

Abstract

Oxidative stress (OS) plays a major role in the pathogenesis of various diseases in humans. OS is a result of an imbalance between reactive oxygen species (ROS) and the biologically available antioxidants that prevent or repair damage that ROS inflict on the host cells. ROS are naturally generated during normal mitochondrial respiration and by oxidative burst during the immune response. Many factors may influence OS, including genetics, diet, exercise, and exposure to environmental toxicants (e.g., tobacco smoke). A nonenzymatic peroxidation product of arachidonic acid (AA), 8-iso-PGF2α (8-isoprostane), is a validated biomarker of OS that is present in urine as both glucuronide conjugate and free acid. Previous studies report that the conjugated forms of 8-isoprostane can vary between 30 and 80% of the total 8-isoprostane levels. By hydrolyzing the conjugated forms, it is possible to obtain a total (free + conjugated) measurement of 8-isoprostane in urine samples. Here, we describe a robust, automated, and high-throughput method for measuring total urinary 8-isoprostane using a polymeric weak anion-exchange solid-phase extraction (SPE) and isotope-dilution ultrahigh performance liquid chromatography electrospray ionization–tandem mass spectrometry (UHPLC–MS/MS). This method, using a 96-well plate platform, showed good sensitivity (8.8 pg/mL LOD) and used only 400 μL of the sample volume with a cycle time of 11 min. The inter- and intraday precision, calculated from 20 repeated measurements of two quality control pools, varied from 4 to 10%. Accuracy, calculated from the recovery percentage at three spiking levels, ranged from 92.7 to 106.7%. We modified this method to allow for the exclusive measurement of free 8-isoprostane by removing the hydrolysis step. We measured both free and total 8-isoprostane in urine collected from 30 cigarette smokers (free: 460 ± 78.8 pg/mL; total: 704 ± 108 pg/mL) and 30 nonusers of tobacco products (free: 110 ± 24.2 pg/mL; total: 161 ± 38.7 pg/mL). This method is robust, accurate, and easily adaptable for large population studies.
Introduction
Oxidative stress (OS) has been linked to several human pathologies, including cancer,1−3 cardiovascular diseases,4,5 respiratory diseases,6 and neurodegenerative disorders.7 Furthermore, OS may be an important part of the aging process.8,9 OS is characterized as an imbalance between pro-oxidant and antioxidant defenses caused by overproduction of reactive oxygen species (ROS) or the loss of effectiveness of antioxidants. ROS are short-lived chemical species generated endogenously during mitochondrial metabolism and immune response and exogenously produced by radiation or exposure to environmental toxicants, such as tobacco smoke.10 Being highly reactive and short lived, ROS are difficult to monitor directly; therefore, it is more reasonable to monitor biomarkers produced by ROS reaction with biological molecules.11 In 1990, Morrow et. al. discovered a series of prostaglandin-like compounds, F2-isoprostanes, that were formed nonenzymatically by free radical peroxidation of arachidonic acid.8,12 Since then, multiple researchers have monitored isoprostanes in human biospecimens and found higher levels in patients with a myriad of human diseases compared to healthy controls.6,12−14
One of the most abundant and stable F2-isoprostanes, 8-isoprostane (CAS #27415-26-5), has been quantified in urine and plasma using immunoassays, gas chromatography–mass spectrometry (GC–MS), GC–tandem mass spectrometry (GC–MS/MS), and liquid chromatography–tandem mass spectrometry (LC–MS/MS).8,11,12,15 Immunoassays offer a cost-effective high-throughput process for analyzing 8-isoprostane; however, they are less reliable due to cross-reactivity of molecules possessing similar structures, such as the COX-derived prostaglandin F2α (PGF2α), and biological impurities interfering with antibody binding.8,16 Several methods have been developed using GC–MS or GC–MS/MS to quantify 8-isoprostane; however, extensive sample preparation is usually necessary before analysis, greatly limiting the throughput. Conversely, LC–MS/MS analysis of 8-isoprostane is both sensitive and selective compared to immunoassays, and the sample preparation generally requires fewer steps than GC–MS methods.13,16
Autoxidation of lipids can occur in plasma samples, requiring special precautions to avoid artifactual production of 8-isoprostane. However, urinary 8-isoprostane is extremely stable existing in the body both as the free (nonconjugated) form and conjugates, primarily glucuronide, and does not suffer from artifactual formation in vitro.17 This makes urine a more suitable matrix for the detection of 8-isoprostane. However, there is significant variability, 30–80%, in the amount of glucuronide conjugation that occurs between individuals.18−21 Some commercially available immunoassays attempt to measure the sum of conjugated and free levels (total) of urinary 8-isoprostane; however, most reported GC–MS or LC–MS methods only measure free 8-isoprostane in urine.
In this study, we developed and validated an analytical method that measures the free and total concentrations of urinary 8-isoprostane by SPE-UHPLC–MS/MS analysis. This method uses an automated liquid handling platform to streamline the analytical process to support large population studies.
Materials and Methods
Materials
Chemicals
Acetonitrile (HPLC grade), methanol (HPLC grade), formic acid (≥ 99.5%), and ammonium hydroxide (certified ACS plus) were purchased from Fisher Scientific (Fair Lawn, NJ). We purchased water (HPLC grade) from JT Baker (Phillipsburg, NJ). We purchased β-glucuronidase, type IX-A from Escherichia coli, from Sigma-Aldrich (St. Louis, MO). 8-Isoprostane and 8-isoprostane-d4 (>99%) were obtained from Cayman Chemical (Ann Arbor, MI). We purchased potassium phosphate and monobasic crystals (Reagent ACS) from Acros (NJ). Phosphate buffer was prepared using a Mettler Toledo S220 pH meter (Greifensee, Switzerland).
Standard Solutions
Certified materials used as the original standard solutions were obtained from Cayman Chemical (Ann Arbor, MI): 8-isoprostane (Catalog No. 16350; CAS #27415-26-5) and 8-isoprostane-d4 (Catalog No. 316350; CAS #211105-40-7). Equivalent sources may be used for standards. The materials were prepared gravimetrically, and the mass results were reported on the conventional basis for weighing in air. The initial native stock solution was prepared by weighing the dry powder into a 200 mL volumetric flask and then adding 50% methanol in HPLC water. Working solutions were prepared for standards from serial dilutions of initial stock solutions with methanol and HPLC water (v/v 1:1). Standards were prepared at 10 concentrations ranging from 0 to 1410 pg/mL by serial dilution of working solutions with methanol and HPLC water (v/v 1:9) and stored in a polypropylene cryogenic, screw cap vial at ≤−20 °C. Aliquots of 10 μL of each calibration standard were injected in the LC–MS/MS for analysis. Calibration curves were constructed by plotting the peak area ratios of the standards and internal standards (IS) against the concentration ratio of standards and IS using weighted linear regression (weight = 1/X).
Internal Standard (ISTD) Spiking Solution
The initial stock d4-labeled internal standard solution was received as 50 μg in 0.5 mL of methyl acetate. The initial solution was diluted to 100 mL in the volumetric flask using 50% methanol in HPLC water, bringing the concentration to 500 ng/mL. The working labeled internal standard solution was prepared by placing 60 mL of the initial stock d4 internal standard stock solution into a 2000 mL volumetric flask using 10% methanol in HPLC water, bringing the final working concentration to 15 ng/mL.
Enzyme Solution
An enzyme solution containing 12.5 units/μL was prepared by adding β-glucuronidase, type IX-A from Escherichia coli, to 0.5 M potassium phosphate buffer. Ammonium hydroxide was added to the buffer solution to bring the pH to 6.1. The amount of the enzyme should be calculated accordingly based on the specific product information. The enzyme solution was stored at or below −65 °C if not used immediately.
Instrumentation and Operation
Robotic Liquid Handling
Sample aliquots, internal standard addition, enzyme, and solvent dilution were performed by a Hamilton Microlab Star liquid handling workstation with Hamilton CO-RE tips (Reno, NV).
Solid-Phase Extraction (SPE)
SPE cleanup was done using the Strata-X-AW 33 μm Polymeric Weak Anion, 60 mg/96-well plates from Phenomenex (Torrance, CA). A Biotage Pressure +96 positive pressure manifold (Biotage, Charlotte, NC) using nitrogen gas generated in-house with a NM20ZA Peak generator. Sample collection and injection were done with an Advantage Series (Analytical Sales and Services Inc., Flanders, NJ) SiliGuard-coated 2 mL 96-deep-square well, with a tapered V-bottom collection plate.
UHPLC
Chromatographic separation was achieved using a Waters Acquity reversed-phase column (150 mm × 2.1 mm, particle size 1.8 μm, C18) and a Waters Acquity reversed-phase precolumn (5 mm ×1 mm, particle size 1.7 μm, C18) (Milford, MA). We used an ultrahigh performance liquid chromatographic system from Shimadzu Corp. (Columbia, MD): Nexera X2 SIL-30ACMP autosampler, two Nexera X2 LC-30AD pumps for programmed analytical gradient, LC-10ADvp for isocratic post-column infusion, CTO-20AC column oven, and CBM-20A system controller.
A flow rate of 0.65 mL/min and a column temperature of 60 °C were maintained during the entire analysis. The gradient program (Table 1) contained 0.15% formic acid in water (mobile phase A) and acetonitrile in 0.15% formic acid in water (v/v 1:1) (mobile phase B). A post-column infusion of acetonitrile, mobile phase C, at a flow rate of 0.15 mL/min was used to enhance ionization in the MS source.
Table 1. LC Gradient Program for Chromatographic Separation of 8-Isoprostanea.
| time | module | event | % mobile phase B by volume |
|---|---|---|---|
| 0.01 | controller | solenoid valve | |
| 0.02 | controller | start | |
| 0.06 | autosampler | inject | |
| 0.30 | pumps | %B | 40 |
| 0.31 | pumps | %B | 55 |
| 7.00 | pumps | %B | 65 |
| 7.01 | pumps | %B | 100 |
| 9.00 | pumps | %B | 100 |
| 9.01 | pumps | %B | 40 |
| 11.00 | controller | stop |
Mobile phase A is 0.15% formic acid in water and mobile phase B is acetonitrile/0.15% formic acid in water (v/v, 1:1)
Tandem Mass Spectrometer (MS/MS)
MS/MS analysis was performed using an AB Sciex 6500 triple quadrupole with a Turbo IonSpray source (Foster City, CA). Negative electrospray ionization (ESI–) mode was used to obtain MRM transition data. All LC–MS data were generated and processed using Analyst 1.6.2 (Sciex).
For the native 8-isoprostane, the mass transitions from m/z 353.3/193 were monitored for quantification and the mass transitions m/z 353.3/291 were monitored for confirmation. The mass transition m/z 357.3/197 was monitored for the deuterated 8-isoprostane-d4 internal standard. The MS source parameters were curtain gas, 30 psi; collision gas, 8 psi; IonSpray voltage, −4000 V; source temperature, 600 °C; ion source gas 1, 60 psi; ion source gas 2, 70 psi. (Table 2) All LC–MS data were generated and processed using Analyst 1.6.2 (Sciex).
Table 2. Optimized Mass Spectrometry Parametersa.
| ion-transition (m/z) |
||||||
|---|---|---|---|---|---|---|
| analyte | quantitation | confirmation | DP | EP | CE | CXP |
| 8-isoprostane | 353.3 → 193 | –124 | –10 | –33 | –10 | |
| 353.3 → 291 | –35 | |||||
| 8-isoprostane-d4 | 357.3 → 197 | –124 | –10 | –33 | –10 | |
DP = declustering potential; EP = entrance potential; CE = collision energy; CXP = cell exit potential.
Sample Preparation
Total 8-Isoprostane
Water blanks, quality control (QC) samples, and urine samples were all prepared following the same procedure. High QC and low QC urine pools were prepared from human urine and native 8-isoprostane was spiked to form the high QCs with appropriate concentrations. Samples, stored at −20 °C or below, were gradually thawed and manually shaken at room temperature. To quantify total 8-isoprostane, 40 μL of the isotopically labeled internal standard working solution, 800 μL of HPLC water, 160 μL of enzyme solution (2000 units Escherichia coli, type IX-A glucuronidase—dissolved in 0.5 M phosphate buffer pH 6.1), and 400 μL of urine specimen were dispensed into glass test tubes (12 mm × 75 mm), capped, and incubated overnight in a water bath for about 21 h at 37 °C. After incubation, 400 μL of methanol was added to each sample tube and the contents were transferred to a 96-well weak anion exchange SPE plate. Liquid transfers were performed using black conductive pipette tips from Hamilton for all liquids except the initial urine transfers. Clear, nonconductive 1000 μL tips from Hamilton were used to transfer all urine samples from the original source tubes. Nitrogen gas was used to push the samples through the SPE plate while the analyte, 8-isoprostane, was retained on the resin. The SPE plate was washed with 1.8 mL of HPLC water, then 3.6 mL (2 washes of 1.8 mL) of methanol in water (v/v 1:3), followed by 1.8 mL of acetonitrile. 8-isoprostane was eluted from the resin with 1.8 mL of methanol into a 96-well collection plate. Subsequently, the methanol was evaporated under nitrogen flow at 37 °C (Figure 1). The sample was then reconstituted using 50 μL of methanol in water (v/v 1:3) and vortexed before placing the plate in the LC autosampler. An aliquot of 10 μL was injected into the LC–MS/MS for analysis
Figure 1.
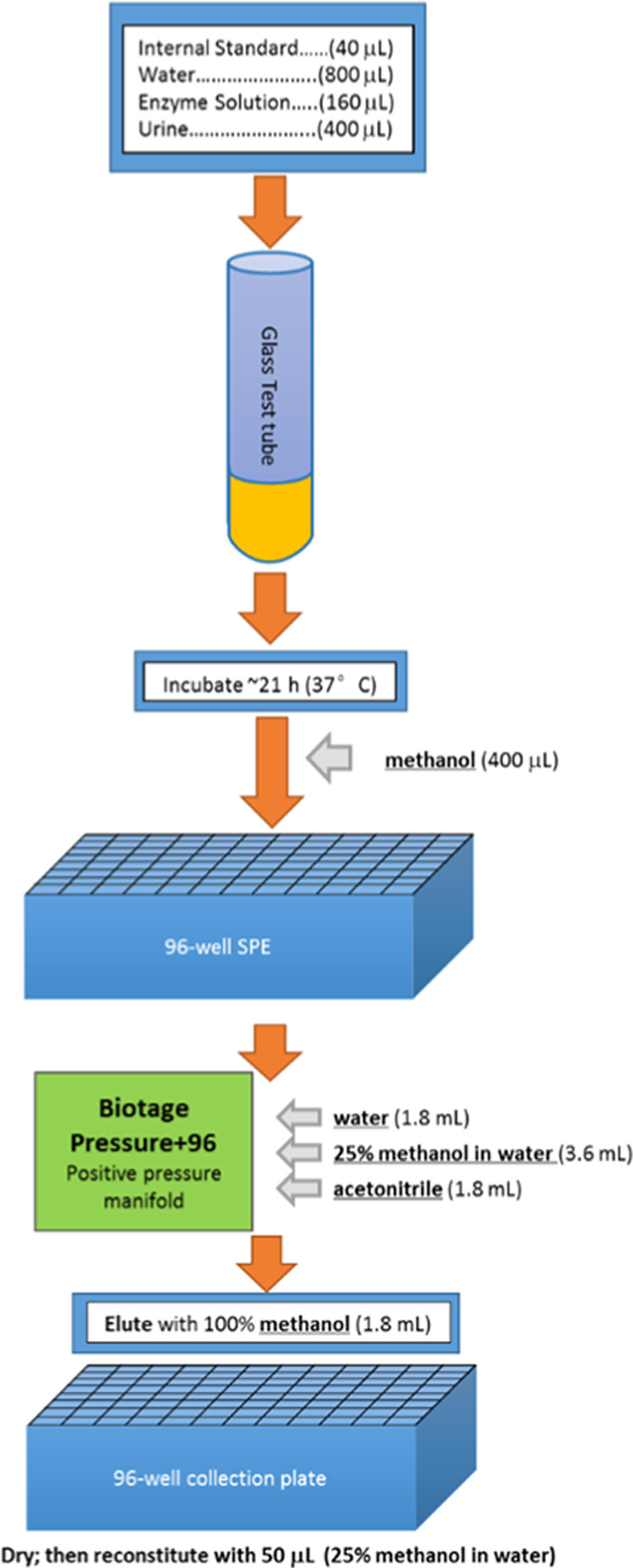
Sample preparation schematic.
Free 8-Isoprostane
To measure free urinary 8-isoprostane, we followed the same protocol listed above for total 8-isoprostane but eliminated the hydrolysis step. We found that the 0.5 M phosphate buffer at pH 6.1 was a critical component for adequate recovery from the SPE. Therefore, in our preparation for free 8-isoprostane, we added 160 μL of the buffer without the enzyme and skipped the incubation period. All other steps were kept identical to the total measurement.
Human Subjects
This method was applied to the measurement of free and total 8-isoprostane for urine samples from 30 cigarette smokers and 30 nontobacco users. No dietary restrictions were applied on the participants. Spot urine samples were collected from anonymous cigarette smokers by Tennessee Blood Services (Memphis, TN). This group of smokers on average used 12 cigarettes per day (CPD). Spot urine samples were collected anonymously from self-described nontobacco users at the Centers for Disease Control and Prevention (CDC, Atlanta, GA). This study was reviewed and approved by the Institutional Review Board (IRB protocol 3994) at the CDC.
Results and Discussion
Automation
Preliminary experiments were conducted to evaluate potential adsorption of 8-isoprostane to the selected laboratory consumables. Samples containing 8-isoprostane were exposed to various pipette tips that could be used for liquid transfers. We observed that 8-isoprostane reversibly adsorbs to the surface of the Hamilton capacitive liquid level detection (cLLD) tips, but no adhesion was seen when using the Hamilton nonconductive pressure-based liquid level detection (pLLD) tips. Based on this finding, clear, nonconductive pLLD 1000 μL tips were used for all transfer and mix steps involving contact with urine samples, and black cLLD tips were used for all other liquid transfers on the Hamilton Star. For this assay, two automation methods were created. The first method handled liquids before incubation, and the second method handled liquids after incubation. In the first method, the Hamilton Star was programmed to transfer 40 μL of the isotopically labeled internal standard, 800 μL of HPLC water, 160 μL of enzyme solution, and 400 μL of urine specimens into glass culture tubes. The tubes were manually capped and placed in a 37 °C water bath overnight (21 h) for incubation. After incubation, the tubes were manually uncapped and placed back onto the Hamilton Star. The second Hamilton method added 400 μL of methanol to each sample, mixed contents thoroughly, and then transferred the resulting mixtures to a 96-well weak anion exchange SPE plate for cleanup.
Hydrolysis
For determination of total 8-isoprostane, urine samples were enzymatically hydrolyzed using β-glucuronidase prior to LC–MS/MS analysis. The reported ratio of conjugated 8-isoprostane to free 8-isoprostane varies greatly from person to person (from 30 to 80% conjugation).18−21 Since both free and conjugated 8-isoprostane are products of nonenzymatic lipid peroxidation, it is important to measure both free and conjugated forms (i.e., total 8-isoprostane) to accurately estimate oxidative stress. One limitation to note, the hydrolysis experiments were performed using real urine samples since we could not find commercially available conjugated 8-isoprostane standards at the time of experimentation. Hydrolysis of 8-isoprostane was evaluated using increasing concentrations of the enzyme to determine the necessary conditions required to take hydrolysis to completion. Complete hydrolysis was observed after 6 h using 1000 units per 400 μL of the sample. Samples incubated greater than 6 h exhibited no change in the measured concentrations of 8-isoprostane, therefore an incubation period of 21 h was selected to adapt to an 8 h workday. To ensure complete hydrolysis in all samples, we doubled the concentration of enzyme to 2000 units per 400 μL of the sample. Results are shown in Table S2 in the Supporting Information file.
SPE
Purification prior to analysis is essential for accurately quantifying 8-isoprostane with SPE being generally accepted as the most efficient practice for purification prior to LC–MS/MS analysis.22 To determine the most suitable SPE plate for the assay, various sorbent materials were tested. The highest recovery and best reproducibility were observed with the Strata-X-AW SPE plate. The samples were loaded onto the SPE plate and underwent a series of washes with 1.8 mL of water, 3.6 mL of 25% methanol in water (1.8 mL twice), and 1.8 mL of acetonitrile to remove interfering compounds. Following the sample cleanup, the SPE plate was rinsed a final time with methanol to elute the 8-isoprostane into a 96-well collection plate. The methanol was evaporated under nitrogen flow at 37 °C and then reconstituted with 50 μL of methanol in water (v/v 1:3) prior to injection into the UHPLC. The pH of the phosphate buffer is critical for SPE recovery for this method. The manufacturer of the SPE plate recommends pH 6.0–7.0 for adequate recovery. For this method, we determined the optimal pH of the phosphate buffer to be 6.1. For this SPE procedure, we calculated the average extraction recovery to be 60%. We evaluated SPE carryover by placing 16 water blanks in random positions throughout the 96-well plate. We observed no contaminants in any of the water blanks and determined that no carryover was taking place during the SPE procedure.
Linearity
Calibration curves were constructed using urine, synthetic urine, and water as matrices. The calibration standards were all prepared using the same procedure and provided comparable analytical results. Strong linearity was observed in urine (r2 = 0.9971), synthetic urine (r2 = 0.9980), and water (r2 = 0.9999) using 8-isoprostane concentrations of 0 to 1410 pg/mL. A representative linear regression plot of standards prepared in water is shown in Figure 2. The concentrations measured in each matrix displayed deviations of less than 5%. The initial accuracy of the standard curve was evaluated by preparing a set of fortified synthetic urine pools. The pools also served as proficiency testing pools that ensured the accuracy of the method over extended time intervals. The results from the matrix comparison test can be found in Table S1 in the Supporting Information file.
Figure 2.

Representative linear regression of calibration standards prepared in water. 1/x weighting: y = 0.00307(x) – 2.4e – 009 (r2 = 0.9999).
A small negative bias was observed when the synthetic pools were analyzed. In-house experiments have demonstrated that the 8-isoprostane analyte adheres reversibly to plastic surfaces in aqueous solution and the adhesion was enhanced when synthetic urine was used as the solvent. Adhesion was attenuated when methanol or acetone was added. The standard curve used for this method was prepared in a 1:1 solution of methanol and water.
Accuracy
Accuracy for this assay was assessed through recovery analyses of blank and spiked urine at known concentrations. All accuracy studies were performed using only the total 8-isoprostane assay. The pools were made in the following manner: Two original blank pools were spiked with three different concentrations in the reportable range, creating four levels for each pool, 0 pg/mL or blank, 206.8, 441.9, and 1305.0 pg/mL. For each of these concentrations, spiking was done in triplicate resulting in a total of 12 samples in each pool. The 24 samples were processed twice in separate analytical runs on separate days resulting in a total of 48 results. The recovery of the added analyte was calculated as [(final concentration-initial concentration)/added concentration]. The recovery of the added analyte ranged from 92.7 to 106.7% with a mean recovery of 99.7%, as shown in Table 3 below.
Table 3. Accuracy for the Measurement of Total 8-Isoprostane through Spiking Known Concentrations.
| low pool (n = 6) |
|||
|---|---|---|---|
| mean | spike concentration | % recovery | |
| pool | 99.1 | 0.0 | |
| pool + spike 1 | 319.8 | 206.8 | 106.7 |
| pool + spike 2 | 554.2 | 441.9 | 103.0 |
| pool + spike 3 | 1345.0 | 1305.0 | 95.5 |
| medium pool (n = 6) |
|||
|---|---|---|---|
| mean | spike concentration | % recovery | |
| pool | 187.7 | 0.0 | |
| pool + spike 1 | 379.3 | 206.8 | 92.7 |
| pool + spike 2 | 650.0 | 441.9 | 104.6 |
| pool + spike 3 | 1433.0 | 1305.0 | 95.5 |
The accuracy of standards was based on the manufacturer’s “Certificate of Analysis” for the 8-isoprostane purchased from Cayman Chemical Company. For verification of accuracy, three standard verification synthetic urine pools (400, 800, and 1200 pg/mL) were prepared using 8-isoprostane purchased from an alternative source (Santa Cruz Biotechnology). The solutions prepared using 8-isoprostane from Santa Cruz Biotechnology yielded calculated values within the RSD of the assay, supporting the accuracy of the standard curve prepared using 8-isoprostane from Cayman Chemical Company.
Precision
The precision of this assay was determined by assessing the closeness of repeat individual measurements of 8-isoprostane in two separate QC pools, as shown in Table 4. All precision experiments were performed using only the total 8-isoprostane assay. The concentration of 8-isoprostane was measured in two samples from the 2 QC pools in 10 different analytical runs for a total of 20 measurements per QC pool.
Table 4. Inter-run, Intrarun, and Total Precision of Total 8-Isoprostane.
| QC high pool (n = 20) |
QC low pool (n = 20) |
||||||
|---|---|---|---|---|---|---|---|
| mean (pg/mL) | inter-run (CV%) | intrarun (CV%) | method (CV%) | mean (pg/mL) | inter-run (CV%) | intrarun (CV%) | method (CV%) |
| 621 | 4 | 0 | 4 | 82.4 | 9 | 4 | 10 |
The measured concentrations were used to calculate the inter-run, intrarun, and total precision. The high QC pool had a within-run coefficient of variation (CV), 4%; intrarun, 0%; and total CV of 4%. The low QC pool had inter-run CV, 9%; intrarun CV, 4%; and total CV of 10%. As expected, the CV increases as analyte concentration approaches the LOD.
Stability
Stability was assessed through a series of four experiments that compared the concentrations of 8-isoprostane before and after the samples were exposed to conditions that a sample may encounter during routine analysis. Each experiment was conducted by measuring two pools (QC high and QC low) in triplicate. Freeze and thaw stability was tested by freezing (at −70 °C) and thawing (passively at room temperature) samples three times before analysis. The concentration of 8-isoprostane was measured before the initial freeze and after the final thaw; the calculated CVs of the QC low and QC high pools were 4.8 and 0.5%, respectively. Benchtop stability was tested by allowing the samples to sit at room temperature for 24 h before being processed. The resulting concentrations were compared to the initial measurements; the calculated CVs of the QC low and QC high pools were 3.9 and 0.1%, respectively. Processed sample stability was tested by allowing the samples to sit at room temperature for 24 h after being processed. The samples were analyzed on the second day and the measured concentrations were compared to the initial measurements; the calculated CVs of the QC low and QC high pools were 4.7 and 2.9%, respectively. The long-term stability was assessed by measuring the concentration of 8-isoprostane in samples after they were stored at −70 °C for >2 years and comparing the results to the initial concentrations of 8-isoprostane in samples from the same pool. The calculated CVs of the QC low and QC high pools were 6.9 and 0.8%, respectively.
Limit of Detection (LOD) and Lower Limit of Quantitation (LLOQ)
The limit of detection (LOD) was determined by preparing and analyzing four low-concentration urine pools over a 3 month period. The standard deviation (SD) of each pool’s concentration was determined, and then the SD of each pool was plotted against the concentration to obtain the S0, given as the Y-intercept.23 The LOD was defined as the highest of three times S0, where S0 was the standard deviation at zero analyte concentration or the concentration of the lowest standard. For this assay, three times S0 was calculated to be 4.77 pg/mL, and the lowest external standard concentration was 8.8 pg/mL, therefore, 8.8 pg/mL was determined to be the LOD. The lower limit of quantitation was defined as three times LOD. For this assay, the LLOQ was calculated to be 26.4 pg/mL.
Selectivity
The analytical selectivity of this method was assessed by the following measures: (1) The monitoring of ion transitions selective to the analyte through the use of MS/MS; (2) Ensuring the target native analyte co-elutes with the corresponding isotopically labeled internal standard analog; (3) Checking that both native analyte and ISTD elute at a specific retention time that is consistent throughout the run; (4) The native analytes have specific ratios of the quantitation transition’s response to the confirmation transition’s response, which was used to confirm the analyte determined in unknown samples. Potential interferences with 8-isoprostane were investigated in over 50 human samples and none was observed. Representative chromatograms of real urine samples and calibration standards in 50% methanol and water are shown in Supporting Information, Figures S1 and S2.
Application of the Method to Human Urine
Figure 3 shows the comparison of free and total urinary 8-isoprostane in smokers and nonusers. The smoker group has a higher geometric mean than the nonuser group for both the free and total measurements. Interestingly, we observed no changes in our results after adjusting for creatinine (Figures S3 and S4 in the Supporting Information file); the smoker group had a higher geometric mean than the nonuser group. Additionally, the order of samples from highest to lowest concentration was identical before and after creatinine adjustment. We observed a 100% detection rate for quantifying both free and total 8-isoprostane in the 60 spot urines. There is no apparent correlation between CPD and 8-isoprostane levels in this small study (data not shown). The percent conjugation ({[total – free]/total} × 100) ranges from 15 to 61% across the entire group of 60 people with an average of 33%. The average percent conjugation of the smoker group was 34%, ranging from 20 to 60%. Similarly, the average percent conjugation of the nonuser group was 31%, ranging from 15 to 53%. The order of samples from highest to lowest concentration was not identical for free 8-isoprostane and total 8-isoprostane. We calculated the ratio of free/total 8-isoprostane concentrations with and without creatinine adjustment and found the ratios to be the same (Table S3). Since the conjugated form is variable and can account for over half of the 8-isoprostane present in collected spot urine, the measurement of the total (free plus conjugated forms) gives the more complete estimate of oxidative stress present in the test samples over measuring the free form.
Figure 3.

Comparison of geometric means ± standard errors of free and total urinary 8-isoprostane in smokers (free: 460 ± 78.8 pg/mL; total: 704 ± 108 pg/mL) and nonusers (free: 110 ± 24.2 pg/mL; total: 161 ± 38.7 pg/mL).
We also applied the total assay to 7141 urine samples from Wave 1 of the Population Assessment of Tobacco and Health (PATH) study. In this study, the total method returned a detection rate of 99.3%, providing results for 7091 out of 7141 samples. We were unable to report results for 49 out of 7141 (0.7%) samples because of interfering substances present, and only 1 out of 7141 samples was found to be below our LOD of 8.8 pg/mL.24 Although several LCMS methods for measuring urinary 8-isoprostane have been published in the last 10 years,21,25−27 none of them provide the combination of high sensitivity, selectivity, accuracy, and precision with the ability to analyze large sample loads. Most of the published methods only measure free 8-isoprostane and do not measure total 8-isoprostane.25−27 Further, previously reported methods have lower sensitivity compared to this assay,21,25 lower chromatographic resolution,26,27 and some require extensive manual sample preparation.25 These results provide robust support for the excellent selectivity and sensitivity of this assay. A laboratory with a single UHPLC–MS/MS system could process 360 samples in a week (8 h a day for 5 days) with this method, making this an ideal assay for large population studies.
Conclusions
We present a partially automated, sensitive, and robust analytical method for detecting either free or total urinary 8-isoprostane. This method uses isotope-dilution UHPLC–MS/MS with a LOD of 8.8 pg/mL and a between-run CV below 10%. With a cycle time of 11 min, this method is adequate for use in large population studies, which can provide accurate baseline levels of 8-isoprostane in a population especially if a total measurement is used. Application of this method confirms higher urinary 8-isoprostane in urine collected from cigarette smokers compared with nonusers. Subsequent applications of this method may better characterize oxidant exposure and the role of oxidative stress in the pathologies of the disease.
Acknowledgments
The authors would like to thank the Center for Tobacco Products, Food and Drug Administration, for providing funding for this project. This project was supported in part by an appointment to the Research Participation Program at the Centers for Disease Control and Prevention administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the U.S. Department of Energy and the Centers for Disease Control and Prevention. We would like to thank Pam Olive and Uliana Danilenko for performing creatinine analysis for this study. We also thank Jim McGuffey, Li Zhang, Olivia Cote, Rachel Lippens, Claire Allison, Andrew Puetz, Brittany Pine, and Caroline Luiken for their suggestions and support during this study.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c00661.
Matrix effect on calibration curves; hydrolysis optimization, free to total 8-isoprostane ratio for smokers and nonusers; representative chromatograms of free and total 8-isoprostane in smokers; representative standard chromatogram; free and total 8-isoprostane geometric means in smokers and nonusers adjusted for creatinine (ng/g creatinine and nmol/g creatinine) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Reuter S.; Gupta S. C.; Chaturvedi M. M.; Aggarwal B. B. Oxidative stress, inflammation, and cancer: how are they linked?. Free Radical Biol. Med. 2010, 49, 1603–1616. 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valko M.; Rhodes C. J.; Mocoi J.; Izakovic M.; Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem.-Biol. Interact. 2006, 160, 1–40. 10.1016/j.cbi.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Halliwell B. Oxidative stress and cancer: have we moved forward?. Biochem. J. 2007, 401, 1–11. 10.1042/BJ20061131. [DOI] [PubMed] [Google Scholar]
- Wang B.; Pan J.; Wang L.; Zhu H.; Yu R.; Zou Y. Associations of plasma 8-isoprostane levels with the presence and extent of coronary stenosis in patients with coronary artery disease. Atherosclerosis 2006, 184, 425–430. 10.1016/j.atherosclerosis.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Griendling K. K.; FitzGerald G. A. Oxidative stress and cardiovascular injury: Part II: animal and human studies. Circulation 2003, 108, 2034–2040. 10.1161/01.CIR.0000093661.90582.c4. [DOI] [PubMed] [Google Scholar]
- Montuschi P.; Collins J. V.; Ciabattoni G.; Lazzeri N.; Corradi M.; Kharitonov S. A.; Barnes P. J. Exhaled 8-isoprostane as an in vivo biomarker of lung oxidative stress in patients with COPD and healthy smokers. Am. J. Respir. Crit. Care Med. 2000, 162, 1175–1177. 10.1164/ajrccm.162.3.2001063. [DOI] [PubMed] [Google Scholar]
- Lin M. T.; Beal M. F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- Milne G. L.; Yin H.; Hardy K. D.; Davies S. S.; Roberts L. J. II Isoprostane generation and function. Chem. Rev. 2011, 111, 5973–5996. 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal R. S.; Weindruch R. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray P. D.; Huang B.-W.; Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell. Signaling 2012, 24, 981–990. 10.1016/j.cellsig.2012.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsikas D.; Theodoridis G. J. Analytical tools and protocols in oxidative stress. J. Chromatogr. B. 2016, 1019, 1–3. 10.1016/j.jchromb.2016.03.015. [DOI] [PubMed] [Google Scholar]
- Morrow J. D.; Hill K. E.; Burk R. F.; Nammour T. M.; Badr K. F.; Roberts L. J. II A series of prostaglandin F2-like compounds are produced in vivo in humans by a non-cyclooxygenase, free radical-catalyzed mechanism. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 9383–9387. 10.1073/pnas.87.23.9383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne G. L.; Morrow J. D.. Isoprostanes. In Wiley Encyclopedia of Chemical Biology; Wiley, 2008; pp 1–9. [Google Scholar]
- Praticò D.; Tangirala R. K.; Rader D. J.; Rokach J.; FitzGerald G. A. Vitamin E suppresses isoprostane generation in vivo and reduces atherosclerosis in ApoE-deficient mice. Nat. Med. 1998, 4, 1189–1192. 10.1038/2685. [DOI] [PubMed] [Google Scholar]
- Xiao Y.; Fu X.; Pattengale P.; Bard J. D.; Xu Y.-K.; O’Gorman M. R. A sensitive LC-MS/MS method for the quantification of urinary 8-iso-prostaglandin F2α (8-iso-PGF2α) including pediatric reference interval. Clin. Chim. Acta 2016, 460, 128–134. 10.1016/j.cca.2016.06.034. [DOI] [PubMed] [Google Scholar]
- Milne G. L.; Dai Q.; Roberts L. J. II The isoprostanes–25 years later. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2015, 1851, 433–445. 10.1016/j.bbalip.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracowski J.-L.; Durand T.; Bessard G. Isoprostanes as a biomarker of lipid peroxidation in humans: physiology, pharmacology and clinical implications. Trends Pharmacol. Sci. 2002, 23, 360–366. 10.1016/S0165-6147(02)02053-9. [DOI] [PubMed] [Google Scholar]
- Morrow J. D.; Zackert W. E.; Yang J. P.; Kurhts E. H.; Callewaert D.; Dworski R.; Kanai K.; Taber D.; Moore K.; Oates J. A.; Roberts L. J. II Quantification of the major urinary metabolite of 15-F2t-isoprostane (8-iso-PGF2alpha) by a stable isotope dilution mass spectrometric assay. Anal. Biochem. 1999, 269, 326. 10.1006/abio.1999.4008. [DOI] [PubMed] [Google Scholar]
- Yan Z.; Mas E.; Mori T. A.; Croft K. D.; Barden A. E. A significant proportion of F2-isoprostanes in human urine are excreted as glucuronide conjugates. Anal. Biochem. 2010, 403, 126–128. 10.1016/j.ab.2010.04.016. [DOI] [PubMed] [Google Scholar]
- Tsikas D.; Suchy M.-S. Protocols for the measurement of the F2-isoprostane, 15(S)-8-iso-prostaglandin F2α, in biological samples by GC-MS or GC-MS/MS coupled with immunoaffinity column chromatography. J. Chromatogr. B 2016, 1019, 191–201. 10.1016/j.jchromb.2014.12.019. [DOI] [PubMed] [Google Scholar]
- Langhorst M. L.; Hastings M. J.; Yokoyama W. H.; Hung S.-C.; Cellar N.; Kuppannan K.; Young S. A. Determination of F2-isoprostanes in urine by online solid phase extraction coupled to liquid chromatography with tandem mass spectrometry. J. Agric. Food Chem. 2010, 58, 6614–6620. 10.1021/jf101146q. [DOI] [PubMed] [Google Scholar]
- Medina S.; Dominguez-Perles R.; Gil J. I.; Ferreres F.; Garcia-Viguera C.; Martinez-Sanz J. M.; Gil-Izquierdo A. A ultra-pressure liquid chromatography/triple quadrupole tandem mass spectrometry method for the analysis of 13 eicosanoids in human urine and quantitative 24 hour values in healthy volunteers in a controlled constant diet. Rapid Commun. Mass Spectrom. 2012, 26, 1249–1257. 10.1002/rcm.6224. [DOI] [PubMed] [Google Scholar]
- McGuffey J. E.; Wei B.; Bernert J. T.; Morrow J. C.; Xia B.; Wang L.; Blount B. C. Validation of a LC-MS/MS method for quantifying urinary nicotine, six nicotine metabolites and the minor tobacco alkaloids--anatabine and anabasine--in smokers’ urine. PLoS One 2014, 9, e101816 10.1371/journal.pone.0101816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- United States Department of Health and Human Services; National Institutes of Health; National Institute on Drug Abuse, and United States Department of Health and Human Services; Food and Drug Administration; Center for Tobacco Products; Population Assessment of Tobacco and Health (PATH) Study [United States] Biomarker Restricted-Use Files; Inter-University Consortium for Political and Social Research, May 11, 2019.
- Teng Y.-H.; Wang C.-W.; Liao Y.-T.; Yang M.-W.; Liu T.-Y. Quantification of Urinary 8-iso-Prostaglandin F2α Using Liquid Chromatography-Tandem Mass Spectrometry During Cardiac Valve Surgery. J. Clin. Lab. Anal. 2010, 24, 237–245. 10.1002/jcla.20392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klawitter J.; Haschke M.; Shokati T.; Klawitter J.; Christians U. Quantification of 15-F2t-isoprostane in human plasma and urine: results from enzyme-linked immunoassay and liquid chromatography/tandem mass spectrometry cannot be compared. Rapid Commun. Mass Spectrom. 2011, 25, 463–468. 10.1002/rcm.4871. [DOI] [PubMed] [Google Scholar]
- Mizuno K.; Kataoka H. Analysis of urinary 8-isoprostane as an oxidative stress biomarker bystable isotope dilution using automated online in-tube solid-phase microextraction coupled with liquid chromatography–tandem massspectrometry. J. Pharm. Bio. Anal. 2015, 112, 36–42. 10.1016/j.jpba.2015.04.020. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.