

Abstract
To establish the molecular mechanism of ginsenoside Rg1 in nonalcoholic fatty liver disease (NAFLD), Sprague Dawley (SD) rats (180–220 g) were randomly divided into a control group, model group, ginsenoside Rg1 low-dose group (30 mg/(kg day)), high-dose (60 mg/(kg day)) group, and simvastatin group (1 mg/(kg day)), with 10 SD rats in each group. The control group was given a normal diet. The model group rats were given high-sugar and high-fat diets for 14 weeks. After the model of NAFLD was established successfully, ginsenoside Rg1 was administered orally for 4 or 8 weeks. The results showed that ginsenoside Rg1 decreased the levels of glucose (GLU), insulin (INS), triglyceride (TG), and total cholesterol (TC) and improved liver function. Meanwhile, ginsenoside Rg1 inhibited the secretion of interleukin-1 (IL-1), IL-6, IL-8, IL-18, and tumor necrosis factor-α (TNF-α) and improved hepatocyte morphology and lipid accumulation in the liver. Furthermore, ginsenoside Rg1 promoted the expression of peroxisome proliferator-activated receptor-α (PPAR-α), carnitine palmitoyl transferase 1α (CPT1A), carnitine palmitoyl transferase 2 (CPT2), and cholesterol 7α-hydroxylase (CYP-7A) and inhibited the expression of sterol regulatory element binding proteins-1C (SREBP-1C). In conclusion, ginsenoside Rg1 can inhibit inflammatory reaction, regulate lipid metabolism, and alleviate liver injury in NAFLD model rats.
1. Introduction
Nonalcoholic fatty liver disease (NAFLD) is the most commonly occuring liver disease without the history of drinking. In China, the prevalence rate of NAFLD is about 20–30%, and in recent years, with the improvement of people’s living standards, the incidence rate has significantly increased and tends to be common in the younger population.1 The pathogenesis of NAFLD is complex, involving oxidative stress, lipid metabolism disorders, inflammatory response, and so on, and shares the same pathophysiological characteristics as those of metabolic syndrome (MS) and type 2 diabetes (T2D).2,3 Severe nonalcoholic steatohepatitis may occur with cirrhosis or even hepatocellular carcinoma.4 From the perspective of morphology, NAFLD is mainly characterized by diffuse hepatocyte steatosis in the hepatic lobule. In clinical manifestations, moderate and severely affected patients may be present with a dull pain in the liver area, systemic fatigue, diarrhea, and other symptoms.5−7 As the disease is not easy to be detected early in clinical practice, the early diagnosis and treatment of the disease becomes the key to prevent its further occurrence and development. Meanwhile, looking for natural drugs with less side effects and definite effects to alleviate NAFLD has become a hot topic in current research works.
More than 80% of the country’s Panax notoginseng is planted in Yunnan, China. P. notoginseng has many active components, such as ginsenoside Rb1, Rg1, Rg2, and Rh. Ginsenoside Rg1 has anti-inflammatory, antioxidant, antifibrosis, antiapoptosis, and neuroprotective effects.8 Most importantly, ginsenoside Rg1 has a good protective effect on the liver.9
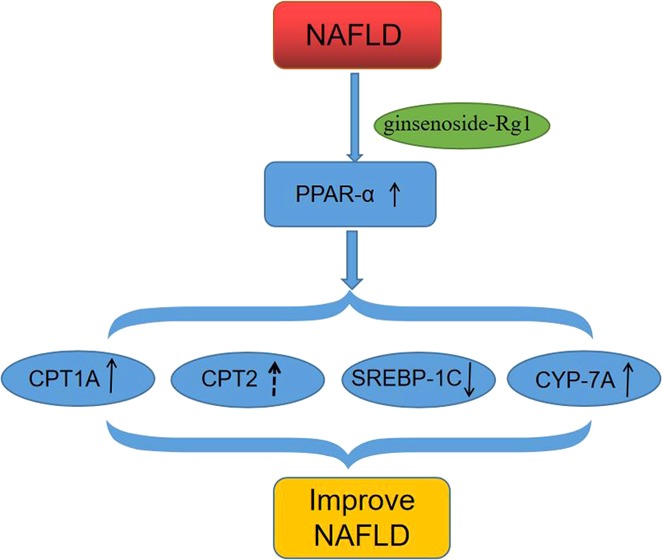
Previous studies have shown that ginsenoside Rg1 protects against nonalcoholic fatty liver disease by upregulating the expression of peroxisome proliferator-activated receptor-α (PPARα), which stimulates fatty acid β oxidation and promotes the metabolism of free fatty acids (FFAs) and triglyceride (TG).8 However, the effects of ginsenoside on PPAR-related molecules carnitine palmitoyl transferase 1α (CPT1A), carnitine palmitoyl transferase 2 (CPT2), sterol regulatory element binding proteins-1C (SREBP-1C), and cholesterol 7α-hydroxylase (CYP-7A) have been rarely reported.
In the present study, we further investigate the effects of ginsenoside Rg1 on PPARα, as well as the underlying mechanisms that involve CPT1A, CPT2, SREBP-1C, and CYP-7A in vivo.
2. Results and Discussion
2.1. Ginsenoside Rg1 Inhibits Insulin Resistance in the Animal Models of NAFLD
To detect whether the model of NAFLD was built successfully, we detected the pathological change of a rat’s liver after 14 weeks of modeling by HE and oil red “O” staining. HE staining results showed that there is an abnormal arrangement of hepatocytes, small vacuoles in some cells, and a large number of vacuole-like structures around liver cells in the model group compared with the control group. In addition, Oil red O staining results showed a large amount of oil red O precipitation with a large number of lipid droplets in the liver of the model group compared with the control group (Figure 1A). The above results indicated that the animal model of NAFLD was established successfully. Next, we examined the effects of ginsenoside Rg1 on glucose (GLU) and insulin (INS) in NAFLD models. The results showed that ginsenoside Rg1 inhibited the increase of GLU, INS, and HOMA-IR induced by NAFLD in a dose-dependent manner after ginsenoside Rg1 treatment. The high-dose ginsenoside Rg1 group was more effective than the simvastatin group at 8 weeks (P < 0.05) (Figure 1B). Simvastatin, a lipid-lowering drug, was used as a positive control. The molecular structure of ginsenoside Rg1 is shown in Figure 1C.
Figure 1.

Ginsenoside Rg1 inhibits insulin resistance in animal models of NAFLD. (A) The morphology of liver tissues in the control group or model group was detected by HE staining and oil red O staining. (B) Ginsenoside Rg1 inhibits insulin resistance after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are the mean ± standard error of the mean (SEM) of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05. (C) The molecular formula of ginsenoside Rg1.
2.2. Ginsenoside Rg1 Improves Liver Function in NAFLD Models
Next, we examined the effects of ginsenoside Rg1 on the liver of NAFLD rat models. At first, we measured the ratio of liver weight and body weight at week 4 and week 8 after the ginsenoside Rg1 treatment. The results showed that the liver weight and liver/body weight ratio increased significantly in the model group compared with the control group. Ginsenoside Rg1 inhibited them in a dose-dependent manner (P < 0.05). High-dose ginsenoside Rg1 was more effective than simvastatin at week 8 (P < 0.05) (Figure 2A,B). Then, we measured the factors that reflect liver function. The results showed that the concentrations of alanine aminotransferase (ALT), aspartate aminotransferase (AST), and alkaline phosphatase (ALP) decreased significantly in a dose-dependent manner after treatment with different concentrations of ginsenoside Rg1 and simvastatin for 4 and 8 weeks. High-dose ginsenoside Rg1 has a better therapeutic effect than simvastatin in reducing ALT, AST, and ALP at 8 weeks (P < 0.05) (Figure 2C). The results showed that chronic liver injury can be caused by NAFLD induced by a high-sugar and high-fat diet and ginsenoside Rg1 can alleviate chronic liver injury.
Figure 2.

Ginsenoside Rg1 improves liver function in NAFLD models. (A, B) Ginsenoside Rg1 decreases the body weight, liver weight, and their ratio induced by high fat and high sugar after ginsenoside Rg1 treatment for 4 or 8 weeks. C. Ginsenoside Rg1 improves the function of the liver under the condition of NAFLD after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05. *Significant compared with the model group alone, P < 0.05.
2.3. Ginsenoside Rg1 Alleviates Liver Inflammation in NAFLD Models
Meanwhile, we observed pathological changes in the liver by HE staining. The results showed that the arrangement of hepatocytes was abnormal and disordered and that some cells had vacuoles of different sizes and vacuole-like degeneration; meanwhile, the number of adipocytes was significantly increased, with a certain degree of steatosis, but no fibrosis or obvious fibrosis changes were found, and inflammatory cell infiltration was observed in the model group compared with the control group. Ginsenoside Rg1 can reverse this change and improve high-fat- and high sugar-induced liver injury in a dose-dependent manner. The high-dose ginsenoside Rg1 group was more effective than the simvastatin group at 8 weeks (P < 0.05) (Figure 3A,B).
Figure 3.

Ginsenoside Rg1 alleviates liver inflammation in NAFLD models. (A, B) Ginsenoside Rg1 attenuates the pathologic change of liver tissues in the NAFLD model in a dose- and time-dependent manner. The sections of liver tissues were detected by HE after ginsenoside Rg1 treatment for 4 or 8 weeks. (C, D) Ginsenoside Rg1 inhibits the secretion of inflammatory cytokines in the NAFLD model in a dose- and time-dependent manner. Serum concentrations of IL-6, IL-1β, and TNFα were detected by enzyme-linked immunosorbent assay (ELISA) kits after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05; @significant compared with simvastatin alone, P < 0.05.
HE staining revealed a large amount of inflammatory cell infiltration in the model group. Furthermore, we measured the secretion of inflammatory factors with an ELISA kit. The results showed that the concentrations of IL-1, IL-6, IL-8, IL-18, and TNF-α in the model group were significantly increased compared with the control group (P < 0.05). Ginsenoside Rg1 can inhibit the increase of these inflammatory factors in a dose-dependent manner. The effect of the high-dose ginsenoside Rg1 group was better than that of the simvastatin group at 8 weeks (P < 0.05) (Figure 3C,D). These results showed that the high-sugar and high-fat diet could lead to liver lesions of rats and cause inflammatory response, while ginsenoside Rg1 could improve liver damage and have an anti-inflammatory effect.
2.4. Ginsenoside Rg1 Reduces Lipid Accumulation in NAFLD Models
Besides, a large number of vacuoles were found in HE staining. To further determine whether the vacuoles in the model group were fat or not, oil red O staining was performed on the liver tissues. The results showed that a large amount of oil red O accumulated in the model group at 4 and 8 weeks. However, after the treatment with ginsenoside Rg1, the lipid accumulation in the liver tissue was significantly decreased. The positive control group of simvastatin had similar effects with ginsenoside Rg1 (Figure 4A,B).
Figure 4.

Ginsenoside Rg1 reduces lipid accumulation in NAFLD models. (A, B) Ginsenoside Rg1 reduces fat deposition of the liver tissues in a dose- and time-dependent manner. Sections of liver tissues were detected by oil red O staining after ginsenoside Rg1 treatment for 4 or 8 weeks. C. Ginsenoside Rg1 reduces lipid factor accumulation of serum in a dose- and time-dependent manner. The serum concentrations of TG and total cholesterol (TC) were detected by an automated biochemistry analyzer after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05; @significant compared with simvastatin alone, P < 0.05.
To find the reason, we measured lipid factors with a biochemical analyzer. The results showed that the concentrations of TG and TC in the model group were significantly increased compared to those in the control group (P < 0.05). After the treatment with ginsenoside Rg1, the concentrations of TG and TC decreased significantly compared with the model group in a dose-dependent manner (P < 0.05). High -dose ginsenoside Rg1 was more effective than simvastatin at 8 weeks (P < 0.05) (Figure 4C,D).
The results suggested that the lipid metabolism was induced in the model group was induced by the high-glucose and high-fat diet and that ginsenoside Rg1 might improve the lipid metabolism.
2.5. Ginsenoside Rg1 Promoted PPAR-α Expression in the Animal Models of NAFLD
The main function of PPAR-α is to promote the oxidation of fatty acids to reduce the deposition of triglycerides in the liver.9 To explore the effect of ginsenoside Rg1 on PPAR-α, we first detected the mRNA expression of PPAR-α. Q-PCR results showed that the expression of PPAR-α mRNA in the model group was significantly decreased compared with the control group. After the treatment of ginsenoside Rg1, the mRNA expression of PPAR-α increased, respectively, in a dose- and time-dependent manner (P < 0.05), and the effect of the high-dose ginsenoside Rg1 group was obvious than that of the simvastatin group (P < 0.05) (Figure 5A).
Figure 5.

Ginsenoside Rg1 obviously promoted PPAR-α expression in animal models of NAFLD. (A) Ginsenoside Rg1 obviously promoted the mRNA expression of PPAR-α. The mRNA expression was detected by quantitative polymerase chain reaction (q-PCR) after ginsenoside Rg1 treatment for 4 or 8 weeks. (B, C) Ginsenoside Rg1 obviously promoted the protein expression of PPAR-α. The protein expression was detected by immunohistochemical and western blot assay after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05; @significant compared with simvastatin alone, P < 0.05.
We further tested the protein expression of PPAR-α in the liver tissue. Immunohistochemical results showed that PPAR-α expressed in the cytoplasm of liver cells and ginsenoside Rg1 inhibited obviously the protein expression of PPAR-α induced by high fat and high sugar in a dose-dependent manner. The effect of ginsenoside Rg1 was better than that of simvastatin (P < 0.05) (Figure 5B). Besides, western blot analysis showed the same results as immunohistochemistry (Figure 5C). The above results showed that ginsenoside Rg1 upregulated the expression of PPAR-α.
2.6. Ginsenoside Rg1 Promoted the Expression of CPT1A and CPT2
CPT1 and CPT2 are two important regulators of fatty acid oxidation.10,11 Previous studies have shown that PPAR-α enhances the expression of CPT1 and CPT2, which promotes lipid oxidation.12 Therefore, we first detected the mRNA and protein expression of CPT1A by q-PCR, immunohistochemistry, and western blot. The results showed that ginsenoside Rg1 can upregulate the mRNA and protein expression of CPT1A, which was inhibited by the high-fat and high-sugar-induced NAFLD model in a dose-dependent manner after 4 or 8 weeks of ginsenoside Rg1 treatment. The effect of ginsenoside Rg1 was better than that of simvastatin at 8 weeks (P < 0.05) (Figure 6).
Figure 6.

Ginsenoside Rg1 obviously promoted the expression of CPT1A2 in the animal models of NAFLD. (A) Ginsenoside Rg1 obviously promoted the mRNA expression of CPT1A. The mRNA expression was detected by q-PCR after ginsenoside Rg1 treatment for 4 or 8 weeks. (B, C) Ginsenoside Rg1 obviously promoted the protein expression of CPT1A. The protein expression was detected by immunohistochemical and western blot assay after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05; @significant compared with simvastatin alone, P < 0.05.
CPT1 mainly exists in the outer mitochondrial membrane, while CPT2 is a membrane-bound protein in the inner mitochondrial membrane.13,14 Furthermore, we detected the expression of CPT2. q-PCR results showed that the mRNA expression of CPT2 in the low- and high-dose ginsenoside Rg1 treatment groups increased gradually compared with the model group, but there was no statistical significance (P > 0.05). Simvastatin and ginsenoside Rg1 have similar effects (Figure 7A) (P > 0.05). The protein expression level of CPT2 showed similar results to its mRNA expression by immunohistochemical and western blot assay (Figure 7B,C). We suspected that the above results may be due to the modeling time not being long enough or the mitochondrial membrane playing a protective role to CPT2. The above results showed that ginsenoside Rg1 can upregulate the expression of CPT1 and has little effect on CPT2.
Figure 7.

Ginsenoside Rg1 promoted the expression of CPT2 in the animal models of NAFLD. (A) Ginsenoside Rg1 promoted the mRNA expression of CPT2, P > 0.05. The mRNA expression was detected by q-PCR after ginsenoside Rg1 treatment for 4 or 8 weeks. (B, C) Ginsenoside Rg1 promoted the protein expression of CPT2, P > 0.05. The protein expression was detected by immunohistochemical and western blot assay after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments.
2.7. Ginsenoside Rg1 Inhibited the Expression of SREBP-1C
Previous studies have shown that the upregulation of PPAR-α can inhibit the expression of SREBP-1C, which promotes the oxidation of fatty acids, and the output of TG in the liver, which in turn alleviates the process of NAFLD.15 Our results showed that the expression of SREBP-1C in the model group was significantly higher than that in the control group and then it goes down dramatically in a dose-dependent manner after ginsenoside Rg1 treatment for 4 or 8 weeks. The effect of the high-dose ginsenoside Rg1 group was better than that of the simvastatin group (P < 0.05) (Figure 8). The above results showed that ginsenoside Rg1 inhibited the expression of SREBP-1C and promoted the oxidation of fatty acids.
Figure 8.

Ginsenoside Rg1 obviously inhibited the expression of SREBP-1C. (A) Ginsenoside Rg1 obviously inhibited the mRNA expression of SREBP-1C. The mRNA expression was detected by q-PCR after ginsenoside Rg1 treatment 4 or 8 weeks. (B, C) Ginsenoside Rg1 obviously inhibited the protein expression of SREBP-1C. The protein expression was detected by immunohistochemical and western blot after ginsenoside Rg1 treatment 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05. *Significant compared with the model group alone, P < 0.05. @Significant compared with simvastatin alone, P < 0.05.
2.8. Ginsenoside Rg1 Promoted the Expression of CYP-7A
CYP7 is a rate-limiting enzyme for the synthesis of bile acids and catalyzes the decomposition of cholesterol into bile acids in the liver.16 Therefore, promoting the expression of CYP7 can reduce liver fat deposition. Previous studies have shown that activation of PPAR-α in a mouse promotes the expression of CYP-7A.17 We detected the expression of CYP7 by q-PCR, immunohistochemistry, and western blot assay. Our results showed that high fat and high sugar inhibited the expression of CYP7 and that ginsenoside Rg1 can reverse this change in a dose-dependent manner in 4 or 8 weeks, similar to the effect of simvastatin. In addition, the effect of high-dose ginsenoside Rg1 was better than that of simvastatin (P < 0.05) (Figure 9). The results showed that ginsenoside Rg1 increased the expression of CYP-7A and promoted the decomposition of cholesterol into bile acids.
Figure 9.

Ginsenoside Rg1 obviously promoted the expression of CYP-7A. (A) Ginsenoside Rg1 obviously promoted the mRNA expression of CYP-7A. The mRNA expression was detected by q-PCR after ginsenoside Rg1 treatment for 4 or 8 weeks. (B, C). Ginsenoside Rg1 obviously promoted the protein expression of CYP-7A. The protein expression was detected by immunohistochemistry and western blot assay after ginsenoside Rg1 treatment for 4 or 8 weeks. The values shown are mean ± SEM of the data from three independent experiments. #Significant compared with the control group alone, P < 0.05; *significant compared with the model group alone, P < 0.05; @significant compared with simvastatin alone, P < 0.05.
2.9. Discussion
Currently, the complex pathogenesis of NAFLD is not fully understood. In 1998, Day et al.18 proposed the so-called “two-hit” theory, which became the theoretical basis to explain NAFLD pathogenesis. On the basis of the hypothesis, the synthesis or accumulation of fat in hepatocytes caused by excessive calorie intake constitutes the first “hit” in the development of NAFLD. In addition, insulin resistance that arises from elevated levels of FFAs in liver has also been suggested to play an essential role.19,20 The second hit includes oxidative stress and inflammation caused by dysregulation of proinflammatory cytokines, which can lead to nonalcoholic steatohepatitis and even liver fibrosis.18 The development of inflammation, fibrosis, and even necrosis of the liver caused by oxidative stress is the key to the second hit.
In addition, many important proteins, such as PPAR-α, CPT, SREBPs, and CYP-7A, are involved in the second hit, leading to liver function damage.21,22 PPAR, a ligand-activated transcription factor, is a member of the nuclear hormone receptor superfamily that can be divided into three subtypes: PPAR-α, PPAR-β, and PPAR-γ. Among them, PPAR-α is mainly expressed in the liver, skeletal muscle, brown adipose tissue, and other tissues with strong lipid metabolism. It is closely related to the body’s lipid metabolism, inflammatory response, immune regulation, cell proliferation and differentiation, and cell apoptosis.23−25
The specific mechanism of the oxidation of intrahepatic fatty acids is as follows: (1) Fatty acid oxidase such as carnitine palmitoyl transferase-1 (CPT1) and carnitine palmitoyl transferase-2 (CPT2) can convert acyl-carnitine into acyl CoA. Lipid acyl CoA enters the mitochondrial matrix and becomes the substrate of the fatty acid β-oxidase system, while PPAR-α can enhance the expression of CPT1 and CPT2, thus promoting the lipid oxidation process.14 (2) Activated PPAR-α can promote the expression of CYP-7A, thus promoting the excretion of cholesterol.26 (3) The upregulated expression of PPAR-α could inhibit the expression of SREBPs, promote the oxidation of fatty acids and the output of TG in the liver, and alleviate the process of NAFLD.27 To validate the utility of ginsenoside Rg1 for NAFLD patient improvement, more detailed investigation regarding the molecular mechanism of ginsenoside Rg1 in the fatty acid oxidase in mitochondria needs to be further carried out.
3. Conclusions
Our studies showed that ginsenoside Rg1 can inhibit the inflammatory response and alleviate liver damage in NAFLD. Meanwhile, ginsenoside Rg1 may promote the expression of CPT1A, CPT2, and CYP-7A and inhibit the expression of SREBP-1C by increasing the expression of PPAR-α and regulating lipid metabolism, thereby alleviating NAFLD.
4. Experimental Section
4.1. Compound Extraction
Air-dried P. notoginseng material was extracted with 95% ethanol under reflux three times, for 2 h each time. The extract was concentrated in a rotary vacuum evaporator to give a residue. The residue was dissolved in H2O and then extracted successfully with EtOAC (2 L). The combined EtOAc extracts was subjected to silica column chromatography (200–300 mesh) and eluted with petroleum ether/EtOAc (90:10, 80:20, 50:50, 25:75) followed by CHCl3/MeOH in a linear gradient (90:10, 80:20, 70:30, 60:40, 0:100) to obtain 6 fractions on the basis of thin-layer chromatography (TLC) profiles. Fraction 4 was further chromatographed on sephadex LH-20 eluted with (CHCl3/MeOH 1:1) to furnish four subfractions (fractions 4-1 to 4-4). Fraction 4-2 was further purified by preparatory TLC to obtain the pure compound ginsenoside Rg1. The purity of ginsenoside Rg1 was identified by high-performance liquid chromatography tandem mass spectrometry (HPLC-MS/MS).
4.2. Animals and Experimental Design
Adult Sprague Dawley (SD) rats (180–220 g) were purchased from the Animal Zoology Department (Kunming Medical University). The rats were housed at 22–26 °C temperature, 40–60% humidity, and a 12 h light/12 h dark cycle in the animal facility with free access to food and water. Ginsenoside Rg1 and simvastatin were dissolved in 40 °C distilled water to form a suspension and then administered by gavage.
SD rats were randomly divided into a control group, model group, ginsenoside Rg1 low-dose (30 mg/(kg day)) and high-dose (60 mg/(kg day)) treatment groups, and simvastatin control group (1 mg/(kg day)), with 10 SD rats in each group. The control group was given a basic diet. The model group was given a high-sugar and high-fat diet (54% basic diet, 15% lard, 25% saccharose, 1% cholesterol, and 5% extract egg yolk powder) for 14 weeks. Two rats were randomly euthanized by intraperitoneal injection of sodium pentobarbital (200 mg/kg body weight). Meanwhile, HE staining was used to detect the arrangement of rat liver cells and oil red “O staining” was used to detect the fat accumulation in the rat liver to determine whether the animal model of NAFLD was successfully established. Then, some of the NAFLD animals were administered ginsenoside Rg1 or simvastatin orally by gavage for 4 or 8 weeks; meanwhile, the remaining animals continued to be intragastrically administered high fat and high sugar for 4 or 8 weeks.
All animal procedures were approved by the Institutional Animal Care and Use Committee of Kunming Medical University, and the protocol of the experiments was approved by the Animal Care and Use Committee of Yunnan University.
4.3. Glucose, Insulin, Lipid Factor, Inflammatory Factor, and Liver Function Test
The levels of serum GLU, TG, TC, ALT, AST, and ALP were detected by an automated biochemistry analyzer (Hitachi 7060, Japan). The levels of serum INS, IL-1, IL-6, IL-8, IL-18, and TNF-α were detected by ELISA kits (Navy Medical Institute, Shanghai, China) according to the manufacturer’s instructions. The insulin resistance index (HOMA-IR) was calculated by the formula INS*GLU/22.5.
4.4. Real-Time PCR
Total RNA was extracted from the liver tissues using the Total RNA Extractor (Trizol) kit. The cDNA of each RNA sample was reverse transcribed with the M-MLV RTase cDNA kit according to the manufacturer’s instructions. Real-time PCR was performed using the SYBR Premix EX Tap 2x kit in the CFX 96 PT-PCR system. The reaction conditions were 95 °C for 30 s, 95 °C for 5 s, 60 °C for 30 s, followed by 30 cycles and then 72 °C for 10 min according to our previous research methods.28,29 The primer sequences are shown in Table 1.
Table 1. Primer Parameters.
| name | sequence | Tm (°C) | amplicon length |
|---|---|---|---|
| PPAR-α | F: ACAGGAGAGCAGGGATTT | 60 | 141 |
| R: CACCATTTCAGTAGCAGGA | |||
| CPT1A | F: CGAGAAGGGAGGACAGAG | 60 | 199 |
| R: ACACCACATAGAGGCAGAAG | |||
| CPT2 | F: CCAACAAAACTAATCCCAAG | 60 | 123 |
| R: CCAAACCCTATCTCCTGAA | |||
| SREBP-1C | F: CTGCTTGGCTCTTCTCTTT | 60 | 89 |
| R: CTTGTTTGCGATGTCTCC | |||
| CYP-7A | F: GGAAAGCAAAGACCACCT | 60 | 150 |
| R: GTTCAAAGCAGGAGAGCA | |||
| β-actin | F: GGAAATCGTGCGTGACATTAAA | 60 | 111 |
| R: GGCAGCTCATAGCTCTTCTC | 62 |
4.5. HE, Oil Red O Staining, and Immunohistochemistry
The tissue sections (5 μm) were subjected to antigen retrieval by microwave after deparaffinization and rehydration for 10 min in sodium citrate buffer. Sections were cooled to room temperature, treated with 3% H2O2 for 10 min, and blocked with 5% goat serum for 40 min at room temperature. One part of the sections was stained with hematoxylin-eosin and oil red O ethanol dye, and the other part was stained with immunohistochemistry. For immunohistochemistry staining, the sections were incubated at 4 °C overnight with the first antibody against PPAR-α, CPT1A, CPT2, SREBP-1C, and CYP-7A (diluted 1:200, Cell Signaling Technology, Danvers, NA). Then, the sections were washed in phosphate-buffered saline (PBS) and incubated with the secondary antibody (biotinylated goat anti-rabbit, 1:150; Vector Laboratories, Burlingame, CA) for 30 min. The sections were counterstained with hematoxylin after diaminobenzidine staining. All of the slices were dehydrated with ethanol for 2 min, followed by xylene transparency for 5 min, and then the tablets were quickly sealed with neutral gum and an ultrathin cover glass. After sealing the film, it was observed under an ordinary optical microscope according to our previous research methods.28
4.6. Western Blotting
Total protein of the liver samples was extracted using the radio immunoprecipitation assay (RIPA) lysate added with PMSF(Solarbio, China), then quantified by BCA (PPLYGEN, China), and finally the protein concentration was pulled to 5 μg/μL. The protein was loaded at 40 μg, separated by 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) gel and transferred to a poly(vinylidene fluoride) (PVDF, Millipore, China) membrane, and blocked with 5% milk for 120 min at room temperature. The membrane was incubated with a specific primary antibody against PPAR-α, CPT1A, CPT2, SREBP-1C, CYP-7A, and β-actin (1:1000 dilution; Cell Signaling Technology, Danvers, NA) at 4 °C overnight. The secondary antibody was a horseradish peroxidase (HRP)-conjugated anti-rabbit or mouse IgG (1:5000 dilution; Santa Cruz Biotech). The membrane was visualized using the enhanced chemiluminescence (ECL) detection system. The signal intensity was quantified using ImageJ software. The experimental process was the same as our previous research methods.28
4.7. Ethics Statement
This study was carried out abiding by the rules of the Laboratory Animal Center of Kunming Medical University. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Kunming Medical University. All surgeries were performed under sodium pentobarbital anesthesia, and all efforts were made to minimize suffering.
4.8. Statistical Analysis
Data were expressed as the mean ± standard deviation (SD) of three independent experiments. Statistical differences between the groups were analyzed using a post-hoc analysis of a randomized controlled trial. Values of P < 0.05 were considered of statistically significant.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China [81360128], Scientific and Technological Development Project of Yunnan Province [2018FE001-162,2015FB046], Education Department Fund of Yunnan Province [2017YJS076], and Research fund of Guilin Medical University [20501020002]. This work was also supported by the Yunnan Province Key Laboratory for Nutrition and Food Safety in Universities.
Author Contributions
Y.H. and D.G. did most of the experiments and contributed equally to this work; J.P., K.J., Z.L., and J.S. helped with the experiments; X.F. analyzed the data and wrote the main manuscript; and S.Y., S.L., and X.F. designed the research.
The authors declare no competing financial interest.
References
- Duvnjak M.; Lerotić I.; Baršić N.; Tomašić V.; Virović Jukić L.; Velagić V. Pathogenesis and management issues for non-alcoholic fatty liver disease. World J. Gastroenterol. 2007, 13, 4539–4550. 10.3748/wjg.v13.i34.4539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grander C.; Grabherr F.; Moschen A. R.; Tilg H. Non-Alcoholic Fatty Liver Disease: Cause or Effect of Metabolic Syndrome. Visc. Med. 2016, 32, 329–334. 10.1159/000448940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa D.; Patel V.; Sanyal A. J. Future therapy for non-alcoholic fatty liver disease. Liver Int. 2018, 38, 56–63. 10.1111/liv.13676. [DOI] [PubMed] [Google Scholar]
- Caballería L.; Pera G.; Rodríguez L.; Auladell M. A.; Bernad J.; Canut S.; Torán P. Metabolic syndrome and nonalcoholic fatty liver disease in a Spanish population: influence of the diagnostic criteria used. Eur. J. Gastroenterol. Hepatol. 2012, 24, 1007–1011. 10.1097/MEG.0b013e328355b87f. [DOI] [PubMed] [Google Scholar]
- Lau J. K. C.; Zhang X.; Yu J. Animal models of non-alcoholic fatty liver disease: current perspectives and recent advances. J. Pathol. 2017, 241, 36–44. 10.1002/path.4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumida Y.; Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J. Gastroenterol. 2018, 53, 362–376. 10.1007/s00535-017-1415-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuschwander-Tetri B. A. Non-alcoholic fatty liver disease. BMC Med. 2017, 15, 45 10.1186/s12916-017-0806-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Yang C.; Zhang S.; Li J.; Xiao Q.; Huang W. Ginsenoside Rg1 Protects against Non-alcoholic Fatty Liver Disease by Ameliorating Lipid Peroxidation, Endoplasmic Reticulum Stress, and Inflammasome Activation. Biol. Pharm. Bull. 2018, 41, 1638–1644. 10.1248/bpb.b18-00132. [DOI] [PubMed] [Google Scholar]
- Renu K.; Sruthy K. B.; Parthiban S.; Sugunapriyadharshini S.; George A.; Pichiah P. B.; Suman S.; Abilash V. G.; Arunachalam S. Elevated lipolysis in adipose tissue by doxorubicin via PPARalpha activation associated with hepatic steatosis and insulin resistance. Eur. J. Pharmacol. 2019, 843, 162–176. 10.1016/j.ejphar.2018.11.018. [DOI] [PubMed] [Google Scholar]
- Du Q.; Tan Z.; Shi F.; Tang M.; Xie L.; Zhao L.; Li Y.; Hu J.; Zhou M.; Bode A.; Luo X.; Cao Y. PGC1alpha/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. 2019, 110, 2050–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q.; Pan R.; Ding L.; Zhang F.; Hu L.; Ding B.; Zhu L.; Xia Y.; Dou X. Rutin exhibits hepatoprotective effects in a mouse model of non-alcoholic fatty liver disease by reducing hepatic lipid levels and mitigating lipid-induced oxidative injuries. Int. Immunopharmacol. 2017, 49, 132–141. 10.1016/j.intimp.2017.05.026. [DOI] [PubMed] [Google Scholar]
- Ding J.; Li M.; Wan X.; Jin X.; Chen S.; Yu C.; Li Y. Effect of miR-34a in regulating steatosis by targeting PPARalpha expression in nonalcoholic fatty liver disease. Sci. Rep. 2015, 5, 13729 10.1038/srep13729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houten S. M.; Wanders R. J. A.; Ranea-Robles P. Metabolic interactions between peroxisomes and mitochondria with a special focus on acylcarnitine metabolism. Biochim. Biophys. Acta, Mol. Basis Dis. 2020, 1866, 165720 10.1016/j.bbadis.2020.165720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adeva-Andany M. M.; Carneiro-Freire N.; Seco-Filgueira M.; Fernández-Fernández C.; Mouriño-Bayolo D. Mitochondrial β-oxidation of saturated fatty acids in humans. Mitochondrion 2019, 46, 73–90. 10.1016/j.mito.2018.02.009. [DOI] [PubMed] [Google Scholar]
- Ren T.; Zhu J.; Zhu L.; Cheng M. The Combination of Blueberry Juice and Probiotics Ameliorate Non-Alcoholic Steatohepatitis (NASH) by Affecting SREBP-1c/PNPLA-3 Pathway via PPAR-alpha. Nutrients 2017, 9, E198. 10.3390/nu9030198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K.; Wang F.; Zeng Y.; Chen X.; Xu X. Salusin-alpha attenuates hepatic steatosis and atherosclerosis in high fat diet-fed low density lipoprotein receptor deficient mice. Eur. J. Pharmacol. 2018, 830, 76–86. 10.1016/j.ejphar.2018.04.026. [DOI] [PubMed] [Google Scholar]
- Engin A. Non-Alcoholic Fatty Liver Disease. Adv. Exp. Med. Biol. 2017, 960, 443–467. 10.1007/978-3-319-48382-5_19. [DOI] [PubMed] [Google Scholar]
- Day C. P.; James O. F. Steatohepatitis: a tale of two “hits”?. Gastroenterology 1998, 114, 842–845. 10.1016/S0016-5085(98)70599-2. [DOI] [PubMed] [Google Scholar]
- Xing L.-J.; Zhang L.; Liu T.; Hua Y.-Q.; Zheng P.-Y.; Ji G. Berberine reducing insulin resistance by up-regulating IRS-2 mRNA expression in nonalcoholic fatty liver disease (NAFLD) rat liver. Eur. J. Pharmacol. 2011, 668, 467–471. 10.1016/j.ejphar.2011.07.036. [DOI] [PubMed] [Google Scholar]
- Zheng H.; Li S.; Ma L.; Cheng L.; Deng C.; Chen Z.; Xie C.; Xiang M.; Jiang W.; Chen L. A novel agonist of PPAR-gamma based on barbituric acid alleviates the development of non-alcoholic fatty liver disease by regulating adipocytokine expression and preventing insulin resistance. Eur. J. Pharmacol. 2011, 659, 244–251. 10.1016/j.ejphar.2011.03.033. [DOI] [PubMed] [Google Scholar]
- Gross B.; Pawlak M.; Lefebvre P.; Staels B. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. 10.1038/nrendo.2016.135. [DOI] [PubMed] [Google Scholar]
- Zhao X.-J.; Yang Y.-Z.; Zheng Y.-J.; Wang S.-C.; Gu H.-M.; Pan Y.; Wang S.-J.; Xu H.-J.; Kong L.-D. Magnesium isoglycyrrhizinate blocks fructose-induced hepatic NF-kappaB/NLRP3 inflammasome activation and lipid metabolism disorder. Eur. J. Pharmacol. 2017, 809, 141–150. 10.1016/j.ejphar.2017.05.032. [DOI] [PubMed] [Google Scholar]
- Montagner A.; Polizzi A.; Fouche E.; Ducheix S.; Lippi Y.; Lasserre F.; Barquissau V.; Regnier M.; Lukowicz C.; Benhamed F.; Iroz A.; Bertrand-Michel J.; Al Saati T.; Cano P.; Mselli-Lakhal L.; Mithieux G.; Rajas F.; Lagarrigue S.; Pineau T.; Loiseau N.; Postic C.; Langin D.; Wahli W.; Guillou H. Liver PPARalpha is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. 10.1136/gutjnl-2015-310798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S.; Stienstra R. The role and regulation of the peroxisome proliferator activated receptor alpha in human liver. Biochimie 2017, 136, 75–84. 10.1016/j.biochi.2016.12.019. [DOI] [PubMed] [Google Scholar]
- Poirier H.; Niot I.; Monnot M.-C.; Braissant O.; Meunier-Durmort C.; Costet P.; Pineau T.; Wahli W.; Willson T. M.; Besnard P. Differential involvement of peroxisome-proliferator-activated receptors alpha and delta in fibrate and fatty-acid-mediated inductions of the gene encoding liver fatty-acid-binding protein in the liver and the small intestine. Biochem. J. 2001, 355, 481–488. 10.1042/bj3550481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Chen Y.; Zhao L.; Chen Y.; Mei M.; Li Q.; Huang A.; Varghese Z.; Moorhead J. F.; Ruan X. Z. Inflammatory stress exacerbates hepatic cholesterol accumulation via disrupting cellular cholesterol export. J. Gastroenterol. Hepatol. 2012, 27, 974–984. 10.1111/j.1440-1746.2011.06986.x. [DOI] [PubMed] [Google Scholar]
- Ferré P.; Foretz M.; Azzout-Marniche D.; Bécard D.; Foufelle F. Sterol-regulatory-element-binding protein 1c mediates insulin action on hepatic gene expression. Biochem. Soc. Trans. 2001, 29, 547–552. 10.1042/bst0290547. [DOI] [PubMed] [Google Scholar]
- Fan X.; Zhang C.; Niu S.; Fan B.; Gu D.; Jiang K.; Li R.; Li S. Ginsenoside Rg1 attenuates hepatic insulin resistance induced by high-fat and high-sugar by inhibiting inflammation. Eur. J. Pharmacol. 2019, 854, 247–255. 10.1016/j.ejphar.2019.04.027. [DOI] [PubMed] [Google Scholar]
- Fan X.; Tao J.; Zhou Y.; Hou Y.; Wang Y.; Gu D.; Su Y.; Jang Y.; Li S. Investigations on the effects of ginsenoside-Rg1 on glucose uptake and metabolism in insulin resistant HepG2 cells. Eur. J. Pharmacol. 2019, 843, 277–284. 10.1016/j.ejphar.2018.11.024. [DOI] [PubMed] [Google Scholar]