

Abstract
Cancer immunotherapy is fast becoming one of the most promising means of treating malignant disease. Cancer vaccines, adoptive cell transfer therapies, and immune checkpoint blockade have all shown varying levels of success in the clinical management of several cancer types in recent years. However, despite the clinical benefits often achieved by these regimens, an ongoing problem for many patients is the inherent or acquired resistance of their cancer to immunotherapy. It is now appreciated that dendritic cells and T lymphocytes both play key roles in antitumor immune responses and that the tumor microenvironment presents a number of barriers to the function of these cells that can ultimately limit the success of immunotherapy. In particular, the engagement of several immunologic and metabolic checkpoints within the hostile tumor microenvironment can severely compromise the antitumor functions of these important immune populations. This review highlights work from both preclinical and clinical studies that has shaped our understanding of the tumor microenvironment and its influence on dendritic cell and T cell function. It focuses on clinically relevant targeted and immunotherapeutic strategies that have emerged from these studies in an effort to prevent or overcome immune subversion within the tumor microenvironment. Emphasis is also placed on the potential of next‐generation combinatorial regimens that target metabolic and immunologic impediments to dendritic cell and T lymphocyte function as strategies to improve antitumor immune reactivity and the clinical outcome of cancer immunotherapy going forward.
Keywords: adoptive cell transfer, cancer, checkpoint blockade, dendritic cell, immune suppression, metabolism, T cell, tumor microenvironment, vaccine
1. BACKGROUND
Over the past decade, the emergence of immunotherapy as a viable and promising treatment option for both solid and blood‐based malignancies has revolutionized the therapeutic landscape of cancer, resulting in unprecedented achievements in the clinical management of this disease. During this short period, significant improvements over traditional forms of cancer therapy have been realized in terms of both the rate and duration of clinical responses, and immunotherapeutic regimens have now become standard‐of‐care treatment for many cancer types. 1 , 2 However, as has often been the case with traditional approaches to cancer treatment, many patients who initially respond to immune‐based therapies eventually experience disease relapse, and still there are others who fail to respond to immunotherapy at all. In light of these disparate clinical outcomes, significant efforts have been made to better understand factors that influence the quality of antitumor immune responses and the clinical efficacy of regimens that rely on their induction and maintenance. In this regard, several forms of innate and adaptive immune resistance have now been described and are linked to tumor cell‐intrinsic and extrinsic factors that ultimately interfere with the complex cross‐talk between cancer cells and the diverse immune/non‐immune cell populations that participate in and regulate antitumor immune responses. Central to many of these resistance mechanisms is the role played by the hostile tumor microenvironment (TME) in preventing or dampening antitumor immunity. In particular, dendritic cells (DC) and T lymphocytes, two immune cell populations critical to the efficacy of antitumor immune responses, are subject to regulation by various immunologic and metabolic checkpoints within the TME. This review highlights our current understanding of these checkpoints and the limitations they place on the immune functions of DC and T cells, and it describes combinatorial strategies for overcoming these barriers to immune function that have the potential to enhance the quality and longevity of anticancer immune responses and to improve the clinical outcome of cancer immunotherapy going forward.
2. DC/T CELL COLLABORATION IN ANTITUMOR IMMUNITY
The contribution of T lymphocytes to immune protection from cancer has long been appreciated. At the turn of the century, seminal studies in murine models first revealed the importance of these cells and effector molecules derived from them in the immunologic control of various tumors. 3 , 4 Prior to and during this time, evidence supporting T cell reactivity against human cancers also emerged from clinical observations of spontaneous tumor regression in patients experiencing T cell‐driven autoimmune disease in the same tissue from which their cancer was derived (ie, vitiligo in melanoma patients) as well as in some patients exhibiting antigen (Ag)‐specific T cell infiltration of their tumors. 5 , 6 While the role of DC in regulating T cell tolerance and immunity to pathogens has also long been appreciated, 7 the ways in which these cells influence antitumor immune responses have only more recently become apparent. Following significant efforts to better understand DC function in the context of cancer, these cells are now known to contribute to various stages of the antitumor T cell response, from induction of an Ag‐specific response in secondary lymphoid organs to recruitment of effector cells into tumor tissue to maintenance/restimulation of cytotoxic T lymphocytes (CTL) within the TME. 8
In most cases, successful priming of antitumor T cell responses requires migration of tumor‐infiltrating DC (TIDC) to regional lymph nodes, where they are able to cross‐present Ag from phagocytosed tumor cells to naïve CD8+ T lymphocytes. Type 1 conventional DC (cDC1), marked by expression of CD103 in mice and CD141 in humans, are particularly relevant to this cross‐priming, 9 , 10 which depends not only on DC access to tumor tissue for Ag acquisition but also appropriate maturation and activation of these cells into potent immunostimulatory Ag‐presenting cells (APC). In addition to priming CD8+ T cell responses within tumor‐draining lymph nodes, the cDC1 subset of DC also regulates effector CTL trafficking to tumors, as those DC that remain within tumor tissue can secrete CXCL9/CXCL10 chemokines to attract CXCR3+ effector CTL, 11 , 12 a process relevant to both endogenous antitumor T cell responses as well as therapeutic regimens that rely on administration of exogenously activated CTL. Finally, cDC1 as well as other DC subsets have the potential to produce high levels of IL‐12 and type I IFN, 13 , 14 , 15 immunostimulatory cytokines that not only promote DC‐mediated cross‐priming of antitumor T cell responses but that also likely help to maintain CTL effector function within the TME when produced by intratumoral DC. While this complex interplay between DC and T lymphocytes over the course of an antitumor immune response therefore enables the initiation, direction, and maintenance of a T cell response to cancer, it also provides multiple opportunities for tumors to circumvent immune‐mediated destruction. Indeed, several immunologic and metabolic checkpoints that restrict the functions of DC and T lymphocytes within the TME have recently been discovered and found to limit the efficacy of antitumor immune responses. Despite the significance of these barriers to successful antitumor immune reactivity, though, advances in our understanding of these immune‐disrupting pathways and the mechanisms underlying their immunoregulatory functions are now paving the way for therapeutic strategies that aim to: (a) restore proper communication between DC and T lymphocytes in the context of cancer and (b) promote the robust antitumor activities capable of being mediated by these cells.
3. INNATE IMMUNE CHECKPOINTS INFLUENCING DC FUNCTION IN THE TUMOR MICROENVIRONMENT
3.1. CD47 and SIRPα
CD47 is a cell surface glycoprotein expressed on nearly all cell types that serves as a marker of self to the innate immune system. Though first recognized for its role in inhibiting host cell phagocytosis by macrophages, 16 CD47 has since emerged as a prominent “don't eat me” signal to various phagocytic cell populations that express the signal regulatory protein‐α (SIRPα) receptor. 17 , 18 , 19 Importantly, CD47 expression is upregulated on tumor cells of several cancer types 20 and has been shown to promote evasion of phagocytosis by both macrophages and DC. 21 , 22 Based on these findings, CD47 has become an attractive target for cancer therapy, and several studies have now confirmed the antitumor efficacy of its blockade, 23 , 24 , 25 , 26 which not only promotes tumor cell clearance by macrophages and DC but also supports therapeutic regimens linked to adaptive antitumor immunity. 27 Indeed, the therapeutic efficacy of CD47 blockade has been shown to be T cell‐dependent, 28 and DC in particular are critical to the enhanced antitumor T cell response resulting from this therapy. Whereas both macrophages and DC benefit from CD47 blockade in terms of phagocytic uptake of tumor cells, Xu et al recently found that CD47 blockade also selectively enhances innate immune sensing of tumor mitochondrial DNA (mtDNA) in DC by activating NOX2 and limiting the phagosomal acidification that otherwise degrades this DNA within macrophages. The increased stability of phagocytosed tumor mtDNA within DC following CD47 blockade enables its subsequent release into the cytosol and triggers activation of the cGAS‐STING pathway, which in turn promotes type I IFN production necessary for efficient cross‐priming of antitumor T cells. 29 Taken together, these findings highlight the significance of the CD47‐SIRPα signaling axis to tumor immune evasion, as this pathway not only limits innate immune clearance of tumor cells but also acts as a barrier to DC‐driven adaptive immunity to cancer as well.
3.2. The leukocyte immunoglobulin‐like receptor B family of phagocytosis inhibitors
Though the CD47‐SIRPα axis is the most extensively studied phagocytosis checkpoint to date, additional pathways influencing this process have also been uncovered in the last decade. While these pathways have primarily been studied in the context of macrophages, given the shared expression of certain phagocytic receptors between these cells and DC, it is likely that tumoral influences on these signaling systems contribute to alterations in DC function as well. One such pathway that has been shown to impair macrophage‐mediated phagocytosis of tumor cells involves MHC class I signaling through the leukocyte immunoglobulin‐like receptor family member leukocyte immunoglobulin‐like receptor B1 (LILRB1). Specifically, inhibition of phagocytosis is driven by interaction between LILRB1 on macrophages and the MHC class I‐associated β2M subunit expressed by tumor cells, 30 highlighting the potential universality of this innate checkpoint as a means for tumor immune evasion in cancer patients regardless of their HLA haplotype. Although tumor cell loss of MHC class I expression is a well‐described mechanism of immune escape, the selective pressure for such downregulation is applied only in the face of an effective CTL response, and tumor cell maintenance of MHC class I and its subversion of innate immune recognition and phagocytosis may actually explain the poor immunogenicity of many cancers. In this regard, LILRB1 expression is not restricted to macrophages – it is also expressed on DC, and its engagement on these cells is therefore likely to interfere with tumor uptake and immune stimulation by this innate population as well. Indeed, work in nontumor models has shown that LILRB1 signaling in DC inhibits Ca++ flux shortly after stimulation and impairs IL‐12 production and T cell activation by these cells. 31 , 32 Though the mechanism for LILRB1‐mediated inhibition of DC function has not been thoroughly investigated in fully differentiated DC, it is interesting that engagement of LILRB1 during DC differentiation from monocytic precursors led to retention of NF‐κB in the cytosol via an ABIN1/TINP1‐dependent mechanism. This interference with NF‐κB nuclear translocation led to impaired phagocytosis, decreased expression of MHC class I and II molecules, and reduced secretion of IL‐12 and IFN‐α by the resulting DC, which were poor stimulators of T cells. 33 As tumors are frequently infiltrated by myeloid precursor populations, this pathway may be particularly relevant to the development of poorly immunogenic DC within the TME. With evidence accumulating that other members of the LILRB family also act as negative regulators of DC function, 34 it will be important going forward to investigate how each of these family members impacts DC activity in the context of cancer, as these receptors may represent multiple targets for therapeutic interventions aiming to prevent tumor subversion of DC‐mediated immunity.
3.3. The CD24/Siglec‐10/G axis and other sialoglycan/siglec receptor interactions
The linkage of sialic acids to glycoproteins/glycolipids on the surface of mammalian cells is a distinguishing feature of the host that is often used by innate immune cells to differentiate self from non‐self. As such, the presence of sialoglycans on self cells can inhibit the activation of innate immune cells when recognized by members of the sialic acid‐binding immunoglobulin‐like lectin (Siglec) family of receptors. 35 In this regard, the most recently discovered “don't eat me” signal found to confer tumor cell resistance to phagocytosis is CD24, a heavily glycosylated cell surface protein known as heat stable antigen. Weissman and colleagues reported upregulation of CD24 gene expression in nearly all tumor types analyzed from TARGET and TCGA datasets and found that CD24 expression on breast and ovarian cancer cells inhibited phagocytosis by macrophages through engagement of Siglec‐10. 36 Though the impact of tumor cell‐associated CD24 on DC was not investigated in this study, previous work has shown that the CD24/Siglec‐10/G axis limits DC responsiveness to damage‐associated molecular patterns (DAMPs) including HMGB1, HSP70, and HSP90. 37 Similar to the effect of LILRB1 signaling in DC described above, the negative regulation of DC responsiveness to DAMPs by CD24/Siglec‐10/G signaling was associated with cytoplasmic retention of NF‐κB. Based on CD24's inhibition of macrophage function in the context of cancer and the inhibitory signaling mediated via Siglec‐10/G that can occur in DC, it will be important going forward to explore how the CD24/Siglec‐10/G axis might disrupt DC function within the TME, as this pathway might also serve as a significant barrier to the induction and/or maintenance of antitumor T cell responses by DC.
In addition to Siglec‐10/G, DC express a number of other inhibitory Siglecs, all of which share ITIM or ITIM‐like cytoplasmic motifs that drive negative regulatory signaling, typically via the SHP‐1 or SHP‐2 phosphatases. 38 These receptors may also be particularly relevant to DC function within the TME. For instance, Siglec‐9 engagement by a cancer‐specific MUC1 mucin with sialylated O‐linked glycans during DC differentiation from monocytic precursors has been shown to impair costimulatory molecule expression by the resulting DC. 39 Other tumor‐derived gangliosides have also been shown to impair DC differentiation and survival, 40 , 41 , 42 , 43 though particular Siglec receptor/ganglioside interactions were not evaluated in these earlier studies. Based on the diverse pattern of Siglec receptor expression on DC and the production of elevated levels or unique forms of cell‐bound and soluble glycans by cancer cells during tumor progression, 44 a systematic evaluation of specific sialoglycan‐Siglec interactions and how they influence DC function in the TME may ultimately prove useful in the development of novel agents that aim to disrupt negative signaling pathways in DC and in turn enhance the quality of DC‐mediated antitumor immune responses.
3.4. CTLA‐4 and PD‐1: More than immune checkpoints for T lymphocytes
CTLA‐4 and PD‐1, the most‐well‐characterized immune checkpoints to date, have been studied extensively as negative regulators of T lymphocyte activation (see below). Recently, each of these immunoglobulin superfamily members has also been shown to negatively regulate the activity of DC through mechanisms driven by tumor cells as well as other cell populations frequently enriched in the TME. Regulatory T cells (Tregs), for example, can limit costimulatory molecule expression by DC through two distinct CTLA‐4‐mediated mechanisms: (a) transendocytosis and degradation of CD80/CD86 45 , 46 and (b) suppression of DC maturation by reverse signaling through CTLA‐4′s high affinity CD80 ligand. 47 CTLA‐4 is also expressed on multiple tumor cell types, 48 and breast cancer cell‐associated CTLA‐4 was found to suppress DC maturation, most likely through its ability to promote ERK and STAT3 signaling in these cells. 49 Finally, DC themselves can express CTLA‐4 and are subject to both cell‐intrinsic and cell‐extrinsic mechanisms of suppression mediated by this immune checkpoint. Halpert et al recently found that intracellular CTLA‐4 stores were secreted by DC as part of microvesicles that could in turn interact with costimulatory molecules on bystander DC and trigger vesicular uptake, leading to loss of CD80/CD86 expression on the recipient DC. 50 Whether such a mechanism occurs within DC in the TME is currently unknown, but the authors of this study did find that silencing CTLA‐4 in bone marrow‐derived DC (BMDC) prior to electroporation with B16 melanoma mRNA improved the antitumor efficacy of BMDC vaccination against established tumors. While this effect might have been due to the prevention of microvesicle‐mediated endocytosis of costimulatory molecules as described in their in vitro experiments, it is also possible that silencing CTLA‐4 in BMDC prevented intrinsic inhibitory signaling through this receptor, a phenomenon that has been demonstrated in human monocyte‐derived DC that display impaired T cell stimulatory activity and skewed IL‐10high/IL‐12low secretion ratios following CTLA‐4 ligation. 51 , 52 Regardless of the mechanism(s) by which CTLA‐4 regulates DC function, these data highlight this inhibitory receptor as a relevant innate checkpoint in DC that can be targeted to enhance the immunostimulatory activity of these cells in the context of cancer.
Like CTLA‐4, PD‐1 has primarily been viewed as a critical immune checkpoint in T lymphocytes. However, PD‐1 is also expressed by innate immune cell populations and can negatively regulate their function as well. Indeed, the PD‐1/PD‐L1 axis has been shown to function as another phagocytic checkpoint in tumor‐associated macrophages. In another recent study by Weissman's group, it was demonstrated that whereas PD‐1‐deficient macrophages engulf PD‐L1+ and PD‐L1 KO CT26 colorectal cancer cells equally well, PD‐1+ macrophages engulfed PD‐L1 KO tumor cells significantly better that PD‐LI‐expressing tumor cells. 53 How PD‐1 expression on DC might impact tumor cell phagocytosis has not been studied to date, but it is known that PD‐1 compromises the immunostimulatory function of DC. First reported in an L. monocytogenes bacterial infection model, it was found that PD‐1 expression by splenic DC limits production of the proinflammatory cytokines IL‐12 and TNF‐α during ex vivo restimulation, a phenomenon that could be reversed by addition of PD‐L1 blocking Ab to splenic cultures. 54 In the context of cancer, immunosuppressive PD‐1+ DC have been isolated from tumors and ascites of mice challenged with ID8 ovarian cancer cells, and ligation of PD‐1 on these DC suppressed NF‐κB activation, Ag presentation, costimulatory molecule expression, and proinflammatory cytokine secretion. 55 , 56 PD‐1‐expressing DC have also been recovered from tumors in a murine model of liver cancer, and a population of PD‐1+, CD3−, CD11c+ cells also likely to be DC were detected in tumor tissue of hepatocellular carcinoma patients. 57 Though the impact of PD‐1 signaling in these endogenous TIDC was not evaluated in this study, the authors did demonstrate in the murine liver cancer model that intratumoral delivery of PD‐1 KO BMDC suppressed tumor growth more efficiently than PD‐1‐expressing BMDC, and the improved tumor control in PD‐1‐deficient DC recipients correlated with an increased frequency of tumor‐infiltrating CD8+ T cells expressing perforin and granzyme B. Together, these data highlight the immune limiting effect of PD‐1 on DC, and they reveal the PD‐1/PD‐L1 axis as a checkpoint that can be targeted in these cells to improve antitumor immunity.
3.5. T cell Ig and mucin domain containing molecule‐3
T cell Ig and mucin domain containing molecule‐3 (TIM‐3) was first identified as a negative regulator of T lymphocytes nearly two decades ago, 58 but it is now known to be expressed on many immune cell populations and can alter the function of multiple DC subsets. TIM‐3 negatively regulates IFN‐α production by plasmacytoid DC (pDC), potentially by mediating lysosomal degradation of the IRF7 transcription factor. 59 TIM‐3 also promotes inhibitory signaling in splenic DC and BMDC, as agonistic crosslinking anti‐Tim‐3 Ab suppresses IL‐12 secretion by these cells in response to LPS‐stimulation. 60 Importantly, upregulation of TIM‐3 on TIDC has been reported in lung, bladder, and breast cancer patients, 61 , 62 , 63 and multiple mechanisms of DC immunosuppression are mediated by TIM‐3 within the TME. In murine models of breast cancer, TIM‐3 expression on intratumoral cDC1 cells suppressed production of the chemokine CXCL9, a known chemoattractant for CXCR3+ T lymphocytes, and the antitumor efficacy mediated by blocking either TIM‐3 or its ligand galectin‐9 was both CD8+ T cell‐ and CXCR3‐dependent, 63 suggesting a suppressive role for TIM‐3 in TIDC‐mediated recruitment of T cells into tumor tissue. Additionally, Chiba et al reported a galectin‐9‐independent mechanism by which DC‐associated TIM‐3 suppresses antitumor immunity. In their study, interaction between HMGB1 and TIM‐3 on DC interfered with nucleic acid recruitment to endosomal compartments and impaired innate immune sensing of nucleic acids released from dying tumor cells. 61 Based on HMGB1's known role as an alarmin that can induce danger signaling through RAGE and various TLR, it is interesting to speculate that TIM‐3‐mediated sequestration of HMGB1 might also prevent direct immunostimulatory signaling from this molecule in tumor‐associated DC. Though this possibility has yet to be investigated experimentally, it is clear that there are diverse immune regulatory effects mediated by TIM‐3 in tumor‐associated DC, and these pathways, as well as factors that drive TIM‐3 expression by DC, offer multiple points for therapeutic intervention to relieve suppression of these cells within the TME.
3.6. Other inhibitory receptors that negatively regulate tumor‐associated DC
A number of other inhibitory receptors that limit the function of DC are known to exist. Though less is known about the role of these receptors in the context of the TME as compared to the innate checkpoints described above, there is evidence that these other receptors can also be co‐opted by tumors as a means of subverting DC‐mediated immunity. The TAM receptor tyrosine kinases (RTK) are a family of receptors that include Tyro3, Axl, and Mer, all of which function to restrain inflammatory responses following phagocytic clearance of dying cells. 64 These receptors bind two primary ligands complexed to phosphatidylserine on the surface of apoptotic cells, PROS1 and Gas6, both of which are expressed at high levels in many cancers. 65 , 66 Upon engagement on BMDC and monocyte‐derived DC, TAM receptors dampen inflammatory responses by upregulating expression of the SOCS1 and SOCS3 suppressors of cytokine signaling and by inhibiting the PI3K/Akt/NF‐κB pathway, respectively. 67 , 68 Though these specific mechanisms have not yet been demonstrated for TIDC in vivo, a recent study did demonstrate that TAM RTK signaling in DC negatively affects the accumulation of these cells within tumors, and a pan‐TAM RTK inhibitor directly enhanced MHC II expression on intratumoral DC and led to improved immunologic control of established tumors. 69
Another inhibitory receptor, B‐ and T‐lymphocyte attenuator (BTLA), was found to be upregulated on DC from tumor tissue (as compared to those from normal adjacent tissue) of bladder cancer patients, and in vitro studies demonstrated that the BTLA ligand HVEM suppressed proinflammatory cytokine secretion by both cDC1 and pDC obtained from PBMC of healthy donors. 62 Others have shown that the HVEM‐BTLA pathway is also critical for maintaining DC homeostasis, 70 suggesting that BTLA may limit not only the activation but also the expansion of tumor‐associated DC.
Tumor‐infiltrating DC, as well as other myeloid populations, can also express high levels of V‐domain Ig suppressor of T‐cell activation (VISTA), a negative immune regulator that shares some structural features with PD‐L1. Mechanistically, VISTA restrains TLR/MyD88‐mediated signaling in several TIDC subsets and renders these cells tolerogenic. 71 Though a ligand for VISTA has yet to be identified, antibody blockade of this regulatory protein has been shown to enhance the immunostimulatory functions of TIDC and to improve T cell effector function and tumor control in murine models of melanoma and bladder cancer. 72 Immunohistochemial analyses of primary cutaneous melanomas have also shown significant correlation between VISTA expression and myeloid cell infiltrates, with VISTA expression being a poor prognostic indicator of disease‐specific survival. 73 Interfering with VISTA signaling in TIDC may therefore be an effective strategy for improving tumor immunity and clinical outcome in cancer patients as well.
Together, these data highlight the diversity of inhibitory receptors expressed by DC and, in turn, the complexity of DC immunoregulation that ultimately occurs in the TME. As the expression and function of these and other innate checkpoints in DC are more fully elucidated within the context of the TME (Figure 1), new targets for personalized cancer therapies will surely emerge, and combinatorial regimens that neutralize multiple inhibitory pathways in DC are likely to become the most effective means of reversing/preventing tumor‐associated dysfunction in these cells. Such approaches have the potential to significantly improve the efficacy of DC‐mediated antitumor immune responses going forward.
FIGURE 1.
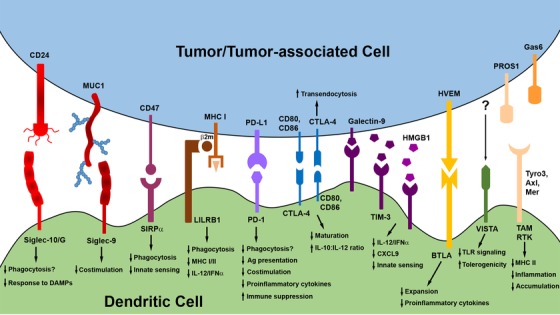
Innate immune checkpoints influencing DC function in the TME. A variety of innate immune checkpoints may limit DC function within the TME when engaged by ligands expressed on or secreted by tumor cells or other tumor‐associated populations, such as Tregs. Signaling through these innate checkpoint receptors compromises several DC functions that are critical to the induction, stimulation, and maintenance of antitumor immune responses. Mechanisms indicated with “?” represent those that have yet to be directly demonstrated in DC but that have been identified as outcomes of checkpoint receptor engagement in other innate populations (ie, macrophages) regulated by the same checkpoint pathway. Many of these checkpoint pathways are now targets for therapeutic interventions that aim to enhance the antitumor immune functions of DC, as described in detail in the main text
4. METABOLIC CHECKPOINTS INFLUENCING DC FUNCTION IN THE TUMOR MICROENVIRONMENT
In addition to engaging the aforementioned innate checkpoints that negatively regulate DC function, both tumor and tumor‐associated cells also release a variety of factors that can either interfere with DC recruitment to tumor tissue or disrupt DC differentiation, maturation, and activation within the TME. While a number of immunosuppressive factors (TGF‐β1, IL‐10, VEGF‐A, etc) frequently enriched in the TME have recently been reviewed elsewhere, 8 , 74 , 75 metabolically suppressive factors within the TME have also emerged as critical regulators of DC function (Figure 2). Though the discovery of these metabolic checkpoints has added increasing complexity to the various mechanisms of DC immunoregulation within the TME, insight into metabolic suppression of DC has also significantly expanded the repertoire of potential therapeutic targets that otherwise limit the immunostimulatory capacity of these cells in the context of cancer.
FIGURE 2.
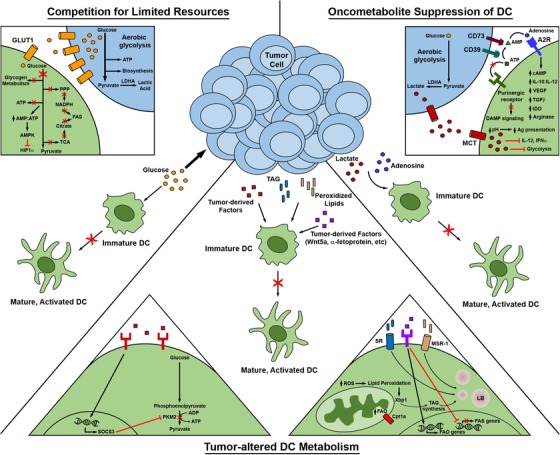
Metabolic regulation of DC function within the TME. DC metabolism, and in turn function, may be compromised within the TME in a number of ways. Tumor cells may outcompete DC for limited resources necessary to support metabolic function, tumor‐derived factors and metabolic products may directly alter metabolism within DC, and immunosuppressive oncometabolites generated by tumor cells may trigger a shift in DC from antitumor to pro‐tumor functionality. Therapeutic strategies that disrupt these mechanisms by which tumors regulate DC metabolism can restore the immunostimulatory and antitumor activity of DC, as described in detail in the main text. Abbreviations in this figure not defined in the main body of the text: TAG, triacylglycerol; SR, scavenger receptor; LB, lipid body; A2R, A2 adenosine receptor (A2A/A2B receptors)
4.1. Tumor‐imposed limitations on DC carbohydrate/energy metabolism
The diverse functions of DC are tightly linked to the maturation and activation status of these cells. In recent years, it has become apparent that the phenotypic and functional plasticity of DC during their progression from an immature to a mature, activated state is driven by metabolic plasticity designed to support the specific functions of these cells. Immature DC fulfill their bioenergetic demands by generating ATP through oxidative phosphorylation (OXPHOS), and this process is driven primarily by fatty acid oxidation (FAO), 76 consistent with the need of these highly phagocytic cells to catabolize fatty acid substrates generated from lipolysis of apoptotic cell membranes acquired during engulfment. Shortly after stimulation, DC maintain OXPHOS activity to generate ATP but undergo a metabolic shift toward glycolysis in order to yield substrates to support the tricarboxylic acid (TCA) cycle and pentose‐phosphate pathway (PPP), which yield citrate and NADPH for the de novo lipogenesis needed to support expansion of endoplasmic reticulum (ER) and Golgi apparatus membrane mass. 77 It is believed that the dedication of resources toward these organelles supports the protein synthesis and trafficking needs of DC that are transitioning from phagocytic cells to functional APC that express/secrete newly synthesized cell surface costimulatory molecules and immunostimulatory cytokines. It has also been shown that early glycolytic flux is required for CCR7 oligomerization and DC migration to draining lymph nodes, 78 where mature, activated DC typically prime T cell responses. Ultimately, genetic reprogramming of activated DC triggers a long‐term commitment to glycolysis to fuel ATP production in the face of the lost mitochondrial respiratory function that accompanies nitric oxide or type I IFN production by these cells. 79 , 80 Interestingly, recent metabolic tracing studies demonstrated that the glycolytic switch in newly activated DC relies heavily on the metabolism of glycogen. This work supports a model in which intracellular glycogen stores present in DC prior to activation drive the early glycolytic burst of these cells following stimulation. Additionally, extracellular glucose taken up by DC following glycolytic reprogramming is utilized not only for direct catabolism but also for rerouting into a pathway of glycogen synthesis‐glycogenolysis known as the glycogen shunt. 81 Though the functional outcomes of these distinct pathways for extracellular glucose utilization by DC during activation remain to be explored, it is clear that both glucose and glycogen metabolism are critical to achieving immunostimulatory functions of DC.
In order to support the biosynthetic and energy demands of rapid cell division, tumor cells also frequently undergo rewiring to a primarily glycolytic mode of metabolism, not only in hypoxic environments where OXPHOS is naturally limited but also under normoxic conditions, a phenomenon known as the Warburg effect (aerobic glycolysis). While the aforementioned studies on glycolytic switching in activated DC have primarily been performed on BMDC and monocyte‐derived DC in vitro (due to technical limitations that preclude analysis of this process in DC in vivo), there are a number of potential and documented effects of glycolytically active tumor cells on DC function within the TME. First, competition between tumor cells and DC for limited resources may simply restrict glucose availability to TIDC. In this regard, a recent comparison of progressor versus regressor tumors revealed diminished glucose concentration in the extracellular milieu of progressor tumors, 82 suggesting that glucose depletion in the TME may limit DC uptake of extracellular glucose needed to sustain a glycolytic switch. As long‐term commitment to glycolysis is needed to support ATP production in activated DC, a failure to maintain glycolytic activity is likely to lead to elevated AMP:ATP ratios in TIDC, an outcome that could in turn trigger AMPK‐mediated downregulation of HIF‐1α, 83 , 84 a transcriptional regulator of several enzymes essential to glycolytic metabolism. 85 HIF‐1α also regulates expression of enzymes necessary for glycogen synthesis, 86 suggesting that interference with the aforementioned glycogen shunt for any glucose that is acquired by DC in the TME may also impair the metabolic and immunologic functions of these cells, a possibility that remains to be investigated. In addition to these passive mechanisms of tumor‐altered carbohydrate metabolism by DC that could result from glucose depletion in the TME, multiple tumor types have been shown to actively inhibit glycolysis in these cells as well. In murine models of melanoma as well as lung and ovarian cancers, tumor‐associated DC exhibited increased expression of SOCS3, which was found to suppress activity of the pyruvate kinase M2 (PKM2) enzyme responsible for catalyzing the final step of glycolysis. 87 In this study, SOCS3 was also found to limit the PKM2‐driven antitumor efficacy of a DC‐based vaccine against established LLC tumors. Together, these data suggest that tumor‐associated regulators of glycolytic metabolism in DC could therefore be useful targets for therapies relying on the activity of either endogenous or exogenous DC.
4.2. Tumor‐altered lipid metabolism in DC
As alluded to above, de novo lipogenesis plays a key role in supporting membrane mass for increased ER and Golgi activity during DC maturation and activation. At the same time, lipid accumulation in tumor‐associated DC is a known hallmark of immune dysfunction in these cells. This apparent dichotomy may be explained by the nature of lipid content found in appropriately activated versus tumor‐altered DC, the latter of which could be compromised by either: (a) tumor‐associated suppression of lipid synthesis needed to support organelle membrane mass and vesicular transport of molecules for T cell stimulation or (b) tumor‐induced uptake/generation of lipids detrimental to DC function. In support of the former, tumor‐derived α‐fetoprotein has been shown to suppress expression of several genes involved in fatty acid synthesis (FAS) in DC, 88 and the defects in differentiation and T cell stimulatory capacity of these cells 89 may therefore ultimately arise from limitations in lipid‐mediated trafficking of proteins otherwise needed for potent APC activity. On the other hand, DC functionality can also be compromised by the uptake of lipids enriched within the TME, a process that can be influenced by lipid metabolism in tumor cells themselves. In a murine model of ovarian cancer, fatty acid synthase (FASN) expression in tumor cells correlates with TME lipid content, including both saturated and unsaturated fatty acids as well as triacylglycerols, and TIDC isolated from tumors with high FASN expression exhibit elevated lipid levels and poor T cell stimulating activity when compared to those recovered from tumors in which FASN is silenced. Importantly, lipid accumulation and DC dysfunction in this model could be reversed therapeutically by treatment with a FASN inhibitor, 90 demonstrating the potential of metabolic interventions to support the antitumor immune functions of DC.
In addition to increasing the lipid content available to DC within the TME, tumors can also actively promote lipid acquisition by DC by inducing their expression of scavenger receptors such as MSR‐1. 91 In particular, the uptake and accumulation of peroxidized lipids in TIDC has been found to interfere with Ag cross‐presentation through a mechanism mediated by lipid bodies containing oxidatively truncated lipids covalently bound to the HSP70 chaperone protein, an interaction that prevents peptide:MHC complex trafficking from late endosomes/lysosomes to the cell surface. 92 , 93 Elevated reactive oxygen species (ROS) in tumor‐associated DC also contribute to intracellular lipid peroxidation, leading to activation of the ER stress sensor XBP1 that in turn drives triglyceride biosynthesis and further accumulation of lipids that blunt Ag presentation. 94 Beyond these lipid‐associated defects in Ag presentation, DC maturation and proinflammatory cytokine secretion have also been found to be negatively influenced by oxidized fatty acids and polyunsaturated fatty acids, respectively. 95 , 96 Finally, the Wnt‐β‐catenin pathway has been shown to drive the oxidation of fatty acids in tolerogenic tumor‐associated DC that not only fail to support CD8+ T cell activation but that also promote Treg induction. 97 , 98 Mechanistically, tumor‐derived Wnt5a activates β‐catenin in DC, leading to PPAR‐γ‐mediated induction of the CPT1A mitochondrial fatty acid transporter and a shift to FAO metabolism. FAO in turn suppresses expression of IL‐6 and IL‐12, and diversion of TCA products to the heme biosynthesis pathway leads to increased production of the protoporphyrin IX prosthetic group necessary for full enzymatic activity of indoleamine 2,3‐dioxygenase‐1 (IDO1), a potent inducer of Tregs. 98 Although these studies collectively highlight diverse mechanisms by which altered lipid metabolism compromises the function of tumor‐associated DC, they also reveal a number of therapeutic strategies for regulating fatty acid metabolic programs in both DC and tumor cells that have the potential to significantly enhance DC function in the context of cancer.
4.3. Suppression of DC by tumor‐derived metabolites
In addition to limiting the availability of nutrients and other key resources for use by immune cell populations infiltrating the TME, the extreme metabolic demands of rapidly growing tumor cells also result in the accumulation of toxic by‐products that are detrimental to immune function. In this regard, another consequence of the glycolytic switch that often occurs in tumor cells is TME accumulation of lactic acid, an oncometabolite that impairs DC function in a variety of ways. Lactate‐driven DC dysfunction was first described in monocyte‐derived DC differentiated in the context of tumor spheroids, where tumor suppression of IL‐12 secretion by DC could be reversed by blocking lactic acid production with a lactate dehydrogenase A (LDHA) inhibitor. 99 Likewise, LDHA inhibition of ex vivo‐cultured lung tumor cells reduced the suppressive effects of tumor‐conditioned media on IFN‐α production by Flt3L‐differentiated BMDC. 100 This study also found that lactic acid triggered endosomal acidification and Ag degradation in DC and reduced their ability to prime CD8+ T cells and confer antitumor immune protection in vivo. Most recently, lactic acid was also found to suppress IFN‐α production by pDC via two distinct mechanisms. First, lactate signaling through the GPR81 receptor on pDC mobilized intracellular Ca++ stores, which in turn dampened IFN‐α induction through activation of the phosphatase calcineurin. Second, lactic acid import into pDC through monocarboxylate transporters (MCT) increased intracellular lactate and suppressed the glycolytic switch needed to induce efficient IFN‐α expression. 101 That intratumoral pDC were found to express higher levels of both GPR81 and MCT than their circulating counterparts underscores the significance of these mechanisms for compromising pDC function within the TME. Importantly, in addition to the potential consequences of reduced IFN‐α expression on the maintenance of CTL effector function within the TME, it was found that lactate conditioning of pDC also reprograms these cells toward increased tryptophan metabolism, a pathway that promotes Treg induction through release of kynurenine, 101 as discussed in more detail below.
Another oncometabolite that has adverse consequences for antitumor immunity in the TME is adenosine, which often accumulates to high levels as a result of the metabolism of extracellular ATP. Extracellular ATP itself is frequently elevated in the TME, 102 and while its release from tumor cells can promote immunogenic signaling as a DAMP through purinergic receptors on DC, 103 it is often converted into adenosine by the ectonucleotidases CD39 and CD73, 104 , 105 leading instead to immunosuppressive signaling through A2A and A2B adenosine receptors on various immune cell populations. In addition to its well‐documented role in the regulation of T cell responses (see below), adenosine has also been shown to repress the immunostimulatory functions of DC. In a murine melanoma model, tumor‐associated DC function was improved by deletion of the A2A receptor on DC, which resulted in significantly reduced Il10 gene expression and slightly improved IL‐12 production by these cells, 106 a finding consistent with data from in vitro studies evaluating the effects of adenosine on human monocyte‐derived DC. 107 Adenosine signaling through the A2B receptor has also been shown to drive gene expression for several tumor‐promoting factors (VEGF, TGF‐β, IDO, arginase, etc) in DC, and intratumoral injection of adenosine‐conditioned DC was found to enhance the vascularization and growth of LLC tumors. 108 Mechanistically, adenosine signaling promotes accumulation of cAMP in DC, leading to activation of PKA and Epac pathways that polarize these cells to a tumor‐promoting phenotype (IL‐10high/IL‐12low) by increasing expression of NF‐κB pathway regulators. 109 Together, these studies reveal a number of possible strategies for interfering with adenosine‐mediated suppression of DC in the TME, some of which have already shown efficacy in preclinical systems. These approaches include targeting the CD39/CD73 ectonucleotidases that yield adenosine from ATP, blocking the expression or activity of A2A/A2B adenosine receptors on DC, and inhibiting intracellular signaling components that mediate the suppressive effects of adenosine on DC. 106 , 110 , 111
5. IMMUNE CHECKPOINTS INFLUENCING T CELL FUNCTION IN THE TUMOR MICROENVIRONMENT
Similar to DC, T lymphocytes also express a number of receptors that function to restrict aberrant immune reactivity. While necessary to prevent autoimmunity and hyperactive responses to acute infections that are cleared quickly, engagement of these adaptive checkpoints in the context of cancer can lead to tumor immune escape by limiting the duration and quality of antitumor T cell responses. Insight into the most well‐studied of these negative regulatory pathways has paved the way for immune checkpoint blockade (ICB), a therapeutic approach to “release the brakes” on the immune system that has achieved unprecedented clinical success against many cancer types. 1 With the discovery of new T lymphocyte checkpoints in recent years and an improved understanding of the mechanisms underlying tumor cell resistance to the first generation of immune checkpoint inhibitors, there is great promise for next‐generation and combinatorial checkpoint blockade therapies to unleash even more potent antitumor T cell responses and further improve patient response to ICB therapy in the future.
5.1. CTLA‐4
The first immune checkpoint found to negatively regulate the function of T lymphocytes was the co‐inhibitory receptor CTLA‐4. 112 Upregulated on T cells shortly after stimulation, CTLA‐4 binds to the same CD80/CD86 ligands as the CD28 costimulatory receptor, but with higher affinity. In addition to limiting costimulation by outcompeting CD28 for ligand binding, CTLA‐4 engagement also transmits inhibitory signals to T lymphocytes, with cell type‐specific consequences. In CD4+ T cells, CTLA‐4 engagement limits T cell activation and differentiation during the priming phase of a response, ultimately inducing anergy. 113 , 114 , 115 , 116 In CD8+ T cells, on the other hand, CTLA‐4 does not impair priming but instead regulates the magnitude of recall responses to secondary stimulation, 117 , 118 which could limit the efficacy of both natural and adoptively transferred CTL following Ag re‐exposure within the TME. Indeed, it was recently reported that TGF‐β‐driven CD80 expression on tumor‐initiating stem cells promotes resistance to adoptive cell transfer (ACT) therapy via a CTLA‐4‐dependent mechanism. 119 Though it remains to be explored, such a process means that lymph node‐invasive tumor cells, in addition to cross‐presenting APC, might also limit the priming of naïve helper T cell responses following direct presentation of tumor Ag within draining lymph nodes. Finally, CTLA‐4 is critical to the immunosuppressive activity of Tregs, driving transendocytosis of costimulatory molecules expressed on DC and thereby limiting their capacity to support the activation of both CD4+ and CD8+ T cells. 45 , 46
Following preclinical studies demonstrating that CTLA‐4 blockade diminishes Treg activity and restores antitumor T cell effector function, 120 a subsequent clinical trial showing the therapeutic efficacy of an anti‐CTLA‐4 antibody in melanoma patients 121 led to FDA approval of ipilimumab as the first immune checkpoint inhibitor for cancer treatment. In both murine models and cancer patients, anti‐CTLA‐4 monotherapy is associated with an expansion of ICOS+ CD4+ Th1 effectors. 122 , 123 A concomitant increase in CD8+ T cells with an exhausted phenotype is also observed following CTLA‐4 blockade, and combinatorial inhibition of other immune checkpoints (see below) can harness the potential of this expanded population for improved tumor immune control. As such, ipilimumab has since been approved in combination with nivolumab (anti‐PD‐1) therapy for certain cases of melanoma, renal cell carcinoma, hepatocellular carcinoma, and microsatellite instability‐high or mismatch repair deficient colorectal cancer. Another CTLA‐4 checkpoint inhibitor, tremelimumab, is also currently under evaluation as part of combinatorial regimens for various other malignancies. Though both ipilimumab and tremelimumab have been shown not to deplete FOXP3+ Tregs in cancer patients, it is possible that modification of the Fc regions of these antibodies may confer this ability shared by antibodies against murine CTLA‐4 and further improve the therapeutic benefit of CTLA‐4 blockade in humans. 124
5.2. PD‐1
The early observation that CTLA‐4 blockade could enhance tumor Ag‐specific CD4+ T cell priming but could not overcome the eventual tolerization of these cells highlighted the contribution of alternative pathways to the regulation of T lymphocyte function. 125 Indeed, several other co‐inhibitory checkpoints for T lymphocytes have now been identified, and of these, the PD‐1/PD‐L1 axis is the most‐well studied. PD‐1 is another member of the immunoglobulin superfamily and is upregulated on both CD4+ and CD8+ T lymphocytes shortly after stimulation. 126 It shares two ligands, PD‐L1 and PD‐L2, the latter of which is expressed primarily on hematopoietic cell populations and the former of which is expressed on both hematopoietic cells as well as non‐hematopoietic cells from many tissue types. 127 Upon engagement of PD‐1, both PD‐L1 and PD‐L2 have been shown to suppress T cell effector function, 128 , 129 though PD‐L2's role in immunosuppression is controversial, as it has also been shown to protect T cells from PD‐L1‐mediated suppression in certain contexts. 130
The PD‐1/PD‐L1 axis negatively regulates T lymphocyte activity in a variety of ways. In addition to driving the differentiation and immunosuppressive activity of inducible Tregs, 131 this pathway plays a significant role in the exhaustion of peripheral T cells during the effector phase of a response, particularly in cases of repeated Ag exposure, such as that encountered during chronic viral infection or in the context of progressing tumors. 122 , 132 , 133 , 134 , 135 There is also accumulating evidence that PD‐1/PD‐L1 interaction during the induction phase of a T cell response can impact clonal expansion and effector cell differentiation as well. 136 , 137 , 138 Indeed, PD‐L1 expressed on either tumor cells or tumor‐associated APC is sufficient to blunt antitumor T cell responses, with negative regulation of T cell function being mediated by PD‐L1 both within the TME and within tumor‐draining lymph nodes. 139 , 140 , 141 Mechanistically, PD‐1 engagement by PD‐L1 promotes recruitment of the SHP‐2 phosphatase to the immunological synapse, leading to dephosphorylation of both TCR and CD28 signaling components and downregulation of the PKC and MAPK signaling pathways. 131 , 142 , 143 , 144 , 145 PD‐1/PD‐L1 interaction also drives CD3 internalization and downregulation of the TCR, 146 a phenomenon frequently observed in tumor‐infiltrating lymphocytes (TIL). Another consequence of PD‐1 engagement on T cells is its impact on metabolism, as PD‐1‐mediated inhibition of glycolysis and OXPHOS leads to bioenergetic insufficiencies that predispose T cells toward exhaustion. 147 , 148 Considering the constraints on T cell metabolism that are frequently encountered within the TME (described in detail below), this mechanism may be particularly relevant to tumor immune escape as it is likely to exacerbate metabolic deficiencies in tumor‐infiltrating T cells.
Like CTLA‐4 blockade, therapeutic targeting of the PD‐1/PD‐L1 pathway also enhances antitumor T cell reactivity, and immune checkpoint inhibitors against both PD‐1 (nivolumab, pembrolizumab, and cemiplimab) and PD‐L1 (atezolizumab, avelumab, and durvalumab) are now standard‐of‐care therapy for many cancer types. 1 Unlike CTLA‐4 blockade, however, PD‐1 inhibitors do not enhance CD4+ effector T cell frequency within tumors but instead primarily drive expansion of phenotypically exhausted CD8+ T cells. 122 , 149 Though these expanded CD8+ T cells retain expression of cell surface markers indicative of exhaustion, likely due to epigenetic maintenance of co‐inhibitory molecule expression, 150 the accumulation of less exhausted non‐terminally differentiated (PD‐1+ TIM3low TBET+ EOMES−) and fully exhausted terminally differentiated (PD‐1high TIM3+ TBET+ EOMES+) CD8+ T cells is associated with improved tumor control. These data suggest that PD‐1 inhibition may improve tumor immunity by temporarily reinvigorating the antitumor reactivity of cells that had already been rendered functionally exhausted and/or by enhancing the activity of partially exhausted cells and preventing their transition to a completely exhausted state.
Interestingly, while combinatorial blockade of both CTLA‐4 and PD‐1 has additive effects on the frequency of many of the specific T cell subsets stimulated by each monotherapy, dual CTLA‐4/PD‐1 blockade also elicits responses distinct from those achieved with monotherapy. Namely, combination therapy decreases the intratumoral frequency of phenotypically exhausted CD8+ T cells that are otherwise expanded by anti‐PD‐1 monotherapy and instead drives the accumulation of activated terminally differentiated effector CD8+ T cells within tumors. 123 It was also recently shown that responders to dual CTLA‐4/PD‐1 checkpoint blockade are enriched for effector memory CD8+ T cells within tumor biopsies. 151 The differential responses to combination therapy versus individual anti‐CTLA‐4 and anti‐PD‐1 monotherapies are in keeping with unique transcriptional signatures identified in CD4+ and CD8+ TIL following treatment with these regimens, 122 , 152 , 153 and these data support combination therapy as the most effective means of programming T cells for robust antitumor reactivity. Indeed, recent clinical trials have highlighted the success of combination ICB therapy against CTLA‐4 and PD‐1. In the CheckMate067 trial, the 5‐year overall survival rate for advanced melanoma patients receiving combination ipilimumab + nivolumab as frontline therapy exceeded 50%, whereas this rate was 44% for patients on single‐agent nivolumab and only 26% for those on ipilimumab alone. 154 Though not as dramatic, a significant improvement in patient survival was also reported for NSCLC patients treated with this same combination therapy versus nivolumab monotherapy or chemotherapy. 155 Despite the promise of these and related trials, though, an ongoing challenge in the field is the need to better understand mechanisms of innate and acquired resistance to CTLA‐4, PD‐1, and PD‐L1 checkpoint blockade. To this end, significant efforts are now being focused on targeting additional regulatory pathways that are known to compromise the quality of antitumor T cell responses, as discussed in more detail below.
5.3. Lymphocyte activation gene 3
The observation that many patients do not respond to CTLA‐4 and PD‐1/PD‐L1 checkpoint blockade despite harboring TIL‐enriched tumors underscores the relevance of additional T cell‐regulating mechanisms within the TME. Indeed, tumor gene expression signatures from many anti‐CTLA‐4/anti‐PD‐1 non‐responders indicate that other co‐inhibitory receptors should also be evaluated when considering optimal combinatorial checkpoint blockade strategies. 151 One such co‐inhibitor that is upregulated in a large percentage of patients who fail to respond to anti‐CTLA‐4/anti‐PD‐1 therapy is lymphocyte activation gene 3 (LAG‐3). Named as a result of its upregulation on activated T lymphocytes, LAG‐3 actually functions to suppress T cell activation, impairing cell cycle progression and expansion of both CD4+ and CD8+ T lymphocytes. 156 , 157 In the context of cancer, LAG‐3 has been shown to limit both the accumulation and effector function of CD8+ T cells in tumor tissue, 158 and its elevated expression on tumor‐infiltrating Tregs correlates with enhanced immunosuppressive activity by these cells. 159 , 160
LAG‐3‐mediated suppression of T cell responses can be achieved in a number of ways. First, inhibitory signaling via LAG‐3 is transmitted by its cytoplasmic KIEELE domain when the receptor binds with high affinity to MHC class II, 161 highlighting the potential for both tumor‐associated APC and MHC class II‐expressing cancer cells to suppress T cell function via this pathway. In addition to direct inhibitory signaling within T cells, LAG‐3/MHC class II interactions can also indirectly interfere with T cell activation by reverse signaling through the MHC, a mechanism that enables LAG‐3‐expressing Tregs to suppress DC maturation. 162 Other LAG‐3 ligands can also negatively regulate T cell function within the TME. Galectin‐3 is a soluble galactoside‐binding lectin secreted by various tumor types that binds to LAG‐3 and suppresses antitumor CD8+ T cells. 163 Similarly, the LSECTin lectin expressed on melanoma cells has also been shown to suppress antitumor T cell activity via a LAG‐3‐dependent mechanism. 164
In keeping with the hypothesis that LAG‐3 may limit clinical responses to currently approved immune checkpoint inhibitors, co‐expression of LAG‐3 and PD‐1 was reported on both CD4+ and CD8+ TIL isolated from primary tumors of renal cell carcinoma patients, and when compared to PD‐1 blockade alone, dual blockade of both LAG‐3 and PD‐1 enhanced IFN‐γ production by these cells following ex vivo stimulation. 165 Though the same benefit of dual LAG‐3/PD‐1 blockade over PD‐1 blockade alone was not observed in double positive tumor Ag‐specific CD8+ TIL isolated from ovarian cancer patients, it was achieved when both co‐inhibitors were targeted during the initial priming of Ag‐specific T cells isolated from PBL. 166 Differences in these studies may reflect the extent to which TIL had become exhausted in each setting and/or expression of yet other co‐inhibitory receptors on ovarian cancer‐derived TIL that continued to repress T cell effector function despite dual LAG‐3/PD‐1 blockade. Still, both studies demonstrate that targeting LAG‐3 at various stages of an antitumor immune response does have the potential to improve tumor‐specific T cell reactivity. In this regard, work in the MC38 murine tumor model has demonstrated that LAG‐3 synergizes with PD‐1 in the suppression of antitumor T cell responses and that dual blockade is often curative for established tumors resistant to either monotherapy. 167 It is also worth noting that a number of factors in the TME have now been implicated in the induction of LAG‐3 expression on T cells, including IL‐6, IL‐10, and tumor‐associated APC, 166 and targeting these LAG‐3 inducers may also prevent suppression of antitumor T cell immunity by this checkpoint pathway.
5.4. T cell Ig and mucin domain containing molecule‐3
Another co‐inhibitory receptor frequently upregulated on exhausted TIL and intratumoral Tregs is TIM‐3. 168 , 169 , 170 Though TIM‐3 has multiple ligands, most studies in tumor models and cancer patients to date have implicated galectin‐9 as the major trigger for TIM‐3‐mediated suppression of T cells. 171 , 172 , 173 Current evidence supports a model whereby this suppression arises as Bat3 is released from TIM‐3′s cytoplasmic tail during receptor engagement by galectin‐9, thereby enabling inhibitory signaling that is otherwise repressed when Bat3 is bound. 174 In this regard, it is worth noting that Bat3 gene expression is significantly reduced in exhausted TIM‐3+ TIL isolated from murine mammary adenocarcinomas 174 and that the long noncoding RNA lnc‐Tim3 binds to TIM‐3′s cytoplasmic tail and promotes release of Bat3 in exhausted CD8+ TIL from hepatocellular carcinoma patients. 175 In addition, TIM‐3 is typically expressed at higher levels on intratumoral Tregs as compared to peripheral Tregs, and its upregulation is associated with more robust immunosuppressive activity. 168 , 169 , 176 , 177 Based on these findings, it is not surprising that TIM‐3 blockade has been found to augment antitumor immunity in multiple ways, with evidence for both enhanced CD8+ and CD4+ T cell effector activity and reduced Treg frequency emerging as mechanisms of therapeutic efficacy. 178 , 179
Considering TIM‐3′s role in inhibiting antitumor immune responses, it is worth noting that its co‐expression with PD‐1 marks CD8+ T cells that are more heavily exhausted than single‐positive PD‐1‐expressing cells. 174 , 180 , 181 , 182 Moreover, TIM‐3 expression on TIL is upregulated following PD‐1 blockade and has been linked with adaptive resistance to anti‐PD‐1 monotherapy, 183 , 184 suggesting that TIM‐3 might function as a failsafe to overcome loss of PD‐1‐mediated inhibition of T cell responses. Indeed, sequential or combinatorial blockade of TIM‐3 and PD‐1/PD‐L1 has shown enhanced antitumor efficacy in several murine models 172 , 180 , 183 , 184 and dual blockade of these checkpoints augments the effector activity of ex vivo‐stimulated CD8+ TIL from hepatocellular carcinoma patients. 185 That similar data have also been reported for tumor‐bearing mice undergoing dual TIM‐3/CTLA‐4 blockade 178 and for TIL treated with other combinations of checkpoint inhibitors 185 underscores the potential utility of combinatorial approaches to checkpoint blockade therapy as a means of eliciting robust antitumor immunity.
5.5. TIGIT and related PVR/nectin family members
The T cell immunoreceptor with Ig and ITIM domains (TIGIT) is an inhibitory receptor belonging to the poliovirus receptor (PVR)/nectin family. Related members of this family include CD96, CD112R, and DNAM‐1 (CD226), which together with TIGIT comprise a complex immunoregulatory system for T lymphocytes and other immune cell populations. With respect to T cells, TIGIT can be highly expressed on Tregs and is upregulated on both activated CD4+ and CD8+ T lymphocytes, where it functions to dampen immune reactivity. 186 , 187 Its ligands include CD112, CD113, and CD155, many of which are expressed on various tumor types as well as tumor‐associated DC. 188 , 189 , 190 , 191 Though these ligands are shared between TIGIT and related PVR family members, their impact on T cell function is receptor‐dependent. For instance, while CD155 can costimulate T cells when signaling through DNAM‐1, such signaling is often limited by competition for ligand binding with the higher affinity TIGIT and CD96 receptors, both of which suppress T cells through their ITIM and other inhibitory signaling motifs. 192 , 193 Similarly, interference with CD112‐mediating signaling through DNAM‐1 has been reported for CD112R, which also has higher affinity for its shared ligand and, like TIGIT, transmits inhibitory signals to T cells. 194 Finally, immunostimulatory DNAM‐1 signaling can also be compromised by a mechanism unrelated to ligand competition, as cis interaction with TIGIT has been shown to prevent the homodimerization necessary for DNAM‐1 function. 195
In addition to its influence over DNAM‐1 signaling, TIGIT has been found to drive T cell dysfunction by a variety of cell‐intrinsic and cell‐extrinsic mechanisms. Inhibitory signaling through TIGIT has been shown to impact T cell metabolism, reprogramming cells away from the glycolytic flux needed to support T cell activation. When compared to TIGIT− CD8+ T cells isolated from gastric cancer patients, functionally exhausted, glycolytically deficient TIGIT+ CD8+ T cells exhibited reduced expression of the GLUT1 glucose importer and the hexokinase 1/2 glycolysis‐initiating enzymes. Importantly, glycolytic metabolism could be restored in CD8+ T cells co‐cultured with gastric cancer cell lines when TIGIT‐CD155 interactions were disrupted. 196 Cell‐intrinsic signaling through TIGIT also supports immunoregulatory functions of Tregs that correlate with the enhanced capacity of TIGIT+ Tregs to suppress TH1/TH17 differentiation. 197 Moreover, elegant studies in which various combinations of TIGIT+ versus TIGIT‐deficient CD8+ T cells and Tregs were co‐transferred into Rag −/− mice prior to tumor challenge have also highlighted the significance of TIGIT signaling in Tregs to the suppression of CD8+ TIL effector function and overall antitumor immunity. 198 Finally, with regard to its cell‐extrinsic functions, TIGIT can also drive reverse signaling through CD155, and TIGIT‐mediated signaling through this ligand on DC augments IL‐10/IL‐12p40 ratios and drives DC‐dependent inhibition of T cell activation. 199
TIGIT expression has been reported on T cells isolated from tumor tissue of a variety of cancer types, including follicular lymphoma, multiple myeloma, melanoma, gastric cancer, NSCLC, and HNSCC, among others. 191 , 195 , 200 , 201 , 202 , 203 As with other T lymphocyte checkpoints, in vivo blockade of TIGIT in murine tumor models has been shown to reduce Treg frequency, enhance CD8+ T cell effector function, and improve antitumor immunity. 200 , 203 Additional murine studies reporting co‐expression of TIGIT with other inhibitory checkpoint receptors on T cells have also shown augmented antitumor immunity following combination checkpoint blockade. 195 , 204 The potential clinical relevance of this work is highlighted by evidence that combinatorial blockade of TIGIT and other checkpoint receptors also enhances the effector function of ex vivo‐stimulated CD8+ TIL from melanoma patients as well as CD4+ and CD8+ T cells from acute lymphocytic leukemia patients, suggesting that such regimens are likely to show improvements over monotherapies in cancer patients as well. 201 , 205 Additionally, alternative approaches for interfering with TIGIT‐mediated suppression of T cells are also emerging. One recent study found that co‐administration of a PD‐1 blocking antibody and an agonistic anti‐glucocorticoid‐induced tumor necrosis factor receptor‐related protein (GITR) antibody improved effector function in CD8+ TIL by restoring proper balance to the DNAM‐1/TIGIT signaling axis. Specifically, PD‐1 blockade enhanced DNAM‐1 activity by preventing SHP‐2‐mediated dephosphorylation of its cytoplasmic tail, and GITR agonism reduced TIGIT expression on these cells. 206 A costimulatory switch receptor that exploits TIGIT to the immune system's advantage was also recently investigated for efficacy in the context of ACT therapy against a xenograft model of established human melanoma. Generated by fusion of the TIGIT exodomain to the immunostimulatory signaling domain of CD28, this costimulatory switch receptor conferred better antitumor immune control by adoptively transferred T cells that had also been transduced to express a MART‐1 Ag‐specific TCR. 207 Together, these studies highlight multiple strategies for improving antitumor immunity through manipulation of the TIGIT checkpoint in T lymphocytes. With data emerging that CD96 blockade also supports antitumor T lymphocyte function, 193 it is likely that several members of the PVR family will become important targets for therapeutic intervention in the near future.
5.6. Other emerging T lymphocyte checkpoints
Following the successful preclinical and clinical outcomes of ICB regimens targeting the well‐characterized T lymphocyte checkpoints described above, efforts have been under way to identify and elucidate the functional roles of other regulators of T cell activation that might also be targeted in the context of cancer. A number of additional negative regulators have indeed been found to control T lymphocyte function, and insight into these checkpoint pathways has revealed new potential targets for therapeutic intervention. For instance, though it is most highly expressed on DC and other myeloid cell populations, VISTA is also expressed on T lymphocytes and can suppress proliferation and effector cytokine production by both CD4+ and CD8+ T cells. 208 Unlike many other checkpoint molecules that are expressed following T cell activation, VISTA is also expressed on naïve T lymphocytes, where it maintains T cell quiescence and immune tolerance and also drives the differentiation of CD4+ T cells into FOXP3‐expressing Tregs. 208 , 209 Importantly, multiple studies have now highlighted a role for VISTA as a negative regulator of T cell responses to cancer. In a murine glioma model, VISTA‐deficient hosts exhibited enhanced control of implanted tumors that was dependent on CD4+ T cells, 210 and antibody blockade of VISTA in mice bearing transplanted melanomas reduced induction of Tregs within tumors, enhanced CD8+ TIL effector function, and delayed tumor progression. 72 This latter study also reported similar results in an inducible melanoma model and in the MB49 bladder tumor model.
BTLA is another inhibitory checkpoint receptor shared by both DC and T lymphocytes that can contribute to the dysfunction of tumor‐specific T cell responses. In melanoma patients, BTLA is expressed at high levels on Ag‐specific CD8+ T cells, and its ligand, HVEM, is expressed on melanoma cells in situ. The potential clinical significance of these findings is underscored by in vitro observations that HVEM‐expressing melanoma cell lines inhibit IFN‐γ production by Melan‐AMART‐1 Ag‐specific CD8+ T cell clones in a BTLA‐dependent manner. 211 Indeed, in patients with diffuse large B cell lymphoma, BTLA+ T cells from the tumor microenvironment exhibit less cytolytic activity than their BTLA‐deficient counterparts. 212 Additional evidence for BTLA‐mediated suppression of antitumor T cells comes from a study reporting that BTLA blockade enhances the proliferation, effector cytokine production, and degranulating activity of minor histocompatibility Ag‐specific CD8+ T cells isolated from PBMC of patients with hematological malignancies that were treated by allogeneic stem cell transplantation. 213 BTLA blockade also bolsters the stimulatory effects of PD‐1 blockade on alloreactive CD8+ T cells, 214 suggesting that combinatorial interference with BTLA and PD‐1 could be a useful approach for augmenting antitumor T cell responses in cancer patients. Moreover, in addition to signaling through BTLA, HVEM also binds to the co‐inhibitory receptor CD160, which drives CD8+ T cell dysfunction in the context of certain viral infections. 215 As the frequency of CD160‐expressing CD8+ T cells in PBMC populations is higher in esophageal cancer patients than in normal donors 216 and CD160 expression levels are increased on CD8+ T cells from bone marrow of multiple myeloma versus healthy patients, 217 this checkpoint receptor might also be a useful target for ICB therapy against certain cancer types.
2B4 (CD244) is a member of the signaling lymphocyte activation molecule (SLAM) family of immunoreceptors. Though activation signals can indeed be transmitted through 2B4 via its CD48 ligand, the ultimate outcome of receptor engagement appears to be controlled by the ratio of 2B4 to SLAM adaptor protein (SAP) content in a given cell. According to the current model, under conditions where 2B4 expression is limited, the intracellular adaptor protein SAP can bind to 2B4's cytoplasmic ITSM signaling motifs and prevent the binding of inhibitory phosphatases, thus enabling delivery of activation signals. On the other hand, elevated expression of 2B4 on exhausted T cells, as has been reported in many cancers, 217 , 218 , 219 shifts the 2B4:SAP balance such that intracellular SAP levels are insufficient to prevent phosphatase‐mediated inhibitory signaling. 220 Although the antitumor activity of 2B4 blockade has yet to be investigated, blockade of this checkpoint receptor does improve overall T cell effector function and survival in an animal model of sepsis with preexisting malignancy, 221 and blockade of either CD48 or 2B4 reverses CD8+ T cell exhaustion in the context of chronic viral infection. 222 , 223 Therefore, as the regulation of antitumor T cell responses by the CD48/2B4 axis continues to be investigated, checkpoint inhibitors targeting this pathway may soon be added to the repertoire of ICB agents available for cancer therapy.
NKG2A, an inhibitory checkpoint classically associated with natural killer (NK) cells, has also been found to negatively regulate T lymphocytes. The receptor for the nonclassical MHC class I molecules Qa‐1 in mice and HLA‐E in humans, NKG2A mediates inhibitory signaling via its cytoplasmic ITIM domains. In murine models, two groups have recently shown that NKG2A blockade enhances CD8+ T cell‐dependent control of tumors, both in the context of naturally occurring and vaccine‐induced immune responses. 224 , 225 NKG2A expression on TIL was also recently found to be a poor prognostic factor for overall survival of colorectal cancer patients. 226 An NKG2A blocking antibody, monolizumab, is currently under investigation as an ICB therapeutic, and an interim report of a Phase II trial has shown clinical benefit of monolizumab in combination with cetuximab for SCCHN patients. 227
With the discovery of such a vast array of inhibitory checkpoints for T lymphocytes over the last two decades, there now exists a multitude of realized and potential therapeutic targets for promoting/restoring T cell reactivity against cancer (Figure 3). As these and newly discovered checkpoint pathways continue to be investigated, the challenge going forward will lie in identifying the particular checkpoints that should be targeted by combinatorial or sequential ICB therapies in specific cancer patients. To this end, tumor immune profiling, 228 not only prior to treatment but also in cases of disease relapse, will become critical to the success of ICB‐based therapies, as it will reveal the specific cohort of immune checkpoint receptors and ligands expressed by an individual's T lymphocytes and other cell populations in the TME. Based on the successes already seen with initial approaches to combination ICB therapy, it is very likely that incorporating additional inhibitors of targetable checkpoints into ICB regimens will further improve the efficacy and durability of antitumor immune responses in many cancer patients.
FIGURE 3.
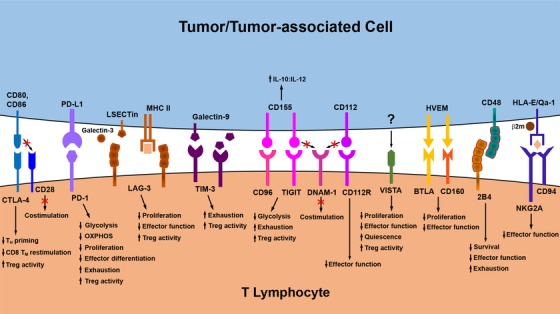
Immunologic checkpoints influencing T lymphocyte function in the TME. Several inhibitory checkpoint pathways that negatively regulate antitumor T lymphocyte function have now been identified. Engagement of these checkpoint receptors by ligands expressed on/by tumor cells and other populations within the TME may compromise antitumor immunity by interfering with the expansion, effecter differentiation, and survival of tumor‐specific T cells. Checkpoint inhibitors and other therapeutic strategies that disrupt engagement of these pathways in T lymphocytes can improve antitumor immunity by negating the signals delivered through these inhibitory receptors, as discussed in more detail in the main text. Abbreviations in this figure not defined in the main body of the text: TM, memory T cell
6. METABOLIC CHECKPOINTS INFLUENCING T CELL FUNCTION IN THE TUMOR MICROENVIRONMENT
Just as DC undergo metabolic reprogramming to support the acquisition of immune‐stimulating functions during their maturation and activation, T lymphocytes also modulate their metabolism following activation so that they may fulfill the biosynthetic and bioenergetic requirements of clonal expansion, effector differentiation, and memory formation. Shifts in carbohydrate, lipid, and amino acid metabolism all contribute to the progression of a T cell through its various stages of an immune response, and interference with any of these metabolic pathways can therefore have negative consequences on the immunoreactivity of these cells. In this regard, similar to its impact on DC metabolism, the TME also poses metabolic constraints on T lymphocytes, with competition for limited resources, regulation of T cell metabolism, and accumulation of immunosuppressive metabolic by‐products all acting as significant barriers to antitumor T cell function (Figure 4). Nevertheless, with recent insights into the metabolic dysfunction of T lymphocytes in the context of cancer, therapeutic strategies that restore immune‐supporting metabolic pathways in T cells and that overcome the deleterious effects of immunosuppressive metabolites on T cells within the TME are now emerging as viable options for improving the quality of antitumor T cell immunity.
FIGURE 4.
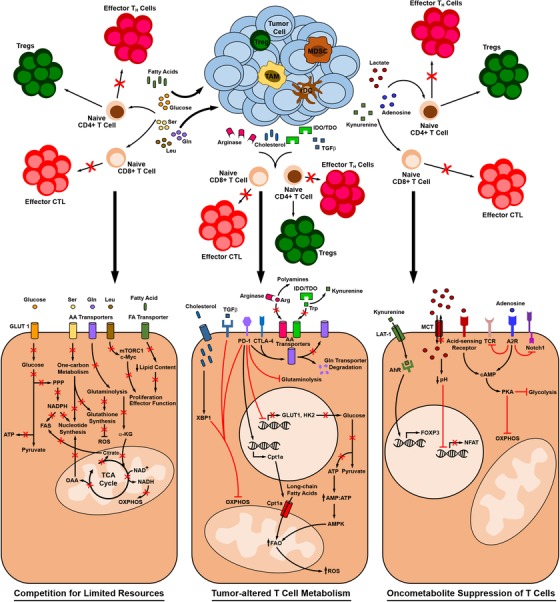
Metabolic regulation of T lymphocyte function within the TME. T lymphocyte metabolism is tightly linked with immune function and may be dysregulated within the TME in several ways. Competition between T lymphocytes and tumor cells for limiting resources can result in a lack of nutrient support for metabolic pathways essential to T cell activation and effector differentiation. Immunosuppressive cytokines, enzymes, and cholesterol derived from tumors and tumor‐associated cells can all impede metabolic pathways that support antitumor T cell function while also driving alternative pathways associated with immune tolerance. Finally, accumulation of suppressive oncometabolites produced by metabolically active tumors and tumor‐associated cells suppress metabolic pathways essential to T cell activation and drive the differentiation of tumor‐supporting Tregs. As described in more detail in the main text, insight into the metabolic regulation of T lymphocyte function within the TME has yielded several targets for therapeutic strategies aiming to improve the metabolic fitness and immune function of antitumor T cells. Note that the magnified intracellular signaling and metabolic pathways shown reflect events that have been reported in either CD4+ or CD8+ T cells. Abbreviations in this figure not defined in the main body of the text or in other figure legends: rDC , regulatory DC; TAM, tumor‐associated macrophage; FA, fatty acid; AA, amino acid, α‐KG, α‐ketoglutarate; OAA, oxaloacetate; HK2, hexokinase 2; AhR, aryl hydrocarbon receptor
6.1. Tumor‐imposed limitations on T lymphocyte carbohydrate/energy metabolism
Though naïve T lymphocytes are relatively inactive in terms of their metabolism, achieving their modest energy needs with low rates of OXPHOS fueled by intermediates derived from glucose, glutamine, and fatty acid metabolism, 229 T cells undergoing activation and effector differentiation are metabolically reprogrammed toward aerobic glycolysis, utilizing glucose as a substrate for various biosynthetic pathways more so than as a source for TCA‐driven OXPHOS. 230 In particular, recently activated T lymphocytes upregulate expression of both GLUT1 and LDHA, which enhance glucose uptake and divert pyruvate away from the TCA cycle, respectively. 231 , 232 The resulting commitment to glycolytic metabolism enables ATP production while also allowing intermediates from this pathway to be directed to the PPP for increased nucleotide biosynthesis and NADPH‐driven FAS, both of which are necessary to support rapid cell division during clonal expansion. In addition to meeting these biosynthetic requirements for T cell proliferation, glycolytic metabolism during T cell differentiation is also essential for acquisition of effector function. In this regard, Peng et al found that LDHA upregulation and reduced TCA cycle activity in stimulated T lymphocytes contribute to maintenance of high levels of acetyl‐coenzyme A, a substrate for histone acetylation needed to activate Ifng gene expression. 232 More recently, activation of glycolysis was also found to occupy LDHA and inhibit its binding to the 3′‐UTR of mRNAs encoding IFN‐γ, TNF‐α, and IL‐2, thereby preventing an interaction that otherwise represses translation of these cytokines. 233 A similar mechanism has also been reported by Chang et al, who found that glycolysis supports IFN‐γ‐based effector activity in T lymphocytes by maintaining glucose‐driven engagement of the glycolytic enzyme GAPDH and in turn preventing its own translational repression of IFN‐γ mRNA. 234 Importantly, in addition to enhancing effector cytokine production, glycolytic metabolism also supports expression of genes encoding the perforin and granzyme molecules essential for cytolytic effector function. 235
It has also become apparent that increased glycolytic metabolism during T cell activation does not come at the expense of OXPHOS, which is also upregulated in these cells in order to meet the bioenergetic demands that accompany their activation. In addition to supporting ATP production in activated T cells, mitochondrial metabolism also yields ROS important for activating specific cell signaling pathways that lead to clonal expansion. 236 With glucose serving as the substrate for aerobic glycolysis in activated T lymphocytes, OXPHOS is instead driven primarily by metabolism of glutamine in these cells, 230 , 237 , 238 as discussed in more detail below. Finally, though the transition from effector to memory T cells is associated with a shift from aerobic glycolysis to FAO‐based metabolism, glucose is still necessary to support the de novo synthesis of lipids for subsequent oxidation. 239 , 240 In memory cells, FAO enhances spare respiratory capacity, supporting mitochondrial function that not only promotes the long‐term survival of these cells but that is also key to the rapid induction and maintenance of both aerobic glycolysis and OXPHOS, which are again upregulated during recall responses. 241 , 242
Because glucose is such an essential nutrient for T cell activation and differentiation, competition for this resource between T lymphocytes and glycolytically active tumor cells poses significant limitations on the efficacy of antitumor T cell responses. To this point, experimental work in a progressor versus regressor tumor model has highlighted the relevance of this competition to antitumor T cell function. In in vitro studies, activated T lymphocytes co‐cultured with highly glycolytic progressor tumor cells exhibited reduced effector activity as compared to T cells co‐cultured with regressor tumor cells that consume less glucose, and glucose supplementation was sufficient to restore the effector function of T cells in progressor tumor co‐cultures. 82 Similarly, in vivo studies in this model demonstrated that glucose levels were reduced in the TME of progressor versus regressor tumors, and TIL isolated from progressor tumors displayed a lower extracellular acidification rate (indicative of reduced glycolytic metabolism) and produced less IFN‐γ than TIL from regressor tumors. Selection of glycolytically active cells from regressor tumor populations or retroviral transduction of regressor tumor cells with glycolysis‐promoting genes were each also sufficient to confer escape from T lymphocyte‐mediated control of tumor outgrowth. Interestingly, this and several other studies have also documented a link between various immunologic checkpoint pathways and glycolytic dysfunction in T cells, 82 , 147 , 148 , 196 demonstrating that competition for glucose is not the only mechanism by which glycolysis can be compromised in T cells within the TME. Indeed, glycolysis can be inhibited in T lymphocytes in a variety of ways. For instance, PD‐1 signaling in T cells downregulates expression of the GLUT1 and hexokinase 2 proteins needed to import and subsequently initiate breakdown of glucose. 147 On the other hand, in CD8+ TIL from renal cell carcinoma patients, reduced glucose uptake was associated not with reduced expression of GLUT1 but instead with a loss of phosphorylation of this transporter, a phenomenon that authors attributed to a probable failure of dephosphorylated GLUT1 to traffic to the cell surface. 243 This same study also reported downregulation of the glycolytic enzyme GAPDH in CD8+ TIL, highlighting another potential means of compromising this pathway downstream of glucose uptake. Another glycolysis‐regulating enzyme, enolase 1, is dysregulated by multiple checkpoint pathways, and reduced glycolytic metabolism of CD8+ TIL from B16 melanomas was associated with a loss of enolase 1 enzymatic activity when compared to acute T effector cells. 244 Interestingly, enolase 1 catalyzes formation of phosphoenolpyruvate, a glycolytic metabolite that sustains TCR‐mediated Ca++/NFAT signaling and effector function in both CD4+ and CD8+ T cells, 245 highlighting the significance of glycolysis not only as a means of achieving bioenergetic and biosynthetic needs in activated T lymphocytes but also as a mediator of stimulatory signaling in these cells.
Mitochondrial dysfunction has also been reported in TIL isolated from multiple cancer types. In addition to the aforementioned glycolytic defects in CD8+ TIL from renal cell carcinoma patients, mitochondria in these cells also exhibited morphological abnormalities (small and fragmented) and were hyperpolarized, producing elevated levels of ROS that limited TIL effector function. 243 Mitochondrial mass and function were also found to be reduced in CD8+ TIL isolated from murine melanomas, colon adenocarcinomas, and lung carcinomas, as well as from patients with HNSCC. 246 These defects were associated with reduced T lymphocyte expression of PGC1α, a protein critical for mitochondrial biogenesis. Others have shown that tumor‐derived TGF‐β impairs mitochondrial respiration in T cells 247 and that PD‐1 signaling alters mitochondrial structure and limits this organelle's capacity for OXPHOS. 248
Based on the glycolytic and mitochondrial deficiencies observed to date in tumor‐associated T lymphocytes, there has been great interest in exploring strategies to metabolically reprogram these cells in order to improve their function within the TME. T cell activation markers and cytokine secretion have been at least partially restored by metabolic reprogramming of TIL ex vivo, either by provision of pyruvate or phosphoenolpyruvate to bypass glycolytic defects or by treatment with mitochondrial ROS scavengers, 243 , 244 suggesting that interventions targeting these metabolic abnormalities might indeed have therapeutic benefit. To this point, overexpressing the PCK1 enzyme that converts oxaloacetate into phosphoenolpyruvate in adoptively transferred tumor‐specific CD4+ or CD8+ T cells improved the control of established B16 melanomas. 245 Likewise, overexpression of PGC1α in tumor‐specific CD8+ T cells enhanced mitochondrial function, effector activity, and the ability of adoptively transferred cells to control established tumors. 246 Together, these studies highlight the significance of biosynthetic and bioenergetic pathways as regulators of T cell function in the TME, and they offer great promise for metabolic manipulation of T lymphocytes as a means of enhancing the antitumor immune reactivity of these cells.
6.2. Lipid metabolism as a regulator of T cell function within the TME
As described above, the metabolic shift toward aerobic glycolysis during T cell activation is accompanied by a shift away from FAO as a major driver of OXPHOS. Rather than catabolizing lipids through lipolysis and subsequent FAO, these activated T cells instead switch to FAS in order to increase cell and organelle membrane biomass as they undergo clonal expansion. Increased fatty acid content is also likely to support effector differentiation of these cells as well, as lipid‐dependent posttranslational modifications have been found to enhance the activity of signaling pathways associated with this process. 249 , 250 In recently activated T cells, the increase in lipid content is driven not only by de novo lipogenesis but also by the uptake of exogenous fatty acids. 230 , 240 , 251 , 252 Likewise, tissue‐resident memory T cells also take up exogenous fatty acids to support their long‐term survival. 253 At the same time, it is well‐established that tumor cells upregulate expression of fatty acid transporters and scavenger receptors such as CD36 so that they too may increase uptake of exogenous fatty acids and lipoproteins to support cell growth, survival, energy production, and a variety of oncogenic functions associated with tumor progression. 254 , 255 , 256 , 257 , 258 , 259 , 260 , 261 Therefore, competition between T lymphocytes and tumor cells for lipids within the TME may limit fatty acid acquisition by T cells, thus compromising the expansion and differentiation of recently activated cells as well as the long‐term persistence of more fully differentiated cells.
While tumor cells may deplete the TME of particular lipids important for T lymphocyte function, they may also promote accumulation of other lipids that suppress T cell responses. For instance, in several murine tumor models, it has been reported that cholesterol levels are enriched in tumor tissue as compared to normal tissues and that the cholesterol content of exhausted CD8+ TIL isolated from both murine tumors and cancer patients is elevated compared to that in T cells isolated from non‐cancerous tissue. 262 Cholesterol accumulation in TIL drives exhaustion by promoting expression of the ER stress sensor XBP1, a transcription factor that directly upregulates expression of PD‐1 and 2B4. Importantly, this and a related study highlighted several interventions that could improve antitumor immune function by interfering with cholesterol metabolism or its downstream effects, including treatment of tumor‐bearing mice with an ER stress inhibitor, adoptive transfer of XBP1‐knockdown tumor‐specific CD8+ T cells, treatment of CD8+ T cells with β‐cyclodextrin to deplete cholesterol prior to adoptive transfer, and pharmacologic reduction of cholesterol content in the TME. 262 , 263 Another group has also shown that cholesterol esterification, rather than cholesterol uptake or biosynthesis, is the primary factor influencing T lymphocyte dysfunction, and treatment of tumor‐bearing mice with an inhibitor of the ACAT1 enzyme that promotes cholesterol esterification increased the number of effector and effector memory CD8+ TIL and resulted in delayed tumor progression. 264
In addition to regulating lipid content within the TME, tumor cells can also exert more direct influence over lipid metabolism by T cells. Glucose depletion by tumor cells can yield higher AMP:ATP ratios within T lymphocytes and therefore activate signaling via the nutrient sensor AMPK, a known driver of FAO. 265 While such metabolic plasticity may enable T lymphocytes to generate ATP and survive in glucose‐depleted environments, FAO does not support the effector differentiation of these cells necessary for antitumor reactivity. Even in a TME where glucose is not limiting, the engagement of multiple checkpoint pathways can also suppress glycolytic metabolism in T lymphocytes, thus requiring these cells to adopt alternative metabolic pathways, including FAO, to meet their bioenergetic demands. Indeed, PD‐1 signaling impedes effector differentiation in part by upregulating expression of CPT1 mitochondrial fatty acid transporters and directly reprogramming T cells toward FAO. 147 , 248 , 266 Of significance, while such reprogramming is detrimental to the differentiation of CD8+ T cells and the TH1, TH2, and TH17 subsets of CD4+ T cells, FAO actually favors the induction of Tregs, which may utilize glycolysis but rely on it less heavily for their differentiation. 267 , 268 Interestingly, lipid availability to Tregs was recently shown to control mitochondrial integrity and suppressive activity, with insufficient lipid uptake driving increased IL‐10 production via mtDNA release from damaged mitochondria and subsequent activation of the cGAS‐STING‐type 1 IFN pathway. 269 Based on these findings, it is interesting to speculate that tumors may not only foster the “development” of Tregs by promoting FAO in these cells at early stages of tumor progression but that they may also drive the “suppressive activity” of these cells as they continue to proliferate and deplete lipids from the TME over time.
The shift from FAO to glycolytic metabolism is clearly important during early stages of T lymphocyte activation and is therefore significant to the induction of natural antitumor T cell responses. Likewise, the antitumor efficacy of ex vivo‐generated CTL used for ACT therapy is also dependent on glycolysis and can therefore be restricted by the glycolytic activity of the tumors they target. 270 As discussed above, metabolic interventions that support glycolytic metabolism by T cells are therefore attractive strategies for promoting T cell effector function within the hostile TME. Alternatively, while FAO fails to support potent T cell effector differentiation in vivo, insights into the role of this pathway in promoting T lymphocyte survival and memory development have spearheaded efforts to manipulate FAO as a means of generating long‐lived cells for ACT‐ and CAR‐T‐based cancer therapies. In this regard, Sukumar et al found that inhibiting glycolysis during ex vivo expansion of Ag‐specific T cells resulted in acquisition of a memory phenotype and led to greater accumulation of these cells in tumors following adoptive transfer. 271 In keeping with the ability of FAO to support memory T cell engagement of glycolysis and OXPHOS during recall responses, 242 when these adoptively transferred cells were re‐isolated from B16 melanomas 5 days post‐transfer, they expressed high levels of glycolytic enzymes and displayed augmented effector functions, consistent with their improved ability to control tumor outgrowth. Similar data in the B16 melanoma model was reported by Crompton et al, who also showed that human TIL expanded under glycolysis‐inhibiting conditions exhibited greater survival following transfer into a humanized mouse model, suggesting this approach might also improve the antitumor efficacy of ACT therapy in cancer patients. 272 In contrast to these studies that have employed inhibitors of glycolysis to modulate T cell metabolism during ex vivo production of T lymphocytes for ACT therapy, others have shown more direct evidence that maneuvers to enhance FAO improve the persistence and antitumor reactivity of adoptively transferred T cells. 273 , 274 It has also been shown that the design of CAR‐T cells influences metabolic activity and that FAO‐promoting CARs enhance survival of these cells in vitro. Specifically, chimeric TCRs fused to 4‐1BB signaling domains are stimulated to undergo FAO and survive longer than those with TCRs fused to CD28 signaling domains, which instead preferentially undergo glycolysis upon stimulation. 275 While this study did not examine T cell survival or antitumor activity in vivo, these data suggest that conditions favoring FAO metabolism in CAR‐T cells might indeed translate to improved therapeutic efficacy post‐adoptive transfer. Future studies will indeed be necessary to confirm this hypothesis and to identify the optimal conditions for generating robust CAR‐T cells, but at this point it is clear that metabolic considerations will play a key role in the design of these as well as non‐chimeric ACT‐based cancer therapies going forward.
6.3. Deficiencies in T lymphocyte amino acid metabolism in the TME
As interest in T cell and cancer metabolism has increased in recent years, a number of amino acids have emerged as critical regulators of the outcome of T lymphocyte/tumor cell interactions within the TME. Along with the metabolic switch to aerobic glycolysis during early stages of T lymphocyte activation, T cells also increase their metabolism of glutamine, upregulating expression of membrane transport proteins that promote glutamine uptake and increasing the activity of enzymes that drive subsequent glutaminolysis. 237 Like glycolysis, glutaminolysis also supports biosynthetic pathways in activated T cells and is critical to both proliferation and cytokine production by these cells. Additionally, glutamine metabolism plays an equally important role in the generation of α‐ketoglutarate, a TCA cycle intermediate that supports additional ATP production through oxidative metabolism 230 , 237 and that drives effector, rather than Treg, differentiation. 238 , 276 Glutaminolysis further supports effector T cell differentiation and survival by driving glutathione synthesis and in turn allowing T lymphocytes to maintain redox homeostasis. 277 , 278 That glutaminolysis is enhanced by AMPK signaling during glucose deprivation highlights in particular the utility of this pathway as a means of maintaining T cell biosynthetic and bioenergetic programs in a glucose‐limiting environment, 279 such as that often encountered in the TME. However, the same benefits that glutamine provides to T lymphocytes also exist for cancer cells themselves, which likewise increase glutamine uptake and metabolism as part of their own metabolic reprogramming and therefore compete with T cells for use of this resource. 280 In addition to this competition, engagement of T lymphocyte checkpoints can also directly interfere with glutamine metabolism in much the same way as described above for other metabolic pathways. Both PD‐1 and CTLA‐4 signaling inhibit expression of glutamine transporters on T cells, and PD‐1 signaling also suppresses glutaminolysis within these cells. 147
Because of its role in supporting cancer progression, there has been interest in targeting glutamine metabolism in cancer cells therapeutically. While the evidence that T cells rely on glutamine uptake and metabolism during their activation suggests that such interventions might also compromise the metabolic fitness of T cells within the TME, recent studies have revealed that at least in certain contexts the metabolic plasticity of T cells may be greater than that of cancer cells, thereby enabling survival and maintenance of effector activity in the face of glutaminolytic inhibition. Though chronic or complete inhibition of glutaminolysis impaired T lymphocyte function in vivo, transient inhibition in vitro enhanced the survival and effector function of adoptively transferred TH1 and CD8+ T cells, 281 suggesting that these cells might be able to provide more durable antitumor immune reactivity. Pharmacologic blockade of glutamine metabolism in MC38 tumor‐bearing mice also supported antitumor T cell function, suppressing not only glutaminolysis but also glutamine‐driven glycolytic metabolism in tumor cells. In addition to these direct antitumor effects, inhibiting these pathways in cancer cells also restored nutrient availability within the TME, allowing metabolically flexible T cells to exploit alternative pathways that enhanced their proliferation, survival, and antitumor effector function. 282
Competition between tumor cells and T lymphocytes for serine and leucine can also compromise the metabolic fitness of T cells within the TME. Acquisition of extracellular serine is necessary to support the proliferation of activated T cells, which rely on this amino acid as a source of one‐carbon metabolism for de novo nucleotide biosynthesis. 283 However, uptake of exogenous serine is utilized for this same purpose by cancer cells, which otherwise exhibit defects in both proliferation and survival under conditions of serine deprivation. 284 , 285 T lymphocytes also upregulate expression of the Slc7a5 leucine transporter following activation, and sustained uptake of leucine is necessary to maintain mTORC1 activation and c‐Myc expression, both of which drive clonal expansion and effector differentiation. 286 However, as Slc7a5 and another leucine transporter, Slc43a1, are also upregulated on many tumor cell types, 287 , 288 leucine uptake by these cells could ultimately disrupt the antitumor functionality of T cells by starving them of this important amino acid within the TME.
In addition to tumor cells, other cell types that frequently accumulate within the TME also interfere with metabolism of amino acids important for T cell function. Myeloid‐derived suppressor cells (MDSC), M2‐like tumor‐associated macrophages, and immunoregulatory DC have all been shown to produce high levels of arginase I within tumor tissue 289 , 290 , 291 , 292 , 293 and can therefore deplete the TME of arginine, an amino acid critical to the survival and antitumor reactivity of T lymphocytes. 294 Arginase activity also promotes the induction of Tregs, further contributing to the immunosuppressive environment within a tumor. 295 Moreover, though the extent to which arginine is utilized by DC during their activation is currently unknown, it has been reported that polyamine by‐products of arginine metabolism do condition DC to acquire an immunosuppressive phenotype, 296 and this pathway may also contribute to T cell dysfunction within a TME where arginine is heavily metabolized. Interestingly, this study revealed that arginine‐derived polyamines promoted DC expression of enzymatically active IDO‐1, a potent inducer of tryptophan catabolism. Tryptophan is another amino acid critical to antitumor T cell function, and IDO‐1 expression by tumor‐associated MDSC and regulatory pDC is a frequently reported mechanism of tumor immune escape. 297 , 298 , 299 Another tryptophan‐catabolizing enzyme, tryptophan‐2,3‐dioxygenase (TDO), is also expressed in several human tumor types and has been shown to limit the immunologic control of murine tumors. 300 , 301
A number of small molecule inhibitors targeting IDO and TDO have now been developed and are in clinical trials for various cancer types, 302 and a first‐in‐class arginase inhibitor (CB‐1158) that has been shown to enhance antitumor T cell reactivity in murine models is also now in early phase clinical trials. 303 Though data has yet to be released for the arginase inhibitor trials, the outcomes of trials for IDO inhibitors administered either as single agents or as part of combinatorial regimens with checkpoint blockade therapy have unfortunately been met with limited success. 304 , 305 Off‐target effects and tumor‐acquired resistance have both compromised the efficacy of various IDO inhibitors developed to date and are issues that will need to be addressed in the future. To overcome the issue of off‐target effects, strategies to interfere with IDO‐mediated immune suppression that do not involve small molecule inhibitors have been devised. In this regard, targeted silencing of IDO in endogenous APC improves antitumor T cell function in a murine melanoma model, 306 and silencing IDO in exogenous DC prior to vaccination improves T cell function and tumor control in a murine breast cancer model. 307 A case report describing this latter strategy in a melanoma patient has revealed that this approach may indeed show immunologic and antitumor efficacy in humans as well. 308 In addition, for IDO and TDO inhibitors that do have tolerable safety profiles, it is hoped that the issue of tumor‐acquired resistance may be overcome by combinatorial administration of these inhibitors or the use of dual‐targeting IDO/TDO inhibitors, both of which might prevent tumor escape from more selective drugs that fail to inhibit an alternative tryptophan‐catabolizing pathway when administered as single agents.
6.4. Suppression of T lymphocytes by oncometabolites in the TME
The same oncometabolites that compromise DC function within the TME also suppress the function of T lymphocytes. First reported in the context of tumor spheroid and other in vitro settings, the accumulation of extracellular lactic acid was found to disrupt lactate export by glycolytically active T cells and to promote its buildup intracellularly in these cells, leading to suppressed proliferation, cytokine secretion, and cytotoxic function. 309 Subsequent work has since shown that lactic acid secretion by highly glycolytic tumors also blunts antitumor T cell functions in vivo. LDHA expression and lactic acid production by tumor cells correlates with suppression of TIL effector function, likely as a result of the poor lactate efflux and resulting intracellular acidification that interferes with NFAT expression in T cells. 310 Evidence has also emerged that T cell function can be compromised by lactate signaling via acid‐sensing receptors. 311 The significance of lactate accumulation to antitumor T cell dysfunction is highlighted by studies exploring approaches to interfere with acidification in the TME. Proton pump inhibitors, nonsteroidal anti‐inflammatory drugs that block lactate secretion by tumors, and bicarbonate therapy have all been shown to neutralize TME acidity and enhance either natural or therapy‐associated antitumor T cell responses. 311 , 312 , 313
As discussed above, lactic acid also indirectly influences T cell function within the TME by reprogramming intratumoral pDC to support Treg induction via increased tryptophan metabolism. 101 In addition to depleting this essential amino acid necessary for antitumor T cell function, another consequence of IDO/TDO activity in the TME is the release of kynurenine as a by‐product of tryptophan metabolism. Kynurenine interacts with the aryl hydrocarbon receptor on CD4+ T cells and promotes expression of FOXP3, 314 which in turn programs Treg metabolism to support survival and maintenance of immune regulatory function in low‐glucose, high‐lactate environments. 315 In this way, oncometabolites derived from both carbohydrate and amino acid metabolism act in concert to create an effector T cell/Treg imbalance that ultimately fosters immune escape and tumor progression.
Adenosine is another oncometabolite enriched in the TME that represses T lymphocyte function and promotes Treg accumulation, primarily via the A2A receptor. Engagement of this receptor promotes intracellular accumulation of cAMP and interferes with membrane proximal TCR signaling and activation of the Notch1 pathway, leading to reduced proliferation, cytokine secretion, and cytotoxic activity. 316 , 317 , 318 Underscoring the role of metabolism in the regulation of these various T cell functions, it was recently shown that adenosine‐driven cAMP accumulation also activates PKA signaling, which in turn suppresses mTORC1‐driven glycolysis and OXPHOS. 319 Together, these studies offer mechanistic explanations for the well‐documented link between adenosine in the TME and dysfunctional TIL. 320 , 321 , 322 Based on these and other reports, a number of strategies for interfering with adenosine‐mediated suppression of antitumor T cells have been investigated, including some that have made their way into clinical trials. Pharmacologic blockade of the A2A receptor reduces the frequency of peripheral and intratumoral Tregs and enhances not only tumor infiltration by CD8+ T cells but also the effector function of these cells. 323 , 324 Likewise, genetic ablation of the A2A receptor enhances the antitumor efficacy of both traditional and CAR‐T‐based ACT therapies. 322 , 325 Consistent with the role of tumor hypoxia in the generation of extracellular adenosine and upregulation of the A2A receptor, 326 hyperoxic therapy can also reverse hypoxia‐adenosinergic immunosuppression within the TME, leading to improved antitumor reactivity by both endogenous and adoptively transferred CD8+ T cells. 327 Finally, blockade of CD39 and CD73, as well as another adenosine‐generating ectoenzyme belonging to the ADP ribosyl cyclase family, CD38, improves the functional quality of antitumor T cells. 328 , 329 These latter approaches in particular may support antitumor T cell function in multiple ways, as evidenced by a recent study demonstrating that CD39 blockade not only reduces the level of adenosine available for direct suppression of T cells but also promotes immunogenic signaling by extracellular ATP through P2X7 purinergic receptors on myeloid cell populations. 330 This finding may also explain the synergistic effects of CD39 blockade in combination with oxaliplatin chemotherapy, 329 which itself promotes ATP release from apoptotic tumor cells and may therefore support purinergic receptor signaling under conditions in which released ATP is not quickly converted to adenosine.
Collectively, the studies described herein reveal great promise for metabolic interventions as a strategy to support the induction and maintenance of antitumor immune responses. They also highlight the significance of tumor metabolomic profiling as a useful tool for designing appropriate treatment plans in cancer patients. As such profiling continues to become more commonplace in clinical settings, 331 , 332 , 333 , 334 our understanding of how particular metabolic signatures are likely to influence antitumor immune responses will have increasing prognostic value in predicting patient response to immunotherapy, and it will therefore allow for more effective patient stratification toward particular treatment regimens most likely to achieve clinical benefit. As discussed in more detail below, these metabolomic insights are also capable of informing the design of combinatorial regimens that may ultimately increase the frequency of patients who benefit from immunotherapy, as such regimens can ultimately yield a TME that is more metabolically permissive to antitumor immune reactivity.
7. COMBINATORIAL APPROACHES TO CANCER THERAPY: TAKING AN OLD IDEA TO NEW HEIGHTS
There is nothing new about the idea of combinatorial therapy for cancer. Indeed, the history of oncology is replete with examples of combinatorial approaches designed to attack cancer from multiple angles. However, a recurring theme over the years has been that while such approaches have achieved success in some cases, their initial promise has never been fully realized, typically due to a poor understanding of cancer biology. Now more than ever, with advances in our understanding of the basic biology of both the immune system and the cancer cell, the field of oncology seems truly poised for significant breakthroughs in both targeted and immunotherapeutic approaches to treatment in the years ahead, and the potential for achieving durable clinical benefits from combinatorial regimens that support the antitumor functions of both DC and T cells is perhaps the most promising for cancer therapy to date.
Based on their success in the clinic, a number of checkpoint inhibitors have received FDA approval for use as cancer therapeutics (Table 1). In particular, recent trials of combinatorial ICB therapy have been extremely promising, leading to unprecedented outcomes in the clinical management of multiple cancer types. Phase III clinical trials for ipilimumab + nivolumab combination therapy have produced staggering overall survival statistics, with a 5‐year survival rate of 52% for advanced melanoma patients 154 and a 2‐year survival rate of 40% for stage IV or recurrent NSCLC patients whose tumors are characterized by PD‐L1 expression levels ≥1%. 155 With the discovery of additional immunologic checkpoints beyond CTLA‐4 and PD‐1/PD‐L1 and ongoing development of novel checkpoint inhibitors targeting these pathways in both DC and T lymphocytes (Figures 1 and 3), it is reasonable to expect in the near future that new combinations will further increase either the percentage of patients responding to ICB therapy or the duration of responses elicited in the subset of patients who do benefit from these regimens.
TABLE 1.
Summary of FDA‐approved immunologic and metabolic checkpoint inhibitors
| Target | Therapeutic | Class of Drug |
|---|---|---|
| CTLA‐4 | Ipilimumab (Yervoy®) | Humanized IgG1 mAb |
| PD‐1 | Nivolumab (Opdivo®) | Fully human IgG4 mAb |
| Pembrolizumab (Keytruda®) | Humanized IgG4 mAb | |
| Cemiplimab (Libtayo®) | Fully human IgG4 mAb | |
| PD‐L1 | Atezolizumab (Tecentriq®) | Humanized IgG1 mAb |
| Avelumab (Bavencio®) | Fully human IgG1 mAb | |
| Durvalumab (Imfinzi®) | Fully human IgG1 mAb | |
| AXL (TAM RTK) | Cabozantinib (Cabometyx®) | Small molecule inhibitor (also inhibits c‐MET, VEGFR2, and MET) |
| Gilteritinib (Xospata®) | Small molecule inhibitor (also inhibits FLT3) | |
| CD38 | Daratumumab (Darzalex®) | Fully human IgG1 mAb |
| Isatuxumab (Sarclisa®) | Chimeric murine/human IgG1 mAb |
Despite the success of ICB therapy to date, there remains a significant population of patients who receive no clinical benefit from treatment with immune checkpoint inhibitors. One major factor that has been associated with response to checkpoint blockade is the presence of a preexisting immune response to the tumor, such that there is a population of T lymphocytes amenable to the effects of checkpoint inhibition. 335 , 336 , 337 , 338 This prerequisite means that patients who do not mount natural immune responses to their tumors are inherently poor candidates for ICB therapy. Pre‐immunization of such immunologically naïve individuals with DC‐based vaccines that induce T cell responses to tumor Ag has therefore become an appealing strategy for increasing the percentage of patients likely to benefit from ICB regimens. Though exogenous DC vaccines and DNA/peptide/protein‐based vaccines that rely on Ag processing and presentation by endogenous DC have had minimal efficacy as monotherapies in cancer patients to date, 75 these approaches have also likely been reciprocally limited by checkpoint inhibition of the T cell responses they induce. Therefore, combining DC‐based and ICB therapies may indeed yield synergistic antitumor and clinical benefits, as vaccination could induce an antitumor T cell response capable of being supported by checkpoint blockade. To this point, a recent study found that autologous DC vaccination of metastatic melanoma patients yielded a T cell‐inflamed TME from tumors that had previously been characterized as immunologically “cold.” 339 Of note, concurrent with the vaccine‐induced transition from “cold” to “hot” tumors, PD‐L1 expression was increased on tumor cells, and intratumoral CD8+ T cells were found not to express granzyme B. These findings support the aforementioned notion that DC‐based therapy alone may be compromised by tumor‐associated suppression of vaccine‐induced T cells but that checkpoint blockade might prevent tumor escape from such a vaccine‐induced response. As proof of principle, studies in murine models have shown that combining DC vaccination with checkpoint blockade therapy enhances TIL effector function and control of tumors. 340 , 341 A recent report from a Phase I clinical trial of gastric cancer patients also documented a reversal of dysfunction in vaccine‐induced CD8+ T cells when these cells were re‐stimulated in the presence of blocking antibodies against various checkpoint receptors, suggesting that a combination of DC vaccination and checkpoint blockade could also have therapeutic benefit in patients. 342 Similar results have been obtained with therapeutic strategies that rely on the activity of endogenous DC. Peptide vaccination of advanced melanoma patients elicits CD8+ T cell responses that are restrained by PD‐1 and TIM‐3, and blockade of these receptors during ex vivo restimulation enhances their proliferative capacity and effector cytokine production. 343 Likewise, regimens that promote cDC1 expansion and activation in tumors 9 , 344 , 345 , 346 or that target tumor Ag to this particular DC subset 347 are also supported by checkpoint blockade.
As described herein, a handful of checkpoint pathways typically associated with the regulation of T cell immunity are also shared by DC. It is therefore likely that currently approved checkpoint inhibitors targeting these particular pathways improve antitumor T cell responses not only via direct influences on T cells but also by indirect mechanisms that involve enhanced DC function, as has recently been shown in a murine tumor model. 348 In addition to the benefits of targeting these pathways, the identification of other innate‐specific immunologic checkpoints now offers a new array of targets for inhibitors that specifically engage innate populations such as DC. Targeting these innate checkpoints in conjunction with what have classically been viewed as T lymphocyte checkpoints is emerging as an attractive way of supporting both the induction and maintenance of antitumor T cell responses. For instance, several preclinical studies have demonstrated the therapeutic efficacy of regimens that concomitantly target CD47 and the PD‐1/PD‐L1 or CTLA‐4 axes. 349 , 350 , 351 , 352 , 353 , 354 As anticipated, mechanistic insights from many of these studies have shown that the enhanced phagocytosis and immune sensing of tumor cells exhibited by DC following CD47 blockade promotes tumor Ag cross‐presentation and the activation of antitumor T cells whose effector functions are then bolstered by interference with the CTLA‐4 and PD‐1 pathways. With the multitude of phagocytic and innate immune sensing checkpoints discovered to date (Figure 1), targeting other combinations of innate and adaptive checkpoints pathways may also broaden the reach and efficacy of ICB therapy for cancer patients.
Though combination therapies targeting both DC and T lymphocytes can yield more robust immune responses than monotherapies, metabolic suppression within the TME remains a significant barrier to the antitumor immune function of these cells and can limit the efficacy of immune responses achieved by vaccination, ICB, adoptive transfer regimens, or any combination thereof. Therefore, therapeutic strategies that also consider ways of improving the metabolic fitness of DC and T lymphocytes are also emerging as effective approaches to support cancer immunotherapies. While many of the checkpoint pathways described in this review regulate immunometabolism themselves, it is also possible to combine ICB or other therapies with more direct approaches to improve metabolic conditions for tumor‐associated DC and T cells. As an example, accumulation of serum kynurenine as a result of the IDO/TDO‐tryptophan‐kynurenine pathway is a poor prognostic factor for the overall survival of melanoma and renal cell carcinoma patients treated with nivolumab, 355 suggesting that interference with this pathway might improve patient response to PD‐1 and possibly other checkpoint inhibitors. To this point, inhibitors of both IDO and TDO have both recently been shown to improve the antitumor efficacy of ICB therapies in murine models, 356 , 357 , 358 as has pharmacologic inhibition of either signaling or metabolic pathways that promote IDO expression in DC. 98 , 359 Likewise, multiple approaches to augment immunotherapy by interfering with adenosine‐mediated immune suppression have also been explored. In murine models, antibody blockade and pharmacological inhibition of either ectonucleotidases that generate adenosine from extracellular ATP or A2A receptors that transmit suppressive signals from adenosine have both been shown to enhance the antitumor efficacy of checkpoint blockade and ACT therapies. 328 , 330 These strategies are currently being investigated in clinical settings as well, and their potential is highlighted by results from a recent Phase I trial demonstrating immunological and clinical benefits of combination therapy with the A2AR antagonist ciforadenant and the PD‐L1 inhibitor atezolizumab in treatment‐refractory renal cell carcinoma patients. 324 Similar combinatorial approaches that aim to offset the immunosuppressive effects of other oncometabolites such as lactate or that drive immune‐promoting metabolic pathways in DC and T cells are also being investigated in preclinical and clinical settings. Examples of these and other types of combination therapies designed to support the immunologic functions of DC and/or T lymphocytes and that have reached clinical stages of development are highlighted in Table 2. Though by no means a comprehensive list, the select trials described here provide a snapshot of the types of combinatorial approaches made possible by recent advances in our understanding of immune regulation within the TME, and it will be exciting to follow the outcomes of these and related trials in the months and years ahead.
TABLE 2.
Select clinical trials investigating combinatorial therapies that target/interfere with immunologic/metabolic checkpoints
| Innate + Adaptive Immune Checkpoint Blockade | |||||
|---|---|---|---|---|---|
| Treatment | Cancer(s) | Trial | Phase | Status | Rationale |
|
TTI‐621 + α‐PD‐1/PD‐L1 |
Various solid tumors | NCT02890368 | I | Recruiting | TTI‐621 is a SIRPα‐IgG1 recombinant fusion protein that binds CD47. May enhance DC‐mediated T cell responses that can be supported by PD‐1/PD‐L1 blockade. |
|
ALX148 + Pembrolizumab |
Advanced solid tumors, Non‐Hodgkin lymphoma |
NCT03013218 | I | Recruiting | ALX148 is a SIRPα‐IgG1 recombinant fusion protein that binds CD47. May enhance DC‐mediated T cell responses that can be supported by PD‐1 blockade. |
|
BI 765063 + BI 754091 |
Advanced solid tumors | NCT03990233 | I | Recruiting | BI 765063 is an α‐SIRPα mAb, and BI 754091 is an α‐PD‐1 mAb. May enhance DC‐mediated T cell responses that can be supported by PD‐1 blockade. |
| Immunologic + Metabolic Checkpoint Blockade | |||||
|---|---|---|---|---|---|
| Treatment | Cancer(s) | Trial | Phase | Status | Rationale |
|
Ciforadenant + Atezolizumab |
RCC, Metastatic castration‐resistant prostate cancer | NCT02655822 | I | Recruiting | Ciforadenant is a small molecule antagonist of A2AR that may prevent adenosine‐mediated suppression of T cells supported by PD‐L1 blockade. |
|
Daratumumab + Nivolumab |
Pancreatic cancer, NSCLC, TNB | NCT03098550 | I/II |
Active, not recruiting |
Daratumumab is an α‐CD38 mAb that blocks conversion of ATP to adenosine. May enhance immunogenic ATP signaling in DC and prevent adenosine‐mediated suppression of T cells supported by PD‐1 blockade. |
|
TTX‐030 + Pembrolizumab |
Solid tumors, Lymphoma |
NCT03884556 | I | Recruiting | TTX‐030 is an α‐CD39 mAb that blocks conversion of ATP to adenosine. May enhance immunogenic ATP signaling in DC and prevent adenosine‐mediated suppression of T cells supported by PD‐1 blockade. |
|
Oleclumab + Durvalumab + Chemotherapy |
TNBC | NCT03616886 | I/II | Recruiting | Oleclumab is an α‐CD73 mAb that may prevent adenosine‐mediated suppression of T cells supported by PD‐L1 blockade. |
|
INCB001158 + Pembrolizumab |
Advanced/metastatic solid tumors | NCT02903914 | I/II | Recruiting | INCB001158 is an arginase inhibitor that may prevent arginine metabolism‐associated suppression of T cells supported by PD‐1 blockade. |
|
BMS‐986205 + Nivolumab + Radiation ± Chemotherapy |
Glioblastoma | NCT04047706 | I | Recruiting | BMS‐986205 is an IDO1 inhibitor that may prevent tryptophan metabolism‐associated suppression of T cells supported by PD‐1 blockade. |
|
Indoximod + Ipilimumab, Nivolumab, or Pembrolizumab |
Unresectable stage III/IV melanoma |
NCT02073123 | I/II |
Active, not recruiting |
Indoximod is a tryptophan mimetic that inhibits IDO/TDO activity and may therefore prevent tryptophan metabolism‐associated suppression of T cells supported by CTLA‐4 or PD‐1 blockade. |
|
BMS986205 + Nivolumab + Relatlimab |
Advanced or metastatic solid cancers | NCT03459222 | I/II | Recruiting | BMS986205 is an IDO1 inhibitor that may prevent tryptophan metabolism‐associated suppression of T cells supported by combination PD‐1/LAG‐3 blockade. |
| Vaccination + Immunologic/Metabolic Checkpoint Blockade | |||||
|---|---|---|---|---|---|
| Treatment | Cancer(s) | Trial | Phase | Status | Rationale |
|
Peptide‐pulsed autologous DC + Pembrolizumab + Cycolphosphamide |
Stage III/IV melanoma | NCT03092453 | I | Recruiting | Cyclophosphamide to deplete Tregs, followed by DC vaccine to induce a melanoma Ag‐specific T cell response that can be supported by PD‐1 blockade. |
|
Tumor lysate‐pulsed autologous DC + Poly ICLC + Pembrolizumab |
Recurrent glioblastoma | NCT04201873 | I | Recruiting | Poly ICLC may enhance the immunogenicity of DC vaccine, inducing a tumor Ag‐specific T cell response that can be supported by PD‐1 blockade. |
|
mRNA vaccine (BI 1361849) + Durvalumab ± Tremelimumab |
NSCLC | NCT03164772 | I/II | Recruiting | BI 1361489 is a self‐adjuvanted mRNA vaccine that may elicit T cell responses capable of being supported by PD‐L1 blockade (durvalumab) or a combination of PD‐L1 and CTLA‐4 blockade (tremelimumab). |
|
TLPLDC Vaccine + Standard‐of‐care ICB |
Stage IV melanoma | NCT02678741 | I/II |
Active, not recruiting |
TLPLDC (tumor lysate, yeast particle‐loaded DC) vaccine may induce T cell responses whose antitumor efficacy may be enhanced by an approved immune checkpoint inhibitor. |
|
Tergenpumatucel‐L vaccine + Docetaxel + Indoximod |
NSCLC | NCT02460367 | I |
Active, not recruiting |
Tergenpumatucel‐L is a vaccine comprised of allogeneic NSCLC cells genetically modified to express a carbohydrate to which humans have pre‐existing immunity. The tryptophan mimetic indoximod may improve the efficacy of standard vaccination and chemotherapy by preventing IDO/TDO‐mediated immune suppression. |
| Adoptive Cell Transfer Therapy + Immune Checkpoint Blockade/Interference | |||||
|---|---|---|---|---|---|
| Treatment | Cancer(s) | Trial | Phase | Status | Rationale |
|
Autologous TIL/IL‐2 + Nivolumab |
Stage IIIb/IIIc/IV melanoma | NCT03374839 | I/II | Recruiting | α‐PD‐1 ICB therapy may enhance the antitumor efficacy of adoptively transferred TIL by preventing PD‐1‐mediated immune suppression. |
|
Autologous TIL/IL‐2 + Nivolumab + Lymphodepleting chemotherapy |
NSCLC, SCC, Adenosquamous carcinoma, Adenocarcinomas | NCT03215810 | I |
Active, not recruiting |
α‐PD‐1 ICB therapy may enhance the antitumor efficacy of adoptively transferred TIL by preventing PD‐1‐mediated immune suppression. |
|
Autologous TIL/IL‐2 + Ipilimumab + Nivolumab |
Metastatic melanoma | NCT03526185 | I | Recruiting | The antitumor efficacy of adoptively transferred TIL may be improved by combinatorial blockade of the CTLA‐4 and PD‐1 checkpoints. |
|
NYCE T cells with CRISPR‐edited TCR and PD‐1 + Lymphodepleting chemotherapy |
Multiple myeloma, Melanoma, Synovial sarcoma, Myxoid/round cell liposarcoma |
NCT03399448 | I |
Active, not recruiting |
NYCE T cells are autologous NY‐ESO‐1 redirected T cells that have been CRISPR‐edited to eliminate expression of endogenous TCR and PD‐1. May improve efficacy of adoptively transferred tumor Ag‐specific T cells by preventing PD‐1‐mediated suppression. |
|
EGFRvIII‐redirected CAR‐T cells + Pembrolizumab |
EGFRvIII+, MGMT‐unmethylated glioblastoma | NCT03726515 | I |
Active, not recruiting |
PD‐1 blockade may enhance the antitumor activity of autologous CAR‐T cells engineered to recognize EGFRvIII+, MGMT‐unmethylated glioblastomas that are poorly responsive to standard‐of‐care alkylating chemotherapy. |
| MUC1‐redirected CAR‐T cells with CRISPR‐mediated PD‐1 KO | Advanced NSCLC | NCT03525782 | I | Recruiting | Interference with PD‐1‐mediated immune suppression by CRISPR gene editing may improve the antitumor efficacy of adoptively transferred MUC1‐redirected CAR‐T cells. |
Abbreviations: KO, knockout; NSCLC, non‐small cell lung cancer; RCC, renal cell carcinoma; SCC, squamous cell carcinoma; TNBC, triple negative breast cancer.
8. CONCLUSIONS
Over the past two decades, the field of oncology has been witness to an unparalleled transformation in our understanding of the immune system in the context of cancer. Insights into the basic immunobiology of DC and T lymphocytes have revealed an exquisite cooperation between these cells over the course of an antitumor immune response, and understanding how the activity of these cells is controlled by both immunologic and metabolic factors has shaped our appreciation for the diverse mechanisms regulating immune function within the TME. The complexity of this regulation cannot be overstated, with an array of immunologic and metabolic checkpoints now being recognized as potential barriers to antitumor immune function. At the same time, this complexity has also shed light on a multitude of potential targets for therapeutic interventions that aim to promote metabolic fitness and antitumor immune reactivity by both DC and T cells. The therapeutic success of many of these interventions in preclinical settings has paved the way for significant advances in the treatment of cancer patients, yielding never‐before‐seen rates, and duration, of clinical response. Moreover, insights into both innate and acquired mechanisms of resistance to these approaches have pointed towards a new age of combinatorial therapies that is already paying significant dividends in the clinical management of diverse cancer types. Though there will certainly be no one‐size‐fits‐all approach to these next‐generation combinatorial regimens, thorough biomarker analyses at the time of initial cancer diagnosis will in many cases reveal the ideal cohort of targets necessary to inform optimal therapeutic design. With this information in hand, the broad armamentarium of drugs and strategies either already available or currently in development will soon enable implementation of perhaps the most sophisticated forms of precision medicine ever seen in the field of immuno‐oncology, and it is expected that these approaches will continue to revolutionize cancer therapy in both the near and distant future.
CONFLICT OF INTEREST
The author has declared no conflict of interest.
ACKNOWLEDGMENTS
Research in the Hargadon laboratory is funded by a Mary Louise Andrews Award for Cancer Research from the Virginia Academy of Science, a Hampden‐Sydney College Research Grant from the Arthur Vining Davis endowment, and generous donations from Mr. Michael Hargadon and Mrs. Patricia Hargadon to support the involvement of undergraduate students in research at Hampden‐Sydney College. These funding sources were not involved in the preparation of this manuscript or in the decision to publish this review article.
Hargadon KM. Tumor microenvironmental influences on dendritic cell and T cell function: a focus on clinically relevant immunologic and metabolic checkpoints. Clin Transl Med. 2020;10:374–411. 10.1002/ctm2.37
REFERENCES
- 1. Hargadon KM, Johnson CE, Williams CJ. Immune checkpoint blockade therapy for cancer: an overview of FDA‐approved immune checkpoint inhibitors. Int Immunopharmacol. 2018;62:29‐39. [DOI] [PubMed] [Google Scholar]
- 2. Jacoby E, Shahani SA, Shah NN. Updates on CAR T‐cell therapy in B‐cell malignancies. Immunol Rev. 2019;290(1):39‐59. [DOI] [PubMed] [Google Scholar]
- 3. Smyth MJ, Thia KY, Street SE, MacGregor D, Godfrey DI, Trapani JA. Perforin‐mediated cytotoxicity is critical for surveillance of spontaneous lymphoma. J Exp Med. 2000;192(5):755‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410(6832):1107‐1111. [DOI] [PubMed] [Google Scholar]
- 5. Smith JL, Stehlin JS. Spontaneous regression of primary malignant melanomas with regional metastases. Cancer. 1965;18(11):1399‐1415. [DOI] [PubMed] [Google Scholar]
- 6. Saleh FH, Crotty KA, Hersey P, Menzies SW. Primary melanoma tumour regression associated with an immune response to the tumour‐associated antigen melan‐A/MART‐1. Int J Cancer. 2001;94(4):551‐557. [DOI] [PubMed] [Google Scholar]
- 7. Reis e Sousa C. Activation of dendritic cells: translating innate into adaptive immunity. Curr Opin Immunol. 2004;16(1):21‐25. [DOI] [PubMed] [Google Scholar]
- 8. Böttcher JP, Reis e Sousa C. The role of type 1 conventional dendritic cells in cancer immunity. Trends in Cancer. 2018;4(11):784‐792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Salmon H, Idoyaga J, Rahman A, Leboeuf M, Remark R, Jordan S, et al. Expansion and activation of CD103+ dendritic cell progenitors at the tumor site enhances tumor responses to therapeutic PD‐L1 and BRAF inhibition. Immunity. 2016;44(4):924‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical role for CD103(+)/CD141(+) dendritic cells bearing CCR7 for tumor antigen trafficking and priming of T cell immunity in melanoma. Cancer Cell. 2016;30(2):324‐336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Spranger S, Dai D, Horton B, Gajewski TF. Tumor‐residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31(5):711‐723.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, et al. Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti‐PD‐1 therapy. Immunity. 2019;50(6):1498‐1512.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ruffell B, Chang‐Strachan D, Chan V, Rosenbush A, Ho CM, Pryer N, et al. Macrophage IL‐10 blocks CD8+ T cell‐dependent responses to chemotherapy by suppressing IL‐12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26(5):623‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garris CS, Arlauckas SP, Kohler RH, Trefny MP, Garren S, Piot C, et al. Successful anti‐PD‐1 cancer immunotherapy requires T cell‐dendritic cell crosstalk involving the cytokines IFN‐γ and IL‐12. Immunity. 2018;49(6):1148‐1161.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Andzinski L, Spanier J, Kasnitz N, Kröger A, Jin L, Brinkmann MM, et al. Growing tumors induce a local STING dependent Type I IFN response in dendritic cells. Int J Cancer. 2016;139(6):1350‐1357. [DOI] [PubMed] [Google Scholar]
- 16. Oldenborg PA, Zheleznyak A, Fang YF, Lagenaur CF, Gresham HD, Lindberg FR. Role of CD47 as a marker of self on red blood cells. Science. 2000;288(5473):2051‐2054. [DOI] [PubMed] [Google Scholar]
- 17. Blazar BR, Lindberg FP, Ingulli E, Panoskaltsis‐Mortari A, Oldenborg PA, Iizuka K, et al. CD47 (integrin‐associated protein) engagement of dendritic cell and macrophage counterreceptors is required to prevent the clearance of donor lymphohematopoietic cells. J Exp Med. 2001;194(4):541‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meinderts SM, Oldenborg PA, Beuger BM, Klei TRL, Johansson J, Kujipers TW, et al. Human and murine splenic neutrophils are potent phagocytes of IgG‐opsonized red blood cells. Blood Adv. 2017;1(14):875‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wu J, Wu H, An J, Ballantyne CM, Cyster JG. Critical role of integrin CD11c in splenic dendritic cell capture of missing‐self CD47 cells to induce adaptive immunity. Proc Natl Acad Sci USA. 2018;115(26):6786‐6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Willingham SB, Volkmer JP, Gentles AJ, Sahoo D, Dalerba P, Mitra SS, et al. The CD47‐signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc Natl Acad Sci USA. 2012;109(17):6662‐6667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vermeer DW, Spanos WC, Vermeer PD, Bruns AM, Lee KM, Lee JH. Radiation‐induced loss of cell surface CD47 enhances immune‐mediated clearance of human papillomavirus‐positive cancer. Int J Cancer. 2013;133(1):120‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Noman MZ, Van Moer K, Marani V, Gemmill RM, Tranchevent LC, Azuaje F, et al. CD47 is a direct target of SNAI1 and ZEB1 and its blockade activates the phagocytosis of breast cancer cells undergoing EMT. Oncoimmunology. 2018;7(4):e1345415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chao MP, Alizadeh AA, Tang C, Myklebust JH, Varghese B, Gill S, et al. Anti‐CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non‐Hodgkin lymphoma. Cell. 2010;142(5):699‐713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chao MP, Alizadeh AA, Tang C, Jan M, Weissman‐Tsukamoto R, Zhao F, et al. Therapeutic antibody targeting of CD47 eliminates human acute lymphoblastic leukemia. Cancer Res. 2011;71(4):1374‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiskopf K, Jahchan NS, Schnorr PJ, Cristea S, Ring AM, Maute RL, et al. CD47‐blocking immunotherapies stimulate macrophage‐mediated destruction of small‐cell lung cancer. J Clin Invest. 2016;126(7):2610‐2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang X, Chen W, Fan J, Wang S, Xian Z, Luan J, et al. Disrupting CD47‐SIRPα axis alone or combined with autophagy depletion for the therapy of glioblastoma. Carcinogenesis. 2018;39(5):689‐699. [DOI] [PubMed] [Google Scholar]
- 27. Sockolosky JT, Dougan M, Ingram JR, Ho CC, Kauke MJ, Almo SC, et al. Durable antitumor responses to CD47 blockade require adaptive immune stimulation. Proc Natl Acad Sci U S A. 2016;113(19):E2646‐E2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu X, Pu Y, Cron K, Deng L, Kline J, Frazier WA, et al. CD47 blockade triggers T cell–mediated destruction of immunogenic tumors. Nat Med. 2015;21(10):1209‐1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xu MM, Pu Y, Han D, Shi Y, Cao X, Liang H, et al. Dendritic cells but not macrophages sense tumor mitochondrial DNA for cross‐priming through signal regulatory protein α signaling. Immunity. 2017;47(2):363‐373.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY, et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy article. Nat Immunol. 2018;19(1):76‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tenca C, Merlo A, Merck E, Bates EE, Saverino D, Simone R, et al. CD85j (leukocyte Ig‐like receptor‐1/Ig‐like transcript 2) inhibits human osteoclast‐associated receptor‐mediated activation of human dendritic cells. J Immunol. 2005;174(11):6757‐6763. [DOI] [PubMed] [Google Scholar]
- 32. Young NT, Waller ECP, Patel R, Roghanian A, Austyn JM, Trowsdale J. The inhibitory receptor LILRB1 modulates the differentiation and regulatory potential of human dendritic cells. Blood. 2008;111(6):3090‐3096. [DOI] [PubMed] [Google Scholar]
- 33. Khanolkar RC, Kalogeropoulos M, Lawrie A, Roghanian A, Vickers MA, Young NT. Leukocyte Ig‐Like receptor B1 restrains dendritic cell function through increased expression of the NF‐κB regulator ABIN1/TNIP1. J Leukoc Biol. 2016;100(4):737‐746. [DOI] [PubMed] [Google Scholar]
- 34. van der Touw W, Chen HM, Pan PY, Chen SH. LILRB receptor‐mediated regulation of myeloid cell maturation and function. Cancer Immunol Immunother. 2017;66(8):1079‐1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Paulson JC, Macauley MS, Kawasaki N. Siglecs as sensors of self in innate and adaptive immune responses. Ann N Y Acad Sci. 2012;1253:37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, et al. CD24 signalling through macrophage Siglec‐10 is a target for cancer immunotherapy. Nature. 2019;572(7769):392‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec‐10 selectively repress tissue damage‐induced immune responses. Science. 2009;323(5922):1722‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bärenwaldt A, Läubli H. The sialoglycan‐Siglec glyco‐immune checkpoint – a target for improving innate and adaptive anti‐cancer immunity. Expert Opin Ther Targets. 2019;23(10):839‐853. [DOI] [PubMed] [Google Scholar]
- 39. Beatson R, Tajadura‐Ortega V, Achkova D, Picco G, Tsourouktsoglou TD, Klausing S, et al. The mucin MUC1 modulates the tumor immunological microenvironment through engagement of the lectin Siglec‐9. Nat Immunol. 2016;17(11):1273‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shurin GV, Shurin MR, Bykovskaia S, Shogan J, Lotze MT, Jr BarksdaleEM. Neuroblastoma‐derived gangliosides inhibit dendritic cell generation and function. Cancer Res. 2001;61(1):363‐369. [PubMed] [Google Scholar]
- 41. Péguet‐Navarro J, Sportouch M, Popa I, Berthier O, Schmitt D, Portoukalian J. Gangliosides from human melanoma tumors impair dendritic cell differentiation from monocytes and induce their apoptosis. J Immunol. 2003;170(7):3488‐3494. [DOI] [PubMed] [Google Scholar]
- 42. Bennaceur K, Popa I, Chapman JA, Migdal C, Péguet‐Navarro J, Touraine JL, et al. Different mechanisms are involved in apoptosis induced by melanoma gangliosides on human monocyte‐derived dendritic cells. Glycobiology. 2009;19(6):576‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bennaceur K, Popa I, Portoukalian J, Berthier‐Vergnes O, Péguet‐Navarro J. Melanoma‐derived gangliosides impair migratory and antigen‐presenting function of human epidermal Langerhans cells and induce their apoptosis. Int Immunol. 2006;18(6):879‐886. [DOI] [PubMed] [Google Scholar]
- 44. Adams OJ, Stanczak MA, von Gunten S, Läubli H. Targeting sialic acid–Siglec interactions to reverse immune suppression in cancer. Glycobiology. 2018;28(9):640‐647. [DOI] [PubMed] [Google Scholar]
- 45. Wing K, Onishi Y, Prieto‐Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA‐4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271‐275. [DOI] [PubMed] [Google Scholar]
- 46. Ovcinnikovs V, Ross EM, Petersone L, Edner NM, Heuts F, Ntavli E, et al. CTLA‐4‐mediated transendocytosis of costimulatory molecules primarily targets migratory dendritic cells. Sci Immunol. 2019;4(35):eaaw0902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kowalczyk A, D'Souza CA, Zhang L. Cell‐extrinsic CTLA4‐mediated regulation of dendritic cell maturation depends on STAT3. Eur J Immunol. 2014;44(4):1143‐1155. [DOI] [PubMed] [Google Scholar]
- 48. Contardi E, Palmisano GL, Tazzari PL, Martelli AM, Falà F, Fabbi M, et al. CTLA‐4 is constitutively expressed on tumor cells and can trigger apoptosis upon ligand interaction. Int J Cancer. 2005;117(4):538‐550. [DOI] [PubMed] [Google Scholar]
- 49. Chen X, Shao Q, Hao S, Zhao Z, Wang Y, Guo X, et al. CTLA‐4 positive breast cancer cells suppress dendritic cells maturation and function. Oncotarget. 2017;8(8):13703‐13715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Halpert MM, Konduri V, Liang D, Chen Y, Wing JB, Paust S, et al. Dendritic cell‐secreted cytotoxic T‐lymphocyte‐associated protein‐4 regulates the T‐cell response by downmodulating bystander surface B7. Stem Cells Dev. 2016;25(10):774‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laurent S, Carrega P, Saverino D, Piccioli P, Camoriano M, Morabito A, et al. CTLA‐4 is expressed by human monocyte‐derived dendritic cells and regulates their functions. Hum Immunol. 2010;71(10):934‐941. [DOI] [PubMed] [Google Scholar]
- 52. Wang XB, Fan ZZ, Anton D, Vollenhoven AV, Ni ZH, Chen XF, et al. CTLA4 is expressed on mature dendritic cells derived from human monocytes and influences their maturation and antigen presentation. BMC Immunol. 2011;12:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD‐1 expression by tumour‐associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495‐499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Yao S, Wang S, Zhu Y, Luo L, Zhu G, Flies S, et al. PD‐1 on dendritic cells impedes innate immunity against bacterial infection. Blood. 2009;113(23):5811‐5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Krempski J, Karyampudi L, Behrens MD, Erskine CL, Hartmann L, Dong H, et al. Tumor‐infiltrating programmed death receptor‐1+ dendritic cells mediate immune suppression in ovarian cancer. J Immunol. 2011;186(12):6905‐6913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Karyampudi L, Lamichhane P, Krempski J, Kalli KR, Behrens MD, Vargas DM, et al. PD‐1 blunts the function of ovarian tumor‐infiltrating dendritic cells by inactivating NF‐κB. Cancer Res. 2016;76(2):239‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lim TS, Chew V, Sieow JL, Goh S, Yeong JP, Soon AL, et al. PD‐1 expression on dendritic cells suppresses CD8+ T cell function and antitumor immunity. Oncoimmunology. 2015;5(3):e1085146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, et al. Th1‐specific cell surface protein Tim‐3 regulates macrophage activation and severity of an autoimmune disease. Nature. 2002;415(6871):536‐541. [DOI] [PubMed] [Google Scholar]
- 59. Schwartz JA, Clayton KL, Mujib S, Zhang H, Rahman AK, Lie J, et al. Tim‐3 is a marker of plasmacytoid dendritic cell dysfunction during HIV infection and is associated with the recruitment of IRF7 and p85 into lysosomes and with the submembrane displacement of TLR9. J Immunol. 2017;198(8):3181‐3194. [DOI] [PubMed] [Google Scholar]
- 60. Maurya N, Gujar R, Gupta M, Yadav V, Verma S, Sen P. Immunoregulation of dendritic cells by the receptor T cell Ig and mucin protein‐3 via Bruton's tyrosine kinase and c‐Src. J Immunol. 2014;193(7):3417‐3425. [DOI] [PubMed] [Google Scholar]
- 61. Chiba S, Baghdadi M, Akiba H, Yoshiyama H, Kinoshita I, Dosaka‐Akita H, et al. Tumor‐infiltrating DCs suppress nucleic acid‐mediated innate immune responses through interactions between the receptor TIM‐3 and the alarmin HMGB1. Nat Immunol. 2012;13(9):832‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Chevalier MF, Bohner P, Pieraerts C, Lhermitte B, Gourmaud J, Nobile A, et al. Immunoregulation of dendritic cell subsets by inhibitory receptors in urothelial cancer. Eur Urol. 2017;71(6):854‐857. [DOI] [PubMed] [Google Scholar]
- 63. de Mingo Pulido Á GardnerA, Hiebler S, Soliman H, Rugo HS, Krummel MF, et al. TIM‐3 regulates CD103+ dendritic cell function and response to chemotherapy in breast cancer. Cancer Cell. 2018;33(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Akalu YT, Rothlin C V, Ghosh S. TAM receptor tyrosine kinases as emerging targets of innate immune checkpoint blockade for cancer therapy. Immunol Rev. 2017;276(1):165‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wium M, Paccez J, Zerbini L. The dual role of TAM receptors in autoimmune diseases and cancer: an overview. Cells. 2018;7(10):E166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Wu G, Ma Z, Cheng Y, Hu W, Deng C, Jiang S, et al. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol Cancer. 2018;17(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rothlin CV, Ghosh S, Zuniga EI, Oldstone MB, Lemke G. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131(6):1124‐1136. [DOI] [PubMed] [Google Scholar]
- 68. Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE, et al. Apoptotic cells induce Mer tyrosine kinase‐dependent blockade of NF‐κB activation in dendritic cells. Blood. 2007;109(2):653‐660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Yokoyama Y, Lew ED, Seelige R, Tindall EA, Walsh C, Fagan PC, et al. Immuno‐oncological efficacy of RXDX‐106, a novel TAM (TYRO3, AXL, MER) family small‐molecule kinase inhibitor. Cancer Res. 2019;79(8):1996‐2008. [DOI] [PubMed] [Google Scholar]
- 70. De Trez C, Schneider K, Potter K, Droin N, Fulton J, Norris PH, et al. The inhibitory HVEM‐BTLA pathway counter regulates lymphotoxin β receptor signaling to achieve homeostasis of dendritic cells. J Immunol. 2008;180(1):238‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xu W, Dong J, Zheng Y, Zhou J, Yuan Y, Ta HM, et al. Immune‐checkpoint protein VISTA regulates antitumor immunity by controlling myeloid cell‐mediated inflammation and immunosuppression. Cancer Immunol Res. 2019;7(9):1497‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Le Mercier I, Chen W, Lines JL, Day M, Li J, Sergent P, et al. VISTA regulates the development of protective antitumor immunity. Cancer Res. 2014;74(7):1933‐1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kuklinski LF, Yan S, Li Z, Fisher JL, Cheng C, Noelle RJ, et al. VISTA expression on tumor‐infiltrating inflammatory cells in primary cutaneous melanoma correlates with poor disease‐specific survival. Cancer Immunol Immunother. 2018;67(7):1113‐1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Hargadon KM. Tumor‐altered dendritic cell function: implications for anti‐tumor immunity. Front Immunol. 2013;4:192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hargadon KM. Strategies to improve the efficacy of dendritic cell‐based immunotherapy for melanoma. Front Immunol. 2017;8:1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Krawczyk CM, Holowka T, Sun J, Balgih J, Amiel E, DeBerardinis RJ, et al. Toll‐like receptor‐induced changes in glycolytic metabolism regulate dendritic cell activation. Blood. 2010;115(23):4742‐4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Everts B, Amiel E, Huang SC, Smith AM, Chang CH, Lam WY, et al. TLR‐driven early glycolytic reprogramming via the kinases TBK1‐IKKɛ supports the anabolic demands of dendritic cell activation. Nat Immunol. 2014;15(4):323‐332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Guak H, Al Habyan S, Ma EH, Aldossary H, Al‐Masri M, Won SY, et al. Glycolytic metabolism is essential for CCR7 oligomerization and dendritic cell migration. Nat Commun. 2018;9(1):2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Everts B, Amiel E, van der Windt GJ, Freitas TC, Chott R, Yarasheski KE, et al. Commitment to glycolysis sustains survival of NO‐producing inflammatory dendritic cells. Blood. 2012;120(7):1422‐1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Pantel A, Teixeira A, Haddad E, Wood EG, Steinman RM, Longhi MP. Direct type I IFN but not MDA5/TLR3 activation of dendritic cells is required for maturation and metabolic shift to glycolysis after poly IC stimulation. PLoS Biol. 2014;12(1):e1001759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Thwe PM, Pelgrom LR, Cooper R, Beauchamp S, Reisz JA, D'Alessandro A, et al. Cell‐intrinsic glycogen metabolism supports early glycolytic reprogramming required for dendritic cell immune responses. Cell Metab. 2017;26(3):558‐567.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Chang CH, Qiu J, O'Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229‐1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Auciello FR, Ross FA, Ikematsu N, Hardie DG. Oxidative stress activates AMPK in cultured cells primarily by increasing cellular AMP and/or ADP. FEBS Lett. 2014;588(18):3361‐3366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shackelford DB, Vasquez DS, Corbeil J, Wu S, Leblanc M, Wu CL, et al. mTOR and HIF‐1alpha‐mediated tumor metabolism in an LKB1 mouse model of Peutz‐Jeghers syndrome. Proc Natl Acad Sci USA. 2009;106(27):11137‐11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Xie H, Simon MC. Oxygen availability and metabolic reprogramming in cancer. J Biol Chem. 2017;292(41):16825‐16832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Semenza GL. HIF‐1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest. 2013;123(9):3664‐3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhang Z, Liu Q, Che Y, Yuan X, Dai L, Zeng B, et al. Antigen presentation by dendritic cells in tumors is disrupted by altered metabolism that involves pyruvate kinase M2 and its interaction with SOCS3. Cancer Res. 2010;70(1):89‐98. [DOI] [PubMed] [Google Scholar]
- 88. Santos PM, Menk AV, Shi J, Tsung A, Delgoffe GM, Butterfield LH. Tumor‐derived α‐fetoprotein suppresses fatty acid metabolism and oxidative phosphorylation in dendritic cells. Cancer Immunol Res. 2019;7(6):1001‐1012. [DOI] [PubMed] [Google Scholar]
- 89. Pardee AD, Shi J, Butterfield LH. Tumor‐derived α‐fetoprotein impairs the differentiation and T cell stimulatory activity of human dendritic cells. J Immunol. 2014;193(11):5723‐5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Jiang L, Fang X, Wang H, Li D, Wang X. Ovarian cancer‐intrinsic fatty acid synthase prevents anti‐tumor immunity by disrupting tumor‐infiltrating dendritic cells. Front Immunol. 2018;9:2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Herber DL, Cao W, Nefedova Y, Novitskiy SV, Nagaraj S, Tyurin VA, et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat Med. 2010;16(8):880‐886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Ramakrishnan R, Tyurin VA, Veglia F, Condamine T, Amoscato A, Mohammadyani D, et al. Oxidized lipids block antigen cross‐presentation by dendritic cells in cancer. J Immunol. 2014;192(6):2920‐2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Veglia F, Tyurin VA, Mohammadyani D, Blasi M, Duperret EK, Donthireddy L, et al. Lipid bodies containing oxidatively truncated lipids block antigen cross‐presentation by dendritic cells in cancer. Nat Commun. 2017;8(1):2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Cubillos‐Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales‐Puchalt A, Song M, et al. ER stress sensor XBP1 controls anti‐tumor immunity by disrupting dendritic cell homeostasis. Cell. 2015;161(7):1527‐1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Coutant F, Agaugué S, Perrin‐Cocon L, André P, Lotteau V. Sensing environmental lipids by dendritic cell modulates its function. J Immunol. 2004;172(1):54‐60. [DOI] [PubMed] [Google Scholar]
- 96. Zeyda M, Säemann MD, Stuhlmeier KM, Mascher DG, Nowotny PN, Zlabinger GJ, et al. Polyunsaturated fatty acids block dendritic cell activation and function independently of NF‐κB activation. J Biol Chem. 2005;280(14):14293‐14301. [DOI] [PubMed] [Google Scholar]
- 97. Liang X, Fu C, Cui W, Ober‐Blöbaum JL, Zahner SP, Shrikant PA, et al. β‐Catenin mediates tumor‐induced immunosuppression by inhibiting cross‐priming of CD8+ T cells. J Leukoc Biol. 2014;95(1):179‐190. [DOI] [PubMed] [Google Scholar]
- 98. Zhao F, Xiao C, Evans KS, Theivanthiran T, , DeVito N, Holtzhausen A, et al. Paracrine Wnt5a‐β‐catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity. 2018;48(1):147‐160.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Gottfried E, Kunz‐Schughart LA, Ebner S, Mueller‐Klieser W, Hoves S, Andreesen R, et al. Tumor‐derived lactic acid modulates dendritic cell activation and antigen expression. Blood. 2006;107(5):2013‐2021. [DOI] [PubMed] [Google Scholar]
- 100. Caronni N, Simoncello F, Stafetta F, Guarnaccia C, Ruiz‐Moreno JS, Opitz B, et al. Downregulation of membrane trafficking proteins and lactate conditioning determine loss of dendritic cell function in lung cancer. Cancer Res. 2018;78(7):1685‐1699. [DOI] [PubMed] [Google Scholar]
- 101. Raychaudhuri D, Bhattacharya R, Sinha BP, Liu CSC, Ghosh AR, Rahaman O, et al. Lactate induces pro‐tumor reprogramming in intratumoral plasmacytoid dendritic cells. Front Immunol. 2019;10:1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Pellegatti P, Raffaghello L, Bianchi G, Piccardi F, Pistoia V, Di Virgilio F. Increased level of extracellular ATP at tumor sites: in vivo imaging with plasma membrane luciferase. PLoS One. 2008;3(7):e2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Ghiringhelli F, Apetoh L, Tesniere A, Aymeric L, Ma Y, Ortiz C. Activation of the NLRP3 inflammasome in dendritic cells induces IL‐1β‐dependent adaptive immunity against tumors. Nat Med. 2009;15(10):1170‐1178. [DOI] [PubMed] [Google Scholar]
- 104. Bastid J, Regairaz A, Bonnefoy N, Déjou C, Giustiniani J, Laheurte C. Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res. 2015;3(3):254‐265. [DOI] [PubMed] [Google Scholar]
- 105. Häusler SF, Montalbán Del Barrio I, Strohschein J, Chandran PA, Engel JB, Hönig A, et al. Ectonucleotidases CD39 and CD73 on OvCA cells are potent adenosine‐generating enzymes responsible for adenosine receptor 2A‐dependent suppression of T cell function and NK cell cytotoxicity. Cancer Immunol Immunother. 2011;60(10):1405‐1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Cekic C, Day YJ, Sag D, Linden J. Myeloid expression of adenosine A2A receptor suppresses T and NK cell responses in the solid tumor microenvironment. Cancer Res. 2014;74(24):7250‐7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Panther E, Corinti S, Idzko M, Herouy Y, Napp M, Ia Sala A, et al. Adenosine affects expression of membrane molecules, cytokine and chemokine release, and the T‐cell stimulatory capacity of human dendritic cells. Blood. 2003;101(10):3985‐3990. [DOI] [PubMed] [Google Scholar]
- 108. Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, et al. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood. 2008;112(5):1822‐1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Kayhan M, Koyas A, Akdemir I, Savas AC, Cekic C. Adenosine receptor signaling targets both PKA and Epac pathways to polarize dendritic cells to a suppressive phenotype. J Immunol. 2019;203(12):3247‐3255. [DOI] [PubMed] [Google Scholar]
- 110. Young A, Ngiow SF, Barkauskas DS, Sult E, Hay C, Blake SJ, et al. Co‐inhibition of CD73 and A2AR adenosine signaling improves anti‐tumor immune responses. Cancer Cell. 2016;30(3):391‐403. [DOI] [PubMed] [Google Scholar]
- 111. Young A, Ngiow SF, Madore J, Reinhardt J, Landsberg J, Chitsazan A, et al. Targeting adenosine in BRAF‐mutant melanoma reduces tumor growth and metastasis. Cancer Res. 2017;77(17):4684‐4696. [DOI] [PubMed] [Google Scholar]
- 112. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA‐4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405‐413. [DOI] [PubMed] [Google Scholar]
- 113. Krummel MF, Allison JP. CD28 and CTLA‐4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182(2):459‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Greenwald RJ, Boussiotis VA, Lorsbach RB, Abbas AK, Sharpe AH. CTLA‐4 regulates induction of anergy in vivo. Immunity. 2001;14(2):145‐155. [DOI] [PubMed] [Google Scholar]
- 115. Wells AD, Walsh MC, Bluestone JA, Turka LA. Signaling through CD28 and CTLA‐4 controls two distinct forms of T cell anergy. J Clin Invest. 2001;108(6):895‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Wei SC, Sharma R, Anang NAS, Levine JH, Zhao Y, Mancuso JJ, et al. Negative co‐stimulation constrains t cell differentiation by imposing boundaries on possible cell states. Immunity. 2019;50(4):1084‐1098.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Bachmann MF, Waterhouse P, Speiser DE, McKall‐Faienza K, Mak TW, Ohashi PS. Normal responsiveness of CTLA‐4‐deficient anti‐viral cytotoxic T cells. J Immunol. 1998;160(1):95‐100. [PubMed] [Google Scholar]
- 118. Chambers CA, Sullivan TJ, Truong T, Allison JP. Secondary but not primary T cell responses are enhanced in CTLA‐4‐deficient CD8+ T cells. Eur J Immunol. 1998;28(10):3137‐3143. [DOI] [PubMed] [Google Scholar]
- 119. Miao Y, Yang H, Levorse J, Yuan S, Polak L, Sribour M, et al. Adaptive immune resistance emerges from tumor‐initiating stem cells. Cell. 2019;177(5):1172‐1186.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Peggs KS, Quezada SA, Chambers CA, Korman AJ, Allison JP. Blockade of CTLA‐4 on both effector and regulatory T cell compartments contributes to the antitumor activity of anti–CTLA‐4 antibodies. J Exp Med. 2009;206(8):1717‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, et al. Distinct cellular mechanisms underlie anti‐CTLA‐4 and anti‐PD‐1 checkpoint blockade. Cell. 2017;170(6):1120‐1133.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Wei SC, Anang NAS, Sharma R, Andrews MC, Reuben A, Levine AH, et al. Combination anti–CTLA‐4 plus anti–PD‐1 checkpoint blockade utilizes cellular mechanisms partially distinct from monotherapies. Proc Natl Acad Sci USA. 2019;116(45):22699‐22709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti‐CTLA‐4 immunotherapy does not deplete FOXP3+ regulatory T cells (Tregs) in human cancers. Clin Cancer Res. 2019;25(4):1233‐1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Sotomayor EM, Borrello I, Tubb E, Allison JP, Levitsky HI. In vivo blockade of CTLA‐4 enhances the priming of responsive T cells but fails to prevent the induction of tumor antigen‐specific tolerance. Proc Natl Acad Sci USA. 1999;96(20):11476‐11481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD‐1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liang SC, Latchman YE, Buhlmann JE, Tomczak MF, Horwitz BH, Freeman GJ, et al. Regulation of PD‐1, PD‐L1, and PD‐L2 expression during normal and autoimmune responses. Eur J Immunol. 2003;33(10):2706‐2716. [DOI] [PubMed] [Google Scholar]
- 128. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD‐1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192(7):1027‐1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD‐L2 is a second ligand for PD‐1 and inhibits T cell activation. Nat Immunol. 2001;2(3):261‐268. [DOI] [PubMed] [Google Scholar]
- 130. Karunarathne DS, Horne‐Debets JM, Huang JX, Faleiro R, Leow CY, Amante F, et al. Programmed death‐1 ligand 2‐mediated regulation of the PD‐L1 to PD‐1 axis is essential for establishing CD4(+) T cell immunity. Immunity. 2016;45(2):333‐345. [DOI] [PubMed] [Google Scholar]
- 131. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD‐L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206(13):3015‐3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Blank C, Brown I, Peterson AC, Spiotto M, Iwai Y, Honjo T, et al. PD‐L1/B7H‐1 inhibits the effector phase of tumor rejection by T cell receptor (TCR) transgenic CD8+ T cells. Cancer Res. 2004;64(3):1140‐1145. [DOI] [PubMed] [Google Scholar]
- 133. Iwai Y, Terawaki S, Ikegawa M, Okazaki T, Honjo T. PD‐1 inhibits antiviral immunity at the effector phase in the liver. J Exp Med. 2003;198(1):39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439(7077):682‐687. [DOI] [PubMed] [Google Scholar]
- 135. Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen‐specific CD8 T cells infiltrating the tumor express high levels of PD‐1 and are functionally impaired. Blood. 2009;114(8):1537‐1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Konkel JE, Frommer F, Leech MD, Yagita H, Waisman A, Anderton SM. PD‐1 signalling in CD4+ T cells restrains their clonal expansion to an immunogenic stimulus, but is not critically required for peptide‐induced tolerance. Immunology. 2010;130(1):92‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Goldberg M V, , Maris CH, Hipkiss EL, Flies AS, Zhen L, Tuder RM, et al. Role of PD‐1 and its ligand, B7‐H1, in early fate decisions of CD8 T cells. Blood. 2007;110(1):186‐192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Ahn E, Araki K, Hashimoto M, Li W, Riley JL, Cheung J, et al. Role of PD‐1 during effector CD8 T cell differentiation. Proc Natl Acad Sci USA. 2018;115(18):4749‐4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Juneja VR, McGuire KA, Manguso RT, LeFleur MW, Collins N, Haining WN, et al. PD‐L1 on tumor cells is sufficient for immune evasion in immunogenic tumors and inhibits CD8 T cell cytotoxicity. J Exp Med. 2017;214(4):895‐904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tang H, Liang Y, Anders RA, Taube JM, Qui X, Mulgaonkar A, et al. PD‐L1 on host cells is essential for PD‐L1 blockade–mediated tumor regression. J Clin Invest. 2018;128(2):580‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Lin H, Wei S, Hurt EM, Green MD, Zhao L, Vatan L, et al. Host expression of PD‐L1 determines efficacy of PD‐L1 pathway blockade. J Clin Invest. 2018;128(2):805‐815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD‐1 inhibits T‐cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004;574(1‐3):37‐41. [DOI] [PubMed] [Google Scholar]
- 143. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD‐1‐mediated inhibition. Science. 2017;355(6332):1428‐1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Mizuno R, Sugiura D, Shimizu K, Maruhashi T, Watada M, Okazaki IM, et al. PD‐1 primarily targets TCR signal in the inhibition of functional t cell activation. Front Immunol. 2019;10:630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Yokosuka T, Takamatsu M, Kobayashi‐Imanishi W, Hashimoto‐Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J Exp Med. 2012;209(6):1201‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Karwacz K, Bricogne C, MacDonald D, Arce F, Bennett CL, Collins M, et al. PD‐L1 co‐stimulation contributes to ligand‐induced T cell receptor down‐modulation on CD8 + T cells. EMBO Mol Med. 2011;3(10):581‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Patsoukis N, Bardhan K, Chatterjee P, Sari D, Liu B, Bell LN, et al. PD‐1 alters T‐cell metabolic reprogramming by inhibiting glycolysis and promoting lipolysis and fatty acid oxidation. Nat Commun. 2015;6:6692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Bengsch B, Johnson AL, Kurachi M, Odorizzi PM, Pauken KE, Attanasio J, et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD‐1 are an early driver of CD8+ T cell exhaustion. Immunity. 2016;45(2):358‐373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Daud AI, Loo K, Pauli ML, Sanchez‐Rodriguez R, Sandoval PM, Taravati K, et al. Tumor immune profiling predicts response to anti‐PD‐1 therapy in human melanoma. J Clin Invest. 2016;126(9):3447‐3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Pauken KE, Sammons MA, Odorizzi PM, Manne S, Godec J, Khan O, et al. Epigenetic stability of exhausted T cells limits durability of reinvigoration by PD‐1 blockade. Science. 2016;354(6316):1160‐1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Gide TN, Quek C, Menzies AM, Tasket AT, Shang P, Holst J, et al. Distinct immune cell populations define response to anti‐PD‐1 monotherapy and anti‐PD‐1/anti‐CTLA‐4 combined therapy. Cancer Cell. 2019;35(2). [DOI] [PubMed] [Google Scholar]
- 152. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour‐specific mutant antigens. Nature. 2014;515(7528):577‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti–CTLA‐4 and anti–PD‐1 leads to distinct immunologic changes in vivo. J Immunol. 2015;194(3):950‐959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Larkin J, Chiarion‐Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lau CD, et al. Five‐year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2019;381(16):1535‐1546. [DOI] [PubMed] [Google Scholar]
- 155. Hellmann MD, Paz‐Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus ipilimumab in advanced non‐small‐cell lung cancer. N Engl J Med. 2019;38(21):2020‐2031. [DOI] [PubMed] [Google Scholar]
- 156. Workman CJ, Cauley LS, Kim IJ, Blackman MA, Woodland DL, Vignali DA. Lymphocyte activation gene‐3 (CD223) regulates the size of the expanding T cell population following antigen activation in vivo. J Immunol. 2004;172(9):5450‐5455. [DOI] [PubMed] [Google Scholar]
- 157. Macon‐Lemaitre L, Triebel F. The negative regulatory function of the lymphocyte‐activation gene‐3 co‐receptor (CD223) on human T cells. Immunology. 2005;115(2):170‐178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Grosso JF, Kelleher CC, Harris TJ, Maris CH, Hipkiss EL, De Marzo A, et al. LAG‐3 regulates CD8+ T cell accumulation and effector function in murine self‐ and tumor‐tolerance systems. J Clin Invest. 2007;117(11):3383‐3392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Wei T, Zhang J, Qin Y, Wu Y, Zhu L, Lu L, et al. Increased expression of immunosuppressive molecules on intratumoral and circulating regulatory T cells in non‐small‐cell lung cancer patients. Am J Cancer Res. 2015;5(7):2190‐2201. [PMC free article] [PubMed] [Google Scholar]
- 160. Camisaschi C, Casati C, Rini F, Perego M, De Filippo A, Triebel F, et al. LAG‐3 expression defines a subset of CD4+ CD25high Foxp3+ regulatory T cells that are expanded at tumor sites. J Immunol. 2010;184(11):6545‐6551. [DOI] [PubMed] [Google Scholar]
- 161. Workman CJ, Dugger KJ, Vignali DA. Cutting edge: molecular analysis of the negative regulatory function of lymphocyte activation gene‐3. J Immunol. 2002;169(10):5392‐5395. [DOI] [PubMed] [Google Scholar]
- 162. Liang B, Workman C, Lee J, Chew C, Dale BM, Colonna L, et al. Regulatory T cells inhibit dendritic cells by lymphocyte activation gene‐3 engagement of MHC class II. J Immunol. 2008;180(9):5916‐5926. [DOI] [PubMed] [Google Scholar]
- 163. Kouo T, Huang L, Pucsek AB, Cao M, Solt S, Armstrong T, et al. Galectin‐3 shapes antitumor immune responses by suppressing CD8+ T cells via LAG‐3 and inhibiting expansion of plasmacytoid dendritic cells. Cancer Immunol Res. 2015;3(4):412‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Xu F, Liu J, Liu D, Liu B, Wang M, Hu Z, et al. LSECtin expressed on melanoma cells promotes tumor progression by inhibiting antitumor T‐cell responses. Cancer Res. 2014;74(13):3418‐3428. [DOI] [PubMed] [Google Scholar]
- 165. Zelba H, Bedke J, Hennenlotter J, Mostböck S, Zettl M, Zichner T, et al. PD‐1 and LAG‐3 dominate checkpoint receptor‐mediated T‐cell inhibition in renal cell carcinoma. Cancer Immunol Res. 2019;7(11):1891‐1899. [DOI] [PubMed] [Google Scholar]
- 166. Matsuzaki J, Gnjatic S, Mhawech‐Fauceglia P, Beck A, Miller A, Tsuji T, et al. Tumor‐infiltrating NY‐ESO‐1‐specific CD8+ T cells are negatively regulated by LAG‐3 and PD‐1 in human ovarian cancer. Proc Natl Acad Sci USA. 2010;107(17):7875‐7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Woo SR, Turnis ME, Goldberg MV, Bankoti J, Selby M, Nirschi CJ, et al. Immune inhibitory molecules LAG‐3 and PD‐1 synergistically regulate T‐cell function to promote tumoral immune escape. Cancer Res. 2010;72(4):917‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Gao X, Zhu Y, Li G, Huang H, Zhang G, Wang F, et al. TIM‐3 expression characterizes regulatory T cells in tumor tissues and is associated with lung cancer progression. PLoS One. 2012;7(2):e30676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yan J, Zhang Y, Zhang JP, Liang J, Li L, Zheng L. Tim‐3 expression defines regulatory t cells in human tumors. PLoS One. 2013;8(3):e58006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Cai C, Xu YF, Wu ZJ, Dong Q, Li MY, Olson JC, et al. Tim‐3 expression represents dysfunctional tumor infiltrating T cells in renal cell carcinoma. World J Urol. 2016;34(4):561‐567. [DOI] [PubMed] [Google Scholar]
- 171. Li H, Wu K, Tao K, Chen L, Zheng Q, Lu X, et al. Tim‐3/galectin‐9 signaling pathway mediates T‐cell dysfunction and predicts poor prognosis in patients with hepatitis B virus‐associated hepatocellular carcinoma. Hepatology. 2012;56(4):1342‐1351. [DOI] [PubMed] [Google Scholar]
- 172. Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, et al. Coexpression of Tim‐3 and PD‐1 identifies a CD8+ T‐cell exhaustion phenotype in mice with disseminated acute myelogenous leukemia. Blood. 2011;117(17):4501‐4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Kang CW, Dutta A, Chang LY, Mahalingam J, Lin YC, Chiang JM, et al. Apoptosis of tumor infiltrating effector TIM‐3+CD8+ T cells in colon cancer. Sci Rep. 2015;5:15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Rangachari M, Zhu C, Sakuishi K, Siao S, Karman J, Chen A, et al. Bat3 promotes T cell responses and autoimmunity by repressing Tim‐3–mediated cell death and exhaustion. Nat Med. 2012;18(9):1394‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ji J, Yin Y, Ju H, Xu X, Liu W, Fu Q, et al. Long non‐coding RNA Lnc‐Tim3 exacerbates CD8 T cell exhaustion via binding to Tim‐3 and inducing nuclear translocation of Bat3 in HCC. Cell Death Dis. 2018;9(5):478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Bu M, Shen Y, Seeger WL, An S, Qi R, Sanderson JA, et al. Ovarian carcinoma‐infiltrating regulatory T cells were more potent suppressors of CD8+ T cell inflammation than their peripheral counterparts, a function dependent on TIM3 expression. Tumor Biol. 2016;37(3):3949‐3956. [DOI] [PubMed] [Google Scholar]
- 177. Sakuishi K, Ngiow SF, Sullivan JM, Teng MW, Kuchroo VK, Smyth MJ, et al. TIM3 + FOXP3 + regulatory T cells are tissue‐specific promoters of T‐cell dysfunction in cancer. Oncoimmunology. 2013;2(4):e23849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178. Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ. Anti‐TIM3 antibody promotes T cell IFN‐γ‐mediated antitumor immunity and suppresses established tumors. Cancer Res. 2011;71(10):3540‐3551. [DOI] [PubMed] [Google Scholar]
- 179. Liu JF, Wu L, Yang LL, Deng WW, Mao L, Wu H, et al. Blockade of TIM3 relieves immunosuppression through reducing regulatory T cells in head and neck cancer. J Exp Clin Cancer Res. 2018;37(1):44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC. Targeting Tim‐3 and PD‐1 pathways to reverse T cell exhaustion and restore anti‐tumor immunity. J Exp Med. 2010;207(10):2187‐2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Fourcade J, Sun Z, Benallaoua M, Guillaume P, Luescher IF, Sander C, et al. Upregulation of Tim‐3 and PD‐1 expression is associated with tumor antigen–specific CD8 + T cell dysfunction in melanoma patients. J Exp Med. 2010;207(10):2175‐2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Granier C, Dariane C, Combe P, Verkarre V, Urien S, Badoual C, et al. Tim‐3 expression on tumor‐infiltrating PD‐1+CD8+ T cells correlates with poor clinical outcome in renal cell carcinoma. Cancer Res. 2017;77(5):1075‐1082. [DOI] [PubMed] [Google Scholar]
- 183. Koyama S, Akbay EA, Li YY, Herter‐Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic PD‐1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL. Adaptive resistance to anti‐PD1 therapy by Tim‐3 upregulation is mediated by the PI3K‐Akt pathway in head and neck cancer. Oncoimmunology. 2016;6(1):e1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Zhou G, Sprengers D, Boor PPC, Doukas M, Schutz H, Mancham S, et al. Antibodies against immune checkpoint molecules restore functions of tumor‐infiltrating T cells in hepatocellular carcinomas. Gastroenterology. 2017;153(4):1107‐1119.e10. [DOI] [PubMed] [Google Scholar]
- 186. Levin SD, Taft DW, Brandt CS, Bucher C, Howard ED, Chadwick EM, et al. Vstm3 is a member of the CD28 family and an important modulator of T‐cell function. Eur J Immunol. 2011;41(4):902‐915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Lozano E, Dominguez‐Villar M, Kuchroo V, Hafler DA. The TIGIT/CD226 axis regulates human T cell function. J Immunol. 2012;188(8):3869‐3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Masson D, Jarry A, Baury B, Blachardie P, Laboisse C, Lustenberger P, et al. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49(2):236‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Casado JG, Pawelec G, Morgado S, Sanchez‐Correa B, Delgado E, Gayoso I, et al. Expression of adhesion molecules and ligands for activating and costimulatory receptors involved in cell‐mediated cytotoxicity in a large panel of human melanoma cell lines. Cancer Immunol Immunother. 2009;58(9):1517‐1526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Oshima T, Sato S, Kato J, Ito Y, Watanabe T, Tsuji I, et al. Nectin‐2 is a potential target for antibody therapy of breast and ovarian cancers. Mol Cancer. 2013;12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Josefsson SE, Huse K, Kolstad A, Beiske K, Pende D, Steen CB, et al. T cells expressing checkpoint receptor TIGIT are enriched in follicular lymphoma tumors and characterized by reversible suppression of T‐cell receptor signaling. Clin Cancer Res. 2018;24(4):870‐881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Harjunpää H, Guillerey C. TIGIT as an emerging immune checkpoint. Clin Exp Immunol. 2020;200(2):108‐119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Mittal D, Lepletier A, Madore J, Aguilera AR, Stannard K, Blake SJ, et al. CD96 is an immune checkpoint that regulates CD8+ T‐cell antitumor function. Cancer Immunol Res. 2019;7(4):559‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Zhu Y, Paniccia A, Schulick AC, Chen W, Koenig MR, Byers JT, et al. Identification of CD112R as a novel checkpoint for human T cells. J Exp Med. 2016;213(2):167‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Johnston RJ, Comps‐Agrar L, Hackney J, Yu X, Huseni M, Yang Y, et al. The immunoreceptor TIGIT regulates antitumor and antiviral CD8(+) T cell effector function. Cancer Cell. 2014;26(6):923‐937. [DOI] [PubMed] [Google Scholar]
- 196. He W, Zhang H, Han F, Chen X, Lin R, Wang W, et al. CD155T/TIGIT signaling regulates CD8+ T‐cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res. 2017;77(22):6375‐6388. [DOI] [PubMed] [Google Scholar]
- 197. Joller N, Lozano E, Burkett PR, Patel B, Xiao S, Zhu C, et al. Treg cells expressing the coinhibitory molecule TIGIT selectively inhibit proinflammatory Th1 and Th17 cell responses. Immunity. 2014;40(4):569‐581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Kurtulus S, Sakuishi K, Ngiow SF, Joller N, Tan DJ, Teng MW, et al. TIGIT predominantly regulates the immune response via regulatory T cells. J Clin Invest. 2015;125(11):4053‐4062. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 199. Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, et al. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat Immunol. 2009;10(1):48‐57. [DOI] [PubMed] [Google Scholar]
- 200. Guillerey C, Harjunpää H, Carrié N, Kassem S, Teo T, Miles K, et al. TIGIT immune checkpoint blockade restores CD81 T‐cell immunity against multiple myeloma. Blood. 2018;132(16):1689‐1694. [DOI] [PubMed] [Google Scholar]
- 201. Chauvin JM, Pagliano O, Fourcade J, Sun Z, Wang H, Sander C, et al. TIGIT and PD‐1 impair tumor antigen‐specific CD8+ T cells in melanoma patients. J Clin Invest. 2015;125(5):2046‐2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Tang W, Pan X, Han D, Rong D, Zhang M, Yang L, et al. Clinical significance of CD8 + T cell immunoreceptor with Ig and ITIM domains+ in locally advanced gastric cancer treated with SOX regimen after D2 gastrectomy. Oncoimmunology. 2019;8(6):e1593807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Wu L, Mao L, Liu JF, Chen L, Yu GT, Yang LL, et al. Blockade of TIGIT/CD155 signaling reverses t‐cell exhaustion and enhances antitumor capability in head and neck squamous cell carcinoma. Cancer Immunol Res. 2019;7(10):1700‐1713. [DOI] [PubMed] [Google Scholar]
- 204. Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksis AS, et al. TIGIT and PD‐1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Zhang X, Zhang H, Chen L, Feng Z, Gao L, Li Q. TIGIT expression is upregulated in T cells and causes T cell dysfunction independent of PD‐1 and Tim‐3 in adult B lineage acute lymphoblastic leukemia. Cell Immunol. 2019;344:103958. [DOI] [PubMed] [Google Scholar]
- 206. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer immunotherapy targeting PD‐1 and GITR can rescue CD8+ T cell dysfunction and maintain memory phenotype. Sci Immunol. 2018;3(29):eaat7061. [DOI] [PubMed] [Google Scholar]
- 207. Hoogi S, Eisenberg V, Mayer S, Shamul A, Barliya T, Cohen CJ. A TIGIT‐based chimeric co‐stimulatory switch receptor improves T‐cell anti‐tumor function. J Immunother Cancer. 2019;7(1):243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Lines JL, Pantazi E, Mak J, Sempere LF, Wang L, O'Connell S, et al. VISTA is an immune checkpoint molecule for human T cells. Cancer Res. 2014;74(7):1924‐1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. ElTanbouly MA, Zhao Y, Nowak E, Schaafsma E, Le Mercier I, Ceeraz S, et al. VISTA is a checkpoint regulator for naïve T cell quiescence and peripheral tolerance. Science. 2020;367(6475):eaay0524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Flies DB, Han X, Higuchi T, Zheng L, Sun J, Ye JJ, et al. Coinhibitory receptor PD‐1H preferentially suppresses CD4+ T cell‐mediated immunity. J Clin Invest. 2014;124:1966‐1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Derré L, Rivals JP, Jandus C, Pastor S, Rimoldi D, Romero P, et al. BTLA mediates inhibition of human tumor‐specific CD8+ T cells that can be partially reversed by vaccination. J Clin Invest. 2010;120(1):157‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Quan L, Lan X, Meng Y, Guo X, Guo Y, Zhao L, et al. BTLA marks a less cytotoxic T‐cell subset in diffuse large B‐cell lymphoma with high expression of checkpoints. Exp Hematol. 2018;60:47‐56.e1. [DOI] [PubMed] [Google Scholar]
- 213. Hobo W, Norde WJ, Schaap N, Fredrix H, Maas F, Schellens K, et al. B and T lymphocyte attenuator mediates inhibition of tumor‐reactive CD8+ T cells in patients after allogeneic stem cell transplantation. J Immunol. 2012;189(1):39‐49. [DOI] [PubMed] [Google Scholar]
- 214. Stecher C, Battin C, Leitner J, Zettl M, Grabmeier‐Pfistershammer K, Höller C, et al. PD‐1 blockade promotes emerging checkpoint inhibitors in enhancing T cell responses to allogeneic dendritic cells. Front Immunol. 2017;8:572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Viganò S, Banga R, Bellanger F, Pellaton C, Farina A, Comte D, et al. CD160‐associated CD8 T‐cell functional impairment is independent of PD‐1 expression. PLoS Pathog. 2014;10(9):e1004380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Xie J, Wang J, Cheng S, Zheng L, Ji F, Yang L, et al. Expression of immune checkpoints in T cells of esophageal cancer patients. Oncotarget. 2016;7(39):63669‐63678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217. Zelle‐Rieser C, Thangavadivel S, Biedermann R, Brunner A, Stoitzner P, Willenbacher E, et al. T cells in multiple myeloma display features of exhaustion and senescence at the tumor site. J Hematol Oncol. 2016;9(1):116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218. Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor‐specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121(6):2350‐2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219. Tan J, Chen S, Lu Y, Yao D, Xu L, Zhang Y, et al. Higher PD‐1 expression concurrent with exhausted CD8+ T cells in patients with de novo acute myeloid leukemia. Chinese J Cancer Res. 2017;29(5):463‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220. Agresta L, Hoebe KHN, Janssen EM. The emerging role of CD244 signaling in immune cells of the tumor microenvironment. Front Immunol. 2018;9:2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. wen ChenC, M Xue, Zhang W, Xie J, Coopersmith CM, Ford ML. 2B4 but not PD‐1 blockade improves mortality in septic animals with preexisting malignancy. JCI Insight. 2019;4(22):127867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Blackburn SD, Shin H, Haining WN, Zou T, Workman CJ, Polley A, et al. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat Immunol. 2009;10(1):29‐37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223. Raziorrouh B, Schraut W, Gerlach T, Nowack D, Grüner NH, Ulsenheimer A, et al. The immunoregulatory role of CD244 in chronic hepatitis B infection and its inhibitory potential on virus‐specific CD8+ T‐cell function. Hepatology. 2010;52(6):1934‐1947. [DOI] [PubMed] [Google Scholar]
- 224. André P, Denis C, Soulas C, Bourbon‐Caillet C, Lopez J, Arnoux T, et al. Anti‐NKG2A mAb is a checkpoint inhibitor that promotes anti‐tumor immunity by unleashing both T and NK Cells. Cell. 2018;175(7):1731‐1743.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225. van Montfoort N, Borst L, Korrer MJ, Sluijter M, Marjit KA, Santegoets SJ, et al. NKG2A blockade potentiates CD8 T cell immunity induced by cancer vaccines. Cell. 2018;175(7):1744‐1755.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226. Eugène J, Jouand N, Ducoin K, Dansette D, Oger R, Deleine C, et al. The inhibitory receptor CD94/NKG2A on CD8+ tumor‐infiltrating lymphocytes in colorectal cancer: a promising new druggable immune checkpoint in the context of HLAE/β2m overexpression. Mod Pathol. 2020;33(3):466‐482. [DOI] [PubMed] [Google Scholar]
- 227. Van Hall T, André P, Horowitz A, Ruan DF, Borst L, Zerbib R, et al. Monalizumab: inhibiting the novel immune checkpoint NKG2A. J Immunother Cancer. 2019;7(1):263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Li X, Wang R, Fan P, Yao X, Qin L, Peng Y, et al. A comprehensive analysis of key immune checkpoint receptors on tumor‐infiltrating T cells from multiple types of cancer. Front Oncol. 2019;9:1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Buck MD, O'Sullivan D, Pearce EL. T cell metabolism drives immunity. J Exp Med. 2015;212(9):1345‐1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Wang R, Dillon CP, Shi LZ, Milasta S, Carter R, Finkelstein D, et al. The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity. 2011;35(6):871‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231. Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16(6):769‐777. [DOI] [PubMed] [Google Scholar]
- 232. Peng M, Yin N, Chhangawala S, Xu K, Leslie CS, Li MO. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science. 2016;354(6311):481‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Menk A V, , Scharping NE, Moreci RS, Zeng X, Guy C, Salvatore S, et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018;22(6):1509‐1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Chang CH, Curtis JD, Jr MaggiLB, Faubert B, Villarino AV, et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153(6):1239‐1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Cham CM, Driessens G, O'Keefe JP, Gajewski TF. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur J Immunol. 2008;38(9):2438‐2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, et al. Mitochondria are required for antigen‐specific T cell activation through reactive oxygen species signaling. Immunity. 2013;38(2):225‐236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Carr EL, Kelman A, Wu GS, Gopaul R, Senkevitch E, Aghvanyan A, et al. Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol. 2010;185(2):1037‐1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Klysz D, Tai X, Robert PA, Craveiro M, Cretenet G, Oburoglu L, et al. Glutamine‐dependent α‐ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal. 2015;8(396):ra97. [DOI] [PubMed] [Google Scholar]
- 239. Pearce EL, Walsh MC, Cejas PJ, Harms GM, Shen H, Wang LS, et al. Enhancing CD8 T‐cell memory by modulating fatty acid metabolism. Nature. 2009;460(7251):103‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. O'Sullivan D, van der Windt GJ, Huang SC, Curtis JD, Chang CH, Buck MD, et al. Memory CD8+ T cells use cell‐intrinsic lipolysis to support the metabolic programming necessary for development. Immunity. 2014;41(1):75‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, et al. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity. 2012;36(1):68‐78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242. van der Windt GJ, O'Sullivan D, Everts B, Huang SC, Buck MD, Curtis JD, et al. CD8 memory T cells have a bioenergetic advantage that underlies their rapid recall ability. Proc Natl Acad Sci U S A. 2013;110(35):14336‐14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243. Siska PJ, Beckermann KE, Mason FM, Andrejeva G, Greenplate AR, Sendor AB, et al. Mitochondrial dysregulation and glycolytic insufficiency functionally impair CD8 T cells infiltrating human renal cell carcinoma. JCI Insight. 2017;2(12):93411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Gemta LF, Siska PJ, Nelson ME, Gao X, Liu X, Locasale JW, et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor‐infiltrating CD8+ T cells. Sci Immunol. 2019;4(31):eaap9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245. Ho P‐C, Bihuniak JD, Macintyre AN, Staron M, Lie X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti‐tumor T cell responses. Cell. 2015;162(6):1217‐1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246. Scharping NE, Menk AV, Moreci RS, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. 2016;45(2):374‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247. Dimeloe S, Gubser P, Loeliger J, Frick C, Develioglu L, Fischer M, et al. Tumor‐derived TGF‐β inhibits mitochondrial respiration to suppress IFN‐γ production by human CD4+ T cells. Sci Signal. 2019;12(599):eaav3334. [DOI] [PubMed] [Google Scholar]
- 248. Ogando J, Sáez ME, Santos J, Nuevo‐Tapioles C, Gut M, et al. PD‐1 signaling affects cristae morphology and leads to mitochondrial dysfunction in human CD8+ T lymphocytes. J Immunother Cancer. 2019;7(1):151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Rubio I, Grund S, Song SP, Biskup C, Bandemer S, Fricke M, et al. TCR‐induced activation of Ras proceeds at the plasma membrane and requires palmitoylation of N‐Ras. J Immunol. 2010;185(6):3536‐3543. [DOI] [PubMed] [Google Scholar]
- 250. Rampoldi F, Bonrouhi M, Boehm ME, Lehmann WD, Popovic ZV, Kaden S, et al. Immunosuppression and aberrant T cell development in the absence of N‐myristoylation. J Immunol. 2015;195(9):4228‐4243. [DOI] [PubMed] [Google Scholar]
- 251. Lee J, Walsh MC, Hoehn KL, James DE, Wherry EJ, Choi Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J Immunol. 2014;192(7):3190‐3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252. Muroski ME, Miska J, Chang AL, Zhang P, Rashidi A, Moore H, et al. Fatty acid uptake in T cell subsets using a quantum dot fatty acid conjugate. Sci Rep. 2017;7(1):5790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253. Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, et al. Survival of tissue‐resident memory T cells requires exogenous lipid uptake and metabolism. Nature. 2017;543(7644):252‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, et al. Fatty acid uptake and lipid storage induced by HIF‐1α contribute to cell growth and survival after hypoxia‐reoxygenation. Cell Rep. 2014;9(1):349‐365. [DOI] [PubMed] [Google Scholar]
- 255. Mika A, Kobiela J, Pakiet A, Czumaj A, Sokolowska E, Makarewicz W, et al. Preferential uptake of polyunsaturated fatty acids by colorectal cancer cells. Sci Rep. 2020;10(1):1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. van der Mijn JC, Fu L, Khani F, Zhang T, Molina AM, Barbieri CE, et al. Combined metabolomics and genome‐wide transcriptomics analyses show multiple HIF1α‐induced changes in lipid metabolism in early stage clear cell renal cell carcinoma. Transl Oncol. 2020;13(2):177‐185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, et al. Adipocyte‐induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene. 2018;37(17):2285‐2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Watt MJ, Clark AK, Selth LA, Haynes VR, Lister N, Rebello R, et al. Suppressing fatty acid uptake has therapeutic effects in preclinical models of prostate cancer. Sci Transl Med. 2019;11(478):eaau5758. [DOI] [PubMed] [Google Scholar]
- 259. Zhao J, Zhi Z, Wang C, Xing H, Song G, Yu X, et al. Exogenous lipids promote the growth of breast cancer cells via CD36. Oncol Rep. 2017;38(4):2105‐2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Nath A, Li I, Roberts LR, Chan C. Elevated free fatty acid uptake via CD36 promotes epithelial‐mesenchymal transition in hepatocellular carcinoma. Sci Rep. 2015;5:14752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Lupien LE, Bloch K, Dehairs J, Traphagen NA, Feng WW, Davis WL, et al. Endocytosis of very low‐density lipoproteins: an unexpected mechanism for lipid acquisition by breast cancer cells. J Lipid Res. 2020;61(2):205‐218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. 2019;30(1):143‐156.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Ma X, Bi E, Huang C, Lu Y, Xue G, Guo X, et al. Cholesterol negatively regulates IL‐9‐producing CD8+ T cell differentiation and antitumor activity. J Exp Med. 2018;215(6):1555‐1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 2016;531(7596):651‐655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265. Ren Y, Shen HM. Critical role of AMPK in redox regulation under glucose starvation. Redox Biol. 2019;25:101154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266. Zhang C, Yue C, Herrmann A, Song J, Egelston C, Wang T, et al. STAT3 activation‐induced fatty acid oxidation in CD8+ T effector cells is critical for obesity‐promoted breast tumor growth. Cell Metab. 2020;31(1):148‐161.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267. Michalek RD, Gerriets VA, Jacobs SR, Macintyre AN, MacIver NJ, Mason EF, et al. Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol. 2011;186(6):3299‐3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268. Pacella I, Procaccini C, Focaccetti C, Miacci S, Timperi E, Faicchia D, et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc Natl Acad Sci USA. 2018;115(28):E6546‐E6555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269. Field CS, Baixauli F, Kyle RL, Puleston DJ, Cameron AM, Sanin DE, et al. Mitochondrial integrity regulated by lipid metabolism is a cell‐intrinsic checkpoint for Treg suppressive function. Cell Metab. 2020;31(2):422‐437.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270. Cascone T, McKenzie JA, Mbofung RM, Punt S, Wang Z, Xu C, et al. Increased Tumor glycolysis characterizes immune resistance to adoptive T cell therapy. Cell Metab. 2018;27(5):977‐987.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest. 2013;123(10):4479‐4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Crompton JG, Sukumar M, Roychoudhuri R, Clever D, Gros A, Eli RL, et al. Akt inhibition enhances expansion of potent tumor‐specific lymphocytes with memory cell characteristics. Cancer Res. 2015;75(2):296‐305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Buck MD, O'Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, et al. Mitochondrial dynamics controls T cell fate through metabolic programming. Cell. 2016;166(1):63‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Zhang Y, Kurupati R, Liu L, Zhou XY, Zhang G, Hudaihed A, et al. Enhancing CD8+ T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell. 2017;32(3):377‐391.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Jr PoseyAD, et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. 2016;44(2):380‐390. [DOI] [PubMed] [Google Scholar]
- 276. Nakaya M, Xiao Y, Zhou X, Chang JH, Chang M, Cheng X, et al. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity. 2014;40(5):692‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277. Lian G, Gnanaprakasam JR, Wang T, Wu R, Chen X, Liu L, et al. Glutathione de novo synthesis but not recycling process coordinates with glutamine catabolism to control redox homeostasis and directs murine T cell differentiation. Elife. 2018;7:e36158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278. Chang WK, Yang KD, Chuang H, Jan JT, Shaio MF. Glutamine protects activated human T cells from apoptosis by up‐regulating glutathione and Bcl‐2 levels. Clin Immunol. 2002;104(2):151‐160. [DOI] [PubMed] [Google Scholar]
- 279. Blagih J, Coulombe F, Vincent EE, Dupuy F, Galicia‐Vázquez G, Yurchenko E, et al. The energy sensor AMPK regulates T cell metabolic adaptation and effector responses in vivo. Immunity. 2015;42(1):41‐54. [DOI] [PubMed] [Google Scholar]
- 280. Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123(9):3678‐3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281. Johnson MO, Wolf MM, Madden MZ, Andrejeva G, Sugiura A, Contreras DC, et al. Distinct regulation of Th17 and Th1 cell differentiation by glutaminase‐dependent metabolism. Cell. 2018;175(7):1780‐1795.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Leone RD, Zhao L, Englert JM, Sun IM, Oh MH, Sun IH, et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science. 2019;366(6468):1013‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, et al. Serine is an essential metabolite for effector T cell expansion. Cell Metab. 2017;25(2):345‐357. [DOI] [PubMed] [Google Scholar]
- 284. Labuschagne CF, van den Broek NJF, Mackay GM, Vousden KH, Maddocks OD. Serine, but not glycine, supports one‐carbon metabolism and proliferation of cancer cells. Cell Rep. 2014;7(4):1248‐1258. [DOI] [PubMed] [Google Scholar]
- 285. Maddocks ODK, Berkers CR, Mason SM, Zheng L, Blyth K, Gottlieb E, et al. Serine starvation induces stress and p53‐dependent metabolic remodelling in cancer cells. Nature. 2013;493(7433):542‐546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286. Sinclair L V, Rolf J, Emslie E, Shi YB, Taylor PM, Cantrell DA. Control of amino‐acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol. 2013;14(5):500‐508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Saito Y, Li L, Coyaud E, Luna A, Sander C, Raught B, et al. LLGL2 rescues nutrient stress by promoting leucine uptake in ER+ breast cancer. Nature. 2019;569(7755):275‐279. [DOI] [PubMed] [Google Scholar]
- 288. Zhang BK, Moran AM, Bailey CG, Rasko JEJ, Holst J, Wang Q. EGF‐activated PI3K/Akt signalling coordinates leucine uptake by regulating LAT3 expression in prostate cancer. Cell Commun Signal. 2019;17(1):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, et al. Arginase I in myeloid suppressor cells is induced by COX‐2 in lung carcinoma. J Exp Med. 2005;202(7):931‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, et al. Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T‐cell receptor expression and antigen‐specific T‐cell responses. Cancer Res. 2004;64(16):5839‐5849. [DOI] [PubMed] [Google Scholar]
- 291. Norian LA, Rodriguez PC, O'Mara LA, Zabaleta J, Ochoa AC, Cella M, et al. Tumor‐infiltrating regulatory dendritic cells inhibit CD8+ T cell function via L‐arginine metabolism. Cancer Res. 2009;69(7):3086‐3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292. Liu Q, Zhang C, Sun A, Zheng Y, Wang L, Cao X. Tumor‐educated CD11bhigh Ialow regulatory dendritic cells suppress T cell response through arginase I. J Immunol. 2009;182(10):6207‐6216. [DOI] [PubMed] [Google Scholar]
- 293. Mahadevan NR, Anufreichik V, Rodvold JJ, Chiu KT, Sepulveda H, Zanetti M. Cell‐extrinsic effects of tumor ER stress imprint myeloid dendritic cells and impair CD8+ T cell priming. PLoS One. 2012;7(12):e51845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. L‐arginine modulates T cell metabolism and enhances survival and anti‐tumor activity. Cell. 2016;167(3):829‐842.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295. Serafini P, Mgebroff S, Noonan K, Borrello I. Myeloid‐derived suppressor cells promote cross‐tolerance in B‐cell lymphoma by expanding regulatory T cells. Cancer Res. 2008;68(13):5439‐5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296. Mondanelli G, Bianchi R, Pallotta MT, Orabona C, Albini E, Iacono A, et al. A relay pathway between arginine and tryptophan metabolism confers immunosuppressive properties on dendritic cells. Immunity. 2017;46(2):233‐244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297. Munn DH, Sharma MD, Hou D, Baban B, Lee JR, Antonia SJ, et al. Expression of indoleamine 2,3‐dioxygenase by plasmacytoid dendritic cells in tumor‐draining lymph nodes. J Clin Invest. 2004;114(2):280‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298. Sharma MD, Hou DY, Liu Y, Koni PA, Metz R, Changler P, et al. Indoleamine 2,3‐dioxygenase controls conversion of Foxp3+ Tregs to TH17‐like cells in tumor‐draining lymph nodes. Blood. 2009;113(24):6102‐6111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299. Yu J, Du W, Yan F, Wang Y, Li H, Cao S, et al. Myeloid‐derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783‐3797. [DOI] [PubMed] [Google Scholar]
- 300. Pilotte L, Larrieu P, Stroobant V, Colau D, Dolusic E, Frédérick R, et al. Reversal of tumoral immune resistance by inhibition of tryptophan 2,3‐dioxygenase. Proc Natl Acad Sci U S A. 2012;109(7):2497‐2502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301. van Baren N, Van den Eynde BJ. Tryptophan‐degrading enzymes in tumoral immune resistance. Front Immunol. 2015;6:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Ye Z, Yue L, Shi J, Shao M, Wu T. Role of IDO and TDO in cancers and related diseases and the therapeutic implications. J Cancer. 2019;10(12):2771‐2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303. Steggerda SM, Bennett MK, Chen J, Emberley E, Huang T, Janes JR, et al. Inhibition of arginase by CB‐1158 blocks myeloid cell‐mediated immune suppression in the tumor microenvironment. J Immunother Cancer. 2017;5(1):101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304. Komiya T, Huang CH. Updates in the clinical development of epacadostat and other indoleamine 2,3‐dioxygenase 1 inhibitors (IDO1) for human cancers. Front Oncol. 2018;8:423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305. Muller AJ, Manfredi MG, Zakharia Y, Prendergast GC. Inhibiting IDO pathways to treat cancer: lessons from the ECHO‐301 trial and beyond. Semin Immunopathol. 2019;41(1):41‐48. [DOI] [PubMed] [Google Scholar]
- 306. Chen D, Koropatnick J, Jiang N, Zheng X, Zhang X, Wang H, et al. Targeted siRNA silencing of indoleamine 2, 3‐dioxygenase in antigen‐presenting cells using mannose‐conjugated liposomes. J Immunother. 2014;37(2):123‐134. [DOI] [PubMed] [Google Scholar]
- 307. Zheng X, Koropatnick J, Chen D, Velenosi T, Ling H, Zhang X, et al. Silencing IDO in dendritic cells: a novel approach to enhance cancer immunotherapy in a murine breast cancer model. Int J Cancer. 2013;132(4):967‐977. [DOI] [PubMed] [Google Scholar]
- 308. Sioud M, Nyakas M, Sæbøe‐Larssen S, Mobergslien A, Aamdal S, Kvalheim G. Diversification of antitumour immunity in a patient with metastatic melanoma treated with ipilimumab and an IDO‐silenced dendritic cell vaccine. Case Rep Med;2016:9639585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, Edinger M, et al. Inhibitory effect of tumor cell‐derived lactic acid on human T cells. Blood. 2007;109(9):3812‐3819. [DOI] [PubMed] [Google Scholar]
- 310. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. LDHA‐associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24(5):657‐671. [DOI] [PubMed] [Google Scholar]
- 311. Pilon‐Thomas S, Kodumudi KN, El‐Kenawi AE, Russel S, Weber AM, Luddy K, et al. Neutralization of tumor acidity improves antitumor responses to immunotherapy. Cancer Res. 2016;76(6):1381‐1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312. Renner K, Bruss C, Schnell A, Koehl G, Becker HM, Fante M, et al. Restricting glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell Rep. 2019;29(1):135‐150.e9. [DOI] [PubMed] [Google Scholar]
- 313. Calcinotto A, Filipazzi P, Grioni M, Iero M, De Milito A, Ricupito A, et al. Modulation of microenvironment acidity reverses anergy in human and murine tumor‐infiltrating T lymphocytes. Cancer Res. 2012;72(11):2746‐2756. [DOI] [PubMed] [Google Scholar]
- 314. Mezrich JD, Fechner JH, Zhang X, Johnson BP, Burlingham WJ, Bradfield CA. An Interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J Immunol. 2010;185(6):3190‐3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315. Angelin A, Gil‐de‐Gómez L, Dahiya S, Jiao J, Guo L, Levine MH, et al. Foxp3 reprograms T cell metabolism to function in low‐glucose, high‐lactate environments. Cell Metab. 2017;25(6):1282‐1293.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316. Lappas CM, Rieger JM, Linden J. A2A adenosine receptor induction inhibits IFN‐γ production in murine CD4+ T cells. J Immunol. 2005;174(2):1073‐1080. [DOI] [PubMed] [Google Scholar]
- 317. Linnemann C, Schildberg FA, Schurich A, Diehl L, Hegenbarth SI, Endl E, et al. Adenosine regulates CD8 T‐cell priming by inhibition of membrane‐proximal T‐cell receptor signalling. Immunology. 2009;128(1 Suppl):e728‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318. Sorrentino C, Hossain F, Rodriguez PC, Sierra RA, Pannuti A, Osborne BA, et al. Adenosine A2A receptor stimulation inhibits TCR‐induced Notch1 activation in CD8+T‐cells. Front Immunol. 2019;10:162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319. Mastelic‐Gavillet B, Navarro Rodrigo B, Décombaz L, Wang H, Ercolano G, Ahmed R, et al. Adenosine mediates functional and metabolic suppression of peripheral and tumor‐infiltrating CD8+ T cells. J Immunother Cancer. 2019;7(1):257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320. Ohta A, Gorelik E, Prasad SJ, Ronchese F, Lukashev D, Wong MK, et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc Natl Acad Sci U S A. 2006;103(35):13132‐13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321. Mediavilla‐Varela M, Castro J, Chiappori A, Noyes D, Hernandez DC, Allard B, et al. A novel antagonist of the immune checkpoint protein adenosine A2a receptor restores tumor‐infiltrating lymphocyte activity in the context of the tumor microenvironment. Neoplasia. 2017;19(7):530‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Kjaergaard J, Hatfield S, Jones G, Ohta A, Sitkovsky M. A2A adenosine receptor gene deletion or synthetic A2A antagonist liberate tumor‐reactive CD8 + T cells from tumor‐induced immunosuppression. J Immunol. 2018;201(2):782‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Ma SR, Deng WW, Liu JF, Mao L, Yu GT, Bu LL, et al. Blockade of adenosine A2A receptor enhances CD8+ T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol Cancer. 2017;16(1):99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK, et al. Adenosine 2A receptor blockade as an immunotherapy for treatment‐refractory renal cell cancer. Cancer Discov. 2020;10(1):40‐53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Beavis PA, Henderson MA, Giuffrida L, Mills JK, Sek K, Cross RS, et al. Targeting the adenosine 2A receptor enhances chimeric antigen receptor T cell efficacy. J Clin Invest. 2017;127(3):929‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Li Y, Patel SP, Roszik J, Qin Y. Hypoxia‐driven immunosuppressive metabolites in the tumor microenvironment: new approaches for combinational immunotherapy. Front Immunol. 2018;9:1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Hatfield SM, Kjaergaard J, Lukashev D, Schreiber TH, Belikoff B, Abbott R, et al. Immunological mechanisms of the antitumor effects of supplemental oxygenation. Sci Transl Med. 2015;7(277):277ra30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Chen L, Diao L, Yang Y, Yi X, Rodriguez BL, Li Y, et al. CD38‐mediated immunosuppression as a mechanism of tumor cell escape from PD‐1/PD‐L1 blockade. Cancer Discov. 2018;8(9):1156‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329. Perrot I, Michaud HA, Giraudon‐Paoli M, Augier S, Docquier A, Gros L, et al. Blocking antibodies targeting the CD39/CD73 immunosuppressive pathway unleash immune responses in combination cancer therapies. Cell Rep. 2019;27(8):2411‐2425.e9. [DOI] [PubMed] [Google Scholar]
- 330. Li XY, Moesta AK, Xiao C, Nakamura K, Casey M, Zhang H, et al. Targeting CD39 in cancer reveals an extracellular ATP‐ and inflammasome‐driven tumor immunity. Cancer Discov. 2019;9(12):1754‐1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Hirayama A, Kami K, Sugimoto M, Sugawara M, Toki N, Onozuka H, et al. Quantitative metabolome profiling of colon and stomach cancer microenvironment by capillary electrophoresis time‐of‐flight mass spectrometry. Cancer Res. 2009;69(11):4918‐4925. [DOI] [PubMed] [Google Scholar]
- 332. Reznik E, Luna A, Aksoy BA, Liu EM, La K, Ostrovnaya I, et al. A landscape of metabolic variation across tumor types. Cell Syst. 2018;6(3):301‐313.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Choi H, Na KJ. Pan‐cancer analysis of tumor metabolic landscape associated with genomic alterations. Mol Cancer. 2018;17(1):150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334. Ortmayr K, Dubuis S, Zampieri M. Metabolic profiling of cancer cells reveals genome‐wide crosstalk between transcriptional regulators and metabolism. Nat Commun. 2019;10(1):1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Ji R‐R, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, et al. An immune‐active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother. 2012;61(7):1019‐1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337. Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, et al. Dynamic changes in PD‐L1 expression and immune infiltrates early during treatment predict response to PD‐1 blockade in melanoma. Clin Cancer Res. 2017;23(17):5024‐5033. [DOI] [PubMed] [Google Scholar]
- 338. Yang S, Wu Y, Deng Y, Zhou L, Yang P, Zheng Y, et al. Identification of a prognostic immune signature for cervical cancer to predict survival and response to immune checkpoint inhibitors. Oncoimmunology. 2019;8(12):e1659094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339. Bulgarelli J, Tazzari M, Granato AM, Ridolfi L, Maiocchi S, de Rosa F, et al. Dendritic cell vaccination in metastatic melanoma turns “non‐T cell inflamed” into “T‐cell inflamed” tumors. Front Immunol. 2019;10:2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340. Ge Y, Xi H, Ju S, Zhang X. Blockade of PD‐1/PD‐L1 immune checkpoint during DC vaccination induces potent protective immunity against breast cancer in hu‐SCID mice. Cancer Lett. 2013;336(2):253‐259. [DOI] [PubMed] [Google Scholar]
- 341. Nagaoka K, Hosoi A, Iino T, Morishita Y, Matsushita H, Kakimi K. Dendritic cell vaccine induces antigen‐specific CD8+ T cells that are metabolically distinct from those of peptide vaccine and is well‐combined with PD‐1 checkpoint blockade. Oncoimmunology. 2017;7(3):e1395124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Lu X, Liu J, Cui P, Liu T, Piao C, Xu X, et al. Co‐inhibition of TIGIT, PD1, and Tim3 reverses dysfunction of Wilms tumor protein‐1 (WT1)‐specific CD8+ T lymphocytes after dendritic cell vaccination in gastric cancer. Am J Cancer Res. 2018;8(8):1564‐1575. [PMC free article] [PubMed] [Google Scholar]
- 343. Fourcade J, Sun Z, Pagliano O, Chauvin JM, Sander C, Janjic B, et al. PD‐1 and Tim‐3 regulate the expansion of tumor antigen‐specific CD8+ T cells induced by melanoma vaccines. Cancer Res. 2014;74(4):1045‐1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344. Sánchez‐Paulete AR, Cueto FJ, Martínez‐López M, Labiano S, Morales‐Kastresana A, Rodríguez‐Ruiz ME, et al. Cancer immunotherapy with immunomodulatory anti‐CD137 and anti‐PD‐1 monoclonal antibodies requires BATF3‐dependent dendritic cells. Cancer Discov. 2016;6(1):71‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Sánchez‐Paulete AR, Á Teijeira, Quetglas JI, Quetglas JI, Rodríguez‐Ruiz ME, Á Sánchez‐Arráez, et al. Intratumoral immunotherapy with XCL1 and sFlt3L encoded in recombinant semliki forest virus‐derived vectors fosters dendritic cell‐mediated T‐cell cross‐priming. Cancer Res. 2018;78(23):6643‐6654. [DOI] [PubMed] [Google Scholar]
- 346. Hammerich L, Marron TU, Upadhyay R, Svennson‐Arvelund J, Dhainaut M, Hussein S, et al. Systemic clinical tumor regressions and potentiation of PD1 blockade with in situ vaccination. Nat Med. 2019;25(5):814‐824. [DOI] [PubMed] [Google Scholar]
- 347. Mizumoto Y, Hemmi H, Katsuda M, Miyazawa M, Kitahata Y, Miyamoto A, et al. Anticancer effects of chemokine‐directed antigen delivery to a cross‐presenting dendritic cell subset with immune checkpoint blockade. Br J Cancer. 2020;122:1185‐11933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348. Sun NY, Chen YL, Lin HW, Chiang YC, Chang CF, Tai YJ, et al. Immune checkpoint Ab enhances the antigen‐specific anti‐tumor effects by modulating both dendritic cells and regulatory T lymphocytes. Cancer Lett. 2019;444:20‐34. [DOI] [PubMed] [Google Scholar]
- 349. Kauder SE, Kuo TC, Harrabi O, Chen A, Sangalang E, Doyle L, et al. ALX148 blocks CD47 and enhances innate and adaptive antitumor immunity with a favorable safety profile. PLoS One. 2018;13(8):e0201832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Liu X, Liu L, Ren Z, Yang K, Xu H, Luan Y, et al. Dual targeting of innate and adaptive checkpoints on tumor cells limits immune evasion. Cell Rep. 2018;24(8):2101‐2111. [DOI] [PubMed] [Google Scholar]
- 351. Liu B, Guo H, Xu J, Qin T, Guo Q, Gu N, et al. Elimination of tumor by CD47/PD‐L1 dual‐targeting fusion protein that engages innate and adaptive immune responses. MAbs. 2018;10(2):315‐324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Lian S, Xie R, Ye Y, Xie X, Li S, Lu Y, et al. Simultaneous blocking of CD47 and PD‐L1 increases innate and adaptive cancer immune responses and cytokine release. EBioMedicine. 2019;42:281‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Tao H, Qian P, Wang F, Yu H, Guo Y. Targeting CD47 enhances the efficacy of anti‐PD‐1 and CTLA‐4 in an esophageal squamous cell cancer preclinical model. Oncol Res. 2017;25(9):1579‐1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354. Ngo M, Han A, Lakatos A, Sahoo D, Hachey SJ, Weiskopf K, et al. Antibody therapy targeting CD47 and CD271 effectively suppresses melanoma metastasis in patient‐derived xenografts. Cell Rep. 2016;16(6). [DOI] [PubMed] [Google Scholar]
- 355. Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bossé D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun. 2019;10(1):4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Gomes B, Driessens G, Bartlett D, Cai D, Cauwenberghs S, Crosignani S, et al. Characterization of the selective indoleamine 2,3‐dioxygenase‐1 (IDO1) catalytic inhibitor EOS200271/PF‐06840003 supports IDO1 as a critical resistance mechanism to PD‐(L)1 blockade therapy. Mol Cancer Ther. 2018;17(12):2530‐2542. [DOI] [PubMed] [Google Scholar]
- 357. Schramme F, Crosignani S, Frederix K, Hoffmann D, Pilotte L, Stroobant V, et al. Inhibition of tryptophan‐dioxygenase activity increases the antitumor efficacy of immune checkpoint inhibitors. Cancer Immunol Res. 2020;8(1):32‐45. [DOI] [PubMed] [Google Scholar]
- 358. Kim C, Kim JH, Kim JS, Chon HJ, Kim J‐H. A novel dual inhibitor of IDO and TDO, CMG017, potently suppresses the kynurenine pathway and overcomes resistance to immune checkpoint inhibitors. J Clin Oncol. 2019;37(15 Suppl):e14228. [Google Scholar]
- 359. Holtzhausen A, Zhao F, Evans KS, Tsutsui M, Orabona C, Tyler DS, et al. Melanoma‐derived Wnt5a promotes local dendritic‐cell expression of IDO and immunotolerance: opportunities for pharmacologic enhancement of immunotherapy. Cancer Immunol Res. 2015;3(9):1082‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]