

Mutations in glycosyltransferases have been implicated in the pathogenesis of neurodegenerative diseases, including ALS and Alzheimer’s disease. Moll et al. review the existing literature, with a focus on mechanisms connecting glycosyltransferase dysfunction to neurodegeneration. They also provide new evidence that mutations in the glycosyltransferase EOGT are associated with sporadic ALS.
Keywords: glycosyltransferase, neurodegeneration, gangliosides, O-linked β-N-acetylglucosamine
Abstract
Glycosyltransferases represent a large family of enzymes that catalyse the biosynthesis of oligosaccharides, polysaccharides, and glycoconjugates. A number of studies have implicated glycosyltransferases in the pathogenesis of neurodegenerative diseases but differentiating cause from effect has been difficult. We have recently discovered that mutations proximal to the substrate binding site of glycosyltransferase 8 domain containing 1 (GLT8D1) are associated with familial amyotrophic lateral sclerosis (ALS). We demonstrated that ALS-associated mutations reduce activity of the enzyme suggesting a loss-of-function mechanism that is an attractive therapeutic target. Our work is the first evidence that isolated dysfunction of a glycosyltransferase is sufficient to cause a neurodegenerative disease, but connection between neurodegeneration and genetic variation within glycosyltransferases is not new. Previous studies have identified associations between mutations in UGT8 and sporadic ALS, and between ST6GAL1 mutations and conversion of mild cognitive impairment into clinical Alzheimer’s disease. In this review we consider potential mechanisms connecting glycosyltransferase dysfunction to neurodegeneration. The most prominent candidates are ganglioside synthesis and impaired addition of O-linked β-N-acetylglucosamine (O-GlcNAc) groups to proteins important for axonal and synaptic function. Special consideration is given to examples where genetic mutations within glycosyltransferases are associated with neurodegeneration in recognition of the fact that these changes are likely to be upstream causes present from birth.
Introduction
Glycosyltransferases represent a large family of enzymes that catalyse biosynthesis of oligosaccharides, polysaccharides, and glycoconjugates. Sugar moieties are transferred from activated sugar donors to specific acceptor molecules via the formation of glycosidic bonds (Chuh et al., 2016). Acceptor molecules include other sugars, nucleic acids, lipids, and proteins. Glycosyltransferases reside predominantly within the Golgi apparatus of eukaryotes as type II transmembrane proteins. Over 90 glycosyltransferase families have been described (www.cazy.org/GlycosylTransferases.html). Sequence alignment tools have been useful for predicting glycosyltransferase function, including a metal-binding motif important for configuration of substrate within the active site (Lairson et al., 2008). However, even closely related sequences have been shown to exhibit different catalytic activity (Breton et al., 2006). Glycosyltransferases are classified as either ‘retaining’ or ‘inverting’ enzymes according to whether the anomeric bond within the donor substrate is retained or inverted during the sugar transfer.
Neurodegenerative diseases are increasing in frequency, in part due to an ageing population. Despite this, neurodegenerative diseases represent a significant unmet health need without effective treatments or clearly delineated pathogenic mechanisms. Changes in expression levels of glycosyltransferases have been strongly linked with neurodegeneration (Ludemann et al., 2005; Desplats et al., 2007; Schneider 2018), but determining whether these effects are upstream of neurotoxicity is difficult. Two distinct glycosyltransferase-associated mechanisms are prominent: ganglioside synthesis and addition of O-linked β-N-acetylglucosamine to proteins (O-GlcNAcylation). Major gangliosides are sialic acid-containing glycosphingolipids. Within the mammalian brain they are synthesized in the endoplasmic reticulum from a lactosylceramide precursor before remodelling during transit from the cis- to the trans-Golgi network by a series of glycosyltransferase enzymes (Fig. 1). Mature gangliosides are expressed on the plasma membrane of most vertebrate cells and within bodily fluids. They are particularly abundant on neuronal and glial cells within the CNS where they are thought to function prominently in cell signalling (Vajn et al., 2013). Altered levels of gangliosides have been reported in animal models of amyotrophic lateral sclerosis (ALS) and in post-mortem CNS tissue from ALS patients (Ariga, 2014; Dodge et al., 2015); similar findings have been reported in Parkinson’s disease (Wu et al., 2012) and Alzheimer’s disease (Gylys et al., 2007). O-GlcNAcylation occurs predominantly in the brain and is regulated by the glycosyltransferases O-linked N-acetylglucosamine transferase (OGT) and EGF domain-specific O-linked N-acetylglucosamine transferase (EOGT), which attach the O-GlcNAc moiety to acceptor proteins at specific serine/threonine residues via an O-linked glycosidic bond. OGT acts intracellularly whereas EOGT acts extracellularly on secreted and membrane proteins (Fig. 2). O-GlcNAcylation of CNS proteins important for axonal and synaptic function is significantly reduced in animal models of neurodegenerative diseases and in patient tissue from diseases including Parkinson’s disease, Huntington’s disease, Alzheimer’s disease and ALS (Liu et al., 2004; Ludemann et al., 2005; Kumar et al., 2014; Frenkel-Pinter et al., 2017).
Figure 1.

Schematic overview of the biosynthesis and function of major gangliosides within the mammalian brain. Lactosylceramide is synthesized at the cytoplasmic leaflet of the endoplasmic reticulum membrane from its ceramide precursor. De novo ceramide is transported to the Golgi apparatus and is converted to glycosphingolipids and sphingomyelin through the addition of saccharides and phosphocholine, respectively. Glycosphingolipids are transported in vesicles to the outer leaflet of the plasma membrane. Sialic acid-enriched glycosphingolipids form gangliosides which are anchored to the membrane via their ceramide-lipid moiety. Four major gangliosides comprise >90% of total gangliosides within the brain. A-series gangliosides (red) are derived from GM3. B-series gangliosides (purple) are synthesized from GM3 by GD3 synthase (St8sia1). G = the ‘ganglioside’ core; the second letter designates the quantity of sialic acid residues; M = mono; D = di; T = tri. Gangliosides are essential to maintaining neuronal integrity with functions including, but not limited to, increasing the neuroprotective properties of astrocytes, stabilizing interactions between neurons and glia, enhancing neurite outgrowth and negatively regulating neuroinflammation through activation of the complement pathway.
Figure 2.
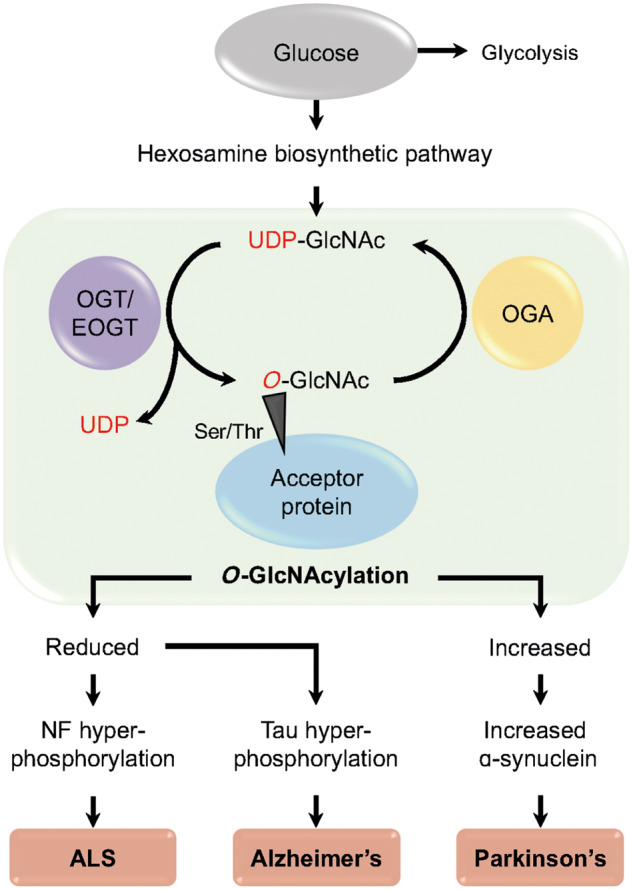
O-GlcNAcylation is implicated the pathophysiology of neurodegenerative disease. An overview of O-GlcNAcylation, a post-translational modification of O-GlcNAc, which has been implicated in neurodegenerative diseases Huntington’s disease, Alzheimer’s disease, Parkinson’s disease and ALS. O-GlcNAcylation occurs predominantly in the brain and is regulated by the glycosyltransferases OGT and EOGT, which attach the O-GlcNAc moiety to acceptor proteins at specific serine/threonine residues via an O-linked glycosidic bond; OGT acts intracellularly whereas EOGT acts extracellularly on secreted and membrane proteins.
Neurodegenerative diseases exhibit late age of onset and it is therefore assumed that genetic mutations are upstream of disease pathogenesis. As a result, the discovery of neurodegenerative disease-associated DNA mutations is a significant step towards identification of upstream therapeutic targets. We have recently discovered that mutations proximal to the substrate-binding site of glycosyltransferase 8 domain-containing 1 (GLT8D1) disrupt enzyme activity and are associated with familial ALS (Cooper-Knock et al., 2019). Our work is the first evidence that dysfunction of a glycosyltransferase is sufficient to cause a neurodegenerative disease. Our data are consistent with an effect of GLT8D1 mutations on ganglioside synthesis. In support of this mechanism, we have demonstrated by immunocytochemistry that ALS-associated GLT8D1 mutations reduce membrane expression of glycosphingolipids, which include gangliosides, in human cells (unpublished data). Moreover, in this review we summarize previous literature linking genetic changes within glycosyltransferases to neurodegeneration, and provide new evidence that genetic mutations within EOGT are significantly associated with sporadic ALS making this another upstream therapeutic target.
Impaired ganglioside synthesis is linked to neurodegeration
Parkinson’s disease
Reduced glycosyltransferase expression and lowered ganglioside synthesis has been implicated in the pathogenesis of Parkinson’s disease. A recent report described a reduction in gene expression of the glycosyltransferases B3GALT4 and ST3GAL2 in neuromelanin-containing neurons in the substantia nigra of patients with Parkinson’s disease compared to control subjects (Schneider, 2018). These genes are key players in the ganglioside biosynthesis pathway (Fig. 1). It is proposed that reduced B3GALT4 and ST3GAL2 expression leads to vulnerability of dopaminergic neurons via aberrant ganglioside synthesis. Consistent with this hypothesis, the number of GM1 ganglioside-expressing cells in the Parkinson’s disease substantia nigra are reduced (Wu et al., 2012), and levels of the major brain gangliosides—GM1, GD1a, GD1b and GT1b—are decreased in whole substantia nigra homogenates from patients with Parkinson’s disease (Seyfried et al., 2018). Model systems provide evidence that dysfunction of ganglioside synthesis is a cause and not just an association of typical Parkinson’s disease pathology: genetically engineered mice lacking major brain gangliosides display overt motor impairment with increasing age, which is accompanied by loss of dopaminergic neurons from the substantia nigra pars compacta and aggregation of α-synuclein (Wu et al., 2012).
Huntington’s disease
In a similar manner to Parkinson’s disease, reduced expression of glycosyltransferases involved in ganglioside synthesis has also been described in the R6/1 mouse model of Huntington’s disease and in human Huntington’s disease patients (Desplats et al., 2007). In this study >80% of gene expression changes observed in the striatum of R6/1 mice were also observed in the post-mortem caudate of human Huntington’s disease subjects. Overlapping genes were significantly enriched with glycosyltransferases involved in ganglioside synthesis including ST3GAL5, ST8SIA3, B4GALNT1 and ST3GAL2 (Fig. 1). Consistent with impaired ganglioside synthesis, the same study reported reduced ganglioside concentrations within both the diseased human caudate and the mouse striatum. It should be noted that despite significant homology to ST8SIA1, which has a well described role in ganglioside biosynthesis (Fig. 1), ST8SIA3 is traditionally associated with N-glycosylation of secreted/membrane proteins within the CNS (Lin et al., 2019). Like gangliosides, N-glycosylated proteins are important for cell signalling.
Alzheimer’s disease
There is good evidence for perturbed ganglioside metabolism in patients with Alzheimer’s disease, and in the development of amyloid-β pathology in particular (Barrier et al., 2007). In contrast to the findings in Parkinson’s disease and Huntington’s disease, the key observation appears to be increased ganglioside synthesis. Elevated GM1, GM2 and GM3 levels have been reported in the cerebral cortices of Alzheimer’s disease patients (Kracun et al., 1992; Gylys et al., 2007). Development of amyloid-β deposition is the defining pathology of Alzheimer’s disease and within brains exhibiting early Alzheimer’s disease pathology, a significant proportion of amyloid-β is bound to ganglioside species (Yanagisawa and Ihara, 1998). It has even been suggested that insoluble GM1-bound amyloid-β is the key toxin leading to neuronal death (Hayashi et al., 2004), as a result of high affinity binding between GM1 and amyloid-β, which facilitates formation of insoluble β-pleated sheets (Yamamoto et al., 2007). With increasing age GM1 is localized to presynaptic nerve terminals and this may have a role in directing amyloid-β deposition to the same locations (Yamamoto et al., 2008). Unlike evidence regarding gangliosides, reports of altered glycosyltransferase expression in Alzheimer’s disease are more limited. There is evidence that glycosyltransferase activity may modify Alzheimer’s disease pathology: overexpression of the glycosyltransferase B4GALNT1 leads to increased ganglioside expression but also increases APP cleavage to form amyloid-β pathology through suppression of lysosomal degradation of BACE1 (Yamaguchi et al., 2016). Currently, transgenic mouse models of Alzheimer’s disease do not mirror changes in ganglioside distribution seen in human post-mortem tissue (Barrier et al., 2007).
Amyotrophic lateral sclerosis
ALS has been linked to abnormal lipid metabolism (Desport et al., 2005) and in particular, gangliosides and their ceramide precursors are thought to be modulators of disease progression (Salazargrueso et al., 1990; Stevens et al., 1993). Whether ganglioside production is increased or decreased is controversial. As early as 1985 a 10% reduction in b-series gangliosides was identified within the motor cortex of ALS brains compared to non-ALS controls (Rapport et al., 1985). More recently elevated levels of gangliosides GM1 and GM3 were reported within ALS post-mortem spinal cords compared to age-matched controls; findings were corroborated in the SOD1-G93A transgenic ALS mouse model (Dodge et al., 2015). Interestingly, autoantibodies against specific gangliosides produce an inflammatory disease of spinal motor neurons known as multifocal motor neuropathy with conduction block (Harschnitz et al., 2014), which is a frequent differential diagnosis of ALS.
ALS specifically inflicts pathology on the upper and lower motor neurons, the neuromuscular junction and muscle. The accessibility of this system in disease models facilitates the differentiation of up- and downstream disease associations. For example, increased expression of glycosphingolipids is observed in muscle tissue from end-stage mutant SOD1-ALS mice compared to controls, but similar changes were observed in response to surgically-induced muscle denervation suggesting a downstream effect (Henriques et al., 2015). Moreover, neurotransmission at the neuromuscular junction is unchanged in aged GM2 and GD3-deficient mice compared to controls (Zitman et al., 2011). However, our discovery that mutations in the glycosyltransferase GLT8D1 are a cause of familial ALS is a step forward, which places glycosyltransferase activity irrefutably upstream in the development of disease.
Genetic mutations in glycosyltransferases cause neurodegeneration
Genetic mutations in the development of an age-associated neurodegenerative disease are, by definition, upstream causes or risk factors rather than secondary to the disease process. Mutations discovered to date are included in Table 1 and described below.
Table 1.
Defects affecting specific glycosyltransferase enzymes observed in neurodegenerative disease
| Glycosyltransferase | Functional consequence | Neurodegenerative disorder | Defect observed | Reference |
|---|---|---|---|---|
| ST6GAL1 | Disrupted cell surface signalling | Alzheimer’s disease | DNA mutations | Lee et al., 2017 |
| B3GALT4 | Reduced ganglioside biosynthesis (GD1b) | Parkinson’s disease | Reduced gene expression | Schneider, 2018 |
| ST3GAL2 | Reduced ganglioside biosynthesis (GT1b) |
|
Reduced gene expression | |
| B4GALNT1 | Reduced ganglioside biosynthesis | Huntington’s disease | Reduced gene expression | Desplats et al., 2007 |
| ST8SIA3 | Implicated in ganglioside biosynthesis but described role in N-glycosylation | Huntington’s disease | Reduced gene expression | Desplats et al., 2007 |
| ST3GAL5 | Reduced ganglioside biosynthesis | Huntington’s disease | Reduced gene expression | Desplats et al., 2007 |
| GLT8D1 | Reduced membrane expression of glycosphingolipids | ALS | DNA mutations | Cooper-Knock et al., 2019 |
| UGT8 | Disruption of myelin synthesis | ALS | DNA mutations | Pamphlett et al., 2011 |
| EOGT | Disruption of O-GlcNAcylation | ALS | DNA mutations | This article |
| OGT | Impaired O-GlcNAcylation |
|
Reduced concentration of O-GlcNAcylated proteins | |
| OGT | Excessive O-GlcNAcylation | Parkinson’s disease | Increased concentration of O-GlcNAcylated proteins | Wani et al., 2017 |
GLT8D1
A recent study from our lab demonstrated that mutations within the glycosyltransferase domain of GLT8D1 are associated with familial ALS (Cooper-Knock et al., 2019). The function of GLT8D1 is unknown, but it is ubiquitously expressed and localized to the Golgi apparatus. Based on sequence homology, GLT8D1 is a member of glycosyltransferase family 8 and is expected to catalyse the transfer of a glycosyl group from a donor to an acceptor via a ‘retaining’ mechanism. Mutated GLT8D1 carrying ALS-associated amino acid changes is toxic to neuronal and non-neuronal cell lines, and induces motor deficits in zebrafish embryos; these observations are consistent with a role in motor neuron degeneration. Interestingly, relative toxicity of ALS-associated mutations in model systems mirrors the clinical severity. Glycosyltransferase enzyme activity is reduced in the mutated form of GLT8D1 commensurate with an increase in substrate affinity, which is predicted to impair cycling of substrate through the enzyme and thus reduce overall velocity (Cooper-Knock et al., 2019). Taken together, these data are consistent with loss-of-function toxicity. Our study is the first time inherited mutations that diminish glycosyltransferase enzyme activity have been associated with ALS. We have recently demonstrated by immunocytochemistry that ALS-associated mutations reduce membrane expression of glycosphingolipids in human cells (unpublished data). Glycosphingolipids include gangliosides and this would be consistent with disruption of ganglioside signalling within the CNS. GLT8D1 was recently identified as a risk gene for schizophrenia (Yang et al., 2018), and while schizophrenia is not a neurodegenerative disorder, it is noteworthy that ALS and schizophrenia share common genetic risk (McLaughlin et al., 2017).
UDP glycosyltransferase 8 (UGT8)
Like GLT8D1, UGT8 is a member of glycosyltransferase family 8. UGT8 functions in the biosynthesis of galactocerebroside, a sphingolipid that forms the myelin membrane in the central and peripheral nervous systems. Rare and potentially pathogenic copy number variants have been identified in the promotor region of UGT8 following in an unbiased genome-wide screen for de novo DNA mutations in 12 trios including sporadic ALS patients and unaffected parents (Pamphlett et al., 2011). Abnormal lipid biosynthesis and metabolism is a pathological hallmark of ALS (Dupuis et al., 2008; Dorst et al., 2011), therefore it is possible that UGT8 plays a role in the hypolipidaemia observed in ALS patients and the SOD1-G93A ALS mouse model (Kim et al., 2011; Yang et al., 2013). Mice lacking Ugt8a, the orthologue of UGT8, exhibit impaired locomotor activity and disruption in nerve conduction followed by degeneration of the myelin sheath (Bosio et al., 1996; Coetzee et al., 1996), which is rescued following transgenic expression of UGT8A (Zoller et al., 2005). Interestingly the rescue occurred with expression of UGT8A under a promoter exclusively expressed within oligodendrocytes, which is consistent with other evidence implicating these cells in ALS-associated neurodegeneration (Morrison et al., 2013).
ST6 β-galactoside α-2,6-sialyltransferase 1 (ST6GAL1)
ST6GAL1 is an ‘inverting’ enzyme and a member of glycosyltransferase family 29. ST6GAL1 catalyses the transfer of sialic acid onto galactose-containing substrates including cell-surface signalling lipids and proteins (Garnham et al., 2019). A genome-wide association study implicated polymorphisms within ST6GAL1 in the conversion of mild cognitive impairment into clinical Alzheimer’s disease (Lee et al., 2017). Interestingly ST6GAL1 is cleaved and occurs in a soluble form; this cleavage is mediated by BACE1 (Kitazume et al., 2001), which is also involved in the cleavage of APP to form amyloid-β. Indeed, overexpression of ST6GAL1 increases APP secretion (Nakagawa et al., 2006) suggesting that the activity of ST6GAL1 can directly modify the central pathway in the development of Alzheimer’s pathology.
Glycosyltransferase O-GlcNAcylation: a key regulator of neurodegeneration?
Protein glycosylation and more specifically the addition of O-GlcNAc groups to CNS proteins important for axonal and synaptic function, is significantly reduced in animal models of neurodegenerative diseases and in patient tissue from diseases including Huntington’s disease, Alzheimer’s disease and ALS (Liu et al., 2004; Ludemann et al., 2005; Kumar et al., 2014; Frenkel-Pinter et al., 2017) (Table 1). O-GlcNAcylation is reported to negatively regulate tau phosphorylation (Liu et al., 2004), which is key in the pathogenesis of a number of neurodegenerative diseases, including Alzheimer’s disease. In contrast, an increase in O-GlcNAcylation is observed in the post-mortem temporal cortex of patients with Parkinson’s disease and is postulated to contribute to neurodegeneration through the inhibition of autophagy leading to an increase in α-synuclein accumulation (Wani et al., 2017). Neurofilaments are critical components of the neuronal cytoskeleton that can undergo O-GlcNAcylation (Yuan et al., 2012). Neurofilament levels are significantly higher in the serum and CSF of ALS patients compared to control subjects (Benatar et al., 2018). This increase is thought to be a consequence of axonal damage. However, there is evidence that neurofilament damage may be upstream in the pathogenesis of ALS including the observation that increased phosphorylation of neurofilaments is associated with neurotoxicity (Julien, 1997). It is thought that phosphorylation and O-GlcNAcylation are reciprocal, meaning that reduced O-GlcNAcylation could precipitate harmful phosphorylation; indeed this has been observed in a transgenic rat model of SOD1-ALS (Ludemann et al., 2005).
O-GlcNAcylation occurs predominantly in the brain and is regulated by the glycosyltransferases OGT and EOGT; the reverse reaction is catalysed by O-GlcNAcase (OGA). Together these reactions constitute a dynamic and reversible process (Fig. 2). OGT is an inverting enzyme and a member of glycosyltransferase family 41; OGT is highly enriched in the brain, where it is 10 times more active than in peripheral tissue (Okuyama and Marshall, 2003). OGT is localized to the nucleus, soma, dendrites and presynaptic terminals of neurons (Akimoto et al., 2003). Removal of postsynaptic OGT from primary neurons inhibits both synapse formation and the development of dendritic spines (Lagerlof et al., 2017). This highlights the importance of OGT in maintaining synaptic stability, and notably loss of synaptic stability is a unifying feature of neurodegenerative disease. EOGT is an inverting enzyme and a member of glycosyltransferase family 61. Despite distinct sites of action, OGT and EOGT are both regulated via the hexosamine biosynthetic pathway (Ogawa et al., 2015). EOGT activity is involved in Notch signalling, which is important for neurodevelopment. Indeed, homozygous loss-of-function mutations in EOGT produce Adams-Oliver syndrome, a congenital developmental disorder associated with actin cytoskeleton defects.
ALS-associated genetic variants within O-GlcNAcylation pathway enzymes
While homozygous EOGT mutations affect neurodevelopment, we hypothesized that heterozygous mutations within EOGT might negatively impact on the maintenance of axon integrity and increase risk of developing ALS. To test this hypothesis we performed rare-variant burden testing (Cirulli et al., 2015) within EOGT to check for a genetic association with ALS. We used whole genome sequencing data from 4493 sporadic ALS patients and 1924 control subjects (van der Spek et al., 2019); we identified 32 missense rare (MAF < 1%) variants within EOGT that were exclusively or predominantly found in ALS cases (Table 2). When considering all rare missense variants found in cases and controls across all exons of EOGT, there was a significant enrichment of such mutations in ALS patients (Firth logistic regression, P = 0.007). Similar testing did not identify an enrichment of ALS-associated mutations within OGT, indeed we only identified two rare missense mutations within OGT in 4493 sporadic ALS patients. It should be noted that OGT is encoded on the X chromosome and therefore males are necessarily hemizygous, which may predispose to a neurodevelopmental phenotype rather than a late age-of-onset disease: for example mutations within N-terminal tetratricopeptide repeats of OGT are associated with X-linked intellectual disability (Gundogdu et al., 2018). There was no significant burden of ALS-associated mutations within OGA (P = 0.91).
Table 2.
Mutations in EOGT found in ALS patients
| DNA change | Protein change | Allele frequency | Exon | |
|---|---|---|---|---|
| ALS | Controls | |||
| c.1575T>G | p.Asp525Glu | 0.001 | 0.0005 | 15 |
| c.1546C>T | p.Pro516Ser | 0.0001 | 0 | 15 |
| c.1466C>T | p.Pro489Leu | 0.0001 | 0 | 15 |
| c.1459dupG | p.Glu487fs | 0.0001 | 0 | 15 |
| c.1456G>T | p.Gly486Trp | 0.0001 | 0 | 15 |
| c.1432G>A | p.Asp478Asn | 0.0001 | 0 | 14 |
| c.1417A>T | p.Lys473* | 0.0001 | 0 | 14 |
| c.1355G>A | p.Arg452His | 0.0001 | 0 | 14 |
| c.1342T>A | p.Cys448Ser | 0.0001 | 0 | 14 |
| c.1256C>T | p.Thr419Met | 0.0001 | 0 | 13 |
| c.1213A>G | p.Arg405Gly | 0.01 | 0.006 | 12 |
| c.1210T>A | p.Tyr404Asn | 0.0001 | 0 | 12 |
| c.1129C>T | p.Arg377Trp | 0.0001 | 0 | 11 |
| c.1114C>T | p.Arg372Trp | 0.0001 | 0 | 11 |
| c.1108C>T | p.Leu370Phe | 0.0001 | 0 | 11 |
| c.829A>G | p.Thr277Ala | 0.0001 | 0 | 10 |
| c.692T>C | p.Ile231Thr | 0.0001 | 0 | 9 |
| c.674C>T | p.Ala225Val | 0.0001 | 0 | 9 |
| c.647A>G | p.Gln216Arg | 0.0001 | 0 | 9 |
| c.563A>T | p.Lys188Ile | 0.0002 | 0 | 8 |
| c.562A>T | p.Lys188* | 0.0002 | 0 | 8 |
| c.430A>G | p.Ser144Gly | 0.0002 | 0 | 7 |
| c.314C>T | p.Thr105Met | 0.0001 | 0 | 6 |
| c.208A>G | p.Lys70Glu | 0.0001 | 0 | 4 |
| c.202C>G | p.Pro68Ala | 0.0001 | 0 | 4 |
| c.192C>G | p.Asp64Glu | 0.0001 | 0 | 4 |
| c.176C>G | p.Thr59Ser | 0.0005 | 0.0003 | 4 |
| c.169A>G | p.Ile57Val | 0.0001 | 0 | 4 |
| c.155A>G | p.His52Arg | 0.0002 | 0 | 4 |
| c.122G>T | p.Arg41Leu | 0.0002 | 0 | 4 |
| c.71C>G | p.Pro24Arg | 0.0007 | 0.0003 | 4 |
| c.9G>A | p.Met3Ile | 0.0001 | 0 | 4 |
ALS-associated missense changes found within EOGT in 4493 sporadic ALS patients and 1924 controls. Mutations are listed 5’ to 3’; EOGT has 15 exons and is encoded on the reverse strand of chromosome 3; exons 1 to 3 are non-coding.
Conclusions
Overall there is substantial evidence for dysfunction of glycosyltransferases in neurodegenerative diseases including ALS, Alzheimer’s disease, Huntington’s disease and Parkinson’s disease. There are diverse functions associated with glycosyltransferase activity and for many of the enzymes the biological pathway associated with their activity is not yet clear. However, in our analysis, dysfunction associated with neurodegenerative disease can be seen to converge on the ganglioside synthesis pathway and altered O-GlcNAcylation. The exact nature of the defect appears to be variable in different diseases; for example ganglioside concentrations are reduced in Parkinson’s disease and Huntington’s disease, increased in Alzheimer’s disease and there is evidence for change in both directions in ALS. Similarly, increased O-GlcNAcylation is associated with the development of Parkinson’s disease pathology but reduced O-GlcNAcylation is associated with the development of tau pathology. We suggest that consensus will arise via efforts to position glycosyltransferase dysfunction within the cascade of pathogenesis leading to neuronal death—it is not glycosyltransferase dysfunction per se that is interesting, but rather upstream changes in glycosyltransferase function that initiate toxicity. With this in mind we have highlighted genetic associations between mutations in glycosyltransferases and neurodegenerative disease. Most prominently we have recently discovered autosomal dominant mutations in GLT8D1 to be a monogenic cause of ALS. Disease-associated mutations have also been discovered in UGT8 and ST6GAL1; and we have revealed a new association between ALS and mutations in EOGT. Glycosyltransferases are likely to be an important therapeutic target in the effort of develop disease-modifying therapies for neurodegenerative disease.
Acknowledgements
The authors would like to thank the Project MinE GWAS Consortium. We are very grateful to those ALS patients and control subjects who donated biosamples.
Funding
We acknowledge grants from the Academy of Medical Sciences, EU Framework 7 (Euro-Motor), and the JPND/MRC SOPHIA, STRENGTH and ALS-CarE projects. T.M. is supported by the University of Sheffield Lee Newton PhD studentship. J.C.-K. holds a NIHR Clinical Lectureship and P.J.S. is supported as an NIHR Senior Investigator. This work was also supported by the NIHR Sheffield Biomedical Research Centre for Translational Neuroscience and the Sheffield NIHR Clinical Research Facility.
Competing interests
The authors report no competing interests.
Glossary
Abbreviations
- ALS =
amyotrophic lateral sclerosis
- O-GlcNAc =
O-linked β-N-acetylglucosamine
References
- Akimoto Y, Comer FI, Cole RN, Kudo A, Kawakami H, Hirano H, et al. Localization of the O-GlcNAc transferase and O-GlcNAc-modified proteins in rat cerebellar cortex. Brain Res 2003; 966: 194–205. [DOI] [PubMed] [Google Scholar]
- Ariga T. Pathogenic role of ganglioside metabolism in neurodegenerative diseases. J Neurosci Res 2014; 92: 1227–42. [DOI] [PubMed] [Google Scholar]
- Barrier L, Ingrand S, Damjanac M, Bilan AR, Hugon J, Page G. Genotype-related changes of ganglioside composition in brain regions of transgenic mouse models of Alzheimer's disease. Neurobiol Aging 2007; 28: 1863–72. [DOI] [PubMed] [Google Scholar]
- Benatar M, Wuu J, Andersen PM, Lombardi V, Malaspina A. Neurofilament light: a candidate biomarker of presymptomatic amyotrophic lateral sclerosis and phenoconversion. Ann Neurol 2018; 84: 130–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosio A, Binczek E, LeBeau MM, Fernald AA, Stoffel W. The human gene CGT encoding the UDP-galactose ceramide calactosyl transferase (cerebroside synthase): cloning, characterization, and assignment to human chromosome 4, band q26. Genomics 1996; 34: 69–75. [DOI] [PubMed] [Google Scholar]
- Breton C, Snajdrova L, Jeanneau C, Koca J, Imberty A. Structures and mechanisms of glycosyltransferases. Glycobiology 2006; 16: 29r–37r. [DOI] [PubMed] [Google Scholar]
- Chuh KN, Batt AR, Pratt MR. Chemical methods for encoding and decoding of posttranslational modifications. Cell Chem Biol 2016; 23: 86–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015; 347: 1436–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coetzee T, Fujita N, Dupree J, Shi R, Blight A, Suzuki K, et al. Myelination in the absence of galactocerebroside and sulfatide: normal structure with abnormal function and regional instability. Cell 1996; 86: 209–19. [DOI] [PubMed] [Google Scholar]
- Cooper-Knock J, Jenkins T, Shaw PJ. Clinical and molecular aspects of motor neuron disease. Colloq Ser Genom Mol Med 2013; 2: 1–60. [Google Scholar]
- Cooper-Knock J, Moll T, Ramesh T, Castelli L, Beer A, Robins H, et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep 2019; 26: 2298–306.e2295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats PA, Denny CA, Kass KE, Gilmartin T, Head SR, Sutcliffe JG, et al. Glycolipid and ganglioside metabolism imbalances in Huntington's disease. Neurobiol Dis 2007; 27: 265–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desport JC, Torny F, Lacoste M, Preux PM, Couratier P. Hypermetabolism in ALS: correlations with clinical and paraclinical parameters. Neurodegener Dis 2005; 2: 202–7. [DOI] [PubMed] [Google Scholar]
- Dodge JC, Treleaven CM, Pacheco J,, Cooper S, Bao C, Abraham M, et al. Glycosphingolipids are modulators of disease pathogenesis in amyotrophic lateral sclerosis. Proc Natl Acad Sci U S A 2015; 112: 8100–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorst J, Kuhnlein P, Hendrich C, Kassubek J, Sperfeld AD, Ludolph AC. Patients with elevated triglyceride and cholesterol serum levels have a prolonged survival in amyotrophic lateral sclerosis. J Neurol 2011; 258: 613–7. [DOI] [PubMed] [Google Scholar]
- Dupuis L, Corcia P, Fergani A, De Aguilar JLG, Bonnefont-Rousselot D, Bittar R, et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008; 70: 1004–9. [DOI] [PubMed] [Google Scholar]
- Frenkel-Pinter M, Shmueli MD, Raz C, Yanku M, Zilberzwige S, Gazit E, et al. Interplay between protein glycosylation pathways in Alzheimer's disease. Sci Adv 2017; 3: e1601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnham R, Scott E, Livermore KE, Munkley J. ST6GAL1: a key player in cancer. Oncol Lett 2019; 18: 983–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundogdu M, Llabres S, Gorelik A, Ferenbach AT, Zachariae U, van Aalten DMF. The O-GlcNAc transferase intellectual disability mutation L254F distorts the TPR helix. Cell Chem Biol 2018; 25: 513–8.e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gylys KH, Fein JA, Yang F, Miller CA, Cole GM. Increased cholesterol in Abeta-positive nerve terminals from Alzheimer's disease cortex. Neurobiol Aging 2007; 28: 8–17. [DOI] [PubMed] [Google Scholar]
- Harschnitz O, Jongbloed BA, Franssen H, Straver DC, van der Pol WL, van den Berg LH. MMN: from immunological cross-talk to conduction block. J Clin Immunol 2014; 34 Suppl 1: S112–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H, Kimura N, Yamaguchi H, Hasegawa K, Yokoseki T, Shibata M, et al. A seed for Alzheimer amyloid in the brain. J Neurosci 2004; 24: 4894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques A, Croixmarie V, Priestman DA, Rosenbohm A, Dirrig-Grosch S, D'Ambra E, et al. Amyotrophic lateral sclerosis and denervation alter sphingolipids and up-regulate glucosylceramide synthase. Hum Mol Genet 2015; 24: 7390–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Julien JP. Neurofilaments and motor neuron disease. Trends Cell Biol 1997; 7: 243–9. [DOI] [PubMed] [Google Scholar]
- Kim SM, Kim H, Kim JE, Park KS, Sung JJ, Kim SH, et al. Amyotrophic lateral sclerosis is associated with hypolipidemia at the presymptomatic stage in mice. PLoS One 2011; 6: e17985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitazume S, Tachida Y, Oka R, Shirotani K, Saido TC, Hashimoto Y. Alzheimer's beta-secretase, beta-site amyloid precursor protein-cleaving enzyme, is responsible for cleavage secretion of a Golgi-resident sialyltransferase. Proc Natl Acad Sci U S A 2001; 98: 13554–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kracun I, Kalanj S, Talan-Hranilovic J, Cosovic C. Cortical distribution of gangliosides in Alzheimer's disease. Neurochem Int 1992; 20: 433–8. [DOI] [PubMed] [Google Scholar]
- Kumar A, Singh PK, Parihar R, Dwivedi V, Lakhotia SC, Ganesh S. Decreased O-linked GlcNAcylation protects from cytotoxicity mediated by Huntingtin Exon1 protein fragment. J Biol Chem 2014; 289: 13543–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerlof O, Hart GW, Huganir RL. O-GlcNAc transferase regulates excitatory synapse maturity. Proc Natl Acad Sci U S A 2017; 114: 1684–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lairson LL, Henrissat B, Davies GJ, Withers SG. Glycosyltransferases: structures, functions, and mechanisms. Annu Rev Biochem 2008; 77: 521–55. [DOI] [PubMed] [Google Scholar]
- Lee E, Giovanello KS, Saykin AJ, Xie F, Kong D, Wang Y, et al. Single-nucleotide polymorphisms are associated with cognitive decline at Alzheimer's disease conversion within mild cognitive impairment patients. Alzheimers Dement (Amsterdam) 2017; 8: 86–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CY, Lai HL, Chen HM, Siew JJ, Hsiao CT, Chang HC, et al. Functional roles of ST8SIA3-mediated sialylation of striatal dopamine D2 and adenosine A2A receptors. Transl Psychiatry 2019; 9: 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer's disease. Proc Natl Acad Sci U S A 2004; 101: 10804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludemann N, Clement A, Hans VH, Leschik J, Behl C, Brandt R. O-glycosylation of the tail domain of neurofilament protein M in human neurons and in spinal cord tissue of a rat model of amyotrophic lateral sclerosis (ALS). J Biol Chem 2005; 280: 31648–58. [DOI] [PubMed] [Google Scholar]
- McLaughlin RL, Schijven D, van Rheenen W, van Eijk KR, O'Brien M, Kahn RS, et al. Genetic correlation between amyotrophic lateral sclerosis and schizophrenia. Nat Commun 2017; 8: 14774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison BM, Lee Y, Rothstein JD. Oligodendroglia: metabolic supporters of axons. Trends Cell Biol 2013; 23: 644–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakagawa K, Kitazume S, Oka R, Maruyama K, Saido TC, Sato Y, et al. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J Neurochem 2006; 96: 924–33. [DOI] [PubMed] [Google Scholar]
- Ogawa M, Sawaguchi S, Kawai T, Nadano D, Matsuda T, Yagi H, et al. Impaired O-linked N-acetylglucosaminylation in the endoplasmic reticulum by mutated epidermal growth factor (EGF) domain-specific O-linked N-acetylglucosamine transferase found in Adams-Oliver syndrome. J Biol Chem 2015; 290: 2137–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuyama R, Marshall S. UDP-N-acetylglucosaminyl transferase (OGT) in brain tissue: temperature sensitivity and subcellular distribution of cytosolic and nuclear enzyme. J Neurochem 2003; 86: 1271–80. [DOI] [PubMed] [Google Scholar]
- Pamphlett R, Morahan JM, Yu B. Using case-parent trios to look for rare de novo genetic variants in adult-onset neurodegenerative diseases. J Neurosci Methods 2011; 197: 297–301. [DOI] [PubMed] [Google Scholar]
- Rapport MM, Donnenfeld H, Brunner W, Hungund B, Bartfeld H. Ganglioside patterns in amyotrophic lateral sclerosis brain-regions. Ann Neurol 1985; 18: 60–7. [DOI] [PubMed] [Google Scholar]
- Salazargrueso EF, Routbort MJ, Martin J, Dawson G, Roos RP. Polyclonal Igm anti-Gm1 ganglioside antibody in patients with motor-neuron disease and variants. Ann Neurol 1990; 27: 558–63. [DOI] [PubMed] [Google Scholar]
- Schneider JS. Altered expression of genes involved in ganglioside biosynthesis in substantia nigra neurons in Parkinson's disease. PLoS One 2018; 13: e0199189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seyfried TN, Choi H, Chevalier A, Hogan D, Akgoc Z, Schneider JS. Sex-related abnormalities in substantia nigra lipids in Parkinson's disease. ASN Neuro 2018; 10: 1759091418781889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A, Weller M, Wietholter H. A characteristic ganglioside antibody pattern in the CSF of patients with amyotrophic-lateral-sclerosis. J Neurol Neurosurg Psychiatry 1993; 56: 361–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vajn K, Viljetic B, Degmecic IV, Schnaar RL, Heffer M. Differential distribution of major brain gangliosides in the adult mouse central nervous system. PLoS One 2013; 8: e75720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Spek RAA, van Rheenen W, Pulit SL, Kenna KP, van den Berg LH, Veldink JH. The project MinE databrowser: bringing large-scale whole-genome sequencing in ALS to researchers and the public. Amyotroph Lateral Scler Frontotemporal Degener 2019; 20: 432–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wani WY, Ouyang XS, Benavides GA, Redmann M, Cofield SS,, Shacka JJ, et al. O-GlcNAc regulation of autophagy and alpha-synuclein homeostasis; implications for Parkinson's disease. Mol Brain 2017; 10: 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GS, Lu ZH, Kulkarni N, Ledeen RW. Deficiency of ganglioside GM1 correlates with Parkinson's disease in mice and humans. J Neurosci Res 2012; 90: 1997–2008. [DOI] [PubMed] [Google Scholar]
- Yamaguchi T, Yamauchi Y, Furukawa K, Ohmi Y, Ohkawa Y, Zhang Q, et al. Expression of B4GALNT1, an essential glycosyltransferase for the synthesis of complex gangliosides, suppresses BACE1 degradation and modulates APP processing. Sci Rep 2016; 6: 34505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Fukata Y, Fukata M, Yanagisawa K. GM1-ganglioside-induced Abeta assembly on synaptic membranes of cultured neurons. Biochim Biophys Acta 2007; 1768: 1128–37. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Matsubara T, Sato T, Yanagisawa K. Age-dependent high-density clustering of GM1 ganglioside at presynaptic neuritic terminals promotes amyloid beta-protein fibrillogenesis. Biochim Biophys Acta Biomembr 2008; 1778: 2717–26. [DOI] [PubMed] [Google Scholar]
- Yanagisawa K, Ihara Y. GM1 ganglioside-bound amyloid beta-protein in Alzheimer's disease brain. Neurobiol Aging 1998; 19: S65–7. [DOI] [PubMed] [Google Scholar]
- Yang CP, Li X, Wu Y, Shen Q, Zeng Y, Xiong Q, et al. Comprehensive integrative analyses identify GLT8D1 and CSNK2B as schizophrenia risk genes. Nat Commun 2018; 9: 838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JW, Kim SM, Kim HJ, Kim JE, Park KS, Kim SH, et al. Hypolipidemia in patients with amyotrophic lateral sclerosis: a possible gender difference? J Clin Neurol 2013; 9: 125–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan AD, Rao MV, Nixon RA. Neurofilaments at a glance. J Cell Sci 2012; 125: 3257–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitman FMP, Todorov B, Verschuuren JJ,, Jacobs BC, Furukawa K, Furukawa K, et al. Neuromuscular synaptic transmission in aged ganglioside-deficient mice. Neurobiol Aging 2011; 32: 157–67. [DOI] [PubMed] [Google Scholar]
- Zoller I, Bussow H, Gieselmann V, Eckhardt M. Oligodendrocyte-specific ceramide galactosyltransferase (CGT) expression phenotypically rescues CGT-deficient mice and demonstrates that CGT activity does not limit brain galactosylceramide level. Glia 2005; 52: 190–8. [DOI] [PubMed] [Google Scholar]