

SUMMARY
A novel strategy employing cyclohexyl spectator ligands in Stille cross-coupling reactions has been developed as a general solution to the long-standing challenge of conducting stereospecific cross-coupling reactions at nitrogen-containing stereocenters. This method enables direct access to enantioenriched products that are difficult (or impossible) to obtain via alternative preparative methods. Selective and predictable transfer of a single secondary alkyl unit can be achieved under reaction conditions that exploit subtle electronic differences between activated and unactivated alkyl units. Through this approach, enantioenriched α-stannylated nitrogen-containing stereocenters undergo Pd-catalyzed arylation and acylation reactions with exceptionally high stereofidelity in all instances investigated. We demonstrate this process by using α-stannylated pyrrolidine, azetidine, and open-chain (benzylic and non-benzylic) nucleophiles in stereospecific reactions. This process will facilitate rapid and reliable access to enantioenriched compounds possessing nitrogen-substituted stereocenters, which constitute ubiquitous structural motifs in biologically active compounds emerging from the drug-discovery process.
Graphical Abstract
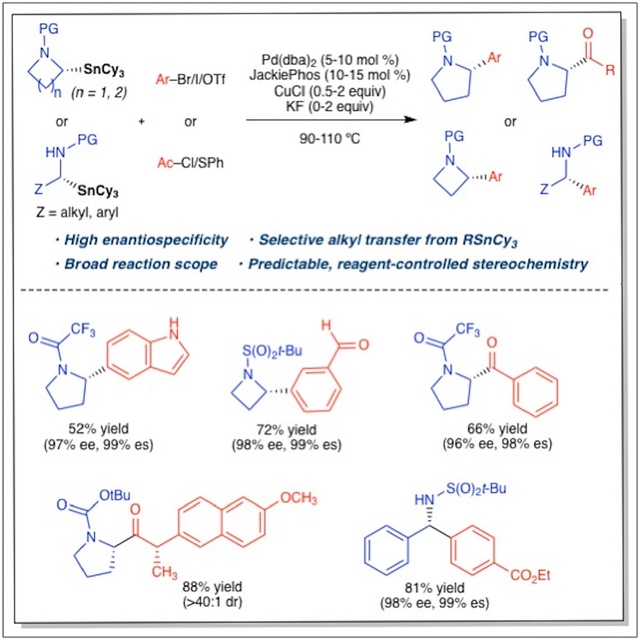
A general approach to the stereospecific cross-coupling of enantioenriched nitrogen-containing stereocenters has been developed. With cyclohexyl spectator ligands on organotin nucleophiles, selective and stereospecific transfer of a nitrogen-containing alkyl unit can be readily achieved in palladium-catalyzed cross-coupling reactions. This new reaction will enable the rapid generation of stereochemically defined nitrogen-containing carbon centers, which are common components of biologically active molecules emerging from the drug-discovery process.
INTRODUCTION
The biological properties of organic molecules are greatly influenced by the presence of nitrogen atoms within their molecular architectures.1 Nitrogen-containing stereocenters are particularly common structural motifs within biologically active molecules that emerge from the drug-discovery process. Indeed, four of the top five most commonly encountered nitrogen-containing heterocycles in FDA-approved drugs contain saturated rings and therefore the possibility of stereoisomers.2 When preparing such molecules, control of the absolute and relative stereochemistry of the nitrogen-containing stereocenter is a vital concern. Thus, the development of general synthetic strategies that enable precise stereochemical control of nitrogen-containing stereocenters constitutes an essential goal in organic chemistry.
Over the past decade, stereospecific cross-coupling strategies have emerged as viable synthetic options to achieve precise stereocontrol of carbon-carbon bonds.3–9 We, and others, have demonstrated that configurationally stable, enantioenriched organotin10–22 and organoboron23–37 may be employed in Pd-catalyzed cross-coupling reactions where transmetallation proceeds primarily through a stereoretentive or stereoinvertive mechanism, leading to predictable stereochemical outcomes (Figure 1A). Commonly, the use of alkyl nucleophiles that bear specific modes of activation, such as α-C(sp2) groups, α-heteroatoms, ring strain, and/or strongly coordinating substituents,38 are required to facilitate transmetallation of the intrinsically hindered, secondary alkyl centers (Figure 1B). The use of a highly electron-deficient electrophilic coupling partner (e.g., acyl electrophiles) can also be employed to render the palladium (Pd) catalyst more electrophilic, thereby accelerating transmetallation.10,21 These activation effects are additive such that nucleophiles bearing multiple activation modes undergo more rapid transmetallation than singly activated comparative systems. Commonly, specific combinations of these activation modes are required to facilitate transfer of secondary alkyl groups from alkyltin or alkylboron reagents to Pd.3 As a result, viable substrates are limited to those that possess the requisite structural features necessary for transmetallation in each specific system. Importantly, the stereochemical outcome of transmetallation (retentive versus invertive) may also vary unpredictably between differently activated systems, resulting in eroded stereochemical transfer. In Suzuki cross-coupling reactions, minor electronic or structural perturbations of alkylboron nucleophiles have particularly unpredictable effects on stereochemical transfer.4 In contrast, studies of analogous alkylstannane reactions have revealed more consistently predictable stereochemical outcomes and broader substrate scopes, which suggests that the use of alkyltin nucleophiles may be more conducive to development of a broadly general method for the stereospecific cross coupling of nitrogen-containing stereocenters.3 Here, we report a new approach to stereospecific cross-coupling reactions involving only marginally activated alkyl nucleophiles. Using cyclohexyl spectator ligands in place of n-butyl spectator ligands on alkylstannane nucleophiles (e.g., RSnCy3 instead of RSnnBu3), we have developed conditions that promote the stereospecific transfer and cross coupling of nitrogen-containing carbon stereocenters. We demonstrate this process by using α-stannylated pyrrolidine and azetidine heterocyclic nucleophiles, as well using α-stannylated open-chain (benzylic and non-benzylic) nucleophiles in Pd-catalyzed cross-coupling reactions. In these reactions, the electronic properties of the nitrogen-protecting group greatly influence the selectivity of alkyl transfer from the organostannane nucleophile. Under our conditions, nitrogen-containing carbon stereocenters undergo stereospecific arylation and acylation reactions with net stereoretention of absolute configuration. This process enables the first cross-coupling reaction using an azetidine nucleophile, and constitutes the first general cross-coupling method to enable stereospecific transfer of nitrogen-containing stereocenters in a highly reliable and predictable manner. These results also suggest that the use of cylclohexyl spectator ligands will be broadly applicable in stereospecific coupling reactions where the n-butyl groups of an analogous RSnnBu3 nucleophile undergo competitive alkyl transfer to Pd.
Figure 1. Stereospecific Cross-Coupling Reactions of Enantioenriched α-Stannylated Amines.
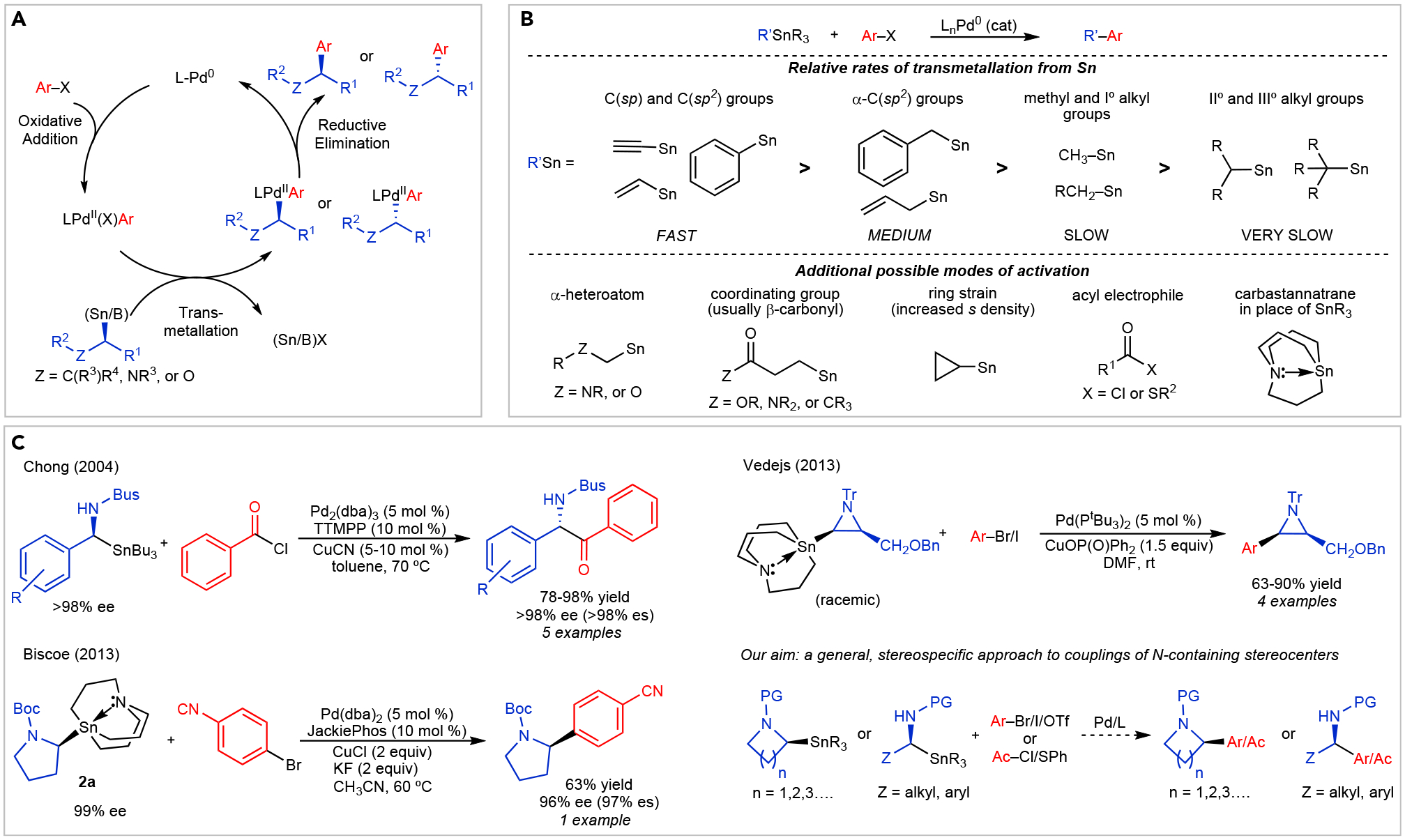
(A) Catalytic cycle for stereospecific Pd-catalyzed cross-coupling reactions involving enantioenriched alkyltin and alkylboron nucleophiles.
(B) Relative rates of group transfer from organotin compounds, as well as activation strategies.
(C) Prior examples of Pd-catalyzed Stille cross-coupling reactions involving the stereospecific transfer of a nitrogen-containing stereocenter.
RESULTS AND DISCUSSION
Development of a General Cross-Coupling Reaction Employing α-Stannyl Pyrrolidine Derivatives
Whereas inclusion of an α-oxygen substituent on a secondary alkyltin nucleophile is an effective strategy for promoting selective alkyl transmetallation,3,39,40 far fewer examples of activation via an α-nitrogen substituent have been demonstrated.13,18,19,41 This possibly arises from the lower electronegativity of nitrogen, which results in a muted propensity to promote alkyl transfer. Cross-coupling reactions involving the transmetallation of nitrogen-substituted alkyltin nucleophiles reported by Kells and Chong,13 Theddu and Vedejs,19 and Li et al.18 have been limited to cases that utilize multiple modes of activation and thereby offered limited potential scope (Figures 1B and 1C). In an attempt to overcome the dependence of transmetallation on remote activating groups, we introduced the use of enantioenriched carbastannatrane42,43 nucleophiles in stereospecific cross-coupling reactions.18 The use of carbastannatranes enabled, for the first time, stereospecific cross-coupling reactions of completely unactivated secondary alkyl nucleophiles when the electron-deficient biarylphosphine JackiePhos44 (1) (Figure 2) was employed as a supporting ligand. Unfortunately, ensuing studies on the generality of this process revealed that α-carbastannatranyl pyrrolidine 2a (Figure 3A) performs poorly in cross-coupling reactions. Combined activation from the α-nitrogen and use of the carbastannatrane unit render 2a over-activated, leading to competitive decomposition through protodestannylation when all but the most reactive aryl electrophiles are employed. Indeed, use of 4-bromoanisole in this reaction resulted in only a 16% yield of arylation product (Figure 3A, entry 1). Attempting to dampen the reactivity and increase the stability of the α-stannyl pyrrolidine nucleophile, we investigated the use of compound 2b, which contains n-butyl units as spectator groups in place of the carbastannatrane. Unfortunately, the use of 2b resulted exclusively in n-butyl coupling product without any evidence of selective pyrrolidine transfer (Figure 3A, entry 2). Because secondary alkyl groups undergo significantly slower transmetallation from tin than primary alkyl groups (Figure 1B), we exchanged the n-butyl groups of compound 2c for cyclohexyl groups.45 Using 2c in methanol, with CuCl as a co-transmetallating agent,46 pyrrolidine transfer predominated, though ca. 20% cyclohexyl transfer was still observed (Figure 3A, entry 5). The ability of JackiePhos to support the transmetallation of unactivated cyclohexyl units was surprising and underscores its unique activity in cross-coupling reactions of alkyltin species. At this point, we reasoned that installation of a more electron-deficient nitrogen-protecting group might address this problem by enhancing the activating effect of the nitrogen atom. Installation of a t-butylsulfonyl (Bus) (2d) or benzoyl protecting group (2e) resulted in improved selectivity. However, a trifluoroacetyl protecting group (2f) was found to promote the optimal selectivity and yield for the pyrrolidine coupling product (Figure 3B, entry 8) with only trace evidence of cyclohexyl transfer.
Figure 2. Molecular Structure of JackiePhos (1).
Figure 3. . Selective Transfer of Nitrogen-Containing Stereocenters from Alkyltin Nucleophiles.
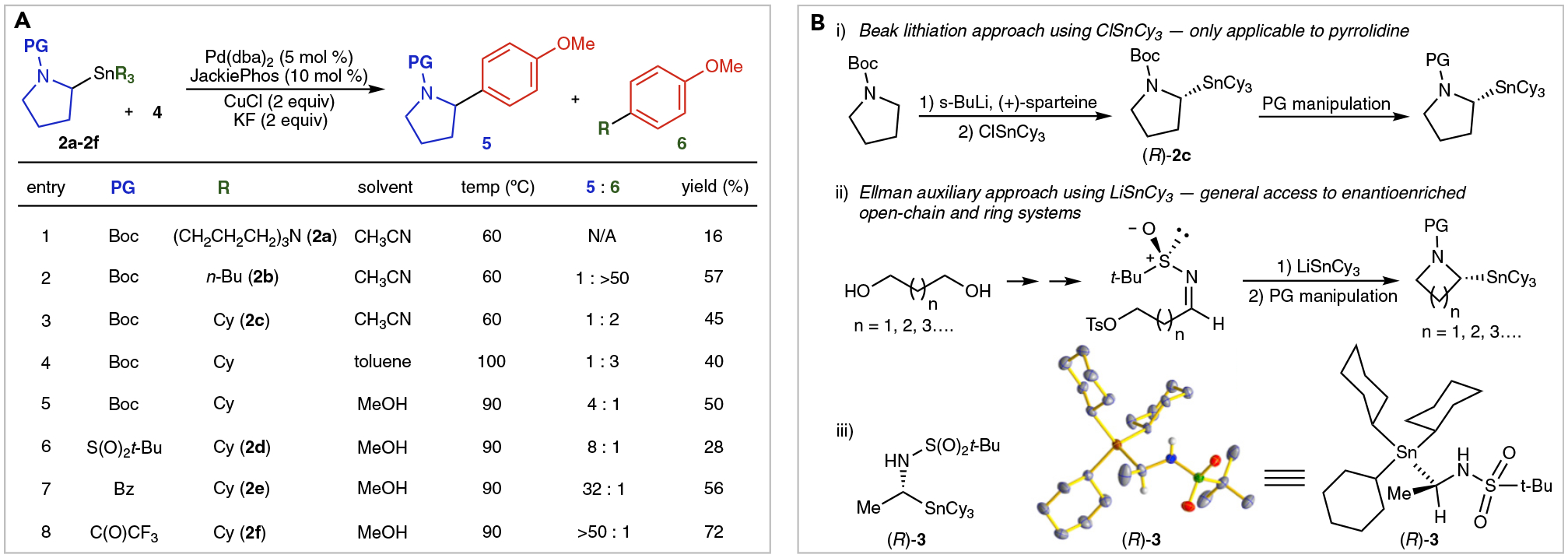
(A) Influence of tin spectator ligands and nitrogen protecting groups on selectivity of α-pyrrolidine transfer in cross-coupling reactions with 4-bromoanisole (4).
(B) Synthetic strategies employed in the preparation of enantioenriched alkyltin nucleophiles bearing an α-nitrogen group. X-ray crystal structure (R)-3 was used to confirm stereochemistry of LiSnCy3 addition to imine bearing Ellman auxiliary (thermal ellipsoids at 50% probability).
Preparation of Enantioenriched Alkylstannanes Bearing Nitrogen-Containing Stereocenters
Few methods for the preparation of enantioenriched alkyltin nucleophiles exist. This is unsurprising as enantioenriched alkyltin nucleophiles lack significant utility without methods to promote selective alkyl transfer. Though the sparteine-mediated asymmetric lithiation chemistry devised by Beak et al.47 works extremely well for the preparation of α-stannylated N-Boc-protected pyrrolidine compounds, it is not applicable to other nitrogen-containing heterocycles or open-chain amines. As a more general alternative to the Beak protocol, we devised a cyclization strategy using Ellman’s auxiliary,48 starting from terminal diols (Figure 3B). Using this strategy, we prepared enantioenriched α-stannylated pyrrolidines and azetidines. Though we have focused on the pyrrolidine and azetidine ring systems for this initial study, larger rings should be readily accessible using an analogous strategy alongside the appropriate diol precursors. As previously demonstrated by Kells and Chong,49 this route also enables the preparation of enantioenriched, α-stannylated derivatives of open-chain secondary amines. Thus, in principle, use of the Ellman auxiliary approach should provide universal access to α-stannylated, enantioenriched amine derivatives. Protecting-group manipulation can be readily accomplished following installation of the tin unit for these products (see Supplemental Information). In contrast to RSnnBu3 compounds, RSnCy3 compounds generally exhibit high crystallinity and have no odor (ClSnCy3 is also odorless and crystalline). Additionally, toxicity of RSnCy3 compounds has been reported to be lower than that of the corresponding RSnBu3 compounds.50–52 Such practical benefits should facilitate the broad implementation of our method, as well as the general use of RSnCy3 compounds in organic synthesis.
Scope of Stereospecific Cross-Coupling Reactions Using Enantioenriched α-Stannyl Pyrrolidine Derivatives
Using the reaction conditions shown in entry 8 of Figure 3A, we employed enantioenriched 2f in cross-coupling reactions with different aryl electrophiles (Figure 4). No observable transfer of the cyclohexyl group occurred in these reactions. For all reactions, transmetallation proceeded primarily through a stereoretentive pathway, which enabled isolation of α-arylated pyrrolidine derivatives in high % e.e. and with predictable stereochemistry. Electron-rich, electron-neutral, electron-deficient, and ortho-substituted aryl electrophiles all underwent cross-coupling reaction with high enantiospecificity (% e.s.). Heteroaryl electrophiles and aryl electrophiles bearing protic functional groups were also well tolerated in these reactions. Aryl bromides, iodides, and triflates all proved to be viable substrates for this method. These examples showcase the breadth of substrate scope compatible with this process. Additionally, the use of benzoyl chloride as the electrophilic component enabled a stereospecific acylation reaction under standard conditions in the absence of the fluoride additive. Highlighting the operational simplicity of this process, all reactions were conducted on the benchtop with disposable test tubes and screw-top septum caps without the need for an inert atmosphere glovebox.
Figure 4.
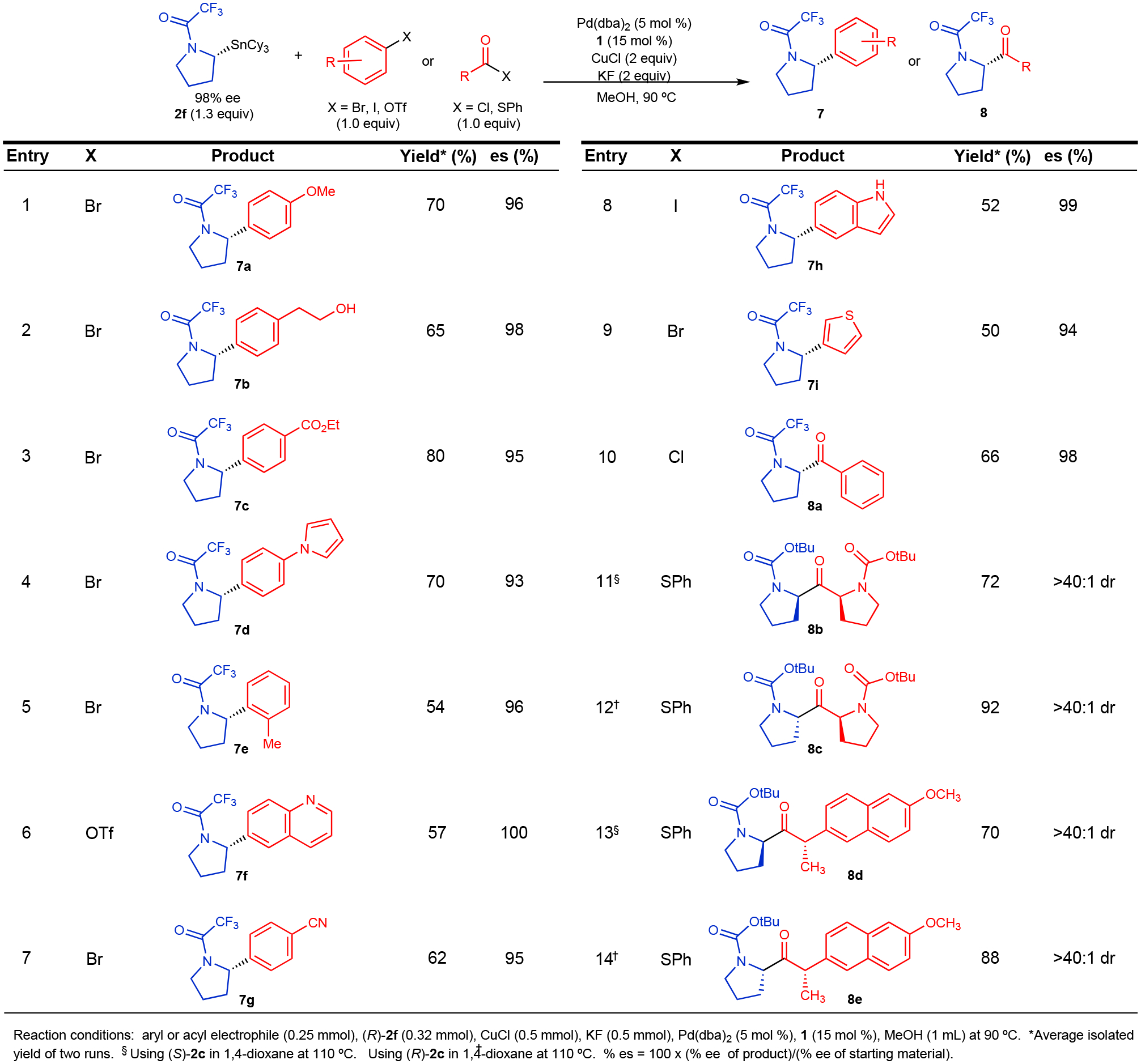
Stereospecific Cross-Coupling Reactions of Enantioenriched N-Trifluoroacetyl-2-tricyclohexylstannyl Pyrrolidine (2f)
Thioesters are valuable synthons for use in diastereoselective processes because enantioenriched thioesters can be readily prepared from α-stereogenic carboxylic acids. Stereospecific cross-coupling reactions of α-stereogenic thioesters and enantioenriched nucleophiles would enable the preparation of individual diastereomers through a completely reagent-controlled process. To expand the scope of our cross-coupling process, we employed thioesters as acyl electrophiles (Figure 4, entries 11–14). Although reactions of 2f with thioesters did provide the desired cross-coupling product with excellent diastereoselectivity, we found that higher yields could be more consistently achieved with Boc-protected pyrrolidine analog 2c. In these reactions, the presence of an acyl ligand on Pd accelerates transmetallation, which enables use of a less electron-deficient protecting group on the pyrrolidine nucleophile, though the SnCy3 group is still essential. Using the thioesters derived from L-proline and (S)-naproxen, we demonstrated that exceptionally high diastereoselectivity could be achieved for both enantiomers of 2c. Thus, selective incorporation of new stereocenters could be readily achieved in a highly rational and predictable manner without influence from existing stereocenters on a chiral substrate. This shows that our approach is not limited to enantioselective processes and can also be employed as a general strategy for achieving diastereocontrol in cross-coupling reactions.
Extension to Stereospecific Cross-Coupling Reactions Using Enantioenriched α-Stannyl Azetidines and Open-Chain Derivatives
Use of the Ellman auxiliary approach to the preparation of enantioenriched α-stannylated amines enables access to numerous potential enantioenriched scaffolds. We employed the cyclization strategy shown in Figure 3B to prepare α-stannylated azetidine 9 in high enantiopurity. Although the t-butylsulfonyl (Bus) protecting group did not enable selective pyrrolidine transfer from 2d, we found that the additional ring strain of the 4-member azetidine ring facilitated selective azetidine transfer from RSnCy3 in the presence of the t-butylsulfonyl protecting group, which was readily formed via oxidation of the Ellman auxiliary. Under the conditions in Figure 4 with only nominal modification, arylation reactions of 9 proceeded with high enantiofidelity for electron-deficient and electron-neutral aryl bromides (Figure 5). Ketone, aldehyde, ester, and ortho substituents were well tolerated in this reaction. This constitutes the first example of a Pd-catalyzed cross-coupling reaction involving α-metallated azetidine nucleophiles (racemic or stereospecific) and provides a simple route to accessing enantioenriched α-aryl azetidine derivatives for potential applications in drug discovery. The unprecedented success of this approach using azetidine and pyrrolidine ring systems suggests that other ring systems (e.g., piperidine, piperazine, and azepane) will also be viable nucleophilic components in stereospecific cross-coupling reactions.
Figure 5.
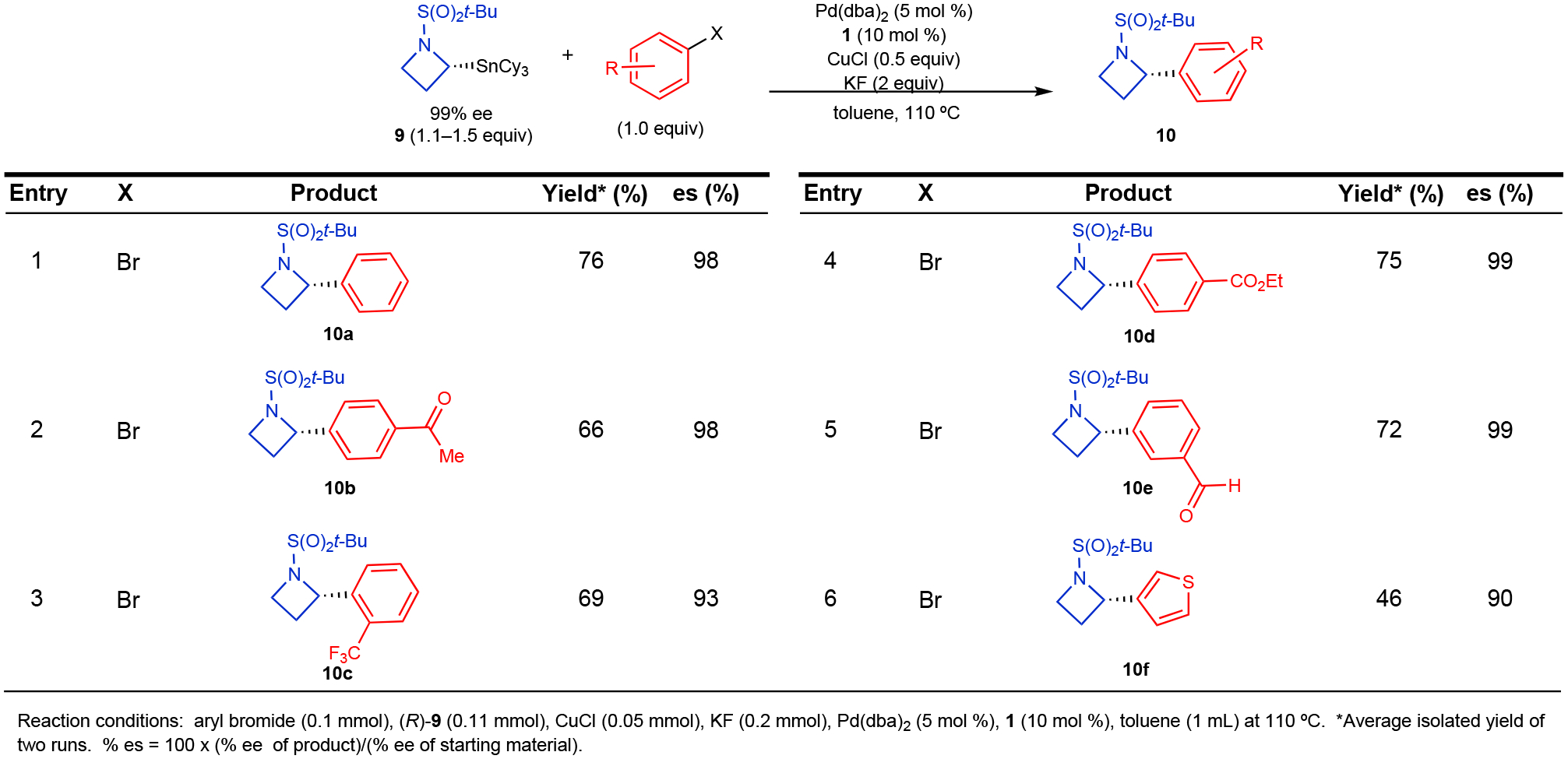
Stereospecific Cross-Coupling Reactions of Enantioenriched N-Bus-2-tricyclohexylstannyl Azetidine (9)
To further demonstrate the generality of this system, we prepared enantioenriched open-chain α-stannylated amine derivatives by using the Ellman auxiliary approach. We found that α-stannylated nucleophiles of secondary amines (11) underwent more facile transmetallation than unactivated tertiary amine nucleophiles, such as pyrrolidine derivatives. As observed for the azetidine nucleophiles, activation imparted by the t-butylsulfonyl protecting group was sufficient to enable selective transfer of the open-chain alkylamine unit from RSnCy3, enabling the first stereospecific cross-coupling reactions of non-benzylic α-stannylated secondary amines (Figure 6). The protecting group, however, can be easily varied if desired.49 For the open-chain nucleophiles, the use of n-butyl groups as spectator ligands resulted in ca. 20% n-butyl transfer during the transmetallation step. Excellent enantiospecificity was observed with non-benzyl and benzylamine derivatives under conditions analogous to those in Figures 4 and 5. These results are particularly noteworthy because they suggest that additional modes of activation, such as the inclusion of an α-C(sp2) substituent, do not affect the mechanism of transmetallation in these cross-coupling reactions, which highlights the truly general scope of this transformation. The inclusion of a branched alkyl substituent (12c) was also tolerated in this reaction, although slightly decreased enantiospecificity was obtained in the cross-coupling product.
Figure 6.
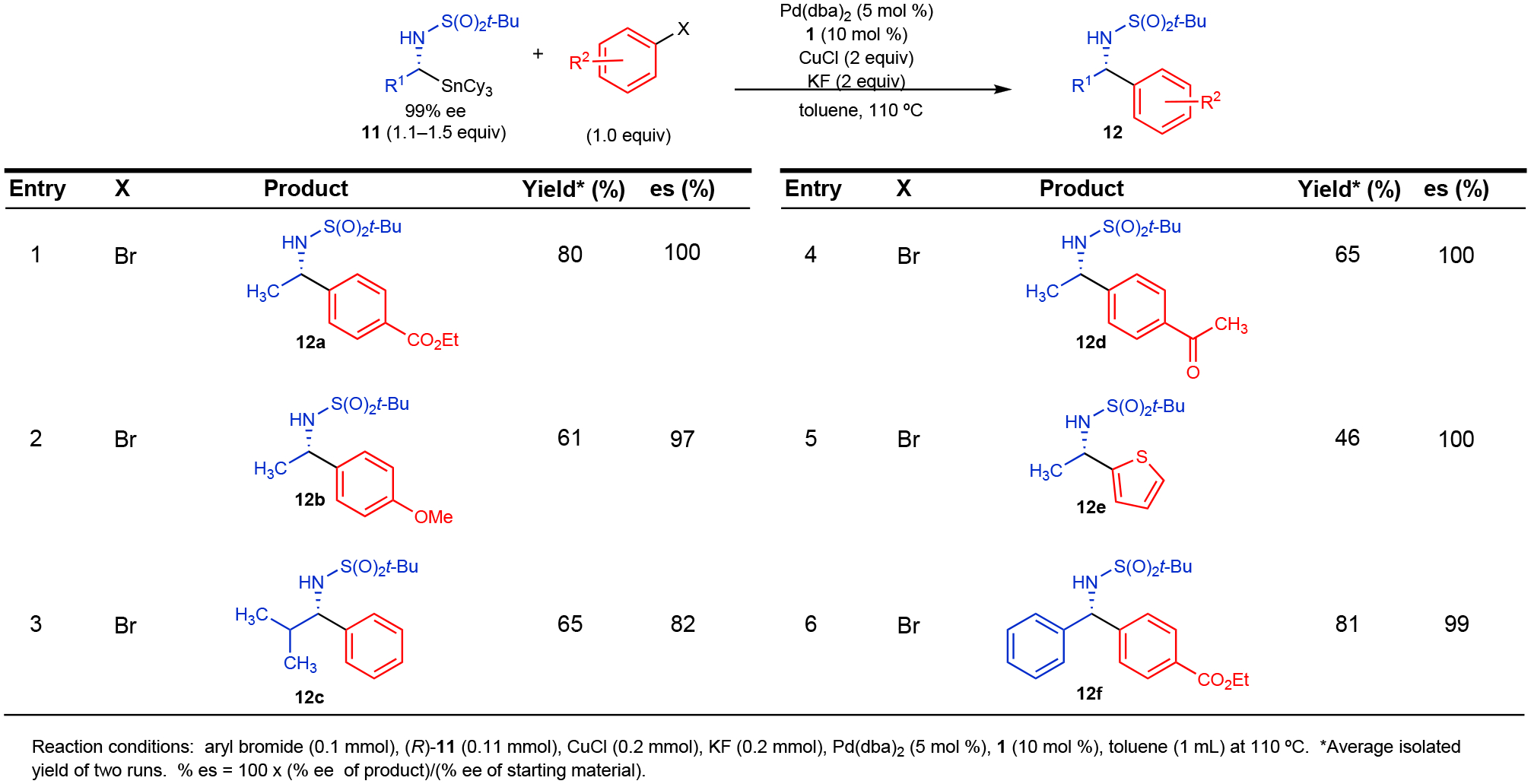
Stereospecific Cross-Coupling Reactions of Enantioenriched Open-Chain α-Amino Tricyclohexylstannanes (11)
Preparation of CDK8 Inhibitors through Stereospecific Cross-Coupling Reactions
Cyclin-dependent kinase CDK8 has been proposed to act as an oncogene in the development of colorectal cancers.53–55 Additionally, increased CDK8 expression has been linked to breast and ovarian cancers.51 High-throughput screening alongside systematic structural modification has recently been employed in the development of compound 15a, a potent inhibitor of CDK8.56 These studies suggested that derivatives of 15a with pyrrolidine units that bear a mono-halogenated aryl group at the stereogenic a position display particularly favorable potency, selectivity, bioavailability, and safety profiles in preclinical in vivo studies. To demonstrate the application of stereospecific cross-coupling reactions to the preparation of enantioenriched drug candidates, we prepared analogs of compound 15a through the stereospecific variation of its mono-halogenated aryl substituent (Figure 7). Stereospecific cross-coupling reactions using the method shown in Figure 4 enabled the direct preparation of highly enantioenriched 13, which could then be elaborated to 15 through simple deprotection and amidation reactions. Through this strategy, we prepared 15a as well as previously unreported analogs 15b–15d. Therefore, the use of stereospecific cross-coupling reactions enables a more streamlined synthetic approach to the preparation of potential CDK8 inhibitors than previously applied strategies that require resolution of the desired enantiomer from the corresponding racemic mixtures.56
Figure 7.
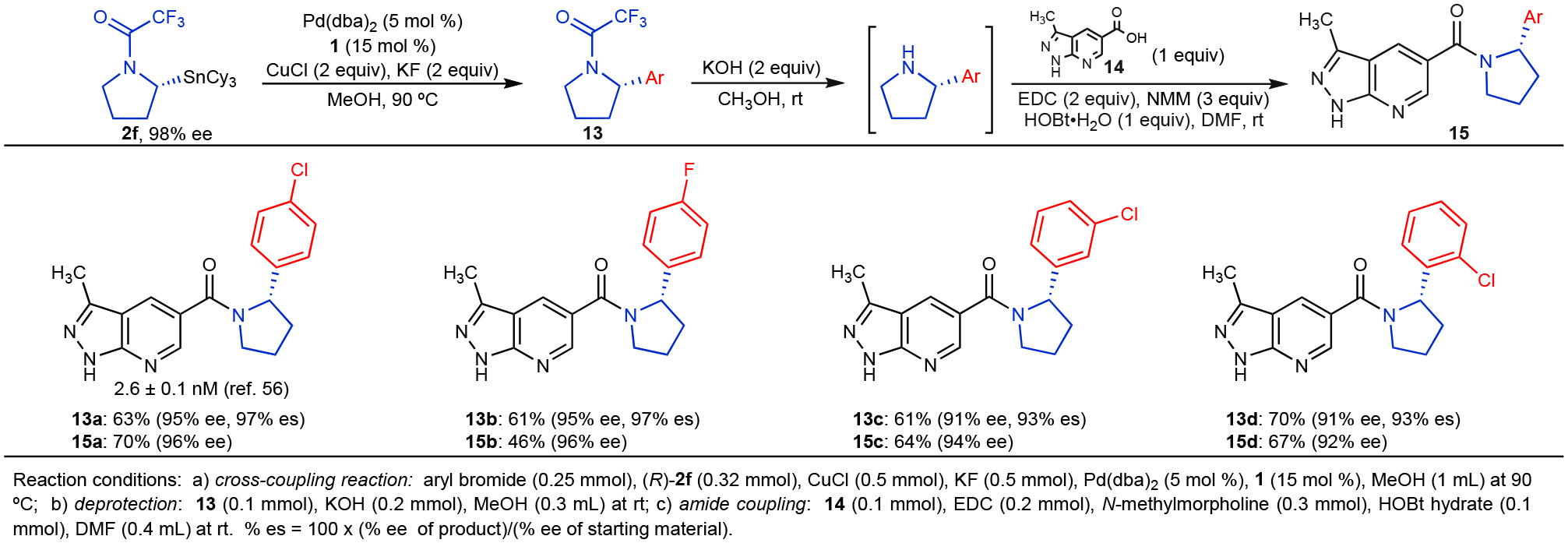
Preparation of New CDK8 Inhibitor Derivatives (15) via a Stereospecific Cross-Coupling Strategy
In summary, we have found that the synergistic use of cyclohexyl spectator ligands on tin (selectively slows transmetallation of undesired units) and the biphenylphosphine ligand JackiePhos (general acceleration of transmetallation) enables the selective transfer of an enantioenriched alkyl unit from tin when a minor structural perturbation electronically differentiates that unit. Thus, competitive transfer of n-Bu ligands from RSnnBu3 reagents can be circumvented in stereospecific Stille reactions of enantioenriched alkylstannanes. Using this strategy, we have developed a general approach to stereospecific Pd-catalyzed cross-coupling reactions of nitrogen-containing stereocenters. This process was demonstrated with α-stannylated pyrrolidine and azetidine nucleophiles, as well as α-stannylated open-chain (benzylic and non-benzylic) nucleophiles, in stereospecific arylation and acylation reactions. The uniformity of reaction conditions employed, the predictability of stereochemical outcomes achieved, and the breadth of α-stannylated amines tolerated in these reactions will facilitate the broad use of this method in organic synthesis. These results suggest that the use of carbastannatrane nucleophiles will be necessary only when transmetallation of completely unactivated nucleophiles is desired and that high stereofidelity could likewise be achievable in cross-coupling reactions using other enantioenriched RSnCy3 nucleophiles bearing an electronically differentiated alkyl unit. Additionally, RSnCy3 compounds offer the practical benefits of lower toxicity and significantly higher crystallinity than their commonly used RSnnBu3 counterparts, which should enhance the general attractiveness of this protocol.
EXPERIMENTAL PROCEDURES
General Procedure for Cross-Coupling Reactions Using Enantioenriched α-Stannylated Amines
Pd(dba)2 (5 mol %), JackiePhos (10–15 mol %),RSnCy3 nucleophile (1.1–1.3 equiv), CuCl (0.5–2 equiv), and KF (for aryl electrophiles, 2 equiv) were weighed out on the benchtop and transferred to an oven-dried 8 mL screw-top testtube with stir bar. The testtube was sealed with a septum screw cap and electrical tape. Using a needle attached to a Schlenk line, the reaction tubewas evacuated(100mTorr) and back filled with argon. This process was repeated three times. The liquid aryl or acyl electrophile (1 equiv), followed by degassed methanol, dioxane, ortoluene (0.5–1.0 mL), was then added to the reaction tube via microsyringe. If the aryl or acyl electrophile was a solid, it was weighed on the benchtop alongside the other solids. The reaction tube was sealed with additional electrical tape and heated to 90°C or 110°C for 12 h (unoptimized reaction time) with a heating block. The cooled reaction mixture was transferred to a separatory funnel, diluted with water, and extracted with diethyl ether (3 × 10 mL). The combined organic layers were washed with brine (10 mL) and then dried over Na2SO4. The dried organic layer was filtered, concentrated, and purified by column chromatography on silica gel.
Supplementary Material
HIGHLIGHTS.
N-Substituted alkyl groups undergo selective transfer from cyclohexyl-substituted tin
Enantioenriched N-substituted alkyl groups transfer from tin with high stereofidelity
Stereospecific couplings work broadly for heterocyclic and open-chain nucleophiles
Stereospecific couplings can be used to prepare a library of CDK8 inhibitors
The Bigger Picture.
Two molecules that have the same structure and composition but are mirror images of each other can produce very different biological responses. Therefore, control of the 3D atomic arrangement in molecules is critical in the drug-discovery process. Because biologically active molecules tend to contain C–N bonds, a general method to control the 3D structure of nitrogen-substituted carbon atoms would be particularly useful toward facilitating the discovery and development of new-medicines. In this report, we detail a general method to modify the 3D structure of nitrogen-containing carbon centers. We employ palladium-catalyzed cross-coupling reactions in C–C bond-forming reactions where the initial 3D structure of the reactant is predictably transferred to the final product. With this new method, scientists will now be able to use cross-coupling reactions to rapidly generate libraries of new compounds while controlling the 3D architecture of the compounds.
ACKNOWLEDGMENTS
We thank the City College of New York, the National Institutes of Health (R01GM131079), and the National Science Foundation (CHE-1665189) for supporting this work. The authors are grateful to Chunhua Hu for assistance with the X-ray analysis and acknowledge the Molecular Design Institute of NYU for purchase of the single-crystal diffractometer.
Footnotes
DATA AND CODE AVAILABILITY
The crystallographic data for compound 3 are available under accession number CCDC: 1903639 and can be obtained free of charge from the Cambridge Crystallo-graphic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chempr.2020.02.002.
DECLARATION OF INTERESTS
M.R.B. has filed provisional patent US 62/876,293 for the use of RSnCy3 in cross-coupling reactions.
REFERENCES
- 1.Lipinski CA, Lombardo F, Dominy BW, and Feeney PJ (2001). Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev 46, 3–26. [DOI] [PubMed] [Google Scholar]
- 2.Vitaku E, Smith DT, and Njardarson JT (2014). Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 3.Wang CY, Derosaa J, and Biscoe MR (2015). Configurationally stable, enantioenriched organometallic nucleophiles in stereospecific Pd-catalyzed cross-coupling reactions: an alternative approach to asymmetric synthesis. Chem. Sci 6, 5105–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leonori D, and Aggarwal VK (2015). Stereospecific couplings of secondary and tertiary boronic esters. Angew. Chem. Int. Ed 54, 1082–1096. [DOI] [PubMed] [Google Scholar]
- 5.Rygus JPG, and Crudden CM (2017). Enantiospecific and iterative Suzuki-Miyaura cross-couplings. J. Am. Chem. Soc 139, 18124–18137. [DOI] [PubMed] [Google Scholar]
- 6.Cherney AH, Kadunce NT, and Reisman SE (2015). Enantioselective and enantiospecific transition-metal-catalyzed cross-coupling reactions of organometallic reagents to construct C–C bonds. Chem. Rev 115, 9587–9652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Swift EC, and Jarvo ER (2013). Asymmetric transition metal-catalyzed cross-coupling reactions for the construction of tertiary stereocenters. Tetrahedron 69, 5799–5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pound SM, and Watson MP (2018). Asymmetric synthesis via stereospecific C–N and C–O bond activation of alkyl amine and alcohol derivatives. Chem. Commun 54, 12286–12301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Campos KR, Klapars A, Waldman JH, Dormer PG, and Chen CY (2006). Enantioselective, palladium-catalyzed α-arylation of N-boc-pyrrolidine. J. Am. Chem. Soc 128, 3538–3539. [DOI] [PubMed] [Google Scholar]
- 10.Ye J, Bhatt RK, and Falck JR (1994). Stereospecific palladium/copper cocatalyzed cross-coupling of.alpha.-alkoxy-and.alpha.-aminostannanes with acyl chlorides. J. Am. Chem. Soc 116, 1–5. [Google Scholar]
- 11.Falck JR, Bhatt RK, and Ye J (1995). Tin-copper transmetalation: cross-coupling of.alpha-heteroatom-substituted alkyltributylstannanes with organohalides. J. Am. Chem. Soc 117, 5973–5982. [Google Scholar]
- 12.Mohapatra S, Bandyopadhyay A, Barma DK, Capdevila JH, and Falck JR (2003). Chiral α, β-dialkoxy- and α-alkoxy-β-aminostannanes: preparation and copper-mediated cross-coupling. Org. Lett 5, 4759–4762. [DOI] [PubMed] [Google Scholar]
- 13.Kells KW, and Chong JM (2004). Stille coupling of stereochemically defined α-sulfonamidoorganostannanes. J. Am. Chem. Soc 126, 15666–15667. [DOI] [PubMed] [Google Scholar]
- 14.Kalkofen R, and Hoppe D (2006). First example of an enantiospecific sp3–sp2 Stille coupling of a chiral allylstannane with aryl halides. ChemInform 37, 1959–1961. [Google Scholar]
- 15.Lange H, Fröhlich R, and Hoppe D (2008). Cu(I)-catalyzed stereospecific coupling reactions of enantioenriched α-stannylated benzyl carbamates and their application. Tetrahedron 64, 9123–9135. [Google Scholar]
- 16.Falck JR, Patel PK, and Bandyopadhyay A (2007). Stereospecific cross-coupling of α-(thiocarbamoyl)organostannanes with alkenyl, aryl, and heteroaryl iodides. J. Am. Chem. Soc 129, 790–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li H, He A, Falck JR, and Liebeskind LS (2011). Stereocontrolled synthesis of α-amino-α’-alkoxy ketones by a copper-catalyzed cross-coupling of peptidic thiol esters and α-alkoxyalkylstannanes. Org. Lett 13, 3682–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li L, Wang C-Y, Huang R, and Biscoe MR (2013). Stereoretentive Pd-catalysed Stille cross-coupling reactions of secondary alkyl azastannatranes and aryl halides. Nat. Chem 5, 607–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Theddu N, and Vedejs E (2013). Stille coupling of an aziridinyl stannatrane. J. Org. Chem 78, 5061–5066. [DOI] [PubMed] [Google Scholar]
- 20.Jia T, Cao P, Wang D, Lou Y, and Liao J (2015). Copper-catalyzed asymmetric three-component borylstannation: enantioselective formation of C–Sn bond. Chemistry 21, 4918–4922. [DOI] [PubMed] [Google Scholar]
- 21.Wang C-Y, Ralph G, Derosa J, and Biscoe MR (2017). Stereospecific palladium-catalyzed acylation of enantioenriched alkylcarbastannatranes: a general alternative to asymmetric enolate reactions. Angew. Chem. Int. Ed 56, 856–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ma X, Diane M, Ralph G, Chen C, and Biscoe MR (2017). Stereospecific electrophilic fluorination of alkylcarbastannatrane reagents. Angew. Chem. Int. Ed 56, 12663–12667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imao D, Glasspoole BW, Laberge VS, and Crudden CM (2009). Cross coupling reactions of chiral secondary organoboronic esters with retention of configuration. J. Am. Chem. Soc 131, 5024–5025. [DOI] [PubMed] [Google Scholar]
- 24.Sandrock DL, Jean-Gérard L, Chen CY, Dreher SD, and Molander GA (2010). Stereospecific cross-coupling of secondary alkyl β-trifluoroboratoamides. J. Am. Chem. Soc 132, 17108–17110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ohmura T, Awano T, and Suginome M (2010). Stereospecific Suzuki-Miyaura coupling of chiral α-(acylamino)benzylboronic esters with inversion of configuration. J. Am. Chem. Soc 132, 13191–13193. [DOI] [PubMed] [Google Scholar]
- 26.Awano T, Ohmura T, and Suginome M (2011). Inversion or retention? Effects of acidic additives on the stereochemical course in enantiospecific Suzuki-Miyaura coupling of α-(acetylamino)benzylboronic esters. J. Am. Chem. Soc 133, 20738–20741. [DOI] [PubMed] [Google Scholar]
- 27.Lee JCH, McDonald R, and Hall DG (2011). Enantioselective preparation and chemoselective cross-coupling of 1,1-diboron compounds. Nat. Chem 3, 894–899. [DOI] [PubMed] [Google Scholar]
- 28.Partridge BM, Chausset-Boissarie L, Burns M, Pulis AP, and Aggarwal VK (2012). Enantioselective synthesis and cross-coupling of tertiary propargylic boronic esters using lithiation-borylation of propargylic carbamates. Angew. Chem. Int. Ed 51, 11795–11799. [DOI] [PubMed] [Google Scholar]
- 29.Molander GA, and Wisniewski SR (2012). Stereospecific cross-coupling of secondary organotrifluoroborates: potassium 1-(benzyloxy)alkyltrifluoroborates. J. Am. Chem. Soc 134, 16856–16868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li L, Zhao S, Joshi-Pangu A, Diane M, and Biscoe MR (2014). Stereospecific Pd-catalyzed cross-coupling reactions of secondary alkylboron nucleophiles and aryl chlorides. J. Am. Chem. Soc 136, 14027–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sun C, Potter B, and Morken JP (2014). A catalytic enantiotopic-group-selective Suzuki reaction for the construction of chiral organoboronates. J. Am. Chem. Soc 136, 6534–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matthew SC, Glasspoole BW, Eisenberger P, and Crudden CM (2014). Synthesis of enantiomerically enriched triarylmethanes by enantiospecific Suzuki-Miyaura cross-coupling reactions. J. Am. Chem. Soc 136, 5828–5831. [DOI] [PubMed] [Google Scholar]
- 33.Lou Y, Cao P, Jia T, Zhang Y, Wang M, and Liao J (2015). Copper-catalyzed enantioselective 1,6-boration of para-quinone methides and efficient transformation of gem-diarylmethine boronates to triarylmethanes. Angew. Chem. Int. Ed 54, 12134–12138. [DOI] [PubMed] [Google Scholar]
- 34.Hoang GL, and Takacs JM (2017). Enantioselective γ-borylation of unsaturated amides and stereoretentive Suzuki-Miyaura cross-coupling. Chem. Sci 8, 4511–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ohmura T, Miwa K, Awano T, and Suginome M (2018). Enantiospecific Suzuki-Miyaura coupling of nonbenzylic α-(acylamino) alkylboronic acid derivatives. Chem. Asian J 13, 2414–2417. [DOI] [PubMed] [Google Scholar]
- 36.Zhao S, Gensch T, Murray B, Niemeyer ZL, Sigman MS, and Biscoe MR (2018). Enantiodivergent Pd-catalyzed C–C bond formation enabled through ligand parameterization. Science 362, 670–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehmann JW, Crouch IT, Blair DJ, Trobe M, Wang P, Li J, and Burke MD (2019). Axial shielding of Pd(II) complexes enables perfect stereoretention in Suzuki-Miyaura cross-coupling of Csp3 boronic acids. Nat. Commun 10, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Itami K, Kamei T, and Yoshida J (2001). Unusually accelerated silylmethyl transfer from tin in stille coupling: implication of coordination-driven transmetalation. J. Am. Chem. Soc 123, 8773–8779. [DOI] [PubMed] [Google Scholar]
- 39.Zhu F, Rourke MJ, Yang T, Rodriguez J, and Walczak MA (2016). Highly stereospecific cross-coupling reactions of anomeric stannanes for the synthesis of C-aryl glycosides. J. Am. Chem. Soc 138, 12049–12052. [DOI] [PubMed] [Google Scholar]
- 40.Dakarapu R, and Falck JR (2018). Stereospecific stille cross-couplings using Mn(II)Cl2. J. Org. Chem 83, 1241–1251. [DOI] [PubMed] [Google Scholar]
- 41.Malova Krizkova PM, and Hammerschmidt F (2013). On the configurational stability of chiral heteroatom-substituted [D1]methylpalladium complexes as intermediates of Stille and Suzuki-Miyaura cross-coupling reactions. Eur. J. Org. Chem 2013, 5143–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jurkschat K, Tszchach A, and Meunier-Piret J (1986). Crystal and molecular structure of 1-aza-5-stanna-methyltricyclo[3.3.3.01,5] undecane.evidence for a transannular donor–acceptor interaction in a tetraorganotin compound. J. Organomet. Chem 315, 45–49. [Google Scholar]
- 43.Vedejs E, Haight AR, and Moss WO (1992). Internal coordination at tin promotes selective alkyl transfer in the Stille coupling reaction. J. Am. Chem. Soc 114, 6556–6558. [Google Scholar]
- 44.Hicks JD, Hyde AM, Cuezva AM, and Buchwald SL (2009). Pd-catalyzed N-arylation of secondary acyclic amides: catalyst development, scope, and computational study. J. Am. Chem. Soc 131, 16720–16734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Labadie JW, and Stille JK (1983). Mechanisms of the palladium-catalyzed couplings of acid chlorides with organotin reagents. J. Am. Chem. Soc 105, 6129–6137. [Google Scholar]
- 46.Mee SPH, Lee V, and Baldwin JE (2004). Stille coupling made easier–the synergic effect of copper(I) salts and the fluoride ion. Angew. Chem. Int. Ed 43, 1132–1136. [DOI] [PubMed] [Google Scholar]
- 47.Beak P, Kerrick ST, Wu S, and Chu J (1994). Complex induced proximity effects: enantioselective syntheses based on asymmetric deprotonations of N-bocpyrrolidines. J. Am. Chem. Soc 116, 3231–3239. [Google Scholar]
- 48.Robak MT, Herbage MA, and Ellman JA (2010). Synthesis and applications of tert-butanesulfinamide. Chem. Rev 110, 3600–3740. [DOI] [PubMed] [Google Scholar]
- 49.Kells KW, and Chong JM (2003). Addition of Bu3SnLi to tert-butanesulfinimines as an efficient route to chiral, nonracemic α-aminoorganostannanes. Org. Lett 5, 4215–4218. [DOI] [PubMed] [Google Scholar]
- 50.Egorova KS, and Ananikov VP (2017). Toxicity of metal compounds: knowledge and myths. Organometallics 36, 4071–4090. [Google Scholar]
- 51.Kimbrough RD (1976). Toxicity and health effects of selected organotin compounds: a review. Rev. Environ 14, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sunday AO, Alafara BA, and Oladele OG (2012). Toxicity and speciation analysis of organotin compounds. Chem. Speciation Bioavailab 24, 216–226. [Google Scholar]
- 53.Adler AS, McCleland ML, Truong T, Lau S, Modrusan Z, Soukup TM, Roose-Girma M, Blackwood EM, and Firestein R (2012). CDK8 maintains tumor dedifferentiation and embryonic stem cell pluripotency. Cancer Res. 72, 2129–2139. [DOI] [PubMed] [Google Scholar]
- 54.Firestein R, Bass AJ, Kim SY, Dunn IF, Silver SJ, Guney I, Freed E, Ligon AH, Vena N, Ogino S, et al. (2008). CDK8 is a colorectal cancer oncogene that regulates β-catenin activity. Nature 455, 547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Firestein R, Shima K, Nosho K, Irahara N, Baba Y, Bojarski E, Giovannucci EL, Hahn WC, Fuchs CS, and Ogino S (2010). CDK8 expression in 470 colorectal cancers in relation to β-catenin activation, other molecular alterations and patient survival. Int. J. Cancer 126, 2863–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Czodrowski P, Mallinger A, Wienke D, Esdar C, Pöschke O, Busch M, Rohdich F, Eccles SA, Ortiz-Ruiz MJ, Schneider R, et al. (2016). Structure-based optimization of potent, selective, and orally bioavailable CDK8 inhibitors discovered by high-throughput screening. J. Med. Chem 59, 9337–9349. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.