

Abstract
In recent years, there has been a rapid and sustained increase in the development and use of one-pot chemoenzymatic reaction processes for the efficient synthesis of high-value molecules. This strategy can provide a number of advantages over traditional synthetic methods, including high levels of selectivity in reactions, mild and sustainable reaction conditions, and the ability to rapidly build molecular complexity in a single reaction vessel. Here, we present several examples of chemoenzymatic one-pot reaction sequences that demonstrate the diversity of transformations that can be incorporated in these processes.
Keywords: chemoenzymatic, biocatalysis, one-pot process
Graphical Abstract
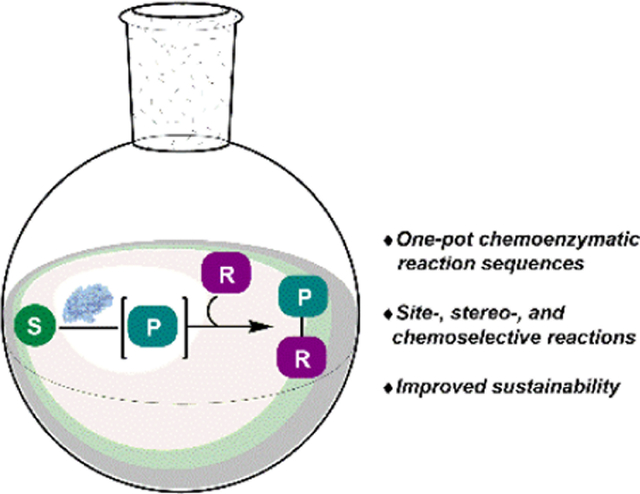
Introduction:
Groundbreaking discoveries in protein engineering and the development of non-native enzymatic reactions, coupled with an ever-expanding database of natural protein sequences, have positioned biocatalysis to be increasingly impactful in synthetic chemistry.1–3 In particular, chemoenzymatic cascades and one-pot reaction sequences are powerful tools for the efficient synthesis of chemical building blocks, natural products, and pharmaceutical agents.1–3 These reaction sequences are often inspired by Nature’s methods for generating natural products, wherein multiple transformations are performed in a single vessel (for example, a cell) to quickly generate molecular complexity. Biocatalysis can provide an array of advantages over traditional synthetic methods, offering improved site-, stereo-, and chemoselectivity, in addition to mild reaction conditions.1–6 Chemoenzymatic, one-pot reaction sequences combine the benefits of biocatalytic methods with the potential to perform multiple transformations in a single reaction vessel with minimal side-reactivity.1–3 Unlike many traditional synthetic transformations, chemoenzymatic reactions are typically performed at ambient temperatures and under mild, aqueous conditions which are amenable to the function of enzymes.4–5 Using enzymes, transformations which traditionally require incompatible reaction conditions can often be performed in a single vessel without issue. Combining traditional chemical methods and biocatalytic reactions is currently underexploited but is poised to expand the repertoire of transformations possible compared to what can be achieved with biocatalysis alone.
Chemoenzymatic processes have been applied to the synthesis of an array of high value molecules, including bioactive natural products, drugs and pharmaceutical intermediates.4–6 A variety of enzyme classes have been used in cascade or sequential one-pot processes including oxygenases, halogenases, oxidases, reductases, and lipases among others.4–5 These reactions are designed to operate in a complementary fashion, where a chemical step generates a substrate for a subsequent enzymatic reaction or a chemical step is used to modify the product of an enzymatic process (Scheme 1). The chemoenzymatic sequences designed by Gröger and coworkers for the one-pot stereo-divergent synthesis of enantioenriched 1,3-diols demonstrate this concept.7 After an initial organo-catalyzed asymmetric aldol reaction (1 to 2, Scheme 2A), the resulting ketone (2) was reduced stereo-specifically by a nicotinamide adenine dinucleotide phosphate hydride (NADPH)-dependent alcohol dehydrogenase (ADH). By selecting an appropriate aldol catalyst and ADH, the configuration of each stereocenter could be controlled. This allowed access to all four possible stereoisomers for the resulting 1,3-diol in good yields and with high levels of enantio- and diastereoselectivity. In comparison, traditional synthetic approaches to selectively access each of the possible stereoisomers require numerous steps and are typically accomplished by modular asymmetric synthesis of each stereocenter using transition metal-catalyzed processes.8–9 Aoki and coworkers developed a similar one-pot stereodivergent synthesis of enantioenriched 1,3-diols through a Zn-catalyzed enantioselective aldol reaction, followed by an analogous enantioselective reduction with complementary NADPH-dependent ADHs (6-8, Scheme 2B).10 These conditions offered improved enantioselectivity in the aldol step, translating to improved diastereomeric ratios of products following the enzyme-catalyzed reduction. These examples highlight the utility of cascade biocatalysis for performing enantioselective reductions, providing selective access to any of the desired possible stereoisomers in a one-pot process.
Scheme 1.

(A) Chemical functionalization of an enzymatic product. (B) Chemical generation of substrate for an enzymatic reaction. S: substrate, P: product, R: chemical modification.
Scheme 2.

Stereodivergent chemoenzymatic one-pot synthesis of 1,3-diols.8–10
Chemoenzymatic one-pot reactions offer significant advantages for the selective oxidation of molecules, particularly for those with redox-sensitive functional groups. The ability to perform selective oxidations without protecting group manipulations allows for one-pot cascade sequences that would not be possible using traditional synthetic methods alone. The opportunities afforded by selective biocatalytic functionalization has inspired new routes to synthetically challenging target molecules. The Turner group has pioneered a number of chemoenzymatic processes for the selective synthesis of nitrogen-containing heterocycles. They have established a variety of approaches, including the chemoenzymatic kinetic resolution of cyclic amines and cascades for cyclization to form an imine, followed by selective reduction using imine reductases (Scheme 3a).11–14 Renata and coworkers recently developed a one-pot chemoenzymatic method that processed through an intermediate imine to generate L-proline derivatives (17-21), a motif common to a number of acyldepsipeptide antibiotics (Scheme 3b).15 Their approach leverages the ability of GriE, an α-ketoglutarate-dependent non-heme iron dioxygenase, to perform perform two successive C–H oxidations on the terminal methyl group of L-leucine, to generate an aldehyde intermediate.15 In situ intramolecular condensation to form imine intermediate 15 is followed by borazane reduction to afford L-proline derivatives 17-21 in good yields. This method demonstrates the efficiency of performing complementary chemoenzymatic transformations in one-pot without the need for isolation of reaction intermediates.15 In comparison, an established synthetic route to access 4-methylproline (17) involves multiple steps, including protection of L-4-hydroxyproline, oxidation to the corresponding ketone, olefination with Petasis reagent, hydrogenation with Crabtree’s catalyst and, finally, global deprotection to yield 14 with moderate diastereomeric ratios.16 Thus, the chemoenzymatic approach employing GriE represents a more efficient, sustainable and selective route for the synthesis of proline derivatives.
Scheme 3.

Significant effort has been devoted to the development of high utility chemoenzymatic sequences. For example, the combination of transition metal-catalyzed and biocatalytic reactions in a single vessel allows chemists to perform transformations that are generally not possible using biocatalysts alone. For example, cross-coupling reactions or olefin metathesis can be used to generate substrates in situ for a biocatalyst.17–20 Innovative work by Gröger and coworkers demonstrated that palladium-catalyzed cross-couplings could be paired in a modular fashion with the asymmetric reduction of aryl ketones for the generation of enantioenriched biaryl benzylic alcohols (29 and 33, Scheme 4).17 Either of the desired enantiomers could be accessed using complementary ADHs, giving products with the targeted configuration with nearly perfect selectivity and in good yields.17 This was the first demonstration of a cross-coupling reaction used in a one-pot fashion with a biocatalytic reaction, expanding the scope of transformations that are possible through chemoenzymatic processes and inspiring future innovation in combining transition-metal catalysis and biocatalysis.
Scheme 4.

Chemoenzymatic synthesis of enantioenriched benzylic alcohols.17
One limitation in the development of one-pot chemoenzymatic processes is the frequent incompatibility of conditions for the chemocatalytic and biocatalytic reactions.18–21 For example, some biocatalysts are sensitive to organic solvents and many chemical catalysts cannot operate in aqueous or aerobic conditions. These contradictory requirements can make some desirable chemoenzymatic transformations logistically difficult or impossible.21 However, recent innovations have allowed the combination of ordinarily incompatible transformations.21 In one example, Hartwig and Zhao developed a tandem chemoenzymatic reaction for the conversion of simple alkenes to substituted, enantioenriched epoxides through an initial olefin metathesis with Hoveyda-Grubbs second generation catalyst, followed by epoxidation with P450BM3 variants (see Scheme 5A and 5B).18–19 In this case, the seemingly incompatible processes could be accomplished in a common reaction vessel by using a biphasic system of isooctane and aqueous buffer.18–19 This solution was effective, generating enantioenriched epoxides in yields that were often greater than those achieved for each standalone reaction.18–19 This increase in yield is likely the result of a change in equilibrium for the metathesis reaction caused by the consumption of the metathesis product in the subsequent biocatalytic reaction.19 Collaborative efforts by Hartwig and Zhao have developed additional novel chemoenzymatic methods that utilize transition metals. In particular, they have demonstrated that rhodium-catalyzed diazocoupling reactions and iridium-catalyzed alkene isomerization can be productively paired with stereoselective reduction by ene-reductases.22–23 Similarly, Turner and Castagnolo developed a system for the chemoenzymatic synthesis of substituted pyrroles (Scheme 5C).20 This reaction sequence featured an olefin metathesis mediated by the second generation Grubbs catalyst, followed by monoamine oxidase-N (MAO-N)-catalyzed oxidation to generate pyrrole products (see 42-44). This method used whole cells containing the biocatalyst as an alternative to purified protein or crude cell lysate, providing a simplified platform for the oxidation of the cyclic amine to the final pyrrole product.20 Traditional methods for this oxidation require stoichiometric oxidants, such as hypervalent iodide,24 or transition-metal catalysts with TEMPO.25 In comparison, the biocatalytic process presented here is selective for the oxidation of amines and uses molecular oxygen as the stoichiometric oxidant. Thus, this biphasic chemoenzymatic system provides several advantages over traditional approaches to pyrrole synthesis.20
Scheme 5.

Chemoenzymatic reactions involving transition metal catalysis.18–20
Synthetic approaches to natural products often take inspiration from biosynthetic pathways, attempting to mimic the elegance of Nature’s ability to rapidly generate complexity from simple building blocks.26 Toward this goal, biosynthetic intermediates are often targeted as intermediates in the total synthesis of natural products.26 However, accessing biosynthetic intermediates using traditional methods can prove challenging, particularly for highly reactive molecules. For example, ortho-quinone methides (o-QMs) are high energy intermediates which have been implicated as biosynthetic intermediates in numerous natural product pathways.27 o-QMs, such as 47, are derived from ortho-phenolic compounds and typically react with nucleophiles or dienophiles in 1,4-additions or cycloaddition reactions, respectively. These reactive intermediates have been used extensively in the total synthesis of natural products and can be accessed through a variety of approaches.28–29 One common method for the generation of o-QMs involves the incorporation of a leaving group at the benzylic position ortho to the phenolic hydroxyl group, followed by heating in the presence of an acid or base to promote elimination.28–29 Alternatively, oxidative conditions can be employed for the generation of o-QMs; however, harsh conditions are typically required and can be difficult to couple with further functionalization of o-QMs due to decomposition of the intermediate.28–29 This can be further complicated by the presence of redox-sensitive functional groups, which may require blocking groups to survive the strong oxidative conditions required for o-QM precursor synthesis.28–29 Recently, our group investigated the α-ketoglutarate-dependent non-heme iron dioxygenase CitB, for the selective benzylic C–H hydroxylation of ortho-phenolic substrates to generate benzylic alcohols (see 46).30 We anticipated that a biocatalyst-initiated approach to o-QM generation would provide the mild conditions required for one-pot o-QM formation and diversification with nucleophiles or dienophiles without the requirement for blocking group manipulations.30 Upon gentle heating of the resulting benzylic alcohol in the presence of nucleophiles or dienophiles, a variety of benzylic ether, thioether and chroman products could be generated (50-59, Scheme 6).30 This could be carried out as a one-pot chemoenzymatic process in which an ortho-phenolic compounds, such as 45, were treated with CitB, followed by the addition of a nucleophile or dienophile and gentle heating to diversify the o-QM in moderate to good conversions.30 Thus, o-QMs could be accessed and elaborated under mild conditions. We anticipate that this method can be executed with a variety of biocatalysts capable of benzylic hydroxylation of ortho-phenolic compounds.
Scheme 6.

Biocatalyst-initiated ortho-quinone methide generation and diversification.30 *Yield from reactions with purified benzyl alcohol (46)
Highly reactive biosynthetic intermediates often represent a branching point for diversification to a variety of natural products. Thus, access to these molecules can set the stage for a divergent approach toward a suite of natural products. One such intermediate, 61, is involved in the biosynthesis of sorbicillinoid natural products, a structurally diverse class of secondary metabolites from fungi which have demonstrated a number of biological activities (Scheme 7).29 Cox and coworkers identified the enzyme responsible for generation of this intermediate in the fungus Penicillium chrysogenum.31 This enzyme, SorbC, is a flavin-dependent monooxygenase that natively performs an enantioselective oxidative dearomatization on sorbicillin (60) or dihydrosorbicillin to generate reactive diene products sorbicillinol (61) or dihyrosorbicillinol, respectively.31 These biosynthetic intermediates can be further elaborated through subsequent transformations, including Diels-Alder cycloadditions and Michael addition reactions, to generate bisvertinolone (63), rezishanone B (72) and C (65), (+)-epoxysorbicillinol (70), and urea sorbicillinoid (67) among others.31 Developing a synthetic route that incorporates oxidative dearomatization has great potential for the divergent synthesis of this class of molecules and synthetic analogs.32–34 However, oxidative dearomatization is a challenging transformation to achieve chemically with site- and stereoselectivity, making it difficult to produce a single desired isomer and reducing the utility of this strategy for selective synthesis of complex molecules.30–32 Furthermore, the intermediates generated by oxidative dearomatization are highly reactive, making isolation and characterization difficult. To overcome these challenges, we35 and others36–37 have developed chemoenzymatic one-pot approaches toward sorbicillinoid natural products. In particular, SorbC-catalyzed oxidative dearomatization generates reactive diene 61 which can be further elaborated to sorbicillinoid natural products in a cascade or sequential fashion (Scheme 7). We performed extensive characterization of the substrate scope of SorbC, demonstrating the structural requirements for productive binding and catalysis and also developed a one-pot chemoenzymatic route to urea sorbicillinoid (67) through biocatalytic generation of 61, followed by cycloaddition with biacylated urea 58 and deprotection to yield 57.35 Gulder and coworkers have also demonstrated the power of this chemoenzymatic approach to access numerous sorbicillinoid natural products.36–37 For example, bisvertinolone (63) was synthesized by [4+2] cycloaddition with the SorbC-generated dienophile 62, using DMF as a cosolvent. Diels-Alder reactions with vinyl ethers 64 and 71, as well as styrene 68, led to the synthesis of rezishanone C (65), rezishanone B (72) and sorbicatechol A (69), respectively.37 In addition, Gulder and coworkers performed the first enantioselective synthesis of (+)-epoxysorbicillinol (70) through the epoxidation of intermediate 61 in situ using Weitz-Scheffer conditions.37 They note that pre-complexation of the potassium tert-butoxide species allows for the addition of the nucleophilic peroxide to the same face as the SorbC-installed hydroxyl group, thus promoting the selective epoxidation of 61.37 These chemoenzymatic approaches offer significant advantages over existing synthetic routes, including improved enantioselectivity, reduced step count and improved yields.36–37 Efforts such as these are critical for improving access to these compounds to enable biological studies and the determination of structure-activity relationships.25–27
Scheme 7.

As the field of biocatalysis continues to expand and play a greater role in synthetic chemistry, it is reasonable to expect that the development of innovative one-pot chemoenzymatic processes will likewise see continued growth. In particular, the combination of biocatalytic and transition metal-catalyzed transformations is representative of this new frontier in biocatalysis. Efforts in this area will likely continue to increase in order to exploit the breadth of transition-metal catalyzed reactions that have been established, broadening the chemical space that can be accessed through chemoenzymatic approaches. Furthermore, the ever-growing repository of annotated natural product biosynthetic pathways will similarly lead to increased strategic use of biosynthetic enzymes to directly generate intermediates toward the synthesis of natural products.6, 26 A chemoenzymatic approach to the synthesis of complex molecules can provide selectivity, efficiency and sustainability and, thus, stands to exert a significant influence on the field of complex molecule synthesis.6 The example methods highlighted here demonstrate the power of pairing the selectivity of biocatalytic reactions with subsequent transformations to improve the efficiency and sustainability of a synthesis. It is worth noting that these are only a brief sampling of the breadth of work that has been performed in this area.1–3, 21 As the volume of genome sequences available continues to grow and new enzymes and transformations are discovered, it is reasonable to expect that the influence of biocatalysis will continue to expand. Chemoenzymatic strategies will play a central role in this expansion, improving the synthetic utility of these transformations and bridging the gap between the traditional synthetic and biocatalysis communities.
Acknowledgment
T.J.D thanks the National Science Foundation for a Graduate Research Fellowship.
Funding Information
This work was supported by funds from the University of Michigan Life Sciences Institute and the Department of Chemistry.
References and Notes
- (1).García-Junceda E; Lavandera I; Rother D; Schrittwieser JH, (Chemo)enzymatic cascades—Nature’s synthetic strategy transferred to the laboratory. J. Mol. Catal. B: Enzym 2015, 114, 1–6. [Google Scholar]
- (2).Ricca E; Brucher B; Schrittwieser JH, Multi-Enzymatic Cascade Reactions: Overview and Perspectives. Adv. Synth. & Catal 2011, 353 (13), 2239–2262. [Google Scholar]
- (3).Sperl JM; Sieber V, Multienzyme Cascade Reactions—Status and Recent Advances. ACS Catal 2018, 8 (3), 2385–2396. [Google Scholar]
- (4).Sun H; Zhang H; Ang EL; Zhao H, Biocatalysis for the synthesis of pharmaceuticals and pharmaceutical intermediates. Bioorg. Med. Chem 2018, 26 (7), 1275–1284. [DOI] [PubMed] [Google Scholar]
- (5).Patel RN, Chemo-enzymatic synthesis of pharmaceutical intermediates. Expert Opin. Drug Discov 2008, 3 (2), 187–245. [DOI] [PubMed] [Google Scholar]
- (6).Li F; Zhang X; Renata H, Enzymatic CH functionalizations for natural product synthesis. Curr. Opin. Chem. Biol 2019, 49, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Baer K; Krausser M; Burda E; Hummel W; Berkessel A; Groger H, Sequential and modular synthesis of chiral 1,3-diols with two stereogenic centers: access to all four stereoisomers by combination of organo- and biocatalysis. Angew. Chem. Int. Ed. Engl 2009, 48 (49), 9355–8. [DOI] [PubMed] [Google Scholar]
- (8).Binder JT; Kirsch SF, Iterative approach to polyketide-type structures: stereoselective synthesis of 1,3-polyols utilizing the catalytic asymmetric Overman esterification. Chem. Commun. (Camb) 2007, (40), 4164–6. [DOI] [PubMed] [Google Scholar]
- (9).Kumar P; Tripathi D; Sharma BM; Dwivedi N, Transition metal catalysis-a unique road map in the stereoselective synthesis of 1,3-polyols. Org. Biomol. Chem 2017, 15 (4), 733–761. [DOI] [PubMed] [Google Scholar]
- (10).Sonoike S; Itakura T; Kitamura M; Aoki S, One-pot chemoenzymatic synthesis of chiral 1,3-diols using an enantioselective aldol reaction with chiral Zn2+ complex catalysts and enzymatic reduction using oxidoreductases with cofactor regeneration. Chem. Asian. J 2012, 7 (1), 64–74. [DOI] [PubMed] [Google Scholar]
- (11).Carr R; Alexeeva M; Dawson MJ; Gotor-Fernández V; Humphrey CE; Turner NJ, Directed Evolution of an Amine Oxidase for the Preparative Deracemisation of Cyclic Secondary Amines. ChemBioChem 2005, 6 (4), 637–639. [DOI] [PubMed] [Google Scholar]
- (12).Dunsmore CJ; Carr R; Fleming T; Turner NJ, A Chemo-Enzymatic Route to Enantiomerically Pure Cyclic Tertiary Amines. J. Am. Chem. Soc 2006, 128 (7), 2224–2225. [DOI] [PubMed] [Google Scholar]
- (13).Ghislieri D; Houghton D; Green AP; Willies SC; Turner NJ, Monoamine Oxidase (MAO-N) Catalyzed Deracemization of Tetrahydro-β-carbolines: Substrate Dependent Switch in Enantioselectivity. ACS Catal 2013, 3 (12), 2869–2872. [Google Scholar]
- (14).Zawodny W; Marshall JR; Finnigan JD; Turner NJ; Clayden J; Montgomery SL, Chemoenzymatic synthesis of substituted azepanes by sequential biocatalytic reduction and organolithium-mediated rearrangement. J. Am. Chem. Soc 2018. [DOI] [PubMed] [Google Scholar]
- (15).Zwick CR 3rd; Renata H, Remote C-H Hydroxylation by an alpha-Ketoglutarate-Dependent Dioxygenase Enables Efficient Chemoenzymatic Synthesis of Manzacidin C and Proline Analogs. J. Am. Chem. Soc 2018, 140 (3), 1165–1169. [DOI] [PubMed] [Google Scholar]
- (16).Murphy AC; Mitova MI; Blunt JW; Munro MHG, Concise, Stereoselective Route to the Four Diastereoisomers of 4-Methylproline. J. Nat. Prod 2008, 71 (5), 806–809. [DOI] [PubMed] [Google Scholar]
- (17).Burda E; Hummel W; Groger H, Modular chemoenzymatic one-pot syntheses in aqueous media: combination of a palladium-catalyzed cross-coupling with an asymmetric biotransformation. Angew. Chem. Int. Ed. Engl 2008, 47 (49), 9551–4. [DOI] [PubMed] [Google Scholar]
- (18).Denard CA; Huang H; Bartlett MJ; Lu L; Tan Y; Zhao H; Hartwig JF, Cooperative tandem catalysis by an organometallic complex and a metalloenzyme. Angew. Chem. Int. Ed. Engl 2014, 53 (2), 465–9. [DOI] [PubMed] [Google Scholar]
- (19).Denard CA; Bartlett MJ; Wang Y; Lu L; Hartwig JF; Zhao H, Development of a One-Pot Tandem Reaction Combining Ruthenium-Catalyzed Alkene Metathesis and Enantioselective Enzymatic Oxidation To Produce Aryl Epoxides. ACS Catal 2015, 5 (6), 3817–3822. [Google Scholar]
- (20).Scalacci N; Black GW; Mattedi G; Brown NL; Turner NJ; Castagnolo D, Unveiling the Biocatalytic Aromatizing Activity of Monoamine Oxidases MAO-N and 6-HDNO: Development of Chemoenzymatic Cascades for the Synthesis of Pyrroles. ACS Catal 2017, 7 (2), 1295–1300. [Google Scholar]
- (21).Denard CA; Hartwig JF; Zhao H, Multistep One-Pot Reactions Combining Biocatalysts and Chemical Catalysts for Asymmetric Synthesis. ACS Catal 2013, 3 (12), 2856–2864. [Google Scholar]
- (22).Litman ZC; Wang Y; Zhao H; Hartwig JF, Cooperative asymmetric reactions combining photocatalysis and enzymatic catalysis. Nature 2018, 560 (7718), 355–359. [DOI] [PubMed] [Google Scholar]
- (23).Wang Y; Bartlett MJ; Denard CA; Hartwig JF; Zhao H, Combining Rh-Catalyzed Diazocoupling and Enzymatic Reduction To Efficiently Synthesize Enantioenriched 2-Substituted Succinate Derivatives. ACS Catal 2017, 7 (4), 2548–2552. [Google Scholar]
- (24).Nicolaou KC; Mathison CJ; Montagnon T, New reactions of IBX: oxidation of nitrogen- and sulfur-containing substrates to afford useful synthetic intermediates. Angew. Chem. Int. Ed. Engl 2003, 42 (34), 4077–82. [DOI] [PubMed] [Google Scholar]
- (25).Zhang E; Tian H; Xu S; Yu X; Xu Q, Iron-Catalyzed Direct Synthesis of Imines from Amines or Alcohols and Amines via Aerobic Oxidative Reactions under Air. Organic Letters 2013, 15 (11), 2704–2707. [DOI] [PubMed] [Google Scholar]
- (26).Bulger PG; Bagal SK; Marquez R, Recent advances in biomimetic natural product synthesis. Nat. Prod. Rep 2008, 25 (2), 254–297. [DOI] [PubMed] [Google Scholar]
- (27).Fan J; Liao G; Kindinger F; Ludwig-Radtke L; Yin WB; Li SM, Peniphenone and Penilactone Formation in Penicillium crustosum via 1,4-Michael Additions of ortho-Quinone Methide from Hydroxyclavatol to gamma-Butyrolactones from Crustosic Acid. J. Am. Chem. Soc 2019. [DOI] [PubMed] [Google Scholar]
- (28).Bai WJ; David JG; Feng ZG; Weaver MG; Wu KL; Pettus TR, The domestication of ortho-quinone methides. Acc. Chem. Res 2014, 47 (12), 3655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Singh MS; Nagaraju A; Anand N; Chowdhury S, ortho-Quinone methide (o-QM): a highly reactive, ephemeral and versatile intermediate in organic synthesis. RSC Adv 2014, 4 (99), 55924–55959. [Google Scholar]
- (30).Doyon TJ; Perkins JC; Baker Dockrey SA; Skinner KC; Zimmerman PM; Narayan ARH, Biocatalyst-Initiated Ortho-Quinone Methide Formation and Derivatization. ChemRxiv, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Fahad AA; Abood A; Fisch KM; Osipow A; Davison J; Avramovic M; Butts CP; Piel J; Simpson TJ; Cox RJ, Oxidative dearomatisation: the key step of sorbicillinoid biosynthesis. Chem. Sci 2014, 5 (2), 523–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bosset C; Coffinier R; Peixoto PA; El Assal M; Miqueu K; Sotiropoulos J-M; Pouységu L; Quideau S, Asymmetric Hydroxylative Phenol Dearomatization Promoted by Chiral Binaphthylic and Biphenylic Iodanes. Angew. Chem. Int. Ed 2014, 53 (37), 9860–9864. [DOI] [PubMed] [Google Scholar]
- (33).Volp KA; Harned AM, Chiral aryl iodide catalysts for the enantioselective synthesis of para-quinols. Chem. Commun 2013, 49 (29), 3001–3003. [DOI] [PubMed] [Google Scholar]
- (34).Zhu J; Grigoriadis NP; Lee JP; Porco JA, Synthesis of the Azaphilones Using Copper-Mediated Enantioselective Oxidative Dearomatization. J. Am. Chem. Soc 2005, 127 (26), 9342–9343. [DOI] [PubMed] [Google Scholar]
- (35).Baker Dockrey SA; Lukowski AL; Becker MR; Narayan ARH, Biocatalytic site- and enantioselective oxidative dearomatization of phenols. Nat. Chem 2017, 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sib A; Gulder TAM, Stereoselective Total Synthesis of Bisorbicillinoid Natural Products by Enzymatic Oxidative Dearomatization/Dimerization. Angew. Chem.. Int. Ed 2017, 56 (42), 12888–12891. [DOI] [PubMed] [Google Scholar]
- (37).Sib A; Gulder TAM, Chemo-enzymatic Total Synthesis of Oxosorbicillinol, Sorrentanone, Rezishanones B and C, Sorbicatechol A, Bisvertinolone, and (+)-Epoxysorbicillinol. Angew. Chem. Int. Ed 2018, 57 (44), 14650–14653. [DOI] [PubMed] [Google Scholar]