

Summary
Background
Benzodiazepine-refractory, or established, status epilepticus is thought to be of similar pathophysiology in children and adults, but differences in underlying aetiology and pharmacodynamics might differentially affect response to therapy. In the Established Status Epilepticus Treatment Trial (ESETT) we compared the efficacy and safety of levetiracetam, fosphenytoin, and valproate in established status epilepticus, and here we describe our results after extending enrolment in children to compare outcomes in three age groups.
Methods
In this multicentre, double-blind, response-adaptive, randomised controlled trial, we recruited patients from 58 hospital emergency departments across the USA. Patients were eligible for inclusion if they were aged 2 years or older, had been treated for a generalised convulsive seizure of longer than 5 min duration with adequate doses of benzodiazepines, and continued to have persistent or recurrent convulsions in the emergency department for at least 5 min and no more than 30 min after the last dose of benzodiazepine. Patients were randomly assigned in a response-adaptive manner, using Bayesian methods and stratified by age group (<18 years, 18–65 years, and >65 years), to levetiracetam, fosphenytoin, or valproate. All patients, investigators, study staff, and pharmacists were masked to treatment allocation. The primary outcome was absence of clinically apparent seizures with improved consciousness and without additional antiseizure medication at 1 h from start of drug infusion. The primary safety outcome was life-threatening hypotension or cardiac arrhythmia. The efficacy and safety outcomes were analysed by intention to treat. This study is registered in ClinicalTrials.gov, NCT01960075.
Findings
Between Nov 3, 2015, and Dec 29, 2018, we enrolled 478 patients and 462 unique patients were included: 225 children (aged <18 years), 186 adults (18–65 years), and 51 older adults (>65 years). 175 (38%) patients were randomly assigned to levetiracetam, 142 (31%) to fosphenyltoin, and 145 (31%) were to valproate. Baseline characteristics were balanced across treatments within age groups. The primary efficacy outcome was met in those treated with levetiracetam for 52% (95% credible interval 41–62) of children, 44% (33–55) of adults, and 37% (19–59) of older adults; with fosphenytoin in 49% (38–61) of children, 46% (34–59) of adults, and 35% (17–59) of older adults; and with valproate in 52% (41–63) of children, 46% (34–58) of adults, and 47% (25–70) of older adults. No differences were detected in efficacy or primary safety outcome by drug within each age group. With the exception of endotracheal intubation in children, secondary safety outcomes did not significantly differ by drug within each age group.
Interpretation
Children, adults, and older adults with established status epilepticus respond similarly to levetiracetam, fosphenytoin, and valproate, with treatment success in approximately half of patients. Any of the three drugs can be considered as a potential first-choice, second-line drug for benzodiazepine-refractory status epilepticus.
Introduction
Status epilepticus is one of the most common neurological emergencies treated in emergency departments, affecting patients of all ages. An estimated 120 000–180 000 episodes occur annually in the USA.1–5 Benzodiazepines are widely recognised as an effective first-line therapy.6,7 No optimal second-line therapy has been agreed, but prospective open-label paediatric trials in the UK and Australasia found approximately equivalent response rates comparing phenytoin with levetiracetam.8,9 The success of second-line drugs is important because longer durations of status epilepticus itself leads to increasing likelihood of further seizure activity through positive feedback mechanisms.10 Failure to stop status epilepticus early is associated with irreversible neuronal injury and the complications caused by metabolic and respiratory derangements of status epilepticus.10,11
The Established Status Epilepticus Treatment Trial (ESETT) was a double-blind, Bayesian re sponse-adaptive, randomised clinical trial of levetiracetam, fosphenytoin, and valproate in adults and children aged 2 years and older with benzodiazepine-refractory convulsive status epilepticus.12 A planned interim analysis after enrolment of 400 participants determined that the chance of showing that one study drug was superior or inferior to the other two was less than 1%, which met a predetermined futility stopping criterion for the overall cohort.12 The same stopping criterion was also met in the adult cohorts, but we could not exclude the possibility that one drug might be more effective in children. The Data and Safety Monitoring Board for the trial allowed continued enrolment of children to enrich the planned secondary age analysis. We report this analysis of the comparative effectiveness and safety of the study medications in the three age groups: children, adults, and older adults.
Methods
Study design and participants
ESETT was a multicentre, randomised, double-blind, comparative effectiveness study of levetiracetam, fosphenytoin, and valproate to treat patients with benzodiazepine-refractory status epilepticus in emergency departments. The study was run by the Neurological Emergencies Treatment Trials network and the Pediatric Emergency Care Applied Research Network. Patients were enrolled at 58 hospital emergency departments across the USA, of which 14 sites enrolled both adults and children, 25 enrolled only adults, and 19 enrolled only children.
Patients were eligible if they were aged 2 years or older, had been treated for a generalised convulsive seizure of longer than 5 min duration with adequate doses of benzodiazepines, and continued to have persistent or recurrent convulsions in the emergency department for at least 5 min and no more than 30 min after the last dose of benzodiazepine. The minimal adequate cumulative doses of benzodiazepines were defined as: diazepam 10 mg intravenously, lorazepam 4 mg intravenously, or midazolam 10 mg intravenously or intramuscularly for all adults and children with bodyweight of 32 kg or heavier; and diazepam 0.3 mg/kg intravenously, loraze pam 0.1 mg/kg intravenously, or midazolam 0.3 mg/kg intramuscularly or 0.2 mg/kg intravenously for children with bodyweight of less than 32 kg. Patients were excluded if they were known to be pregnant, a prisoner, or postanoxic, or if their seizures were precipitated by trauma, hypoglycaemia or hyperglycaemia. Individuals who pre-emptively opted out, or who had already been treated for this episode of status epilepticus with non-benzodiazepine anticonvulsant medications were also excluded. Individuals with known allergies or contraindications to any of the study drugs were not enrolled.
The study protocol was approved by the Institutional Review Boards for all study sites, and is available online at the University of Michigan. The trial was done using exception from informed consent for emergency research under US Food and Drug Administration (FDA) regulation 21 CFR 50.24.13 The Institutional Review Boards for all study sites engaged in this research approved local community consultation and public disclosure activities. Patients or their legally authorised representatives were notified about enrolment in the trial by the study team as soon as possible, and were asked for their consent for continued data collection until the participants’ planned completion of their last study visit.
Randomisation and masking
In this Bayesian response-adaptive comparative effectiveness trial, patients with established status epilepticus were randomly assigned in a response-adaptive manner to receive levetiracetam, fosphenytoin, or valproate. The study design has been published previously.14 Briefly, randomisation was response-adaptive and stratified by age group: younger than 18 years, 18–65 years, and older than 65 years. The randomisation scheme was equal allocation (1:1:1) for the first 300 patients, then the target allocation ratio was updated every 100 patients. Response-adaptive randomisation was used primarily to allocate more patients to the treatment group most likely to be the most effective. Allocation was concealed with web-based randomisation of non-sequentially numbered drug vials of identical appearance to an age-stratified use-next sequence, and were kept in close proximity to patient care in the emergency department. All investigators, patients, clinical and study teams, and pharmacists were masked to study drug allocation. Unmasking for the purposes of patient care, after determination of the primary outcome at 60 min, was allowed per protocol if deemed necessary by the treating team.
Procedures
Vials of study drug containing either levetiracetam 50 mg/mL, fosphenytoin 16.66 mg phenytoin equivalents (PE) per mL, or valproate 33.33 mg/mL and were produced, packaged, and labelled by the University of California (University of California Davis, Davis, CA, USA) at UC Davis Good Manufacturing Practice facility. The study drugs were identical in appearance, formulation, packaging, and administration, including volume and rate of infusion. The weight-based infusion rate provided treatment doses of levetiracetam 60 mg/kg (maximum 4500 mg), fosphenytoin 20 mg PE per kg (maximum 1500 mg PE), or valproate 40 mg/kg (maximum 3000 mg) infused over 10 min.
Eligible patients were identified by the treating clinical team and immediately enrolled on an emergent basis. After determining eligibility, the clinical team accessed an age-stratified ESETT medication box in the use-next sequence. The medication box was opened, an enclosed protocol assist device was activated, and the prerandomised vial of study drug was directly accessed with an administration set, inverted, and used to prime an intravenous infusion line. The protocol assist device was a mobile electronic device that would automatically alert the study team, and remind treatment teams about eligibility criteria and protocol interventions before enrolment. It also provided timed alerts after enrolment, and facilitated unmasking as required for patient care. A weight-based infusion rate was determined from an enclosed dose administration chart by use of a measured, stated, or estimated weight. Alternatively, the infusion rate could be determined from an enclosed length-based weight estimation tool for children in whom an accurate weight was not known. Trial drug was administered with an infusion pump that was programmed with the determined rate over a 10-min interval. Blood was not sampled as part of the primary protocol, but a small subset of patients had blood drawn to assess study drug pharmacodynamics as part of an ancillary study (to be reported elsewhere).
Outcomes
The primary efficacy outcome was absence of clinically apparent seizures with improving responsiveness, at 60 min after the start of study drug infusion, without additional antiseizure medication, including medications required for endotracheal intubation. A clinically apparent seizure was defined as visually determined focal or generalised tonic-clonic movements, nystagmoid or rhythmic eye movements, or generalised or segmental myoclonus. Improvement in responsiveness was defined by purposeful responses to noxious stimuli, or the ability to follow commands or verbalisation. Clinically apparent seizures were determined by the treating clinician, who was masked to study drug assignment. Patients were followed up until hospital discharge or 30 days, whichever occurred first.
The primary safety outcome was a composite of life-threatening hypotension or life-threatening cardiac arrhythmia. Secondary safety outcomes were need for endotracheal intubation within 60 min of the start of study drug infusion, acute seizure recurrence 60 min to 12 h after the start of study drug infusion, acute respiratory depression at any time during the study period, and mortality. Life-threatening cardiac arrhythmia was defined as any arrhythmia that persists despite reducing the rate of infusion of study drug, and that requires termination with chest compressions, pacing, defibrillation, or use of an antiarrhythmic drug or procedure. Respiratory depression was defined as impairment of ventilation or oxygenation necessitating definitive endotracheal intubation and mechanical ventilation, and is distinct from intubations done only for airway protection in those with decreased levels of consciousness. Those who were given only supraglottic airways or transient bag-valve-mask support were not included in this outcome. Need for endotracheal intubation within 60 min of start of study drug infusion included any placement or attempt at placement of a definitive tracheal airway (orotracheal, nasotracheal, cricothyroidotomy, or tracheostomy) for support of respirations or protection of airway. The use of non-definitive or non-tracheal airways (oral or nasal airways, laryngeal mask airways, or other supraglottic airways), or both, is not included in this outcome if the patient is not subsequently intubated unless specifically deemed to have been used in lieu of tracheal intubation.
Statistical analysis
The total planned sample size was a maximum of 795 enrolments (with participants able to enrol more than once), which was to include a minimum of 336 children. A sample size of 795 provided 90% power to identify the most effective treatment when one treatment group has a true response rate of 65% and the true response rate is 50% in the other two groups, and a sample size of 336 children would have 80% power to detect a 20% difference in response rates between drugs. Interim analyses were planned after 400, 500, 600, and 700 patients were enrolled, with early stopping allowed if the best treatment could be identified with high probability or for futility.
Secondary analyses of the primary outcome, secondary efficacy outcomes, and safety outcomes are reported by age group. Although the type I error rate for the primary analysis was well controlled,14 we did not correct for multiple comparisons for the age subgroup analyses. In each age group, we used a Bayesian approach to calculate the probability that each treatment is the most or least effective. In each age group, each treatment group was modelled independently, assuming a non-informative Uniform(0,1) distributionprior, and updated with the observed binomial primary outcome data using a conjugate beta-binomial model. From these posterior distributions, we calculated the probability that a given treatment was the most effective, and the pre-specified criterion for success was a probability of 0.975 or higher.11 The primary efficacy outcome is given by age group for the intention-to-treat sample, which included all unique patients who were randomly assigned to treatment, regardless of the amount of treatment actually received. The primary outcome is presented with 95% credible intervals. Patients who were enrolled more than once had only their first enrolment included in the primary analysis, but both enrolments were included in the safety analysis. We also did a per-protocol analysis of the primary outcome, excluding those with eligibility deviations or who did not receive the intervention.
Pre-specified secondary analyses included logistic regressions of the primary outcome to formally assess whether an interaction exists between age (either as <18 years vs >18 years and in years) and treatment groups. We used p values of less than 0.05 to assess the evidence of an interaction for the overall test of interaction terms (a χ2 test with two degrees of freedom). In a post-hoc analysis, we examined the primary outcome response rate for a number of smaller age groups subgroups (0–5, 6–10, 11–17, 18–40, 41–65, >65 years).
We calculated the frequency of safety outcomes for each predefined age group and difference was tested at a significance level of α=0.01 via Fisher’s exact test. We compared baseline characteristics across treatment groups within predefined age groups via χ2 and Kruskall-Wallis tests for lorazepam equivalents and p values of less than 0.05 were used for evidence of statistical significance.
We used SAS (version 9.4) and R (version 3.5.2) for all analyses. This study is registered in ClinicalTrials.gov, NCT01960075. This study was done under FDA approval, 119 756.
Role of the funding source
The funder of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report. All authors had full access to all the data in the study and final responsibility for the decision to submit for publication.
Results
Between Nov 3, 2015, and Dec 29, 2018, we enrolled 462 patients from a total of 478 enrolments. The 462 patients enrolled included 225 children (220 aged 2–17 years and five aged 1 year who were inadvertently enrolled despite eligibility criterion), 186 adults (aged 18–65 years), and 51 older adults (aged >65 years). This cohort is enriched with an additional 78 patients (76 children and two adults) who were recruited between Nov 29, 2017, and Dec 29, 2018, after a predefined futility stopping boundary specific to the paediatric cohort was crossed. The paediatric specific stopping boundary was assessed at a single planned interim analysis that was to occur after 500 enrolments, but was done early as part of a response to an adverse event. The baseline characteristics of patients are provided by age group in table 1 and by treatment group in the appendix (p 9). As expected, the disease differed by age group, with children having the highest proportion of unprovoked seizures, and febrile illness was the most common cause of provoked seizures in children, which rarely was the precipitant in adults (table 1). A higher proportion of children than adults and older adults were of Hispanic ethnicity or white. Although differences did exist among the age groups, we did not detect differences in baseline charac teristics between treatment groups within age groups (appendix p 9). Randomisation, group assignments, and analysis populations are shown in figure 1.
Table 1:
Baseline characteristics of the study population by age group
| Children (aged <18 years; n=225) | Adults (aged 18–65 years; n=186) | Older adults (aged >65 years; n=51) | |
|---|---|---|---|
| Age | |||
| Mean (SD) | 6.1 (4.3) | 42.6 (14.1) | 73.8 (7.2) |
| Range | 1–17* | 18–65 | 66–94 |
| Treatment allocation | |||
| Levetiracetam | 85 (38%) | 71 (38%) | 19 (37%) |
| Fosphenytoin | 71 (32%) | 54 (29%) | 17 (33%) |
| Valproate | 69 (31%) | 61 (33%) | 15 (29%) |
| Gender | |||
| Male | 124 (55%) | 108 (58%) | 30 (59%) |
| Female | 101 (45%) | 78 (42%) | 21 (41%) |
| Race | |||
| Black | 75 (33%) | 90 (48%) | 28 (55%) |
| White | 109 (48%) | 78 (42%) | 14 (27%) |
| Other, mixed race, or unknown | 41 (18%) | 18 (10%) | 9 (18%) |
| Ethnicity | |||
| Hispanic | 50 (22%) | 18 (10%) | 8 (16%) |
| Non-Hispanic | 166 (74%) | 159 (85%) | 41 (80%) |
| Unknown | 9 (4%) | 9 (5%) | 2 (4%) |
| History of epilepsy | 149 (66%) | 128 (69%) | 29 (57%) |
| Aetiology: precipitant of enrolling episode | |||
| Unprovoked | 106 (47%) | 43 (23%) | 12 (24%) |
| Febrile illness | 90 (40%) | 2 (1%) | 0 |
| Othert† | 11 (5%) | 43 (23%) | 12 (24%) |
| Antiseizure drug withdrawal or non-compliance | 9 (4%) | 43 (23%) | 6 (12%) |
| Toxic (eg, alcohol or drug withdrawal, poisoning) | 1 (0%) | 25 (13%) | 2 (4%) |
| Insufficient information to determine or idiopathic | 2 (1%) | 12 (6%) | 5 (10%) |
| Acute stroke or haemorrhage | 1 (0%) | 9 (5%) | 7 (14%) |
| CNS tumour | 0 | 5 (3%) | 3 (6%) |
| CNS infection | 4 (2%) | 2 (1%) | 1 (2%) |
| Metabolic (eg, hypoglycaemia, hyponatraemia) | 1 (0%) | 2 (1%) | 3 (6%) |
| Lorazepam equivalentst‡ | |||
| In mg for those weighing ≥32 kg | 6.0 (4.0–8.4) | 5.0 (4.0–6.4) | 4.0 (4.0–6.0) |
| In mg/kg forthose weighing <32 kg | 0.2 (0.1–0.2) | NA | NA |
Data are n (%) or median (IQR), unless otherwise indicated.
Five participants enrolled when aged younger than 2 years are eligibility deviations.
Most often included afebrile and non-CNS infections, combinations of aetiology, subacute stroke or haemorrhage, vasculitis, other encephalopathy, ventricular-peritoneal shunt failure, or sleep deprivation. Age groups differed by precipitant of enrolling episode, race, or ethnicity, but no baseline differences among treatment groups within age groups were detected.
Includes all 478 enrolments.
Figure 1: Study profile.
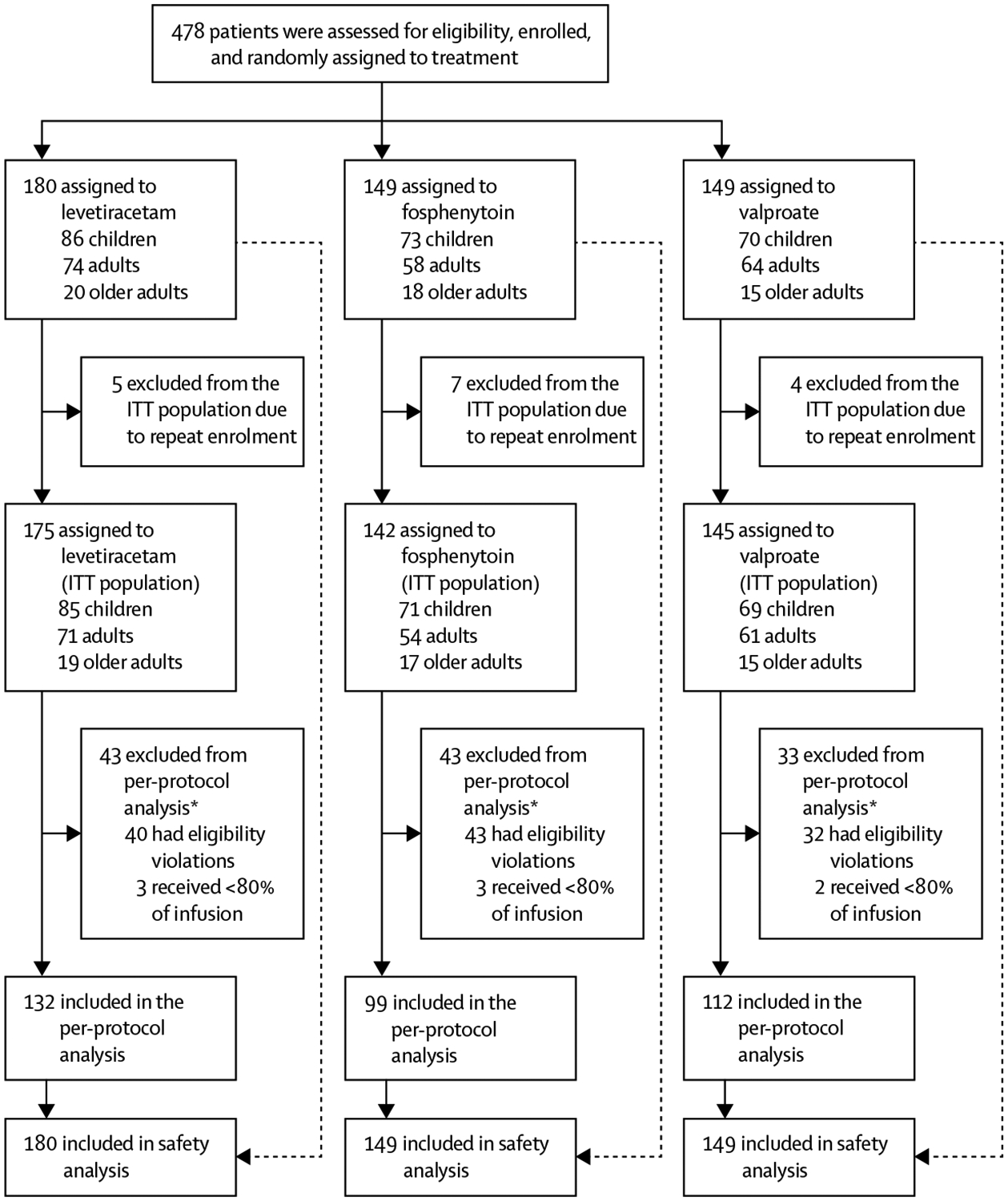
ITT=intention-to-treat. *Reasons for exclusion are non-exclusive.
The primary efficacy outcome results are in table 2. The probabilities of whether one treatment was better than another are shown in table 2 and figure 2. None of these probabilities met the pre-specified criteria for identifying the best (or worst) drug. We found no difference in the primary outcome between treatment groups within each age group (≤18 years and >18 years; p=0.93). Although children overall had numerically higher response rates, the probability of treatment success did not vary as a function of age in years (p=0.69 for the main effect of continuous age and p=0.88 for interaction of age [in years] by treatment). In our post-hoc analysis, efficacy was also similar between age subgroups (figure 3). Results of the per-protocol analysis were consistent with those of the intention-to-treat analysis (appendix p 10).
Table 2:
Efficacy analysis by age group
| Children (aged <18 years; n=225) | Adults (aged 18–65 years; n=l86) | Older adults (aged >65 years; n=51) | |
|---|---|---|---|
| Levetiracetam | 85 | 71 | 19 |
| Primary outcome | 44 (52%; 41–62) | 31 (44%; 33–55) | 7 (37%; 19–59) |
| Probability treatment is the most effective | 0.37 | 0.22 | 0.22 |
| Probability treatment is the least effective | 0.27 | 0.44 | 0.40 |
| Fosphenytoin | 71 | 54 | 17 |
| Primary outcome | 35 (49%; 38–61) | 25 (46%; 34–59) | 6 (35%; 17–59) |
| Probability treatment is the most effective | 0.22 | 0.41 | 0.19 |
| Probability treatment is the least effective | 0.47 | 0.27 | 0.46 |
| Valproate | 69 | 61 | 15 |
| Primary outcome | 36 (52%; 41–63) | 28 (46%; 34–58) | 7 (47%; 25–70) |
| Probability treatment is the most effective | 0.41 | 0.37 | 0.59 |
| Probability treatment is the least effective | 0.26 | 0.29 | 0.14 |
Data are n, n (%; 95% credible interval), or probability.
Figure 2: Posterior probabilities of success by age and treatment groups for the primary outcome.
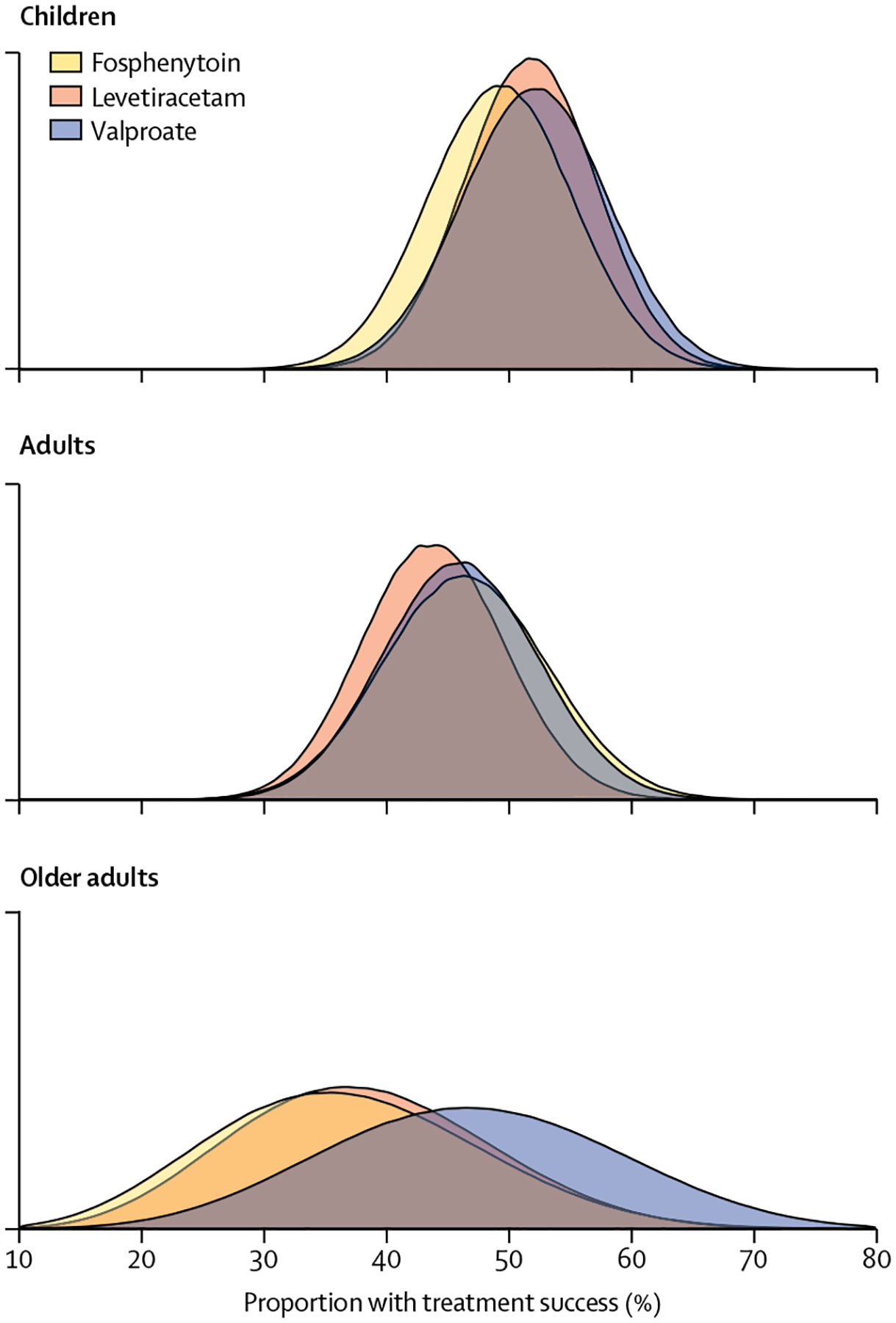
Figure 3: Forest plot of proportion of treatment success for the primary outcome by age ranges, post hoc.
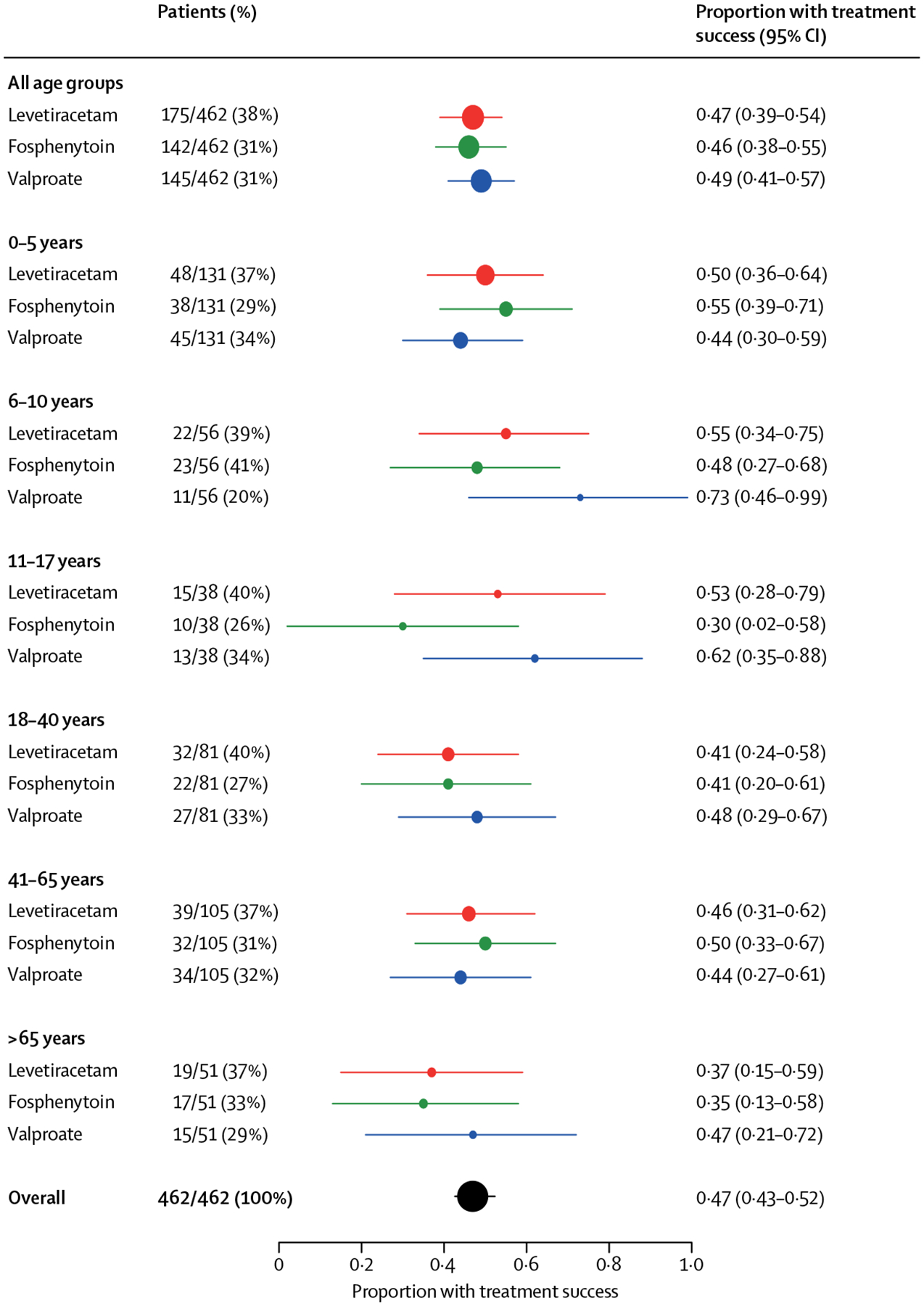
The size of the circle in each row is representative of the number of patients in that group and the error bars show 95% CIs. These narrower age groupings were determined post hoc.
Safety outcomes by age group are shown in table 3. The primary safety outcome of life threatening hypotension or life threatening cardiac arrhythmia was rare and did not differ by treatment group in any age group. Endotracheal intubation of children occurred more frequently in the fosphenytoin group (24 [33%] in fosphenytoin group, seven [8%] in levetiracetam group, and eight [11%] in valproate group; Fisher’s exact test p=0.0001), but did not differ by treatment group in either adult age group. No other differences in safety outcomes were detected.
Table 3:
Secondary efficacy outcomes and safety analysis by age group
| Children (aged <18 years) | Adults (aged 18–65 years) | Older adults (aged >65 years) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Levetiracetam | Fosphenytoin | Valproate | Levetiracetam | Fosphenytoin | Valproate | Levetiracetam | Fosphenytoin | Valproate | |
| ITT population | 85 | 71 | 69 | 71 | 54 | 61 | 19 | 17 | 15 |
| Admission to ICU | 53 (62%) | 45 (63%) | 43 (62%) | 38 (54%) | 26 (48%) | 34 (56%) | 15 (79%) | 13 (76%) | 8 (53%) |
| Length of ICU stay, days | 1 (0–2) | 1 (0–2) | 1 (0–2) | 1 (0–3) | 0 (0–3) | 1 (0–3) | 3 (1–6) | 2 (1–7) | 2 (0–6) |
| Length of hospital stay, days | 2 (1–3) | 2 (1–3) | 2 (1–4) | 3 (1–6) | 3 (1–7) | 2 (1–6) | 7 (5–12) | 5 (3–16) | 5 (3–16) |
| Safety population | 86 | 73 | 70 | 74 | 58 | 64 | 20 | 18 | 15 |
| Life-threatening hypotension within 60 min of start of study drug infusion | 0 | 2 (3%) | 3 (4%) | 0 | 2 (3%) | 0 | 1 (5%) | 1 (6%) | 0 |
| Life-threatening cardiac arrhythmia within 60 min of start of study drug infusion | 0 | 0 | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 |
| Acute respiratory depression | 5 (6%) | 13 (18%) | 7 (10%) | 10 (14%) | 6 (10%) | 5 (8%) | 0 | 2 (11%) | 0 |
| Endotracheal intubation within 60 min of start of study drug infusion | 7 (8%) | 24 (33%) | 8 (11%) | 19 (26%) | 13 (22%) | 14 (22%) | 7 (35%) | 5 (28%) | 2 (13%) |
| Acute seizure recurrence 60 min-12 h after start of study drug infusion | 8 (9%) | 11 (15%) | 6 (9%) | 5 (7%) | 3 (5%) | 5 (8%) | 4 (20%) | 4 (22%) | 3 (20%) |
| Death | 1 (1%) | 0 | 1 (1%) | 2 (3%) | 2 (3%) | 1 (2%) | 4 (20%) | 1 (6%) | 0 |
Data are n, n (%), or median (IQR). No patients had anaphylaxis or purple glove syndrome. ITT=intention-to-treat. ICU=intensive care unit.
Discussion
In this large trial of three treatments of benzodiazepine-refractory status epilepticus, we did not find differences in effectiveness among the three age groups: children, adults, and older adults. In all age groups, effectiveness at stopping status epilepticus within 1 h was approximately 50% for each of the three study medications. The frequency of safety events did not differ by age group and study drug, except endotracheal intubation, but, notably, the study was underpowered to detect small differences in uncommon safety events.
The comparative effectiveness of the study drugs is consistent with two recent large studies in children that studied phenytoin and levetiracetam.8,9 The higher response rates in these studies than in our study were likely due to the allowance of additional anticonvulsant medications, whereas our study considered administration of additional anticonvulsant to be a treatment failure.
We found no differences in the primary safety outcome in any age group. This finding is surprising because fosphenytoin has been previously associated with reports of bradycardia and hypotension, especially in older adults. Notably, the incidence of endotracheal intubation among children allocated to fosphenytoin was significantly higher than that in children allocated to either of the other groups. However, this result is difficult to interpret because it is not consistent with other observations in this and other trials. We did not see this effect in other age groups in our trial, and this difference was not seen in children in the recent EcLiPSE and ConSEPT trials.8,9 Furthermore, this finding was not supported by differences in other safety outcomes and adverse events expected to be related to endotracheal intubation, such as respiratory depression, decreased level of consciousness, or length of intensive care unit or hospital stay. The effect of this observation on clinical decision making is unclear because it was an isolated and unanticipated finding, with a previously low likelihood.
This study has two important limitations. First, we did not confirm the presence or absence of seizures with an electroencephalogram (EEG). This approach is consistent with clinical practice because an EEG is generally not available on an emergency basis. Patients who met the primary outcome are unlikely to have had ongoing seizures because we required improving mental status to meet this outcome. Conversely, some patients who did not reach the primary outcome might have done so because of the sedative effects of treatments rather than subclinical persistence of status epilepticus. This effect is unlikely to have differed by treatment group and therefore should not affect the findings of the study. Finally, few older adults were enrolled in the study, so it is difficult to make meaningful inferences about this age group, especially with uncommon safety events.
In summary, in this large randomised controlled trial we showed that approximately half of patients with established status epilepticus respond to high doses of levetiracetam, fosphenytoin, or valproate. These results were consistent across the three age groups: children, adults, and older adults. The primary safety outcome did not differ by study drug or age group. Any of the three drugs can be considered as potential first-choice, second-line drugs for benzodiazepine-refractory status epilepticus.
Supplementary Material
Research in context.
Evidence before this study
Previous observational data about the optimal treatment for patients with persistent status epilepticus despite treatment with benzodiazepines has been inconsistent and unable to identify if treatment efficacy varies with patient age. By contrast, two recent open-label randomised trials in children with established status epilepticus, EcLiPSE and ConSEPT, consistently showed no difference in outcomes between those treated with fosphenytoin or levetiracetam. ESETT was a double-blind trial of these two drugs and valproate in both adults and children with established status epilepticus, that also found no difference in outcomes overall, but did not determine if efficacy was affected by age.
Added value of this study
In this extension of the ESETT study, enrolment of additional children with status epilepticus enriched the overall cohort, allowing comparisons of efficacy and safety of fosphenytoin, levetiracetam, and valproate by age group in children, adults, and older adults. This double-blind clinical trial confirmed the findings of the open-label trials of fosphenytoin and levetiracetam in children with established status epilepticus, extended these findings to valproate, and showed that the lack of difference in outcomes between drugs is similar across age groups.
Implications of all the available evidence
Taken together, this and other recent randomised trials indicate that any of three anticonvulsants—fosphenytoin, levetiracetam, or valproate—can be considered a reasonable and appropriate first-choice, second-line drug in the treatment of both children and adults with established, benzodiazepine-refractory, status epilepticus. Dosing of these medications in clinical practice should follow the guidelines used in the trials to replicate the efficacy seen.
For Archived Clinical Research Datasets website see https://www.ninds.nih.gov/Current-Research/Research-Funded-NINDS/Clinical-Research/Archived-Clinical-Research-Datasets
Acknowledgments
This study was funded by the National Institute of Neurological Disorders and Stroke (awards U01NS088034, U01NS088023, U01NS056975, U01NS059041, and U01NS073476). We thank Gerhard Bauer and Brian Fury of the University of California at Davis Good Manufacturing Practice Laboratory for manufacturing of the investigational drug products; Jessica Munson of ARL Bio Pharma for testing the quality and stability of the investigational drugs; Henry Wang and Edward Jauch for serving as independent medical safety monitors; the members of the data and safety monitoring board (Barbara Dworetzky [chair], Gail Anderson, Jeffrey Buchhalter, Elizabeth Sugar, Alexis Topjian, and Peter Gilbert [NINDS liaison]); and especially all our patients and the emergency department nurses, pharmacists, and physicians who made the trial possible.
Funding
National Institute of Neurological Disorders and Stroke, National Institutes of Health.
Data sharing
The data from National Institute of Neurological Disorders and Stroke (NINDS)-supported clinical trials are an important scientific resource, made available to the wider scientific community, while ensuring that the confidentiality and privacy of study participants are protected. NINDS requires all investigators seeking access to data from archived NINDS-supported trials to agree to specific terms and conditions. A complete de-identified dataset, including individual participant data and a data dictionary defining each field in the dataset, will be made available within 1 year after publication of the primary results. Access to the Established Status Epilepticus Treatment Trial (ESETT) data will be through the NINDS repository of Archived Clinical Research Datasets. To request a dataset, please complete the NINDS Data Request Form and send it to the NINDS Clinical Research Liaison at CRLiaison@ninds.nih.gov. Please indicate the types of analyses for which the data will be made available (eg, for any purpose or for a specified purpose). The data will be made available for any purpose consistent with the conditions and terms established by the NINDS. Trial results will also be posted to ClinicalTrials.gov. The data will be made available without investigator support through the mechanisms established by the NINDS. The investigators welcome queries and collaboration with external users of the data as resources and time permit.
Footnotes
Declaration of interests
JC reports holding a patent (US9629797B2) on intravenous carbamazepine and holding intellectual property on intravenous topiramate, licensed to Ligand, on intravenous baclofen licensed to Allaysis, and on water-soluble benzodiazepine prodrugs for intranasal administration. DL reports serving on an advisory board for Bloom Science. SS reports receiving fees for serving on data and safety monitoring boards from Eisai, INSYS Therapeutics, and UCB Biosciences. HC reports receiving consulting fees from BIAL Pharma UK and Sage Therapeutics, and her institution has received funds for her work on other trials from UCB Pharma, Eisai Europe, Novartis, and GW Pharmaceuticals. NBF reports receiving grant support, paid to the University of Virginia Rectors and Visitors, from Cerebral Therapeutics, GW Pharmaceuticals, Medtronic, Neurelis, NeuroPace, SK Life Science, Takeda California, and UCB Biosciences. All other authors declare no competing interests.
Contributor Information
James M Chamberlain, Division of Emergency Medicine Children’s National Hospital, Washington, DC, USA;.
Jaideep Kapur, Department of Neurology, University of Virginia Health Sciences Center, Charlottesville, VA, USA;.
Shlomo Shinnar, Neurology, Pediatrics and Epidemiology and Population Health Montefiore Medical Center, Albert Einstein College of Medicine, Bronx, NY, USA;.
Jordan Elm, Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA;.
Maija Holsti, Division of Pediatric Emergency Medicine, University of Utah, Salt Lake City, UT, USA;.
Lynn Babcock, Division of Pediatric Emergency Medicine, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, University of Cincinnati, Cincinnati, OH, USA;.
Alex Rogers, Department of Emergency Medicine,; Department of Pediatrics,
William Barsan, Department of Emergency Medicine, Neuro Emergencies Research,.
James Cloyd, University of Michigan, Ann Arbor, MI, USA;; Center for Orphan Drug Research, College of Pharmacy, University of Minnesota, Minneapolis, MN, USA;
Daniel Lowenstein, Department of Neurology, University of California San Francisco, San Francisco, CA, USA;.
Thomas P Bleck, Division of Stroke and Neurocritical Care, Northwestern University Feinberg School of Medicine, Chicago, IL USA;.
Robin Conwit, National Institute of Neurological Disorders and Stroke, National Institutes of Health Neuroscience Center, Bethesda, MD, USA;.
Caitlyn Meinzer, Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA;.
Hannah Cock, Institute of Molecular and Clinical Sciences, St George’s University of London, London, UK;.
Nathan B Fountain, Department of Neurology, University of Virginia Health Sciences Center, Charlottesville, VA, USA;.
Ellen Underwood, Department of Public Health Sciences, Medical University of South Carolina, Charleston, SC, USA;.
Jason T Connor, ConfluenceStat LLC and University of Central Florida College of Medicine, Cooper City, FL, USA.
Robert Silbergleit, Department of Emergency Medicine, Neuro Emergencies Research,.
References
- 1.DeLorenzo RJ, Pellock JM, Towne AR, Boggs JG. Epidemiology of status epilepticus. J Clin Neurophysiol 1995; 12: 316–25. [PubMed] [Google Scholar]
- 2.DeLorenzo RJ, Hauser WA, Towne AR, et al. A prospective, population-based epidemiologic study of status epilepticus in Richmond, Virginia. Neurology 1996; 46: 1029–35. [DOI] [PubMed] [Google Scholar]
- 3.Shinnar S, Pellock JM, Moshé SL, et al. In whom does status epilepticus occur: age-related differences in children. Epilepsia 1997; 38: 907–14. [DOI] [PubMed] [Google Scholar]
- 4.Towne AR. Epidemiology and outcomes of status epilepticus in the elderly. Int Rev Neurobiol 2007; 81: 111–27. [DOI] [PubMed] [Google Scholar]
- 5.Betjemann JP, Lowenstein DH. Status epilepticus in adults. Lancet Neurol 2015; 14: 615–24. [DOI] [PubMed] [Google Scholar]
- 6.Glauser T, Shinnar S, Gloss D, et al. Evidence-based guideline: treatment of convulsive status epilepticus in children and adults: report of the guideline committee of the American Epilepsy Society. Epilepsy Curr 2016; 16: 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trinka E, Höfler J, Leitinger M, Rohracher A, Kalss G, Brigo F. Pharmacologic treatment of status epilepticus. Expert Opin Pharmacother 2016; 17: 513–34. [DOI] [PubMed] [Google Scholar]
- 8.Dalziel SR, Borland ML, Furyk J, et al. Levetiracetam versus phenytoin for second-line treatment of convulsive status epilepticus in children (ConSEPT): an open-label, multicentre, randomised controlled trial. Lancet 2019; 393: 2135–45. [DOI] [PubMed] [Google Scholar]
- 9.Lyttle MD, Rainford NEA, Gamble C, et al. Levetiracetam versus phenytoin for second-line treatment of paediatric convulsive status epilepticus (EcLiPSE): a multicentre, open-label, randomised trial. Lancet 2019; 393: 2125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trinka E, Kälviäinen R. 25 years of advances in the definition, classification and treatment of status epilepticus. Seizure 2017; 44: 65–73. [DOI] [PubMed] [Google Scholar]
- 11.Gaínza-Lein M, Sánchez Fernández I, Jackson M, et al. Association of time to treatment with short-term outcomes for pediatric patients with refractory convulsive status epilepticus. JAMA Neurol 2018; 75: 410–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kapur J, Elm J, Chamberlain JM, et al. Randomized trial of three anticonvulsant medications for status epilepticus. N Engl J Med 2019; 381: 2103–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.CFR-code of federal regulations title 21. 50.24. Silver Spring, MD: US Food and Drug Administration, 2019. https://www.accessdata.fda.gov/scripts/cdrh/cfdocs/cfcfr/cfrsearch.cfm?fr=50.24 (accessed April 21, 2019). [Google Scholar]
- 14.Connor JT, Elm JJ, Broglio KR. Bayesian adaptive trials offer advantages in comparative effectiveness trials: an example in status epilepticus. J Clin Epidemiol 2013; 66 (suppl): S130–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.