

Abstract
The historical view of β-lactams as ineffective antimycobacterials has given way to growing interest in the activity of this class against Mycobacterium tuberculosis (Mtb) in the presence of a β-lactamase inhibitor. However, most antimycobacterial β-lactams kill Mtb only or best when the bacilli are replicating. Here, a screen of 1904 β-lactams led to the identification of cephalosporins substituted with a pyrithione moiety at C3’ that are active against Mtb under both replicating and nonreplicating conditions, neither activity requiring a β-lactamase inhibitor. Studies showed that activity against nonreplicating Mtb required the in situ release of the pyrithione, independent of the known class A β-lactamase, BlaC. In contrast, replicating Mtb could be killed both by released pyrithione and by the parent β-lactam. Thus, the antimycobacterial activity of pyrithione-containing cephalosporins arises from two mechanisms that kill mycobacteria in different metabolic states.
Keywords: Mycobacterium, tuberculosis, cephalosporin, pyrithione, β-lactamase, antimycobacterial
Graphical Abstract
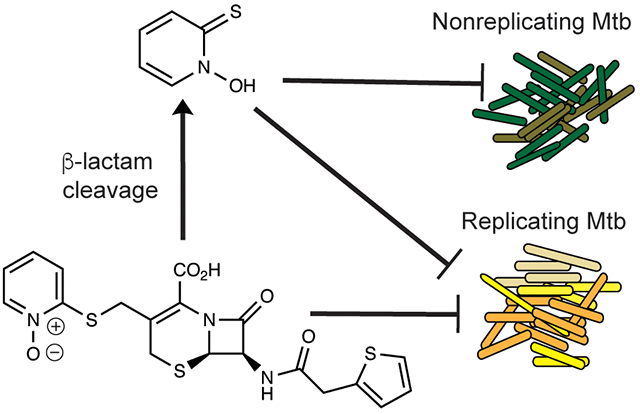
Mycobacterium tuberculosis (Mtb) is a leading infectious cause of death in the world.1 Active tuberculosis (TB) is characterized by heterogeneous populations of mycobacteria in diverse microenvironments, ranging from cavities, necrotic granulomas, non-necrotic granulomas, and pneumonitic infiltrates to macrophages in otherwise healthy-appearing tissues.2 In a given subject at a given time, some Mtb bacilli may be replicating and other bacilli may be replicating slowly or not at all. Non- or slowly replicating bacteria display phenotypic tolerance to most antibiotics selected for their ability to kill replicating bacteria.3–5
The cure of drug-sensitive TB involves prolonged treatment with isoniazid, rifampicin, pyrazinamide, and ethambutol. This may be necessary both to prevent selection for heritable antibiotic resistance and to overcome phenotypic tolerance.6 Except for pyrazinamide, known antimycobacterials are most effective on replicating cells. In contrast, Mtb becomes phenotypically tolerant toward most TB drugs when subjected to hypoxia, nutrient starvation, acidity, reactive nitrogen intermediates, or combinations of these stresses.7–9 Given the rising global burden of TB resistance to front-line drugs and the toxicity, cost, and limited efficacy of second-line agents, a major goal for TB drug development is to find safe compounds that inhibit new targets in Mtb, thereby avoiding pre-existent resistance, and that kill Mtb in diverse metabolic and replicative states, including states associated with phenotypic tolerance to other agents.
β-Lactams are among the oldest antibiotics and have been so pervasive in infectious disease treatment that their historical absence from TB therapy is striking. Classic β-lactam classes have been considered ineffective against Mtb due to natural resistance of its predominant peptidoglycan transpeptidases, efflux pumps, production of a class A β-lactamase (BlaC), and limited permeability of its thick, waxy cell wall.10–13 The best-known targets of β-lactams are penicillin-binding proteins (PBPs) that catalyze the final D,D-transpeptidation steps in peptidoglycan biosynthesis. However, mycobacterial peptidoglycan formation relies largely on L,D-transpeptidases that are structurally distinct from PBPs,14,15 catalyze transpeptidation via an active site cysteine rather than serine,16 and are resistant to most β-lactams.17
In 1983, Cynamon et al. challenged the view that β-lactams could not be used to kill Mtb by demonstrating the antimycobacterial activity of amoxicillin combined with the β-lactamase inhibitor clavulanate.18 More recently, a clinical trial indicated strong early bactericidal activity of meropenem in combination with amoxicillin and clavulanate.19 Most such studies have been carried out on replicating Mtb. Howeer, Hugonnet et al. reported a ca. 1 order of magnitude reduction in colony-forming units (CFUs) after a 14-day exposure of hypoxic Mtb to meropenem plus clavulanate.20 In 2016, using a combination of hypoxia, mild acidity, a flux of reactive nitrogen species, and a fatty acid carbon source to simulate conditions in the host and to impose nonreplication,21 we reported a new class of cephalosporins that cause multilog killing of Mtb but only when the Mtb is not replicating.22 The above results inspired us to seek β-lactams that were more broadly active against Mtb. Herein, we report the discovery of cephalosporins that kill both replicating and nonreplicating Mtb without dependence on clavulanate.
■ RESULTS AND DISCUSSION
High Throughput Screen for β-Lactams Active against Replicating and Nonreplicating Mtb.
Working under the auspices of the Tres Cantos Open Lab Foundation at GlaxoSmithKline’s (GSK’s) Tres Cantos Campus, we selected for testing 1904 compounds representative of the >14000 β-lactams in the GSK collection. This focused set included ca. 1330 cephalosporins, 370 monobactams, 150 penems, and 30 carbapenems. Seeking compounds that would be active under both replicating (R) and nonreplicating (NR) conditions, we tested the β-lactams against Mtb at 100 and 10 μM without a β-lactamase inhibitor under standard R culture conditions and three NR conditions that combined hypoxia, mild acidity, and a fatty acid carbon source. In two of the NR conditions, we included a flux of reactive nitrogen species generated from nitrite at the low pH.4,23 Because the extent of killing by β-lactams is often inversely correlated with the initial bacterial concentration,24–26 we tested Mtb at a low initial inoculum (OD580 of 0.01, where OD is optical density) in three of the four conditions and one NR condition at an OD580 of 0.1 (Figure 1A and Table S1).
Figure 1.
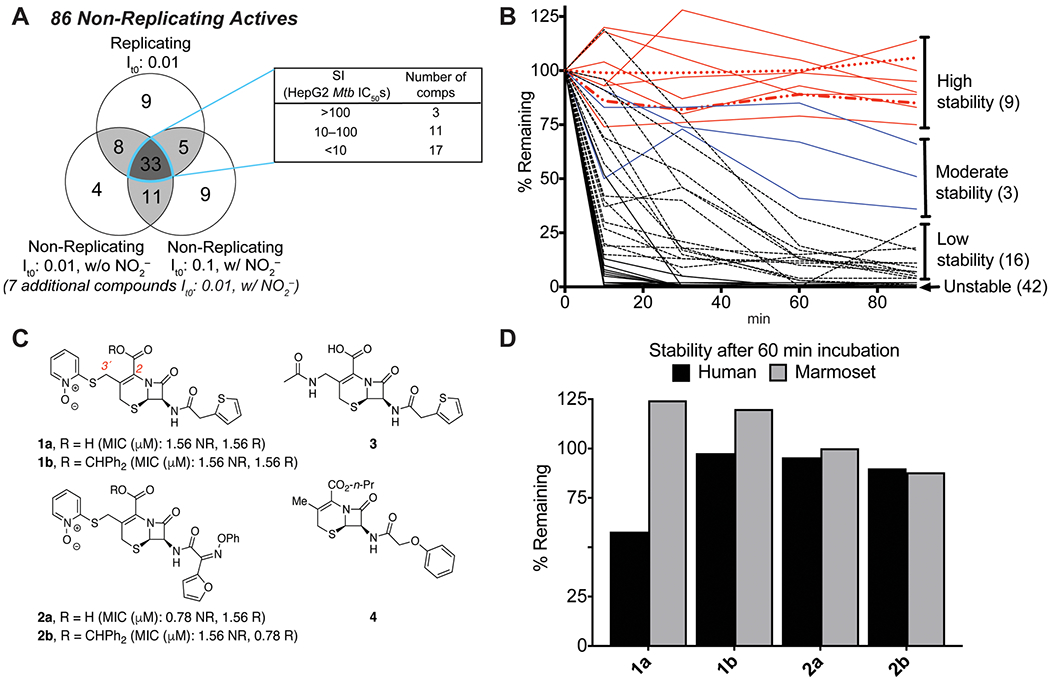
A whole cell screen resulted in the identification of 86 β-lactams active under NR conditions. (A) Analysis of Table S1 according to compound activity under replicating (R) or nonreplicating (NR) conditions with or without 0.5 mM NaNO2 and with a starting inoculum of 0.1 or 0.01. Seven compounds were active only under NR conditions with NaNO2 and low inoculum. (B) Stability of 70 out of the 86 active compounds in mouse plasma. Compound 1a is represented as a broken red line (− .. −); compound 3 is represented as a dotted red line (…). This large-scale experiment was conducted once. (C) Structure and MIC90 values of selected compounds. (D) Stability in human or marmoset plasma after a 60 min incubation at 37 °C of the dual-active compounds selected for further studies.
After eliminating compounds that were only active under R conditions, 86 compounds remained that had an IC50 < 100 μM in one or more of the three NR conditions, and 55 of these were also active against R cells (Figure 1A and Table S1). Of the 1904 β-lactams tested, 4.1% were active in at least two conditions tested, a high hit rate compared to the <1.5% hit rates reported in other whole cell Mtb screens (and noteworthy given the above-noted history of β-lactams in the Mtb area).23,27 Of these dual-active compounds, 33 were active against NR cells whether or not nitrite was included in the medium and whether or not the inoculum was high or low. Fourteen of these 33 most broadly active compounds had a selectivity index (SI) ≥ 10 when their cytotoxicity was tested against HepG2 human hepatoma cells (Figure 1A, inset table). Unfortunately, two of the broadly active compounds were not available for HepG2 assays, and the SI could not be determined. We tested the stability of 70 of the 86 active compounds in mouse plasma and eliminated 58 because of their plasma instability (Figure 1B).
Of the 9 dual-active compounds with high stability in mouse plasma, five were selected for the present study. The previously reported compound 1a28 was active under all four conditions tested. Three other compounds were structurally related to 1a and shared its activity profile, while a representative inactive compound, 3, was selected for comparison purposes. All four actives contained a pyrithione (PYR) heterocycle attached at C3’ (Figure 1C). PYR, derived from the natural product aspergillic acid, has a long history in medicinal chemistry, having been reported to have antimicrobial activity against A. niger, C. neoformans, M. bovis Bacille Calmette-Guèrin (BCG), S. aureus, and K. pneumoniae, among other fungi and bacteria.29,30 It has recently been studied, as the copper complex, in Mtb.31
We also noted that compounds 1a and 2a bear a carboxylic acid at C2, as is typical of antibacterial cephalosporins, while 1b and 2b have a diphenylmethane ester at this position. These β-lactams were stable in marmoset and human plasma (Figure 1D), although compound 1a was slightly less stable in human plasma. All four compounds retained activity over at least 72 h at 37 °C in both acidified nitrite-containing NR or R media (data shown for 2a only; Figure S1A,B).
The cephalosporins were subjected to further anti-Mtb characterization under NR conditions that included nitrite and used an initial inoculum of OD580 of 0.01. Activity was not limited to growth inhibition but represented cidality. After only 7 days of exposure at 1.6 μM, 2a and 2b reduced the CFU of R Mtb by 5 orders of magnitude and of NR Mtb by >3 orders of magnitude (Figure 2A). Compound 1b required a higher concentration, 6.26 μM, to reduce the CFU of both R and NR Mtb by 5 orders of magnitude (i.e., below the limit of detection) (Figure 2B).
Figure 2.
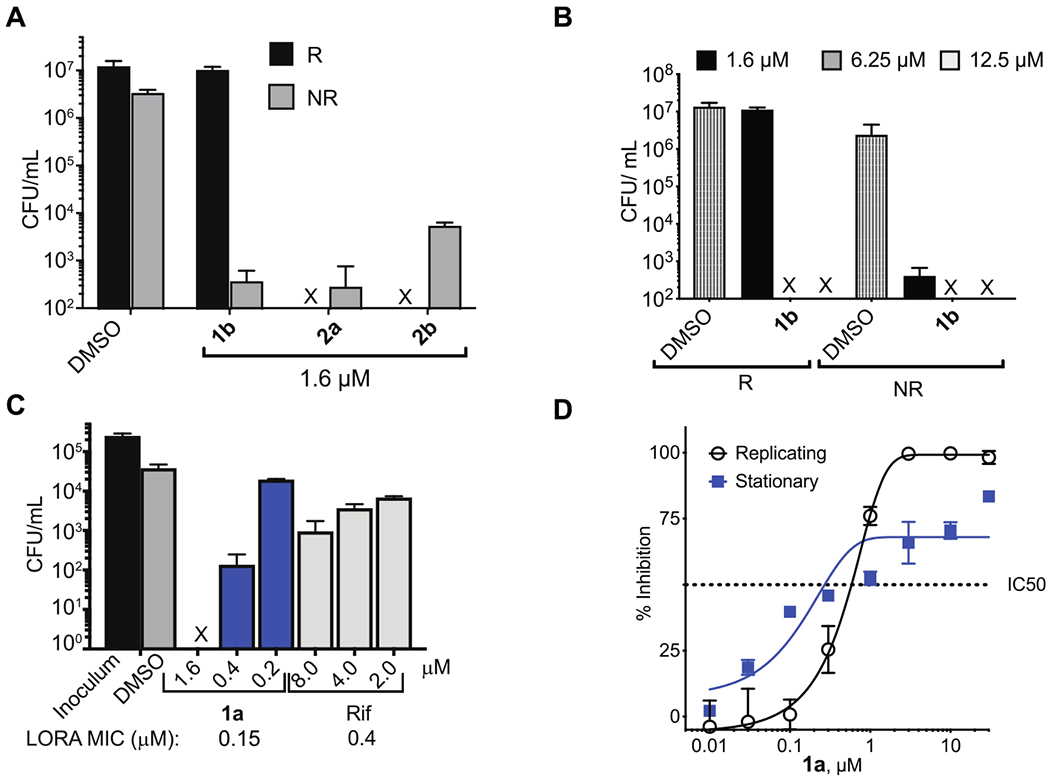
Activity of PYR-containing cephalosporins. (A) Quantification of the change in CFU/mL of 1b, 2a, and 2b after 7 days of drug exposure. “X” indicates the CFU was below the limit of detection. (B) Concentration dependence of 1b activity, indicating its cidality at higher concentrations. (C, D) NR activity of 1a as assessed by the LORA and BCG stationary phase assays, respectively. Graphs represent the mean ± SD of triplicate samples. Experiments were conducted ≥2 times.
Cephalosporin 1a was assessed in three different models of nonreplication. The low oxygen recovery assay (LORA)32 assesses activity of bacteria placed in a state of phenotypic tolerance to most antimycobacterial agents by hypoxic conditions. In this assay, 1a reduced the Mtb CFU/mL by at least 4 orders of magnitude at 1.6 μM, retaining its potency in contrast to the rifampicin control, which lost activity under similar conditions (Figure 2C). In the M. bovis BCG stationary phase model, cells were grown in nutrient rich media for 40 days. Stationary phase BCG cells were frozen for storage and then resuspended in PBS to test for compound sensitivity. Here, 1a partially lost activity (but not completely) compared to R cells (Figure 2D). Additional experiments confirmed that NR activity was not due to carryover of compound from the R to the NR phase of the assay. This was accomplished using the charcoal-agar resazurin assay (CARA),7 which controls for carryover of compound by absorbing residual compound found in the NR media. In this assay, the action of 1a and 2a was nitrite independent (Figure S2A,B), unlike a representative NR-selective cephalosporin 422 (Figure S2C), and was not due to the cephalosporins inhibiting Mtb’s ability to transition from R to NR conditions (Figure S2D).
Role and Mechanisms of PYR Release.
An established feature of cephalosporins is their ability to release a C3’ nucleofuge upon opening of the β-lactam ring (Figure 3A). First noted by Abraham and co-workers in 196533–35 and mechanistically examined biochemically36–38 and theoretically39 some decades later,33,36–38 this concept has been extensively applied to anti-infective agent design,42–51 including recently against Mtb.52,53 This aspect of cephalosporin chemistry has also been leveraged to deliver other classes of bioactive molecules, such as antibody conjugates against cancer54–59 or cephalosporins bearing chromophores for β-lactamase detection.60–63 Most relevant to the present work is the incorporation of a PYR moiety to cephalosporin derivatives by O’Callaghan and co-workers40 and Greenwood and O’Grady41 in 1976. To investigate the contribution of PYR to the antimycobacterial activity of the cephalosporins, we tested the activity of PYR sodium salt as a function of concentration and time. We found that PYR was equipotent on a molar basis with cephalosporin 1a under both R and NR conditions and its activity did not depend on the inclusion of nitrite in the assay (Figure 3B–D). In terms of the rate of killing, at 2.5 μM (1.5× the CARAMBC for R Mtb, where MBC is minimum bactericidal concentration), PYR reduced the CFU of R Mtb by >5 orders of magnitude within 24 h. In contrast, an equimolar concentration of 1a took 72 h to reduce the CFU/mL of R Mtb to the same extent (Figure 3E). However, under NR conditions, both compounds at 2.5 μM killed Mtb at the same rate and to the same extent (Figure 3F). For comparison, killing was much slower when rifampin was tested against R Mtb (Figure 3E) or 4 was tested against NR Mtb at their 1.5× CARAMBC concentrations (Figure 3F).
Figure 3.
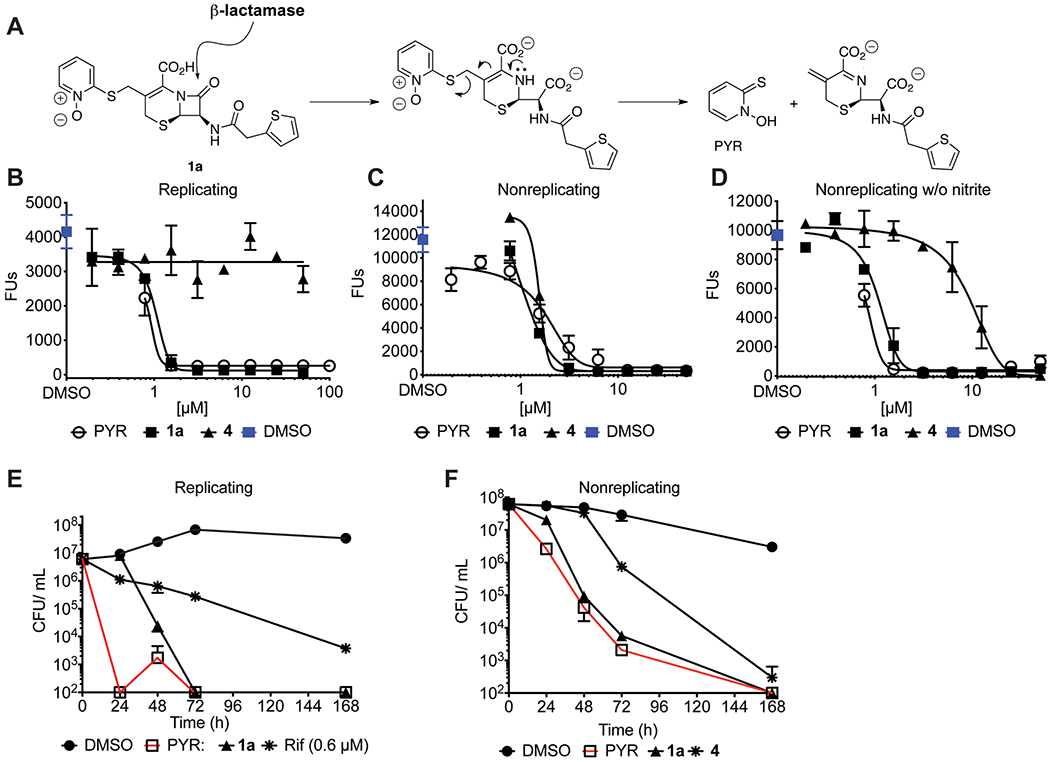
Studies of the mode of action of the PYR containing-cephalosporins. (A) Hypothetical release of PYR from the cephalosporin backbone following β-lactamase cleavage. (B−D) CARAs were used to compare the activity profile of 1a, 4, and PYR under R conditions and NR conditions with and without nitrite. (E, F) CFU/mL reduction over time when cells are exposed to 1.5× CARAMBC concentrations of the 1a and PYR sodium salt under R and NR conditions as compared to rifampicin under R conditions or 4 under NR conditions. Results are means ± SD of triplicates. Experiments were conducted at least twice.
The antibacterial activity of PYR has been proposed to arise from its ability to chelate metals64,65 or disrupt disulfide bonds in certain proteins.66,67 For PYR to act by such mechanisms, it must first be liberated from the cephalosporin molecule.52 The foregoing results suggested that PYR release could account quantitatively for the activity of the dual-active cephalosporins under both R and NR conditions but could account kinetically for killing only if PYR is released slowly under R conditions and quickly under NR conditions. In previous studies, O’Callaghan et al. and Zaengle-Barone et al. ascribed the activity of a pyrithione-containing cephalosporin against E. coli in part or completely to the expulsion and subsequent activity of the pyrithione40,65
A series of analogues was synthesized to address the necessity of PYR ejection for activity (Figure 4). Compound 5 was previously shown to lack activity against Mtb,22 but the analogue containing PYR at C3’ is active under both R and NR conditions (Figure 4A). This is not surprising insofar as 6 differs from 1a by only the replacement of a side-chain thiophene with a phenyl group. Compound 6 was also recently reported by Zaengle-Barone et al. to be potently active against E. coli strains, particularly when the organisms had low levels of β-lactamases.65 To test the role of PYR release, we prepared and tested the carbon-linked analogue 7, which was, on its own, inactive against Mtb under both R and NR conditions. However, with the addition of clavulanate, ca. 99% of the Mtb was killed in R, but not NR, assays. Similarly, meropenem in combination with clavulanic acid has been reported to kill 2 orders of magnitude of replicating Mtb cells.68 This highlights the importance of activity of the parent cephalosporin backbone in situations where the bacterial expression of β-lactamase is low. Additional evidence was obtained by comparing the activity of the known40 compound 8 with its Δ3,4 isomer 9, which was obtained as a byproduct of deprotecting the N-Boc precursor of 8 (compound 9 is a single isomer of unknown stereochemistry at C2; see the Supporting Information for details).69 While the former can easily open and eject a C3’ leaving group analogously to the mechanism in Figure 3A, 9 cannot because the nitrogen atom revealed by the β-lactam opening is insulated from the leaving thiol (Figure 4C). The addition of 25 μM clavulanate partially restored activity to 7 under R but not NR conditions (Figure 4D).
Figure 4.
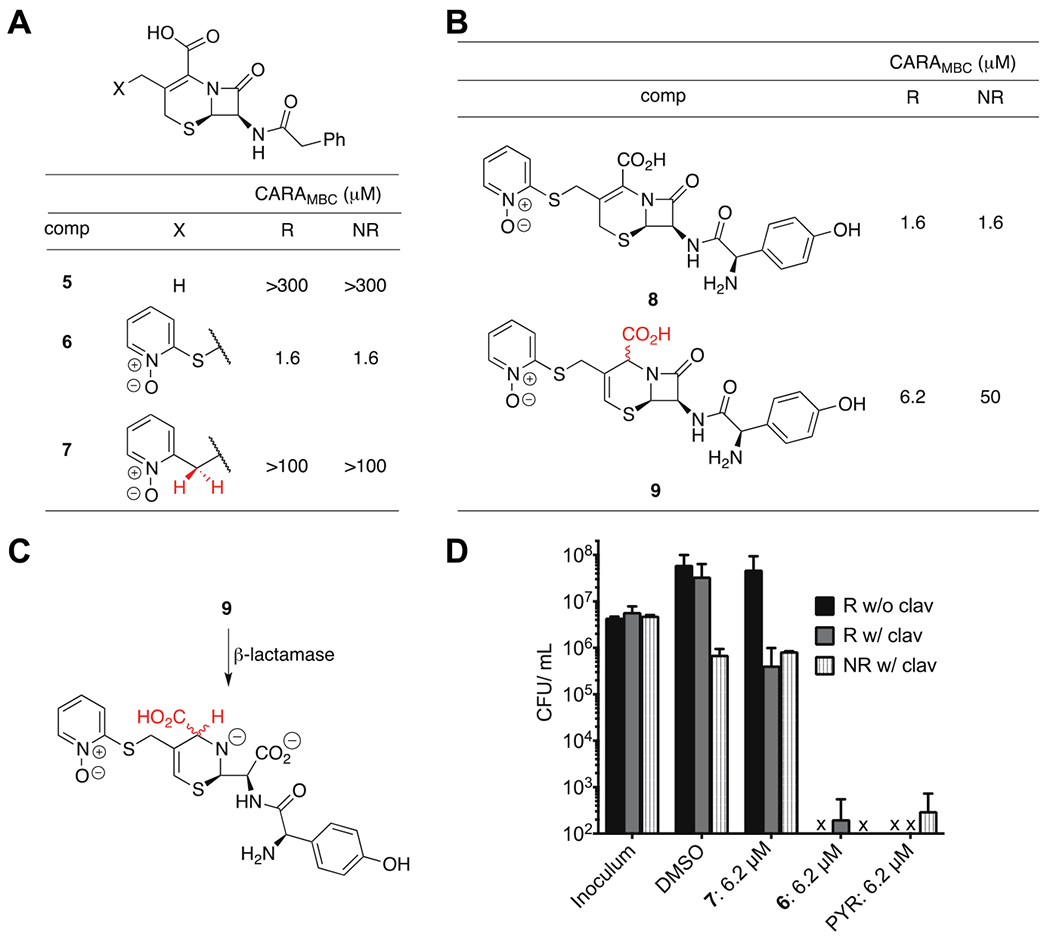
(A) Comparison of a compound able to release PYR (6) with analogues lacking a PYR moiety (5 and 7). Activity was measured as CARAMBC. (B) Comparison of a compound able to release PYR (8) with an analogue unable to do so (9). (C) Ring-opened form of compound 9. (D) CFU/mL reduction after 7 days of compound 6 and 7 exposure under R conditions without 25 μM clavulanate (black bar) or with clavulanate (gray bar) or NR conditions with clavulanate (striped bar). CARAMBC represents the concentrations at which compounds reduced the fluorescent signal to or below 1% of the signal seen with 1% DMSO as a control. CFU/mL reduction experiments were conducted in triplicate, and bars represent mean ± SD.
Taken together, the above experiments suggest substantial activity against Mtb under R conditions in the presence of clavulanate independent of PYR release. Although this may represent a classic β-lactam mechanism entailing the inhibition of the final transpeptidation step in peptidoglycan synthesis, we have not yet done any experiments that inform this point and alternative targets are possible. In the case of compound 9, we also cannot exclude the possibility that the remaining activity arises from isomerization to 8 in the course of the experiment. However, since this is not possible for carbon-linked analogue 7, which also retains clavulanate-dependent activity against R Mtb, we conclude that direct action of the β-lactam is likely a contributor to anti-Mtb activity in R organisms.
As noted earlier, the PYR-containing cephalosporins retained activity after incubating in cell-free medium. However, this could be due to either survival of the parent β-lactam over the time course of the experiment or persistence of released PYR in the media. To address this, we confirmed that PYR is not released from 1a or a simple alkylated PYR analogue (2-(carboxymethylthio)pyridine-N-oxide, 10) in cell-free medium using an assay that detects the formation of an Fe3+–PYR complex visually or by absorbance at 600 nm (Figure S3).70 In contrast, we detected PYR in the medium of R or NR Mtb exposed to PYR-releasing 1a or 6 but not 10 or 7 (Figure 5). The amount of PYR detected in the medium of Mtb cultures was about one-half the starting concentration of the cephalosporin under R conditions and one-quarter under NR conditions. However, while free PYR was stable over at least 24 h in the medium used for R conditions, it largely disappeared over 48 h from the medium used for NR conditions. We did not determine the fate of PYR in these experiments; it might disappear due to oxidative dimerization or conjugation. The proportion of PYR detected by the assay is the proportion remaining in the media or released by the bacterial cell, and thus, the amount detected over longer time points does not reflect PYR taken up by cells, released inside the cells, or degraded.
Figure 5.
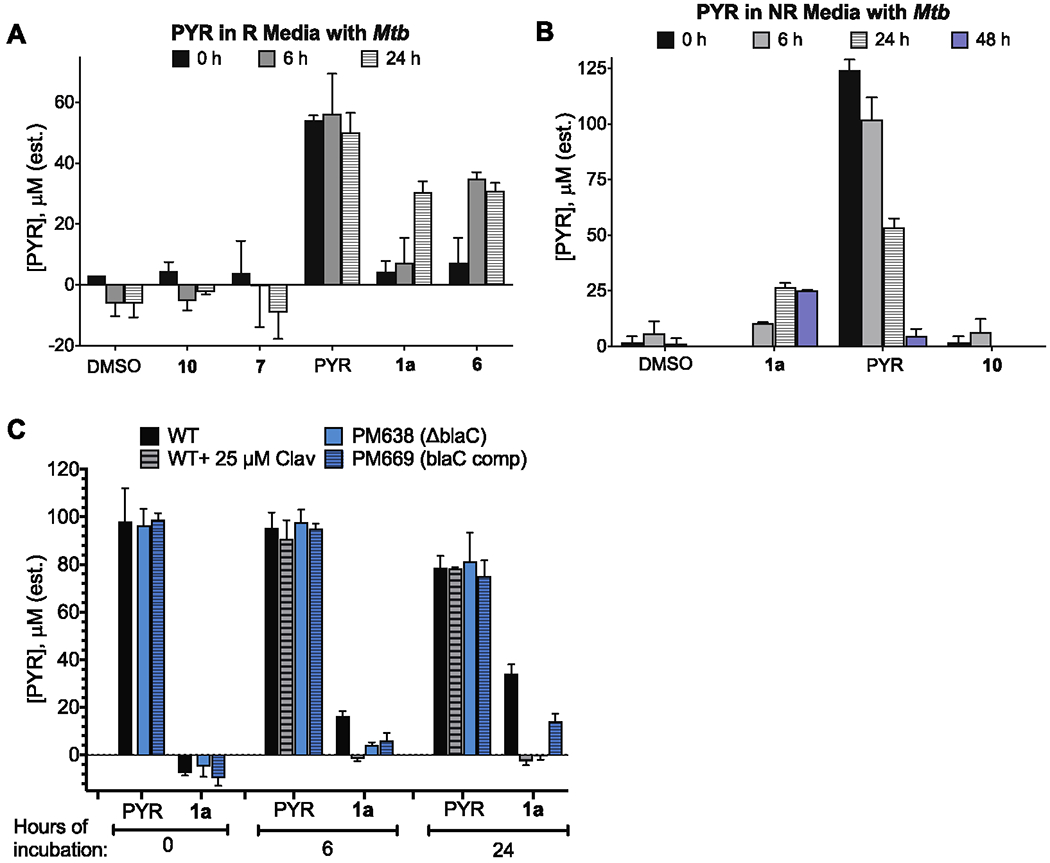
PYR found in media after incubation with whole cells. (A) Estimated levels of PYR in R medium after 6 or 24 h in the presence of an estimated starting 106 CFU and an initial compound concentration of 50 μM. (B) Estimated levels of PYR in NR medium after 6, 24, and 48 h in the presence of an estimated starting 106 CFU and an initial compound concentration of 100 μM. (C) Estimated levels of PYR release over 24 h in R conditions by the ΔblaC strain or complemented strain or WT Mtb cells alone or in the presence of 25 μM clavulanic acid. Results are means ± SD of triplicates.
BlaC, a chromosomally encoded class A serine β-lactamase, is Mtb’s only functionally defined β-lactamase.71 In previous work, it was shown that deletion of the gene encoding BlaC and/or addition of clavulanate, a BlaC inhibitor, rendered Mtb susceptible to the cephalosporin cefoxitin, multiple penicillins such as piperacillin and mezlocillin, and the carbapenems imipenem72 and meropenem.20 Other studies showed that Mtb can use BlaC to release a fluorophore or quencher from the C3’ of a cephalosporin within Mtb.73,74 In accordance with these studies, the levels of PYR in the media were undetectable when WT cells were coincubated with 1a and clavulanic acid or when BlaC was knocked out (Figure 5C). We verified the ability of BlaC to release PYR from the cephalosporin backbone by purifying a truncated, soluble form of BlaC as described.71 Within 10 min of incubation of 100 μM β-lactam with 1 μM BlaC at 37 °C, PYR was quantitatively released from the cephalosporins 1a, 6, and 8 but, as expected, not from 7 or 9 (Figure 6A). When 1a and cephalothin, which bears an acetoxy group at C3’ (see Table 1 for the structure), were incubated with BlaC, the UV-visible absorption spectrum of the solution containing 1a converted to that of PYR while the spectrum of that containing cephalothin did not change (Figure 6B).
Figure 6.
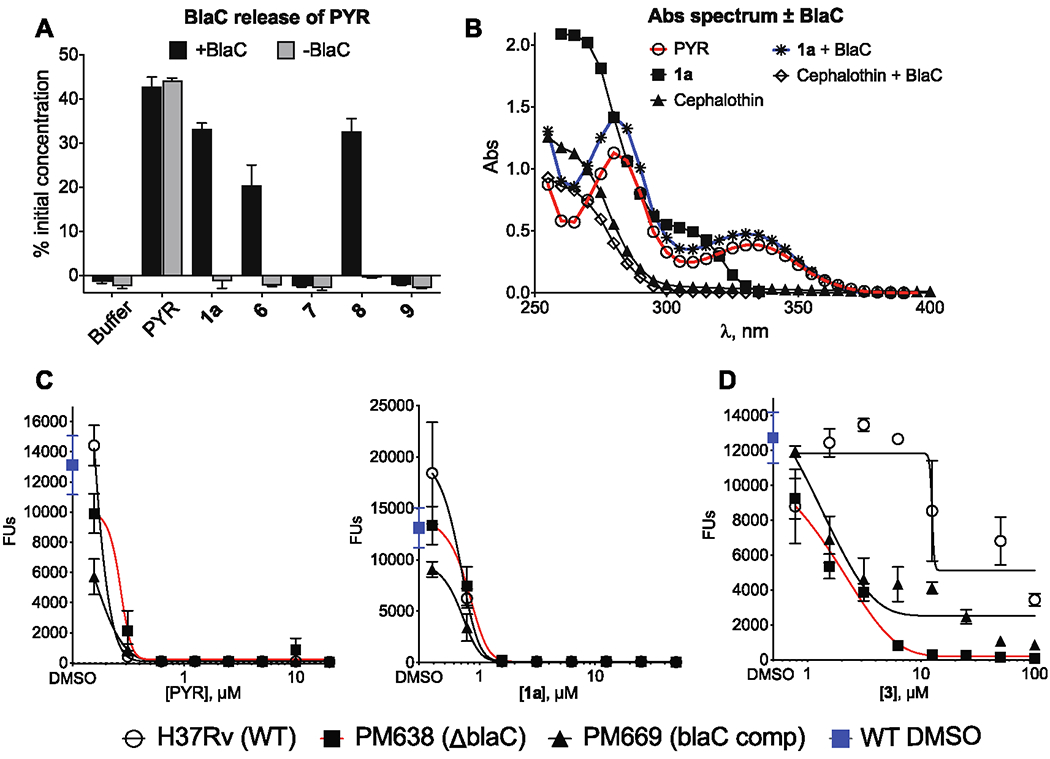
Role of BlaC in PYR release. (A) Estimated levels of PYR recovered after incubating with 1 μM BlaC for 10 min represented as the percent of the initial concentration of parent compound. (B) The absorption spectrum of 100 μM of cephalothin and 1a before and after 10 min of incubation with 1 μM BlaC. (C) The efficacy of PYR and 1a in M. tuberculosis WT (open circles), BlaC KO (PM638, squares), and complemented strain (PM669, triangles). (D) The efficacy of clavulanate dependent 3 in M. tuberculosis WT (open circles), BlaC KO (PM638, squares), and complemented strain (PM669, triangles). Results are means ± SD of triplicates
Table 1.
Clavulanate Dependence of Selected Cephalosporins and PYR
| MBC(μM) |
|||||
|---|---|---|---|---|---|
| entry | compound | R CARA | R CARA + 25 μM Clav | NR CARA | NR CARA + 25 μM Clav |
| 1 | 1a | 1.6 | 1.6 | 3.1 | 1.6 |
| 2 | 3 | 100 | 3.1 | >100 | >100 |
| 3 | 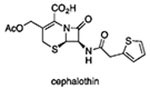 |
>100 | 12.5 | >100 | >100 |
| 4 | 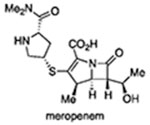 |
12.5 | 3.1 | >100 | >100 |
| 5 | PYR | 1.25 | 1.25 | 1.25 | 1.25 |
Thus, both Mtb and recombinant BlaC could release PYR from a PYR-containing cephalosporin. However, the mycobactericidal activity of these PYR-containing cephalosporins was unaffected by clavulanate at concentrations that inhibit BlaC within Mtb (Table 1). Moreover, the Mtb ΔblaC strain showed no change in 1a’s CARAMBC under R conditions (Figure 6C) and, at the CFU/mL level, showed a greater sensitivity than WT to 1a (Figure S4A). No change in 1a activity was also noted under NR conditions (Figure S4B). In contrast, 3, the antimycobacterial activity of which is clavulanate dependent, was 10-fold more potent against the Mtb ΔblaC strain than against WT Mtb (Figure 6D) but had no gain in activity under NR conditions (Figure S4B). Complementation of the ΔblaC strain with the wild-type allele partly restored Mtb’s resistance to 3 in the absence of clavulanate (Figure 6D). Taken together, these observations suggest that the antimycobacterial activity of the compounds described here does not rely on either the action or inhibition of BlaC, Mtb’s functionally defined β-lactamase, despite the observation that the PYR–cephalosporins can be a substrate for BlaC. These results indicate that PYR can be released from the cephalosporins by an unknown, clavulanate-resistant, non-BlaC β-lactamase, an alternative covalent reaction with unknown cellular targets, or a nonenzymatic hydrolysis reaction. Voladri et al. presented evidence for the existence of a non-BlaC mycobacterial cephalosporinase,75 and at least 12 proteins in Mtb besides BlaC are annotated to have sequence similarity to β-lactamases. Indeed, in the nonpathogenic Mycobacterium smegmatis, an appreciable loss of 1a activity was detected in the double β-lactamases knockout but not the single knockout (Figure S4C). An alternative explanation would be that these cephalosporins have a second, non-PYR-dependent mechanism of action, most likely related to “normal” β-lactam reactivity as suggested by the activity profile of compound 7.
Spectrum of Activity, Structure-Activity, and Preliminary Pharmacokinetics Studies.
The hit cephalosporin 1a was tested against 11 clinical isolates of Mtb. The MIC90’s (where MIC is minimum inhibitory concentration) ranged from 0.8 to 1.9 μM whether the isolates were drug-sensitive, monoresistant, or multidrug resistant (Table S2). In contrast, when selected PYR–cephalosporins were tested for activity against ESKAPE pathogens, methicillin-sensitive S. aureus was sensitive to the cephalosporins (MIC90 range: 0.2–0.78 μM), including the carbon-linked cephalosporin 7, but not to PYR (MIC90 25 μM) (Table S3) indicating that this activity is most likely due to the cephalosporin backbone rather than the PYR. The PYR-bearing cephalosporins (and PYR) were poorly active against E. faecium, K. pneumoniae, E. cloacae, and P. aeruginosa (MIC90 12.5->100 μM, Table S3). Consistent with these observations, we have now shown that outfitting selected cephalosporins with PYR payloads at C3’ can confer relatively narrow bactericidal activity against Mtb. The released PYR may be solely responsible for bactericidal activity, although we cannot exclude other β-lactam mechanisms at this stage.
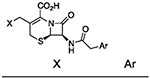
We briefly examined the structure–activity relationships of dual-active cephalosporins. As expected from the evidence that the PYR contributes to the activity of these cephalosporins against Mtb, replacement of the PYR moiety with alternative C3’ moieties abolished activity against Mtb (Table 2, entries 1–3). In contrast, placing a PYR onto a compound previously shown22 to have only activity against NR Mtb conferred dual activity (entry 4). However, like other compounds containing an ester in that series, this material was unstable in mouse plasma. Two analogues containing substitution on C6 of the PYR were also potently active, especially in NR assays (entries 5 and 6). Finally, selected analogues were found to be nontoxic to mammalian cells (Table S4).
Table 2.
Activity of Additional Analogues
| MIC90 (μM) | |||||
|---|---|---|---|---|---|
| entry | compound | R | NR | ||
 |
|||||
| 1 | 11 | SPh | Ph | 74.0 | >100 |
| 2 | 12 | 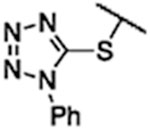 |
Ph | >100 | >100 |
| 3 | 13 | 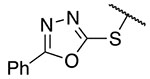 |
Ph | >100 | >100 |
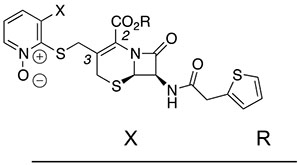 |
|||||
| 4 | 14 | H | n-Pr | 1.14 | 0.57 |
| 5 | 15 | Me | H | 0.82 | 0.059 |
| 6 | 16 | Br | H | 1.27 | 0.042 |
Although compound 1a was stable in mouse and primate plasma (Figure 1D) and in mouse and human liver microsomes (Figure S5A), orally administered 1a was cleared rapidly from the blood of mice after oral or parenteral administration (Figure S6A). In contrast, compound 8, which was reported 40 years ago,76 was both stable to microsomes of various animals and measurable in mouse plasma for more than 5 h after an oral dose of 20 mg per kg or even longer when administered by intraperitoneal (IP) dosing (Figure S6B). The bioavailability of 8 was 5.2% after oral gavage and 31.8% after intraperitoneal injection. At 150 mg/kg, compound 8 was well tolerated by mice for at least 24 h after oral administration.
■ CONCLUSIONS
The established use of cephalosporins to release a PYR payload upon β-lactam cleavage is here shown to lead to agents active against Mtb in culture under both R and NR conditions. Although the spectrum of the reported compounds is relatively narrow, some of the agents reported were also active against methicillin-sensitive S. aureus. Mechanistic studies, which include time-course measurements of PYR release and control agents bearing a nonreleasable pyridine N-oxide, suggest that the activity of these compounds arises both from PYR release, although additional β-lactam-dependent mechanisms cannot be excluded at this time. The mechanism of PYR-mediated killing of Mtb is as yet unknown. Metal chelation, of copper in particular, and nonspecific electrophilic reactions with bacterial nucleophiles have been proposed in studies in refs 31 and 77 and other bacteria.64,65 A recent study, using a close analogue of PYR, found an upregulation of genes associated with metal toxicity in Mtb cells exposed for 2 h to the PYR analogue.31 Furthermore, the metal chelator EDTA partially reduced the efficacy of free PYR against C. albicans but not against K. pneumonia.78 No direct mechanistic inference can be drawn from the observations that PYR led to a marked drop in ATP levels in fungi66 and bacteria79 and that PYR salts induced membrane depolarization in Neurospora crassa.80
In conclusion, a hybrid molecule of this nature could open a new chemical space in antibiotic development by allowing the use of moieties that would otherwise be dismissed as potentially toxic on their own, provided that the β-lactam targets the warhead to bacterial cells.
■ EXPERIMENTAL SECTION
Mycobacterial Strains and Culture Methods.
Wild-type Mtb strain H37Rv (Mtb, ATCC 27294) was grown under standard replicating (R) conditions of Middlebrook 7H9 broth (supplemented with 10% oleic acid, dextrose, and catalase, OADC) (BD Difco, USA), 0.02% tyloxapol (Sigma), 5% CO2, and 20% O2. M. tuberculosis strains PM638 (ΔblaC) and PM669 (PM638 attB::blaC) were grown as previously described.42 Colony forming units (CFUs) were plated on 7H11 (BD Difco, NJ, USA) agar supplemented with 10% OADC. Minimum inhibitory concentrations (MIC90) were all conducted in tissue culture treated flat bottom 96-well plates (Corning, USA). The MIC90 value was determined as the concentration of compound that inhibited at least 90% of the bacterial growth measure in the DMSO treated wells. Growth was measured by the optical density at 580 nm. Our studies predominantly used the 4-stress model of nonreplication conditions. Other nonreplicating conditions described in our studies include the BCG stationary phase model and the low oxygen recovery model. The BCG stationary phase model involved Mycobacterium bovis BCG (IP173) grown for 40 days at 37 °C without agitation in Middlebrook 7H9 broth medium supplemented with 10% OADC and 0.05% Tween 80 (Sigma). Stocks were quantified and stored at −80 °C. On the day of the experiments, BCG stationary phase stock solutions were thawed in PBS without vortexing and the suspension was diluted and dispensed into the plates at a final concentration of 104 to 5 X 104 CFU per well. Compounds were then added. Plates were incubated at 37 °C and ambient oxygen levels. After 7 days, viability was determined using ATP quantification using BacTiter-Glo (Promega) as per the manufacturer’s recommendations. Plates were incubated for 10 min in the dark, and luminescence was read using Envision (PerkinElmer). The low oxygen recovery assay (LORA) was conducted as previously described32 in which a luxAB (luciferase) containing strain of Mtb H37Rv was diluted in Middlebrook 7H12 broth (Middlebrook 7H9 containing Casitone, palmitic acid, BSA, and catalase) and dispensed into 96-well plates containing antimicrobial agents. Microtiter plates were placed under anaerobic conditions consisting of oxygen levels of less than 0.16% and incubated for 10 days at 37 °C. Plates were transferred to replicating growing conditions, and CFU/mL were enumerated after a 28 h recovery period. LORA MIC90’s were determined after incubating the microtiter plates for 7 additional days under replicating condition. 100 μL of cultures was then used to measure luminescence, and MIC90 was determined as the lowest compound concentration inhibiting at least 90% of untreated control growth.
Compounds, Materials, and Synthesis.
Compounds for the high throughput screen were obtained from the GSK compound collection (Harlow, UK). Hit analogues 1 and 2 and all other compounds subjected to biological evaluation were synthesized in house and had measured purities of >90% (HPLC; see the Supporting Information for details and experimental procedures). Resazurin, PYR, and cephalothin were purchased from Sigma-Aldrich (MO, USA) while 10 (2-[(carboxymethyl)sulfanyl]pyridine-1-ium-1-olate) was purchased from Enamine (NJ, USA).
Small Molecule Whole Cell Screening.
Whole cell screening was conducted at the GSK Diseases of the Developing World campus in Tres Cantos, Spain. At the outset, 1904 compounds were selected on the basis of structural diversity and compound availability from the GSK β-lactam library. Structural families were clustered using the Sphere Exclusion method, and representative compounds from each family were selected on the basis of availability. The assays were conducted as described23 in which the compounds were dissolved in DMSO and dispensed onto 384-well plates. Test conditions followed those described21,23 with a few modifications. Briefly, virulent H37Rv Mtb cells were exposed to compounds under 4 conditions: replicating (R) and NR in the 4-stress model with the addition of 0.5 mM sodium nitrite or without the addition of sodium nitrite. R cells were diluted to an initial inoculum OD580 of 0.01 while NR cells were diluted to an OD580 of 0.01 or 0.1. R cells were incubated at 37 °C with 20% O2 and 5% CO2 in the presence of β-lactams at 10 or 100 μM (note: the MIC90’s of the clinical isolates shown in Table S2 were carried out in the absence of CO2). The OD580 of the plates of R cells were read after 7 days of compound exposure. NR cells were exposed to compounds at 37 °C for 3 days at 1% O2 and 5% CO2 followed by a 1:5 dilution in R medium. The cells were then allowed to recover for 5 days at 37 °C at 20% O2 and 5% CO2, and then, the OD580 was determined. Compounds active at 100 μM were confirmed with fresh stock in a full (100–0.4 μM) dose response curve. Percent growth inhibition data was analyzed using ActivityBase program, and IC50’s and IC90’s were calculated using nonlinear regression analysis. Initial cytotoxicity assays for establishing the SI were conducted at the GSK facility using library compounds and HepG2 cells in a 384-well plate format. Cytotoxicity assays for the de novo synthesized compounds were carried out at WCM using a similar protocol but in a 96-well format and two mammalian cell lines.
Preliminary Cidality Estimation Using Charcoal Agar Resazurin Assays (CARAs).
Minimum inhibitory concentrations (MIC90’s) for compounds progressed for further characterization were performed in a 96-well plate; for NR conditions, this MIC90 value was described as the two plate assay23 in which, after 7 days of compound exposure under NR conditions, 10 μL of cells from these assay plates was diluted 1:21 into R medium. With dual-active compounds, this NR MIC90 value can reflect the activity of the compound under NR conditions and the R activity of compound carried over in the 10 μL. Therefore, cidality was estimated using CARA plates. 96-well CARA plates were prepared, and assays were carried out as described.7,81 Briefly, 4 g/L of activated charcoal was added to 7H11 agar with glycerol, and 200 μL/well was dispensed into a 96-well plate. After mycobacterial cells were exposed to a dose range of compounds or vehicle control, 10 μL of cells was added to the surface of each well. The level of cidality was estimated by measuring the level of growth on each well. Growth was assessed after 10 days for R cells and 12–15 days for NR cells. Thus, to each well, we added 50 μL of phosphate buffered saline solution containing 50 μL of 0.02% tyloxapol and 0.01% resazurin in phosphate buffered saline. Plates were developed for 1 h at 37 °C, and the level of fluorescence was measured at an excitation of 530 nm and emission of 590 nm. The CARAMBC was measured as the compound concentration at which the fluorescence drops to <1% of the average DMSO value.
PYR in Vitro Detection Assay.
A colorimetric ferric nitrate method was used to estimate the concentration of PYR in solution as described40 with some modifications. In a 96-well plate, test solution (e.g., bacterial media) and 0.01 M ferric nitrate (dissolved in 0.1 N sulfuric acid) were mixed 1:1 and incubated with shaking for 5–10 min at room temperature. The absorbance of the solution was measured at 600 nm. PYR concentrations were determined using a linear standard curve of sodium PYR.
BlaC Purification and Activity Assay.
The pET28 vector containing the truncated form of the Mtb blaC gene was kindly shared by Dr. John Blanchard (Albert Einstein College of Medicine) and was expressed and purified in a similar fashion to the described protocol.10,71 After BL21 (DE3) E. coli cells were induced overnight with 0.5 mM IPTG, cells were lysed in 50 mM Tris (pH 8.0) containing 500 mM NaCl and 30 mM imidazole. The soluble extract was mixed with a Ni-NTA bead slurry (Qiagen) and incubated overnight at 4 °C. Beads were then washed twice with the lysis buffer (10× bed volume), and BlaC was eluted with lysis buffer containing 250 mM imidazole. After dialysis in tris buffered saline (TBS), BlaC was concentrated and gel filtration was performed using Superdex 16/200 (GE) to exclude aggregates. Activity assays were performed in 50 mM 2-(N-morpholino)ethanesulfonic acid (MES) buffer, pH 6.4, containing 20 μg/mL of BSA.
Mammalian Cell Toxicity Assay.
Compound toxicity of compounds synthesized in house was measured using HepG2, a human liver hepatocellular carcinoma cell line, and Vero cells, a green monkey kidney epithelial cell line. HepG2 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS), 1 mM pyruvate, 2 mM glutamine, 10 mM HEPES, and 1% nonessential amino acids. Vero cells were grown in DMEM containing 2% FBS, 1 mM pyruvate, 2 mM glutamine, and 10 mM HEPES. Using a TC-treated 96-well plate, 1 × 104 cells/well were seeded and incubated at 37 °C with 5% CO2 for 24 h before cells were washed with PBS and fresh medium containing compound was added. Cells were then incubated for an additional 2 days in the presence of each of the compounds. HepG2 cell viability was determined using CellTiterGlo (Promega). Vero cell viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) (Promega)/phenazine methosulfate (PMS) (Sigma) colorimetric assay. For the MTS/PMS assay, culture medium was removed and replaced with phenol-free DMEM culture medium. A fresh mixture of MTS and PMS was added and incubated for 2 h, and absorption was determined at 570 nm. An aliquot of the cell medium was used to estimate PYR levels using the PYR detection assay described above.
Antimicrobial Spectrum.
Staphylococcus aureus MSSA (ATCC25923), Staphylococcus aureus MRSA (ATCC43300), Klebsiella pneumoniae SHV-1 (ATCC700603), K. pneumoniae (ATCC35657), Enterobacter cloacae (ATCC13047), and Pseudomonas aeruginosa (ATCC27853) were grown in Mueller-Hinton medium (Sigma). Enterococcus faecium VSE (ATCC19434) and E. faecium VRE (ATCC51559) were grown in brain heart infusion media (Sigma). MIC90 values were determined using cells at an OD600 of 0.001 in a 96-well format in which drug was tested in 2-fold dilutions. OD600 of the cultures was read after 24 h of compound exposure. The MIC90 value was calculated as the concentration that inhibited at least 90% of growth of the DMSO treated cells. All MIC90 analyses were conducted in triplicate and reproduced in two to three independent experiments.
Plasma Stability.
The stability of 73 compounds from the screen was carried out using plasma from female CD-1 mice (Bioreclamation). Stability samples consisted of 5 μL of stock compound solution in 100% DMSO and 95 μL of plasma to a final 10 μg/mL concentration. The samples were incubated at 37 °C with shaking. Ten μL was removed at each time point and combined with 100 μL of 1:1 acetonitrile/methanol, 10 ng/mL of verapamil (Toronto Research Chemicals, Inc.), and 10 μL of acetonitrile/water. Samples were analyzed by LC-MS/MS using the Q-Extractive high-resolution mass spectrometer (Themo Fisher Scientific). Stability in plasma from other species was similarly carried out. Mouse, human, and nonhuman primate plasma stability for compound 4 was independently verified by Wuxi Biologics (Wuxi, China). Stability was calculated as the percent remaining of the parent compound compared to the initial concentration; otherwise, assays for each species were conducted once.
Microsomal Stability.
Liver microsomes from the indicated species (0.5 mg/mL) were suspended in 50 mM phosphate buffer at pH 7.4 containing 4 mM MgCl2 and 1 mM NADPH. Compounds were added to a final concentration of 0.5 μM, and the percent of parent compound remaining was monitored by LC-MS/MS.
In Vivo Pharmacokinetics in CD-1 Mice.
Initial PK data was gathered by administering a single dose of 1a or 8 to two mice per compound via oral gavage at 25 and 20.8 mg/kg, respectively, and plasma levels of parent compound were monitored with LC-MS/MS. Compounds were suspended in 5% N,N-dimethylacetamide/60% PEG300/35% dextrose in water. PK studies for compound 8 were repeated using additional mice and with some modifications. For further oral (po) and peritoneal (ip) studies, compound 8 was dissolved in 0.5% carboxymethyl cellulose/0.5% Tween 80. For intravenous administration (iv), the compound was administered in 0.9% saline solution. Mouse bleeds were conducted via tail snips. Mice were active and showed no abnormal behavior during the course of the studies. Bioavailability (F) was calculated as
All studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
Supplementary Material
■ ACKNOWLEDGMENTS
We thank Laura Via and Clif Barry at the National Institute of Health for the gift of marmoset plasma, Martin Pavelka (University of Rochester Medical Center) for the generous donation of the β-lactamase knockout strains, John Blanchard (Albert Einstein College of Medicine) for the BlaC expression plasmids, Kristin Burns-Huang for her helpful input, and Wasima Shinwari and Madeleine Wood for their technical assistance. This work was supported by the Tres Cantos Open Lab Foundation, the Tri-Institutional TB Research Unit via NIH grants U19 AI109748 and AI111143, and the Abby and Howard Milstein Program in Chemical Biology and Translational Medicine. The Department of Microbiology and Immunology at Cornell Medicine is supported by the William Randolph Hearst Trust.
■ ABBREVIATIONS
- BCG
Bacille Calmette-Guérin
- BlaC
class A β-lactamase
- CARA
charcoal-agar resazurin assay
- MBC
minimum bactericidal concentration
- CFU
colony-forming unit
- GSK
GlaxoSmithKline
- I
inoculum
- Ldt
L,D-transpeptidase
- LORA
low oxygen recovery assay
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- MTS
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfo-phenyl)-2H-tetrazolium
- PMS
phenazine methosulfate
- TB
tuberculosis
- OADC
oleic acid, dextrose, and catalase
- OD
optical density
- PBP
penicillin-binding protein
- PYR
pyrithione
- SI
selectivity index
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsinfecdis.9b00112.
Stability of 2a in R and NR media and inoculating effect; activity of 1a and 2a against R and NR media; quantification of PYR release in media; BlaC effect on compound 1a activity; stability of 1a, 1b, 2a, and 8 in microsomes; preliminary pharmacokinetics of 8; activities from high-throughput screen; activity of PYR-containing cephalosporins against drug-sensitive and drug-resistant clinical isolates of Mtb; activity of select compounds against bacterial pathogens other than Mtb; mammalian cell cytotoxicity of selective agents; synthetic schemes; experimental procedures; characterization of new compounds (PDF)
The authors declare no competing financial interest.
■ REFERENCES
- (1).World Health Organization (2017) Global tuberculosis report 2017. [Google Scholar]
- (2).Lenaerts A, Barry CE, and Dartois V (2015) Heterogeneity in tuberculosis pathology, microenvironments and therapeutic responses. Immunol. Rev 264, 288–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Gold B, and Nathan C (2017) Targeting phenotypically tolerant Mycobacterium tuberculosis. Microbiol. Spectrum 5, TBTB2-0031-2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Bryk R, Gold B, Venugopal A, Singh J, Samy R, Pupek K, Cao H, Popescu C, Gurney M, Hotha S, Cherian J, Rhee K, Ly L, Converse PJ, Ehrt S, Vandal O, Jiang X, Schneider J, Lin G, and Nathan C (2008) Selective killing of nonreplicating mycobacteria. Cell Host Microbe 3, 137–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Aldridge BB, Keren I, and Fortune SM (2014) The spectrum of drug susceptibility in mycobacteria. Microbiol. Spectrum 2, MGM2-0031-2013. [DOI] [PubMed] [Google Scholar]
- (6).Koul A, Arnoult E, Lounis N, Guillemont J, and Andries K (2011) The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. [DOI] [PubMed] [Google Scholar]
- (7).Gold B, Roberts J, Ling Y, Quezada LL, Glasheen J, Ballinger E, Somersan-Karakaya S, Warrier T, Warren JD, and Nathan C (2015) Rapid, semiquantitative assay to discriminate among compounds with activity against replicating or nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother 59, 6521–6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Gengenbacher M, Rao SP, Pethe K, and Dick T (2010) Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiology 156, 81–87. [DOI] [PubMed] [Google Scholar]
- (9).Wayne LG, and Hayes LG (1996) An in vitro model for sequential study of shiftdown of Mycobacterium tuberculosis through two stages of nonreplicating persistence. Infect. Immun 64, 2062–2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wang F, Cassidy C, and Sacchettini JC (2006) Crystal structure and activity studies of the Mycobacterium tuberculosis betalactamase reveal its critical role in resistance to beta-lactam antibiotics. Antimicrob. Agents Chemother 50, 2762–2771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kurz SG, and Bonomo RA (2012) Reappraising the use of beta-lactams to treat tuberculosis. Expert Rev. Anti-Infect. Ther 10, 999–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dinesh N, Sharma S, and Balganesh M (2013) Involvement of efflux pumps in the resistance to peptidoglycan synthesis inhibitors in Mycobacterium tuberculosis. Antimicrob. Agents Chemother 57, 1941–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Jarlier V, Gutmann L, and Nikaido H (1991) Interplay of cell wall barrier and beta-lactamase activity determines high resistance to beta-lactam antibiotics in Mycobacterium chelonae. Antimicrob. Agents Chemother 35, 1937–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Lavollay M, Arthur M, Fourgeaud M, Dubost L, Marie A, Veziris N, Blanot D, Gutmann L, and Mainardi JL (2008) The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J. Bacteriol 190, 4360–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Biarrotte-Sorin S, Hugonnet JE, Delfosse V, Mainardi JL, Gutmann L, Arthur M, and Mayer C (2006) Crystal structure of a novel beta-lactam-insensitive peptidoglycan transpeptidase. J. Mol. Biol 359, 533–538. [DOI] [PubMed] [Google Scholar]
- (16).Mainardi JL, Fourgeaud M, Hugonnet JE, Dubost L, Brouard JP, Ouazzani J, Rice LB, Gutmann L, and Arthur M (2005) A novel peptidoglycan cross-linking enzyme for a beta-lactamresistant transpeptidation pathway. J. Biol. Chem 280, 38146–38152. [DOI] [PubMed] [Google Scholar]
- (17).Dubee V, Triboulet S, Mainardi JL, Etheve-Quelquejeu M, Gutmann L, Marie A, Dubost L, Hugonnet JE, and Arthur M (2012) Inactivation of Mycobacterium tuberculosis L,D-transpeptidase LdtMt1 by carbapenems and cephalosporins. Antimicrob. Agents Chemother 56, 4189–4195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Cynamon MH, and Palmer GS (1983) In vitro activity of amoxicillin in combination with clavulanic acid against Mycobacterium tuberculosis. Antimicrob. Agents Chemother 24, 429–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Diacon AH, van der Merwe L, Barnard M, von Groote-Bidlingmaier F, Lange C, Garcia-Basteiro AL, Sevene E, Ballell L, and Barros-Aguirre D (2016) Beta-lactams against tuberculosis–new trick for an old dog? N. Engl. J. Med 375, 393–394. [DOI] [PubMed] [Google Scholar]
- (20).Hugonnet JE, Tremblay LW, Boshoff HI, Barry CE 3rd, and Blanchard JS (2009) Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Gold B, Warrier T, and Nathan C (2015) A multi-stress model for high throughput screening against non-replicating Mycobacterium tuberculosis. Methods Mol. Biol 1285, 293–315. [DOI] [PubMed] [Google Scholar]
- (22).Gold B, Smith R, Nguyen Q, Roberts J, Ling Y, Lopez Quezada L, Somersan S, Warrier T, Little D, Pingle M, Zhang D, Ballinger E, Zimmerman M, Dartois V, Hanson P, Mitscher LA, Porubsky P, Rogers S, Schoenen FJ, Nathan C, and Aubé J. (2016) Novel cephalosporins selectively active on nonreplicating Mycobacterium tuberculosis. J. Med. Chem 59, 6027–6044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Warrier T, Martinez-Hoyos M, Marin-Amieva M, Colmenarejo G, Porras-De Francisco E, Alvarez-Pedraglio AI, Fraile-Gabaldon MT, Torres-Gomez PA, Lopez-Quezada L, Gold B, Roberts J, Ling Y, Somersan-Karakaya S, Little D, Cammack N, Nathan C, and Mendoza-Losana A (2015) Identification of novel anti-mycobacterial compounds by screening a pharmaceutical small-molecule library against nonreplicating Mycobacterium tuberculosis. ACS Infect. Dis 1, 580–585. [DOI] [PubMed] [Google Scholar]
- (24).Brook I (1989) Inoculum effect. Clin. Infect. Dis 11, 361–368. [DOI] [PubMed] [Google Scholar]
- (25).Wi YM, Choi JY, Lee JY, Kang CI, Chung DR, Peck KR, Song JH, and Ko KS (2017) Antimicrobial effects of beta-lactams on imipenem-resistant ceftazidime-susceptible Pseudomonas aeruginosa. Antimicrob. Agents Chemother 61, e00054–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Harada Y, Morinaga Y, Kaku N, Nakamura S, Uno N, Hasegawa H, Izumikawa K, Kohno S, and Yanagihara K (2014) In vitro and in vivo activities of piperacillin-tazobactam and meropenem at different inoculum sizes of ESBL-producing Klebsiella pneumoniae. Clin. Microbiol. Infect 20, O831–O839. [DOI] [PubMed] [Google Scholar]
- (27).Bonnett SA, Ollinger J, Chandrasekera S, Florio S, O’Malley T, Files M, Jee J-A, Ahn J, Casey A, Ovechkina Y, Roberts D, Korkegian A, and Parish T (2016) A target-based whole cell screen approach to identify potential inhibitors of Mycobacterium tuberculosis signal peptidase. ACS Infect. Dis 2, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ochiai M, Aki O, Morimoto A, Okada T, and Kaneko T (1972) New reaction in cephalosporin. derivatives. Nucleophilic substitution of 2-(4-carboxy-7-acylaminoceph-3-em-3-ylmethylthio)-pyridine N-oxide derivatives in the presence of copper(II) salts. Tetrahedron Lett. 13, 2345–2348. [Google Scholar]
- (29).Pansy FE, Stander H, Koerber WL, and Donovick R (1953) In vitro studies with 1-hydroxy-2(1H) pyridinethione. Exp. Biol. Med 82, 122–124. [DOI] [PubMed] [Google Scholar]
- (30).Shaw E, Bernstein J, Losee K, and Lott W (1950) Analogs of aspergillic acid. IV Substituted 2-bromopyridine-N-oxides and their conversion to cyclic thiohydroxamic acids. J. Am. Chem. Soc 72, 4362–4364. [Google Scholar]
- (31).Salina EG, Huszár S, Zemanová J, Keruchenko J, Riabova O, Kazakova E, Grigorov A, Azhikina T, Kaprelyants A, Mikušová K, and Makarov V. (2018) Copper-related toxicity in replicating and dormant Mycobacterium tuberculosis caused by 1-hydroxy-5-R-pyridine-2(1H)-thiones. Metallomics 10, 992–1002. [DOI] [PubMed] [Google Scholar]
- (32).Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, and Franzblau SG (2007) Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother 51, 1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sabath LD, Jago M, and Abraham EP (1965) Cephalosporinase and penicillinase activities of a β-lactamase from Pseudomonas pyocyanea. Biochem. J 96, 739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Hamilton-Miller JMT, Newton GGF, and Abraham EP (1970) Products of aminolysis and enzymic hydrolysis of the cephalosporins. Biochem. J 116, 371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hamilton-Miller JMT, Richards E, and Abraham EP (1970) Changes in proton-magnetic-resonance spectra during aminolysis and enzymic hydrolysis of cephalosporins. Biochem. J 116, 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Mobashery S, and Johnston M (1986) Reactions of Escherichia coli TEM β-lactamase with cephalothin and with C10-dipeptidyl cephalosporin esters. J. Biol. Chem 261, 7879–7887. [PubMed] [Google Scholar]
- (37).Mobashery S, Lerner SA, and Johnston M (1986) Conscripting β-lactamase for use in Drug delivery. Synthesis and biological activity of a cephalosporin C10-ester of an antibiotic dipeptide. J. Am. Chem. Soc 108, 1685–1686. [Google Scholar]
- (38).Mobashery S, and Johnston M (1987) Inactivation of alanine racemase by β-chloro-L-alanine released enzymically from amino acid and peptide C10-esters of deacetylcephalothin. Biochemistry 26, 5878–5884. [DOI] [PubMed] [Google Scholar]
- (39).Boyd DB, and Lunn W (1979) Electronic structures of cephalosporins and penicillins. 9. Departure of a leaving group in cephalosporins. J. Med. Chem 22, 778–784. [DOI] [PubMed] [Google Scholar]
- (40).O’Callaghan CH, Sykes RB, and Staniforth SE (1976) A new cephalosporin with a dual mode of action. Antimicrob. Agents Chemother 10, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Greenwood D, and O’Grady F (1976) Dual-action cephalosporin utilizing a novel therapeutic principle. Antimicrob. Agents Chemother 10, 249–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Georgopapadakou NH, Bertasso A, Chan KK, Chapman JS, Cleeland R, Cummings LM, Dix BA, and Keith DD (1989) Mode of action of the dual-action cephalosporin RO 23−9424. Antimicrob. Agents Chemother 33, 1067–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Albrecht HA, Beskid G, Chan KK, Christenson JG, Cleeland R, Deitcher KH, Georgopapadakou NH, Keith DD, Pruess DL, et al. (1990) Cephalosporin 3′-quinolone esters with adual mode of action. J. Med. Chem 33, 77–86. [DOI] [PubMed] [Google Scholar]
- (44).Gu JW, and Neu HC (1990) In vitro activity of RO 23− 9424, a dual-action cephalosporin, compared with activities of other antibiotics. Antimicrob. Agents Chemother 34, 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Jones RN (1990) Antimicrobial activity of RO 24−6778, a covalent bonding of desmethylfleroxacin and desacetylcefotaxime. Diagn. Microbiol. Infect. Dis 13, 253–259. [DOI] [PubMed] [Google Scholar]
- (46).Albrecht HA, Beskid G, Christenson JG, Durkin JW, Fallat V, Georgopapadakou NH, Keith DD, Konzelmann FM, Lipschitz ER, et al. (1991) Dual-action cephalosporins: Cephalosporin 3′-quaternary ammonium quinolones. J. Med. Chem 34, 669–675. [DOI] [PubMed] [Google Scholar]
- (47).Albrecht HA, Beskid G, Georgopapadakou NH, Keith DD, Konzelmann FM, Pruess DL, Rossman PL, Wei CC, and Christenson JG (1991) Dual-action cephalosporins: Cephalosporin 3′-quinolone carbamates. J. Med. Chem 34, 2857–2864. [DOI] [PubMed] [Google Scholar]
- (48).Georgopapadakou NH, and McCaffrey C (1994) 3′-Lactamase hydrolysis of cephalosporin 3′-quinolone esters, carbamates, and tertiary amines. Antimicrob. Agents Chemother 38, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Jones RN, and Sanchez ML (1994) Antimicrobial activity of a new antipseudomonal dual-action drug, RO 25−0534. Diagn. Microbiol. Infect. Dis 18, 61–68. [DOI] [PubMed] [Google Scholar]
- (50).Veinberg G, Shestakova I, Petrulanis L, Grigan N, Musel D, Zeile D, Kanepe I, Domrachova I, Kalvinsh I, et al. (1997) Synthesis and evaluation of dual action cephalosporins as elastase inhibitors. Bioorg. Med. Chem. Lett 7, 843–846. [Google Scholar]
- (51).Panunzio M, Malabarba A, and Vicennati P (2004) Synthesis and antibacterial activity of new antibiotics arising from cephalosporin-monobactam coupling. ARKIVOC (Gainesville, FL, U. S.) 2004, 36–47. [Google Scholar]
- (52).Bianchet MA, Pan YH, Basta LAB, Saavedra H, Lloyd EP, Kumar P, Mattoo R, Townsend CA, and Lamichhane G (2017) Structural insight into the inactivation of Mycobacterium tuberculosis non-classical transpeptidase LdtMt2 by biapenem and tebipenem. BMC Biochem. 18, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Evans LE, Krishna A, Ma Y, Webb TE, Marshall DC, Tooke CL, Spencer J, Clarke TB, Armstrong A, and Edwards AM (2019) Exploitation of antibiotic resistance as a novel drug target: Development of a β-lactamase-activated antibacterial prodrug. J. Med. Chem 62, 4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Meyer DL, Law KL, Payne JK, Mikolajczyk SD, Zarrinmayeh H, Jungheim LN, Kling JK, Shepherd TA, and Starling JJ (1995) Site-specific prodrug activation by antibody-β-lactamase conjugates: Preclinical investigation of the efficacy and toxicity of doxorubicin delivered by antibody directed catalysis. Bioconjugate Chem. 6, 440–446. [DOI] [PubMed] [Google Scholar]
- (55).Meyer DL, Jungheim LN, Law KL, Mikolajczk SD, Shepherd TA, Mackensen DG, Briggs SL, and Starling JJ (1993) Site-specific prodrug activation by antibody-β-lactamase conjugates: Regression and long-term growth inhibition of human colon carcinoma xenograft models. Cancer Res. 53, 3956–3963. [PubMed] [Google Scholar]
- (56).Jungheim LN, Shepherd TA, and Kling JK (1993) Synthesis of a cephalosporin-doxorubicin antitumor prodrug: A substrate for an antibody-targeted enzyme. Heterocycles 35, 339–348. [Google Scholar]
- (57).Meyer DL, Jungheim LN, Mikolajczyk SD, Shepherd TA, Starling JJ, and Ahlem CN (1992) Preparation and characterization of a beta-lactamase-Fab′ conjugate for the sitespecific activation of oncolytic agents. Bioconjugate Chem. 3, 42–48. [DOI] [PubMed] [Google Scholar]
- (58).Jungheim LN, Shepherd TA, and Meyer DL (1992) Synthesis of acylhydrazido-substituted. cephems. Design of cephalosporin-vinca alkaloid prodrugs: Substrates for an antibody targeted enzyme. J. Org. Chem 57, 2334–2340. [Google Scholar]
- (59).Shepherd TA, Jungheim LN, Meyer DL, and Starling JJ (1991) A novel targeted delivery system utilizing a cephalosporinoncolytic prodrug activated by an antibody β-lactamase conjugate for the treatment of cancer. Bioorg. Med. Chem. Lett 1, 21–26. [Google Scholar]
- (60).Gao W, Xing B, Tsien RY, and Rao J (2003) Novel fluorogenic substrates for imaging β-lactamase gene expression. J. Am. Chem. Soc 125, 11146–11147. [DOI] [PubMed] [Google Scholar]
- (61).Rukavishnikov A, Gee KR, Johnson I, and Corry S (2011) Fluorogenic cephalosporin substrates for β-lactamase TEM-1. Anal. Biochem 419, 9–16. [DOI] [PubMed] [Google Scholar]
- (62).Mizukami S, Watanabe S, Akimoto Y, and Kikuchi K (2012) No-wash protein labeling with designed fluorogenic probes and application to real-time pulse-chase analysis. J. Am. Chem. Soc 134, 1623–1629. [DOI] [PubMed] [Google Scholar]
- (63).Cheng Y, Xie H, Sule P, Hassounah H, Graviss EA, Kong Y, Cirillo JD, and Rao J (2014) Fluorogenic probes with substitutions at the 2 and 7 positions of cephalosporin are highly BlaC-specific for rapid Mycobacterium tuberculosis detection. Angew. Chem., Int. Ed 53, 9360–9364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Albert A, Rees CW, and Tomlinson AJ (1956) The influence of chemical constitution on antibacterial activity. VIII. 2-Mercaptopyridine-N-oxide, and some general observations on metalbinding agents. Br. J. Exp. Pathol 37, 500–511. [PMC free article] [PubMed] [Google Scholar]
- (65).Zaengle-Barone JM, Jackson AC, Besse DM, Becken B, Arshad M, Seed PC, and Franz KJ (2018) Copper influences the antibacterial outcomes of a beta-lactamase-activated prochelator against drug-resistant bacteria. ACS Infect. Dis 4, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chandler CJ, and Segel IH (1978) Mechanism of the antimicrobial action of pyrithione: Effects on membrane transport, ATP levels, and protein synthesis. Antimicrob. Agents Chemother 14, 60–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Stoyanov S, Petkov I, Antonov L, Stoyanova T, Karagiannidis P, and Aslanidis P (1990) Thione-thiol tautomerism and stability of 2-mercaptopyridines and 4-mercaptopyridines and 2-mercaptopyrimidines. Can. J. Chem 68, 1482–1489. [Google Scholar]
- (68).England K, Boshoff HIM, Arora K, Weiner D, Dayao E, Schimel D, Via LE, and Barry CE (2012) Meropenem-clavulanic acid shows activity against Mycobacterium tuberculosis in vivo. Antimicrob. Agents Chemother 56, 3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Compound 9 was obtained by deprotecting the N-Boc derivative of 8 with TFA (see the Supporting Information for details). Such double bond migration is characteristic of many cephalosporins: Mobashery S, and Johnston M (1986) Preparation of ceph-3-em esters unaccompanied by Δ3 to Δ2 isomerization of the cephalosporin. J. Org. Chem 51, 4723–4726. [Google Scholar]
- (70).Doose CA, Szaleniec M, Behrend P, Muller A, and Jastorff B (2004) Chromatographic behavior of pyrithiones. J. Chromatogr. A 1052, 103–110. [DOI] [PubMed] [Google Scholar]
- (71).Hugonnet JE, and Blanchard JS (2007) Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Flores AR, Parsons LM, and Pavelka MS Jr. (2005) Genetic analysis of the beta-lactamases of Mycobacterium tuberculosis and Mycobacterium smegmatis and susceptibility to beta-lactam antibiotics. Microbiology 151, 521–532. [DOI] [PubMed] [Google Scholar]
- (73).Kong Y, Yao H, Ren H, Subbian S, Cirillo SL, Sacchettini JC, Rao J, and Cirillo JD (2010) Imaging tuberculosis with endogenous beta-lactamase reporter enzyme fluorescence in live mice. Proc. Natl. Acad. Sci. U. S. A 107, 12239–12244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Cheng Y, Xie J, Lee KH, Gaur RL, Song A, Dai T, Ren H, Wu J, Sun Z, Banaei N, Akin D, and Rao J (2018) Rapid and specific labeling of single live Mycobacterium tuberculosis with a dualtargeting fluorogenic probe. Sci. Transl. Med 10, No. eaar4470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Voladri RK, Lakey DL, Hennigan SH, Menzies BE, Edwards KM, and Kernodle DS (1998) Recombinant expression and characterization of the major beta-lactamase of Mycobacterium tuberculosis. Antimicrob. Agents Chemother 42, 1375–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Uri JV, Actor P, Phillips L, and Weisbach JA (1978) Semisynthetic cephalosporins with antifungal activity: Laboratory studies on 2-pyridinethiol 1-oxide cephalosporins. J. Antibiot 31, 580–585. [DOI] [PubMed] [Google Scholar]
- (77).Adamek RN, Credille CV, Dick BL, and Cohen SM (2018) Isosteres of hydroxypyridinethione as drug-like pharmacophores for metalloenzyme inhibition. J. Biol. Inorg. Chem 23, 1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Khattar MM, Salt WG, and Stretton RJ (1988) The influence of pyrithione on the growth of micro-organisms. J. Appl. Bacteriol 64, 265–272. [DOI] [PubMed] [Google Scholar]
- (79).Dinning AJ, Al-Adham IS, Eastwood IM, Austin P, and Collier PJ (1998) Pyrithione biocides as inhibitors of bacterial ATP synthesis. J. Appl. Microbiol 85, 141–146. [DOI] [PubMed] [Google Scholar]
- (80).Ermolayeva E, and Sanders D (1995) Mechanism of pyrithione-induced membrane depolarization in Neurospora crassa. Appl. Environ. Microbiol 61, 3385–3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Gold B, Roberts J, Ling Y, Lopez Quezada L, Glasheen J, Ballinger E, Somersan-Karakaya S, Warrier T, and Nathan C (2016) Visualization of the charcoal agar resazurin assay for semiquantitative, medium-throughput enumeration of mycobacteria. J. Visualized Exp, e54690. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.