

Abstract
Animal silks are built from pure protein components and their mechanical performance, such as strength and toughness, often exceed most engineered materials. The secret to this success is their unique nanoarchitectures that are formed through the hierarchical self-assembly of silk proteins. This natural material fabrication process in sharp contrast to the production of artificial silk materials, which usually are directly constructed as bulk structures from silk fibroin (SF) molecular. In recent years, with the aim of understanding and building better silk materials, a variety of fabrication strategies have been designed to control nanostructures of silks or to create functional materials from silk nanoscale building blocks. These emerging fabrication strategies offer an opportunity to tailor the structure of SF at the nanoscale and provide a promising route to produce structurally and functionally optimized silk nanomaterials. Here, we review the critical roles of silk nanoarchitectures on property and function of natural silk fibers, outline the strategies of utilization of these silk nanobuilding blocks, and we provide a critical summary of state of the art in the field to create silk nanoarchitectures and to generate silk-based nanocomponents. Further, such insights suggest templates to consider for other materials systems.
Keywords: silk, nanomaterials, nanofabrication, biomineralization, carbon nanomaterials
1. Introduction
A ubiquitous feature of animal silks is the presence of sophisticated nanostructures with dimensions ranging from the several to hundreds of nanometers. Typical examples include β-sheet nanocrystals (2-4 nm in crystal size), nanofibrils (2-20 nm in diameter), microfibrils (20-200 nm in width) and microfibril bundles (several hundred nanometers in width). These structures enable the integration of strength, stiffness, and toughness in natural silk fibers, and lead to matching essential biological functions, such as mechanical enhancements, fracture resistance, protection and prey capture.[1]
With the aim of constructing structurally and functionally optimized artificial silk materials, numerous recent studies have focused on directly utilizing these silk nanostructures to create functional materials, such as hydrogels, fibers, membranes.[1b] The applications for these silk nanomaterials has also expanded from biomedicine to other emerging fields, such as for water-treatment, environmentally adaptive materials, and optical and electronic devices.[2]
In addition to important features such as sustainable, biocompatible and biodegradable nanomaterials, silk nanomaterials, silk nanofibrils (SNFs) for example, also provide naturally inspired designs, fabrication and functional materials through unique architectural designs. For instance, fibrillar structures and highly-orientated nanofibrils are ubiquitous in the different animal silks.[1a] These material constructions endow outstanding strength and toughness, as well as superior fracture resistance of silks, when compared to most engineered materials.
Aside from fibrillar nanofeatures highlighted above, other interesting nanoscale features found in biological materials include periodic nanoporous structures; universal in biological photonic crystals such as bird feathers, butterflies, weevils, and longhorns.[3] Nature uses these periodic arrangements of pores to produce structural color for camouflage, avoiding the use of organic dyes. In addition, most natural structural materials are grown by hierarchical self-assembly and in vivo biomineralization[4], usually involving near ambient temperatures and pressure and with water as the key solvent. These material fabrication approaches are in sharp contrast to conventional engineering approaches that tend to be more energy-intensive and often require the use of toxic agents.
Inspired by these natural strategies for material designs and fabrications, a series of advanced fabrication strategies have been reported to generate and tailor the biomimetic nanostructures of silk fibroin (SF). For instance, different lithography techniques[5], such as ion-beam lithography, electron-beam lithography, soft lithography, and multi-photon lithography, have been developed to create silk nanostructures for optical and photoelectric applications. Different fabrication methods, such as in vitro biomineralization, spinning and membrane formation processes, have also been used to integrate bionanominerals (e.g., calcium carbonate, hydroxyapatite, and silica) or carbonic nanomaterials (CNMs, e.g., graphene, carbon nanotubes and graphene oxide) with SFs. These biofriendly, mechanically enhanced and functionally optimized nanocomposites combine the property and functional advantages of both SF and inorganic nanomaterials, with promising applications in biomedical and electronic fields.
This review provides a critical summary of the state of the art in the field of design, fabrication, and function of silk-based nanomaterials. We first discuss the structure-property-function relationships of nanobuilding blocks in silk fibers and review the emerging strategies for fabrication and use of natural and regenerated SF nanomaterials. We also discuss advanced nanotechnologies for the generation of SF nanocomposites, including SF/bionanominerials and SF/CNMs. Finally, we conclude by outlining future challenges for the development of silk-based nanomaterials.
2. Structure-Property-Function Relationship of Silk Nanobuilding Blocks in Nature
Most animal silks, such as spider dragline silks as well as domestic and wild silkworm silks, have exceptional mechanical properties.[1c] They exhibit a unique combination of high tensile strength, modulus, and extensibility (ductility) (Figure 1A and B). These mechanical features are distinct from synthetic fibers and other natural and engineered materials (Table 1). For example, although the Kevlar fibers are twice as strong as spider dragline silk, the extensibility is tenfold weaker.[6] Nylon fiber is another example.[6] Chemically, it is a polyamide, a chemical analog of SF, while its strength and extensibility are approximately half that of wild silkworm and spider dragline silks. The unique combination of strength and extensibility of animal silks enables the fibers to absorb a large amount of energy before breaking. As a result, the toughness of animal silks (the area under a stress-strain curve) is superior to any other synthetic or biological material (Table 1).[1a]
Figure 1.

Mechanical properties of typical biomaterials and tools for characterization. A, Stress–strain curves of A. yamamai silk, B mori silk, and Nephila edulis dragline silk compared with Kevlar 46 and nylon 66 fiber. Reproduced with permission.[11] Copyright 2018, American Chemical Society. B, Comparison of the strength and stiffness (Young’s modulus) for various biomaterials. Reproduced with permission.[118] Copyright 2011, Springer-Nature. C, Comparison of timescale and lengthscale of different tools for the rational design of nanomaterials. Reproduced with permission. [1a] Copyright 2018, Springer-Nature.
Table 1.
Mechanical properties of animal silk fibers, synthetic fibers and biological materials.
| Density (g cm−3) |
Strength (MPa) |
Strain (%) |
Modulus (GPa) |
Toughness (MJ·m−3) |
Refs | ||
|---|---|---|---|---|---|---|---|
| Animal silk fibers | Araneus dragline silk | 1.3 | 1100 | 27 | 10 | 160 | [119] |
| Araneus viscid silk | 1.3 | 500 | 270 | 0.5 | 150 | [119] | |
| Nephila clavipes dragline silk | 1.3 | 880-970 | 0.17-0.18 | 11-13 | 110 | [120] | |
| B. mori silk | 1.3 | 600 | 19 | 7 | 70 | [1c] | |
| A. yamamai silk | 1.3 | 650 | 31 | 9 | 113 | [11] | |
| A. pernyi silk | 1.3 | 460 | 23 | 12 | 65 | [11] | |
| A. mylitta silk | 1.3 | 513 | 26 | 8 | 79 | [121] | |
| A. assamensis silk | 1.307 | 564 | 26.4 | 8.5 | 95 | [121] | |
| P. ricini silk | 1.308 | 400 | 27.5 | 3.6 | 71 | [121] | |
| Synthetic fibers | Nylon fiber | 1.14 | 950 | 18 | 5 | 80 | [1c] |
| Kevlar 49 fiber | 1.44 | 3600 | 2.7 | 130 | 50 | [119] | |
| Silicone rubber | 0.98 | 50 | 850 | 0.001 | 100 | [119] | |
| Polypropylene (UIstron) |
0.89-0.91 | 585 | 17 | 0.9-1.55 | 64 | [122] | |
| PET | 1.29-1.4 | 610 | 15 | 13.8 | 55 | [122] | |
| Polyurethane | 1.12-1.24 | 370 | 540 | 0.0085 | 78 | [122] | |
| PVA | 1.19-1.31 | 210 | 26 | 2.75 | 30 | [122] | |
| PE(Courlene X3) | 0.95-0.96 | 320 | 10 | 4.2 | 18 | [122] | |
| PLA | 1.2-1.3 | 350 | 56 | 5.3 | / | [123] | |
| Carbon fiber | 1.5-2 | 4000 | 1.3 | 300 | 25 | [1c] | |
| High-tensile steel | 7.82 | 1650 | 0.8 | 190-210 | 6 | [1c] | |
| Biological materials | Tendon collagen | 120 | 12 | 1.2 | 6 | [1c] | |
| Wood (longitudinal) | 0.6-0.8 | 60-100 | 6-20 | 5-9 | [124] | ||
| Wood (transverse) | 0.6-0.8 | 4-9 | 0.5-3 | 0.5-0.8 | [124] | ||
| Skin | 1.3 | 10 | 0.04 | 15(KJ/m2) | [125] | ||
| Elastin | 2 | 150 | 0.0011 | 2 | [1c] | ||
| Resilin | 3 | 190 | 0.002 | 4 | [119] | ||
| Wool, 100%RH | 1.3 | 200 | 50 | 0.5 | 60 | [119] | |
| Cellulose fiber | 1.5 | 1000 | 100 | / | [125] | ||
| Bone | 1.8-2.08 | 160 | 3 | 20 | 4 | [126] | |
| Coral | 0.89-1.5 | 22-65 | 0 | 13-21.5 | 3e-3~4e-3 (KJ/m2) |
[126] |
Considerable research has been devoted to understanding the interplay between the molecular structure and mechanical properties of silks, with a goal towards the transfer of these insights into artificial material designs.[1c] On the basis of classic polymer theory, several structure-property models of silk fibers have been established, including the two-phase crosslinking network model[7], the mean field theory-based order/disorder fraction model[8], and the Maxwell model[9]. However, in these models, animal silks are simplified to a uniform polymer fiber, and their mechanical properties are directly related to the amino acid sequences (primary structure) and/or the secondary structures. The β-sheet nanocrystals and amorphous regions in silk fibers are thought to contribute to the strength/stiffness and toughness, respectively. However, animal silks, such as spider dragline silk[10] and Antherea yamamai silk (referred to as tensan silk, a natural fiber produced by the Japanese oak silk moth)[11] are much tougher than B. mori silk, yet with similar β-sheet contents (Table 2). These silks feature outstanding fracture resistance when compared to most synthetic polymer fibers.
Table 2.
Comparison of β-sheet content (%) of domestic silkworm, wild silkworm and spider dragline silk fibers obtained by different methods.
| B. mori silks | Wild silkworm silks | Spider dragline silks | |
|---|---|---|---|
| FTIR spectroscopy | 28a(ref[127]) | 20 -23b(ref[127-128]); 40c (ref[11]) | 17 ± 4e(ref[127]); 36f (ref[129]) |
| Raman spectroscopy | 50a(ref[130]) | 45d(ref[130]) | 36-37e, f(ref[130]) |
| 13C CP-MAS NMR | 60.5-62a(ref [131]) | 41-50b,d(ref[128, 131d, 132]) | 34f(ref [133]); 46.5g(ref[131d]) |
| X-ray diffraction | 37-60a(ref [131d, 134]) | 25-33.2b,d(ref[131d, 134a, 134d]) | 10-15f,h (ref [135]); 31.3g(ref[136]) |
| Ordered fraction [137] | 77a | 57b | 29-31f |
B. mori.
A. pernyi.
A. yamamai
S. c. ricini.
N. edulis.
N. clavipes
A.assamensis
N. Inaurata
Careful examination of these animal silks allows the identification of their universal structural features at the nanoscale: (i) most silks are composed of hierarchical fibrillar structures with a width span from several to hundreds of nanometers[12]; (ii) most silks contain nano-sized defects between nanofibrils, such as cavities and cracks.[11, 13] As an example, Figure 2 presents the characteristic hierarchical structure of tensan silk and the nanosized defects. These weak interfaces between the nanoscale features do not result in brittle failure of silk fibers; in contrast they result in tougher fibers than even defect-free fibers, such as nylon fibers.
Figure 2.

Hierarchical structure of A. yamamai silk fiber. A, schematic of the hierarchical structure of A. yamamai silk. B-C, polarized light microscopy image of A. yamamai silk. D, the cross-sectional SEM image of A. yamamai silk after tensiled to fracture. E, cross-sectional SEM image of A. yamamai silk fiber embedded in epoxy resin. Reproduced with permission. [11] Copyright 2018, American Chemical Society.
The combination of computational modeling and experimental characterization techniques provides synergistic support to understand structure-process-property relationships of animal silk fibers from the atomic scale (Å) to the meter scale (Figure 1C). Some critical evidence from both modeling and experimental characterization have disclosed that nature achieves high toughness through the design of the hierarchical nanostructures in silks (Figure 3).[10a, 14] Figure 4 summarizes the typical impact of β-sheet nanocrystals of silks on their final mechanical properties. The antiparallel β-sheet nanocrystals in spider silks are typically confined to a few nanometres, e.g., 2-4 nanometer in height. This length scale makes the most efficient use of hydrogen bonds and results in the emergence of dissipative molecular stick-slip deformation, achieving higher strength, stiffness, and toughness than found in larger nanocrystals.[14]
Figure 3.

Summary of structural hierarchy of spider silk spanning length scales from angstroms to centimeters, with crucial mechanisms that define its mechanical function.[16a]
Figure 4.

Nanoconfinement in spider silks and their effects on mechanical properties. A, schematic illustrates the relationships between β-sheet nanocrystal dimensions and their corresponding mechanical properties. S=schematic plot of the strength of a β-strand as a function of strand length h, F=strength of nanocrystal as a function of crystal size L, T=toughness of nanocrystal as a function of crystal size L. B, schematic illustrates the mechanism of how hierarchical structure formation in the strand length h and crystal size L of the β-sheet nanocrystals lead to the formation of their high mechanical performance. [14] Copyright 2010, Springer-Nature.
Specifically, when β-sheet nanocrystal size is larger than a critical scale (4 nm in length for spider silk), the deformation mode of the nanocrystals changes from a favorable shear mode into an unfavorable bending mode. In the bending mode, the competing hydrogen bonds are in varying extents of compression and tension across the β-strand, which leads to crack-like flaws. When there are open cracks in large nanocrystals, water molecules enter the crystal regions that are under tension and can disrupt the crystal, leading to earlier catastrophic failure through brittle fracture. In contrast, smaller, shear-dominated crystals feature a self-healing ability unless complete rupture occurs. Hydrogen bonds can reform during stick-slip shear motions, significantly increasing the total dissipated energy and protecting hydrogen bonds from adverse exposure to the surrounding water.
This nanoconfinement is also found along the diameter of nanofibrils (Figure 3), which are confined to a dimension of approximately 20-80 nm[10a]. This size of nanofibrils is necessary to achieve superior toughness in the presence of nanosized defects. In these regions the failure stresses and strains of defect-containing silk fibers (the crack size is 50% the width of the fibers) converge towards defect-free fibers. Also, fibrillar structures in spider dragline silk[10a] and tensan silk[11] inhibited the transverse growth of cracks through longitudinal splitting when the fiber was stretched. In addition, during fibril stretching, the heterogeneous globules/protrusions contribute to the toughness through restricted fibril shearing and controlled slippage.[15] All of the above synergic affections at the nanoscale result in the ability of silk to maintain near-molecular-level strength at the single fiber scale.[16]
The roles of the orientation of SNFs on dynamic mechanical properties of silk fibers are also exposed through inelastic (Brillouin) light scattering experiments under varying applied elastic strains.[17] The uniaxial mechanical anisotropy, nonlinearity, and spatial nonlocality generating from highly oriented SNFs provide new synergy for the formation of hypersonic bandgaps[18]. However, silk fibers without a hierarchical organization or structural orientation follow anisotropic acoustic behavior and do not form hypersonic bandgaps. As the pre-strain of the silk fiber increases from super-contracted to stretched silk, the mechanical anisotropy and the nonlocal nonlinearity increases and results in a smaller bandgap, preventing the transmittance of a smaller range of frequencies.[18]
3. Fabrication of Silk Nanobuilding Blocks
3.1. SF Self-Assembly in Aqueous Solution
Self-assembly of SF in silk glands and in aqueous solution have been intensively studied in the past few decades as a primary route to understand silk fiber formation in both living systems (silk glands) and in regenerated silk solutions.[19] In contrast, exploiting self-assembly as a strategy to understand and fabricate silk nanomaterials is relatively recent. Animal silks, especially silkworm silks, can be dissolved to SF aqueous solution through various solvent systems, such as LiBr/H2O[20] and CaCl2/ethanol/H2O[21]. The dissolved SF molecules are stable as micelles and can further assemble into elongated nanofibrils due to amphipathic features (Figure 5A and B).[19b] Different chemical and physical stimuli, such as ethanol, heat, mechanical, stirring, and ultrasound, have been used to trigger the self-assembly of SF.[22]
Figure 5.

Self-assembly of silk fibroin. A, Illustration of SF fibrils in Aqueous solution. B, AFM image of SF fibrils. Reproduced with permission.[22e] Copyright 2014, Wiley-VCH. C, Illustration of SF assembled with Nanofibril interface. D, AFM image of assembled SF-30 wt% CNF materials. Reproduced with permission. [27] Copyright 2017, American Chemical Society. E, Illustration of SF fibrils assembled with graphene nanosheet. F, AFM image of the hybrid prepared from SF/graphene (8:2) assembly at pH 10.3. Reproduced with permission.[29] Copyright 2017, American Chemical Society. G, Illustration of SF assembly within oil/water interface. H, 3D reconstructions of confocal images of silk micrococoons assembled by SF. Reproduced with permission.[32] Copyright 2017, Springer-Nature.
SNF hydrogels can be generated via self-assembly of SF. One merit of these hydrogels is their extracellular matrix-like structure to guide tissue homeostasis and repair.[23] However, most SNF hydrogels are mechanically weak due to the absence of chemical cross-linking between nanofibrils. Horseradish peroxidase (HRP) cross-linking is a useful route to improve the strength of SNF hydrogels without reducing biocompatibility.[24] Another merit of SNF hydrogels is their outstanding thixotropic character.[25] For example, the storage modulus of SNF hydrogels produced by centrifugation can be recovered to 93% with within 40 seconds after a shear strain of 5,000%. SNF hydrogels with a concentration of silk of 0.5 to 2 wt% can be transformed back into the solution state through the addition of water and mechanical stirring due to the weak interactions between the SNFs.[22g] In addition, the SNF aqueous solution can be directly obtained from SF self-assembly at low SF concentrations (below 0.3 wt%) [22e, 26]. Alcohol [22e] or heat[26] can be used to trigger SF self-assembly into SNFs.
3.2. SF Self-Assembly on Nanointerfaces
In addition to self-assembly in aqueous solution, SF also can self-assemble at one- to three-dimensional nanointerfaces, such as water/cellulose nanofibrils[27], water/inorganic nanosheets [28] [29] [30] [31], and oil/water interfaces[32]. These nanosized interfacial self-assemblies improved the properties (e.g., mechanical and biocompatibility) of the material at the nanoscale, and also created new nanocomposites for construction of nanodevices.
At the one dimensional scale, cellulose nanofibrils guided SF self-assembly along the fibril direction due to modulated interfacial interactions (e.g., hydrogen bonding and hydrophobic interactions) along heterogeneous surfaces of amphiphilic cellulose nanofibrils (Figure 5C and D). The resultant cellulose/SF nanofibril assemblies maintained the advantages of both the cellulose nanofibrils and SNFs and contained unique “shish kebab” nanostructures, allowing further functionalization through protein-based modification methods.[27]
The hydrophobic-hydrophilic surface of graphene oxide (GO), as well as the hydrophobic and pure sp2-hybridized carbon surface of graphene, are two ideal 2D interfaces for the regulation of SF self-assembly (Figure 5E and F).[28-30] Molecular dynamics simulations[28] disclosure that the balance between electrostatic and van der Waals contributions lead to the retention of SF secondary structure on the GO surface, while strong van der Waals interactions were critical for disrupting secondary structure on graphene surfaces. Experimentally, reduced GO nanosheets can direct one-dimensional growth of SF, forming unprecedented types of nanohybrids. [29] For example, under specific experimental conditions (pH 10-11, the mass ratio of SF:GO between 5:5 to 9:1, and temperature of ~60-80 °C), SF can grow preferentially and efficiently on reduced GO nanosheets. The resultant SNFs entirely covered the surface of single nanosheets, thus enhancing the toughness and cytocompatibility of the graphene. Interestingly, fibrillar structures of SF were also observed on the SF/graphene nanocomposites[33], which were produced from HFIP solvent systems, instead of aqueous solutions. The SNF formation mechanism in this process has not been studied. The high β-sheet content and negative charge characteristics of SNFs also enabled the effective exfoliation of graphite into graphene in aqueous conditions. [34] With the assistance of sonication, a concentration of 1.92 mg mL−1 and yield of ~20% were achieved using this approach. Recently, there has been increasing interest in understanding the effects of topological constraints on SF self-assembly (Figure 5G and H).[1a] Spatial constraints can break the symmetry of confined structures, leading to deviation from equilibrium morphology.[35] Spherical topologies, either as droplets or thin shells of liquid, are particularly interesting because they offer isotropic confinement and controlled curvature, usually leading to topological defects in the structure, such as radial declinations and dislocations.[36] These conditions are very useful for fundamental studies of particle packing and anisotropic self-assembly, but also are a valuable method for designing functional materials.
Geometric confinement can be achieved via microfluidic techniques (Figure 6A-D).[32] An inherent advantage of microfluidics is that the spinning channel can be designed to control the flow of the dope and to optimize processing parameters. Also, some important parameters that can alter the mechanical properties of natural and regenerated SF materials, such as the gradient of pH, salt, and shear-flow, can be applied in the microfluidic channel. A simplified schematic to explain the SF self-assembly process during spinning is shown in the bottom right of Figure 6A. At the intersection of three microfluidic channels, the core flow of the SF spinning dope is subjected to shell flows caused by the oil flowing along the spinning channels (Figure 6B), colliding with it from the sides. The shear applied to SF spinning dope further induced the transition of SF from its initial state into highly aggregated β-sheet-rich silk microstructures formed as monodispersed microemulsions (Figure 6C).[32] Polymorphic SNF nanoarchitectures, such as spheres, cylinders, short fibers, thin fibers and thick fibers, have been obtained and are considered aspotential micro-capsules to store and release drugs and antibodies (Figure 6D).
Figure 6.

Confined self-assembly of SF. A, Illustration of microfluidic device for confined self-assembly of SF with oil/water interface. B, Photograph of the microfluidic device, the inset is optical microscopy images of the fluid channel in the microfluidic device. Scale bar, 20 mm. C, Illustration of the sphere assembled with SF by the microfluidic device. D, Bright field microscopy images of various SF micrococoons. Scale bar, 5 mm. Reproduced with permission.[32] Copyright 2017, Springer-Nature. E, Illustration of a groove on a soft substrate with the confined shape. F, Polarization microscopy image of large-scale structures of silk fibrils composed of tri-hexagonal components. Inset: Camera images of the whole structure taken under the diffuse white light. Scale bars, 1 mm. G, Buckling of high-aspect-ratio beams with increasing birefringence/alignment of fibers induced via contraction. Cl, Cm, and Ch indicate samples respectively with low (10–25%), medium (25–40%) and high (>40%) contraction. Reproduced with permission. [37] Copyright 2017, Springer-Nature.
Spatial confinement is also a possible strategy for assembling nanobuilding blocks into macroscopic hierarchical materials (Figure 6E-G). A pattern molding approach can produce centimeter-scale channels with sophisticated geometric constraints, such as anchors, cables, lattices, and webs (Figure 6E and F).[37] These pre-designed structures can tailor the orientation and gradients of SNFs in materials via shear flow and force extension, offering a new route to designing materials with predictable structures and specified mechanical and physical properties. For instance, controlled placement of mechanical constraints in the molds allows for the generation of three-dimensional architectures with tailed alignment of SNFs, thereby achieving hierarchical shapes to adopt to environmental stimuli, such ashumidity-responsive designs, Figure 6G.
Superhydrophobic micropatterned surfaces have also been designed to induce the self-assembly of soluble spider silk protein (referred to as spidroin) into various forms; localized coatings, nanowire arrays, and free standing sheets.[38] Among others, aligned nanowires with diameters of around 300 nm were generated by moving the silk solution droplet laterally with appropriate speed along an array of micropillars with large diameters and small interspaces. These protein nanowires are appealing owing to their biocompatibility and ease of biological functionalization, with potential applications in optical waveguides and other opitical interfaces.
Chiral liquid crystal self-assembly of biopolymer nanofibrils, such as cellulose, chitin and collagen nanofibrils, has been investigated in recent years.[39] These self-assembly processes and resultant structures are critical for several fields, including chemical separation processes, chiral sensors, optical displays, drug delivery, and meso-templates.[40] However, the liquid crystal self-assembly of SNF system has not been studied. This field could be an exciting research direction in the future because it is beneficial for fundamental studies of natural silk spinning, as well as for the design of chiral SF materials.
3.3. Electrospinning
Electrospinning, a fiber production method which uses electric force to draw charged threads of spinning dope into fibers with nanosized diameters[41], is a relatively simple and effective technique for preparing SF nanofibers. This process offers unique capabilities for producing nonwoven fabrics with the controllable surface to volume ratio, pore size, and shape. [42] Electrospun SF nanofibers have been widely reported during the last few decades for uses in tissue engineering and drug delivery.[43] The structure of SF nanofiber depends both on the electrospinning set-up and the characteristic of the SF solution, such as molecular weight of SF, concentration, viscosity, conductivity, surface tension and the aggregation state of SF chains.[44] The application of electrospun SF nanofibers has been extended to environmental fields due to their unique nanoporous structures as well as good optical transparency.[45] These materials can be used as a translucent window screen with air-filtering and chemosensing functions.[45b] Despite considerable progress with SF-based electrospinning, efforts are still needed to improve the mechanical properties of electrospun SF nanofiber scaffolds to meet their practical applications, especially in biomedical fields.
4. Advanced Manufacturing Techniques for Creating SF Nanoarchitectures
Nature often uses periodic arrays of nanostructures to manipulate light for survival by enabling coloration/camouflage and photoprotection. Periodic helicoidal nanostructures (with a mean value of cholesteric pitch of 145 nm) in the epicarp of the fruit of Pollia condensata[46] and a hexagonal array of nanotips (∼250 nm tall and a center-to-center distance of ∼150 nm) in the Cicada wing[47] are examples. These periodic arrays of nanostructures render unique and well-defined optical properties through the efficient manipulation of light through absorption, reflection, scattering, diffraction, interference, transmission (bulk transparency), wave guiding, and lensing, as well as combinations of two or more of these effects. These natural material designs have inspired the fabrication of a variety of biomimetic periodic nanostructures.[48]
In particular, decorating SF materials with two- and three-dimensional nanotopographies has been pursued.[49] The seamless match between SF and these state-of-the-art nanomanufacturing technologies enables the creation of 2D/3D nanopatterned topographies to impart new functionalities and opportunities for SF materials. Nanostructuring of SF materials, combined with their intrinsic biocompatibility, biodegradability, transparency, and robust mechanical properties, allows for interfaces of these optics/electronics with biological systems. Optically functionalized SF films are promising materials for colorimetric biosensing (2D/3D photonic crystal) or diffraction-based biosensing (diffractive optical elements) applications, providing a simple and visual detection approach without having to utilize complex optical systems such as fluorescence detection methods. The facile formation of free-standing, flexible nanopatterned SF matrices is applicable for implantable and bioresorbable optics for biomedical applications, including tracking the biodegradation process and to monitor drug delivery, tracking the growth/remodeling of cells, and for glucose detection. To date, a variety of solid- and liquid- lithography approaches, as well as template-assisted self-assembly, have been utilized to create elaborate architectures of SF on the nanoscale.
4.1. Solid lithography
Solid lithography is a method to generate nanostructures on cured SF films through different nanolithographic techniques, such as nanoimprinting lithography, electron-beam lithography, and ion-beam lithography. Nanoimprinting lithography is a low-cost and straightforward nanolithography process with high throughput and high resolution (down to ~50 nm feature size). In this process, a nanopattern on a hard-master mold was transferred to the surface of a targeted material at elevated temperature and pressure (Figure 7A). For nanoimprinting lithography of SF, the dependence of glass transition temperature (Tg) on water content within SF films enables the execution of nanoimprinting at different temperatures.[50] For instance, SF films prepared at ambient humidity (~35%) have a Tg around 100oC, and thus can be nanoimprinted by a heated hard master pattern at 100oC, using ~50 psi for 5 s. Water-saturated SF films have a Tg around ambient temperature, and therefore can be nanoimprinted at room temperature. However, the durability of the master and the cost for generating the master need to be considered for practical applications.
Figure 7.

Nanoimpriting and electron-beam patterning for the manufacture of SF nanostructures. A, Schematics of two nanoimprinting processes: hot embossing and room-temperature embossing. AFM and SEM images of period patterns in SF film. Reproduced with permission.[50] Copyright 2010, Wiley-VCH. B, Schematics of the protein-protein imprinting process and the corresponding SEM images showing the patterns of crystallized SF master (yellow) and imprinted amorphous SF (blue). Reproduced with permission.[51] Copyright 2013, Wiley-VCH. C, Schematic of all-water-based electron-beam patterning on a SF film. Dark-field and electron microscopy images of SF nanostructures generated on the positive and negative resist. Scale bars, 1 μm. Reproduced with permission.[52] Copyright 2014, Springer-Nature.
A dry crystallized SF film can also use as a master to replace the hard master. As shown in Figure 7B, the nanopattern on dry crystallized SF film can be directly transferred to a wet non-crystallized “blank” SF film. This “protein-protein imprinting” strategy significantly increases throughput and allows for direct conformal imprinting on nonplanar surfaces due to the flexibility of the crystallized SF master.[51]
Electron-beam lithography creates nanopatterns on electron-sensitive films using a focused beam of electrons, during which the electron beam changes the solubility of the resist and makes the exposed/non-exposed regions be removed by a solvent. For SF based electron-beam lithography, water is the most common solvent, and the secondary structure of SF determines processability. Energy inputs, such as electron irradiation, can induce the conformational transition of SF between random coil, helix or β-sheet structure. For these secondary structures, random coil SF is soluble while helical or β-sheet crystalline SF is insoluble in water. Therefore, SF can be utilized as a positive or negative resist in electron-beam lithography (Figure 7C).[52] For positive resist fabrication, crystalline SF exposed to the electron beam undergoes degradation, resulting in the formation of short water-soluble polypeptides, thus creating nanohole arrays after the “water development” process. In contrast, high power electron beam drives the amorphous-to-helix conformational transition of SF during negative resist fabrication. This processing makes SF water-insoluble. In this case, nanoscale pillars are generated by the subsequent “water development” that only removes the amorphous SF in the non-exposed regions. More complex pseudo-3D nanotopographies and layer-by-layer structures also can be generated by precisely controlling the dose of electron irradiation.[53]
Ion irradiation is another stimulus to induce the conformation transition of SF and is widely used to tailor the precise nanostructure of SF. Figure 8A demonstrates an experimental route to create bionanopatterns using ion-beam lithography.[49] In this experiment, genetically engineered spidroin was used as the matrix. When amorphous spidroin was exposed to the ion irradiation, etching occurs on the surface of the spidroin film while crosslinking of protein occurs underneath the areas that are etched away by the ions. As a result, distinct 2D and grayscale patterns were achieved.
Figure 8.

Ion-beam lithography for the manufacture of SF nanostructures. A, Schematic of nanopatterning on genetically engineered spider silk using ion-beam lithography. SEM images of fabricated nanopatterns and grayscale patterns. Scale bars, 1 μm. Reproduced with permission.[49] Copyright 2018, Wiley-VCH. B, Schematic of the creation of 3D nanostructures using ion-beam lithography combined with electron-beam lithography. SEM images of fabricated 3D nanodisks and spider nanowebs. Fluorescent images showing the maintaining of the activity of the dopants in the spider silk. Reproduced with permission.[49] Copyright 2018, Wiley-VCH.
Ion-beam lithography offers faster and higher resolution patterning (~10 nm) than electron-beam lithography (~20 nm) due to the high energy and reduced scattering of the ion beam. However, both electron-beam and ion-beam lithography techniques are only capable of creating 2D and pseudo-3D nanostructures. To fabricate complex 3D structures, two kinds of lithography techniques were combined. As illustrated in Figure 8B, electron-beam lithography was used to construct nanostructures from the bottom, through crosslinking of SF, while ion-beam lithography was applied to create nanostructures from the top, through modulation of structural changes of SF. Following this procedure, 3D nanoarchitectures, e.g., nanodisks and nanowebs, were realized.[49]
4.2. Liquid Lithography
Liquid lithography is an approach to process nanostructures directly from aqueous SF solution. These techniques include soft-lithography and multiphoton lithography. Soft-lithography represents a time-effective, convenient, and low-cost lithography strategy for the manufacturing of nanostructures through self-assembly and replica molding. This technique empowers the formation of mechanically robust and flexible, optically transparent SF films containing intricate 2D and 3D nanopatterns with feature sizes sub-40 nm (Figure 9A).[54]In addition, this strategy avoids the utilization of additional harsh chemicals, salts, or high temperatures and pressures, thus providing a simple, green and versatile route for the fabrication of large-scale SF-based optical and implantable nanodevices.[55]
Figure 9.

Soft lithography and multiphoton lithography for the manufacture of SF nanostructures. A, Schematic of the soft lithography process. SEM and AFM images of the surface of a SF film with a periodic square pattern of air holes. Reproduced with permission.[54a] Copyright 2008, Wiley-VCH. B, Schematic of multiphoton lithography using diversiform SF based aqueous inks. SEM images of all-SF-based 3D micro/nanosculptures. Scale bars: 1 μm. Reproduced with permission.[56] Copyright 2015, Springer-Nature.
Multiphoton lithography, however, uses a femtosecond laser to write a desired 3D nanoconstruct in a photosensitive material based on two-photon-adsorption polymerization. Multiphoton lithography of pure/doped SF inks was realized recently (Figure 9B).[56] In the process, SF was mixed with methylene blue (photosensitizer) and a lase was used to crosslink the SF/photosensitizer system into arbitrary 3D periodic or non-periodic nanostructures. The resultant nanostructures were free-standing and could be further functionalized by adding different metal ions (e.g., silver and gold) in the SF inks.
4.3. Template-Assisted Self-Assembly
Inverse opal is a paradigm of template-assisted self-assembly. Inverse opals are a close-packed array of spherical voids, formed by infiltration of the artificial colloidal crystal template with a high refractive index material and the removal of the colloidal spheres by chemical etching or calcination. SF can infiltrate into the colloidal crystal template and self-assemble through control over the dynamics of water evaporation. After removing the template, amorphous/crystalline SF inverse opal can be generated. Crystalline SF inverse opals with highly-ordered and controllable nanostructures have been obtained by using polymethyl methacrylate opals as templates and acetone as the etching solvent[57], even though the photonic lattice of the resultant materials were difficult to modulate because the SF matrix was physically cross-linked.
Recently, large-scale amorphous SF inverse opals were fabricated using polystyrene colloidal photonic crystal multilayers as templates (Figure 10A).[58] The polystyrene multilayers were prepared through layer-by-layer scooping transfer of the floating monolayers at the water/air interface. Then, SF solution was infiltrated into the PS colloidal crystal multilayers, followed by drying. SF inverse opals with amorphous structures were then obtained by removing the polystyrene spheres using toluene. As displayed in Figure 10B, the silk inverse opal showed vivid structural colors due to the diffraction of incident light induced by the structural periodicity. Structural colors of these films are tunable by bending and knotting because of the angular dependence of the stop-band. Further, the lattice constants (and the resulting structural colors) of the films could be reconfigured with water vapor and UV light exposure, which are two effects that can induce the conformational transition of SF.
Figure 10.

Template-assisted self-assembly for the manufacture of SF nanostructures. A, Schematic of preparation of large-scale patterned SF inverse opal. B, SEM image showing highly-ordered hexagonal arrays and photograph of a large-scale bent SF inverse opal. Floral pattern created by selectively exposing part of SF inverse opal to water vapor or UV for different times, respectively. Reproduced with permission.[58a] Copyright 2017, Wiley-VCH.
5. SF Nanocomposites Synthesized by in vitro Biomineralization
In addition to the architectural designs of the materials, nature also inspires the manufacture of materials with desired structures, properties, and functions. Most natural structures are formed via self-assembly and in vivo biomineralization, such as calcification[4a-c] and silicification[4d, 4e], usually involving conditions near ambient temperatures and pressure and with water as the solvent. This approach to controlled mineralization is in contrast to engineering manufacturing approaches that tend to be more expensive and energy-intensive, and often require the use of toxic agents. In recent years, many attempts have been reported to integrate SF and biomimetic assembly (e.g., in vitro biomineralization) to replicate natural structures and properties with the aim of achieving specific functions, such as for utility in regenerative medicine, optical devices, and drug delivery systems.
The abundant amino acids in SF can be used as the reductant, template, and capping agent to trigger and regulate in vitro biomineralization, such as with calcium carbonate (CaCO3), hydroxyapatite (HAP), silica, ferrous oxide and other inorganics. SF-derived in vitro biomineralization systems offers unique molecular recognition (amino acid residues/chemistry) and sequence-specific self-assembly sites with conformational control, the capacity of functionalization, multiple modes of processability and ease of formation of different solid-state material formats to match various applications.
5.1. In Vitro Calcification
5.1.1. Calcium carbonate
CaCO3 is one of the most abundant biominerals in nature with polymorphs including vaterite, aragonite, calcite, monohydrocalcite, ikaite, and amorphous structure.[59] At ambient conditions, calcite is the thermodynamically most stable CaCO3 polymorph; aragonite is metastable CaCO3 but also widely distributed in nacre.[60] Accordingly, extensive studies have pursued the growth of aragonite through in vitro calcification, with the involvement of a wide range of soluble and insoluble additives.[61] SF is water soluble and thus has been a focus as a soluble additive. The advantages of SF for calcification include: (i) aspartic acid and glutamic acid residues to provide binding sites for calcium ions;[62] (ii) SF contains similar amino acid motifs as that found in the organic components of nacre;[63] (iii) β-sheets of SF present a preference for the formation of the aragonite polymorph of CaCO3.[64]
SF can be directly used as a trigger and molecular template to control the structure and morphology of CaCO3 nanocrystals. This control can be achieved at different structural levels, such as via hydrogen bonds, amino acid sequences, and secondary structures. SF-derived CaCO3 microparticles were successfully fabricated by controlling the nucleation and crystallization of CaCO3 microparticles via hydrogen bonding between the mineral and protein.[65] At higher structural levels, the acidic amino acid residues in SF provide nucleation, while the secondary structures regulate the polymorphs of CaCO3.
Different morphologies (hollow rice grains, hollow spheres) and polymorphs (calcite, aragonite) of CaCO3 were obtained by modulating the secondary structure of SF. The growth of CaCO3 by employing SF as a soluble additive with short-chain silane self-assembled monolayers as matrices were studied.[64b] The conformational transition of SF from silk I (random coil- and helix-dominated structure) to silk II (β-sheet-dominated structure) determined the formation of aragonite. The role of SF for different crystalline stages throughout the growth process of CaCO3 was investigated to determine which steps in calcification relied on the presence of SF,[66] and indicated that SF affected the morphology of vaterite and two morphologies of CaCO3, i.e., lens-like and multilayered vaterite were obtained.
The preparation of CaCO3 particles with homogeneous structures and pure polymorphs has also been a focus. Monodisperse vaterite particles with ordered nanostructures were fabricated using SF as a crystal modifier via compressed CO2.[67] Particles with disc-like, erythrocyte-like, and spherical morphologies were generated under different experimental conditions; SF concentration, reaction temperature, and reaction time. Calcification at the air/water interface mediated by SF was also explored since SF trends to form complex structures at air/water interfaces due to its intrinsic multi-block amphipathy.[68]
Another motivation for SF-derived in vitro calcification is the preference to form aragonite. Direct growth or dissolution and recrystallization can achieve this process.[64a] The relatively low concentrations of Ca2+ and SF allows for the formation of aragonite needles and their aggregates, of which the lattice structure of the precursor is similar to that of the organic matrix in the natural shell (i.e., regularly aligned lattice structure composed of parallel lines). Higher component concentrations led to the preferential deposition of vaterite and thus resulted in the formation of aragonite aggregates via a dissolution and recrystallization process (ref). In addition to the direct use of SF as a soluble additive for the modulation of calcification, SF-based materials, such as fibers,[69] films,[70] hydrogels,[71] and micro-/nanospheres,[72] have been used as templates to control the nucleation or polymorphs of CaCO3. For instance, SF microspheres have been used as templates to regulate the deposition of CaCO3 crystals.[72] Poly- or mono-disperse SF/CaCO3 composite microparticles were synthesized (Figure 11A and B). Degummed B. mori silk fibers and SF solution have been combined into a template/additive mineralization system to regulate the crystallization of CaCO3.[69] Aragonite crystals nucleated and grew on the fiber surfaces and were aligned with the long axis of the fibers. The SF films have also been applied for studies of in vitro calcification[70], and the thickness of films had a significant effect on calcification. Thin films were conducive to oriented aragonite growth from the surface due to the presence of β-sheets or β-strands, while thick films mainly resulted in the formation of calcite due to their random coil structures.
Figure 11.

Biomineralization of CaCO3 using different SF formats. A and B, Schematic (A) and SEM image (B) of CaCO3 with silk nanoparticles as a template. Reproduced with permission.[72b] Copyright 2015, American Chemical Society. C and D, Schematic (C) and SEM image (D) of prismatic-type thin film. Reproduced with permission.[75] Copyright 2017, Nature Publishing Group. E, Fabrication scheme of the synthetic nacre from chitosan/acetic acid solution, F, Photograph of the bulk synthetic nacre. Scale bar, 1cm. G, cross-sectional SEM image of the synthetic nacre. Scale bar, 3 μm. Reproduced with permission.[76] Copyright 2016, AAAS
Studies of biomimetic mineralization of CaCO3 are motivated not only by scientific issues aimed at the elucidation of biomineralization mechanisms evolved in nature, but also by the development of biomimetic nacre with hierarchical architectures and excellent mechanical properties that are comparable (or even superior) to their biogenic counterparts. One virtue of nacre and bone is their organic/inorganic layered nanostructure. Nacre, as an example, consists of about 95 wt% of aragonite and approximately 5 wt% of biopolymers [73]. These organic/inorganic nanocomposites are further assembled into unique “bricks and mortar” structure. This mixture of brittle inorganic platelets and the thin layers of biopolymers makes the material strong and resilient. The adhesion by the “brickwork” arrangement of the platelets further inhibits transverse crack propagation. This structure, at multiple length sizes, dramatically increases itoughness, resulting in materials that are 2-fold stronger and 1,000-fold tougher than he primary constituents.[74]
Highly-oriented, prismatic-type CaCO3 thin films have been fabricated using SF as a soluble additive.[75] As shown in Figure 11C and D, the thin films possessed a three-layered structural heterogeneity and showed comparable hardness and Young’s modulus to their biogenic counterparts. The introduction of SF significantly improved the toughness and under-water superoleophobicity of the prismatic thin films by inducing exterior micro- or nano-textures. However, the calcified crystals in artificial nacre are usually too small in size to be associated with the macroscopic mechanical properties of the materials.
Recently, an “assembly-and-mineralization” method (Figure 11E), which combines preformed laminated matrices and in situ calcification techniques, have been assessed to mimic natural processes to generate macroscale nacre-like materials.[76] The resultant millimeter-thick synthetic nacre consisted of alternating organic layers and aragonite platelet layers (91 weight percent) (Figure 11F and G), and exhibited good ultimate strength (63.8±8.3 MPa) and strain to failure (0.38±0.06%), comparable to natural Anodonta woodiana nacre (strength of 124.2±10.3 MPa and strain of 0.34±0.07%). This key report paves the new way for the design of various composite materials with unique properties. Based on the preference of SF β-sheets to the aragonite phase, and the vertical growth of aragonite needles on thin SF films, repeated spin-coating of SF solution and the deposition of aragonite crystals should be a promising route to fabricate multilayered organic-inorganic hybrid materials with outstanding mechanical properties that match natural nacreous tablets.
5.1.2. Hydroxyapatite
The development of bone substitute materials to mimic the hierarchical structure and properties of natural bone has been a hot topic in the field of bone tissue engineering for many years. As mentioned above, the main components of natural bone are collagen type I and HAP.[77] Artificial collagen/HAP composites bear a similarity to natural bone. However, collagen, which can be extracted from some animals (e.g., skin of pig and cow) or generated through transgenic techniques, may bring about concerns related to cost, quality, purity, immune responses, and contamination risks,[78] along with challenges of the retention or reformation of the key hierarchical structures. SF derived HAP nanocomposites are a promising route to generate “artificial bone” due to their biomedical applicability and robust mechanical properties.[79]
For HAP-based biomineralization, SF also can serve as a soluble regulator to grow nanosized HAPs. The introduction of SF regulated HAP nanocrystals grown along their c-axis, resulting in a structure similar to natural bone.[80] Investigations that separated the hydrophobic crystalline fractions and hydrophilic electronegative amorphous fractions of the SF demonstrated that only the amorphous fractions formed carbonated apatite when immersed in simulated body fluid. These findings support the conclusion that the electronegative amino-acidic sequences of SF direct the in vitro mineralization.[81] Moreover, SF increased the water dispersibility of HAP nanocrystals, since SF in solution adhered on the HAP nanocrystal surfaces and formed a negative charge layer to prevent aggregation of the HAP nanocrystals.[82]
Various water-insoluble SF formats, such as fibers,[83] films,[78, 84] nanofibrils/nanoparticles, [26, 85] hydrogels,[86] and scaffolds,[87] have been utilized as templates for the deposition of HAP nanocrystals. The deposition processes can be conducted through the incubation of SF-based materials in simulated body fluids or supersaturated HAP solution. Alternatively, the SF-based materials can be immersed repetitively in calcium and phosphate-rich solutions. For instance, the incubation of spider dragline silk (collected from Daddy Longlegs spiders) in supersaturated HAP solution led to the oriented nucleation of HAP crystals on the fiber surfaces.[83a]
The synergistic effects on stereochemistry and crystal plane matching, as well as favorable electrostatic interactions between SF and HAP, were found in the production of SF/HAP films.[78] The nucleation of HAP enhanced the molecular orientation and crystallinity of SF, in contrast, SF promoted the oriented growth of HAP along the c-axis. SF with different nanostructures induced the formation of different morphologies of HAP. For example, SF nanoparticles induced the formation of rice-like HAP, while SF nanofibers directed the formation of HAP nanofibers.[85b, 86a] To mimic the three-dimensional (3D) porous structure of bone tissue, pure or hybrid SF hydrogels/scaffolds were used as templates to fabricate regenerative bone-containing HAP through calcification in simulated body fluids. SF/sodium alginate hydrogels induced the formation of HAP nanorods with rectangular column morphologies at room temperature. Arginine-glycine-aspartic acid (RGD) sequence-functionalized SF preferentially induced the nucleation of HAP crystals, compared to non-RGD functionalized fibroin.[88] Additionally, HAP binding peptides, such as dentin matrix protein 1,[89] VTKHLNQISQSY,[90] were fused with SF derivatives by genetic engineering techniques to construct new proteins for site-specific in vitro calcification. The same sequence design strategy used in genetically engineered spider SF-silica fusion proteins was used in the SF-HAP binding peptide chimeras.[89-90] The SF domains offered processing and mechanical support while the HAP binding domains promoted crystallization and enhanced the osteoinductive properties of the final nanocomposites.
SF-based electrospun nanofibers were mixed with HAP nanoparticles for in vitro bone formation.[91] However, the low mechanical strength limited applications. Recently, SNF/HAP nanocomposites with flower-like structures were fabricated through the mediation of the growth of HAP in situ by SNFs.[85c] Further, extracellular matrix (ECM)-like SNF/HAP hydrogels were fabricated via centrifugation. The hydrogels showed improved mechanical properties and remarkable thixotropy, with potential for bone tissue engineering.
When HAP was deposited on the solid SF substrates, the interface bonding between HAP and the matrix directly affected the performance of the composites. Cocoon silk fibers with sericin have shown advantages in combination with HAP, as sericin can induce the nucleation and growth of HAP crystals.[83f] Graft-modified silk fibers are commonly used as the matrix for HAP growth. The coating of sintered HAP on silk fibers grafted with different functional groups has also been investigated. Rod-shaped HAP nanocrystals were used to improve the surface deposition of HAP and thereby increase the interaction between HAP and the organic matrix.[83b-d]
Although SF/HAP nanocomposites showed utility in bone repair, the mismatch in mechanical requirements remains a challenge for many applications. The enhanced strength and toughness of natural bone not only depends on collagen/HAP ratios, but also on the highly-organized arrangement of HAP crystals with the collagen fibrils. Therefore, future studies could focus on the regulation of the 3D organization of the HAP.
SF/HAP nanocomposites also show potential for environmental applications. Recently, comparative computational simulations based on coarse-grained models of SNFs and HAPs revealed that the multilayer structure could only form with weak interactions between nanofibrils and mineral plates. Based on this modeling design (Figure 12A), biomimetic water purification membranes with well-defined multilayer structures were achieved through the self-assembly of SF and in situ growth of HAP (Figure 12B).[26] These membranes exhibited highly-efficient water purification capability for dyes, proteins, metal ions and nanocolloids.
Figure 12.

Preparation of high-performance nanomaterials under computer simulation guidance. A, Coarse-grained computational MD simulations of SNF/HAP assembly and deposition. B, The pathway to fabricate the SNF/HAP membranes with visualization of typical multilayer structures. Reproduced with permission.[26] Copyright 2017, AAAS. C, The pathway to fabricate the SNF/HAP:CNF nacre-like membranes with hierarchical structures and the mechanical performance of those membranes. Reproduced with permission.[92] Copyright 2018, Wiley-VCH.
Most recently, a “in silico design and biomimetic assembly” route (Figure 12C) was designed to achieve the de novo fabrication of biomimetic SNF/HAP/chitin nanofibril self-adaptive materials.[92] This route started from computational modeling design, which provided predictions of the mechanical performance and self-folding behavior of the materials, which were then experimentally achieved by biomimetic fabrication methods. All experimental synthesis was conducted in an aqueous solution with mild and green processing. These pre-designed SNF/HAP/chitin nanofibril materials demonstrated highly ordered nacre-like structures with high mechanical performance (superior to natural nacre and other nacre-like materials) and dynamically interacted with the environment through self-folding deformation.
5.2. In vitro Silicification
Natural and bioengineered SFs are usually mixed or fused with silica-binding protein/peptide, e.g., R5 peptide (a partial sequence of silaffin) and silaffin protein, toward in vitro silicification, in order to maintain mechanical strength and processing of the protein while enhancing the selectivity of silicification. The integration of these protein components was achieved by genetic[93] or chemical approaches[94].
Spidroin derived in vitro silicification was achieved through a family of chimeric proteins that combined the R5 peptide with partial domains from the major ampullate spidroin 1 (MaSp1) of the spider, Nephila clavipes.[93e] In such fusion proteins, the segments from MaSp1 self-assembled into different material formats (e.g., films and fibers) through versatile processing techniques (e.g., solution casting, artificial spinning) and thus provided control over structure and morphology of the composites. Moreover, the R5 peptide domain catalyzed and regulated silica deposition under mild reaction conditions. The resulting mineralized composites generated silica distributions of 0.5-2 μm in diameter.
Besides the silica nanoparticles, silicified fibers and films were generated by regulating processing methods (electrospinning or methanol treatments as examples).[93b, 95]
To understand the synergetic interactions (including protein folding and protein-triggered silicification) between the protein and silica in these hybrid systems, three characteristic domains were required in spidroin-R5 peptide chimeric proteins, i.e., spidroin-derived core silk domain (for material self-assembly), R5 peptide (a silicification domain), and a histidine tag (for purification).[96] Computational modeling revealed that the presence of silica and histidine domains increased the accessible surface area to positively charged amino acids in solvents and reduced the formation of β-sheet structures, thus resulting in higher degrees of silicification. Processing conditions including reaction agents, types of crystallization and temperature were also confirmed to impact the secondary structure of the chimeric proteins and thus silicification. However, the location of silica domains had a minimal effect on β-sheet formation and exposure to the silicon ions in the solvents.[97]
The formation of genetically engineered spidroin-silica fusion proteins helped to understand the self-assembly behavior in SF derived in vitro silicification, while also establishing new material systems for functionalized biomaterials. For example, the genetically engineered spidroin-silica nanocomposites demonstrated osteoinductivity in vitro with potential applications in bone tissue engineering.[93b, 93e, 98] The presence of silica nanoparticles on the protein films promoted hMSCs proliferation, upregulated their osteogenic markers and facilitated apatite formation. Furthermore, biodegradable silica particles in the hybrid systems led to control over the degradation/remodeling rates of the spidroin-silica nanocomposites.[98b, 99]
In addition to bioengineered SFs, R5 peptides also can be chemically coupled to SF to form SF-silica chimera nanocomposites. The tyrosine groups in B. mori SF were modified by diazonium coupling and carbodiimide /N-hydroxysuccinimide (EDC/NHS) activation in preparation for chemical conjugating the silica binding peptide to the SF templates.[93f] Upon silicification under mild conditions (room temperature, ambient pressure), silica spheres with diameters of ~1.1 μm were formed on the proteinic templates. Higher degrees of silicification were observed on the SF-silica chimeras than on genetically engineered spider silk-silica fusion proteins, probably because the surface modification increased the silica binding sites. Also, the silica formation and the resultant morphologies were tunable by variation of the processing parameters, such as the peptide sequences, silk sources and synthesis routes.[93f]
These chemically-synthesized SF-silica nanocomposites displayed osteoinductive properties in 2D films, with biocompatibility.[79, 100] In these systems, the R5 peptide was chemically fused to the SF to control the assembly and deposition of the silica particles for osteogenesis.[93e, 93f] To mimic 3D cellular environments in vivo,[101] the R5 peptide was coupled to the SF in a through HRP crosslinking to create 3D protein/nanomineral composites (Figure 13A and B).[94, 100a] The R5 peptide served as a trigger and regulator for in situ silicification.[100a] The gradient distribution of the R5 peptide in the 3D composites allowed the morphology and distribution of biosilica nanoparticles to be controlled, thus offering gradients of nanocomposites to match mechanical anisotropies at osteochondral interfaces.
Figure 13.
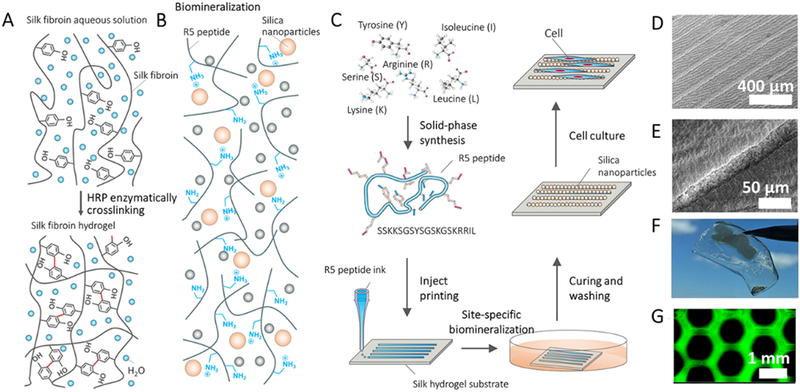
Chemically-synthesized SF-silica fusion proteins. A, Silk hydrogelation mechanism. B, Biomineralization of biosilica particles catalyzed by the biosilica selective peptide R5. C, Design strategy and processing to create silica micropatterns. D-F, SEM (D and E) and visual (F) images show mineralized peptide logo with microscale patterns (1.2 cm width). G, Fluorescence images of FITC-labeled R5 patterns on the silk hydrogels. Reproduced with permission.[102] Copyright 2018, John Wiley and Sons.
Additionly, aqueous-peptide inkjet printing and site-specific silicification were developed to form macroscale anisotropic structures with tailorable silica micropatterns (Figure 13C). In this method, inkjet printing allowed for the direct writing of macroscopic R5 peptide patterns with microscale resolution on the surface of SF hydrogels.[102] Then, in vitro silicification of the R5 peptide was carried out with the site-specific growth of silica nanoparticles on the micropatterns (Figure 13D-F). The dimensions of these micropatterns were initiated at the small scale, while they can be scaled to sub-meters to match the dimensions of human tissues (e.g., centimeter to meter scale). The resulting mineralized micropatterned systems supported the alignment of human mesenchymal stem cells and bovine serum albumin in vitro (Figure 13G).
6. SF/carbon nanomaterials
As discussed in Section 2, animal silks are one of the most robust materials and feature advantages in terms of biocompatibility and tunable in vivo degradability. However, these merits often still can not meet practical applicationrequirements; strength and stiffness of many biological materials, such as bones and teeth, are much higher than that of SF materials [103] [104]. Also, many functional applications, such as biosensors and smart textiles, require conductivity and thermostability. In these respects, carbonic nanomaterials (CNMs), such as graphene, graphene oxide, and carbon nanotubes (CNT), are mechanical and functional complements to SF materials. Owing to their sp2 carbon honeycomb structure, most CNMs, such as graphene and CNTs, are electrically conductive (terms as conductive CNMs) and known as the strongest material with a tensile strength of 130 GPa, about 200 times stronger than steel [105]. Therefore, integration of tough silk and stiff CNMs is a rational route to make mechanical and functional enhanced materials (Figure 14).
Figure 14.
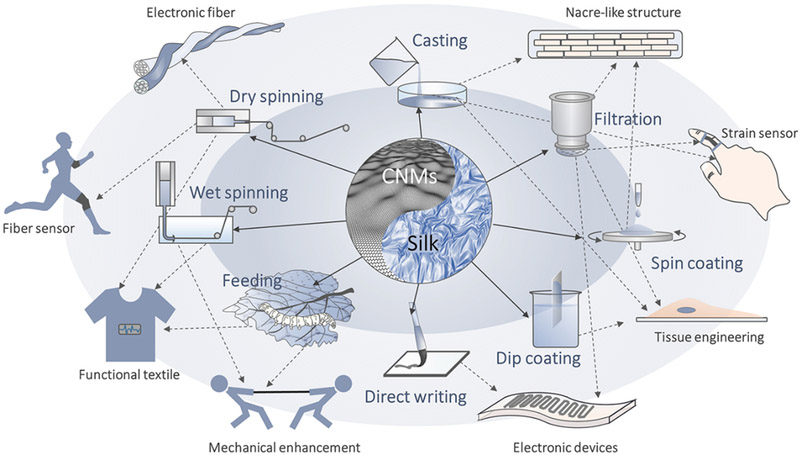
Schematic of fabrication and applications of SF/carbonic nanomaterials.
Among the different material forms, the fibers were particularly interesting in the SF/CNM system, due to advantages in processability and mechanical properties. However, SF was immiscible with conductive CNMs, especially with high volume CNMs fillings, owing to their strong π-π interaction, hydrophobicity, and chemical inertness. Alternatively, graphene oxide (GO), which has epoxy and hydroxyl groups on its graphene basal plane and carboxylic groups along the edges, was often used as a conductive CNM substitute to combine with SF[106]. Different spinning technologies, including electrospinning[107], dry spinning[106b, 108] and wet-spinning[109], have been applied to fabricate SF/GO fibers. Most of these SF/GO nanocomposites demonstrated enhanced mechanical performance when compared to the individual counterparts, which were typically stiffer than pure SF materials and tougher than GO materials (Table 3).
Table 3.
Mechanical properties and conductivity of SF/carbonic nanocomopisties.
| Materials | Methods | Content of GO or CNMs (wt%) |
Strain (%) |
Stress (MPa) |
Modulus (GPa) |
Toughness (MJ/m3) |
Conductivity (S/cm) |
|---|---|---|---|---|---|---|---|
| SF/rGO film[138] | Electrospinning | 0.5% | 11.4±5 | 0.3±0.4 | (0.4±0.5)×10−3 | / | / |
| 1% | 3±0.8 | 0.1±0.04 | (0.2±0.06)×10−3 | / | / | ||
| 2% | 4.2±1 | 0.05±0.01 | (0.08±0.1)×10−3 | / | / | ||
| SF/rGO film[139] | Electrospinning-post coating | / | 7.1±0.9 | 1.6±0.8 | 26.1±6.2 | / | / |
| SF/GO fiber[109] | Wet-spinning | 1% | / | 540 | 0.21 | / | / |
| 3% | / | 700 | 0.22 | / | / | ||
| 5% | / | 550 | 0.23 | / | / | ||
| 7% | / | 380 | 0.25 | / | / | ||
| SF/GO fiber[106b] | Dry-spinning | 0.5% | 35.9±10.9 | 299.1±45 | 4.7±1.1 | / | / |
| 1% | 21±9.5 | 435.5±71.6 | 4.8±1.1 | / | / | ||
| 1.5% | 19.3±7.1 | 321±51.6 | 6.8±1.2 | / | / | ||
| 2% | 26±9.2 | 341.5±60 | 5.8±0.8 | / | / | ||
| SF/GO film[106a] | Solution casting | 85% | 1.8±0.4 | 222±17 | 17±2 | / | / |
| SF/GO film[114] | Spin assisted layer-by-layer | 1.2% | 0.6 | 140 | 25 | 0.65 | / |
| 1.24% | 0.74 | 200 | 65 | 0.9 | / | ||
| 2.5% | 1.1 | 280 | 75 | 2.2 | / | ||
| 5.8% | 1.05 | 300 | 140 | 2.3 | / | ||
| SF/GO film[106c] | Spin-assisted layer-by-layer | 2.6% | 1.05 | 150 | 23 | 1.1 | / |
| 8.8% | 0.8 | 250 | 45 | 1.2 | / | ||
| 26.9% | 0.75 | 275 | 63 | 1.3 | / | ||
| 56.4% | 0.76 | 300 | 75 | 1.45 | / | ||
| 70% | 0.7 | 325 | 90 | 1.5 | / | ||
| SF/GO scaffold [140] | Solution phase separation | 0.5% | 50.96±7.41 | 1.85±0.41 | 3.63±0.62 | / | / |
| 1% | 43.55 ± 4.65 | 2.21±0.19 | 5.07±0.27 | / | / | ||
| 3% | 41.49 ± 9.86 | 2.84±0.51 | 6.85±0.98 | / | / | ||
| 5% | 36.74 ± 14.25 | 3.79±0.33 | 10.32±2.31 | / | / | ||
| SF/ GO/ HAP foam[141] | Solution freeze-dried | 16.7% | 10 | 30 | 0.9 | / | / |
| 10% | 17 | 80 | 1 | / | / | ||
| 5.6% | 12.5 | 70 | 0.5 | / | / | ||
| SF/CNTs film[142] | Electrospinning | 1% | 2.3 ± 0.34 | 25.57 ± 1.38 | 1.3±0.02 | / | / |
| SF/MWCNTs film [143] | Electrospinning | 0.5% | 12.3±0.8 | 7.20±0.6 | 0.06 | / | 8.66×10−6 |
| 1% | 9.25±1.5 | 9.94±1.2 | 0.11 | / | 1.2×10−4 | ||
| 1.5% | 6.67±0.6 | 6.12±0.9 | 0.09 | / | 8.61×10−8 | ||
| 2% | 4.98±0.9 | 4.28±0.4 | 0.086 | / | 7.15×10−9 | ||
| SF/MWCNTs film [144] | Electrospinning | 0.1% | 7.41 | 2.25 | 0.031 | / | / |
| 0.5% | 11.64 | 2.75 | 0.024 | / | / | ||
| 1% | 8.94 | 3.04 | 0.034 | / | / | ||
| 2% | 4.89 | 3.65 | 0.075 | / | / | ||
| SF/SWCNTs film [145] | Electrospinning | 0.5% | 1.7±0.3 | 2.84±1.1 | 0.18±0.108 | / | / |
| 1% | 1.4±0.5 | 7.4±2.5 | 0.705±0.698 | / | / | ||
| 2% | 1.8±0.5 | 3.7±1.3 | 0.285±0.112 | / | / | ||
| SF/MWCNTs film [146] | Solution casting | 0.1% | 2.75 | 33 | 1.98 | / | / |
| 0.5% | 3.75 | 44 | 2.1 | / | / | ||
| 1% | 3.5 | 40 | 2.17 | / | / | ||
| SF/graphene film [107b] | Electrospinning | 3% | 18 | 4.28±1.36 | (56.05±3.42)×10−3 | / | / |
| SF/Graphene film [147] | Self-assembly | 30% | 1.6% | 90 | 4.6 | 0.65 | 10−3 |
| 50% | 0.8% | 70 | 9.5 | 0.3 | 10−2 | ||
| 70% | 0.6% | 15 | 9 | 0.13 | 10−1 | ||
| SF/graphene/Ca2+ ions film[33] | Solution casting | 25% | 8.1 ± 2.7 | 242 ± 66 | 4.0 ± 1.1 | 11.6 ± 3.6 | 3.2×10−4 |
| 50% | 4.4 ± 0.6 | 177 ± 14 | 6.0 ± 1.1 | 4.1 ± 0.8 | 0.05 | ||
| 75% | 2.2 ± 0.5 | 49 ± 6 | 5.0 ± 1.3 | 0.7 ± 0.2 | 0.67 | ||
| Solution casting | 10% | 611 ± 85 | 0.5 ± 0.1 | 0.004 ± 0.001 | 3.3 ± 0.6 | 3.2×10−4 | |
| 20% | 382 ± 59 | 4.2 ± 1.3 | 0.135 ± 0.005 | 13.9 ± 4.7 | 8×10−4 | ||
| 40% | 202 ± 67 | 5.9 ± 1.5 | 0.079 ± 0.069 | 10.3 ± 3.2 | 2.1×10−3 | ||
| Silk fibers/Graphite flakes [148] | Dry-Meyer-rod-coating | / | 15 | / | / | / | / |
| Spider dragline silks/CNTs[149] | Dry-coating and wet-collapsing | / | 35 | 4800 | 70-72 | / | 4300 |
| Carbonized SF film [150] | Electrospinning-carbonization | 50 | / | / | / | / | |
| Spider silk fiber/GO fiber[151] | Solution coating | 45% | / | / | / | / | 0.55-0.6 |
| A. mylitta silk fibroin/carbon fiber film[152] | Solution casting | 0.1% | 20 | 20 | 0.147 | / | / |
| 0.5% | 22 | 19 | 0.178 | / | / | ||
| 1% | 23 | 11 | 0.333 | / | / | ||
| 5% | 27.5 | 6 | 0.74 | / | / | ||
| 10% | 31.5 | 3 | 1.423 | / | / | ||
| Spider silk fibers/MWCNTs fiber [112] | Solution coating | / | 73 | 600 | 7 | 290 | 12-15 |
However, mechanically, GO is much weaker than conductive CNMs, and electronically, it is an insulator.[110] These two critical disadvantages of SF/GO nanocomposites hinder their functional applications, for example, in electronic and energy storage-related fields. To maintain the advantages of conductive CNMs, several groups have attempted to feed conductive CNMs to silkworms and spiders to produce silk/CNMs fibers directly.[111] The advantage of these approaches is the retention of the mechanical superiority of natural silk fibers. The addition of the conductive CNMs can also enhance the strength and modulus of silk fibers. However, in these SF/CNM systems, the content of CNMs was insufficient to reach conductive percolation thresholds, thus limiting this approach for the production of conductive silk fibers.
Dip coating is another useful strategy to build SF/CNM fibers.[10b, 112] The resultant fibers usually are core-shell structures. Thus, the shell SF fiber is a mechanical layer while the shell CNM layer endows conductivity and other functions, such as hydrophobicity and solvent resistance properties. Amine-functionalized CNTs has been used to coat with spider silks due to their strong affinity, assisted by water and mechanical shear (refs). After this process, the CNTs adhered uniformly to the silk surface and produced tough, custom-shaped and electrically conducting fibers (Table 3), which were reversibly sensitive to strain and humidity.[10b] [112] To increase the bonding between silk fibers and conductive CNMs, several solvents, such as HFIP and formic acid/CaCl2, have been used.[10b, 33] These solvents disperse and stabilize the conductive CNMs, and also allow the partial dissolution of the silk fiber surface. A concern with these dip-coated silk/CNM fibers and textiles is washing resistance. Thus far, no detailed standard assessments of the materials for these applications have been addressed.
Membranes (also films and papers) are another material format for SF/CNM nanocomposites. Solution-casting, vacuum filtration, spinning coating, dip coating, transfer printing and direct writing are the most straightforward methods for making homogeneous SF/CNM nanocomposite membranes, as well as patterned SF/CNM membranes. The initial motivation for these studies was to mimic nacre-like structures. Nacre-like SF/GO membranes were assembled through solution-casting[106a] [113], vacuum filtration[29], and layer-by-layer spinning coating [106c, 114]. Benefiting from a dense network of weak interactions between the SF and GO surface, these SF/GO nanocomposites achieved remarkable mechanical properties. For example, spin coated ultrathin SF/GO membranes had a tensile modulus of 145 GPa and ultimate strength of more than 300 MPa (Table 3).[114] In particular, the modulus was by far the highest value that has been reached in SF/GO systems. However, the toughness of these SF/GO systems left room for improvement. For example, the toughness of ultrathin SF/GO nanocomposites with a SF content around 80% was around 2.2 MJ m−3, which was inferior to several polymeric/GO nacre-like membranes, such as polycaprolactone (PCL)/GO and cellulose/GO nanocomposites.[115] Most recently, an ultrahigh stretchable SF/graphene membrane was synthesized by addition of Ca2+ ions. The resultant membrane achieved a strain to failure of 611 ± 85%, which was comparable to natural elastomers and higher than synthetic elastomers (Table 3).[33]
In addition to developing mechanically enhanced membranes, conductive SF/CNM membranes can be utilized for electronic devices. These smart SF/CNM systems can detect and monitor surroundings and communicate the acquired physical data (e.g., force, strain, temperature, and humidity) based on variations in electrical parameters (e.g., resistance, capacitance, piezoelectricity, and triboelectricity).[116] Most electronic SF/CNM materials can be simplified into two-component systems: the conductive composition of CNM and a flexible SF substrate. The conductive CNMs endowed the electrical response with environmental stimuli, while the flexible SF substrate provided the deformability and conformability. Several unique merits of SF, such as optical transparency, biocompatibility, and biodegradability, further promote the applications of electronic SF/CNM devices in biomedical fields. So far, smart SF/CNM systems have been designed as biosensors, strain sensors and e-skins and have been described in a recent review.[117]
5. Conclusions and Outlook
In the first section of this review, we summarized how spiders and silkworms use nanoconstructions to impact the functions of silk fibers. Understanding “structure-property” relationships of animal silks at the nanoscale offers the possibility to enhance the physical properties and function of engineered SF nanomaterials. However, some nanosized structural characters of natural silk fibers require further investigation. For example, (i) the precise dimensions of single SNFs and (ii) the secondary structure organization of SFs in single SNFs remains unknown. The challenge to experimentally examine the nanobuilding blocks of silks is the lack of effective approaches to isolate and retain these nanobuilding blocks for characterization. Ultramicrotome approaches allows slicing silk fibers into thin sections, which are useful for the characterization of fibrillar topology. However, these methods only detect the surface morphologies of the fiber sections and cannot offer the detailed hierarchical nano-architecture information of the fibers [7]. Integration of partial dissolution and physical dispersion has achieved exfoliation of silk fibers at the single SNF level. Comprehensive characterization of these SNFs may provide some insight that relates nanostructure of natural silks to function.
In the second part of the review, we comprehensively discussed engineering routes to design and fabricate functional SF nanomaterials. Significant improvements have been achieved in recent years, while critical questions remain: (i) how to optimize “structure-property” relationships of engineered SF nanomaterials, and (ii) how to maintain the advantages presented at the nanoscale with nanomaterials into the macroscale. For example, SF-derived in vitro biomineralization is still unable to directly generate macroscopic materials with sophisticated structures as displayed in natural materials. Most of the in vitro biomineralization methods can only be used for the synthesis of nanoscale inorganic particles, and it remains a challenge to achieve the macroscale. Similar issues remain in advanced nanomanufacturing techniques. Nanolithography technologies, for instance, generate high-quality and high-resolution SF nanostructures, but these methods remain challenging to create sophisticated 3D nanostructures. Further, the cost and efficiency of these techniques, as well as the mechanical stability of patterned SF nanomaterials, remain essential to consider in future applications.
In practice, the properties and functions of silk-based nanomaterials depend not only on the composition, but also on structure, spatial organization, distribution of each component and the interface between components. Accordingly, the rational design and controlled fabrication of silk-based nanomaterials is important for the realization of improved materials performance and to enhance our understanding of fundamental structure-property relationships of SF materials. In the future, a better understanding the molecular mechanisms by which SFs interplay with other functional nanomaterials can help to guide the fabrication of high-performance macroscopic nanocomposites with desired structures, properties and functions.
Acknowledgments
Y.W. and J.G. contributed equally to this work. We thank the NIH (U01EB014976, R01AR068048, R01DE016525), the AFOSR and the NSF for support of various aspects of this work. Prof. S. Ling acknowledges a starting grant given by ShanghaiTech University.
Contributor Information
Yu Wang, Department of Biomedical Engineering, Tufts University, MA 02155, USA.
Jin Guo, Department of Biomedical Engineering, Tufts University, MA 02155, USA; Department of Chemical and Biological Engineering, Tufts University, MA 02155, USA.
Liang Zhou, Department of Material Science and Engineering, AnHui Agricultural University, Hefei 230036, China.
Chao Ye, School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China.
Fiorenzo G. Omenetto, Department of Biomedical Engineering, Tufts University, MA 02155, USA
David L. Kaplan, Department of Biomedical Engineering, Tufts University, MA 02155, USA
Shengjie Ling, School of Physical Science and Technology, ShanghaiTech University, Shanghai 201210, China.
References
- [1] a).Ling S, Kaplan DL, Buehler MJ, Nature Reviews Materials 2018, 3, 18016; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ling S, Chen W, Fan Y, Zheng K, Jin K, Yu H, Buehler MJ, Kaplan DL, Prog. Polym. Sci 2018, 85, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yarger JL, Cherry BR, van der Vaart A, Nat. Rev. Mater 2018, 3, 18008. [Google Scholar]
- [2].Huang W, Ling S, Li C, Omenetto FG, Kaplan DL, Chem. Soc. Rev 2018, DOI: 10.1039/C8CS00187A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Zhao Y, Xie Z, Gu H, Zhu C, Gu Z, Chem. Soc. Rev 2012, 41, 3297. [DOI] [PubMed] [Google Scholar]
- [4] a).Sudo S, Fujikawa T, Nagakura T, Ohkubo T, Sakaguchi K, Tanaka M, Nakashima K, Takahashi T, Nature 1997, 387, 563; [DOI] [PubMed] [Google Scholar]; b) Barthelat F, Tang H, Zavattieri PD, Li CM, Espinosa HD, J. Mech. Phys. Solids 2007, 55, 306; [Google Scholar]; c) Sanchez C, Arribart H, Guille MMG, Nat Mater 2005, 4, 277; [DOI] [PubMed] [Google Scholar]; d) Wang XH, Wiens M, Schroder HC, Hu SX, Mugnaioli E, Kolb U, Tremel W, Pisignano D, Muller WEG, Adv Eng Mater 2010, 12, B422; [Google Scholar]; e) Sumper M, Science 2002, 295, 2430. [DOI] [PubMed] [Google Scholar]
- [5].Zhitao Z, Shaoqing Z, Yunteng C, Benedetto M, Xiaoxia X, T. H. T, Adv. Mater 0, 1706983. [Google Scholar]
- [6].Omenetto FG, Kaplan DL, Science 329, 528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Termonia Y, Macromolecules 1994, 27, 7378. [Google Scholar]
- [8] a).Porter D, Vollrath F, Shao Z, The European physical journal. E, Soft matter 2005, 16, 199; [DOI] [PubMed] [Google Scholar]; b) Vollrath F, Porter D, Applied Physics A 2006, 82, 205. [Google Scholar]
- [9] a).Krasnov I, Diddens I, Hauptmann N, Helms G, Ogurreck M, Seydel T, Funari SS, Muller M, Physical review letters 2008, 100, 048104; [DOI] [PubMed] [Google Scholar]; b) Von Fraunhofer J, Sichina W, Biomaterials 1992, 13, 715. [DOI] [PubMed] [Google Scholar]
- [10] a).Giesa T, Arslan M, Pugno NM, Buehler MJ, Nano Lett. 2011, 11, 5038; [DOI] [PubMed] [Google Scholar]; b) Ling S, Qin Z, Li C, Huang W, Kaplan DL, Buehler MJ, Nature Communications 2017, 8, 1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang W, Ye C, Zheng K, Zhong J, Tang Y, Fan Y, Buehler MJ, Ling S, Kaplan DL, ACS Nano 2018, DOI: 10.1021/acsnano.8b02430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12] a).Lin N, Liu XY, Chem. Soc. Rev 2015, 44, 7881; [DOI] [PubMed] [Google Scholar]; b) Ruchuan L, Qinqiu D, Zhen Y, Daiwen Y, Ming-Yong H, Yang LX, Adv. Funct. Mater 2016, 26, 5534. [Google Scholar]
- [13].Frische S, Maunsbach A, Vollrath F, Journal of Microscopy 1998, 189, 64. [Google Scholar]
- [14].Keten S, Xu ZP, Ihle B, Buehler MJ, Nat. Mater 2010, 9, 359. [DOI] [PubMed] [Google Scholar]
- [15].Cranford SW, Journal of The Royal Society Interface 2013, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16] a).Tarakanova A, Buehler MJ, JOM 2012, 64, 214; [Google Scholar]; b) Cranford SW, Tarakanova A, Pugno NM, Buehler MJ, Nature 2012, 482, 72. [DOI] [PubMed] [Google Scholar]
- [17].Schneider D, Gomopoulos N, Koh CY, Papadopoulos P, Kremer F, Thomas Edwin L., Fytas G, Nat. Mater 2016, 15, 1079. [DOI] [PubMed] [Google Scholar]
- [18].Su I, Buehler MJ, Nat. Mater 2016, 15, 1054. [DOI] [PubMed] [Google Scholar]
- [19] a).Vollrath F, Knight DP, Nature 2001, 410, 541; [DOI] [PubMed] [Google Scholar]; b) Jin HJ, Kaplan DL, Nature 2003, 424, 1057. [DOI] [PubMed] [Google Scholar]
- [20].Zhou GQ, Shao ZZ, Knight DP, Yan JP, Chen X, Adv. Mater 2009, 21, 366. [Google Scholar]
- [21] a).Luo J, Zhang L, Peng Q, Sun M, Zhang Y, Shao H, Hu X, Int. J. Biol. Macromol 2014, 66, 319; [DOI] [PubMed] [Google Scholar]; b) Wei W, Zhang Y, Zhao Y, Shao H, Hu X, Materials & Design (1980-2015) 2012, 36, 816; [Google Scholar]; c) Wei W, Zhang Y, Zhao Y, Luo J, Shao H, Hu X, Materials Science and Engineering: C 2011, 31, 1602; [Google Scholar]; d) Jin Y, Zhang Y, Hang Y, Shao H, Hu X, J. Mater. Res 2013, 28, 2897. [Google Scholar]
- [22] a).Gong Z, Huang L, Yang Y, Chen X, Shao Z, Chemical Communications 2009, DOI: 10.1039/B914218E7506; [DOI] [PubMed] [Google Scholar]; b) Gong Z, Yang Y, Huang L, Chen X, Shao Z, Soft Matter 2010, 6, 1217; [Google Scholar]; c) Greving I, Cai M, Vollrath F, Schniepp HC, Biomacromolecules 2012, 13, 676; [DOI] [PubMed] [Google Scholar]; d) Bai S, Liu S, Zhang C, Xu W, Lu Q, Han H, Kaplan DL, Zhu H, Acta Biomaterialia 2013, 9, 7806; [DOI] [PubMed] [Google Scholar]; e) Ling S, Li C, Adamcik J, Shao Z, Chen X, Mezzenga R, Adv. Mater 2014, 26, 4569; [DOI] [PubMed] [Google Scholar]; f) Zhong J, Ma M, Li W, Zhou J, Yan Z, He D, Biopolymers 2014, 101, 1181; [DOI] [PubMed] [Google Scholar]; g) Bai S, Zhang X, Lu Q, Sheng W, Liu L, Dong B, Kaplan DL, Zhu H, Biomacromolecules 2014, 15, 3044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hussey GS, Dziki JL, Badylak SF, Nature Reviews Materials 2018, 3, 159. [Google Scholar]
- [24] a).Su D, Yao M, Liu J, Zhong Y, Chen X, Shao Z, ACS Appl. Mater. Interfaces 2017, 9, 17489; [DOI] [PubMed] [Google Scholar]; b) Dong X, Zhao Q, Xiao L, Lu Q, Kaplan DL, Biomacromolecules 2016, 17, 3000. [DOI] [PubMed] [Google Scholar]
- [25].Liu Y, Ling S, Wang S, Chen X, Shao Z, Biomater. Sci 2014, 2, 1338. [DOI] [PubMed] [Google Scholar]
- [26].Ling S, Qin Z, Huang W, Cao S, Kaplan DL, Buehler MJ, Science Advances 2017, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Xiong R, Kim HS, Zhang S, Kim S, Korolovych VF, Ma R, Yingling YG, Lu C, Tsukruk VV, ACS Nano 2017, 11, 12008. [DOI] [PubMed] [Google Scholar]
- [28].Grant AM, Kim HS, Dupnock TL, Hu K, Yingling YG, Tsukruk VV, Adv. Funct. Mater 2016, 26, 6380. [Google Scholar]
- [29].Ling SJ, Li CX, Adamcik J, Wang SH, Shao ZZ, Chen X, Mezzenga R, ACS Macro Lett. 2014, 3, 146. [DOI] [PubMed] [Google Scholar]
- [30].Wang Y, Song Y, Wang Y, Chen X, Xia Y, Shao Z, J. Mater. Chem. A 2015, 3, 773. [Google Scholar]
- [31].Li Z, Yang Y, Yao J, Shao Z, Chen X, Materials Science and Engineering: C 2017, 79, 123. [DOI] [PubMed] [Google Scholar]
- [32].Shimanovich U, Ruggeri FS, De Genst E, Adamcik J, Barros TP, Porter D, Müller T, Mezzenga R, Dobson CM, Vollrath F, Holland C, Knowles TPJ, Nat Commun 2017, 8, 15902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ling S, Wang Q, Zhang D, Zhang Y, Mu X, Kaplan DL, Buehler MJ, Advanced Functional Materials, DOI: 10.1002/adfm.2017052911705291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang X, Wang L, Lu Q, Kaplan DL, ACS Appl. Mater. Interfaces 2018, 10, 22924. [DOI] [PubMed] [Google Scholar]
- [35] a).Parker RM, Frka-Petesic B, Guidetti G, Kamita G, Consani G, Abell C, Vignolini S, ACS Nano 2016, 10, 8443; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Suen J. Jun-Yan, Prince E, Larin EM, Klinkova A, Thérien-Aubin H, Zhu S, Yang B, Helmy AS, Lavrentovich OD, Kumacheva E, Nat Commun 2016, 7, 12520; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Jativa F, Schutz C, Bergstrom L, Zhang X, Wicklein B, Soft Matter 2015, 11, 5374; [DOI] [PubMed] [Google Scholar]; d) Wang P-X, Hamad WY, MacLachlan MJ, Angew. Chem. Int. Ed 2016, 55, 12460. [DOI] [PubMed] [Google Scholar]
- [36] a).Håkansson KMO, Fall AB, Lundell F, Yu S, Krywka C, Roth SV, Santoro G, Kvick M, Prahl Wittberg L, Wågberg L, Söderberg LD, Nat Commun 2014, 5, 4018; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Nyström G, Arcari M, Mezzenga R, Nat. Nanotechnol 2018, 13, 330. [DOI] [PubMed] [Google Scholar]
- [37].Tseng P, Napier B, Zhao S, Mitropoulos AN, Applegate MB, Marelli B, Kaplan DL, Omenetto FG, Nat. Nanotechnol 2017, 12, 474. [DOI] [PubMed] [Google Scholar]
- [38].Gustafsson L, Jansson R, Hedhammar M, van der Wijngaart W, Adv Mater 2018, 30. [DOI] [PubMed] [Google Scholar]
- [39].Kelly JA, Giese M, Shopsowitz KE, Hamad WY, MacLachlan MJ, Acc. Chem. Res 2014, 47, 1088. [DOI] [PubMed] [Google Scholar]
- [40].Meseck GR, Terpstra AS, MacLachlan MJ, Curr Opin Colloid In 2017, 29, 9. [Google Scholar]
- [41].D. L, Y. X, Adv. Mater 2004, 16, 1151. [Google Scholar]
- [42].Bhardwaj N, Kundu SC, Biotechnol Adv 2010, 28, 325. [DOI] [PubMed] [Google Scholar]
- [43] a).Soffer L, Wang XY, Zhang XH, Kluge J, Dorfmann L, Kaplan DL, Leisk G, J Biomat Sci-Polym E 2008, 19, 653; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li HJ, Zhu JX, Chen S, Jia L, Ma YL, Rsc Adv 2017, 7, 56550. [Google Scholar]
- [44].Zhang F, Zuo BQ, Fan ZH, Xie ZG, Lu Q, Zhang XG, Kaplan DL, Biomacromolecules 2012, 13, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45] a).Wang CY, Wu SY, Jian MQ, Xie JR, Xu LP, Yang XD, Zheng QS, Zhang YY, Nano Res 2016, 9, 2590; [Google Scholar]; b) Min K, Kim S, Kim S, Sci Rep-Uk 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Vignolini S, Rudall PJ, Rowland AV, Reed A, Moyroud E, Faden RB, Baumberg JJ, Glover BJ, Steiner U, Proceedings of the National Academy of Sciences 2012, 109, 15712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Huang Y-F, Jen Y-J, Chen L-C, Chen K-H, Chattopadhyay S, ACS Nano 2015, 9, 301. [DOI] [PubMed] [Google Scholar]
- [48].Tadepalli S, Slocik JM, Gupta MK, Naik RR, Singamaneni S, Chem. Rev 2017, 117, 12705. [DOI] [PubMed] [Google Scholar]
- [49].Jiang JJ, Zhang SQ, Qian ZG, Qin N, Song WW, Sun L, Zhou ZT, Shi ZF, Chen L, Li XX, Mao Y, Kaplan DL, Corder SNG, Chen XZ, Liu MK, Omenetto FG, Xia XX, Tao TH, Adv Mater 2018, 30. [DOI] [PubMed] [Google Scholar]
- [50].Amsden JJ, Domachuk P, Gopinath A, White RD, Dal Negro L, Kaplan DL, Omenetto FG, Adv. Mater. 2010, 22, 1746. [DOI] [PubMed] [Google Scholar]
- [51].Brenckle MA, Tao H, Kim S, Paquette M, Kaplan DL, Omenetto FG, Adv. Mater 2013, 25, 2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Kim S, Marelli B, Brenckle MA, Mitropoulos AN, Gil ES, Tsioris K, Tao H, Kaplan DL, Omenetto FG, Nat Nanotechnol 2014, 9, 306. [DOI] [PubMed] [Google Scholar]
- [53].Qin N, Zhang SQ, Jiang JJ, Gilbert Corder S, Qian ZG, Zhou ZT, Lee W, Liu KY, Wang XH, Li XX, Shi ZF, Mao Y, Bechtel HA, Martin MC, Xia XX, Marelli B, Kaplan DL, Omenetto FG, Liu MK, Tao TH, Nat Commun 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54] a).Perry H, Gopinath A, Kaplan DL, Dal Negro L, Omenetto FG, Adv. Mater 2008, 20, 3070; [Google Scholar]; b) Omenetto FG, KapLan DL, Nat Photonics 2008, 2, 641. [Google Scholar]
- [55].Zhou ZT, Shi ZF, Cai XQ, Zhang SQ, Corder SG, Li XX, Zhang YS, Zhang GZ, Chen L, Liu MK, Kaplan DL, Omenetto FG, Mao Y, Tao ZD, Tao TH, Adv Mater 2017, 29. [DOI] [PubMed] [Google Scholar]
- [56].Sun YL, Li Q, Sun SM, Huang JC, Zheng BY, Chen QD, Shao ZZ, Sun HB, Nat Commun 2015, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kim S, Mitropoulos AN, Spitzberg JD, Tao H, Kaplan DL, Omenetto FG, Nat. Photonics 2012, 6, 817. [Google Scholar]
- [58].Wang Y, Aurelio D, Li WY, Tseng P, Zheng ZZ, Li M, Kaplan DL, Liscidini M, Omenetto FG, Adv Mater 2017, 29; [DOI] [PubMed] [Google Scholar]; Wang Y, Li M, Colusso E, Li WY, Omenetto FG, Adv Opt Mater 2018, 6. [Google Scholar]
- [59].Nebel H, Neumann M, Mayer C, Epple M, Inorg Chem 2008, 47, 7874. [DOI] [PubMed] [Google Scholar]
- [60].Treves K, Traub W, Weiner S, Addadi L, Helv Chim Acta 2003, 86, 1101. [Google Scholar]
- [61] a).Song RQ, Colfen H, Crystengcomm 2011, 13, 1249; [Google Scholar]; b) Ren DN, Feng QL, Bourrat X, Micron 2011, 42, 228. [DOI] [PubMed] [Google Scholar]
- [62].Tanaka K, Kajiyama N, Ishikura K, Waga S, Kikuchi A, Ohtomo K, Takagi T, Mizuno S, Bba-Protein Struct M 1999, 1432, 92. [DOI] [PubMed] [Google Scholar]
- [63] a).Weiner S, Talmon Y, Traub W, Int J Biol Macromol 1983, 5, 325; [Google Scholar]; b) Mann S, Nature 1988, 332, 119; [Google Scholar]; c) Zaremba CM, Belcher AM, Fritz M, Li YL, Mann S, Hansma PK, Morse DE, Speck JS, Stucky GD, Chem Mater 1996, 8, 679. [Google Scholar]
- [64] a).Wang T, Porter D, Shao ZZ, Adv Funct Mater 2012, 22, 435; [Google Scholar]; b) An XQ, Cao CB, J Phys Chem C 2008, 112, 15844. [Google Scholar]
- [65].Cheng C, Shao ZZ, Vollrath F, Adv Funct Mater 2008, 18, 2172. [Google Scholar]
- [66].Wang T, Che RC, Li WT, Mi RX, Shao ZZ, Cryst Growth Des 2011, 11, 2164. [Google Scholar]
- [67].Xu SJ, Wu PY, Crystengcomm 2013, 15, 5179. [Google Scholar]
- [68].Hao W, Porter D, Wang XT, Shao ZZ, Crystengcomm 2014, 16, 9176. [Google Scholar]
- [69].Cheng C, Yang YH, Chen X, Shao ZZ, Chem. Commun 2008, DOI: 10.1039/b810337b5511. [DOI] [PubMed] [Google Scholar]
- [70].Hao W, Porter D, Shao ZZ, Rsc Adv 2014, 4, 35258. [Google Scholar]
- [71] a).Keene EC, Evans JS, Estroff LA, Cryst Growth Des 2010, 10, 5169; [Google Scholar]; b) Ma YF, Feng QL, Bourrat X, Mat Sci Eng C-Mater 2013, 33, 2413; [DOI] [PubMed] [Google Scholar]; c) Ming JF, Zuo BQ, J Cryst Growth 2014, 386, 154. [Google Scholar]
- [72] a).Zhang XL, Fan ZH, Lu Q, Huang YL, Kaplan DL, Zhu HS, Acta Biomater. 2013, 9, 6974; [DOI] [PubMed] [Google Scholar]; b) Liu LJ, Zhang XL, Liu X, Liu J, Lu GZ, Kaplan DL, Zhu HS, Lu Q, Acs Appl Mater Inter 2015, 7, 1735; [DOI] [PubMed] [Google Scholar]; c) Liu SS, Liu LJ, Xiao LY, Lu Q, Zhuc HS, Kaplanad DL, J Mater Chem B 2015, 3, 8314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Nudelman F, Gotliv BA, Addadi L, Weiner S, J. Struct. Biol 2006, 153, 176. [DOI] [PubMed] [Google Scholar]
- [74].Proceedings of the Royal Society of London. Series B. Biological Sciences 1988, 234, 415. [DOI] [PubMed] [Google Scholar]
- [75].Xiao CL, Li M, Wang BJ, Liu MF, Shao CY, Pan HH, Lu Y, Xu BB, Li SW, Zhan D, Jiang Y, Tang RK, Liu XY, Colfen H, Nat Commun 2017, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Mao L-B, Gao H-L, Yao H-B, Liu L, Cölfen H, Liu G, Chen S-M, Li S-K, Yan Y-X, Liu Y-Y, Yu S-H, Science 2016, 354, 107. [DOI] [PubMed] [Google Scholar]
- [77].Weiner S, Wagner HD, Annu Rev Mater Sci 1998, 28, 271. [Google Scholar]
- [78].Du CL, Jin J, Li YC, Kong XD, Wei KM, Yao JM, Mat Sci Eng C-Bio S 2009, 29, 62. [Google Scholar]
- [79].Altman GH, Diaz F, Jakuba C, Calabro T, Horan RL, Chen JS, Lu H, Richmond J, Kaplan DL, Biomaterials 2003, 24, 401. [DOI] [PubMed] [Google Scholar]
- [80] a).Wang J, Yu F, Qu LJ, Meng XC, Wen G, Biomed Mater 2010, 5; [DOI] [PubMed] [Google Scholar]; b) Wang JL, Zhou W, Hu W, Zhou L, Wang SQ, Zhang SM, J Biomed Mater Res A 2011, 99A, 327; [DOI] [PubMed] [Google Scholar]; c) Fan CQ, Li JS, Xu GH, He HL, Ye XJ, Chen YY, Sheng XH, Fu JW, He DN, J Mater Sci 2010, 45, 5814; [Google Scholar]; d) Kong XD, Cui FZ, Wang XM, Zhang M, Zhang W, J Cryst Growth 2004, 270, 197. [Google Scholar]
- [81].Marelli B, Ghezzi CE, Alessandrino A, Barralet JE, Freddi G, Nazhat SN, Biomaterials 2012, 33, 102. [DOI] [PubMed] [Google Scholar]
- [82].Huang XW, Liu X, Liu SS, Zhang AL, Lu Q, Kaplan DL, Zhe HS, J Biomed Mater Res B 2014, 102, 1720. [DOI] [PubMed] [Google Scholar]
- [83] a).Cao B, Mao C, Langmuir 2007, 23, 10701; [DOI] [PubMed] [Google Scholar]; Furuzono T, Kishida A, Tanaka J, J Mater Sci-Mater M 2004, 15, 19; [DOI] [PubMed] [Google Scholar]; b) Korematsu A, Furuzono T, Yasuda S, Tanaka J, Kishida A, J Mater Sci 2004, 39, 3221; [Google Scholar]; c) Korematsu A, Furuzono T, Yasuda S, Tanaka J, Kishida A, J Mater Sci-Mater M 2005, 16, 67; [DOI] [PubMed] [Google Scholar]; d) Xu ZP, Shi LY, Hu DD, Hu BH, Yang MY, Zhu LJ, Rsc Adv 2016, 6; [Google Scholar]; e) Takeuchi A, Ohtsuki C, Miyazaki T, Tanaka H, Yamazaki M, Tanihara M, J Biomed Mater Res A 2003, 65A, 283. [DOI] [PubMed] [Google Scholar]
- [84] a).Li YC, Cai YR, Kong XD, Yao JM, Appl. Surf. Sci 2008, 255, 1681; [Google Scholar]; b) Kino R, Ikoma T, Monkawa A, Yunoki S, Munekata M, Tanaka J, Asakura T, J Appl Polym Sci 2006, 99, 2822. [Google Scholar]
- [85] a).Choi Y, Cho SY, Park DJ, Park HH, Heo S, Jin HJ, J Biomed Mater Res B 2012, 100B, 2029; [DOI] [PubMed] [Google Scholar]; b) He XL, Huang XW, Lu Q, Bai SL, Zhu HS, Prog Nat Sci-Mater 2012, 22, 115; [Google Scholar]; c) Mi RX, Liu YX, Chen X, Shao ZZ, Nanoscale 2016, 8, 20096. [DOI] [PubMed] [Google Scholar]
- [86] a).Ming JF, Bie SY, Jiang ZJ, Wang P, Zuo BQ, Mater Lett 2014, 126, 169; [Google Scholar]; b) Jin YS, Kundu B, Cai YR, Kundu SC, Yao JM, Colloid Surface B 2015, 134, 339; [DOI] [PubMed] [Google Scholar]; c) Ming JF, Jiang ZJ, Wang P, Bie SY, Zuo BQ, Mat Sci Eng C-Mater 2015, 51, 287. [DOI] [PubMed] [Google Scholar]
- [87] a).Lin F, Li YC, Jin J, Cai YR, Wei KM, Yao JM, Mater Chem Phys 2008, 111, 92; [Google Scholar]; b) Vachiraroj N, Ratanavaraporn J, Damrongsakkul S, Pichyangkura R, Banaprasert T, Kanokpanont S, Int J Biol Macromol 2009, 45, 470; [DOI] [PubMed] [Google Scholar]; c) Kim HJ, Kim UJ, Kim HS, Li CM, Wada M, Leisk GG, Kaplan DL, Bone 2008, 42, 1226; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jiang XQ, Zhao J, Wang SY, Sun XJ, Zhang XL, Chen J, Kaplan DL, Zhang ZY, Biomaterials 2009, 30, 4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88] a).Sofia S, McCarthy MB, Gronowicz G, Kaplan DL, J Biomed Mater Res 2001, 54, 139; [DOI] [PubMed] [Google Scholar]; b) Meinel L, Karageorgiou V, Hofmann S, Fajardo R, Snyder B, Li CM, Zichner L, Langer R, Vunjak-Novakovic G, Kaplan DL, J Biomed Mater Res A 2004, 71A, 25; [DOI] [PubMed] [Google Scholar]; c) Xu WP, Zhang W, Asrican R, Kim HJ, Kaplan DL, Yelick PC, Tissue Eng. Part A 2008, 14, 549; [DOI] [PubMed] [Google Scholar]; d) Morgan AW, Roskov KE, Lin-Gibson S, Kaplan DL, Becker ML, Simon CG, Biomaterials 2008, 29, 2556. [DOI] [PubMed] [Google Scholar]
- [89].Huang J, Wong C, George A, Kaplan DL, Biomaterials 2007, 28, 2358. [DOI] [PubMed] [Google Scholar]
- [90].Dinjaski N, Plowright R, Zhou S, Belton DJ, Perry CC, Kaplan DL, Acta Biomater 2017, 49, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91] a).Li CM, Jin HJ, Botsaris GD, Kaplan DL, J Mater Res 2005, 20, 3374; [Google Scholar]; b) Li CM, Vepari C, Jin HJ, Kim HJ, Kaplan DL, Biomaterials 2006, 27, 3115; [DOI] [PubMed] [Google Scholar]; c) Cao H, Chen X, Yao JR, Shao ZZ, J Mater Sci 2013, 48, 150; [Google Scholar]; d) Lee MJ, Park JB, Kim HH, Ki CS, Park SY, Kim HJ, Park YH, Macromol Res 2014, 22, 710. [Google Scholar]
- [92].Shengjie Ling KJ, Qin Zhao, Li Chunmei, Wang Qi, Kaaplan David L., Buehler Markus J., Adv. Mater 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93] a).Zhou S, Huang WW, Belton DJ, Simmons LO, Perry CC, Wang XQ, Kaplan DL, Acta Biomater 2015, 15, 173; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Plowright R, Dinjaski N, Zhou S, Belton DJ, Kaplan DL, Perry CC, Rsc Adv 2016, 6, 21776; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Martín‐Moldes Zaira, Ebrahimi Davoud, Plowright Robyn, Dinjaski Nina, Perry Carole C., Buehler Markus J., Kaplan DL, Adv Funct Mater 2017, 1702570; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Mieszawska AJ, Nadkarni LD, Perry CC, Kaplan DL, Chem Mater 2010, 22, 5780; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Foo CWP, Patwardhan SV, Belton DJ, Kitchel B, Anastasiades D, Huang J, Naik RR, Perry CC, Kaplan DL, P Natl Acad Sci USA 2006, 103, 9428; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Canabady-Rochelle LLS, Belton DJ, Deschaume O, Currie HA, Kaplan DL, Perry CC, Biomacromolecules 2012, 13, 683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Guo J, Li CM, Ling SJ, Huang WW, Chen Y, Kaplan DL, Biomaterials 2017, 145, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95] a).Krishnaji ST, Huang WW, Rabotyagova O, Kharlampieva E, Choi I, Tsukruk VV, Naik R, Cebe P, Kaplan DL, Langmuir 2011, 27, 1000; [DOI] [PubMed] [Google Scholar]; b) Rabotyagova OS, Cebe P, Kaplan DL, Biomacromolecules 2009, 10, 229. [DOI] [PubMed] [Google Scholar]
- [96].Dinjaski N, Ebrahimi D, Ling SJ, Shah S, Buehler MJ, Kaplan DL, Acs Biomater Sci Eng 2017, 3, 2877. [DOI] [PubMed] [Google Scholar]
- [97] a).Emami FS, Puddu V, Berry RJ, Varshney V, Patwardhan SV, Perry CC, Heinz H, Chem Mater 2014, 26, 5725; [Google Scholar]; b) Krishnaji ST, Bratzel G, Kinahan ME, Kluge JA, Staii C, Wong JY, Buehler MJ, Kaplan DL, Adv Funct Mater 2013, 23, 241; [Google Scholar]; c) Belton DJ, Mieszawska AJ, Currie HA, Kaplan DL, Perry CC, Langmuir 2012, 28, 4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98] a).Zhou SH, Varughese B, Eichhorn B, Jackson G, McIlwrath K, Angew Chem Int Edit 2005, 44, 4539; [DOI] [PubMed] [Google Scholar]; b) Mieszawska AJ, Fourligas N, Georgakoudi I, Ouhib NM, Belton DJ, Perry CC, Kaplan DL, Biomaterials 2010, 31, 8902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Gomes S, Leonor IB, Mano JF, Reis RL, Kaplan DL, Prog Polym Sci 2012, 37, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100] a).Partlow BP, Hanna CW, Rnjak-Kovacina J, Moreau JE, Applegate MB, Burke KA, Marelli B, Mitropoulos AN, Omenetto FG, Kaplan DL, Adv Funct Mater 2014, 24, 4615; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Meinel L, Hofmann S, Karageorgiou V, Kirker-Head C, McCool J, Gronowicz G, Zichner L, Langer R, Vunjak-Novakovic G, Kaplan DL, Biomaterials 2005, 26, 147. [DOI] [PubMed] [Google Scholar]
- [101].Izadifar Z, Chen X, Kulyk W, J. Funct. Biomater 2012, 3, 799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Guo Jin, Ling Shengjie, Li Wenyi, Chen Ying, Li Chunmei, Omenetto Fiorenzo G., Kaplan DL, Adv Funct Mater 2018, 1800228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Espinosa HD, Rim JE, Barthelat F, Buehler MJ, Prog. Mater Sci 2009, 54, 1059. [Google Scholar]
- [104].Barthelat F, Yin Z, Buehler MJ, Nature Reviews Materials 2016, DOI: 10.1038/natrevmats.2016.716007. [DOI] [Google Scholar]
- [105].Lee C, Wei X, Kysar JW, Hone J, Science 2008, 321, 385. [DOI] [PubMed] [Google Scholar]
- [106] a).Huang L, Li C, Yuan W, Shi G, Nanoscale 2013, 5, 3780; [DOI] [PubMed] [Google Scholar]; b) Zhang C, Zhang Y, Shao H, Hu X, ACS Appl. Mater. Interfaces 2016, 8, 3349; [DOI] [PubMed] [Google Scholar]; c) Wang Y, Ma R, Hu K, Kim S, Fang G, Shao Z, Tsukruk VV, ACS Appl. Mater. Interfaces 2016, 8, 24962. [DOI] [PubMed] [Google Scholar]
- [107] a).Wang S-D, Ma Q, Wang K, Chen H-W, ACS Omega 2018, 3, 406; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yang Y, Ding X, Zou T, Peng G, Liu H, Fan Y, Rsc Adv 2017, 7, 7954. [Google Scholar]
- [108].Zhang C, Zhang Y, Luo J, Shi J, Shao H, Hu X, Rsc Adv 2017, 7, 3108. [Google Scholar]
- [109].Hu X, Li J, Bai Y, Mater. Lett 2017, 194, 224. [Google Scholar]
- [110].Dreyer DR, Park S, Bielawski CW, Ruoff RS, Chem. Soc. Rev 2010, 39, 228. [DOI] [PubMed] [Google Scholar]
- [111] a).Wang Q, Wang C, Zhang M, Jian M, Zhang Y, Nano Lett. 2016, 16, 6695; [DOI] [PubMed] [Google Scholar]; b) Emiliano L, Federico B, Francesco B, Matteo B, Simone T, Giovanni G, Andrea CF, Nicola Maria P, 2D Materials 2017, 4, 031013. [Google Scholar]
- [112].Steven E, Saleh WR, Lebedev V, Acquah SFA, Laukhin V, Alamo RG, Brooks JS, Nat Commun 2013, 4, 2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Zhang C, Shao H, Luo J, Hu X, Zhang Y, Int. J. Biol. Macromol. 2018, 107, 2590. [DOI] [PubMed] [Google Scholar]
- [114].Kesong H, K. GM, D. KD, V. TV, Adv. Mater 2013, 25, 2301. [DOI] [PubMed] [Google Scholar]
- [115].Zhang Y, Gong S, Zhang Q, Ming P, Wan S, Peng J, Jiang L, Cheng Q, Chem. Soc. Rev 2016, 45, 2378. [DOI] [PubMed] [Google Scholar]
- [116].Kenry J Yeo C, Lim CT, Microsyst. Nanoeng 2016, 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Bowen Z, Hong W, Ru LW, Yurong C, Jun LX, Ming-Yong H, Xiaodong C, Adv. Mater 2016, 28, 4250.26684370 [Google Scholar]
- [118].Knowles TPJ, Buehler MJ, Nat. Nanotechnol 2011, 6, 469. [DOI] [PubMed] [Google Scholar]
- [119].Gosline JM, Guerette PA, Ortlepp CS, Savage KN, Journal of Experimental Biology 1999, 202, 3295. [DOI] [PubMed] [Google Scholar]
- [120].M. CP, A. FS, A. AM, W. SJ, L. KD, Wade AW, K. ER, David M, L. VD, Polymers for Advanced Technologies 1994, 5, 401. [Google Scholar]
- [121].R. R, B. GV, K. KV, Journal of Applied Polymer Science 2000, 77, 2418. [Google Scholar]
- [122].Morton WE, Hearle JWS, in Physical Properties of Textile Fibres (Fourth Edition), DOI: 10.1533/9781845694425.274 (Eds: Morton WE, Hearle JWS), Woodhead Publishing; 2008, p. 274. [DOI] [Google Scholar]
- [123].Solarski S, Ferreira M, Devaux E, Polymer 2005, 46, 11187. [Google Scholar]
- [124].in Materials Selection in Mechanical Design (Fourth Edition), DOI: 10.1016/B978-1-85617-663-7.00020-5 (Ed: Ashby MF), Butterworth-Heinemann, Oxford: 2011, p. 559. [DOI] [Google Scholar]
- [125].Ashby MF, Gibson LJ, Wegst U, Olive R, Proceedings Mathematical & Physical Sciences 1995, 450, 123. [Google Scholar]
- [126].Ashby MF, 2008.
- [127].Ling S, Qi Z, Knight DP, Shao Z, Chen X, Biomacromolecules 2011, 12, 3344. [DOI] [PubMed] [Google Scholar]
- [128].Wang Y, Wen J, Peng B, Hu B, Chen X, Shao Z, Biomacromolecules 2018, 19, 1999. [DOI] [PubMed] [Google Scholar]
- [129].Dionne J, Lefèvre T, Bilodeau P, Lamarre M, Auger M, Physical Chemistry Chemical Physics 2017, 19. [DOI] [PubMed] [Google Scholar]
- [130].Lefevre T, Rousseau ME, Pezolet M, Biophys. J 2007, 92, 2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131] a).Asakura T, Yao JM, Yamane T, Umemura K, Ultrich AS, J. Am. Chem. Soc. 2002, 124, 8794; [DOI] [PubMed] [Google Scholar]; b) Demura M, Minami M, Asakura T, Cross TA, J. Am. Chem. Soc 1998, 120, 1300; [Google Scholar]; c) Asakura T, Yao JM, Protein Sci. 2002, 11, 2706; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guo C, Zhang J, Jordan JS, Wang X, Henning RW, Yarger JL, Biomacromolecules 2018, 19, 906. [DOI] [PubMed] [Google Scholar]
- [132].Yang MY, Yao JM, Sonoyama M, Asakura T, Macromolecules 2004, 37, 3497. [Google Scholar]
- [133].Jenkins JE, Creager MS, Lewis RV, Holland GP, Yarger JL, Biomacromolecules 2010, 11, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134] a).Tanaka C, Takahashi R, Asano A, Kurotsu T, Akai H, Sato K, Knight DP, Asakura T, Macromolecules 2008, 41, 796; [Google Scholar]; b) Martel A, Burghammer M, Davies RJ, Riekel C, Biomacromolecules 2007, 8, 3548; [DOI] [PubMed] [Google Scholar]; c) Drummy LF, Farmer BL, Naik RR, Soft Matter 2007, 3, 877; [DOI] [PubMed] [Google Scholar]; d) Guo C, Zhang J, Wang X, Nguyen AT, Liu XY, Kaplan DL, Small 2017, 13. [DOI] [PubMed] [Google Scholar]
- [135] a).Grubb DT, Jelinski LW, Macromolecules 1997, 30, 2860; [Google Scholar]; b) Guinea GV, Elices M, Plaza GR, Perea GB, Daza R, Riekel C, Agulló-Rueda F, Hayashi C, Zhao Y, Pérez-Rigueiro J, Biomacromolecules 2012, 13, 2087; [DOI] [PubMed] [Google Scholar]; c) And DTG, Jelinski LW, Macromolecules 1997, 30, 2860. [Google Scholar]
- [136].Yang Z, G. DT And, *, ‡, Jelinski LW†, Macromolecules 1997, 30, 8254. [Google Scholar]
- [137].Fu CJ, Porter D, Chen X, Vollrath F, Shao ZZ, Adv. Funct. Mater 2011, 21, 729. [Google Scholar]
- [138].Nalvuran H, Elçin AE, Elçin YM, International Journal of Biological Macromolecules 2018, 114. [DOI] [PubMed] [Google Scholar]
- [139].Aznarcervantes S, Pagán A, Martínez JG, Bernabeuesclapez A, Otero TF, Meseguerolmo L, Paredes JI, Cenis JL, Materials Science & Engineering C Materials for Biological Applications 2017, 79, 315. [DOI] [PubMed] [Google Scholar]
- [140].Wang SD, Ma Q, Wang K, Ma PB, International Journal of Biological Macromolecules 2018, 111, 237. [DOI] [PubMed] [Google Scholar]
- [141].Wang Q, Chu Y, He J, Shao W, Zhou Y, Qi K, Wang L, Cui S, Materials Science & Engineering C 2017, 80, 232. [DOI] [PubMed] [Google Scholar]
- [142].Gandhi M, Yang H, Shor L, Ko F, Polymer 2009, 50, 1918. [Google Scholar]
- [143].Zuo LY, Zhang F, Gao B, Zuo BQ, Fibres Text. East. Eur 2017, 25, 40. [Google Scholar]
- [144].Wei K, Xia JH, Kim BS, Kim IS, Journal of Polymer Research 2011, 18, 579. [Google Scholar]
- [145].Ayutsede J, Gandhi M, Sukigara S, Ye H, Hsu CM, Gogotsi Y, Ko F, Biomacromolecules 2006, 7, 208. [DOI] [PubMed] [Google Scholar]
- [146].Pan C, Xie Q, Hu Z, Yang M, Zhu L, Fibers & Polymers 2015, 16, 1781. [Google Scholar]
- [147].Ling S, Li C, Adamcik J, Wang S, Shao Z, Chen X, Mezzenga R, Acs Macro Letters 2014, 3, 146. [DOI] [PubMed] [Google Scholar]
- [148].Zhang M, Wang C, Qi W, Jian M, Zhang Y, Acs Appl Mater Interfaces 2016, 8, 20894. [DOI] [PubMed] [Google Scholar]
- [149].Hou J, Xie Y, Ji A, Cao A, Fang Y, Shi E, Acs Applied Materials & Interfaces 2018. [Google Scholar]
- [150].Wang C, Xia K, Zhang M, Jian M, Zhang Y, Acs Applied Materials & Interfaces 2017, 9. [DOI] [PubMed] [Google Scholar]
- [151].Li X, Zong L, Wu X, You J, Li M, Li C, J. Mater. Chem. C 2018, 6. [Google Scholar]
- [152].Naskar D, Bhattacharjee P, Ghosh AK, Mandal M, Kundu SC, Acs Appl Mater Interfaces 1944, 9. [DOI] [PubMed] [Google Scholar]