribution of ATAC peaks in the absence of drug treatment showed that, as expected, the baseline ATAC peaks were concentrated in promoters (12,917 out of a total of 36,373 peaks = 36% of all peaks) (Figure S1B). CHK1i treatment intensified the ATAC peaks in a SLFN11- and time-dependent manner, which was also observed in response to CPT treatment in a parallel experiment (Figures 1F–1H and S1C–S1F). The chromatin remodeling by SLFN11 occurred exclusively at pre-existing regions of ATAC peaks (Figures 1D, 1F, 1H, and S1C–S1F) (i.e., closed regions are not opened by SLFN11). Additionally, the enhancement of ATAC peaks was more prominent at the promoter than the intergenic regions both for the CHK1i and CPT treatments (Figures 1G). Hence, irrespective of subsequent cell fate (Figure 1C), we conclude that SLFN11 is, to our knowledge, the most effective global chromatin modifier in response to replication stress by enhancing pre-existing ATAC-seq accessible sites.
SLFN11 Selectively Activates the Transcription of IEGs
Because chromatin accessibility at promoter regions is generally associated with transcription activation (Tsompana and Buck, 2014), we tested the potential impact of the enhanced chromatin accessibility induced by SLFN11 on transcription genome wide.
CCRF-CEM SLFN11-positive parental and SLFN11-KO cells were treated either with prexasertib (CHK1i) or CPT, and fold-change transcriptions after the treatments were analyzed by RNA-seq. CHK1 inhibition by prexasertib decreased the transcripts of only a small number of genes both in the parental and SLFN11-KO cells (Figure 2A, CHK1i, down). In contrast, close to 300 genes were upregulated by prexasertib in the parental cells, and 50 genes were upregulated in the SLFN11-KO cells, with 42 overlapping genes (Figure 2A, CHK1i, up). Parallel experiments with CPT showed downregulation of 200 genes regardless of SLFN11 with 90 overlapping genes (Figure 2A, CPT, down). Notably, gene upregulation by CPT was SLFN11 dependent, with 179 genes upregulated in the SLFN11-positive cells, whereas only 17 genes were upregulated in the SLFN11-negative cells (Figure 2A, CPT, up). A comparison of the common genes upregulated in the SLFN11-positive parental cells by both CHK1i and CPT retrieved 104 overlapping genes. Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis revealed that these genes were enriched in MAPK, tumor necrosis factor (TNF), and virus-infection-signaling pathways (Figure 2A). Among those 104 overlapping genes, 36 were categorized as IEGs (Arner et al., 2015), including JUN, FOS, EGR1, ATF3, and NFKB2 (Figure 2B; Tables S1 and S2). Most of these genes were only marginally activated in SLFN11-KO cells (Figure 2B). Although the IEG response generally occurs within minutes of external stimuli, the SLFN11-dependent IEG activation was only apparent after 4 h of CHK1i treatment and 2 h of CPT treatment (Figure 2C). These timings coincide with the timing of chromatin accessibility induced by each drug (Figures 1D and G), suggesting the coupling of chromatin remodeling and the IEG activation through SLFN11.
Figure 2. SLFN11 Activates Immediate Early Gene (IEG) Transcription in Response to Replication Stress.

(A and B) RNA-seq results of CCRF-CEM parental and SLFN11-KO cells treated with prexasertib (CHK1i) (100 nM) for 6 h or CPT (100 nM) for 4 h. Sample number is three for each condition (technical triplicate).
(A) Venn diagrams showing upregulated (fold change > 2, p < 1E-07) and downregulated (fold change < −2, p < 1E-07) genes by CHK1i (left) or by CPT (right). The number of uniquely upregulated or downregulated genes are indicated in or beside each circle. The numbers of overlapping genes are in bold at the circle-overlapping parts. Analyses of KEGG pathways for the 104 overlapping genes upregulated by both drugs in parental cells are shown at right.
(B) Gene lists showing the most highly upregulated genes by CHK1i (left) or by CPT (right) in parental cells. Gene names in red indicate overlapping genes in the parental cells induced by the CHK1i and CPT. Asterisks (*) indicate IEGs listed by Arner et al. (2015). Whole lists are available in Tables S1 and S2.
(C) Quantitative real-time reverse transcription PCR (qRT-PCR) results for the indicated genes and times in response to CHK1i (100 nM) or CPT (100 nM) compared to untreated in CCRF-CEM parent and SLFN11-KO cells. Each transcript was normalized to the expression level of 18S RNA. Results are the average of three independent experiments with ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (two-tailed unpaired t test).
(D and E) DNA microarray analyses of isogenic (parent and SLFN11-KO) leukemia CCRF-CEM, and prostate DU145 in response to CPT (250 nM for 6 and 12 h). Sample number is three for each condition (technical triplicate).
(D) Venn diagrams showing upregulated (fold change > 2, p < 1E-05) and downregulated (fold change < −2, p < 1E-05) genes by CPT in each cell set. The number of uniquely upregulated or downregulated genes is indicated in each circle, and the number of overlapping genes is in bold. Analyses of KEGG pathways for 43 overlapping upregulated genes in both parental cells are shown.
(E) Gene lists showing the most highly upregulated genes in CCRF-CEM parental (left) or in DU145 parental cells (right). Gene names in red indicate overlapping upregulated genes in both DU145 and CCRF-CEM parental cells by CPT. Asterisks (*) indicate IEGs listed by Arner et al. (2015).
To generalize the finding that SLFN11 activates IEG transcription, we tested another isogenic cell set (prostate DU145 SLFN11-positive parent and the SLFN11-KO) (Murai et al., 2016) in parallel with the retesting of the CCRF-CEM isogenic set (parent and SLFN11-KO) by using a different method for quantifying transcripts based on DNA microarrays. Although there exists some concerns about different sensitivity and dynamic range for DNA microarray, we obtained consistent results showing selective transcription activation in response to the CPT treatment in both cell line sets in the SLFN11-positive cells (Figure 2D). The activated genes were similar to the overlapping genes and IEGs shown in Figure 2B (Figure 2E; Tables S3 and S4). Hence, the SLFN11-dependent activation of IEGs by replication stress appears to be general across cell lines of blood (CCRF-CEM) or prostate (DU145) lineage.
Global Chromatin Remodeling and Selective IEG Activation Depend on SLFN11’s Putative ATPase and Helicase Activity
We previously reported that the replication block induced by SLFN11 is abolished by introducing a point mutation at the putative ATPase Walker-B motif of SLFN11 (E669Q) in spite of a similar recruitment of SLFN11 to chromatin (Murai et al., 2018); therefore, we tested whether the putative ATPase activity of SLFN11 was required for chromatin remodeling and IEG activation. To that aim, we used wild-type (WT) SLFN11-overex-pressing or E669Q SLFN11-overexpressing cells in the human leukemia K562 line that endogenously carries a very low level of SLFN11 (Murai et al., 2018; Tang et al., 2018). We refer to the corresponding cell lines as K562-Vector, K562-WT, and K562-E669Q. The K562-WT cells enhanced chromatin accessibility more than the K562-E669Q cells, and the K562-E669Q cells enhanced chromatin accessibility only marginally compared with the K562-Vector cells (Figures 3A and 3B). The global enhancement of chromatin accessibility was similar between the promoter and intergenic regions (Figure 3B).
Figure 3. The ATPase Walker-B Sequence of SLFN11 Is Required for the Global Chromatin Accessibility and Selective IEG Induction by SLFN11.

(A and B) ATAC-seq results of human leukemia K562+Vector, +WT (wild-type SLFN11) and +E669Q (putative ATPase-dead SLFN11) (Murai et al., 2018) without or with drug treatments (no treatment [control], 1,000 nM CPT treatment for 4 h). (A) Dot plots representing ATAC peak quantification (x axis) before and after CPT treatment (y axis). Each dot corresponds to a single ATAC peak (n = 18,088; note that 189, 239, and 212 data points are outside the axis limits for K562+Vector, +WT, and +E669Q, respectively). (B) Bar graphs representing ATAC peak quantification (FPKM) at promoter and intergenic regions (mean ± SEM). The number of ATAC peak of each region is shown in parentheses. ***p ≤ 0.001.
(C) Bar graphs of qRT-PCR for the indicated genes and treatments (250 nM CPT, 10 nM CHK1i, or 100 nM CHK1i) in K562-Vector, -WT, -E669Q cells. Each transcript was normalized to the expression level of 18S RNA. Results are the average of three independent experiments with ± SD. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001 (two-tailed unpaired t test). See also Table S5.
To determine whether the putative ATPase and helicase activity of SLNF11 is required for IEG transcription activation, K562-Vector, K562-WT, and K562-E669Q cells were treated with CPT or the CHK1 inhibitor. The SLFN11-dependent upregulated genes found in CCRF-CEM parental cells (e.g., ATF3, NFKB2, and JUN) were also highly upregulated in K562-WT cells but only marginally in K562-Vector and K562-E669Q cells (Figure 3C). These results indicate that the ATPase domain of SLFN11 is required for the global SLFN11-mediated chromatin accessibility and the selective IEG transcription activation in response to replication stress.
SLFN11-Mediated Transcription Activation Is Neither Dependent of the TP53 Pathway nor Dependent of DHX9, a Binding Partner of SLFN11
Because some downstream genes of the TP53 pathway, such as CDKN1A and NOXA (PMAIP1), were also upregulated in a SLFN11-dependent manner (Tables S1, S2, S3, and S4), we examined whether the activation of those genes was TP53 dependent. First, we found that TP53 transcription was not activated by CPT regardless of SLFN11 (Figure S2A). Second, stabilization of TP53 and phosphorylation of TP53 were observed in CPT-treated cells regardless of SLFN11 in the two different isogenic cell sets (Figure S2B). In addition, K562 cells genetically lack TP53 (Sousa et al., 2015). Nevertheless, the expression of CDKN1A as well as NOXA were further activated in the SLFN11-proficient cells compared to the SLFN11-deficient cells (Figures S2C). The lack of transcription activation of CDKN1A and NOXA in K562-E669Q cells demonstrates that the ATPase activity of SLFN11 is required for the activation of those genes even in the absence of TP53 (Figures S2C).
Because DHX9 (DExH-box helicase 9) was reported as a robust binding partner of SLFN11 (Murai et al., 2018; Nogales et al., 2016), we examined whether DHX9 might impact SLFN11-mediated IEG activation. To test this hypothesis, we depleted DHX9 by small interfering RNA (siRNA) transfection and examined the transcription level of ATF3 in DU145 parental and SLFN11-KO cells. The SLFN11 protein level as well as the transcript level of ATF3 were not altered by DHX9 depletion (Figures S3A and S3B). Overall, we conclude that SLFN11-dependent gene activation is TP53 and DHX9 independent.
SLFN11-Mediated IEG Transcription Activation Occurs in a Different Pathway from Canonical External Stimuli
To determine the mechanisms of SLFN11-mediated IEG activation, first, we examined whether SLFN11 activates the IEGs at the transcription level (i.e., not by stabilization of mRNAs). We labeled nascent RNA with EU (ethynyl uridine) for 1 h before collecting RNA from CPT-treated or nontreated SLFN11-positive cells and performed quantitative RT-PCR for representative IEGs (ATF3, JUN, and NFKB2). Nascent RNA of the IEGs were more enriched in the CPT-treated than the untreated samples (Figure 4A), indicating that the SLFN11-mediated increase of IEG transcripts occurs by transcriptional activation.
Figure 4. SLFN11-Mediated Transcription Activation Occurs in a Different Pathway from Canonical External Stimuli.

(A) Bar graphs of qRT-PCR for the nascent transcripts of the indicated genes without or with CPT (100 nM for 4 h) treatment. CCRF-CEM parental and SLFN11-KO cells were labeled with or without EU (ethynyl uridine). Each transcript was normalized to the expression level of total 18S RNA from the RNA samples before enrichment of nascent RNA. Results are the average of three independent experiments with ± SD.
(B) Bar graphs of qRT-PCR for the ATF3 gene and for the indicated times in DU145 parental and SLFN11-KO cells. Cells were incubated with epithelial growth factor (EGF) (25 ng/ml) for 10 min and released into regular medium (left). Serum induction was performed after 40 h of serum starvation (right). Each transcript was normalized to the expression of 18S RNA. Results are the average of three independent experiments with ± SD (left) or representative of two independent experiments with average ± SD (n = 3) (right). *p ≤ 0.05, (two-tailed unpaired t test).
(C) Western blots with the indicated antibodies and conditions in DU145 parental cells. Cells were incubated with CPT (100 nM) continuously or with EGF (25 ng/ml) for 10 min and released in regular medium.
(D) Bar graphs of qRT-PCR for the ATF3 gene for the indicated times in DU145 parental and SLFN11-KO cells. Cells were incubated with CPT (100 nM) continuously. Each transcript was normalized to the expression of 18S RNA. Results are average of three independent experiments with ± SD. *p ≤ 0.05, ***p ≤ 0.001 (two-tailed unpaired t test).
See also Figures S2 and S3 and Table S5.
As the IEGs are characterized by their rapid activation in response to various external stimuli, we next examined the potential involvement of SLFN11 in IEG activation through epithelial growth factor (EGF) stimulation or serum induction. In the isogenic cell set of DU145 (parent and SLFN11-KO), both stimuli increased the transcription of ATF3, with the peak at 1 h, followed by rapid reduction at 2 h after the stimuli regardless of SLFN11 (Figure 4B). The time course of ATF3 transcription activation by these stimuli was different from SLFN11-dependent ATF3 induction by CPT or CHK1i prexasertib, which was markedly slower and matched chromatin remodeling (Figures 1G and 2C). Furthermore, MAPK pathway activation, which is marked by phosphorylation of MEK1/2, ERK1/2, and RSK, was not activated under the CPT treatment when ATF3 transcription was highly induced in a SLFN11-dependent manner (Figures 4C and 4D). We conclude that SLFN11 activates the IEGs at the transcription level but by a different mechanism from canonical external stimulations.
SLFN11-Dependent IEG Activation Is Conserved in Non-isogenic Cell Lines
To extend our finding of SLFN11-dependent IEG activation over a cell line dataset with a broad endogenous SLFN11 expression range, such as the NCI-60 where approximately 45% of the cell lines do not express SLFN11 (Rajapakse et al., 2018; Tang et al., 2018; Zoppoli et al., 2012), we mined a recently published resource, which analyzed differential gene expression in response to 15 clinical drugs across the NCI-60 cell line (Monks et al., 2018). Genome-wide transcriptome data for 55 of the NCI-60 cell lines treated with the clinical derivative of CPT topotecan for 6 h were examined. Expression of SLFN11, which was not listed in this database (because it used a relatively old microarray platform), was obtained from the genome-wide analyses of the NCI-60 line, which is consistent across the Broad Institute and Sanger Institute databases (https://discover.nci.nih.gov/cellminercdb; Rajapakse et al., 2018).
Ranking the genes according to their fold change (CPT treated/untreated) showed that the IEGs ATF3, FOS, and JUNB were among the most upregulated genes (Figure 5A).Next, we analyzed the correlations between basal SLFN11 expression and the fold change of each gene listed across the 55 cell lines. The fold changes of transcripts for IEGs were significantly correlated with the basal SLFN11 expression levels (Figures 5A and 5B). The significantly correlated genes ATF3, FOS, JUNB, FOSB, EGR1, and GADD45B were also found to be upregulated in the isogenic setting both by CPT and prexasertib (Figures 2 and 3; Tables S1, S2, S3, and S4). Hence, the activation of the IEGs by SLFN11 in response to replication stress can be generalized not only across the three isogenic cell line sets (CCRF-CEM, DU145 and K562) but also across 55 non-isogenic diverse cell lines.
Figure 5. SLFN11-Dependent Activation of the IEGs in Non-Isogenic Cell Lines from the NCI-60.

(A) Gene lists showing the most highly upregulated genes induced by topotecan (1 μM for 6 h). The sum of fold change (FC) of gene expression (log2[treated/untreated]) from 55 cell lines are shown on the 2nd column from the left. IEG transcriptome data were computed from Monks et al. (2018), and SLFN11 transcript levels (average log2) were obtained from the NCI-60 database (https://discover.nci.nih.gov/cellminercdb/; Rajapakse et al., 2018). Pearson’s correlation between SLFN11 expression and the FC in 55 cell lines and corresponding p values are listed in the 3rd and 4th (right) column, respectively.
(B) Dot plots showing the basal SLFN11 expression (x axis) and the CPT-induced transcript FC for the indicated genes (y axis) across the 55 cell lines. Linear regression curves and R square are shown in each graph. Asterisks (*) refer to IEGs listed by Arner et al. (2015).
IEGs Characterized by Pre-existing Accessible Chromatin at Promoters and Short Transcripts Are Transcriptionally Activated by SLFN11-Induced Chromatin Remodeling
To gain insight on the features of genes activated by SLFN11 in response to replication stress, we plotted the fold change of ATAC score versus the fold difference of mRNA expression. Figures 6A and S4A show that enhanced chromatin accessibility occurred in most genes regardless of expression changes, whether their expression was stable, upregulated, or downregulated. Hence, enhanced chromatin accessibility in itself is not sufficient for SLFN11-mediated gene activation. As IEGs are generally short in length (Bahrami and Drabløs, 2016), we next considered the gene length. While genes downregulated by CPT were distributed widely, SLFN11-dependent upregulated genes were overall short in genomic gene length (Figure 6B). Furthermore, genes upregulated by SLFN11 tended to have an elevated basal (pretreatment) chromatin accessibility level relative to their basal mRNA expression level (Figure 6C). These results indicate that the small subset of genes with short genomic gene length and with pre-existing chromatin accessibility but with low basal expression are preferentially upregulated by SLFN11.
Figure 6. Features of the Genes Activated by SLFN11 in Response to Replication Stress.

(A–C) Compilation of the ATAC-seq data (Figure 1) and the RNA-seq data (Figure 2). (A) Dot blots represent the fold difference of mRNA expression (CPT 4 h/0 h) (y axis) and fold difference of ATAC-seq score (CPT 4 h/0 h) (x axis) in CCRF-CEM parent (left) and SLFN11-KO cells (right). Three representative IEGs are annotated. Upregulated (>2-fold and p < 1E-07) and downregulated (< −2-fold and p < 1E-07) genes are colored in red and blue, respectively. (B) Dot blots representing gene length (bp) of the upregulated (up) and downregulated (down) genes compared to all genes (all). (C) Dot blots representing basal mRNA expression level (y axis) and basal ATAC-seq score (x axis) for each gene (left). Each parameter is replotted on the right panels for all the upregulated (up) and downregulated (down) genes.
(D) Dot plots representing H3K27 acetylation levels in basal condition (x axis) and after drug treatment (CPT 100 nM for 4 h, y axis). Each point represents the intensity of ChIP-seq peaks of the 2 conditions. The black, red, and blue lines indicate no difference, 2-fold increase, and 2-fold decrease, respectively. The hyperacetylated IEGs (>2-fold) at their promoters are annotated and highlighted in red.
(E) Sequencing tracks around the NFKB2 locus for the indicated conditions in CCRF-CEM parent and SLFN11-KO.
(F) Enrichment of the active histone mark H3K27Ac around the NFKB2 locus was examined by ChIP assay in CCRF-CEM parental cells with or without CPT treatment (250 nM or 1,000 nM, 4 h). The loci of primer sets for ChIP qPCR are shown at the bottom of (E). Results are representative of three independent experiments and the average of three technical repeats with ± SD. **p ≤ 0.01, ***p ≤ 0.001 (two-tailed unpaired t test).
Although we showed that the H3K27ac level is overall consistent before and after the CPT treatment (Figure 1E), several activated genes listed in Figures 2B and 2E (NFKB2, NEU4, RELB, DUSP8, and PPP1R15A) were also hyperacetylated at their promoters after CPT treatment (Figures 6D and 6E). The hyperacetylation at the promoter of NFKB2 after CPT treatment was confirmed by ChIP qPCR (Figure 6F). Yet, the majority of the upregulated genes, such as JUN and ATF3, were not hyperacetylated by CPT treatment (Figure 6D), which was confirmed by ChIP qPCR (Figure S4B). This implies that H3K27 hyperacetylation after CPT treatment can be observed for some IEGs but is not an essential step for the activation of the IEGs in SLFN11-positive cells. Together, we conclude that SLFN11-mediated chromatin remodeling may provide accessibility for transcription activators or histone modification enzymes to release the transcripts of IEGs.
DISCUSSION
Since the discovery of SLFN11 as a dominant determinant of sensitivity to a broad range of clinically relevant DNA-targeted agents with a broad range of expression in cancers (Barretina et al., 2012; Murai et al., 2019; Tang et al., 2018; Zoppoli et al., 2012) and the discovery of SLFN11 as a restriction factor for viral infections (Kiselinova et al., 2016; Li et al., 2012) and a translation regulator (Li et al., 2018), understanding the molecular functions of SLFN11 is important both from basic cell biology and biomedical viewpoints.
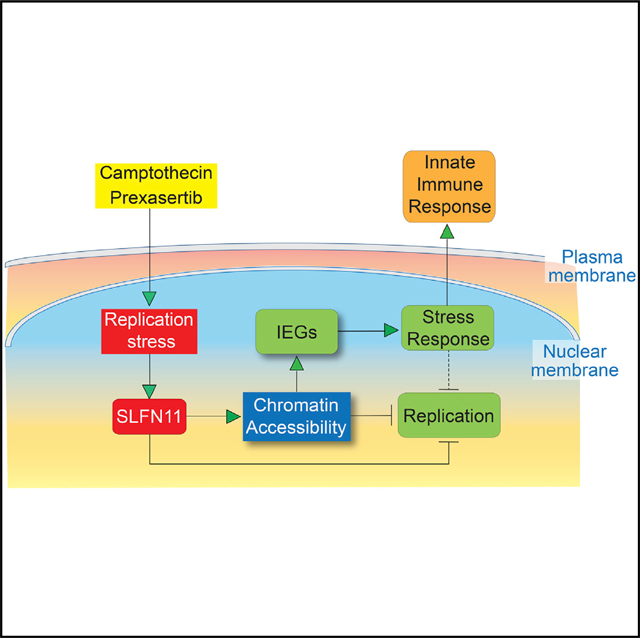
In this study, we demonstrate that, in response to replication stress, SLFN11 increases genome-wide chromatin accessibility and induces the selective activation of IEG transcription (transcriptional stress response). We present evidence that SLFN11 acts as a master regulator of the “intrinsic gateway to the genomic response” (Figure 7, 1–5), leading to IEG activation in response to two mechanistically different inducers of replication stress, namely, TOP1 and CHK1 inhibitors. This intrinsic activation pathway (gateway) is distinct from the canonical IEG activation by extrinsic stimuli (serum induction, EGF stimulation, or MAPK pathway activation), which is commonly referred to as the “extrinsic gateway to the genomic response” (Figure 7, 8). The IEGs are also upregulated in SLFN11-deficient cells to some extent, but their upregulation is magnified several folds in the presence of SLFN11 (Figures 2, 3, and 5). We also show that the putative ATPase and helicase activity of SLFN11 is required for SLFN11-induced chromatin accessibility and activation of the IEGs (Figure 3). The current study demonstrates that SLFN11 acts beyond its recently described activity as a replication fork blocker through its direct binding to RPA and the CMG helicase complexes (Figure 7, 6 and 7; Murai et al., 2018).
Figure 7. Model for SLFN11-Dependent IEG Activation in Cells under Replication Stress.

Scheme of the proposed intrinsic activation pathway by SLFN11. Replication stress activates SLFN11 (1) to enhance global chromatin accessibility (2) at active promoters and in intergenic regions through SLFN11’s ATPase-Walker B consensus motif. SLFN11-induced chromatin accessibility selectively releases the IEGs (3) and induces a cellular stress response (4) (including innate immune response). IEGs as well as CDKN1A (p21WAF1) and GADD45 activation also block replication (5). In addition, SLFN11 blocks replication by altering chromatin structure (6) and binding directly to RPA and the CMG replicative helicase complex (7) (Murai et al., 2018). The classical extrinsic gateway to the genomic response (typically in response to serum or growth factors) is SLFN11 independent (8).
Physiological Significance of SLFN11-Dependent IEG Activation
IEGs, such as CDKN1A and GADD45A/B (Figure S2; Tables S1, S2, S3, and S4), are known to produce cell cycle arrest (Karimian et al., 2016; Salvador et al., 2013). This induction is likely to contribute to the SLFN11-dependent replication block in addition to the local effects of SLFN11 on replication forks through its binding to RPA and the CMG replication helicase complex (Figure 7, 5; Mu et al., 2016; Murai et al., 2018, 2019). Moreover, JUN, FOS, and ATF3, components of the AP-1 transcription factor, have dual functions for survival and apoptosis depending on a complex network of signaling pathway and cell types (Hess et al., 2004). Therefore, it is plausible that IEG activation contributes to the SLFN11-mediated cell cycle arrest and cell death in response to replication stress. Yet, we showed that the IEG activation by SLFN11 occurs irrespective of subsequent cell fate (sensitization in the case of CPT and lack of sensitizing effect in the case of prexasertib) (Figure 1B). This can be interpreted as enforced unscheduled replication origin firings by prexasertib, which determines cell fate independently of the SLFN11-mediated cell death (i.e., the cytotoxic effect of unscheduled origin firings overwhelms the toxicity of SLFN11-dependent replication block). Nevertheless, there remains a possibility that SLFN11 enforces the cytotoxicity of CPT or DNA damaging agents that target DNA replication partially though the activation of the IEGs and growth arrest genes, such as CDKN1A and GADD45.
Chromatin Accessibility and IEG Activation by SLFN11
Global chromatin accessibility induction, both at active promoters and at intergenic pre-existing ATAC-seq sites (Figures 1 and 3), is a striking feature of SLFN11 in response to replication stress. To our knowledge, no other molecule or pathway has yet been reported with a similarly global action. Although SLFN11 accumulates at stressed replication forks through direct binding to RPA (Maréchal and Zou, 2013; Mu et al., 2016; Murai et al., 2018), we observed genome-wide chromatin accessibility, predominantly at active promoter regions independently of notable global change in histone H3K27 acetylation (Figure 1). It is unlikely that the global effects of SLFN11 on promoters and pre-existing chromatin accessible sites are directly physically related to the SLFN11 foci, which are detected at stalled replication forks and the nuclear periphery (Figure 1A; Maréchal and Zou, 2013; Mu et al., 2016; Murai et al., 2018). Indeed, SLFN11 shows a diffuse nuclear staining unless nuclei are pre-extracted with mild detergents (Mu et al., 2016; Murai et al., 2018; Zoppoli et al., 2012). Furthermore, our repeated ChIP and ChIP-seq for SLFN11 failed to detect specific peaks at promoters both before and after drug treatment. Thus, we propose that the effect of SLFN11 on chromatin accessibility is exerted beyond the observed foci detected by immunofluorescence in response to replication stress.
SLFN11 enhances ATAC-seq peaks only at pre-existing chromatin accessible regions, which are a hallmark of active promoters. SLFN11 also binds the RNA helicase DHX9 that resolves R-loops (DNA structures composed of RNA-DNA hybrids and single-stranded DNA) (Lee and Pelletier, 2016; Murai et al., 2018; Nogales et al., 2016). Promoter and terminal regions represent hotspots of R-loop formation, and R-loop-forming promoters are enriched specifically for RNA polymerase pausing and open chromatin (Sanz and Chedin, 2019; Sanz et al., 2016). Hence, the action of SLFN11 on IEG promoters is likely to require additional factors including but not limited to interactions with DHX9 or RNA polymerase. Of note, we found that DHX9 is not necessary for the IEG activation by SLFN11 (Figure S3). Considering the essential role of the ATPase domain of SLFN11 for chromatin remodeling, so far, our data indicate that SLFN11 is a core element for chromatin accessibility in response to replicative stress. Further studies are warranted to elucidate the detailed molecular mechanisms connecting IEG activation with enhanced chromatin accessibility and the potential sensitivity of IEGs to chromatin accessibility as a threshold for their activation.
Generally, ATAC-seq peaks are coupled with transcription activation. However, the induced chromatin accessibility by SLFN11 only upregulated a small subset of genes (Figures 6A and S4). These genes are short, with low baseline transcription, whereas their promoters are readily accessible to ATAC-seq (Figure 6). These features are necessary for IEGs to be activated within minutes without new protein synthesis (Bahrami and Drabløs, 2016). There are several models by which IEGs are rapidly activated. One model for the FOS gene is that transcription-activator proteins are pre-bound to promoters and ready to transcribe under signaling pathways (RhoA-actin, ERK-MAPK, or p38-MAPK) (O’Donnell et al., 2012). However, we excluded the possibility that the MAPK pathway is activated under CPT treatment where SLFN11 activates IEG transcription (Figures 4C and 4D). As IEG activation was also observed at a low level in SLFN11-negative cells (Figures 2, 3, and 5), signaling pathways other than the MAPK pathway may be activated regardless of SLFN11 in response to replication stress. We propose that chromatin relaxation by SLFN11 may provide a favorable environment to pre-bound transcription factors and enhance the pause-release of RNA polymerase complexes. In this scenario, chromatin remodeling and IEG activation are functionally coupled (Figure 7, 3).
Functions of SLFN11 Beyond Replication Block and Sensitization to DNA-Targeted Chemotherapeutic Agents
SLFN11 is likely to have functions involving not only DNA but also RNA. It not only responds to replication stress induced by anticancer drugs but also has been reported to act in antiviral and innate immune responses. SLFN11, as the other Schlafen genes, has been described as an IFN-responsive gene (Mavrommatis et al., 2013; Mezzadra et al., 2019; Murai et al., 2019) and a restriction factor against HIV (Kiselinova et al., 2016; Li et al., 2012; Li et al. (2018)) recently reported that SLFN11 cleaves type II tRNAs in response to DNA damage and suppresses the translation of high TTA (Leu) codon use proteins, such as ATR, and concluded that the suppression of ATR contributes to SLFN11-dependent drug sensitivity. This mechanism can be associated with the recent structural studies demonstrating that SLFN13 cleaves tRNAs through its N-terminal domain, which is conserved in SLFN11 (Mavrommatis et al., 2013; Murai et al., 2019; Yang et al., 2018). So far, the RNA functions of SLFN11 have not been directly linked to our findings reporting the effects of SLFN11 on DNA (chromatin binding, chromatin remodeling, and transcriptional activation of the IEGs). Further studies are warranted to uncover the whole picture of SLFN11 and to elucidate its potential role in the coupling of the innate immune responses with replication stress induced by chemotherapeutic drugs and viral infections (Murai et al., 2019).
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement. Further information for reagents may be directed to and will be fulfilled by Lead Contact Yves Pommier (pommier@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human prostate DU145, human leukemia CCRF-CEM, and human leukemia K562 cell lines were obtained from Developmental Therapeutics Program (NCI/NIH). They were grown in RPMI medium 1640 (1x, GIBCO, 11875–093) added with 10% fetal bovine serum (Gemini, 100–106) and 1% penicillin-streptomycin (GIBCO, 15140–122) at 37 C in 5% CO2. SLFN11-knockout cells in DU145 and CCRF-CEM, genetically modified K562 cell lines (K562-Vec, -wild-type SLFN11, -E669Q SLFN11) were established previously (Murai et al., 2016, 2018)
METHOD DETAILS
Immunofluorescence analysis with pre-extraction
Cells were deposited onto slide glasses (Superfrost Plus Microscope Slides precleaned, Fisher Scientific, 12–550-15) by cytospin (300 rpm, 3 min, slow mode). The deposited cells were pretreated with cold 0.1% Triton X-100/PBS for 1 min on ice, and then fixed with 4% paraformaldehyde in PBS for 10 min. Next, cells were incubated with 4% BSA/PBST for 10 min and incubated for overnight with primary antibody (anti-SLFN11 antibody)/4% bovine serum albumin (BSA)/PBST in moisture chamber at 4 C by 1:1000 dilution. After washing with PBST, the cells were incubated with Alexa Fluor 488 goat anti-mouse IgG/5% BSA/PBST by 1:1000 dilution for 2–4 hours. After washing with PBST, cells were mounted with Vectashield with DAPI (VECTOR, H-1200). Images were captured with a Zeiss LSM 780 confocal microscope.
Viability assay
To measure the sensitivity of cells to drugs, cells were continuously exposed to various concentrations of the drugs. Two thousand CCRF-CEM cells were seeded in 384-well white plates (Perkin Elmer Life Sciences, 6007680) in 40 μL of medium per well. Cellular viability was determined using the ATPlite 1-step kits (PerkinElmer). Luminescence was measured with an EnVision 2104 Multilabel Reader (PerkinElmer) or TECAN infinite M200. The ATP level in untreated cells was defined as 100%. Viability (%) of treated cells was defined as ATP treated cells/ATP untreated cells x 100.
Cell cycle analysis
Untreated or treated cells were labeled with 20 μM BrdU for 30 min before collection. Collected cells were fixed with cold 100% ethanol for 10 min at −20°C. Fixed cells were denatured with 2N HCl for 2 hours followed by intensive wash with PBS. The cells were incubated with anti-BrdU antibody conjugated with FICT in 4% FBS/PBST for overnight, and then incubated with RNase and propidium iodide. Samples were analyzed with flow cytometry (MACSQuant10).
Immunoblotting
To prepare whole cell lysates, cells were lysed with CellLytic™M lysis reagent (C2978, Sigma-Aldrich). After thorough mixing and incubation at 4°C for 30 min, lysates were centrifuged at 15,000 g at 4°C for 10 min, and supernatants were collected. Samples were mixed with tris-glycine SDS sample buffer (Novex, LC2676) and loaded onto Novex tris-glycine gels (Novex). Blotted membrane was blocked with 4% bovine serum albumin (BSA) (Sigma-Aldrich. A9418) in phosphate-buffered saline (PBS) with 0.1% tween-20 (PBST). The primary antibodies were diluted in 5% BSA/PBST by 1:3000 for phospo-p53, total p53, p-MEK1/2, p-ERK1/2, p-RSK, SLFN11 and 1:10000 for Histone H3, Pan-actin, and GAPDH. The secondary antibodies were diluted in 5% non-fat milk/PBST by 1:10000.
RNA extraction and reverse transcription qPCR
Total RNA was extracted using TRIzol reagent (Invitrogen), followed by further purification using PureLink RNA Mini Kit (Life Technology) with DNase treatment (QIAGEN). Complementary DNA (cDNA) was synthesized using SuperScript II Reverse Transcriptase Kit (Invitrogen) or iScript RT supermix for RT-qPCR (Bio-Rad). The primers used to amplify specific genes are listed in Table S5. Real-time qPCR was carried out with FastStart Universal SYBR Green Master (Roche Applied Science) by the 7900HT Fast Real-Time PCR System (Applied Biosystems) or SsoAdvanced Universal SYBR Green supermix (Bio-Rad) by Step One Plus (AB applied biosystem) according to the manufacture’s instruction. The melting curve was generated to confirm the amplification specificity. Ribosomal RNA 18S was used as the internal control. The relative level of gene expression was determined using the 2(−ΔΔCt) method.
Nascent RNA extraction and reverse transctiption qPCR
Untreated or treated cells were labeled or not labeled with 0.5 mM EU for 60 min before collection. After the click reaction, total RNA was collected and EU labeled RNA was enriched by Dyna-beads following the manufacture protocol (Click-iT Nascent RNA Capture Kit, for gene expression analysis, ThermoFisher, C10365). cDNA was synthesized using SuperScript VILO cDNA Synthesis Kit (ThermoFisher). Real-time qPCR was carried out with ssoAdvanced Universal SYBR Green supermix (Bio-Rad) by Step One Plus (AB applied biosystem) according to the manufacture’s instruction. cDNA of ribosomal RNA 18S reverse-transcribed from total RNA was used as the internal control. The relative level of gene expression was determined using the 2(−ΔΔCt) method.
RNA-seq
RNA quality and quantity were analyzed with 2100 Expert (Agilent). RIN (the RNA Integrity Number) of all samples were above 8.0. Sample analyses were done at the Center for Cancer Research Sequence Facility (Frederick, MD). RNA-Seq samples were pooled and sequenced on one paired-end run, which was conducted on HiSeq4000 with HiSeq3000/4000 chemistry. Adapters and low-quality bases were trimmed using Trimmomatic software before alignment with the reference genome (hg19) and the annotated transcripts (Ensembl_v70) using STAR. The mapping statistics are calculated using Picard software. In addition, the gene expression quantification analysis was performed for all samples using STAR/RSEM tools.
DNA Microarray
CCRF-CEM parental and the SLFN11-KO cells were treated/untreated with 250 nM CPT for 6 hours or 12 hours. DU145 parental and the SLFN11-KO cells were treated/untreated with 250 nM CPT for 6 hours or 12 hours. Total RNA was extracted using TRIzol reagent (Invitrogen), followed by further purification using PureLink RNA Mini Kit (Life Technology) with DNase treatment (QIAGEN). RNA quality was checked on Agilent Bioanalyzer. All samples used for microarray analysis have high quality score (RIN > 7). 100 ng of RNA was reverse transcribed, amplified and sense strand cDNA was fragmented and labeled using Affymetrix’s GeneChip WT Plus Reagent Kit following manufacturer’s suggested protocol. Four replicates of each group were hybridized to Affymetrix human Gene ST 2.0 GeneChip in Affymetrix hybridization oven at 45°C, 60rpm for 16 hr. Wash and stain were performed on Affymetrix Fluidics Station 450 and scanned on Affymetrix GeneChip scanner 3000 with AutoLoader. Data were collected using Affymetrix AGCC software. Statistical and clustering analysis was performed with Partek Genomics Suite software using RMA normalization algorithm. Differentially expressed genes were identified with ANOVA analysis. Genes that are up- or downregulated more than 2-fold and with a p < 0.00001 were considered significant. Significant genes were analyzed for enrichment for pathways using DAVID bioinformatics database (https://david.abcc.ncifcrf.gov/) and Ingenuity Pathway Analysis software.
Assay for Transposase-Accessible Chromatin with high throughput sequencing (ATAC-seq)
Genome-wide mapping of chromatin accessibility was done by following the published method (Buenrostro et al., 2013). Briefly, fifty thousand CCRF-CEM parental and SLFN11-KO cells were treated with DMSO or CHK1 inhibitor (prexasertib 100 nM) for 2, 4, and 6 hours, and CPT (100 nM) for 2 and 4 hours. K562-Vector, -WT and –E669Q cells were treated with DMSO or CPT (1 μM) for 4 hours.
To prepare nuclei, cells were lysed using cold lysis buffer (10 mM Tris-Cl, pH 7.4, 10 mM NaCl, 3 mM MgCl2 and 0.1% IGEPAL CA-630). Immediately following the nuclei preparation, the pellet was re-suspended in the transposase reaction mix [25 μL 2x TD buffer, 2.5 μL Transposase (Illumina) and 22.5 μL of nuclease free water], and incubated for 30 minutes at 37°C. Directly following transposition, the sample was purified using a DNA Clean and Concentrator-5 (ZYMO RESEARCH) and eluted with 25 μL of DNA elution buffer. Following purification, we amplified library fragments using 1x NEBnext PCR master mix and 1.25 μM of custom Nextera PCR primers 1 and 2, using the following PCR conditions: 72°C for 5 minutes, 98°C for 30 s, followed by thermo-cycling at 98°C for 10 s, 63°C for 30 s and 72°C for 1 minute. We amplified the full libraries for 5 cycles, after 5 cycles we took an aliquot of the PCR reaction and added 10 μL of the PCR cocktail with Syber Green at a final concentration of 0.6x. We ran this reaction for 20 cycles, to determine the additional number of cycles needed for the remaining 45 μL reaction. Libraries were amplified for a total of 10 cycles and purified using a PCRClean DX yielding a final library in 20 μl. ATAC sequences were obtained by Illumina sequencing and aligned to the genome (hg19) in the form of BAM flies.
Chromatin immunoprecipitation (ChIP)-seq
Twenty-five million CCRF-CEM parental and SLFN11-KO cells were treated or untreated with CPT (100 nM) for 4 hours. ChIP assay was done by following the instruction manual of ChIP-IT Express (Active Motif). Briefly, cells were fixed with 1% formaldehyde in medium for 10 min at room temperature. Fixation was stopped with x1 glycine/PBS. Cells were lysed with lysis buffer with proteinase inhibitor cocktail and PMSF and homogenized 60 times by small tight homogenizer. Cell pellets were re-suspended with sharing buffer, and sonicated 30 minutes by the following settings: 30 s on, 30 s off, level at H, (Bioruptor XL). Five %/volume of each sample was saved as input. The left of the supernatant was incubated with 2 μg antibody (H3K27ac or normal rabbit IgG) and protein G beads for overnight at 4°C. After reversing cross-link and proteinase K treatment, immunoprecipitated DNA was purified with ChIP DNA Clean & concentrator-5 (ZYMO Research) according to the manual. DNA libraries were constructed following standard protocol and submitted to the Center for Cancer Research Sequencing Facility (Frederick).
Chromatin immunoprecipitation (ChIP) assay and ChIP qPCR
Twenty-five million CCRF-CEM parental cells were treated or untreated with CPT (250 nM or 1000 nM) for 4 hours. ChIP assay was done by following the instruction manual of ChIP-IT Express (Active Motif). Briefly, cells were fixed with 1% formaldehyde in medium for 10 min at room temperature. Fixation was stopped with x1 glycine/PBS. Cells were lysed with lysis buffer with proteinase inhibitor cocktail and PMSF and homogenized 30 times by small tight homogenizer. Cell pellets were re-suspended with sharing buffer, and sonicated by the following settings: DNA 200 bp-microtube-130 μL (Covaris M220 Focused-ultrasonicator). Twenty %/volume of each sample was saved as input. The left of the supernatant was incubated with 2 μL antibody (H3K27ac or normal rabbit IgG) and protein G beads for overnight at 4°C. After reversing cross-link and proteinase K treatment, immunoprecipitated DNA was purified with NucleoSpin Gel and PCR Clean-up (MACHEREY-NAGEL) according to the manual. Real-time qPCR was carried out with ssoAdvanced Universal SYBR Green supermix (Bio-Rad) by Step One Plus (AB applied biosystem) according to the manufacture’s instruction. Relative enrichment of ChIP DNA was calculated from the relative amount of input samples obtained with the standard curves of each primer set.
Serum induction
DU145 parental and SLFN11-KO cells were seeded on 6-well plates with ~50% confluency. On the next day, medium was replaced with serum-free medium and cells were incubated for 40 hours. Then, the serum-free medium was replaced with 10% FBS containing medium. Cells were collected before serum induction, and 0.5 h, 1h and 2h after the serum induction. The samples without serum induction but after serum starvation were used as a control to normalize the data.
Epithelial growth factor (EGF) stimulation
Recombinant human EGF was solved with 0.1% BSA/H2O to make 50 mg/ml stock (x2000). DU145 parental and SLFN11-KO cells were seeded on 6-well plates with ~70% confluency one day before the treatment. EGF was added or not added with the final concentration 25 ng/ml and cells were incubated for 10 min, followed by immediate wash with regular medium (RPMI1640+10% FBS) two times. Cell were kept with regular medium for 0h, 0.2h, 0.5h, 1h and 2 hours, and then collected the cells. The non-treated samples were used as a control to normalize the data.
siRNA transfection
siRNA pool targeting DHX9 (final concentration 10 nM) or non-targeting control was transfected to DU145 parental cells using Lipofectamin RNAiMax (Invitrogen) according to the manufacture protocol. Two day after the transfection, cells were used for experiments.
Bioinformatics analysis
Alignment of next-generation sequencing data
The obtained ATAC-seq and RNA-seq fastq files were trimmed using trimmomatic (version 0.39) (Bolger et al., 2014) and trimGalore (version 0.6.2) to remove adaptor sequences and reads with low qualities. ATAC-seq data was aligned using the default settings of the bwa mem aligner (version 0.7.17) (Li and Durbin, 2009) using the hg19 genome version as reference. RNA-seq reads were mapped to the hg19 reference genome with the STAR aligner (version 2.5.4a) (Dobin et al., 2013) using the gencode (v19) gene anno-tation using the two-pass mode and “best recall at base and read level” as shown in Table S37 from (Baruzzo et al., 2017). Aligned reads were sorted and indexed using samtools (version 1.8) (Li et al., 2009). Duplicated reads were marked using picard-tools (version 2.9.2) MarkDuplicates function.
Expression quantification from RNA-seq data
Raw read counts for each gene and condition were calculated with TPMcalculator (version 0.0.1) (Vera Alvarez et al., 2019). Genes with mean counts less than 5 were removed from the analysis. Read count normalization was performed with RUVseq (Risso et al., 2014), followed by differential expression analysis with the DESeq2 (version 1.24.0) (Love et al., 2014) pipeline.
ATAC-seq peak calling
ATAC-seq peaks were identified using the MACS (version 2.1.2) (Zhang et al., 2008) peak caller. Peaks were identified using the –nomodel, as well as –shift 37–extsize 73 parameters in BAMPE mode with false discovery rate (FDR) set to < 0.01. Peaks identified in different conditions were combined using bedtools (version 2.27.1) (Quinlan and Hall, 2010) to generate a uniform peak set. Identified peaks and the combined peaks were annotated using the homer (version 4.10.1) (Heinz et al., 2010) package using the hg19 genome annotation. Peak strength quantification and normalized coverage track creation (for visualization) were performed using the BAMscale (version 0.1) (Pongor et al., 2019) program. Coverage tracks and peaks were visualized using the IGV browser (Robinson et al., 2011).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were carried out using GraphPad prism 7 or 8 software. Test methods are described in figure legends or in this STAR Methods. p < 1E-05 and p < 1E-07 are considered significant in microarray analyses and RNA-seq analyses, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-SLFN11 (D-2) (special request, 200 μg/100 μl) | Santa Cruz | sc-515071X |
| Rabbit polyclonal anti-Histone H3 | Millipore | 07-690; RRID:AB_417398 |
| Mouse monoclonal anti-p53 (DO-7) | Santa Cruz | sc-47698; RRID:AB_628083 |
| Mouse monoclonal anti-p-p53 (D-9) | Santa Cruz | sc-377567 |
| Rabbit polyclonal anti-Histone H3K27Ac | Abcam | Ab4729; RRID:AB_2118291 |
| Mouse monoclonal anti-GAPDH (D4C6R) | Cell Signaling Technology | 97166; RRID:AB_2756824 |
| Rabbit monoclonal anti-p-MEK1/2 (Ser217/221) (41G9) | Cell Signaling Technology | 9154; RRID:AB_2138017 |
| Rabbit monoclonal anti-p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP | Cell Signaling Technology | 4370; RRID:AB_2315112 |
| Rabbit monoclonal anti-p-p90RSK (Ser380) (D3H11) | Cell Signaling Technology | 11989; RRID:AB_2687613 |
| Rabbit monoclonal anti-pan-actin (D18C11) (HRP conjugate) | Cell Signaling Technology | 12748; RRID:AB_2798015 |
| Rabbit polyclonal anti-DHX9 | Bethyl Laboratories | A300-854A; RRID:AB_609441 |
| BrdU monoclonal antibody (MoBU-1), Alexa Fluor 488 | Invitrogen | B35130; RRID:AB_2536434 |
| Alexa Fluor 488 goat anti-mouse IgG | ThermoFisher | A-21121; RRID:AB_2535764 |
| ECL anti-mouse IgG, horseradish peroxidase linked whole antibody (from sheep) | GE Healthcare | NA931V |
| ECL anti-rabbit IgG, horseradish peroxidase linked whole antibody (from donkey) | GE Healthcare | NA934V |
| Normal rabbit IgG | Santa Cruz | sc-2027; RRID:AB_737197 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Camptothecin (CPT) | Developmental Therapeutics Program (NCI/NIH) | NSC#94600 |
| LY2606368 (CHK1 inhibitor) | Developmental Therapeutics Program (NCI/NIH) | NSC#758257 |
| cOmplete Mini, EDTA-free (protease inhibitor cocktail) | Roche | 11836170001 |
| EGF recombinant Human Protein (100 μg) | ThermoFisher | PHG0311 |
| SMARTpool: ON-TARGETplus DHX9 siRNA | Horizon/Dharmacon | L-009950-00-0005 |
| siRNA targeting non-targeting pool | Horizon/Dharmacon | D-001810-10-05 |
| Critical Commercial Assays | ||
| ATPlite 1step luminescence assay system | PerkinElmer | 6016739 |
| Nextera DNA Library Preparation Kit | Illumina | 15028212 |
| ChIP-IT Express | Active Motif | 53008 |
| ChIP DNA Clean & Concentrator | ZYMO RESEARCH | D5205 |
| FastStart Universal SYBR Green Master (Rox) | Roche | 4913850001 |
| SsoAdvanced Universal SYBR Green supermix | Bio-Rad | 1725271B02 |
| TRIzol RNA Isolation Reagents | ThermoFisher | 15596018 |
| Monarch total RNA miniprep kit | NEB | T2010S |
| RNAqueous Total RNA Isolation Kit | ThermoFisher | AM1912 |
| Lipofectamin RNAiMAX | ThermoFisher | 13778150 |
| Click-iT Nascent RNA Capture Kit, for gene expression analysis | ThermoFisher | C10365 |
| RNaseOUT Recombinant Ribonuclease Inhibitor | ThermoFisher | 10777-019 |
| DynaMagTM-2 Magnet | ThermoFisher | 12321D |
| UltraPureTM Glycogen | ThermoFisher | 10814-010 |
| Ammonium Acetate (5 M), RNase-free | ThermoFisher | AM9070G |
| SuperScript VILO cDNA Synthesis Kit | ThermoFisher | 11754050 |
| iScript RT supermix for RT-qPCR | Bio-Rad | 1708841 |
| Human Gene 2.0 ST Array | ThermoFisher | 902112 |
| Deposited Data | ||
| RNA-seq data | This paper | Tables S1 and S2 |
| GSE140938 | ||
| ATAC-seq data for K562 cell lines | (Murai et al., 2018) | GSE140938 |
| ATAC-seq data for CCRF-CEM cell lines | This paper | GSE140938 |
| ChIP-seq data of H3K27ac | This paper | GSE140938 |
| Microarray data of CCRF-CEM and DU145 | This paper | Tables S3 and S4 |
| GSE140175 | ||
| Microarray data of NCI-60 cell lines | (Monks et al., 2018) | GSE116436 |
| All the unprocessed and uncompressed imaging data (microscopy and blots) | This paper | Mendeley Data at: https://doi.org/10.17632/x4vc33kjrv.1 |
| Experimental Models: Cell Lines | ||
| Human: prostate cancer DU145 cells | Developmental Therapeutics Program (NCI/NIH) | N/A |
| Human: leukemia CCRF-CEM cells | Developmental Therapeutics Program (NCI/NIH) | N/A |
| Human: leukemia K562 cells | Developmental Therapeutics Program (NCI/NIH) | N/A |
| DU145 SLFN11 knockout cells | (Murai et al., 2016) | N/A |
| CCRF-CEM SLFN11 knockout cells | (Murai et al., 2016) | N/A |
| K562 vector overexpressing cells | (Murai et al., 2016) | N/A |
| K562 wild-type SLFN11 overexpressing cells | (Murai et al., 2016) | N/A |
| K562 E669Q SLFN11 overexpressing cells | (Murai et al., 2018) | N/A |
| Oligonucleotides | ||
| Primers for reverse transcription qPCR | Table S5 | |
| Primers for ChIP qPCR | Table S6 | |
| Software and Algorithms | ||
| GraphPad Prism 7 (software for drawing graphs and statistics analysis) | GraphPad | N/A |
| ZEN (software for microscopy image analysis) | ZEISS | N/A |
| Integrated Genome Viewer | Broad Institute | http://www.broadinsititeu.org/software/igv/ |
| Partek Genomics Suite (software for analysis of microarray data) | Partek | https://www.partek.com/partek-genomics-suite/ |
| Trimmomatic (v0.39) | (Bolger et al., 2014) | http://www.usadellab.org/cms/?page=trimmomatic |
| trimGalore (v0.6.2) | Babraham Bioinformatics | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Bwa (v0.7.17) | (Li and Durbin, 2009) | https://github.com/lh3/bwa |
| STAR aligner (v2.5.4a) | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| RUVseq (v3.10) | (Risso et al., 2014) | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| Samtools (v1.8) | (Li et al., 2009) | https://github.com/samtools/samtools |
| Picard-tools (v2.9.2) | Broad Institute | https://broadinstitute.github.io/picard/ |
| MACS (v2.1.2) | (Zhang et al., 2008) | https://github.com/taoliu/MACS |
| BAMscale (v0.1) | (Pongor et al., 2019) | https://github.com/ncbi/BAMscale |
| Bedtools (v2.27.1) | (Quinlan and Hall, 2010) | https://bedtools.readthedocs.io/en/latest/ |
| Homer (v4.10.1) | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/index.html |
| TPMcalculator (v0.0.1) | (Vera Alvarez et al., 2019) | https://github.com/ncbi/TPMCalculator |
| DESeq2 (v1.24.0) | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| IGV (v2.4.16) | (Robinson et al., 2011) | https://software.broadinstitute.org/software/igv/ |
| All the unprocessed and uncompressed imaging data (microscopy and blots) | This paper | Mendeley Data at: https://doi.org/10.17632/x4vc33kjrv.1 |
Highlights.
Upon replication stress, SLFN11 induces genome-wide accessibility at promoters
SLFN11 selectively activates the transcription of the immediate early genes (IEGs)
The putative ATPase and helicase activity is required for the functions of SLFN11
SLFN11 activates the innate stress response upon replication stress
ACKNOWLEDGMENTS
We are grateful to Dr. James Doroshow and the NCI Developmental Therapeutics Program group (DTP-DCTD) for providing the dataset of NCI-60 and drug responses, the CCR sequencing facility for sequencing and analyzing ATAC-seq and ChIP-seq data, and the junior technical assistants (Mr. Kennosuke Sato, Tomohiro Watanabe, and Yui Sato) from Tsuruoka Chuo Senior High School. We also thank the DTP-DCTD, NCI/NIH, for providing the drugs and cell lines used in this study. The study used the high-performance computer capabilities of the Biowulf HPC cluster at the NIH. This project was supported by the Intramural Program, Center for Cancer Research of the National Cancer Institute, NIH (Z01 BC 006161 and 006150) (to Y.P.) and AMED (Japan Agency for Medical Research and Development) Project for Cancer Research and Therapeutic Evolution (to J.M.).
Footnotes
DATA AND CODE AVAILABILITY
All RNA-seq, ATAC-seq and ChIP-seq data in this study is accessible at GSE140938. Microarray data of CCRF-CEM and DU145 are accessible at GSE140175. All the unprocessed and uncompressed imaging data are accessible at https://doi.org/10.17632/x4vc33kjrv.1.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.02.117.
REFERENCES
- Abdel-Mohsen M, Raposo RA, Deng X, Li M, Liegler T, Sinclair E, Salama MS, Ghanem Hel.-D., Hoh R, Wong JK, et al. (2013). Expression profile of host restriction factors in HIV-1 elite controllers. Retrovirology 10, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison Stewart C, Tong P, Cardnell RJ, Sen T, Li L, Gay CM, Masrorpour F, Fan Y, Bara RO, Feng Y, et al. (2017). Dynamic variations in epithelial-to-mesenchymal transition (EMT), ATM, and SLFN11 govern response to PARP inhibitors and cisplatin in small cell lung cancer. Oncotarget 8, 28575–28587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arner E, Daub CO, Vitting-Seerup K, Andersson R, Lilje B, Drabløs F, Lennartsson A, Rönnerblad M, Hrydziuszko O, Vitezic M, et al. ; FANTOM Consortium (2015). Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahrami S, and Drabløs F. (2016). Gene regulation in the immediate-early response process. Adv. Biol. Regul 62, 37–49. [DOI] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baruzzo G, Hayer KE, Kim EJ, Di Camillo B, FitzGerald GA, and Grant GR (2017). Simulation-based comprehensive benchmarking of RNA-seq aligners. Nat. Methods 14, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro JD, Giresi PG, Zaba LC, Chang HY, and Greenleaf WJ (2013). Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 10, 1213–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussy F, EI-Botty R, Chateau-Joubert S, Dahmani A, Montaudon E, Leboucher S, Morisset L, Painsec P, Sourd L, Huguet L, et al. (2020). BRCAness, SLFN11, and RB1 loss predict response to topoisomerase I inhibitors in triple-negative breast cancers. Science Translational Medicine 12 10.1126/scitranslmed.aax2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Cai Y, Huang Y, Yang Z, Bai Y, Liu Y, Deng X, and Wang J. (2015). High SLFN11 expression predicts better survival for patients with KRAS exon 2 wild type colorectal cancer after treated with adjuvant oxaliplatin-based treatment. BMC Cancer 15, 833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. (2017). Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 31, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. (2012). Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KL, and Gordon DJ (2016). Gene expression signature based screening identifies ribonucleotide reductase as a candidate therapeutic target in Ewing sarcoma. Oncotarget 7, 63003–63019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess J, Angel P, and Schorpp-Kistner M. (2004). AP-1 subunits: quarrel and harmony among siblings. J. Cell Sci 117, 5965–5973. [DOI] [PubMed] [Google Scholar]
- Kang MH, Wang J, Makena MR, Lee JS, Paz N, Hall CP, Song MM, Calderon RI, Cruz RE, Hindle A, et al. (2015). Activity of MM-398, nanoliposomal irinotecan (nal-IRI), in Ewing’s family tumor xenografts is associated with high exposure of tumor to drug and high SLFN11 expression. Clin. Cancer Res 21, 1139–1150. [DOI] [PubMed] [Google Scholar]
- Karimian A, Ahmadi Y, and Yousefi B. (2016). Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair (Amst.) 42, 63–71. [DOI] [PubMed] [Google Scholar]
- Kiselinova M, De Spiegelaere W, Buzon MJ, Malatinkova E, Lichterfeld M, and Vandekerckhove L. (2016). Integrated and Total HIV-1 DNA Predict Ex Vivo Viral Outgrowth. PLoS Pathog. 12, e1005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, and Pelletier J. (2016). The biology of DHX9 and its potential as a therapeutic target. Oncotarget 7, 42716–42739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, and Durbin R. (2009). Fast and accurate short read alignment with Bur-rows-Wheeler transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kao E, Gao X, Sandig H, Limmer K, Pavon-Eternod M, Jones TE, Landry S, Pan T, Weitzman MD, and David M. (2012). Codon-us-age-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Kao E, Malone D, Gao X, Wang JYJ, and David M. (2018). DNA damage-induced cell death relies on SLFN11-dependent cleavage of distinct type II tRNAs. Nat. Struct. Mol. Biol 25, 1047–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. (2017). PARP Inhibitor Activity Correlates with SLFN11 Expression and Demonstrates Synergy with Temozolomide in Small Cell Lung Cancer. Clin. Cancer Res 23, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maréchal A, and Zou L. (2013). DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol 5, a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mavrommatis E, Fish EN, and Platanias LC (2013). The schlafen family of proteins and their regulation by interferons. J. Interferon Cytokine Res 33, 206–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzadra R, de Bruijn M, Jae LT, Gomez-Eerland R, Duursma A, Scheeren FA, Brummelkamp TR, and Schumacher TN (2019). SLFN11 can sensitize tumor cells towards IFN-γ-mediated T cell killing. PLoS One 14, e0212053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monks A, Zhao Y, Hose C, Hamed H, Krushkal J, Fang J, Sonkin D, Palmisano A, Polley EC, Fogli LK, et al. (2018). The NCI Transcriptional Pharmacodynamics Workbench: A Tool to Examine Dynamic Expression Profiling of Therapeutic Response in the NCI-60 Cell Line Panel. Cancer Res. 78, 6807–6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu Y, Lou J, Srivastava M, Zhao B, Feng XH, Liu T, Chen J, and Huang J. (2016). SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep. 17, 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Feng Y, Yu GK, Ru Y, Tang SW, Shen Y, and Pommier Y. (2016). Resistance to PARP inhibitors by SLFN11 inactivation can be over-come by ATR inhibition. Oncotarget 7, 76534–76550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Tang SW, Leo E, Baechler SA, Redon CE, Zhang H, Al Abo M, Rajapakse VN, Nakamura E, Jenkins LMM, et al. (2018). SLFN11 Blocks Stressed Replication Forks Independently of ATR. Mol. Cell 69, 371–384.e376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murai J, Thomas A, Miettinen M, and Pommier Y. (2019). Schlafen 11 (SLFN11), a restriction factor for replicative stress induced by DNA-targeting anti-cancer therapies. Pharmacol. Ther 201, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales V, Reinhold WC, Varma S, Martinez-Cardus A, Moutinho C, Moran S, Heyn H, Sebio A, Barnadas A, Pommier Y, and Esteller M. (2016). Epigenetic inactivation of the putative DNA/RNA helicase SLFN11 in human cancer confers resistance to platinum drugs. Oncotarget 7, 3084–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell A, Odrowaz Z, and Sharrocks AD (2012). Immediate-early gene activation by the MAPK pathways: what do and don’t we know? Biochem. Soc. Trans 40, 58–66. [DOI] [PubMed] [Google Scholar]
- Paull KD, Shoemaker RH, Hodes L, Monks A, Scudiero DA, Rubinstein L, Plowman J, and Boyd MR (1989). Display and analysis of patterns of differential activity of drugs against human tumor cell lines: development of mean graph and COMPARE algorithm. J. Natl. Cancer Inst 81, 1088–1092. [DOI] [PubMed] [Google Scholar]
- Pietanza MC, Waqar SN, Krug LM, Dowlati A, Hann CL, Chiappori A, Owonikoko TK, Woo KM, Cardnell RJ, Fujimoto J, et al. (2018). Randomized, Double-Blind, Phase II Study of Temozolomide in Combination With Either Veliparib or Placebo in Patients With Relapsed-Sensitive or Refractory Small-Cell Lung Cancer. J. Clin. Oncol 36, 2386–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pongor LS, Gross JM, Alvarez RV, Murai J, Jang S-M, Zhang H, Redon C, Fu H, Huang S-Y, Thakur B, et al. (2019). BAMscale: quantification of DNA sequencing peaks and generation of scaled coverage tracks. bioRxiv. 10.1101/669275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse VN, Luna A, Yamade M, Loman L, Varma S, Sunshine M, Iorio F, Sousa FG, Elloumi F, Aladjem MI, et al. (2018). CellMinerCDB for Integrative Cross-Database Genomics and Pharmacogenomics Analyses of Cancer Cell Lines. iScience 10, 247–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Sunshine M, Varma S, Doroshow JH, and Pommier Y. (2015). Using CellMiner 1.6 for Systems Pharmacology and Genomic Analysis of the NCI-60. Clin. Cancer Res 21, 3841–3852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Thomas A, and Pommier Y. (2017a). DNA-Targeted Precision Medicine; Have we Been Caught Sleeping? Trends Cancer 3, 2–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhold WC, Varma S, Sunshine M, Rajapakse V, Luna A, Kohn KW, Stevenson H, Wang Y, Heyn H, Nogales V, et al. (2017b). The NCI-60 Methylome and Its Integration into CellMiner. Cancer Res. 77, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S. (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat. Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JC, Cortez D, and Cimprich KA (2017). The essential kinase ATR: ensuring faithful duplication of a challenging genome. Nat. Rev. Mol. Cell Biol 18, 622–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvador JM, Brown-Clay JD, and Fornace AJ Jr. (2013). Gadd45 in stress signaling, cell cycle control, and apoptosis. Adv. Exp. Med. Biol 793, 1–19. [DOI] [PubMed] [Google Scholar]
- Sanz LA, and Chedin F. (2019). High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat. Protoc 14, 1734–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz LA, Hartono SR, Lim YW, Steyaert S, Rajpurkar A, Ginno PA, Xu X, and Chédin F. (2016). Prevalent, Dynamic, and Conserved R-Loop Structures Associate with Specific Epigenomic Signatures in Mammals. Mol. Cell 63, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherf U, Ross DT, Waltham M, Smith LH, Lee JK, Tanabe L, Kohn KW, Reinhold WC, Myers TG, Andrews DT, et al. (2000). A gene expression database for the molecular pharmacology of cancer. Nat. Genet 24, 236–244. [DOI] [PubMed] [Google Scholar]
- Shee K, Wells JD, Jiang A, and Miller TW (2019). Integrated pan-cancer gene expression and drug sensitivity analysis reveals SLFN11 mRNA as a solid tumor biomarker predictive of sensitivity to DNA-damaging chemotherapy. PLoS One 14, e0224267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa FG, Matuo R, Tang SW, Rajapakse VN, Luna A, Sander C, Varma S, Simon PH, Doroshow JH, Reinhold WC, and Pommier Y. (2015). Alterations of DNA repair genes in the NCI-60 cell lines and their predictive value for anticancer drug activity. DNA Repair (Amst.) 28, 107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SW, Thomas A, Murai J, Trepel JB, Bates SE, Rajapakse VN, and Pommier Y. (2018). Overcoming Resistance to DNA-Targeted Agents by Epigenetic Activation of Schlafen 11 (SLFN11) Expression with Class I Histone Deacetylase Inhibitors. Clin. Cancer Res 24, 1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Song S, Liu X, Wang Y, Xu X, Hu Y, and Xu J. (2014). Schlafen-11 sensitizes colorectal carcinoma cells to irinotecan. Anticancer Drugs 25, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, and Lukas J. (2013). ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155, 1088–1103. [DOI] [PubMed] [Google Scholar]
- Tsompana M, and Buck MJ (2014). Chromatin accessibility: a window into the genome. Epigenetics Chromatin 7, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera Alvarez R, Pongor LS, Mariño-Ramírez L, and Landsman D. (2019). TPMCalculator: one-step software to quantify mRNA abundance of genomic features. Bioinformatics 35, 1960–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JY, Deng XY, Li YS, Ma XC, Feng JX, Yu B, Chen Y, Luo YL, Wang X, Chen ML, et al. (2018). Structure of Schlafen13 reveals a new class of tRNA/rRNA-targeting RNase engaged in translational control. Nat. Commun 9, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoppoli G, Regairaz M, Leo E, Reinhold WC, Varma S, Ballestrero A, Doroshow JH, and Pommier Y. (2012). Putative DNA/RNA helicase Schla-fen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc. Natl. Acad. Sci. USA 109, 15030–15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.