

ABSTRACT
Pathological angiogenesis is a hallmark of several conditions including eye diseases, inflammatory diseases, and cancer. Stromal cells play a crucial role in regulating angiogenesis through the release of soluble factors or direct contact with endothelial cells. Here, we analysed the properties of the extracellular vesicles (EVs) released by bone marrow mesenchymal stromal cells (MSCs) and explored the possibility of using them to therapeutically target angiogenesis. We demonstrated that in response to pro-inflammatory cytokines, MSCs produce EVs that are enriched in TIMP-1, CD39 and CD73 and inhibit angiogenesis targeting both extracellular matrix remodelling and endothelial cell migration. We identified a novel anti-angiogenic mechanism based on adenosine production, triggering of A2B adenosine receptors, and induction of NOX2-dependent oxidative stress within endothelial cells. Finally, in pilot experiments, we exploited the anti-angiogenic EVs to inhibit tumour progression in vivo. Our results identify novel pathways involved in the crosstalk between endothelial and stromal cell and suggest new therapeutic strategies to target pathological angiogenesis.
KEYWORDS: Angiogenesis, adenosine, extracellular vesicles, endothelial cells, stromal cells
Introduction
Angiogenesis is a finely tuned process occurring throughout life in both health and disease. It consists in the growth of new blood vessels from the existing ones and involves several processes, including endothelial cell proliferation, migration, rearrangement of the basement membrane and tubulogenesis [1–3]. Angiogenesis is directly involved in the pathophysiology of several diseases, including the retinopathy of prematurity (ROP), the age-related macular degeneration (AMD) and the corneal neovascularization [4,5]. Furthermore, it is a hallmark of most inflammatory diseases and solid cancers, as tumour cells require energy and oxygen supply to grow, proliferate and metastasise [6].
The molecular mechanisms and players involved in the regulation of this multistep process, both in health and disease, have been deeply studied although not completely identified [7]. The stromal compartment seems to play a key role in the regulation of angiogenesis by acting through diverse mechanisms involving secretion of soluble factors as well as direct contact with endothelial cells [8]. For instance, in response to a wounding event, fibroblasts enter in a proliferating state and rapidly upregulate genes involved in matrix remodelling [9], tissue repair [10] and angiogenesis [11]. In particular, properly activated fibroblasts secrete pro-angiogenic molecules, cytokines and growth factors including angiopoietin-1 (ANG-1), angiogenin, interleukin-6 (IL-6), IL-8, vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF) and granulocyte colony stimulating factor (G-CSF) [8,12]. On the other hand, pericytes are mural cells in direct contact with endothelial cells (ECs) [13]. Pericytes control endothelial sprouting and proliferation, mainly by enforcing contract strengths [14,15], and regulate vessel plasticity, regression and thereby patterning of remodelling vascular networks [16].
Several reports have demonstrated that mesenchymal stromal cells (MSCs) regulate vascular remodelling and angiogenesis through mechanisms not entirely clarified. MSCs have been primary isolated as a non-haemopoietic, tissue culture plastic adherent sub-fraction of bone marrow cells [17]. Nowadays, they are recognized as key players of the haematopoietic stem cell (HSC) niche where they provide structural and environmental support to HSCs. In the bone marrow, MSCs have been identified as perivascular nestin-expressing cells, closely associated with HSCs [18], localized both into the central area of the marrow and in the proximity of the endosteum [18,19]. Intriguingly, multiple studies have demonstrated that bone marrow MSCs, as also MSCs from other sources, modulate the vascular network [20,21]. Accordingly, by inducing neovascularization, transplanted MSCs have been shown to sustain amelioration of ischaemic hindlimb [22], ischaemic brain [23], myocardial infarction [24] and peripheral artery disease (PAD) [25]. A paracrine mechanism seems to drive the angiogenic MSC potential [26]. Indeed, it has been reported that the MSC-conditioned medium is enriched in numerous pro-angiogenic factors, including ANG-1, placental growth factor (PlGF), IL-6, monocyte chemotactic protein-1 (MCP-1), FGF, VEGF, transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF), thus sustaining the formation of new vessels both in vitro and in vivo [27]. In addition, the pro-angiogenic effects of MSCs seem to involve their extracellular vesicles (EVs) [28].
EVs are defined as heterogeneous plasma membrane vesicles, classified mainly by their size and cargo [29] that can be released from various cell types [30]. It has been reported that MSC-derived EVs enhance, for instance, the neovascularization after ischaemic injury in a rat myocardial infarction model [31]. Similarly, they increased postischemic neuroangiogenesis after focal cerebral ischaemia in mice [32]. In most cases, precise mechanisms by which EVs exert their functions remain to be elucidated. However, a recent proteomic analysis reveals that MSC-derived EVs are strongly enriched in several proangiogenic signalling associated proteins, such as epithelial growth factor (EGF), FGF and PDGF [33]. Furthermore they can transfer pro-angiogenic miRNAs, such as the pro-angiogenic miR-126, miR-130a [34] and miR-125 [35,36], as well as signalling proteins and transcription factors [28].
Intriguingly, MSCs have been described to have anti-angiogenic effects, too [37]. Bone marrow MSCs inhibited angiogenesis in a concentration-dependent manner, when supplemented in in vitro capillary cultures [38]. In vivo, intravenously administered MSCs inhibited angiogenesis through a VE-Cadherin/β-catenin signalling pathway [39]. Additionally, we have recently demonstrated that, once licenced by a pro-inflammatory microenvironment, MSCs exert strong anti-angiogenic effects. In details, we showed that transplanted MSCs suppress leukocyte recruitment to the inflamed lymph nodes by affecting activation and expansion of the lymph node specialized endothelial cells forming high endothelial venules (HEVs) [40,41,42].
However, little is known about the anti-angiogenic effects of EVs released by MSCs in specific microenvironments. In this study, we demonstrated that, in response to pro-inflammatory cytokines, bone marrow MSCs produce EVs that modulate angiogenesis targeting multiple endothelial cell functions. In particular, we identified a novel anti-angiogenic mechanism based on adenosine and oxidative stress. Finally, in pilot experiments, we exploited EVs to control tumour angiogenesis, collecting evidence of their efficacy in counteracting pathological angiogenesis.
Methods
Isolation of MSCs and collection of conditioned media (MSC-CM)
Murine MSCs were isolated as described in [40,41]. The collection of the murine MSC-CM was performed as described in [40,42]. Briefly, murine MSCs were plated in 24 wells plate (50 000 cells/well) with DMEM low glucose supplemented with 20% FBS, 2 mM glutamine, 100 U/mL penicillin/streptomycin for 24 h. Then, MSCs were stimulated in DMEM low glucose supplemented with 10% FBS, 2 mM glutamine, 100 U/ml penicillin/streptomycin with or without 25 ng/mL mIL1ß, 20 ng/mL mIL6, 25 ng/mL mTNFα for 24 h. After three washes with DMEM low glucose to remove cytokines, cells were cultured in DMEM low glucose supplemented with 2 mM glutamine, 100 U/mL penicillin/streptomycin for the following 18 h. Conditioned media were harvested, cellular debris were eliminated by centrifugation (2000 g 10 min), and the MSC-CM were stored at −80°C.
Human MSC were provided by Orbsen Therapeutics Ltd. (Galway, Ireland). Ethical approvals are granted from the NUIG Research Ethics Committee and the Galway University Hospitals Clinical Research Ethics Committee (CREC). All samples were obtained with informed consent. Procurement of the sample conformed to European Parliament and Council directives (2001/20/EC; 2004/23/EC). Human MSCs (25000 cells/well) were plated in MEM-Alpha with Glutamax supplemented with 10% FBS, 2 mM glutamine, 100 U/mL penicillin/streptomycin and grow in a humidified incubator with 5% CO2 and 37°C. At the moment of the confluence, medium was substituted with MEM-Alpha with Glutamax supplemented with 2% FBS, 2mM glutamine, 100 U/mL penicillin/streptomycin, with or without 25 ng/mL hIL1ß, 20 ng/mL hIL6, 25 ng/mL hTNFα. Later than 24 h, after three washes in MEM Alpha with Glutamax, the medium was changed with MEM Alpha with Glutamax supplemented with 2 mM glutamine, 100 U/mL penicillin/streptomycin for the following 18 h. Conditioned medium was harvested and centrifuged at 2000 g for 10 min.
EV isolation and characterization
EVs were isolated from murine MSC-CM by ultrafiltration using Amicon® Ultra 15 mL Filters (Merck Millipore) following manufacturer’s instructions. Briefly, each tube was first sterilised with 70% ethanol and then washed two times by centrifuging it at 4000 g for 10 min. Subsequently, 12 mL of MSC-CM (corresponding to the medium conditioned by 1 × 106 cells) were loaded into the tube and centrifuged at 2800 g per 20 min at RT. This last step was repeated by adding 10 mL of PBS at RT at the obtained EVs. Finally, EVs were collected, concentrated in about 150 µL of PBS (with a protein concentration of about 33 ± 5 µg/mL), and directly stored at −80°C. The flow-through of the first ultrafiltration step was collected and stored at −80°C. It was considered as the MSC-CM deprived of EVs (control CM, cCM or primed CM, pCM). When human EVs were used, we isolated and prepared them with the same protocol used for murine EVs. EVs from ultracentrifugation were obtained as described in [43]. Briefly, supernatants were centrifugated at 10.000 g for 30 min to eliminate cell debris. After that, a 100.000 g for 70 min ultracentrifugation was performed twice to eliminate contaminating proteins and then to concentrate small vesicles. They were resuspended in 120 µL of sterile PBS.
Total proteins of purified EVs were extracted with PBS 0.4% SDS. Total amount of proteins in EVs was quantified by MicroBCA kit (Pierce). About 3 µg of proteins were separated by 10% SDS-PAGE under non-reductive conditions for CD39, CD63 and CD9 and reductive conditions with DTT 5 mM (Sigma-Aldrich) for CD73 and TIMP-1. Gels were transferred onto PVDF membranes, 0.45 µm (Millipore) activated with methanol (Sigma Aldrich), using Transfer Tris-Glycine buffer. Membranes were blocked with 5% of BSA (Sigma Aldrich) in TBS 1X with 0.2% of Tween 20 for 1 h, before an overnight incubation with primary antibodies diluted in TBS1X, 0.2% tween and 1% of BSA. Anti-CD63 (MBL D263-3, clone R5G2), anti-CD9 (eBiosciences, clone KMC8), anti-CD39 (Biolegend, clone Duha59), anti-CD73 (Abcam, ab175396) and TIMP-1 (R&D systems AF980) antibodies were used at the dilution 1:1000. Thus, membranes were incubated with the appropriate peroxidase-conjugated secondary antibody, ECL anti-Rabbit (GE), ECL anti-Rat (GE) and anti-Goat (BioRad). Chemiluminescence was achieved by ECL Prime Western Blot Detection reagent (GE). Images were acquired with ImageQuant LAS 4000 Mini (GE), following the manufacturer’s instructions. Densitometric analysis of the Western blots was performed by ImageJ software.
For, anti-CD63 IP, EVs were lysed with IP Buffer containing 50 mM Hepes, 150 mM NaCl, 2 mM EDTA and 0.5% Triton and protease inhibitors (cOmplete™, EDTA-free Protease Inhibitor Cocktail, Roche 11873580001). Pre-cleaning was performed with Protein G (Thermo Fisher). Samples (30 µg protein) were incubated overnight with 1 µg of anti-CD63 (MBL D263-3, clone R5G2) at 4°C, and the complex was capture with protein G (Thermo Fisher). After washes, the complex was eluted with glycine 0.1 M, pH 2.5. The eluted samples and input were blotted and incubated with the antibody against TIMP-1 (R&D systems AF980) at the dilution 1:1000.
Transmission electron microscopy (TEM)
For the transmission electron microscopy, 20 µL of EVs were dispensed for 2 min on 300 mesh carbon-coated copper grids that were made hydrophilic by a 15 s exposure to a glow discharge. A filterpaper (Whatman, Maidstone, UK) was used to remove the excess of liquid. EVs were then stained with 1% uranyl acetate for 2 min. FEI Tecnai G2 transmission electron microscope operating at 100 kV, with a Veleta (Olympus Soft Imaging System, Münster, Germany) digital camera, was exploited to capture images.
Cell lines
SVEC4-10 (ATCC #CRL-2181 Manassas, VA), an endothelial cell line from murine axillary lymph nodes, were cultured in a humidified incubator with 5% CO2 and 37°C, in DMEM (ATCC 30–2002 Manassas, VA) supplemented with 10% heat-inactivated FBS, 1 mM penicillin and streptomycin (Lonza, Braine-L’Alleud, Belgium). MILE SVEN 1 (MS1, ATCC #CRL-2279 Manassas, VA), and endothelial cell line from pancreas/islet of Langerhans, were cultured in a humidified incubator with 5% CO2 and 37°C, in DMEM (DMEM, Lonza, Braine-L’Alleud, Belgium) supplemented with 10% heat-inactivated FBS, 1 mM penicillin and streptomycin (Lonza, Braine-L’Alleud, Belgium). Human Umbilical Vein Endothelial Cells, Pooled, (HUVEC, Lonza #C2519A, Braine-L’Alleud, Belgium) were cultured in 0,02% gelatin-coated flasks in a humidified incubator with 5% CO2 and 37°C, in EBMTM-2 Basal Medium (Lonza #CC-3156) supplemented with EGMTM-2 SingleQuotsTM Supplements (Lonza #CC-4176). Mouse embryonic fibroblast (ThermoFisher A34960), E0771 mouse mammary adenocarcinoma cells (CH3 Biosystems) [44], and MCA-203 cells (fibrosarcoma cells from C57BL/6 mice H-2b, kindly provided by Prof Vincenzo Bronte) were cultured in a humidified incubator with 5% CO2 and 37°C, in complete Dulbecco modified Eagle medium high glucose (DMEM, Lonza, Braine-L’Alleud, Belgium) supplemented with 10% heat-inactivated FBS, 10 mM HEPES (Lonza, Braine-L’Alleud, Belgium) and 1 mM penicillin and streptomycin (Lonza, Braine-L’Alleud, Belgium). All cells were subcultured using 0.05% trypsin, 0.02% EDTA solution. Cells were routinely tested for Mycoplasma.
Scratch wound healing assay
In total, 105 cells (SVEC4-10, MS-1, E0771) were seeded on a 48-well plate in complete medium (DMEM-ATCC 30-2002 supplemented with 10% heat-inactivated FBS, 1% penicillin and streptomycin) and left 24 h to reach 90% confluence. The endothelial cell monolayer was then scratched by using a pipette tip. Cells were gently washed with PBS without calcium and magnesium and 200 µL of medium were added (DMEM low Glucose, 1% P/S, 1% Glutamine), containing or not 1.75 µg/mL EVs. When indicated, cells were treated with 50 ng/mL recombinant VEGF (450–32 Peprotech), 200 µM ARL 67156 (ARL67156A265 Sigma-Aldrich), 10 µM Adenosine 5′-(α, β-methylene), diphosphate ADP analogue (AMP-CP M3763 Sigma-Aldrich), 1 mM N-acetyl-l-cysteine (NAC A7250 Sigma-Aldrich), 10 nM MRS 1706 (1584 R&D System), 100 nM GSK2795039 (HY-18950 MedChem Express) and 10 nM or 100 nM BAY 60-6583 (4472 Tocris). Three lines for well were drawn on the bottom of the plate. Images of the scratches were acquired using an inverted optical microscope equipped with a 4× objective, at time 0 and after 6 h of culture (incubation at 10% CO2 and 37°C). Image analysis was performed using ImageJ. Migration Index was calculated as the difference between the starting (time 0 h) and the final (time 6 h) distance covered by migrating endothelial cells. HUVEC were seeded at the same concentration but the experiment was performed in EBMTM-2 Basal Medium (Lonza #CC-3156) supplemented with EGMTM-2 SingleQuotsTM Supplements (Lonza #CC-4176), which is already enriched in hVEGF. Thus, no additional pro-angiogenic factor was supplemented to the growth medium.
Tube formation assay
The tube formation assay was performed as described in [40,42]. 1.3 × 104 SVEC4-10 or 2 × 104 HUVEC cells were suspended in 100 µL medium (respectively, DMEM low glucose and EBMTM-2 Basal Medium) supplemented with 10% FBS, in the presence or absence of EVs (1.75 µg/mL) or anti-TIMP-1 5 µg/mL (AF980 R&D). Cells were seeded on the solidified matrix and incubated for 6 h at 37°C 10% CO2. The formation of the tube networks develops in 6 hrs at 37°C 10% CO2, for SVEC4-10, or 5% CO2 for HUVEC. At the end of the incubation, cell tubes were imaged with a phase contrast inverted microscope at 4× objective magnifications and analysis was performed with ImageJ Angiogenesis Analyser.
Proliferation assay
1 × 105 SVEC4-10 or E0771 cells were seeded on a 24-well plate in their culture medium (respectively, DMEM-ATCC 30-2002 supplemented with 10% heat-inactivated FBS, 1% penicillin and streptomycin and complete Dulbecco modified Eagle medium high glucose with 10% heat-inactivated FBS, 10 mM HEPES and 1 mM penicillin and streptomycin). After 2 h of starvation, cells were treated with 10 μM 2-Bromo-deoxyuridine (Sigma) in the presence of VEGF (50 ng/mL) (PeproTech Cat #450-32) for SEVC4-10 or FBS (10%v/v) for E0771 cells. Simultaneously, 1.75 μg/mL EVs were added. Cells were incubated for 6 h (SVEC4-10) or 24 h (E0771) at 37°C 5% CO2. The Brdu internalization was measured by flow cytometry. Briefly, cells were fixed and permeabilized with the BD Cytofix/Cytoperm kit. Then, cells were treated with 300 μg/mL DNase (Sigma Cat#D5025-150KU) for in 1 h at 37°C before the staining with AlexaFluor647 MoBU1-antiBrdU (Cat#B35140 Invitrogen) for 20 min at room temperature. Labelled cells were detected at FACS Canto II. Data analysis was performed with FlowJo software.
Apoptosis assay
1 × 105 SVEC4-10 or E0771 cells were seeded on a 24-well plate in their culture medium (respectively DMEM-ATCC 30-2002 supplemented with 10% heat-inactivated FBS, 1% penicillin and streptomycin and complete Dulbecco modified Eagle medium high glucose with 10% heat-inactivated FBS, 10 mM HEPES and 1 mM penicillin and streptomycin). After 2 h of starvation, 1.75 μg/mL EVs were added to cells. 2 μM Staurosporin (Sigma) was used as a positive control. Cells were incubated for 6 h (SVEC4-10) or 24 h (E0771) at 37°C 5% CO2. Supernatants and cells were collected and stained with Annexin V APC (BD Pharmingen Cat#550475) according to manufacturer’s instruction. Labelled cells were detected at FACS Canto II. Data analysis was performed with FlowJo software.
FITC-gelatin digestion assay
Fluorescently labelled gelatin coverslips were prepared. Briefly, 13 mm-diameter coverslips were coated with Poly-L-Lysine (Sigma-Aldrich) and crosslinked with 0.5% glutharaldehyde (Sigma-Aldrich). Then, they were coated with fluorescently labelled gelatin (BD Cat# 613186) following the manufacturer’s instruction. Fluorescence was quenched in 5 mg/mL Sodium Borohydride (Sigma). 1 × 105 SVEC4-10 or E0771 cells were seeded in complete medium with 50 ng/mL VEGF (PeproTech Cat#450-32) or FBS in the presence, where indicated, of 1.75 μg/mL EVs. Cells were incubated for 6 h (SVEC4-10) or 24 h (E0771) at 37°C 5% CO2. Coverslips were then fixed in Paraformaldehyde 4% in PBS (Sigma-Aldrich) and mounted with ProLongTM Gold Antifade mountant (Thermo Fisher Scientific Cat#P36930). Images were acquired at 40× magnification at confocal microscope Zeiss LSM 800. Data analysis was performed with ImageJ software.
MMP2/MMP9 activity
The EV-mediated alteration of MMP-2/MPP-9 activity was analysed with InnoZyme MMP-2/MMP-9 activity assay kit (Millipore) following manufacturer’s instructions. Briefly, MMP-2/MMP-9 recombinant control was activated with p-Aminophenylmercuric Acetate for 5 min. 200 ng/mL of activated positive control was added in each well. Then, vehicle, gelatinase inhibitor or EVs were added respectively and incubated for 15 min at room temperature. Gelatinase (MMP-2/MMP-9) substrate was added and incubated at 37°C for 2 h. Finally, fluorescence (320 nm ex./405 nm em.) was read with Spark Microplate reader (TECAN).
Measurement of ROS by fluorescence microscopy
ROS production was analysed during the scratch assay. Scratch experiments were performed as previously described, adding 10 µM antimycin (Sigma-Aldrich, A8674) as positive control. After 4 h of incubation at 10% CO2 and 37°C, ROS were detected by using the CM-H2DCFDA probe (2.5 µM; C6827 Thermo Scientific), following the manufacturer’s instructions. Cells were washed with Hank’s Balanced Salt Solution (HBSS, Lonza) supplemented with 2 mM Ca2+. Images were acquired by confocal microscopy (10× objective) using a Leica TCS SP5 microscope (Leica Microsystems) with the LAS-AF (Leica) software. Fluorescence intensity was measured using ImageJ software.
Adenosine quantification
Adenosine production was quantified using the Adenosine Assay Kit (BioVision). The enzymatic reaction was performed adding a 100 μM solution of Adenosine 5-triphosphate (ATP) disodium salt hydrate (Sigma-Aldrich), diluted in water. 1.75 μg of pEVs or cEVs were used. The reaction mix was prepared according to manufacturer’s instruction. The assay was performed in an Optical Plate (Corning). Samples were acquired with a FluoStar Optima plate reader (BMG Labtech), at 5 and 65 min after incubation at 37°C.
Mouse retina neovascularization model
One-day-old C57BL/6 J pups were intraperitoneally injected with 10 µg EVs (three injections). After 4 days, mice were sacrificed for retina collection and measurement of the retinal vascular expansion [45]. For ROS detection in vivo, pups at postnatal day 1 were i.p. injected twice with 2.5 µg EVs at 24 and 4 h before the sacrifice. At the same time, 0.625 µg of MRS 1706 were retro-orbital infused (1.25 µg per pup). Both eyes were enucleated and stained at RT with 0.625 µM Dihydroethidium (DHE-D11347 Thermo Fisher Scientific) for 30 min. Images were acquired with a custom-built multiphoton microscope (Thorlabs) described in [46]. The excitation was set at 800 nm with an average power of 35 mW on the sample. The fluorescent signal was collected with a GaAsP PMT with 525/40 nm bandpass filter, using a 20× water dipping objective. Images analysis was performed by ImageJ.
Matrigel plug assay
Anesthetized, 12-week-old, male, C57BL/6 N mice were subcutaneously injected in the dorsal back with 5 µg EVs, mixed with 400 µL Matrigel (354234 Corning) supplemented with 100 ng/mL VEGF (450–32 Peprotech) and 50 units/mL Heparin. After 7 days, mice were sacrificed and plugs were harvested and weighed. For haemoglobin quantification, plugs were processed by TissueLyser in 250 µL of H2O-milliQ, at the maximal frequency, for 8 min. Haemoglobin content was measured using Drabkin’s reagent kit 525 (Sigma-Aldrich) and normalized to the total protein (Pierce™ BCA Protein Assay Kit, Thermo Fisher Scientific), following the manufacturer’s instructions.
Tumour-bearing mouse models
Syngeneic tumour-bearing mouse model was generated via the orthotropic or the subcutaneous injection of respectively 5 × 105 E0771 or 1.5 × 106 MCA203 cells into female 7-week-old mice. After 7 days, 5 µg EVs were intraperitoneally administered and the injection was repeated every 2 days, using PBS as control. A total of 25 µg EVs were used per mouse. Two days after the last injection, mice were sacrificed, blood and spleen collected, and tumours were harvested, weighed and photographed. The serum was used to assess liver injury, by measuring γ-Glutamyltranspeptidase (γGT) and alanine aminotransferase (ALT), respectively, with the Pchem Gamma-GT Reagent Kit (ADA-R0200000601, Adaltis) and the Pchem ALT-GPT Reagent Kit (ADA-R0200001001, Adaltis). Spleens were mechanically processed for flow cytometry analysis (CD45 BD, clone 30-F11). Tumour masses were stocked at −80°C or processed for flow cytometry and immunofluorescence analysis. For multiparametric analysis, tumours were mechanically and enzymatically digested with 1.5 mg/mL Collagenase II (Gibco) and 0.4 mg/mL DNase I (Roche), before proceeding with the staining. Endothelial cells were labelled with CD45 (BD, clone 30-F11) and CD31 (BD, clone MEC 13.3) antibodies. For immunofluorescence, tumour masses were washed in PBS and immediately fixed in 4% paraformaldehyde overnight at room temperature. Then, they were equilibrated in 15% and 30% of sucrose solution, each step for 48 h at 4°C. Once processed, tumour samples were embedded in O.C.T. compound (Vetrotecnica) on dry ice and stocked at −80°C. Tissue sections (5 μm of thickness) were obtained using a Leica cryostat CM1860 (Leica Biosystems). Sections were rehydrated for 10 min in PBS (P4417, Sigma-Aldrich) and incubated with the blocking solution (1% BSA, 0.3% Triton X-100) for 2 h, at room temperature, before staining. CD31 antibody (MEC 13.3, BD Biosciences) was added in blocking solution at 4°C ON. After washing, the appropriate Alexa-conjugated secondary antibodies were used (Molecular Probes). Nuclei were counterstained with Hoechst 33342 (0.1 μg/mL, Molecular Probes) and mounted with ProLong (Invitrogen). Negative controls included slides incubated with the secondary antibodies alone. Images were acquired using a Leica TCS-SP5 confocal scanning microscope (Leica Microsystems, Wetzlar, Germany) with the software LAS-AF (Leica). Image analysis was performed by ImageJ.
Statistics
The sample size per group was estimated from previous experience with similar experiments. Statistical significance was analysed using Prism Software (GraphPad). Sample comparison was assessed using t-test, Mann–Whitney test or Kruskal–Wallis test with Dunn’s multiple comparisons test and one-way ANOVA according to sample group numerosity and datasets distribution (D’agostino-Pearson and Shapiro–Wilk normality test). Results with a P-value of <0.05 were considered significant.
Study approval
C57BL/6 J mice were purchased from Charles River Laboratories (Calco, Italy). Procedures involving animals and their care conformed to institutional guidelines in compliance with national (4D.L.N.116, G.U., suppl. 40, 18 February 1992) and international (EEC Council Directive 2010/63/UE; National Institutes of Health Guide for the Care and Use of Laboratory Animals) law and policies. The protocols were approved by the Italian Ministry of Health (authorizations n°382/2015-PR, 383/2015-PR and 88/2017-PR). All efforts were made to minimize the number of animals used and their suffering. In all the experiment, mice were sex and age matched, no further randomization was applied.
Results
MSCs primed by pro-inflammatory cytokines secrete anti-angiogenic EVs
EVs were obtained by ultrafiltration of the medium conditioned by murine MSCs. In particular, since the proper licencing of bone marrow MSCs is required to induce an anti-angiogenic phenotype [40,42,47], we isolated EVs released by murine MSCs that were either unstimulated (control-EVs; cEVs) or previously stimulated with a mixture of pro-inflammatory cytokines, following an already published protocol [42] (primed-EVs; pEVs). Validation of the EV isolation procedure was performed by measuring the size of the purified particles by Nanosight and TEM and by analysing the conventional EV markers by western blot [48,49]. Data confirmed that cEVs and pEVs have similar concentration (15 × 108 ± 5 × 106 particles/mL), size, and express the classical exosome markers CD9 and CD63 (Figure S1(a–c)). The vesicles obtained by this protocol were compared to those obtained by ultracentrifugation, and no major differences were identified (Figure S1(d)). cEVs and pEVs were functionally compared for their ability to modulate angiogenesis. We firstly assessed the ability of collected EVs to modify VEGF-induced endothelial cell migration using the scratch wound healing assay [50]. As reported in literature [51–53], 6 h after VEGF stimulation, the murine endothelial cells SVEC4-10 significantly increased their migration of about 30% (Figure 1(a, b)), corresponding to 100 μm (Figure S2(a)). VEGF-induced migration of SVEC4-10 cells was inhibited by pEVs, whereas cEVs had no effect (Figures 1(a, b), S2(a)). However, in the absence of the pro-angiogenic VEGF, EVs did not alter endothelial cell migration (Figure S2(b)). Again, EVs isolated by ultracentrifugation had comparable effects to those purified by ultrafiltration (Figure S2(c)). The specificity of the pEV effect was confirmed by performing the scratch wound healing assay with the conditioned medium deprived of EVs (control conditioned medium-cCM; primed conditioned medium-pCM); results showed that either cCM and pCM did not affect the endothelial migration (Figure S2(d)).
Figure 1.
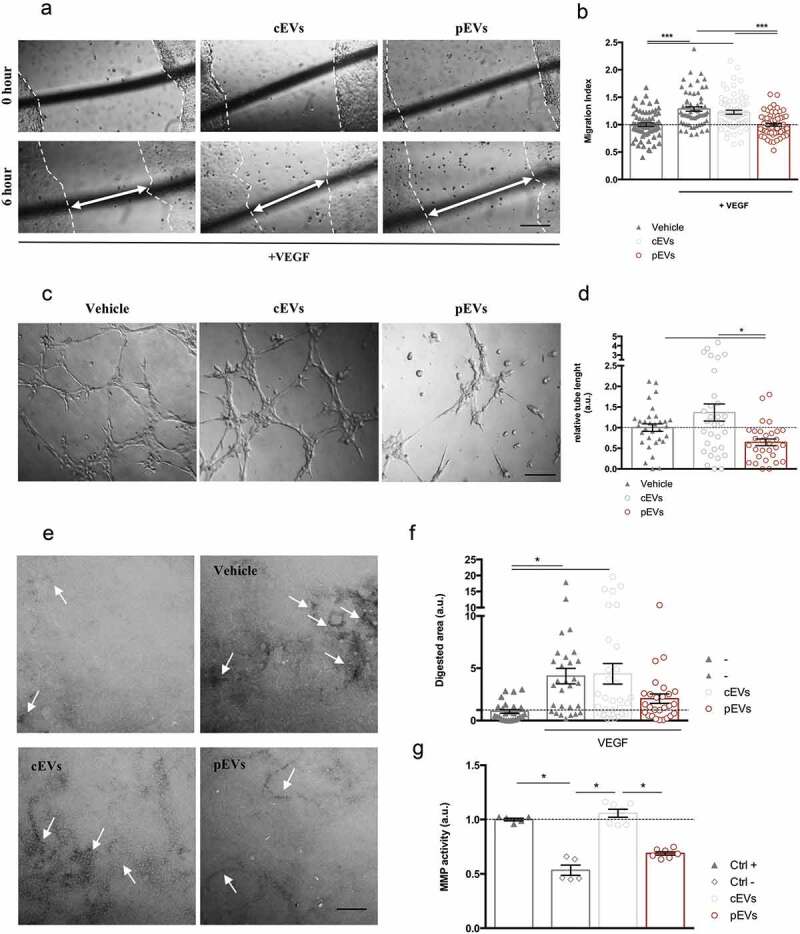
pEVs affect endothelial cell migration and matrix remodelling in vitro. (a, b) Scratch assay was made by scratching a line across the bottom of the dish on a confluent SVEC4-10 monolayer. Cells were treated for 6 h with the medium (Vehicle: DMEM low Glu, 1%P/S, 1% L-Glu; Positive control: Vehicle plus 50 ng/mL VEGF) and 1.75 μg/mL of either cEVs or pEVs. The closure of the wound was quantified with the difference between initial and final scratched areas (Migration Index), calculated by ImageJ and normalized on the vehicle without VEGF. (c, d) For the tube-formation assay SVEC4-10 were seeded in a Matrigel-coated well in the presence of EVs (1.75 μg/mL), using the medium as control (Vehicle: DMEM low Glu, 1%p-s, 1% L-Glu, 10% FBS). After 6 h of incubation, cells were imaged and the quantification of segment length was performed using the ImageJ Angiogenesis Analyze plugin, and normalized on the vehicle. (e, f) The effect on the ECM remodelling was analysed by the FITC-gelatin assay. SVEC4-10 were seeded on the top of the gelatin-coated dishes and cultured for 6hrs with the medium supplemented with 50 ng/mL VEGF in the presence or not of 1.75 μg/mL of either cEVs or pEVs. The vehicle (DMEM low Glu, 1%P/S, 1% L-Glu) was used as control. Cells were then imaged by confocal microscopy and the quantification of the black-digested area (indicated by the with arrows), measured by ImageJ. (g) Analysis of the MMP9/12 activity in the presence of pEVs with InnoZyme MMP2/MMP9 activity assay kit (Millipore) following manufacturer’s instructions. Data are expressed as means ± SEM (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05. Scale bar A-C, 150 μm. Scale bar E, 10 μm.
Data were validated with a different murine endothelial cell line, mile sven 1 (MS1) (Figure S3(a)), and using primary human endothelial cells, treated with EVs derived from human bone marrow derived – MSCs (Figure S3(b)).
We then analysed whether EVs were able to interfere with the ability of endothelial cells to form in vitro capillary-like structures, using the tube formation assay [54]. We found that both murine and human pEVs, but not cEVs, inhibit the formation of tube networks (Figures 1(c, d), S3(c)). Again, the isolation procedure did not influence the EV function (S4A), and cCM and pCM had no effect in the same experimental procedure (Figure S4(b)).
Since alterations of the in vitro tube formation might be related to cell proliferation and survival or to the matrix (Matrigel) digestion, we investigated the direct effect of pEVs on endothelial functions. In particular, we noted that EVs were not able to modify either the EC proliferation (evaluated by BrdU incorporation, Figure S5(a)) or their vitality (Annexin positivity, Figure S5(b)). In contrast, pEVs inhibited the ability of EC to digest the matrix, as assessed by the gelatin degradation assay (Figure 1(e, f)). As expected, SVEC4-10 stimulated by VEGF showed higher propensity to matrix digestion (Figure 1(e, f)), but this was inhibited by pEVs, whereas cEVs had no effect (Figure 1(e, f)). We then analysed the ability of EVs to directly inhibit metalloproteinases (MMPs) activity. In agreement with the previous results, we found that pEVs have a direct, dose-dependent inhibitory effect on MMP activity (Figure 1(g), S6).
On the basis of these in vitro approaches, we concluded that EVs released by primed MSCs affect two important processes required for angiogenesis: VEGF-induced migration and ECM digestion. This hypothesis was then verified exploiting two different in vivo models. We implanted matrigel plugs supplemented with VEGF and EVs in the dorsal back of C57BL/6 mice to analyse induced vascularization [45]. Quantification of haemoglobin content in explanted plugs revealed that pEVs reduce vessel formation in vivo, whereas cEVs have no effects (Figure 2(a)). Furthermore, we analysed the postnatal retinal development in mice treated with EVs [55]. Remarkably, pEVs significantly decreased the retina vascular arborisation, affecting both the relative radial expansion and the relative branching points (Figure 2(b, c)). Again, cEVs had no effects, thus confirming the essential role of the correct MSC licencing to the acquisition of anti-angiogenic property and the crucial role of secreted EVs in controlling the process.
Figure 2.
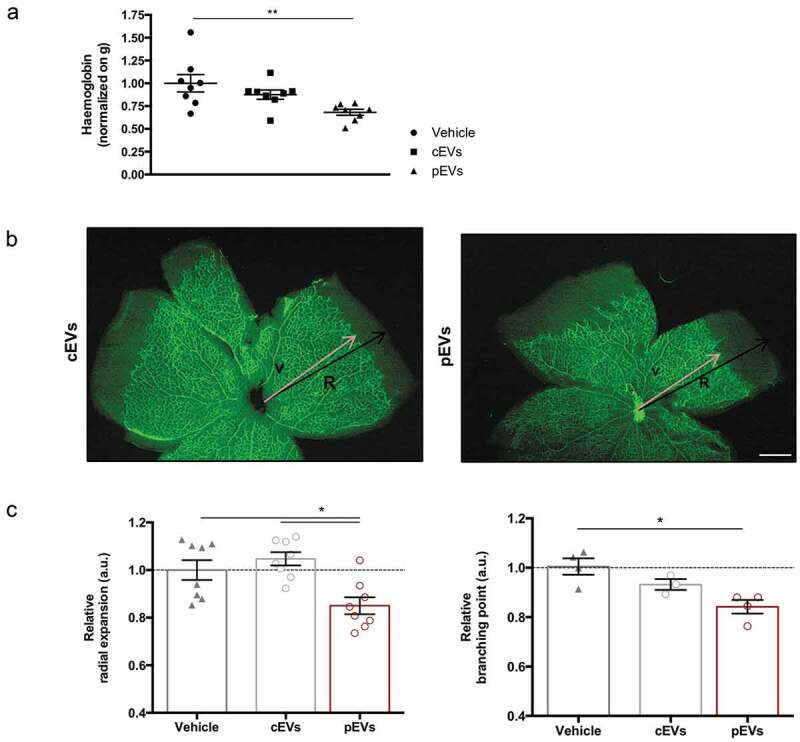
pEVs inhibit angiogenesis in vivo. (a) 12-week-old male C57BL/6J mice were subcutaneously injected with either 5 µg of cEVs and pEVs, mixed with Matrigel Matrix supplemented with VEGF 100 ng/mL (450-32 Peprotech) and Heparin 50 units/mL. After 7 days, plugs were harvested. For haemoglobin quantification, plugs were processed by TissueLyser and the haemoglobin content was measured using Drabkin’s reagent kit 525 (Sigma-Aldrich). Each value was first normalized on the total plug protein quantity, measured by BCA assay, and then on the negative control (plugs with vehicle). (b, c) 1-day-old C57BL/6J mouse pups were intraperitoneally injected with a total of 10 μg of cEVs or pEVs, using PBS as control. Mice were sacrificed for retina collection. Dissected retina were stained with isolectin-b4 (green) and digital images were captured using inverted fluorescence confocal microscope. Analyses of the relative radial expansion and of the relative branching point were performed. All data are expressed as means ± SEM, normalized on control (mice treated with vehicle) (n = 8 mice/group). Ordinary one-way ANOVA; *P < 0.05.
pEVs induce oxidative stress in migrating endothelial cells
We had recently shown that in vivo administration of TIMP-1 inhibits the inflammation-induced angiogenesis within draining lymph nodes [42]. Notably, we observed that TIMP-1 is highly enriched in pEVs (Figures 3(a-b), S1(d)). In particular, co-immunoprecipitation experiments indicated that TIMP-1 interacts with the cell-surface protein CD63, confirming previous findings [56,57] (Figure S7(a)). Consistently with our aforementioned work [42], the use of αTIMP-1 blocking antibody rescued the ability of endothelial cells to form capillary-like structures in the presence of pEVs (Figure 3(c)). In the same line, EVs isolated from the medium conditioned by stimulated MEF, not carrying TIMP-1 (Figure S7(b)), did not affect the tube formation of SVEC4-10 (Figure S7(c)). However, the TIMP-1 blocking antibody could not restore VEGF-induced cell migration (Figure 3(d)), thus suggesting the contribution of at least another, TIMP1-independent, mechanism in the alteration of the migration process by pEVs.
Figure 3.
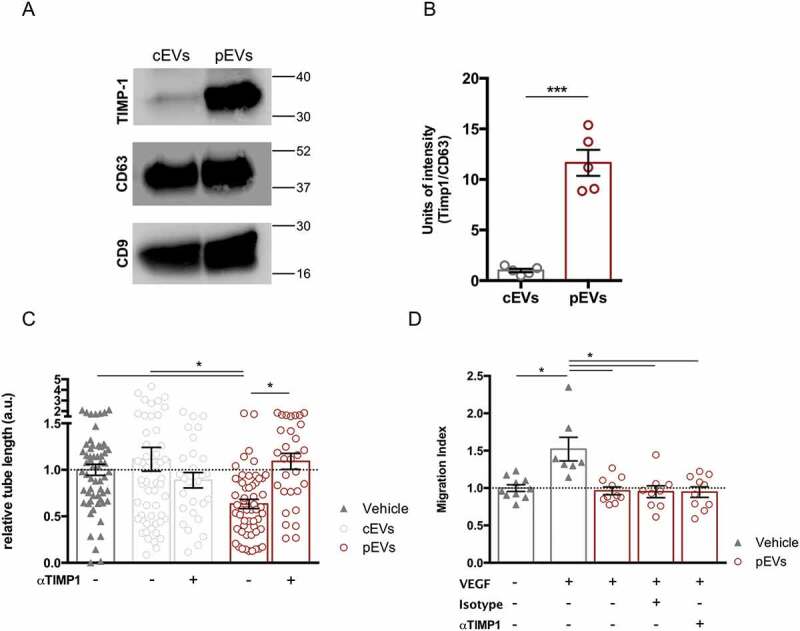
TIMP-1 carried by pEVs affects tube formation, but not endothelial migration in vitro. (a) Western Blot and relative quantification showing TIMP-1 overexpression in pEVs. Unpaired T test; *P < 0.05. (b) Analysis of the relative tube lengths in the tube formation assay. The treatment of SVEC4-10 with the anti-TIMP-1 5 µg/mL (AF980 R&D) rescued the tube formation. (c) Conversely, the analysis of relative migration in the scratch assay in the presence of the same anti-TIMP-1 antibody did not shown any restoration of the endothelial migration ability in the presence of pEVs. Data are expressed as means ± SEM (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05.
Reactive oxygen species (ROS) are critical regulators of endothelial cells. Although controlled ROS production may promote cell migration [58–62], oxidative stress is a well-known inhibitor of endothelial cell motility and function [63–66]. We measured ROS levels in in vitro migrating endothelial cells in the presence of either cEVs or pEVs, using Antimycin A (AA) as positive control (Figure S8(a)). VEGF slightly induced ROS production in endothelial cells and cEVs did not alter this process (Figure 4(a, b)). Conversely, pEVs induced a dramatic accumulation of ROS in migrating endothelial cell (Figure 4(a, b)). Similar results were collected when pEVs were administered in vivo and retinal developing vessels were analysed (Figure 4(c, d)). To identify the source of ROS in pEV-treated endothelial cells, we focused our attention on NADPH oxidase 2 (NOX2), which known to play a key role in the production of endothelial ROS [67–69]. Thus, we selectively inhibited the activity of the NOX2 in the scratch wound-healing assay (Figure 4(e)). Interestingly, NOX2 inhibition in vitro had no effect on VEGF-induced endothelial cell migration, whereas it completely abolished the anti-migratory effect of pEVs (Figure 4(e)).
Figure 4.
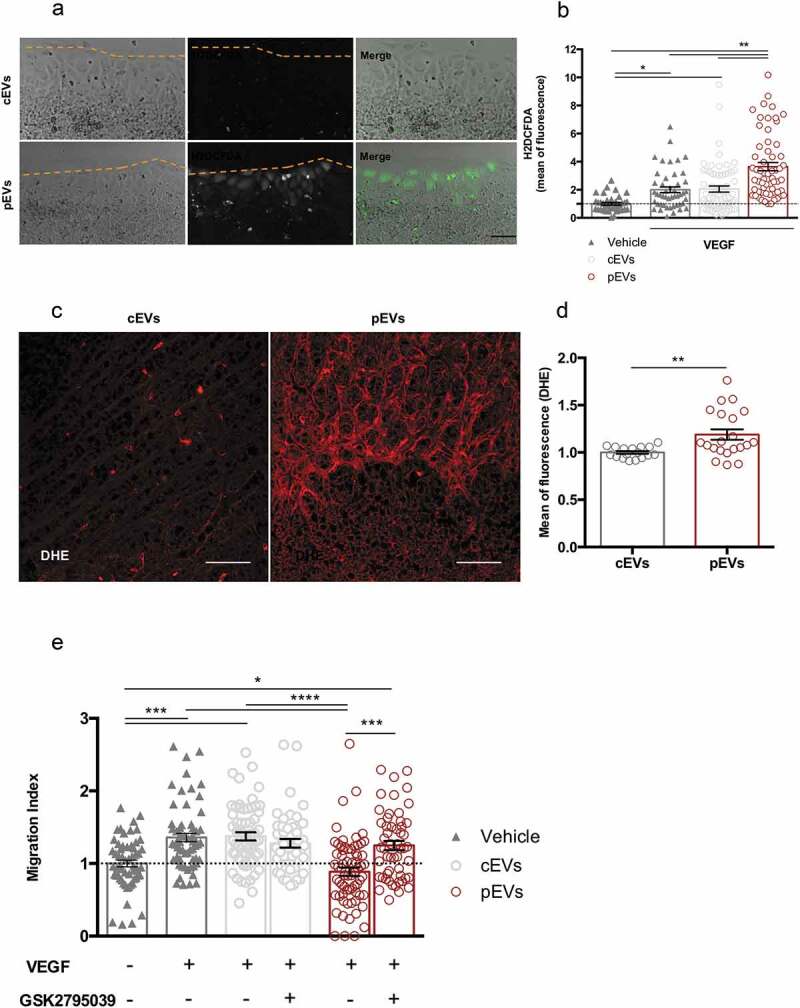
pEVs induce endothelial ROS accumulation both in vitro and in vivo. (a, b) To analyse a potential implication of oxidative stress in the pEV-mediated inhibition of endothelial migration, a scratch assay was performed. After 4 h of incubation with VEGF alone or in combination with cEVs/pEVs, intercellular ROS levels were detected with the 2.5 µM CM-H2DCFDA probe (green). Cells were imaged with Leica fluorescent microscope and the mean of fluorescence calculated with ImageJ. Scale bar 50 μm. Data are expressed as means ± SEM, normalized on the negative control, medium without VEGF (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05. (c, d) To confirm the involvement of oxidative stress in the anti-angiogenic effect of pEVs in vivo, we treated 5-days-old C57BL/6J mouse pups with a total of 5 µg of cEVs or pEVs. After one day, collected retinas were dyed with DHE 0.625 µM for ROS detection (red). Digital images were captured using two-photons microscopy and the mean of fluorescence was calculated with ImageJ. Scale bar 150 μm. Data are expressed as means ± SEM normalized on control, mice treated with cEVs. (n = 5 mice/group). Unpaired t test; *P < 0.05. (e) We assessed the crucial activity of NOX2 enzyme in the ROS-production by performing the scratch wound healing assay with 100nM GSK2795039 (HY-18950 MedChem Express), a NOX2 specific inhibitor. Data are expressed as means ± SEM (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05.
Altogether, there results indicate that pEVs decrease endothelial cell migration by triggering NOX2 and inducing oxidative stress.
pEVs induce endothelial oxidative stress by producing adenosine
Among the stimuli that may induce NOX2 activity in endothelial cells, adenosine receptors seem to play a major role [70,71]. Adenosine is produced by extracellular ATP hydrolysis through the concerted action of two enzymes, the ectonucleotidases CD39 and CD73 [72]. Since EVs derived from different types of cells express ectonucleotidases [73–75], we hypothesized that pEVs accumulate extracellular adenosine that, in turn, stimulates the NOX2 activity responsible for the detrimental ROS accumulation in migrating endothelial cells.
To verify our hypothesis, the expression of CD39 (Figure S9(a)) and CD73 (Figure 5(a), S1(d)) in EVs was analysed by western blot. We found that EVs carry CD39 and CD73; however, pEVs showed higher levels of CD73 (Figure 5(a)), suggesting that these vesicles are more efficient in adenosine production. Indeed, the measurement of the extracellular adenosine amount, produced by the ATP hydrolysis by the same quantity of EVs (1.75 μg), revealed that pEVs generate more adenosine than cEVs (Figure 5(b)). Similar results were obtained with human EVs (Figure S9(b, c)).
Figure 5.
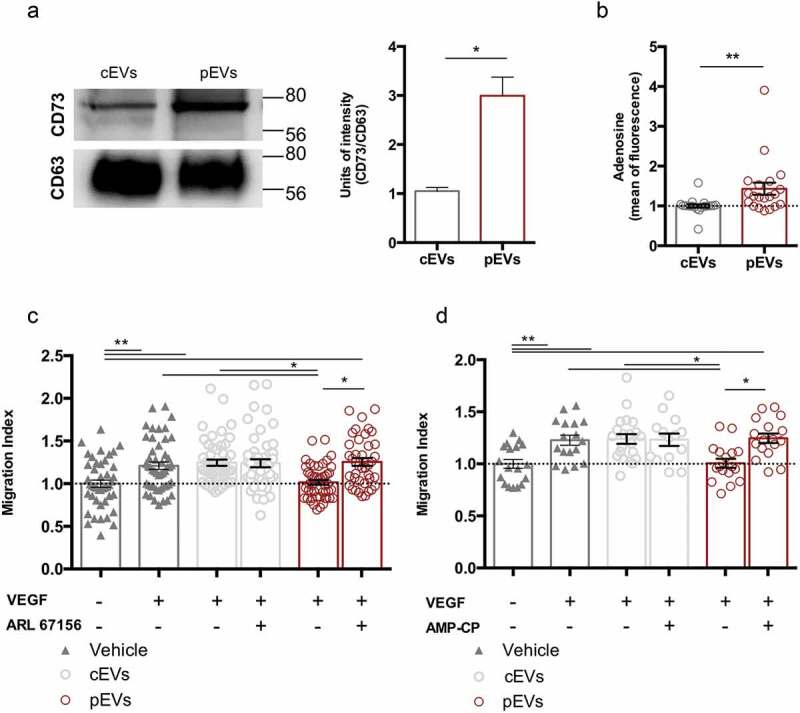
Ectonucleotidases carried by pEVs are crucial for their anti-angiogenic potential in vitro. (a) The expression of ectonucleotidases in EVs was validated by WB, using CD63 as housekeeping. Data are normalized on cEV level and expressed as means ± SEM (3 independent experiments). Unpaired t test, *P < 0.05. (b) The ATP-hydrolyse ability of EVs was checked by using a fluorimetric adenosine assay kit. The mean of fluorescence, proportional to the adenosine amount, was normalized on the cEVs. Data are expressed as means ± SEM (3 independent experiments). Mann–Whitney test, *P < 0.05. (c, d) We demonstrated the causative role of ectonucleotidase expression in the anti-angiogenic effect of pEVs, by exploiting the scratch wound healing assay. Here, we blocked both CD39 and CD73, through, respectively, 200 µM ARL 67156 (ARL67156A265 Sigma-Aldrich) and diphosphate ADP analogue AMP-CP M3763 Sigma-Aldrich). We measured the Migration Index, and normalized it on the medium without VEGF. Data are expressed as means ± SEM (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05.
Then, we performed the scratch wound healing assay in the presence of either CD39 or CD73 inhibitors (respectively, ARL 67156 and AMP-CP) (Figure 5(c-d)). The inhibition of each enzyme fully restored the migration of SVEC4-10 treated with pEVs, but had no effect on untreated or cEV-treated endothelial cells. In agreement, we found that CD73 activity is necessary for pEVs to induce ROS accumulation in SVEC4-10 migrating cells (Figure 6(a-b)).
Figure 6.
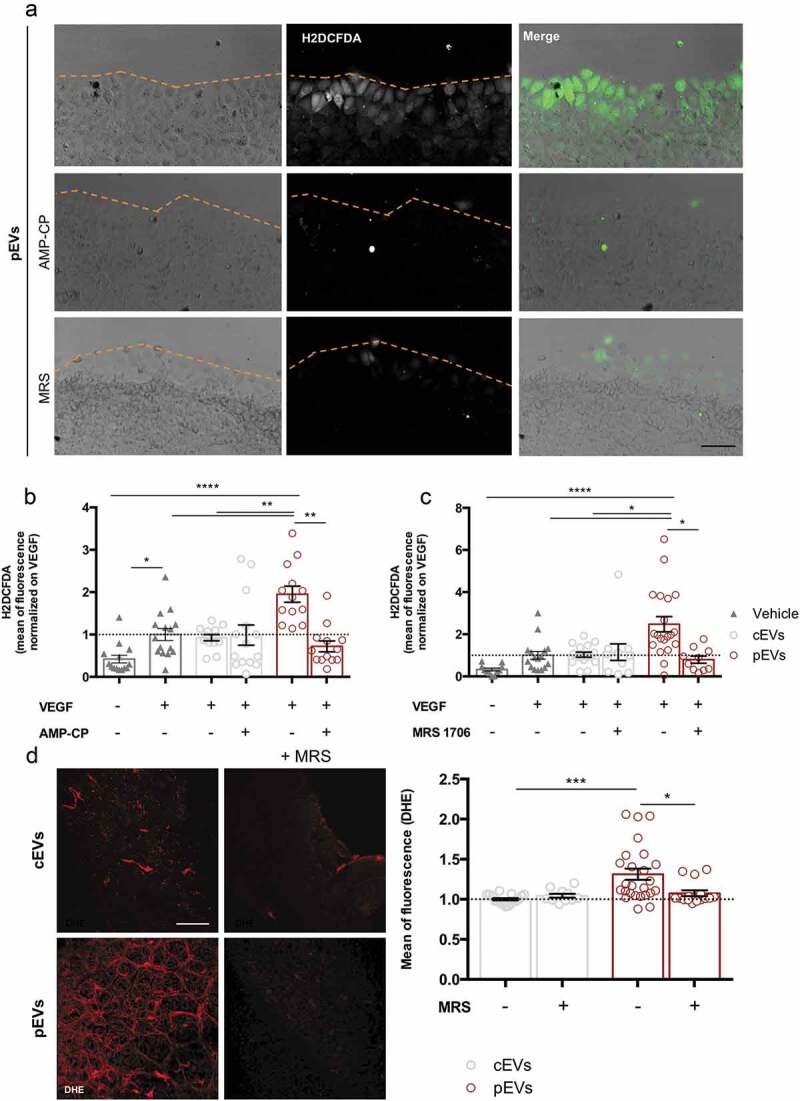
pEV-generated adenosine triggers ROS accumulation in migrating endothelial cells. In a scratch experiments, the employment of specific chemical inhibitors of CD73 (AMP-CP), and of A2BAR (MRS1706) blocked significantly the ROS production, detected with H2DCFDA (green). (a) Representative pictures. Scale bar 50 μm. (b, c) The mean of fluorescence was calculated by ImageJ and normalized on the medium supplemented with VEGF. Data are expressed as means ± SEM (3 independent experiments). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05. (d) The pre-treating of pups with 1.25 µg MRS1706, to block the A2BAR, inhibits pEVs-dependent ROS increase. Retinas were dyed with DHE 0.625µM for ROS detection. Digital images were captured using two-photons microscopy and the mean of fluorescence was calculated with ImageJ. Scale bar 150 μm. Data are expressed as means ± SEM (n = 5) Kruskal-Wallis test with Dunn’s multiple comparisons test; *P < 0.05.
To identify the adenosine receptor responsible for NOX2 activation in pEV-treated endothelial cells, we first analysed SVEC4-10 cells for the expression of A2A and A2B adenosine receptors (A2AAR and A2BAR), the predominantly expressed adenosine receptors in ECs [76]. Western Blot and immunofluorescence analyses revealed that SVEC4-10 express A2BAR only (Figure S10(a, b)). Accordingly, inhibition of A2BAR by its inverse agonist (MRS 1706) prevented ROS accumulation in SVEC4-10 treated with pEVs (Figure 6(a-c)), and completely restored their migration ability (Figure S11(a)). Consistently, A2BAR stimulation through a specific A2BAR agonist (BAY-606583) perfectly mimicked the effects of pEVs on in vitro migrating endothelial cells (Figure S11(B,C)). In vivo, the A2BAR inverse agonist MRS 1706 prevented accumulation of oxidative stress within the retinal endothelium of pups treated with pEVs (Figure 6(d)), thus confirming the major role of A2BAR in the induction of endothelial oxidative stress in response to pEV treatment.
pEVs inhibit tumour associated angiogenesis
Prompted by our results, we performed pilot experiments to assess the angiogenic effects of pEVs in a mouse model of pathological angiogenesis. Thus, we injected murine mammary carcinoma cells, E0771 cells, in syngeneic mice and analysed tumour growth and vascularization. E0771 cells were orthotopically injected in 7-week-old female mice and, one week later, mice were intraperitoneally treated with EVs every 2 days, for a total of five injections. Analyses of the tumour growth (Figure 7(a–c)) and of the tumour vascularization (haemoglobin content and percentage of CD45neg/CD31pos endothelial cells, Figure 7(d,e)) showed that pEV treatment was effective in reducing both these parameters. The effects of pEV administration on the tumour endothelial compartments were confirmed by confocal microscopy analyses on tumour sections immunostained for the endothelial marker CD31 (Figure 7(f,g)). Major systemic alterations were not registered as a consequence of EV treatments (Figure S12(a,b)). Results were further validated in mice bearing a fibrosarcoma, induced by the injection of MCA203 cells. Also in this second tumour model, we confirmed that pEV treatment results in reduced tumour vascularization and growth (Figure S13 (a–c)). A direct effect of pEVs on the survival, proliferation and migration of E0771 cells was analysed by in vitro assays (respectively, Figure S14(a–c)), which showed that pEVs do not affect these parameters. However, in agreement with the results obtained with endothelial cells, pEVs were able to reduce gelatin degradation by E0771 cells (Figure S14(d)).
Figure 7.
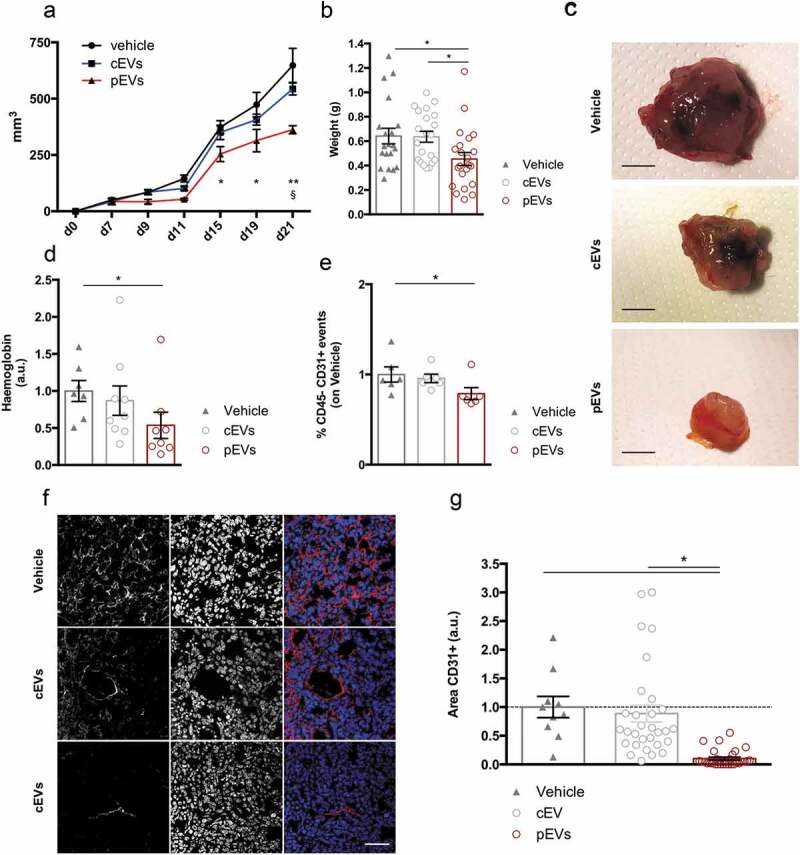
pEVs control pathological tumour-associated angiogenesis. . E0771 tumor cells were subcutaneously injected in 7-weeks old female mice. (a) The mass growth was measured. 2 way ANOVA; *P < 0.05 between PBS and pEVs. $P < 0.05 between cEVs and pEVs. (b, c) At the day of the sacrifice, tumours were pictured and weighted. The vascularization of the tumors was quantified by the analysis of the haemoglobin content, trough the Drabkin’s reagent kit 525 (Sigma-Aldrich (d), and by the calculating the percentage of endothelial cells by flow cytometry (e). Data are expressed as means ± SEM (at least 6 mice/group). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05. (f, g) Tumor cryosections were stained for CD31 (red). The positive areas were calculated with ImageJ and normalized on the vehicle. Scale bar 75 μm. Data are expressed as means ± SEM (5 mice/group). Kruskal–Wallis test with Dunn’s multiple comparisons test; *P < 0.05.
Discussion
Abnormal vascular remodelling causes or contributes to several diseases. In particular, an excessive sprouting of vessels has been reported in a long list of disorders, including cancer, psoriasis, arthritis, blindness, obesity, asthma, atherosclerosis and infectious disease [1]. Conversely, an insufficient vascularization depicts, for instance, heart and brain ischaemia, neurodegeneration, hypertension, pre-eclampsia, respiratory distress and osteoporosis [1].
Recently, the stromal compartment has emerged as a major regulator of endothelial cell functions [77]. Specialised fibroblasts, perycites and MSCs are all involved in the control of the vascular architecture, mainly by recruiting endothelial progenitor cells or releasing pro-angiogenic factors [78–81], and, for what concerns pericytes, by regulating blood vessel diameter, vessel permeability and endothelial cell proliferation [82]. It is therefore not surprising that, in addition to the classical strategies aimed to control angiogenesis by direct targeting of the endothelium, novel therapeutic approaches are being developed focusing on the stromal compartment [83]. For instance, several pre-clinical studies attempted to inhibit the activity of the pro-angiogenic cancer-associated fibroblasts (CAFs) with nonsteroidal anti-inflammatory drugs (NAIDs), in mammary gland involution [84]. With the same aim, inhibitors of FAP, which is a membrane-bound serine protease selectively expressed in fibroblasts, have been used to control lung and colon cancer progression [85]. Conversely, recruited or injected stromal cells have been exploited to sustain a prolonged and local expression of angiogenic growth factors to improve myocardial perfusion [86] and for recovery from ischaemic disease [87]. Despite this sprouting number of studies focused on the development of stroma-related therapies, several issues have to be overcome for an efficient and safe clinical translation [83]. Fibroblasts, pericytes and MSCs are essential components of healthy tissues and a successful therapy requires the identification of the precise target in the specific context. Thus, it is pivotal to understand how stromal cells modulate endothelial cell functions in various physio-pathological conditions.
It is now well established that several effects of stromal cells are mediated by paracrine signals [88], and that these signals are largely convoyed by EVs [89]. EVs are attracting increasing interest as therapeutic tools in place of their cells of origin, because of several reasons. Mostly, they cause less concern regarding the risks of ectopic colonization and tumorigenesis. Moreover, their effect should be more reproducible and less dependent on the environment of the target tissue, compared to living cells [90,91]. In the context of angiogenesis, previous studies have analysed the properties of MSC-derived EVs [92,93], especially those generated under hypoxic conditions [94–96], and found that they promote angiogenesis mainly through the modulation of VEGF [93,97–99]. Conversely, here we demonstrated that licencing of bone marrow MSCs by pro-inflammatory cytokines induces the secretion of EVs with strong anti-angiogenic properties.
This evidence supports the concept of MSCs as “sensors” of the surrounding environment. In accordance with the literature, we can hypothesize that in hypoxic conditions, when the generation of new vessels is required, MSCs secrete EVs promoting angiogenesis. On the opposite way, in an inflammatory microenvironment, MSCs release EVs that inhibit endothelial cell migration and formation of new vessels.
Vessel branching and expansion requires the spatiotemporal integration of several processes including ECM remodelling, cell migration and cell differentiation. Remodelling of the ECM is required for proper migration of endothelial cells and it depends on the balance between metalloproteases (MMPs) and their endogenous inhibitors. We recently demonstrated that TIMP-1 has a key role in determining MSC anti-inflammatory and anti-angiogenic properties [42] and that both human and mouse MSCs secrete TIMP-1 in response to a pro-inflammatory microenvironment [40]. The results presented in this manuscript demonstrate that TIMP-1 is carried by EVs through CD63 and that EVs are responsible for the anti-angiogenic properties of MSCs. However, our results indicate that, in addition to TIMP-1, EVs exert their anti-angiogenic effects through a novel mechanism based on the generation of extracellular adenosine.
Adenosine is a multifunctional signalling molecule able to control inflammation and tissue homoeostasis by activating G-protein-coupled adenosine receptors (A1, A2A, A2B and A3 adenosine receptors) [100]. Extracellular ATP accumulates in the local microenvironment during cell damage, hypoxia, or inflammation and adenosine is produced by the concerted activity of the ectonucleotidases CD39 and CD73. CD39 and CD73 are indeed expressed by several immune cells, including monocytes, dendritic cells, and several T-cell subsets and they have been widely considered key players in the generation of immunosuppressive microenvironments [101,102]. Interestingly, during an inflammatory response, adenosine also regulates endothelial cell activation. Indeed, it has been demonstrated that the accumulation of extracellular adenosine reduces the expression of adhesion molecules in endothelial cell, thus preventing the recruitment of leukocytes into the injured tissue [103,104]. Our data identified a specific upregulation of CD73 in EVs released by MSCs primed by pro-inflammatory cytokines, with a consequent increment in their ability to generate adenosine. Interestingly, our data identify endothelial cells as a crucial target of EV-produced adenosine. Furthermore, these results identify a novel mechanism responsible for adenosine-mediated modulation of angiogenesis involving A2BAR, NOX2 activation and ROS production.
It is largely described that ROS act as a double-edged sword during the vascular remodelling [105]: while low levels of ROS are required to support cell growth, migration, differentiation and gene expression [106,107], high levels are cytotoxic and mutagenic [108]. In particular, sustained endothelial oxidative stress triggers cell death, apoptosis and senescence, thus blocking angiogenesis [109–111]. ROS can be produced by a variety of mechanisms, with NOX2 representing one of the most important during inflammation. Interestingly, several studies connect adenosine receptor stimulation with NOX2 activity [58,71,112]. In our experimental systems, selective inhibition of NOX2 abolished the production of ROS in endothelial cells induced by pEVs, indicating that endothelial NOX2 is triggered by pEVs, both in vitro and in vivo. Moreover, our data indicate that NOX2-dependent ROS production is induced by stimulation of endothelial A2BAR.
Although additional studies are required to elucidate the bio-distribution and pharmacokinetics of EVs, our results, together with previous studies [40,42,113], suggest that the endothelium might represent the first and more attainable target of MSC-products. Therefore, the identification of the molecular mechanisms underlying the angiogenic role of EVs provides novel insights for the development of innovative therapeutic approaches to target pathological angiogenesis. Our results, indeed, prompted us to perform proof-of-concept experiments aimed at exploiting pEVs to target cancer angiogenesis.
A number of approaches have been used to block tumour-associated angiogenesis in cancer patients. Among the approved angiogenesis inhibitors for the treatment of human cancer there are humanized VEGF-specific monoclonal antibody (Bevacizumab), VEGFR-2 monoclonal antibody (Ramucirumab), and several small molecule multi-kinase inhibitors (MKIs), mainly targeting the VEGF receptor (VEGFR) tyrosine kinases, such as sunitinib, sorafenib, pazopanib hydrochloride and axitinib [114]. For instance, the combination of bevacizumab with chemotherapy has been shown to be effective for the treatment of primary peritoneal cancer, metastatic colorectal and breast cancer, advanced nonsquamous non-small cell lung cancer (NSCLC), renal cell carcinoma, and ovarian cancers [115]. In addition, gastrointestinal stromal tumours and hepatocellular carcinomas have been treated with sunitinib and sorafenib, respectively [116]. However, the absence of long-lasting clinical responses and the occurrence of systemic side-effects in most of the treated patients limit the efficacy of the aforementioned inhibitors [117,118].
Although our study provides preliminary results indicating that pEVs are able to control tumour-associated angiogenesis, the effects of pEVs in the context of tumour growth may be much more complex. The gelatin degradation experiment performed with E0771 cells suggested a possible implication of pEVs in the regulation of the tumour invasiveness. Moreover, within the tumour microenvironment, EVs may modulate the activity of infiltrating cells, which could, in turn, affect tumour growth. Further studies have to be performed to deeply investigate these processes.
In conclusion, this study identifies EVs released by stromal cells as a crucial and modifiable regulator of endothelial cell functions and, by the identification of the molecular mechanisms, it paves the way for developing new antiangiogenic therapies.
Supplementary Material
Acknowledgments
We thank Sara Zumerle for the scientific discussion and Fabio Munari for technical advice. We thank Federica Collino for critical reading of the manuscript. We also thank Mattia Albiero for ALT and yGT quantification and A.M. Merino Rodriguez and Martin J. Hoogduijn for the EV measurements.
Authorship
Contributions: RA designed and performed most of the experiments and wrote the manuscript; CL provided technical assistance throughout the project; SH performed part of the in vivo experiments; RS performed all the western blots; GB acquired the two-photons pictures; EM provided technical assistance during the revision; MM provided suggestions and wrote the paper; BC performed part of the experiments and wrote the paper; AV coordinated the study, wrote the manuscript and provided funds.
Declaration of interest statement
AV and MM declare that they are inventors in a patent application for the production of pEVs (W02018109700A1).
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9(6):653–20. [DOI] [PubMed] [Google Scholar]
- [2].Bikfalvi A. History and conceptual developments in vascular biology and angiogenesis research: a personal view. Angiogenesis. 2017;20:463–478. [DOI] [PubMed] [Google Scholar]
- [3].Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–427. [DOI] [PubMed] [Google Scholar]
- [4].Usui Y, Westenskow PD, Murinello S, et al. Angiogenesis and eye disease. Annu Rev Vis Sci. 2015;1:155–184. [DOI] [PubMed] [Google Scholar]
- [5].Cabral T, Mello LGM, Lima LH, et al. Retinal and choroidal angiogenesis: a review of new targets. Int J Retina Vitreous. 2017;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Sagar SM, Yance D, Wong RK. Natural health products that inhibit angiogenesis: a potential source for investigational new agents to treat cancer-Part 1. Curr Oncol Tor Ont. 2006;13:14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Pollina EA, Legesse-Miller A, Haley E, et al. Regulating the angiogenic balance in tissues: a potential role for the proliferative state of fibroblasts. Cell Cycle Georget Tex. 2008;7:2056–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Newman AC, Nakatsu MN, Chou W, et al. The requirement for fibroblasts in angiogenesis: fibroblast-derived matrix proteins are essential for endothelial cell lumen formation. Mol Biol Cell. 2011;22:3791–3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rohani MG, Parks WC. Matrix remodeling by MMPs during wound repair. Matrix Biol. 2015;44–46:113–121. [DOI] [PubMed] [Google Scholar]
- [10].Li B, Wang JH-C. Fibroblasts and myofibroblasts in wound healing: force generation and measurement. J Tissue Viability. 2011;20(4):108–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Iyer VR, Eisen MB, Ross DT, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283(5398):83–87. [DOI] [PubMed] [Google Scholar]
- [12].Darvishi B, Majidzadeh-A K, Ghadirian R, et al. Recruited bone marrow derived cells, local stromal cells and IL-17 at the front line of resistance development to anti-VEGF targeted therapies. Life Sci. 2019;217:34–40. [DOI] [PubMed] [Google Scholar]
- [13].Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell. 2011;21:193–215. [DOI] [PubMed] [Google Scholar]
- [14].Chang WG, Andrejecsk JW, Kluger MS, et al. Pericytes modulate endothelial sprouting. Cardiovasc Res. 2013;100:492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Durham JT, Surks HK, Dulmovits BM, et al. Pericyte contractility controls endothelial cell cycle progression and sprouting: insights into angiogenic switch mechanics. Am J Physiol Cell Physiol. 2014;307:C878–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Simonavicius N, Ashenden M, van Weverwijk A, et al. Pericytes promote selective vessel regression to regulate vascular patterning. Blood. 2012;120(7):1516–1527. [DOI] [PubMed] [Google Scholar]
- [17].Castro-Malaspina H, Gay RE, Resnick G, et al. Characterization of human bone marrow fibroblast colony-forming cells (CFU-F) and their progeny. Blood. 1980;56(2):289–301. [PubMed] [Google Scholar]
- [18].Méndez-Ferrer S, Michurina TV, Ferraro F, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466(7308):829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ono N, Ono W, Mizoguchi T, et al. Vasculature-associated cells expressing nestin in developing bones encompass early cells in the osteoblast and endothelial lineage. Dev Cell. 2014;29:330–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Watt SM, Gullo F, van der Garde M, et al. The angiogenic properties of mesenchymal stem/stromal cells and their therapeutic potential. Br Med Bull. 2013;108(1):25–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lin R-Z, Moreno-Luna R, Zhou B, et al. Equal modulation of endothelial cell function by four distinct tissue-specific mesenchymal stem cells. Angiogenesis. 2012;15:443–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Smadja DM, d’Audigier C, Guerin CL, et al. Angiogenic potential of BM MSCs derived from patients with critical leg ischemia. Bone Marrow Transplant. 2012;47:997–1000. [DOI] [PubMed] [Google Scholar]
- [23].Chen J, Zhang ZG, Li Y, et al. Intravenous administration of human bone marrow stromal cells induces angiogenesis in the ischemic boundary zone after stroke in rats. Circ Res. 2003;92:692–699. [DOI] [PubMed] [Google Scholar]
- [24].Miyahara Y, Nagaya N, Kataoka M, et al. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat Med. 2006;12:459–465. [DOI] [PubMed] [Google Scholar]
- [25].Ransohoff JD, Wu JC. Imaging stem cell therapy for the treatment of peripheral arterial disease. Curr Vasc Pharmacol. 2012;10(3):361–373. Available from: http://www.eurekaselect.com/96299/article [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tao H, Han Z, Han ZC, et al. Proangiogenic Features of Mesenchymal Stem Cells and Their Therapeutic Applications. Stem Cells Int. 2016;2016:1314709. Available from: https://www.hindawi.com/journals/sci/2016/1314709/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kwon HM, Hur S-M, Park K-Y, et al. Multiple paracrine factors secreted by mesenchymal stem cells contribute to angiogenesis. Vascul Pharmacol. 2014;63:19–28. [DOI] [PubMed] [Google Scholar]
- [28].Todorova D, Simoncini S, Lacroix R, et al. Extracellular vesicles in angiogenesis. Circ Res. 2017;120:1658–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].van der Pol E, Böing AN, Harrison P, et al. Classification, functions, and clinical relevance of extracellular vesicles. Pharmacol Rev. 2012;64:676–705. [DOI] [PubMed] [Google Scholar]
- [30].Raposo G, Stoorvogel W. Extracellular vesicles: exosomes, microvesicles, and friends. J Cell Biol. 2013;200:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Teng X, Chen L, Chen W, et al. Mesenchymal Stem Cell-Derived Exosomes Improve the Microenvironment of Infarcted Myocardium Contributing to Angiogenesis and Anti-Inflammation. Cell Physiol Biochem. 2015;37(6):2415–2424. [DOI] [PubMed] [Google Scholar]
- [32].Doeppner TR, Herz J, Görgens A, et al. Extracellular vesicles improve post-stroke neuroregeneration and prevent postischemic immunosuppression. Stem Cells Transl Med. 2015;4:1131–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anderson JD, Johansson HJ, Graham CS, et al. Comprehensive proteomic analysis of mesenchymal stem cell exosomes reveals modulation of angiogenesis via nuclear factor-KappaB signaling. Stem Cells Dayt Ohio. 2016;34:601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sahoo S, Klychko E, Thorne T, et al. Exosomes from human CD34+ stem cells mediate their pro-angiogenic paracrine activity. Circ Res. 2011;109:724–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liang X, Zhang L, Wang S, et al. Exosomes secreted by mesenchymal stem cells promote endothelial cell angiogenesis by transferring miR-125a. J Cell Sci. 2016;129:2182–2189. [DOI] [PubMed] [Google Scholar]
- [36].Ferguson SW, Wang J, Lee CJ, et al. The microRNA regulatory landscape of MSC-derived exosomes: a systems view. Sci Rep. 2018;8(1):1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Marfy‐Smith SJ, Clarkin CE. Are mesenchymal stem cells so bloody great after all? Stem Cells Transl Med. 2017;6:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Otsu K, Das S, Houser SD, et al. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood. 2009;113(18):4197–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Menge T, Gerber M, Wataha K, et al. Human mesenchymal stem cells inhibit endothelial proliferation and angiogenesis via cell–cell contact through modulation of the VE-Cadherin/β-catenin signaling pathway. Stem Cells Dev. 2013;22(1):148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Maffioli E, Nonnis S, Angioni R, et al. Proteomic analysis of the secretome of human bone marrow-derived mesenchymal stem cells primed by pro-inflammatory cytokines. J Proteomics. 2017;166:115–126. [DOI] [PubMed] [Google Scholar]
- [41].Zanotti L, Sarukhan A, Dander E, et al. Encapsulated mesenchymal stem cells for in vivo immunomodulation. Leukemia. 2013;27(2):500–503. [DOI] [PubMed] [Google Scholar]
- [42].Zanotti L, Angioni R, Calì B, et al. Mouse mesenchymal stem cells inhibit high endothelial cell activation and lymphocyte homing to lymph nodes by releasing TIMP-1. Leukemia. 2016;30(5):1143–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Théry C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3. https://www.ncbi.nlm.nih.gov/pubmed/18228490 [DOI] [PubMed]
- [44].Casey AE, Laster WR, Ross GL. Sustained enhanced growth of carcinoma EO771 in C57 black mice. Proc Soc Exp Biol Med Soc Exp Biol Med N Y N. 1951;77:358–362. [DOI] [PubMed] [Google Scholar]
- [45].Herkenne S, Paques C, Nivelles O, et al. The interaction of uPAR with VEGFR2 promotes VEGF-induced angiogenesis [published correction appears in Sci Signal. 2015 Dec 8;8(406):er9]. Sci Signal. 2015;8(403):ra117. [DOI] [PubMed] [Google Scholar]
- [46].Filippi A, Dal Sasso E, Iop L, et al. Multimodal label-free ex vivo imaging using a dual-wavelength microscope with axial chromatic aberration compensation. J Biomed. Opt. 2018;23:1–9. [DOI] [PubMed] [Google Scholar]
- [47].Kaundal U, Bagai U, Rakha A. Immunomodulatory plasticity of mesenchymal stem cells: a potential key to successful solid organ transplantation. J Transl Med. 2018;16(1):31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Rupert DLM, Claudio V, Lässer C, et al. Methods for the physical characterization and quantification of extracellular vesicles in biological samples. Biochim Biophys Acta BBA - Gen Subj. 2017;1861:3164–3179. [DOI] [PubMed] [Google Scholar]
- [49].Kowal J, Arras G, Colombo M, et al. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc Natl Acad Sci USA. 2016;113:E968–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Liang -C-C, Park AY, Guan J-L. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc. 2007;2:329–333. [DOI] [PubMed] [Google Scholar]
- [51].Bhattacharya R, Kwon J, Li X, et al. Distinct role of PLCbeta3 in VEGF-mediated directional migration and vascular sprouting. J Cell Sci. 2009;122:1025–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brem H, Kodra A, Golinko MS, et al. Mechanism of sustained release of vascular endothelial growth factor in accelerating experimental diabetic healing. J Invest Dermatol. 2009;129:2275–2287. [DOI] [PubMed] [Google Scholar]
- [53].Guo D, Wang Q, Li C, et al. VEGF stimulated the angiogenesis by promoting the mitochondrial functions.. Oncotarget. 2017;8(44):77020–77027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Guo S, Lok J, Liu Y, et al. Assays to examine endothelial cell migration, tube formation, and gene expression profiles. Methods Mol Biol. 2014;1135:393–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Stahl A, Connor KM, Sapieha P, et al. The mouse retina as an angiogenesis model. Invest Ophthalmol Vis Sci. 2010;51:2813–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Aaberg-Jessen C, Sørensen MD, Matos ALSA, et al. Co-expression of TIMP-1 and its cell surface binding partner CD63 in glioblastomas. BMC Cancer. 2018;18(1):270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Jung -K-K, Liu X-W, Chirco R, et al. Identification of CD63 as a tissue inhibitor of metalloproteinase-1 interacting cell surface protein. EMBO J. 2006;25(17):3934–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hurd TR, DeGennaro M, Lehmann R. Redox regulation of cell migration and adhesion. Trends Cell Biol. 2012;22(2):107–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Abid MR, Kachra Z, Spokes KC, et al. NADPH oxidase activity is required for endothelial cell proliferation and migration. FEBS Lett. 2000;486:252–256. [DOI] [PubMed] [Google Scholar]
- [60].Wang Y, Zang QS, Liu Z, et al. Regulation of VEGF-induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol - Cell Physiol. 2011;301:C695–C704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Moldovan L, Mythreye K, Goldschmidt-Clermont PJ, et al. Reactive oxygen species in vascular endothelial cell motility. Roles of NAD(P)H oxidase and Rac1. Cardiovasc Res. 2006;71:236–246. [DOI] [PubMed] [Google Scholar]
- [62].Kim Y-W, Byzova TV. Oxidative stress in angiogenesis and vascular disease. Blood. 2014;123:625–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Rezabakhsh A, Ahmadi M, Khaksar M, et al. Rapamycin inhibits oxidative/nitrosative stress and enhances angiogenesis in high glucose-treated human umbilical vein endothelial cells: Role of autophagy. Biomed Pharmacother. 2017;93:885–894. [DOI] [PubMed] [Google Scholar]
- [64].Urbich C, Dernbach E, Aicher A, et al. CD40 ligand inhibits endothelial cell migration by increasing production of endothelial reactive oxygen species. Circulation. 2002;106:981–986. [DOI] [PubMed] [Google Scholar]
- [65].Jiang C, Jiang L, Li Q, et al. Acrolein induces NLRP3 inflammasome-mediated pyroptosis and suppresses migration via ROS-dependent autophagy in vascular endothelial cells. Toxicology. 2018;410:26–40. [DOI] [PubMed] [Google Scholar]
- [66].Davalli P, Mitic T, Caporali A, et al. ROS, cell senescence, and novel molecular mechanisms in aging and age-related diseases. Oxid Med Cell Longev. 2016;2016:3565127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Fan LM, Geng L, Cahill-Smith S, et al. Nox2 contributes to age-related oxidative damage to neurons and the cerebral vasculature. J Clin Invest. 2019;129(8):3374–3386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Du J, Fan LM, Mai A, et al. Crucial roles of Nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice: dietary obesity and vascular oxidative stress. Br J Pharmacol. 2013;170:1064–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. 2017;120:713–735. [DOI] [PubMed] [Google Scholar]
- [70].Thakur S, Du J, Hourani S, et al. Inactivation of adenosine A2A receptor attenuates basal and angiotensin II-induced ROS production by Nox2 in endothelial cells. J Biol Chem. 2010;285:40104–40113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhou Z, Rajamani U, Labazi H, et al. Involvement of NADPH oxidase in A2A adenosine receptor-mediated increase in coronary flow in isolated mouse hearts. Purinergic Signal. 2015;11(2):263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cauwels A, Rogge E, Vandendriessche B, et al. Extracellular ATP drives systemic inflammation, tissue damage and mortality. Cell Death Dis. 2014;5:e1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Ghiringhelli F, Bruchard M, Chalmin F, et al. Production of adenosine by ectonucleotidases: A key factor in tumor immunoescape. J Biomed Biotechnol. 2012;2012:473712.Available from: https://www.hindawi.com/journals/bmri/2012/473712/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Smyth LA, Ratnasothy K, Tsang JY, et al. CD73 expression on extracellular vesicles derived from CD4+ CD25+ Foxp3+ T cells contributes to their regulatory function. Eur J Immunol. 2013;43(9):2430–2440. [DOI] [PubMed] [Google Scholar]
- [75].Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Invest. 2016;126:1216–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sands WA, Palmer TM. Adenosine receptors and the control of endothelial cell function in inflammatory disease. Immunol Lett. 2005;101:1–11. [DOI] [PubMed] [Google Scholar]
- [77].Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887. [DOI] [PubMed] [Google Scholar]
- [78].Nishimura S, Manabe I, Nagasaki M, et al. Adipogenesis in obesity requires close interplay between differentiating adipocytes, stromal cells, and blood vessels. Diabetes. 2007;56:1517–1526. [DOI] [PubMed] [Google Scholar]
- [79].MacDonald IJ, Liu SC, Su CM, et al. Implications of Angiogenesis Involvement in Arthritis. Int J Mol Sci. 2018;19(7):2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Yu QC, Song W, Wang D, et al. Identification of blood vascular endothelial stem cells by the expression of protein C receptor. Cell Res. 2016;26:1079–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Zhang M, Rehman J, Malik AB. Endothelial progenitor cells and vascular repair. Curr Opin Hematol. 2014;21:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Stapor PC, Sweat RS, Dashti DC, et al. Pericyte dynamics during angiogenesis: new insights from new identities. J Vasc Res. 2014;51:163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Xing F, Saidou J, Watabe K. Cancer associated fibroblasts (CAFs) in tumor microenvironment. Front Biosci (Landmark Ed). 2010;15:166–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Lyons TR, O'Brien J, Borges VF, et al. Postpartum mammary gland involution drives progression of ductal carcinoma in situ through collagen and COX-2. Nat Med. 2011;17(9):1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Santos AM, Jung J, Aziz N, et al. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119:3613–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Fitzpatrick JR, Frederick JR, McCormick RC, et al. Tissue-engineered pro-angiogenic fibroblast scaffold improves myocardial perfusion and function and limits ventricular remodeling after infarction. J Thorac Cardiovasc Surg. 2010;140:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Yong KW, Choi JR, Mohammadi M, et al. Mesenchymal Stem Cell Therapy for Ischemic Tissues. Stem Cells Int. 2018;2018:8179075. Available from: https://www.hindawi.com/journals/sci/2018/8179075/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Doorn J, Moll G, Le Blanc K, et al. Therapeutic applications of mesenchymal stromal cells: paracrine effects and potential improvements. Tissue Eng Part B Rev. 2012;18:101–115. [DOI] [PubMed] [Google Scholar]
- [89].Fierabracci A, Del Fattore A, Luciano R, et al. Recent advances in mesenchymal stem cell immunomodulation: the role of microvesicles. Cell Transplant. 2015;24:133–149. [DOI] [PubMed] [Google Scholar]
- [90].Coumans FAW, Brisson AR, Buzas EI, et al. Methodological guidelines to study extracellular vesicles. Circ Res. 2017;120:1632–1648. [DOI] [PubMed] [Google Scholar]
- [91].Rani S, Ryan AE, Griffin MD, et al. Mesenchymal stem cell-derived extracellular vesicles: toward cell-free therapeutic applications. Mol Ther. 2015;23:812–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Gong M, Yu B, Wang J, et al. Mesenchymal stem cells release exosomes that transfer miRNAs to endothelial cells and promote angiogenesis. Oncotarget. 2017;8:45200–45212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Chen J, Liu Z, Hong MM, et al. Proangiogenic compositions of microvesicles derived from human umbilical cord mesenchymal stem cells. PLoS One. 2014;9:e115316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gonzalez-King H, García NA, Ontoria-Oviedo I, et al. Hypoxia Inducible Factor-1α Potentiates Jagged 1-mediated angiogenesis by mesenchymal stem cell-derived exosomes. Stem Cells. 2017;35(7):1747–1759. [DOI] [PubMed] [Google Scholar]
- [95].Liu J, Hao H, Xia L, et al. Hypoxia pretreatment of bone marrow mesenchymal stem cells facilitates angiogenesis by improving the function of endothelial cells in diabetic rats with lower ischemia. PLoS One. 2015;10(5):e0126715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Xue C, Shen Y, Li X, et al. Exosomes derived from hypoxia-treated human adipose mesenchymal stem cells enhance angiogenesis through the PKA signaling pathway. Stem Cells Dev. 2018;27:456–465. [DOI] [PubMed] [Google Scholar]
- [97].Zou X, Gu D, Xing X, et al. Human mesenchymal stromal cell-derived extracellular vesicles alleviate renal ischemic reperfusion injury and enhance angiogenesis in rats. Am J Transl Res. 2016;8:4289–4299. [PMC free article] [PubMed] [Google Scholar]
- [98].Zou X, Yu Y, Lin S, et al. Comprehensive miRNA analysis of human umbilical cord-derived mesenchymal stromal cells and extracellular vesicles. Kidney Blood Press Res. 2018;43:152–161. [DOI] [PubMed] [Google Scholar]
- [99].Ren W, Hou J, Yang C, et al. Extracellular vesicles secreted by hypoxia pre-challenged mesenchymal stem cells promote non-small cell lung cancer cell growth and mobility as well as macrophage M2 polarization via miR-21-5p delivery. J Exp Clin Cancer Res. 2019;38(1):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Haskó G, Linden J, Cronstein B, et al. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nat Rev Drug Discov. 2008;7(9):759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Bono MR, Fernández D, Flores-Santibáñez F, et al. CD73 and CD39 ectonucleotidases in T cell differentiation: beyond immunosuppression. FEBS Lett. 2015;589:3454–3460. [DOI] [PubMed] [Google Scholar]
- [102].Antonioli L, Pacher P, Vizi ES, et al. CD39 and CD73 in immunity and inflammation. Trends Mol Med. 2013;19:355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Bouma MG, van den Wildenberg FA, Buurman WA. Adenosine inhibits cytokine release and expression of adhesion molecules by activated human endothelial cells. Am J Physiol. 1996;270:C522–529. [DOI] [PubMed] [Google Scholar]
- [104].Bours MJL, Swennen ELR, Di Virgilio F, et al. Adenosine 5ʹ-triphosphate and adenosine as endogenous signaling molecules in immunity and inflammation. Pharmacol Ther. 2006;112:358–404. [DOI] [PubMed] [Google Scholar]
- [105].Ushio-Fukai M, Nakamura Y. Reactive oxygen species and angiogenesis: NADPH oxidase as target for cancer therapy. Cancer Lett. 2008;266:37–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Łuczak K, Balcerczyk A, Soszyński M, et al. Low concentration of oxidant and nitric oxide donors stimulate proliferation of human endothelial cells in vitro. Cell Biol Int. 2004;28:483–486. [DOI] [PubMed] [Google Scholar]
- [107].Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;2006(349):re8. [DOI] [PubMed] [Google Scholar]
- [108].Lum H, Roebuck KA. Oxidant stress and endothelial cell dysfunction. Am J Physiol - Cell Physiol. 2001;280:C719–C741. [DOI] [PubMed] [Google Scholar]
- [109].Bhayadia R, Schmidt BM, Melk A, et al. Senescence-Induced Oxidative Stress Causes Endothelial Dysfunction. J Gerontol A Biol Sci Med Sci. 2016;71(2):161–169 [DOI] [PubMed] [Google Scholar]
- [110].Kobayashi N, DeLano FA, Schmid-Schönbein GW. Oxidative stress promotes endothelial cell apoptosis and loss of microvessels in the spontaneously hypertensive rats. Arterioscler Thromb Vasc Biol. 2005;25:2114–2121. [DOI] [PubMed] [Google Scholar]
- [111].Dong LF, Swettenham E, Eliasson J, et al. Vitamin E analogues inhibit angiogenesis by selective induction of apoptosis in proliferating endothelial cells: the role of oxidative stress. Cancer Res. 2007;67(24):11906–11913. [DOI] [PubMed] [Google Scholar]
- [112].El-Awady MS, Ansari HR, Fil D, et al. NADPH oxidase pathway is involved in aortic contraction induced by A3 adenosine receptor in mice. J Pharmacol Exp Ther. 2011;338(2):711–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Luu NT, McGettrick HM, Buckley CD, et al. Crosstalk between mesenchymal stem cells and endothelial cells leads to downregulation of cytokine-induced leukocyte recruitment. Stem Cells. 2013;31(12):2690–2702. [DOI] [PubMed] [Google Scholar]
- [114].Jászai J, Schmidt MHH. Trends and challenges in tumor anti-angiogenic therapies. Cells. 2019;8:1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Keating GM. Bevacizumab: a review of its use in advanced cancer. Drugs. 2014;74:1891–1925. [DOI] [PubMed] [Google Scholar]
- [116].Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Jain RK. Antiangiogenic therapy for cancer: current and emerging concepts. Oncology (Williston Park). 2005;19(4 Suppl 3):7–16. [PubMed] [Google Scholar]
- [118].Saltz LB, Lenz HJ, Kindler HL, et al. Randomized phase II trial of cetuximab, bevacizumab, and irinotecan compared with cetuximab and bevacizumab alone in irinotecan-refractory colorectal cancer: the BOND-2 study. J Clin Oncol. 2007;25(29):4557–4561. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Théry C, Amigorena S, Raposo G, et al. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr Protoc Cell Biol 2006; Chapter 3. https://www.ncbi.nlm.nih.gov/pubmed/18228490 [DOI] [PubMed]