

Abstract
The cyclic nucleotides cyclic adenosine-3′,5′-monophosphate (cAMP) and cyclic guanosine-3′,5′-monophosphate (cGMP) maintain physiological cardiac contractility and integrity. Cyclic nucleotide–hydrolysing phosphodiesterases (PDEs) are the prime regulators of cAMP and cGMP signalling in the heart. During heart failure (HF), the expression and activity of multiple PDEs are altered, which disrupt cyclic nucleotide levels and promote cardiac dysfunction. Given that the morbidity and mortality associated with HF are extremely high, novel therapies are urgently needed. Herein, the role of PDEs in HF pathophysiology and their therapeutic potential is reviewed. Attention is given to PDEs 1–5, and other PDEs are briefly considered. After assessing the role of each PDE in cardiac physiology, the evidence from pre-clinical models and patients that altered PDE signalling contributes to the HF phenotype is examined. The potential of pharmacologically harnessing PDEs for therapeutic gain is considered.
Keywords: Cyclic AMP, Cyclic GMP, Heart failure, Phosphodiesterase
Introduction
Cyclic adenosine-3′,5′-monophosphate (cAMP) and cyclic guanosine-3′,5′-monophosphate (cGMP) are cyclic nucleotides that serve as key second messengers within the heart. As siblings, cAMP and cGMP concurrently preserve cardiac physiology despite frequently signalling in a contrary fashion. The cyclic nucleotide phosphodiesterases (PDEs) facilitate cyclic nucleotide degradation by catalysing the hydrolysis of the phosphodiester bond of cAMP and/or cGMP. Reducing, both spatially and temporally, the intracellular concentration of cyclic nucleotides, PDEs compartmentalise cAMP and cGMP signalling and are vital for mediating crosstalk between the two pathways. The expression and/or activity of multiple PDEs is altered during heart failure (HF). HF affects approximately 37 million people worldwide and is typified by a chronic deterioration of cardiac function during which the heart is unable to sustain a cardiac output (CO) sufficient for the maintenance of tissue homeostasis [1, 2]. Given the influence of cAMP and cGMP on cardiac contractility, hypertrophy, fibrosis and apoptosis, variations in PDE and cyclic nucleotide levels contribute to the cardiac dysfunction characteristic of HF. Understanding the role of PDEs in HF pathophysiology may identify novel targets for pharmacological manipulation and thus tackle the existing shortfall of effective therapies. Considering that nearly half of all HF patients die within 5 years of diagnosis [3, 4], this is certainly a pressing aim.
Cyclic Nucleotides in Cardiac Physiology
Cardiac cAMP synthesis is catalysed by adenylyl cyclases (ACs) principally in response to beta 1- and beta 2-adrenergic receptor (β1/2-AR) stimulation by noradrenaline (NA) [5]. Within the heart, cAMP signals primarily by way of cAMP-dependent protein kinase (PKA) and exchange protein activated by cAMP (EPAC, a guanine nucleotide exchange factor that binds cAMP) [6]. PKA provokes positive chronotropic (i.e. heart rate), inotropic (i.e. contractile force), lusitropic (i.e. diastolic relaxant) and dromotropic (i.e. atrioventricular node depolarisation) cardiac responses by phosphorylating a number of cardiomyocyte-localised proteins. L-type calcium (Ca2+) channel (LTCC) phosphorylation raises the L-type Ca2+ current (ICa,L) [7], and phosphorylation of the sarcoplasmic reticulum (SR) ryanodine receptor (RyR2) [8] increases further the intracellular concentration of Ca2+ ([Ca2+]i; although controversy persists regarding the role of RyR2 phosphorylation in cardiac physiology and HF pathophysiology) [9, 10]. Ca2+ reuptake is simultaneously promoted via phospholamban (PLB) phosphorylation and sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) activation [11]. Additionally, PKA regulates several cardiac excitation-contraction–coupling (ECC) proteins including cardiac troponin I (cTnI) [12], titin [13] and cardiac myosin–binding protein C (cMYBPC) (Fig. 1) [14].
Fig. 1.

Cyclic nucleotide signalling within cardiomyocytes. Cyclic adenosine-3′,5′-monophosphate (cAMP) is synthesised from adenosine-5′-triphosphate (ATP) by the action of adenylyl cyclases (ACs) in response to noradrenaline (NA). Cyclic guanosine-3′,5′-monophosphate (cGMP) is synthesised from guanosine-5′-triphosphate (GTP) by the action of guanylyl cyclases (GCs). GC-1/2 is stimulated by nitric oxide (NO) and is primarily located within the cytosol. Transmembrane GC-A is activated by atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP), and GC-B is triggered by C-type natriuretic peptide (CNP). cAMP and cGMP act predominantly via activation of protein kinase A (PKA) and protein kinase G (PKG), respectfully. cAMP also binds exchange protein activated by cAMP (EPAC). Both PKA and PKG phosphorylate multiple cardiomyocyte proteins to effect changes in chronotropy, inotropy and lusitropy, as well as cardiomyocyte hypertrophy and apoptosis. Arrows indicate stimulation. Blunt lines indicate inhibition. Abbreviations: [Ca2+]i, intracellular concentration of calcium; β-AR, beta-adrenergic receptor; Ca2+, calcium; CaM, calmodulin; cMYBPC, cardiac myosin–binding protein C; cTnI, cardiac troponin I; GPCR, G protein–coupled receptor; LTCC, L-type calcium channel; mTORC1, mammalian target of rapamycin complex 1; NFAT, nuclear factor of activated T cells; NP, natriuretic peptide; PLB, phospholamban; RGS, regulator of G protein signalling; RyR2, ryanodine receptor; SERCA, sarcoplasmic/endoplasmic reticulum calcium ATPase; SR, sarcoplasmic reticulum; TRPC, transient receptor potential cation channel; TSC2, tuberin
Production of cGMP is initiated by guanylyl cyclases (GCs), which are sensitive to either nitric oxide (NO) or natriuretic peptides (NPs). NO activates the largely cytoplasmic or soluble form of GC, denoted GC-1 and GC-2 (formerly termed sGC) [15]. Conversely, the NPs stimulate the transmembrane or particulate GC, with atrial and brain NPs (ANP and BNP, respectively) activating GC-A, whilst C-type NP (CNP) binds GC-B [15]. cGMP-dependent protein kinase (PKG) is the predominant effector of cGMP signalling in the heart. PKG-facilitated phosphorylation modulates PLB [16], as well as myofilament components, for instance cTnI [17], troponin-t [18], titin [19] and cMYBPC [20]. PKG inhibits the transient receptor potential cation channel 6 (TRPC6) to counteract the calcineurin-nuclear factor of activated T cells (NFAT) pathway [21, 22] and activates the regulator of G protein signalling subtype 4 (RGS4) to offset Gi/q activation [23]. Hence, PKG is anti-adrenergic. PKG also phosphorylates tuberin (TSC2), restraining mammalian target of rapamycin complex 1 (mTORC1) activity [24]. In this way, PKG regulates basal cardiac contractility, lowering inotropy and raising lusitropy, and precludes cardiac hypertrophy, fibrosis and apoptosis.
Cyclic Nucleotides in Heart Failure Pathophysiology
Heart failure develops from an initial insult to the myocardium. Cardiomyocyte death following a myocardial infarction (MI) [25], pressure overload during hypertension (HT) [26] and doxorubicin-induced cardiotoxicity [27] are among the most common causes of myocardial damage. The sympathetic nervous system (SNS) responds by stimulating chronotropy, inotropy and lusitropy in a cAMP/PKA-dependent manner. Initially, this conserves cardiac function, sustaining a CO and blood pressure necessary to uphold organ perfusion. However, continual cAMP/PKA signalling elicits maladaptive remodelling, comprising left ventricular hypertrophy (LVH), cardiac fibrosis and cardiomyocyte apoptosis [28–31]. In contrast, cGMP/PKG mitigates the injurious effects of repeated cardiac sympathetic activation, precluding the hypertrophy and fibrosis engendered by NA and angiotensin II (AngII, a central component of the renin-angiotensin-aldosterone system, which is also upregulated in HF) [32, 33]. cGMP also limits adverse remodelling in the wake of ischaemia-reperfusion injury (IRI) [34–36] and guards against arrhythmia induction in the wake of infarction [37]. NO bioavailability and NP signalling are impaired in HF patients, despite greater circulating NP concentrations, which heightens hospitalisation and mortality [38–42], whereas greater plasma concentrations of NA are associated with increased mortality risk in patients with HF [43].
Cardiac Cyclic Nucleotide Phosphodiesterases
The PDE superfamily is made up of eleven closely related isozymes (PDE1–11) that are categorised according to the homology of the amino acid sequence within their C-terminal catalytic domains and distinguished by variations in their N-terminal regulatory regions [44]. Each isozyme is further classified into subtypes (gene products), of which multiple isoforms (splice variants) may exist. All PDEs catalyse the hydrolysis and inactivation of the 3′-cyclic phosphodiester bond of cAMP and/or cGMP. Within the heart, PDEs 1, 2, 3, 4, 5, 8 and 9 are expressed. Because PDEs are the prime regulators of cyclic nucleotides, they are responsible for integrating the often disparate signalling cascades of cAMP and cGMP in the heart (Fig. 2). Moreover, by controlling when and where hydrolysis occurs, PDEs confine cAMP and cGMP to separate subcellular compartments, compartmentalising individual cardiac cyclic nucleotide pools [45]. In view of this, it is not surprising that changes in PDE expression and/or activity are liable, somewhat, for the alterations in cAMP and cGMP during LVH and HF.
Fig. 2.

Phosphodiesterase regulation of cyclic nucleotides within cardiomyocytes. Phosphodiesterases (PDEs) catalyse the hydrolysis and inactivation of cyclic adenosine-3′,5′-monophosphate (cAMP) and cyclic guanosine-3′,5′-monophosphate (cGMP) forming adenosine monophosphate (AMP) and guanosine monophosphate (GMP), respectively. PDEs 1, 2, 3, 4, 5 and 9 are the principal regulators of cAMP and cGMP signalling in cardiomyocytes. PDE1 is calcium/calmodulin (Ca2+/CaM)-dependent and hydrolyses cAMP and cGMP. PDE2 is stimulated by cGMP binding to its GAF-B domain. PDE2 degrades cAMP and cGMP, and there is evidence that PDE2 hydrolyses both the natriuretic peptide (NP) and the nitric oxide (NO) pools of cGMP. PDE3 is a dual esterase but is inhibited by cGMP, and PDE4 is cAMP-selective. PDE5 is stimulated by cGMP binding to its GAF-A domain and largely hydrolyses NO/cGMP. PDE9 metabolises NP/cGMP. Abbreviations: AC, adenylyl cyclase; ANP, atrial natriuretic peptide; ATP, adenosine-5′-triphosphate; β-AR, beta-adrenergic receptor; BNP, brain natriuretic peptide; CNP, C-type natriuretic peptide; GAF, cGMP-stimulated PDE, Anabaena AC and Fhla transcription factor; GC, guanylyl cyclase; GTP, guanosine-5′-triphosphate; NA, noradrenaline
Phosphodiesterase 1
Overview
Members of the PDE1 isozyme family are Ca2+/calmodulin (CaM)-dependent enzymes. Each subtype (PDE1A, PDE1B, PDE1C) contains at their N-termini two CaM binding domains, two phosphorylation sites and an inhibitory region that maintains the protein in an inactive configuration when the [Ca2+]i is low (Fig. 3) [46]. Phosphorylation of PDE1 by either PKA (for PDE1A and PDE1C) [47, 48] or Ca2+/CaM-dependent protein kinase II (CaMKII; for PDE1B) [49] reduces the affinity of each subtype for Ca2+/CaM, thereby limiting enzymatic activity. Conversely, the binding of CaM to its respective sites elevates hydrolytic activity by preventing PKA/CaMKII-mediated phosphorylation, as well as effecting a conformational change that raises the maximal catalytic activity (Vmax) [50, 51]. PDE1 functions as a dual esterase, hydrolysing cAMP and cGMP. Whilst PDE1C hydrolyses each cyclic nucleotide with comparably high affinity (low Michaelis constant, KM), PDE1A and PDE1B display a lower affinity for cAMP, hence favouring cGMP hydrolysis [52, 53]. (Readers interested in exploring the enzymology of PDEs further are directed to the following excellent reviews) [44, 54].
Fig. 3.

Structures of phosphodiesterases 1–5. Phosphodiesterase (PDE) 1 contains two calmodulin (CaM) binding domains, two phosphorylation sites and an inhibitory (I) region. PDE2 and PDE5 possess two cGMP-stimulated PDE, Anabaena AC and Fhla transcription factor (GAF) domains. Binding of cyclic guanosine-3′,5′-monophosphate (cGMP) to GAF-B and GAF-A stimulates the hydrolytic activity of PDE2 and PDE5, respectively. PDE3 can be phosphorylated at multiple regions, and PDE4 contains one phosphorylation site within its first upstream conserved regions (UCRs). Abbreviations: C, carboxyl-terminus; N, amino terminus; P, phosphate
Cardiac Physiology
Both PDE1A and PDE1C messenger RNAs (mRNAs) are present in the human heart [53], with PDE1C serving as the principal subtype [55]. Although the majority of cardiac cyclic nucleotide hydrolysis is mediated by PDE1 in humans [56, 57], its roles in cardiac physiology are largely unknown. PDE1C is transcriptionally regulated by peroxisome proliferator–activated receptor alpha (PPARα) [58]. In cardiomyocytes, PDE1C shows a predominantly cytosolic distribution, localising to the M- and Z-lines of the sarcomere, and is present in microsomal fractions [55]. PDE1A protein is abundant in rabbit sinoatrial (SA) node cells where it is purported to moderate pacemaker activity [59], but whether it functions in an analogous capacity in human hearts is currently unknown. Similarly, whilst PDE1A appears to regulate cell death in vascular smooth muscle cells (VSMCs) [60], a corresponding cardiac-specific role is not established.
Heart Failure Pathophysiology
Phosphodiesterase 1C mRNA and protein are raised in failing mouse and human hearts [61]. Likewise, PDE1A protein expression is increased by AngII and the β-AR agonist isoprenaline (ISO) in isolated cardiomyocytes, as well as following pressure overload (i.e. transverse aortic constriction, TAC) in vivo [62]. cAMP/PKA signalling is maintained in PDE1C−/− cardiomyocytes, which moderates AngII- and ISO-stimulated hypertrophy and apoptosis, and PDE1C−/− mice exhibit an improved phenotype with TAC relative to wild-type (WT) animals [61]. AngII promotes PDE1A levels in isolated rat cardiac myofibroblasts, and PDE1 inhibition (PDE1i) ameliorates the cardiac fibrosis associated with ISO-induced HF via cAMP and cGMP [63]. Although PDE1C is absent from cardiac fibroblasts, PDE1C deletion is anti-fibrotic, which may be a consequence of either diminished cardiomyocyte apoptosis or enhanced protective signalling between the two cell types [61]. Although this remains unclear, multidrug-resistant proteins (MRPs) have been implicated in the efflux of cAMP and cGMP [64, 65]. This could account for intercellular cyclic nucleotide signalling, and MRPs constitute prospective drug targets in HF. The hypertrophic and fibrotic actions of AngII are blunted by the PDE1 inhibitor vinpocetine in vitro and in vivo [66], and PDE1i improves cardiac function in failing mouse hearts through greater proteasomal activity [67]. Indeed, the pharmacological and genetic ablation of PDE1 was recently shown to enhance cAMP signalling through the adenosine A2 receptor (A2R), which is protective in multiple models of HF (including in larger mammals, e.g. rabbits and dogs), enhancing inotropy and vasodilation, as well as limiting apoptosis [68, 69]. It has been proposed that targeting the A2R, rather than the β-AR, pool of cAMP will circumvent the unfavourable actions of positive inotropic agents in HF (see below). However, this awaits further verification, as do the precise effects of raising this particular cAMP pool on hypertrophy and fibrosis. The upcoming clinical trial examining the safety and tolerability of the PDE1 inhibitor, ITI-214, in HF patients will undoubtedly illuminate these issues further (Clinicaltrials.gov: NCT03387215).
Phosphodiesterase 2
Overview
Like PDE1, PDE2 is capable of hydrolysing both cAMP and cGMP (Fig. 2), displaying comparable maximal rates and low KM values for each cyclic nucleotide (with a slight preference for cGMP) [70, 71]. PDE2 is commonly designated as the cGMP-stimulated PDE since the enzyme possesses a regulatory segment at its N-terminus comprising two cGMP-stimulated PDE, Anabaena AC and Fhla transcription factor (GAF) domains (Fig. 3). Designated GAF-A and GAF-B, these domains are so called after the proteins in which they were originally discovered [72]. cGMP selectively binds to the allosteric GAF-B domain with high affinity, altering the conformation of PDE2 and raising esterase activity by a factor of 30 [73, 74]. It is unlikely that cAMP regulates PDE2 activity in vivo in the same way given that the affinity of cAMP for the GAF-B site is approximately 100-fold lower than cGMP [75]. A single isogene product of PDE2 (PDE2A) exists, and alternative gene splicing gives rise to three distinct isoforms that are either soluble (PDE2A1) or particulate (PDE2A2, PDE2A3) in their distribution [76, 77].
Cardiac Physiology
Phosphodiesterase 2 is expressed in human hearts [78] and associates with the sarcomere and the plasma membrane in isolated cardiomyocytes [79]. As is the case for the majority of investigations pertaining to PDE expression and function, much of our knowledge concerning PDE2 has been obtained from in vitro analyses in isolated cardiomyocytes. Though there are evident shortcomings of such techniques (e.g. disparities between neonatal and adult cells, complications in integrating in vitro and in vivo physiology), PDE2 is increasingly recognised as an important moderator of cardiac function despite accounting for only a minor fraction of total cardiac PDE activity [80]. Indeed, given its activation by cGMP and its dual substrates, PDE2 occupies a unique position; not only does it facilitate negative feedback of cGMP signalling, it also mediates cross-communication between the cGMP and cAMP pathways. cGMP-induced activation of PDE2 reduces the ICa,L following greater cAMP hydrolysis and diminished LTCC phosphorylation by PKA [81–84]. PDE2 also contributes to the control of cardiac contractility by opposing the effects of β1/2-AR activation through cAMP degradation [79]. This appears largely dependent upon activation by NO/cGMP and β3-AR signalling, which can generate NO via NO synthase 3 (NOS3; formerly termed endothelial NO synthase/eNOS) stimulation [85]. Although β3-AR expression within the myocardium is contentious [86], this corresponds well with the different actions attributed to NO/cGMP and NP/cGMP on contractility. That is, whilst NO acts, primarily, as a negative inotrope, ANP either reduces or has no effect on inotropy [87, 88]. PDE2 also promotes cardiomyocyte apoptosis [89] and is highly expressed in cardiac fibroblasts where it regulates myofibroblast formation and fibrosis by counteracting the increase in cAMP in response to ISO and β-AR stimulation [90, 91].
Despite sharing a downstream second messenger, NO and NPs exert distinctive effects in both the heart and the vasculature. PDE2 makes a significant contribution to this by compartmentalising the cGMP signal in discrete cells and distinct intracellular regions. For example, in isolated rat cardiomyocytes, NO promotes the synthesis of a cytoplasmic pool of cGMP that is hydrolysed specifically by PDE5, whereas NPs generate a separate juxta-membrane GMP pool that is regulated by PDE2 [92]. Similarly, PDE2 confines the membrane-associated pool of cGMP generated via NP/GC-A signalling within the region of the T-tubules in isolated cardiomyocytes [93]. However, the role of PDE2 in restricting the sphere of activity of NP/cGMP can change with disease [87].
Heart Failure Pathophysiology
Although the activity of myocardial PDE2 is amplified in pre-clinical models of pressure overload and in human HF [94, 95], its precise role in HF pathogenesis remains controversial. During HF, desensitisation of β-AR signalling occurs following prolonged SNS activation. Indeed, in ventricular cardiomyocytes from failing human hearts, β1-AR mRNA and protein expression are severely diminished relative to non-failing hearts, whereas those of β3-AR are tripled [96]. Consequently, the positive inotropic actions of β1-AR stimulation are hampered, whereas the negative inotropic effects of β3-AR signalling are maintained. Whether this is beneficial or detrimental to the failing heart is unclear, and PDE2 may be protective or harmful depending on this context. Firstly, cardiomyocyte-specific overexpression of PDE2 lowers the [Ca2+]i and the ICa,L, guarding against catecholamine-induced hypertrophy and inotropy in vitro [95]. Similarly, mice overexpressing PDE2 are resistant to arrhythmias and their cardiac contractility is maintained following MI [97]. Thus, PDE2 upregulation may well defend against sustained SNS signalling, and its activation could offer therapeutic benefit in HF. On the other hand, since PDE2 activation via the β3-AR/NOS3/NO/cGMP pathway abrogates β-AR-induced cAMP in cardiomyocytes and PDE2i can at least moderately restore β-AR responsiveness, PDE2 signalling might be unfavourable in HF [79]. Thus, inhibition of PDE2 could benefit the failing heart by restoring systolic function; recent work has given credence to this concept.
Whilst the inhibition of PDE3 and PDE4 elevates the concentration of cAMP and fosters hypertrophy in isolated cardiomyocytes, PDE2i is anti-hypertrophic [98]. This is thought to occur via an increase in a confined cAMP pool that promotes PKA activation, limiting pro-hypertrophic NFAT signalling. Likewise, PDE2i curtails the development of LVH, compromised contractility and cardiac fibrosis in pressure overload–induced HF. Rather than amplifying cAMP signalling, inhibition of PDE2 augments cGMP signalling, which is dependent upon endogenous NO activity and stimulation of GC-1 (rather than NP-activated GC-A) [87]. This supports previous suggestions that increases in cGMP produced by PDE2i functions akin to cAMP to curtail fibroblast to myofibroblast conversion and restrain fibrosis [91]. Pharmacological blockade of PDE2 also exerts favourable effects in mice with right ventricular hypertrophy (RVH) and pulmonary HT (PH); although in the right side of the heart, GC-A, rather than GC-1, plays an obligatory role in mediating the actions of PDE2i [99].
These disparate observations regarding PDE2 in HF pathology are likely attributable to differences in the concentrations of cyclic nucleotide generated in the varying models, as well as the different cyclic nucleotide pools that PDE2 regulates. Indeed, it has been postulated that PDE2 favours cGMP hydrolysis when NO signalling is diminished, whilst biasing toward cAMP hydrolysis in the setting of intense β-AR activation [100]. Whilst there are always difficulties translating pre-clinical observations into patients, the insights offered from the studies of PDE2 also afford the potential of stratifying treatment and targeting PDE2 selectively, depending on the nature of pathology. For example, given the positive pharmacodynamic profile of PDE2i during pressure overload, as well as the recent observations that blocking PDE2 activity specifically within cardiac sympathetic neurons may be advantageous during sympathetic hyperactivity [101, 102], inhibiting PDE2 may be effective in the context of HF and HT. Alternatively, enhancing PDE2 activity could perhaps help the failing heart post-MI. Further study of PDE2 as a therapeutic target in HF is certainly warranted.
Phosphodiesterase 3
Overview
Phosphodiesterase 3 is the third dual esterase. It hydrolyses cAMP and cGMP with high affinity, but the Vmax for cGMP is ten times lower than that for cAMP [71]. That is to say, whilst cGMP binds to the catalytic site of PDE3, it is hydrolysed very slowly, which lessens cAMP metabolism. Hence, contrary to PDE2, PDE3 is often termed the cGMP-inhibited PDE (Fig. 2). This positions PDE3 as an important regulator of cAMP/cGMP crosstalk as cGMP serves as a positive regulator of cAMP signalling through PDE3 [103, 104]. However, PDE3 also possess three phosphorylation sites at its N-terminus, and PKA negatively modulates its own signal by phosphorylating PDE3, which raises enzymatic activity and cAMP hydrolysis (Fig. 3) [105, 106]. The two subtypes of PDE3 (PDE3A, PDE3B) are both present in the heart, but PDE3A predominates in the myocardium where (in conjunction with PDE4) it accounts for the majority of cAMP hydrolytic activity [107–109].
Cardiac Physiology
By kerbing cAMP signalling, PDE3 is a key regulator of cardiac contractility. PDE3A associates with intracellular membranes in isolated rat cardiomyocytes and localises to Z-bands in human cardiomyocytes where it forms a scaffold with SERCA and PLB at the SR [80, 110]. PKA-mediated phosphorylation of PDE3 potentiates this interaction, and the degradation of cAMP precludes PLB phosphorylation by PKA and SERCA activation. Hence, PDE3 controls Ca2+ reuptake into the SR [110, 111]. PDE3 also limits contractility by curtailing the ICa,L [112, 113]. PDE3A is largely responsible for this as PDE3A−/− mice, but not PDE3B−/− animals, exhibit increased chronotropy basally compared to WT [114]. Likewise, the inotropic and chronotropic responses to ISO are not potentiated by the PDE3 inhibitor cilostamide in PDE3A−/− mice, but these are in PDE3B−/− animals. PDE3 also reduces spontaneous SA node activity by restricting cAMP-mediated activation of hyperpolarisation-activated cyclic nucleotide (HCN)–activated channels, as well as by reducing the phosphorylation of several pacemaker components (e.g. LTCC, RyR2, PLB) by PKA [115–117]. PDE3B complexes with phosphatidylinositol 3-kinase-gamma (PI3Kγ) in the mouse myocardium, which stimulates PDE3B activity, reducing cAMP levels and cardiac contractility [118]. PDE3B also interacts with PI3Kγ in human arterial endothelial cells and increases endothelial cell proliferation and angiogenesis through cAMP degradation and diminution of PKA [119, 120]. Whether a similar phenomenon occurs within the myocardium is unknown but seems probable given the associations observed in mouse hearts. Additionally, PDE3A reduces cardiomyocyte apoptosis by preventing inducible cAMP early repressor (ICER) expression, which stops the reduction in pro-survival genes [121, 122].
Phosphodiesterase 3 is differentially regulated by NO/cGMP and NP/cGMP. GC-1−/− mice exhibit impaired PDE3 expression and activity [123]. Although NO/cGMP is predominantly negatively inotropic, low concentrations of NO/cGMP can inhibit PDE3 [124]. Whilst this increases a particular cAMP/PKA pool in isolated cardiomyocytes, ANP/cGMP favours activation of PDE2, which decreases cAMP and PKA activation [88]. Thus, whilst NO/cGMP can exert dual effects on cardiac contractility, ANP/cGMP tends to oppose inotropy. CNP/cGMP, however, boosts the positive inotropic and lusitropic responses to β-AR stimulation in isolated rat cardiomyocytes via PDE3 antagonism, whilst BNP/cGMP does not [125]. Therefore, PDE3 is also distinctively controlled by different NPs.
Heart Failure Pathophysiology
Both the expression and activity of PDE3A are diminished in pre-clinical models of pressure overload and in failing human hearts [121, 126]. Pharmacological inhibition of PDE3 has been reported to blunt TAC-stimulated hypertrophy and fibrosis, an effect recapitulated in PDE3A−/−, but not PDE3B−/−, mice [127]. Conversely, genetic ablation of PDE3B, but not PDE3A, counteracts the adverse effects of IRI [128]. This occurs following PKA-mediated opening of mitochondrial Ca2+-activated potassium (K+) channels, which reduces reactive oxygen species formation and protects against apoptosis. Although such findings intimate that PDE3 downregulation could represent a protective mechanism in HF and that selective targeting of PDE3 may provide therapeutic benefit to failing hearts, this is clearly not the case. The diminution of PDE3 activity is associated with β-AR desensitisation in LVH, and PDE3i fosters cardiomyocyte apoptosis [121, 126]. Similarly, greater PDE3A expression protects against IRI and apoptosis in the murine heart by regulating β-AR/cAMP [129, 130], whilst the formation of a complex between PDE3B and PI3Kγ guards against TAC-induced cardiac dysfunction [118].
Similar detrimental outcomes have been observed in patients. It was originally postulated that inotropes would be beneficial in HF. Indeed, the PDE3 inhibitor milrinone acutely increases inotropy, chronotropy and stroke volume, whilst reducing MABP in HF patients [131]. However, prolonged PDE3i is not beneficial to the failing heart since it exacerbates SNS activity, increasing arrhythmias and mortality [132–135]. This may be accounted for by the adverse effects discussed above regarding PDE3 blockade and could be attributable to genetic polymorphism within PDE3 in populations of HF patients [136]. It has been suggested that these side effects may be offset by more tailored therapy targeting particular PDE3 isoforms (milrinone is a non-specific PDE3 inhibitor) or by concomitant administration of PDE3 and β-AR antagonists. Whilst such a tandem therapy is well tolerated in HF patients, it fails to improve exercise capacity and mortality [137]. Therefore, positive inotropic agents (including PDE3 inhibitors and β-agonists) are now only used to improve haemodynamic status in acute decompensated HF or as a bridge to heart transplantation [137, 138]. Milrinone may provide some benefit in the context of HF with preserved ejection fraction (HFpEF) [139], and an extended release formulation exhibits early positive effects in advanced HF [140]. HFpEF may constitute as much as 70% of all HF cases [141, 142], and unlike those with HF with reduced ejection fraction (HFrEF), these patients, the majority of whom are older and female, do not respond as well to routine treatment (e.g. angiotensin-converting enzyme inhibitors) [143]. Presently, there is no licenced medication for HFpEF, and targeting PDE3 may afford a novel therapeutic paradigm. For mature women with HFpEF, this would likely be more efficacious than targeting PDE5, for example, since the menopause is associated with impaired NO bioactivity [144] and PDE5 inhibition fosters NO/cGMP signalling specifically (see below). However, larger, longer-term randomised clinical evaluation of PDE3 blockade in HFpEF is necessary.
Phosphodiesterase 4
Overview
The four subtypes of PDE4 (PDE4A, PDE4B, PDE4C, PDE4D) are highly selective for cAMP, displaying very low KM values for this cyclic nucleotide [145–147]. Alternative gene splicing produces more than twenty PDE4 isoforms, which can exist in short and long forms depending on the presence of so-called upstream conserved regions (UCRs) within the N-terminus (Fig. 3) [148]. Short PDE4 isoforms possess a single UCR (UCR2), whilst long versions contain two (UCR1, UCR2). PDE4 possesses a phosphorylation site within UCR1, and PKA-mediated phosphorylation elevates esterase activity by precluding UCR1-UCR2 interactions [149]. An extracellular signal–regulated kinase (ERK) phosphorylation region at the C-terminus serves as a negative and positive regulator of long and short isoforms, respectfully [150].
Cardiac Physiology
In the heart, PDE4A, PDE4B and PDE4D are expressed whereas PDE4C is not [151]. In mouse and rat cardiomyocytes, PDE4 behaves analogously to PDE3 and perpetuates the main cAMP hydrolytic activity (although PDE4 constitutes a substantially smaller fraction of total PDE activity in human hearts) [152]. PDE4 and PDE3 are the principal PDEs adjusting cAMP levels and ICa,L basally, whilst PDE4 predominantly contributes to the control of β-AR–stimulated elevations in cAMP, ICa,L and cardiac contractility [80, 113]. Indeed, PDE4 is capable of interacting with β1/2-ARs and the LTCC, and the ICa,L is amplified in PDE4−/− cardiomyocytes in response to ISO [153–155]. Additionally, PDE4 associates with RyR2 and PLB/SERCA [156–158]. By curtailing cAMP at the SR, PDE4 limits PKA-induced phosphorylation of these sites, reducing Ca2+ release/reuptake. Like PDE3, PDE4 also regulates the pacemaker activity of the SA node [117, 159, 160].
Heart Failure Pathophysiology
Resembling PDE3A, the expression and activity of PDE4A and PDE4B, but not PDE4D, declines with pressure overload in rats as well as in human HF [126]. Consequently, the control of β-AR–stimulated cAMP by PDE4 is diminished. Moreover, when PDE4 is knocked out, the heart is rendered more susceptible to ventricular tachycardia in response to β-AR agonists, and arrhythmogenesis is exacerbated when PDE4 is inhibited [155, 159, 161, 162]. PDE4−/− mice experience more severe HF with MI due to heightened RyR2 phosphorylation and defective Ca2+ regulation [156]. This likely contributes to the cardiac dysfunction in failing human hearts, also [8].
Phosphodiesterase 5
Overview
Phosphodiesterase 5 is both selective for and activated by cGMP. Akin to PDE2, PDE5 contains GAF-A and GAF-B domains within its N-terminus, and its esterase activity is promoted by cGMP binding to the GAF-A site (Fig. 3) [163–165]. PKG-mediated phosphorylation of PDE5 amplifies the cGMP affinity of the GAF-A domain, thereby stabilising the active enzyme conformation and maintaining hydrolytic activity [166–168]. Thus, cGMP fosters its own degradation through negative feedback (i.e. facilitated hydrolysis).
Cardiac Physiology
The one PDE5 subtype (PDE5A) is alternatively expressed as three isoforms (PDE5A1, PDE5A2, PDE5A3), although dispute persists concerning the physiological relevance of these to cardiac physiology. Whilst some studies have reported either no or minimal cardiac expression of PDE5 [56, 169–171], others have detected it within certain cell types [172–174]. In isolated cardiomyocytes, PDE5 appears restricted to the cytosol where it is anchored to the Z-lines and preferentially hydrolyses NO/cGMP (although this may change with disease) [92, 175, 176]. Inhibition of PDE5 with sildenafil (Viagra) suppresses the contractile response to β-AR stimulation in isolated mouse cardiomyocytes and human hearts [175, 177]. This is dependent upon greater NOS3-derived cGMP and heightened PKG activity and can be enhanced by sGC stimulation [178, 179]. Sildenafil also enhances the cGMP-dependent activation of PDE2, which reduces chronotropy via attenuation of cAMP signalling [180]. PDE5 is expressed in cardiac fibroblasts and likely participates in cardiac fibroblast transformation and proliferation [170, 181]. In vascular endothelial cells, PDE5 localises to caveolae where it negatively modulates NOS3 signalling [182], and PDE5 is abundantly expressed in VSMCs where it plays a central role in regulating vascular tone [56, 183].
Heart Failure Pathophysiology
Since PDE5 knockout models are still lacking, particularly those of a cell-restricted nature, knowledge of its precise physiological role in the heart remains elusive. However, it is clear that PDE5 contributes to the pathophysiology of HF. Greater levels of PDE5 are observed in multiple pre-clinical models of pressure overload and cardiac ischaemia [184, 185] (although exceptions have been reported) [170]. Moreover, PDE5 is upregulated in PH patients with RVH [186, 187] and during human LVH and HF [184, 188]. Blockade of PDE5 with sildenafil reduces infarct size and improves cardiac contractility and survival in mice with MI [189]. This is mediated by PKG, which diminishes cardiomyocyte apoptosis through the inhibition of glycogen synthase kinase 3 beta (GSK3β) [190] and protects the heart via hydrogen sulphide synthesis and the opening of mitochondrial ATP-sensitive K+ channels [191–193]. Sildenafil also bolsters the degradation of damaged proteins by the proteasome [194], which may further benefit the failing heart. Vardenafil and tadalafil, two further selective PDE5 inhibitors, display similar protective profiles to sildenafil. Vardenafil maintains diastolic function in rats with HFpEF associated with diabetic cardiomyopathy [195], and tadalafil attenuates doxorubicin-induced PKG oxidation and cardiac dysfunction in mice [196, 197]. Tadalafil has also been shown to promote contractility in experimental HF by raising the density of transverse tubules in cardiomyocytes, thereby improving β-AR responsiveness [198].
The anti-hypertrophic effects exerted by PDE5i appear dependent upon PKG. Sildenafil attenuates NFAT activation by amplifying PKG-mediated inactivation of TRPC6, which prevents and reverses LVH in mice after TAC [199–201]. Sildenafil also suppresses hypertrophy by promoting the activation of RGS2 by PKG [202]. These effects are lost in mice expressing a dysfunctional form of PKG [203], and the hypertrophic response of isolated cardiomyocytes to phenylephrine is lessened following PDE5 gene silencing [176]. However, the contribution of PDE5 and PKG to cardiac hypertrophy has been contested. PKG deletion within the myocardium does not alter basal cardiac function and hypertrophy, nor does it affect the progression of AngII-mediated LVH- or ISO-induced dysfunction [170, 204, 205]. Yet, the loss of PKG in mice does exacerbate pressure overload-induced HF [204, 206].
Evidently, further research is needed to resolve these conflicting observations. Variations in the cellular distribution of PKG and mechanisms to compensate for the cardiomyocyte-specific loss of PKG probably contribute, as may the differences in the type of HF model employed and the possible concurrent inhibition of PDE1 and PDE5 with high doses of sildenafil [57, 170]. Nevertheless, even if impaired PKG signalling within the myocardium does not innately influence cardiac hypertrophy, it might render the heart more susceptible to fibrosis. PDE5 may not be detectable in cardiomyocytes in the wake of pressure overload, but its expression in cardiac fibroblasts is elevated with TAC [170]. Moreover, the ability of NP/cGMP to moderate collagen production in isolated cardiac fibroblasts is supplemented by PDE5i [207], and sildenafil mitigates fibroblast activation and fibrosis associated with pressure overload in mice [181]. PKG deficiency also negates the anti-fibrotic effects of sildenafil [204, 205]. This evidence establishes that PDE5 can be targeted pharmacologically for therapeutic gain in HF, even if its expression in cardiomyocytes per se is questionable.
Sildenafil lowers systolic pulmonary artery pressure (PAP) in HF patients [208, 209] and increases CO and reduces hospitalisations in HF patients with secondary pulmonary arterial HT (PAH) [210]. Though sildenafil causes a modest (approximately 10%) reduction in mean PAP in PAH patients [210], the salutary effects of PDE5 inhibition appear to occur through additional protection of the RV (e.g. reductions in RVH) [211]. The effects of PDE5 blockade on cardiac function in HFrEF are variable with both positive and negative effects on cardiac function observed [212–214]. Despite displaying early positive signs in HFpEF [215], sildenafil does not significantly improve cardiac structure and function or clinical status in these patients [216–219].
Other Phosphodiesterases
Phosphodiesterases 6 and 11 are not found in the heart [220, 221]. PDE7 mRNA is detectable within the heart [222], but a cardiac-specific role for this PDE has not yet been recognised. PDE8 mRNA and protein are expressed in cardiomyocytes, and this cAMP-selective esterase might play a part in cardiac excitation-contraction coupling [223]. PDE10 has been implicated in RVH, yet this is presumably due to elevated expression within the pulmonary vasculature during PH [224]. PDE9 is surfacing as a decisive regulator of cGMP signalling. Like PDE5, it is a cGMP-specific esterase, though myocardial PDE9 curtails NP-mediated increases in cGMP rather than hydrolysing the cGMP pool generated by NO/GC-1 [225]. PDE9 is also expressed in VSMCs where, contrary to the myocardium, it was recently shown to metabolise NO/cGMP [226]. PDE9 is upregulated during LVH and HF in mice and humans, and its inhibition slows the progression of experimental HF [225, 227] by augmenting NP/cGMP-triggered pathways that are well established to be anti-hypertrophic and anti-fibrotic [228–230].
Conclusion
The evidence discussed here demonstrates that PDEs occupy a pivotal position in cardiac physiology and HF pathophysiology. PDE expression changes in failing hearts and pharmacological manipulation of PDE activity ought to amend the aberrant cardioprotective cyclic nucleotide signalling characteristic of cardiac dysfunction (Fig. 4). Future research will aid in the identification of the interventions that will be most successful in realising the necessary, and often divergent, therapeutic objectives in the distinct types of HF. For example, PDE3 inhibition, whilst deleterious in HFrEF, may represent an effective means of treating HFpEF patients. Moreover, investigations into different combination therapies, such as the inhibition of multiple PDEs, or simultaneously activating GC (e.g. sGC stimulators) and blocking PDEs, should also prove fruitful. In sum, targeting PDEs will doubtless prove essential in rectifying the present dearth of effective HF therapies, thereby curtailing the morbidity and mortality associated with this disease.
Fig. 4.
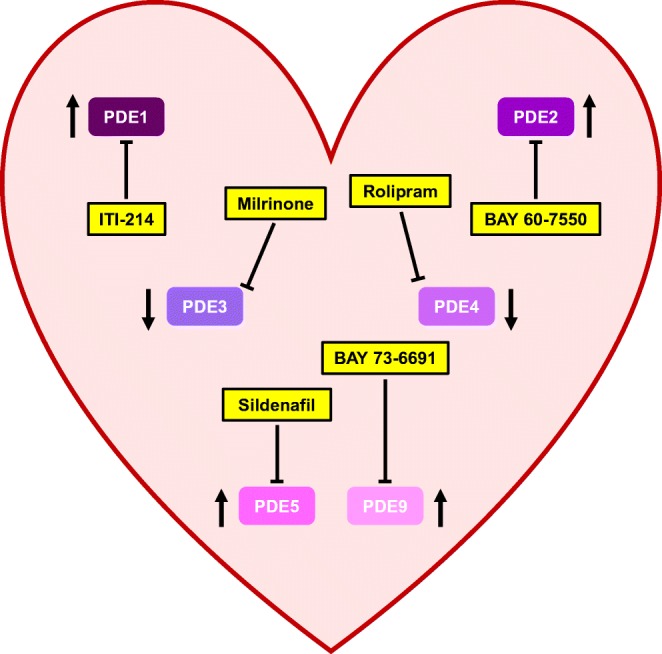
Changes in cardiac phosphodiesterase expression during heart failure and pharmacological modulators. Phosphodiesterases (PDEs) 1, 2, 3, 4, 5 and 9 contribute to the pathophysiology of heart failure (HF). Arrows indicate changes in PDE expression during HF. Blunt lines indicate inhibition with selective pharmacological modulators
Acknowledgements
This article is associated with the award of the Bernard and Joan Marshall Early Career Investigator Prize from the British Society for Cardiovascular Research. Thanks to Professor Adrian Hobbs for his critical reading of the manuscript.
Abbreviations
- [Ca2+]i,
Intracellular concentration of calcium
- A2R
Adenosine A2 receptor
- AC
Adenylyl cyclase
- AngII
Angiotensin II
- ANP
Atrial natriuretic peptide
- β-AR
Beta-adrenergic receptor
- BNP
Brain natriuretic peptide
- Ca2+
Calcium
- CaM
Calmodulin
- CaMKII
-
Calcium/calmodulin-dependent protein kinase II
cAMP
Cyclic adenosine-3′-5′-monophosphate
- cGMP
Cyclic guanosine-3′,5′-monophosphate
- cMYBPC
Cardiac myosin–binding protein C
- CNP
-
C-type natriuretic peptide
CO
Cardiac output
- cTnI
Cardiac troponin I
- ECC
Excitation-contraction coupling
- EPAC
Exchange protein activated by cAMP
- ERK
Extracellular signal–regulated kinase
- GAF
cGMP-stimulated PDE, Anabaena AC and Fhla transcription factor
- GC
Guanylyl cyclase
- GSK3β
Glycogen synthase kinase 3 beta
- HCN
Hyperpolarisation-activated cyclic nucleotide
- HF
Heart failure
- HFpEF
Heart failure with preserved ejection fraction
- HFrEF
Heart failure with reduced ejection fraction
- HT
Hypertension
- ICa,L
L-type calcium current
- ICER
Inducible cAMP early repressor
- IRI
Ischaemia-reperfusion injury
- ISO
Isoprenaline
- K+
Potassium
- KM
Michaelis constant
- LTCC
L-type calcium channel
- LVH
Left ventricular hypertrophy
- MI
Myocardial infarction
- MRP
Multidrug-resistant protein
- mTORC1
Mammalian target of rapamycin complex 1
- NA
Noradrenaline
- NFAT
Nuclear factor of activated T cells
- NO
Nitric oxide
- NOS
Nitric oxide synthase
- NP
Natriuretic peptide
- PAH
Pulmonary arterial hypertension
- PAP
Pulmonary artery pressure
- PH
Pulmonary hypertension
- PDE
Phosphodiesterase
- PI3Kγ
Phosphatidylinositol 3-kinase-gamma
- PKA
cAMP-dependent protein kinase
- PKG
cGMP-dependent protein kinase
- PLB
Phospholamban
- PPARα
Peroxisome proliferator–activated receptor alpha
- RGS
Regulator of G protein signalling
- RVH
Right ventricular hypertrophy
- RyR2
Ryanodine receptor
- SA
Sinoatrial
- SERCA
Sarcoplasmic/endoplasmic reticulum calcium ATPase
- sGC
Soluble guanylyl cyclase
- SNS
Sympathetic nervous system
- SR
Sarcoplasmic reticulum
- TAC
Transverse aortic constriction
- TRPC
Transient receptor potential cation channel
- TSC2
Tuberin
- UCR
Upstream conserved region
- Vmax
Maximal catalytic activity
- VSMC
Vascular smooth muscle cell
- WT
Wild-type
Funding Information
The author is supported by a British Heart Foundation MRes/PhD Scholarship FS/13/58/30648.
Compliance with Ethical Standards
Conflict of Interest
The author declares that he has no conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by the author.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Vos T, Flaxman AD, Naghavi M, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380(9859):2163–2196. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. 2016;13(6):1–11. doi: 10.1038/nrcardio.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Redfield MM, Jacobsen SJ, Burnett JC, Jr, Mahoney DW, Bailey KR, Rodeheffer RJ. Burden of systolic and diastolic ventricular dysfunction in the community: appreciating the scope of the heart failure epidemic. JAMA. 2003;289(2):194–202. doi: 10.1001/jama.289.2.194. [DOI] [PubMed] [Google Scholar]
- 4.Hobbs FDR, Roalfe AK, Davis RC, Davies MK, Hare R. Prognosis of all-cause heart failure and borderline left ventricular systolic dysfunction: 5 year mortality follow-up of the Echocardiographic Heart of England Screening Study (ECHOES) Eur Heart J. 2007;28(9):1128–1134. doi: 10.1093/eurheartj/ehm102. [DOI] [PubMed] [Google Scholar]
- 5.Brodde O, Michel M. Adrenergic and muscarinic receptors in the human heart. Pharmacol Rev. 2006;51(4):651–690. [PubMed] [Google Scholar]
- 6.de Rooij J, Zwartkruis F, Verheijen M, et al. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396(6710):474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- 7.Bünemann M, Gerhardstein B, Gao T, Hosey M. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the beta(2) subunit. J Biol Chem. 1999;274(48):33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 8.Marx S, Reiken S, Hisamatsu Y, et al. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101(4):365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 9.Eschenhagen T. Is ryanodine receptor phosphorylation key to the fight or flight response and heart failure? J Clin Invest. 2010;120(12):4197–4203. doi: 10.1172/JCI45251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dobrev D, Wehrens XHT. Role of RyR2 phosphorylation in heart failure and arrhythmias: controversies around ryanodine receptor phosphorylation in cardiac disease. Circ Res. 2014;114(8):1311–1319. doi: 10.1161/CIRCRESAHA.114.300568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kiss E, Edes I, Sato Y, Luo W, Liggett S, Kranias E. Beat-adrenergic regulation of cAMP and protein phosphorylation in phospholamban-knockout mouse hearts. Am J Phys. 1997;272(2):H785–H790. doi: 10.1152/ajpheart.1997.272.2.H785. [DOI] [PubMed] [Google Scholar]
- 12.Zhang R, Zhao J, Mandveno A, Potter J. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ Res. 1995;76(6):1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 13.Yamasaki R, Wu Y, McNabb M, Greaser M, Labeit S, Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ Res. 2002;90(11):1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 14.Stelzer JE, Patel JR, Moss RL. Protein kinase A-mediated acceleration of the stretch activation response in murine skinned myocardium is eliminated by ablation of cMyBP-C. Circ Res. 2006;99(8):884–890. doi: 10.1161/01.RES.0000245191.34690.66. [DOI] [PubMed] [Google Scholar]
- 15.Alexander SPH, Fabbro D, Kelly E, et al. The concise guide to pharmacology 2017/18: enzymes. Br J Pharmacol. 2017;174(S1):S272–S359. doi: 10.1111/bph.13877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Raeymaekers L, Hofmannt F, Casteels R. Cyclic GMP-dependent protein kinase phosphorylates phospholamban in isolated sarcoplasmic reticulum from cardiac and smooth muscle. Biochem J. 1988;252(1):269–273. doi: 10.1042/bj2520269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blumenthal D, Stull J, Gill G. Phosphorylation of cardiac troponin by guanosine 3′:5′-monophosphate-dependent protein kinase. J Biol Chem. 1978;253(2):324–326. [PubMed] [Google Scholar]
- 18.Yuasa K, Michibata H, Omori K, Yanaka N. A novel interaction of cGMP-dependent protein kinase I with troponin T. J Biol Chem. 1999;274(52):37429–37434. doi: 10.1074/jbc.274.52.37429. [DOI] [PubMed] [Google Scholar]
- 19.Krüger M, Kötter S, Grützner A, et al. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104(1):87–94. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 20.Thoonen R, Giovanni S, Govindan S, et al. Molecular screen identifies cardiac myosin–binding protein-C as a protein kinase G-Iα substrate. Circ Heart Fail. 2015;8(6):1115–1122. doi: 10.1161/CIRCHEARTFAILURE.115.002308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kinoshita H, Kuwahara K, Nishida M, et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase—a signaling in the heart. Circ Res. 2010;106(12):1849–1860. doi: 10.1161/CIRCRESAHA.109.208314. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi S, Lin H, Geshi N, et al. Nitric oxide-cGMP-protein kinase G pathway negatively regulates vascular transient receptor potential channel TRPC6. J Physiol. 2008;586(17):4209–4223. doi: 10.1113/jphysiol.2008.156083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tokudome T, Kishimoto I, Horio T, et al. Regulator of G-protein signaling subtype 4 mediates antihypertrophic effect of locally secreted natriuretic peptides in the heart. Circulation. 2008;117(18):2329–2339. doi: 10.1161/CIRCULATIONAHA.107.732990. [DOI] [PubMed] [Google Scholar]
- 24.Ranek MJ, Kokkonen-Simon KM, Chen A, et al. PKG1-modified TSC2 regulates mTORC1 activity to counter adverse cardiac stress. Nature. 2019;566(7743):264–269. doi: 10.1038/s41586-019-0895-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lloyd-Jones DM, Larson MG, Leip EP, et al. Lifetime risk for developing congestive heart failure: the Framingham Heart Study. Circulation. 2002;106(24):3068–3072. doi: 10.1161/01.cir.0000039105.49749.6f. [DOI] [PubMed] [Google Scholar]
- 26.Haider A, L MG, Franklin S, Levy D. Systolic blood pressure, diastolic blood pressure, and pulse pressure as predictors of risk for congestive heart failure in the Framingham Heart Study. Ann Intern Med. 2003;138(1):10–16. doi: 10.7326/0003-4819-138-1-200301070-00006. [DOI] [PubMed] [Google Scholar]
- 27.Bovelli D, Plataniotis G, Roila F. Cardiotoxicity of chemotherapeutic agents and radiotherapy-related heart disease: ESMO clinical practice guidelines. Ann Oncol. 2010;21(S5):277–282. doi: 10.1093/annonc/mdq200. [DOI] [PubMed] [Google Scholar]
- 28.D’Angelo DD, Sakata Y, Lorenz JN, et al. Transgenic G-alpha-q overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci U S A. 1997;94(15):8121–8126. doi: 10.1073/pnas.94.15.8121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96(12):7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Taigen T, De Windt LJ, Lim HW, Molkentin JD. Targeted inhibition of calcineurin prevents agonist-induced cardiomyocyte hypertrophy. Proc Natl Acad Sci U S A. 2000;97(3):1196–1201. doi: 10.1073/pnas.97.3.1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morel E, Marcantoni A, Gastineau M, et al. cAMP-binding protein Epac induces cardiomyocyte hypertrophy. Circ Res. 2005;97(12):1296–1304. doi: 10.1161/01.RES.0000194325.31359.86. [DOI] [PubMed] [Google Scholar]
- 32.Calderone A, Thaik CM, Takahashi N, Chang DLF, Colucci WS. Nitric oxide, atrial natriuretic peptide, and cyclic GMP inhibit the growth-promoting effects of norepinephrine in cardiac myocytes and fibroblasts. J Clin Invest. 1998;101(4):812–818. doi: 10.1172/JCI119883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wollert K, Fiedler B, Gambaryan S, et al. Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension. 2002;39(1):87–92. doi: 10.1161/hy1201.097292. [DOI] [PubMed] [Google Scholar]
- 34.Burkard N, Williams T, Czolbe M, et al. Conditional overexpression of neuronal nitric oxide synthase is cardioprotective in ischemia/reperfusion. Circulation. 2010;122(16):1588–1603. doi: 10.1161/CIRCULATIONAHA.109.933630. [DOI] [PubMed] [Google Scholar]
- 35.D’Souza SP, Yellon DM, Martin C, et al. B-type natriuretic peptide limits infarct size in rat isolated hearts via KATP channel opening. Am J Physiol Heart Circ Physiol. 2003;284(5):H1592–H1600. doi: 10.1152/ajpheart.00902.2002. [DOI] [PubMed] [Google Scholar]
- 36.Sangawa K, Nakanishi K, Ishino K, Inoue M, Kawada M, Sano S. Atrial natriuretic peptide protects against ischemia-reperfusion injury in the isolated rat heart. Ann Thorac Surg. 2004;77(1):233–237. doi: 10.1016/s0003-4975(03)01493-0. [DOI] [PubMed] [Google Scholar]
- 37.Burger DE, Lu X, Lei M, et al. Neuronal nitric oxide synthase protects against myocardial infarction-induced ventricular arrhythmia and mortality in mice. Circulation. 2009;120(14):1345–1354. doi: 10.1161/CIRCULATIONAHA.108.846402. [DOI] [PubMed] [Google Scholar]
- 38.Burnett JJ, Kao P, Hu D, et al. Atrial natriuretic peptide elevation in congestive heart failure in the human. Science. 1986;231(4742):1145–1147. doi: 10.1126/science.2935937. [DOI] [PubMed] [Google Scholar]
- 39.Hirooka Y, Takeshita A, Imaizumi T, et al. Attenuated forearm vasodilative response to intra-arterial atrial natriuretic peptide in patients with heart failure. Circulation. 1990;82(1):147–153. doi: 10.1161/01.cir.82.1.147. [DOI] [PubMed] [Google Scholar]
- 40.Fischer D, Rossa S, Landmesser U, et al. Endothelial dysfunction in patients with chronic heart failure is independently associated with increased incidence of hospitalization, cardiac transplantation, or death. Eur Heart J. 2005;26(1):65–69. doi: 10.1093/eurheartj/ehi001. [DOI] [PubMed] [Google Scholar]
- 41.Katz SD, Hryniewicz K, Hriljac I, et al. Vascular endothelial dysfunction and mortality risk in patients with chronic heart failure. Circulation. 2005;111(3):310–314. doi: 10.1161/01.CIR.0000153349.77489.CF. [DOI] [PubMed] [Google Scholar]
- 42.Dickey D, Dries D, Margulies K, Potter L. Guanylyl cyclase (GC)-A and GC-B activities in ventricles and cardiomyocytes from failed and non-failed human hearts: GC-A is inactive in the failed cardiomyocyte. J Mol Cell Cardiol. 2012;52(3):727–732. doi: 10.1016/j.yjmcc.2011.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cohn JN, Levine TB, Olivari MT, et al. Plasma norepinephrine as a guide to prognosis in patients with chronic congestive heart failure. N Engl J Med. 1984;311:819–823. doi: 10.1056/NEJM198409273111303. [DOI] [PubMed] [Google Scholar]
- 44.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58(3):488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 45.Fischmeister R, Castro LRV, Abi-Gerges A, et al. Compartmentation of cyclic nucleotide signaling in the heart: the role of cyclic nucleotide phosphodiesterases. Circ Res. 2006;99(8):816–828. doi: 10.1161/01.RES.0000246118.98832.04. [DOI] [PubMed] [Google Scholar]
- 46.Sonnenburg WK, Seger D, Kwak KS, Huang J, Charbonneau H, Beavo JA. Identification of inhibitory and calmodulin-binding domains of the PDE1A1 and PDE1A2 calmodulin-stimulated cyclic nucleotide phosphodiesterases. J Biol Chem. 1995;270(52):30989–31000. doi: 10.1074/jbc.270.52.30989. [DOI] [PubMed] [Google Scholar]
- 47.Sharma R. Phosphorylation and characterization of bovine heart calmodulin-dependent phosphodiesterase. Biochemistry. 1991;30(24):5963–5968. doi: 10.1021/bi00238a021. [DOI] [PubMed] [Google Scholar]
- 48.Ang KL, Antoni FA. Reciprocal regulation of calcium dependent and calcium independent cyclic AMP hydrolysis by protein phosphorylation. J Neurochem. 2002;81(3):422–433. doi: 10.1046/j.1471-4159.2002.00903.x. [DOI] [PubMed] [Google Scholar]
- 49.Sharma RK, Wang JH. Calmodulin and Ca2+-dependent phosphorylation and dephosphorylation of 63-kDa subunit-containing bovine brain calmodulin-stimulated cyclic nucleotide phosphodiesterase isozyme. J Biol Chem. 1986;261(3):1322–1328. [PubMed] [Google Scholar]
- 50.Kincaid RL, Stith-Coleman IE, Vaughan M. Proteolytic activation of calmodulin-dependent cyclic nucleotide phosphodiesterase. J Biol Chem. 1985;260(15):9009–9015. [PubMed] [Google Scholar]
- 51.Sonnenburg WK, Seger D, Beavo JA. Molecular cloning of a cDNA encoding the “61-kDa” calmodulin-stimulated cyclic nucleotide phosphodiesterase. Tissue-specific expression of structurally related isoforms. J Biol Chem. 1993;268(1):645–652. [PubMed] [Google Scholar]
- 52.Yan C, Zhao AZ, Bentley JK, Loughney K, Ferguson K, Beavo JA. Molecular cloning and characterization of a calmodulin-dependent phosphodiesterase enriched in olfactory sensory neurons. Proc Natl Acad Sci U S A. 1995;92(21):9677–9681. doi: 10.1073/pnas.92.21.9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Loughney K, Martins TJ, Harris EAS, et al. Isolation and characterization of cDNAs corresponding to two human calcium, calmodulin-regulated, 3′,5′-cyclic nucleotide phosphodiesterases. J Biol Chem. 1996;271(2):796–806. doi: 10.1074/jbc.271.2.796. [DOI] [PubMed] [Google Scholar]
- 54.Maurice DH, Ke H, Ahmad F, Wang Y, Chung J, Manganiello VC. Advances in targeting cyclic nucleotide phosphodiesterases. Nat Rev Drug Discov. 2014;13(4):290–314. doi: 10.1038/nrd4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vandeput F, Wolda SL, Krall J, et al. Cyclic nucleotide phosphodiesterase PDE1C1 in human cardiac myocytes. J Biol Chem. 2007;282(45):32749–32757. doi: 10.1074/jbc.M703173200. [DOI] [PubMed] [Google Scholar]
- 56.Wallis RM, Corbin JD, Francis SH, Ellis P. Tissue distribution of phosphodiesterase families and the effects of sildenafil on tissue cyclic nucleotides, platelet function, and the contractile responses of trabeculae carneae and aortic rings in vitro. Am J Cardiol. 1999;83(5A):3C–12C. doi: 10.1016/s0002-9149(99)00042-9. [DOI] [PubMed] [Google Scholar]
- 57.Vandeput F, Krall J, Ockaili R, et al. cGMP-hydrolytic activity and its inhibition by sildenafil in normal and failing human and mouse myocardium. J Pharmacol Exp Ther. 2009;330(3):884–891. doi: 10.1124/jpet.109.154468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shete V, Liu N, Jia Y, Viswakarma N, Reddy JK, Thimmapaya B. Mouse cardiac Pde1C is a direct transcriptional target of PPARα. Int J Mol Sci. 2018;19(12):1–14. doi: 10.3390/ijms19123704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lukyanenko YO, Younes A, Lyashkov AE, et al. Ca2+/calmodulin-activated phosphodiesterase 1A is highly expressed in rabbit cardiac sinoatrial nodal cells and regulates pacemaker function. J Mol Cell Cardiol. 2016;98:73–82. doi: 10.1016/j.yjmcc.2016.06.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagel DJ, Aizawa T, Jeon KI, et al. Role of nuclear Ca2+/calmodulin-stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res. 2006;98(6):777–784. doi: 10.1161/01.RES.0000215576.27615.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knight WE, Chen S, Zhang Y, et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci U S A. 2016;113(45):E7116–E7125. doi: 10.1073/pnas.1607728113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller CL, Oikawa M, Cai Y, et al. Role of Ca2+/calmodulin-stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res. 2009;105(10):956–964. doi: 10.1161/CIRCRESAHA.109.198515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miller CL, Cai Y, Oikawa M, et al. Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol. 2011;106(6):1023–1039. doi: 10.1007/s00395-011-0228-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schuetz JD, Connelly MC, Sun D, et al. MRP4: a previously unidentified factor in resistance to nucleoside-based antiviral drugs. Nat Med. 1999;5(9):1048–1051. doi: 10.1038/12487. [DOI] [PubMed] [Google Scholar]
- 65.Jedlitschky G, Burchell B, Keppler D. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J Biol Chem. 2000;275(39):30069–30074. doi: 10.1074/jbc.M005463200. [DOI] [PubMed] [Google Scholar]
- 66.Wu M-p, Zhang Y-s, Xu X, Zhou Q, Li JD, Yan C. Vinpocetine attenuates pathological cardiac remodeling by inhibiting cardiac hypertrophy and fibrosis. Cardiovasc Drugs Ther. 2017;31(2):157–166. doi: 10.1007/s10557-017-6719-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang H, Pan B, Wu P, et al. PDE1 inhibition facilitates proteasomal degradation of misfolded proteins and protects against cardiac proteinopathy. Sci Adv. 2019;5(5):1–15. doi: 10.1126/sciadv.aaw5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y, Knight W, Chen S, Mohan A, Yan C. Multiprotein complex with TRPC (transient receptor potential-canonical) channel, PDE1C (phosphodiesterase 1C), and A2R (adenosine A2 receptor) plays a critical role in regulating cardiomyocyte cAMP and survival. Circulation. 2018;138(18):1988–2002. doi: 10.1161/CIRCULATIONAHA.118.034189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hashimoto T, Kim GE, Tunin RS, et al. Acute enhancement of cardiac function by phosphodiesterase type 1 inhibition. Circulation. 2018;138(18):1974–1987. doi: 10.1161/CIRCULATIONAHA.117.030490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Martins T, Mumby M, Beavo J. Purification and characterization of a cyclic GMP-stimulated cyclic nucleotide phosphodiesterase from bovine tissues. J Biol Chem. 1982;257(4):1973–1979. [PubMed] [Google Scholar]
- 71.Weishaar R, Burrows S, Kobylarz D, Quade M, Evans D. Multiple molecular forms of cyclic nucleotide phosphodiesterase in cardiac and smooth muscle and in platelets: isolation, characterization, and effects of various reference phosphodiesterase inhibitors and cardiotonic agents. Biochem Pharmacol. 1986;35(5):787–800. doi: 10.1016/0006-2952(86)90247-9. [DOI] [PubMed] [Google Scholar]
- 72.Aravind L, Ponting C. The GAF domain: an evolutionary link between diverse phototransducing proteins. Trends Biochem Sci. 1997;22(12):458–459. doi: 10.1016/s0968-0004(97)01148-1. [DOI] [PubMed] [Google Scholar]
- 73.Mumby MC, Martins TJ, Chang ML, Beavo JA. Identification of cGMP-stimulated cyclic nucleotide phosphodiesterase in lung tissue with monoclonal antibodies. J Biol Chem. 1982;257(22):13283–13290. [PubMed] [Google Scholar]
- 74.Martinez SE, Wu AY, Glavas NA, et al. The two GAF domains in phosphodiesterase 2A have distinct roles in dimerization and in cGMP binding. Proc Natl Acad Sci U S A. 2002;99(20):13260–13265. doi: 10.1073/pnas.192374899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wu AY, Tang XB, Martinez SE, Ikeda K, Beavo JA. Molecular determinants for cyclic nucleotide binding to the regulatory domains of phosphodiesterase 2A. J Biol Chem. 2004;279(36):37928–37938. doi: 10.1074/jbc.M404287200. [DOI] [PubMed] [Google Scholar]
- 76.Pyne NJ, Cooper ME, Houslay MD. Identification and characterization of both the cytosolic and particulate forms of cyclic GMP-stimulated cyclic AMP phosphodiesterase from rat liver. Biochem J. 1986;234(2):325–334. doi: 10.1042/bj2340325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rosman GJ, Martins TJ, Sonnenburg WK, Beavo JA, Ferguson K, Loughney K. Isolation and characterization of human cDNAs encoding a cGMP-stimulated 3′,5′-cyclic nucleotide phosphodiesterase. Gene. 1997;191(1):89–95. doi: 10.1016/s0378-1119(97)00046-2. [DOI] [PubMed] [Google Scholar]
- 78.Sadhu K, Hensley K, Florio VA, Wolda SL. Differential expression of the cyclic GMP-stimulated phosphodiesterase PDE2A in human venous and capillary endothelial cells. J Histochem Cytochem. 1999;47(7):895–905. doi: 10.1177/002215549904700707. [DOI] [PubMed] [Google Scholar]
- 79.Mongillo M, Tocchetti CG, Terrin A, et al. Compartmentalized phosphodiesterase-2 activity blunts beat-adrenergic cardiac inotropy via an NO/cGMP-dependent pathway. Circ Res. 2006;98(2):226–234. doi: 10.1161/01.RES.0000200178.34179.93. [DOI] [PubMed] [Google Scholar]
- 80.Mongillo M, McSorley T, Evellin S, et al. Fluorescence resonance energy transfer-based analysis of cAMP dynamics in live neonatal rat cardiac myocytes reveals distinct functions of compartmentalized phosphodiesterases. Circ Res. 2004;95(1):67–75. doi: 10.1161/01.RES.0000134629.84732.11. [DOI] [PubMed] [Google Scholar]
- 81.Méry P, Pavoine C, Pecker F, Fischmeister R. Erythro-9-(2-hydroxy-3-nonyl)adenine inhibits cyclic GMP-stimulated phosphodiesterase in isolated cardiac myocytes. Mol Pharmacol. 1995;48(1):121–130. [PubMed] [Google Scholar]
- 82.Rivet-Bastide M, Vandecasteele G, Hatem S, et al. cGMP-stimulated cyclic nucleotide phosphodiesterase regulates the basal calcium current in human atrial myocytes. J Clin Invest. 1997;99(11):2710–2718. doi: 10.1172/JCI119460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dittrich M, Jurevicius J, Georget M, et al. Local response of L-type Ca(2+) current to nitric oxide in frog ventricular myocytes. J Physiol. 2001;534(1):109–121. doi: 10.1111/j.1469-7793.2001.00109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Vandecasteele G, Verde I, Rücker-Martin C, Donzeau-Gouge P, Fischmeister R. Cyclic GMP regulation of the L-type Ca(2+) channel current in human atrial myocytes. J Physiol. 2001;533(2):329–340. doi: 10.1111/j.1469-7793.2001.0329a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gauthier C, Leblais V, Kobzik L, et al. The negative inotropic effect of β3-adrenoceptor stimulation is mediated by activation of a nitric oxide synthase pathway in human ventricle. J Clin Invest. 1998;102(7):1377–1384. doi: 10.1172/JCI2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Heubach JF, Rau T, Eschenhagen T, Ravens U, Kaumann AJ. Physiological antagonism between ventricular beta(1)-adrenoceptors and alpha(1)-adrenoceptors but no evidence for beta(2)- and beta(3)- adrenoceptor function in murine heart. Br J Pharmacol. 2002;136(2):217–229. doi: 10.1038/sj.bjp.0704700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Baliga RS, Preedy MEJ, Dukinfield MS, et al. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc Natl Acad Sci U S A. 2018;115(31):E7428–E7437. doi: 10.1073/pnas.1800996115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Stangherlin A, Gesellchen F, Zoccarato A, et al. cGMP signals modulate cAMP levels in a compartment-specific manner to regulate catecholamine-dependent signaling in cardiac myocytes. Circ Res. 2011;108(8):929–939. doi: 10.1161/CIRCRESAHA.110.230698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Monterisi S, Lobo MJ, Livie C, et al. PDE2A2 regulates mitochondria morphology and apoptotic cell death via local modulation of cAMP/PKA signalling. eLife. 2017;6:1–20. doi: 10.7554/eLife.21374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gustafsson ÅB, Brunton LL. Attenuation of cAMP accumulation in adult rat cardiac fibroblasts by IL-1beta and NO: role of cGMP-stimulated PDE2. Am J Physiol Cell Physiol. 2002;283(2):463–471. doi: 10.1152/ajpcell.00299.2001. [DOI] [PubMed] [Google Scholar]
- 91.Vettel C, Lämmle S, Ewens S, et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol. 2014;306(8):1246–1252. doi: 10.1152/ajpheart.00852.2013. [DOI] [PubMed] [Google Scholar]
- 92.Castro LRV, Verde I, Cooper DMF, Fischmeister R. Cyclic guanosine monophosphate compartmentation in rat cardiac myocytes. Circulation. 2006;113(18):2221–2228. doi: 10.1161/CIRCULATIONAHA.105.599241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Subramanian H, Froese A, Jönsson P, Schmidt H, Gorelik J, Nikolaev VO. Distinct submembrane localisation compartmentalises cardiac NPR1 and NPR2 signalling to cGMP. Nat Commun. 2018;9(1):1–9. doi: 10.1038/s41467-018-04891-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yanaka N, Kurosawa Y, Minami K, Kawai E, Omori K. cGMP-phosphodiesterase activity is up-regulated in response to pressure overload of rat ventricles. Biosci Biotechnol Biochem. 2003;67(5):973–979. doi: 10.1271/bbb.67.973. [DOI] [PubMed] [Google Scholar]
- 95.Mehel H, Emons J, Vettel C, et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts β-adrenergic responses in cardiomyocytes. J Am Coll Cardiol. 2013;62(17):1596–1606. doi: 10.1016/j.jacc.2013.05.057. [DOI] [PubMed] [Google Scholar]
- 96.Moniotte S, Kobzik L, Feron O, Trochu J, Gauthier C, Balligand J. Upregulation of beta(3)-adrenoceptors and altered contractile response to inotropic amines in human failing myocardium. Circulation. 2001;103(12):1649–1655. doi: 10.1161/01.cir.103.12.1649. [DOI] [PubMed] [Google Scholar]
- 97.Vettel C, Lindner M, Dewenter M, et al. Phosphodiesterase 2 protects against catecholamine-induced arrhythmia and preserves contractile function after myocardial infarction. Circ Res. 2017;120(1):120–132. doi: 10.1161/CIRCRESAHA.116.310069. [DOI] [PubMed] [Google Scholar]
- 98.Zoccarato A, Surdo NC, Aronsen JM, et al. Cardiac hypertrophy is inhibited by a local pool of cAMP regulated by phosphodiesterase 2. Circ Res. 2015;117(8):707–719. doi: 10.1161/CIRCRESAHA.114.305892. [DOI] [PubMed] [Google Scholar]
- 99.Bubb KJ, Trinder SL, Baliga RS, et al. Inhibition of phosphodiesterase 2 augments cGMP and cAMP signaling to ameliorate pulmonary hypertension. Circulation. 2014;130(6):496–507. doi: 10.1161/CIRCULATIONAHA.114.009751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Zhao CY, Greenstein JL, Winslow RL. Roles of phosphodiesterases in the regulation of the cardiac cyclic nucleotide cross-talk signaling network. J Mol Cell Cardiol. 2016;91:215–227. doi: 10.1016/j.yjmcc.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Li D, Lu CJ, Hao G, et al. Efficacy of B-type natriuretic peptide is coupled to phosphodiesterase 2A in cardiac sympathetic neurons. Hypertension. 2015;66(1):190–198. doi: 10.1161/HYPERTENSIONAHA.114.05054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Liu K, Li D, Hao G, et al. Phosphodiesterase 2A as a therapeutic target to restore cardiac neurotransmission during sympathetic hyperactivity. JCI Insight. 2018;3(9):e98694. doi: 10.1172/jci.insight.98694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Mol Pharmacol. 1990;37(5):671–681. [PubMed] [Google Scholar]
- 104.Meacci E, Taira M, Moos M, et al. Molecular cloning and expression of human myocardial cGMP-inhibited cAMP phosphodiesterase. Proc Natl Acad Sci U S A. 1992;89(9):3721–3725. doi: 10.1073/pnas.89.9.3721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shakur Y, Holst L, Landstrom T, Movsesian M, Degerman E, Manganiello V. Regulation and function of the cyclic nucleotide phosphodiesterase (PDE3) gene family. Prog Nucleic Acid Res Mol Biol. 2001;66:241–277. doi: 10.1016/s0079-6603(00)66031-2. [DOI] [PubMed] [Google Scholar]
- 106.Palmer D, Jimmo SL, Raymond DR, Wilson LS, Carter RL, Maurice DH. Protein kinase A phosphorylation of human phosphodiesterase 3B promotes 14-3-3 protein binding and inhibits phosphatase-catalyzed inactivation. J Biol Chem. 2007;282(13):9411–9419. doi: 10.1074/jbc.M606936200. [DOI] [PubMed] [Google Scholar]
- 107.Reinhardt RR, Chin E, Zhou J, et al. Distinctive anatomical patterns of gene expression for cGMP-inhibited cyclic nucleotide phosphodiesterases. J Clin Invest. 1995;95(4):1528–1538. doi: 10.1172/JCI117825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wechsler J, Choi YH, Krall J, Ahmad F, Manganiello VC, Movsesian MA. Isoforms of cyclic nucleotide phosphodiesterase PDE3A in cardiac myocytes. J Biol Chem. 2002;277(41):38072–38078. doi: 10.1074/jbc.M203647200. [DOI] [PubMed] [Google Scholar]
- 109.Hambleton R, Krall J, Tikishvili E, et al. Isoforms of cyclic nucleotide phosphodiesterase PDE3 and their contribution to cAMP hydrolytic activity in subcellular fractions of human myocardium. J Biol Chem. 2005;280(47):39168–39174. doi: 10.1074/jbc.M506760200. [DOI] [PubMed] [Google Scholar]
- 110.Ahmad F, Shen W, Vandeput F, et al. Regulation of sarcoplasmic reticulum Ca2+ ATPase 2 (SERCA2) activity by phosphodiesterase 3A (PDE3A) in human myocardium: phosphorylation-dependent interaction of PDE3A1 with SERCA2. J Biol Chem. 2015;290(11):6763–6776. doi: 10.1074/jbc.M115.638585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Beca S, Ahmad F, Shen W, et al. Phosphodiesterase type 3A regulates basal myocardial contractility through interacting with sarcoplasmic reticulum calcium ATPase type 2a signaling complexes in mouse heart. Circ Res. 2013;112(2):289–297. doi: 10.1161/CIRCRESAHA.111.300003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Weishaar RE, Kobylarz-Singer DC, Steffen RP, Kaplan HR. Subclasses of cyclic AMP-specific phosphodiesterase in left ventricular muscle and their involvement in regulating myocardial contractility. Circ Res. 1987;61(4):539–547. doi: 10.1161/01.res.61.4.539. [DOI] [PubMed] [Google Scholar]
- 113.Verde I, Vandecasteele G, Lezoualc’h F, Fischmeister R. Characterization of the cyclic nucleotide phosphodiesterase subtypes involved in the regulation of the L-type Ca2+ current in rat ventricular myocytes. Br J Pharmacol. 1999;127(1):65–74. doi: 10.1038/sj.bjp.0702506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sun B, Li H, Shakur Y, et al. Role of phosphodiesterase type 3A and 3B in regulating platelet and cardiac function using subtype-selective knockout mice. Cell Signal. 2007;19(8):1765–1771. doi: 10.1016/j.cellsig.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 115.DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351(6322):145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- 116.Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME, Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci U S A. 2009;106(29):12189–12194. doi: 10.1073/pnas.0810332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Galindo-Tovar A, Vargas ML, Kaumann AJ. Phosphodiesterases PDE3 and PDE4 jointly control the inotropic effects but not chronotropic effects of (-)-CGP12177 despite PDE4-evoked sinoatrial bradycardia in rat atrium. Naunyn Schmiedeberg's Arch Pharmacol. 2009;379(4):379–384. doi: 10.1007/s00210-008-0367-7. [DOI] [PubMed] [Google Scholar]
- 118.Patrucco E, Notte A, Barberis L, et al. PI3Kγ modulates the cardiac response to chronic pressure overload by distinct kinase-dependent and -independent effects. Cell. 2004;118(3):375–387. doi: 10.1016/j.cell.2004.07.017. [DOI] [PubMed] [Google Scholar]
- 119.Wilson LS, Baillie GS, Pritchard LM, et al. A phosphodiesterase 3B-based signaling complex integrates exchange protein activated by cAMP 1 and phosphatidylinositol 3-kinase signals in human arterial endothelial cells. J Biol Chem. 2011;286(18):16285–16296. doi: 10.1074/jbc.M110.217026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.MacKeil JL, Brzezinska P, Burke-Kleinman J, et al. Phosphodiesterase 3B (PDE3B) antagonizes the anti-angiogenic actions of PKA in human and murine endothelial cells. Cell Signal. 2019;62:109342. doi: 10.1016/j.cellsig.2019.06.007. [DOI] [PubMed] [Google Scholar]
- 121.Ding B, Abe JI, Wei H, et al. Functional role of phosphodiesterase 3 in cardiomyocyte apoptosis: implication in heart failure. Circulation. 2005;111(19):2469–2476. doi: 10.1161/01.CIR.0000165128.39715.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ding B, Abe JI, Wei H, et al. A positive feedback loop of phosphodiesterase 3 (PDE3) and inducible cAMP early repressor (ICER) leads to cardiomyocyte apoptosis. Proc Natl Acad Sci U S A. 2005;102(41):14771–14776. doi: 10.1073/pnas.0506489102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Dünnes S, Voussen B, Aue A, et al. Phosphodiesterase 3A expression and activity in the murine vasculature is influenced by NO-sensitive guanylyl cyclase. Pflugers Arch. 2018;470(4):693–702. doi: 10.1007/s00424-017-2106-8. [DOI] [PubMed] [Google Scholar]
- 124.Vila-Petroff M, Younes A, Egan J, Lakatta E, Sollott S. Activation of distinct cAMP-dependent and cGMP-dependent pathways by nitric oxide in cardiac myocytes. Circ Res. 1999;84(9):1020–1031. doi: 10.1161/01.res.84.9.1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Meier S, Andressen KW, Aronsen JM, et al. PDE3 inhibition by C-type natriuretic peptide-induced cGMP enhances cAMP-mediated signaling in both non-failing and failing hearts. Eur J Pharmacol. 2017;812:174–183. doi: 10.1016/j.ejphar.2017.07.014. [DOI] [PubMed] [Google Scholar]
- 126.Abi-Gerges A, Richter W, Lefebvre F, et al. Decreased expression and activity of cAMP phosphodiesterases in cardiac hypertrophy and its impact on beta-adrenergic cAMP signals. Circ Res. 2009;105(8):784–792. doi: 10.1161/CIRCRESAHA.109.197947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Polidovitch N, Yang S, Sun H, et al. Phosphodiesterase type 3A (PDE3A), but not type 3B (PDE3B), contributes to the adverse cardiac remodeling induced by pressure overload. J Mol Cell Cardiol. 2019;132:60–70. doi: 10.1016/j.yjmcc.2019.04.028. [DOI] [PubMed] [Google Scholar]
- 128.Chung YW, Lagranha C, Chen Y, et al. Targeted disruption of PDE3B, but not PDE3A, protects murine heart from ischemia/reperfusion injury. Proc Natl Acad Sci U S A. 2015;112(17):E2253–E2262. doi: 10.1073/pnas.1416230112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yan C, Ding B, Shishido T, et al. Activation of extracellular signal-regulated kinase 5 reduces cardiac apoptosis and dysfunction via inhibition of a phosphodiesterase 3A/inducible cAMP early repressor feedback loop. Circ Res. 2007;100(4):510–519. doi: 10.1161/01.RES.0000259045.49371.9c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oikawa M, Wu M, Lim S, et al. Cyclic nucleotide phosphodiesterase 3A1 protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2013;64:11–19. doi: 10.1016/j.yjmcc.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Jaski BE, Fifer MA, Wright RF, Braunwald E, Colucci WS. Positive inotropic and vasodilator actions of milrinone in patients with severe congestive heart failure. Dose-response relationships and comparison to nitroprusside. J Clin Invest. 1985;75(2):643–649. doi: 10.1172/JCI111742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.DiBianco R, Shabetai R, Kostuk W, Moran J, Schlant R, Wright R. A comparison of oral milrinone, digoxin, and their combination in the treatment of patients with chronic heart failure. N Engl J Med. 1989;320(11):677–683. doi: 10.1056/NEJM198903163201101. [DOI] [PubMed] [Google Scholar]
- 133.Packer M, Carver JR, Rodeheffer RJ, DeMets DL. Effect of oral milrinone on mortality in severe chronic heart failure. The PROMISE Study Research Group. N Engl J Med. 1991;325(21):1468–1475. doi: 10.1056/NEJM199111213252103. [DOI] [PubMed] [Google Scholar]
- 134.Cuffe MS, Califf RM, Adams KF, et al. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized controlled trial. JAMA. 2002;287(12):1541–1547. doi: 10.1001/jama.287.12.1541. [DOI] [PubMed] [Google Scholar]
- 135.Amsallem E, Kasparian C, Haddour G, Boissel J-P, Nony P. Phosphodiesterase III inhibitors for heart failure. Cochrane Database Syst Rev. 2005;1:CD002230. doi: 10.1002/14651858.CD002230.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Sucharov CC, Nakano SJ, Slavov D, et al. A PDE3A promoter polymorphism regulates cAMP-induced transcriptional activity in failing human myocardium. J Am Coll Cardiol. 2019;73(10):1173–1184. doi: 10.1016/j.jacc.2018.12.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Metra M, Eichhorn E, Abraham WT, et al. Effects of low-dose oral enoximone administration on mortality, morbidity, and exercise capacity in patients with advanced heart failure: the randomized, double-blind, placebo-controlled, parallel group ESSENTIAL trials. Eur Heart J. 2009;30(24):3015–3026. doi: 10.1093/eurheartj/ehp338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Assad-Kottner C, Chen D, Jahanyar J, et al. The use of continuous milrinone therapy as bridge to transplant is safe in patients with short waiting times. J Card Fail. 2008;14(10):839–843. doi: 10.1016/j.cardfail.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 139.Kaye DM, Nanayakkara S, Vizi D, Byrne M, Mariani JA. Effects of milrinone on rest and exercise hemodynamics in heart failure with preserved ejection fraction. J Am Coll Cardiol. 2016;67(21):2554–2556. doi: 10.1016/j.jacc.2016.03.539. [DOI] [PubMed] [Google Scholar]
- 140.Nanayakkara S, Mak V, Crannitch K, Byrne M, Kaye DM. Extended release oral milrinone, CRD-102, for advanced heart failure. Am J Cardiol. 2018;122(6):1017–1020. doi: 10.1016/j.amjcard.2018.06.009. [DOI] [PubMed] [Google Scholar]
- 141.Hogg K, Swedberg K, McMurray J. Heart failure with preserved left ventricular systolic function: epidemiology, clinical characteristics, and prognosis. J Am Coll Cardiol. 2004;43(3):317–327. doi: 10.1016/j.jacc.2003.07.046. [DOI] [PubMed] [Google Scholar]
- 142.Andersson C, Vasan R. Epidemiology of heart failure with preserved ejection fraction. Heart Fail Clin. 2015;10(3):377–388. doi: 10.1016/j.hfc.2014.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cleland JGF, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27(19):2338–2345. doi: 10.1093/eurheartj/ehl250. [DOI] [PubMed] [Google Scholar]
- 144.Majmudar NG, Robson SC, Ford GA. Effects of the menopause, gender, and estrogen replacement therapy on vascular nitric oxide activity. J Clin Endocrinol Metab. 2000;85(4):1577–1583. doi: 10.1210/jcem.85.4.6530. [DOI] [PubMed] [Google Scholar]
- 145.Wang P, Myers JG, Wu P, Cheewatrakoolpong B, Egan RW, Billah MM. Expression, purification, and characterization of human cAMP-specific phosphodiesterase (PDE4) subtypes A, B, C, and D. Biochem Biophys Res Commun. 1997;234(2):320–324. doi: 10.1006/bbrc.1997.6636. [DOI] [PubMed] [Google Scholar]
- 146.Salanova M, Jin SLC, Conti M. Heterologous expression and purification of recombinant rolipram-sensitive cyclic AMP-specific phosphodiesterases. Methods. 1998;14(1):55–64. doi: 10.1006/meth.1997.0565. [DOI] [PubMed] [Google Scholar]
- 147.Huston E, Lumb S, Russell A, et al. Molecular cloning and transient expression in COS7 cells of a novel human PDE4B cAMP-specific phosphodiesterase, HSPDE4B3. Biochem J. 1997;328(2):549–558. doi: 10.1042/bj3280549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bolger G, Michaeli T, Martins T, et al. A family of human phosphodiesterases homologous to the dunce learning and memory gene product of Drosophila melanogaster are potential targets for antidepressant drugs. Mol Cell Biol. 1993;13(10):6558–6571. doi: 10.1128/mcb.13.10.6558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Beard MB, Olsen AE, Jones RE, Erdogan S, Houslay MD, Bolger GB. UCR1 and UCR2 domains unique to the cAMP-specific phosphodiesterase family form a discrete module via electrostatic interactions. J Biol Chem. 2000;275(14):10349–10358. doi: 10.1074/jbc.275.14.10349. [DOI] [PubMed] [Google Scholar]
- 150.Baillie GS, MacKenzie SJ, McPhee I, Houslay MD. Sub-family selective actions in the ability of Erk2 MAP kinase to phosphorylate and regulate the activity of PDE4 cyclic AMP-specific phosphodiesterases. Br J Pharmacol. 2000;131(4):811–819. doi: 10.1038/sj.bjp.0703636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kostic MM, Erdogan S, Rena G, et al. Altered expression of PDE1 and PDE4 cyclic nucleotide phosphodiesterase isoforms in 7-oxo-prostacyclin-preconditioned rat heart. J Mol Cell Cardiol. 1997;29(11):3135–3146. doi: 10.1006/jmcc.1997.0544. [DOI] [PubMed] [Google Scholar]
- 152.Richter W, Xie M, Scheitrum C, Krall J, Movsesian MA, Conti M. Conserved expression and functions of PDE4 in rodent and human heart. Basic Res Cardiol. 2011;106(2):249–262. doi: 10.1007/s00395-010-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Richter W, Day P, Agrawal R, et al. Signaling from beta1- and beta2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008;27(2):384–393. doi: 10.1038/sj.emboj.7601968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.De Arcangelis V, Liu R, Soto D, Xiang Y. Differential association of phosphodiesterase 4D isoforms with beta2-adrenoceptor in cardiac myocytes. J Biol Chem. 2009;284(49):33824–33832. doi: 10.1074/jbc.M109.020388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leroy J, Richter W, Mika D, et al. Phosphodiesterase 4B in the cardiac L-type Ca2+ channel complex regulates Ca2+ current and protects against ventricular arrhythmias in mice. J Clin Invest. 2011;121(7):2651–2661. doi: 10.1172/JCI44747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lehnart SE, Wehrens XHT, Reiken S, et al. Phosphodiesterase 4D deficiency in the ryanodine-receptor complex promotes heart failure and arrhythmias. Cell. 2005;123(1):25–35. doi: 10.1016/j.cell.2005.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kerfant BG, Zhao D, Lorenzen-Schmidt I, et al. PI3Kgamma is required for PDE4, not PDE3, activity in subcellular microdomains containing the sarcoplasmic reticular calcium ATPase in cardiomyocytes. Circ Res. 2007;101(4):400–408. doi: 10.1161/CIRCRESAHA.107.156422. [DOI] [PubMed] [Google Scholar]
- 158.Beca S, Helli PB, Simpson JA, et al. Phosphodiesterase 4D regulates baseline sarcoplasmic reticulum Ca2+ release and cardiac contractility, independently of L-type Ca2+ current. Circ Res. 2011;109(9):1024–1030. doi: 10.1161/CIRCRESAHA.111.250464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Galindo-Tovar A, Kaumann AJ. Phosphodiesterase-4 blunts inotropism and arrhythmias but not sinoatrial tachycardia of (-)-adrenaline mediated through mouse cardiac beta(1)-adrenoceptors. Br J Pharmacol. 2008;153(4):710–720. doi: 10.1038/sj.bjp.0707631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Christ T, Galindo-Tovar A, Thoms M, Ravens U, Kaumann AJ. Inotropy and L-type Ca2+ current, activated by β1- and β2-adrenoceptors, are differently controlled by phosphodiesterases 3 and 4 in rat heart. Br J Pharmacol. 2009;156(1):62–83. doi: 10.1111/j.1476-5381.2008.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Molina CE, Leroy J, Richter W, et al. Cyclic adenosine monophosphate phosphodiesterase type 4 protects against atrial arrhythmias. J Am Coll Cardiol. 2012;59(24):2182–2190. doi: 10.1016/j.jacc.2012.01.060. [DOI] [PubMed] [Google Scholar]
- 162.Holbrook M, Coker SJ. Effects of zaprinast and rolipram on platelet aggregation and arrhythmias following myocardial ischaemia and reperfusion in anaesthetized rabbits. Br J Pharmacol. 1991;103(4):1973–1979. doi: 10.1111/j.1476-5381.1991.tb12362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Thomas MK, Francis SH, Corbin JD. Substrate- and kinase-directed regulation of phosphorylation of a cGMP-binding phosphodiesterase by cGMP. J Biol Chem. 1990;265(25):14971–14978. [PubMed] [Google Scholar]
- 164.Rybalkin SD, Rybalkina IG, Shimizu-Albergine M, Tang XB, Beavo JA. PDE5 is converted to an activated state upon cGMP binding to the GAF A domain. EMBO J. 2003;22(3):469–478. doi: 10.1093/emboj/cdg051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zoraghi R, Bessay EP, Corbin JD, Francis SH. Structural and functional features in human PDE5A1 regulatory domain that provide for allosteric cGMP binding, dimerization, and regulation. J Biol Chem. 2005;280(12):12051–12063. doi: 10.1074/jbc.M413611200. [DOI] [PubMed] [Google Scholar]
- 166.Corbin JD, Turko IV, Beasley A, Francis SH. Phosphorylation of phosphodiesterase-5 by cyclic nucleotide-dependent protein kinase alters its catalytic and allosteric cGMP-binding activities. Eur J Biochem. 2000;267(9):2760–2767. doi: 10.1046/j.1432-1327.2000.01297.x. [DOI] [PubMed] [Google Scholar]
- 167.Francis SH, Bessay EP, Kotera J, et al. Phosphorylation of isolated human phosphodiesterase-5 regulatory domain induces an apparent conformational change and increases cGMP binding affinity. J Biol Chem. 2002;277(49):47581–47587. doi: 10.1074/jbc.M206088200. [DOI] [PubMed] [Google Scholar]
- 168.Shimizu-Albergine M, Rybalkin SD, Rybalkina IG, et al. Individual cerebellar Purkinje cells express different cGMP phosphodiesterases (PDEs): in vivo phosphorylation of cGMP-specific PDE (PDE5) as an indicator of cGMP-dependent protein kinase (PKG) activation. J Neurosci. 2003;23(16):6452–6459. doi: 10.1523/JNEUROSCI.23-16-06452.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Corbin J, Rannels S, Neal D, et al. Sildenafil citrate does not affect cardiac contractility in human or dog heart. Curr Med Res Opin. 2003;19(8):747–752. doi: 10.1185/030079903125002522. [DOI] [PubMed] [Google Scholar]
- 170.Lukowski R, Rybalkin S, Loga F, Leiss V, Beavo J, Hofmann F. Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci U S A. 2010;107(12):5646–5651. doi: 10.1073/pnas.1001360107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Li EA, Xi W, Han YS, Brozovich FV. Phosphodiesterase expression in the normal and failing heart. Arch Biochem Biophys. 2019;662:160–168. doi: 10.1016/j.abb.2018.12.013. [DOI] [PubMed] [Google Scholar]
- 172.Kotera J, Fujishige K, Imai Y, et al. Genomic origin and transcriptional regulation of two variants of cGMP-binding cGMP-specific phosphodiesterases. Eur J Biochem. 1999;262(3):866–872. doi: 10.1046/j.1432-1327.1999.00450.x. [DOI] [PubMed] [Google Scholar]
- 173.Giordano D, De Stefano ME, Citro G, Modica A, Giorgi M. Expression of cGMP-binding cGMP-specific phosphodiesterase (PDE5) in mouse tissues and cell lines using an antibody against the enzyme amino-terminal domain. Biochim Biophys Acta. 2001;1539(1–2):16–27. doi: 10.1016/s0167-4889(01)00086-6. [DOI] [PubMed] [Google Scholar]
- 174.Campolo F, Zevini A, Cardarelli S, et al. Identification of murine phosphodiesterase 5A isoforms and their functional characterization in HL-1 cardiac cell line. J Cell Physiol. 2018;233(1):325–337. doi: 10.1002/jcp.25880. [DOI] [PubMed] [Google Scholar]
- 175.Takimoto E, Champion HC, Belardi D, et al. cGMP catabolism by phosphodiesterase 5A regulates cardiac adrenergic stimulation by NOS3-dependent mechanism. Circ Res. 2005;96(1):100–109. doi: 10.1161/01.RES.0000152262.22968.72. [DOI] [PubMed] [Google Scholar]
- 176.Zhang M, Koitabashi N, Nagayama T, et al. Expression, activity, and pro-hypertrophic effects of PDE5A in cardiac myocytes. Cell Signal. 2008;20(12):2231–2236. doi: 10.1016/j.cellsig.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Borlaug BA, Melenovsky V, Marhin T, Fitzgerald P, Kass DA. Sildenafil inhibits beta-adrenergic-stimulated cardiac contractility in humans. Circulation. 2005;112(17):2642–2649. doi: 10.1161/CIRCULATIONAHA.105.540500. [DOI] [PubMed] [Google Scholar]
- 178.Takimoto E, Belardi D, Tocchetti CG, et al. Compartmentalization of cardiac B-adrenergic inotropy modulation by phosphodiesterase type 5. Circulation. 2007;115(16):2159–2167. doi: 10.1161/CIRCULATIONAHA.106.643536. [DOI] [PubMed] [Google Scholar]
- 179.Nagayama T, Zhang M, Hsu S, Takimoto E, Kass DA. Sustained soluble guanylate cyclase stimulation offsets nitric-oxide synthase inhibition to restore acute cardiac modulation by sildenafil. J Pharmacol Exp Ther. 2008;326(2):380–387. doi: 10.1124/jpet.108.137422. [DOI] [PubMed] [Google Scholar]
- 180.Isidori AM, Cornacchione M, Barbagallo F, et al. Inhibition of type 5 phosphodiesterase counteracts β2-adrenergic signalling in beating cardiomyocytes. Cardiovasc Res. 2015;106(3):408–420. doi: 10.1093/cvr/cvv123. [DOI] [PubMed] [Google Scholar]
- 181.Gong W, Yan M, Chen J, Chaugai S, Chen C, Wang D. Chronic inhibition of cyclic guanosine monophosphate-specific phosphodiesterase 5 prevented cardiac fibrosis through inhibition of transforming growth factor β-induced Smad signaling. Front Med. 2014;8(4):445–455. doi: 10.1007/s11684-014-0378-3. [DOI] [PubMed] [Google Scholar]
- 182.Gebska MA, Stevenson BK, Hemnes AR, et al. Phosphodiesterase-5A (PDE5A) is localized to the endothelial caveolae and modulates NOS3 activity. Cardiovasc Res. 2011;90(2):353–363. doi: 10.1093/cvr/cvq410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Lugnier C, Schoeffter P, Le Bec A, Strouthou E, Stoclet J. Selective inhibition of cyclic nucleotide phosphodiesterases of human, bovine and rat aorta. Biochem Pharmacol. 1986;35(10):1743–1751. doi: 10.1016/0006-2952(86)90333-3. [DOI] [PubMed] [Google Scholar]
- 184.Pokreisz P, Vandenwijngaert S, Bito V, et al. Ventricular phosphodiesterase-5 expression is increased in patients with advanced heart failure and contributes to adverse ventricular remodeling after myocardial infarction in mice. Circulation. 2009;119(3):408–416. doi: 10.1161/CIRCULATIONAHA.108.822072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Zhang M, Takimoto E, Hsu S, et al. Myocardial remodeling is controlled by myocyte-targeted gene regulation of phosphodiesterase type 5. J Am Coll Cardiol. 2010;56(24):2021–2030. doi: 10.1016/j.jacc.2010.08.612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Nagendran J, Archer SL, Soliman D, et al. Phosphodiesterase type 5 is highly expressed in the hypertrophied human right ventricle, and acute inhibition of phosphodiesterase type 5 improves contractility. Circulation. 2007;116(3):238–248. doi: 10.1161/CIRCULATIONAHA.106.655266. [DOI] [PubMed] [Google Scholar]
- 187.Shan X, Quaile MP, Monk JK, French B, Cappola TP, Margulies KB. Differential expression of PDE5 in failing and nonfailing human myocardium. Circ Heart Fail. 2012;5(1):79–86. doi: 10.1161/CIRCHEARTFAILURE.111.961706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Garcia AM, Nakano SJ, Karimpour-Fard A, et al. Phosphodiesterase-5 is elevated in failing single ventricle myocardium and affects cardiomyocyte remodeling in vitro. Circ Heart Fail. 2018;11(9):e004571. doi: 10.1161/CIRCHEARTFAILURE.117.004571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Salloum FN, Abbate A, Das A, et al. Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol. 2008;294(3):1398–1406. doi: 10.1152/ajpheart.91438.2007. [DOI] [PubMed] [Google Scholar]
- 190.Das A, Xi L, Kukreja RC. Protein kinase G-dependent cardioprotective mechanism of phosphodiesterase-5 inhibition involves phosphorylation of ERK and GSK3beta. J Biol Chem. 2008;283(43):29572–29585. doi: 10.1074/jbc.M801547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ockaili R, Salloum F, Hawkins J, Kukreja R. Sildenafil (Viagra) induces powerful cardioprotective effect via opening of mitochondrial K(ATP) channels in rabbits. Am J Physiol Heart Circ Physiol. 2002;283(3):H1263–H1269. doi: 10.1152/ajpheart.00324.2002. [DOI] [PubMed] [Google Scholar]
- 192.Salloum FN, Takenoshita Y, Ockaili RA, et al. Sildenafil and vardenafil but not nitroglycerin limit myocardial infarction through opening of mitochondrial K(ATP) channels when administered at reperfusion following ischemia in rabbits. J Mol Cell Cardiol. 2007;42(2):453–458. doi: 10.1016/j.yjmcc.2006.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Salloum FN, Chau VQ, Hoke NN, et al. Phosphodiesterase-5 inhibitor, tadalafil, protects against myocardial ischemia/reperfusion through protein-kinase g-dependent generation of hydrogen sulfide. Circulation. 2009;120(11):S31–S36. doi: 10.1161/CIRCULATIONAHA.108.843979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ranek MJ, Terpstra EJM, Li J, Kass DA, Wang X. Protein kinase G positively regulates proteasome-mediated degradation of misfolded proteins. Circulation. 2013;128(4):365–376. doi: 10.1161/CIRCULATIONAHA.113.001971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mátyás C, Németh BT, Oláh A, et al. Prevention of the development of heart failure with preserved ejection fraction by the phosphodiesterase-5A inhibitor vardenafil in rats with type 2 diabetes. Eur J Heart Fail. 2017;19(3):326–336. doi: 10.1002/ejhf.711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Koka S, Das A, Zhu SG, Durrant D, Xi L, Kukreja RC. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther. 2010;334(3):1023–1030. doi: 10.1124/jpet.110.170191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Prysyazhna O, Burgoyne JR, Scotcher J, Grover S, Kass D, Eaton P. Phosphodiesterase 5 inhibition limits doxorubicin-induced heart failure by attenuating protein kinase G Iα oxidation. J Biol Chem. 2016;291(33):17427–17436. doi: 10.1074/jbc.M116.724070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Lawless M, Caldwell JL, Radcliffe EJ, et al. Phosphodiesterase 5 inhibition improves contractile function and restores transverse tubule loss and catecholamine responsiveness in heart failure. Sci Rep. 2019;9(1):1–17. doi: 10.1038/s41598-019-42592-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Takimoto E, Champion HC, Li M, et al. Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med. 2005;11(2):214–222. doi: 10.1038/nm1175. [DOI] [PubMed] [Google Scholar]
- 200.Koitabashi N, Aiba T, Hesketh GG, et al. Cyclic GMP/PKG-dependent inhibition of TRPC6 channel activity and expression negatively regulates cardiomyocyte NFAT activation. Novel mechanism of cardiac stress modulation by PDE5 inhibition. J Mol Cell Cardiol. 2010;48(4):713–724. doi: 10.1016/j.yjmcc.2009.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nishida M, Watanabe K, Sato Y, et al. Phosphorylation of TRPC6 channels at Thr69 is required for anti-hypertrophic effects of phosphodiesterase 5 inhibition. J Biol Chem. 2010;285(17):13244–13253. doi: 10.1074/jbc.M109.074104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Takimoto E, Hsu S, Ketner EA, et al. Regulator of G protein signaling 2 mediates cardiac compensation to pressure overload and antihypertrophic effects of PDE5 inhibition in mice. J Clin Invest. 2009;119(2):408–420. doi: 10.1172/JCI35620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Sasaki H, Nagayama T, Blanton R, et al. PDE5 inhibitor efficacy is estrogen dependent in female heart disease. J Clin Invest. 2014;124(6):2464–2471. doi: 10.1172/JCI70731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Frantz S, Klaiber M, Baba H, et al. Stress-dependent dilated cardiomyopathy in mice with cardiomyocyte-restricted inactivation of cyclic GMP-dependent protein kinase I. Eur Heart J. 2013;34(16):1233–1244. doi: 10.1093/eurheartj/ehr445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Patrucco E, Domes K, Sbroggió M, et al. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc Natl Acad Sci U S A. 2014;111(35):12925–12929. doi: 10.1073/pnas.1414364111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Blanton RM, Takimoto E, Lane AM, et al. Protein kinase g iα inhibits pressure overload-induced cardiac remodeling and is required for the cardioprotective effect of sildenafil in vivo. J Am Heart Assoc. 2012;1(5):1–10. doi: 10.1161/JAHA.112.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Redondo J, Bishop JE, Wilkins MR. Effect of atrial natriuretic peptide and cyclic GMP phosphodiesterase inhibition on collagen synthesis by adult cardiac fibroblasts. Br J Pharmacol. 1998;124(7):1455–1462. doi: 10.1038/sj.bjp.0701994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Guazzi M, Samaja M, Arena R, Vicenzi M, Guazzi MD. Long-term use of sildenafil in the therapeutic management of heart failure. J Am Coll Cardiol. 2007;50(22):2136–2144. doi: 10.1016/j.jacc.2007.07.078. [DOI] [PubMed] [Google Scholar]
- 209.Behling A, Rohde LE, Colombo FC, Goldraich LA, Stein R, Clausell N. Effects of 5′-phosphodiesterase four-week long inhibition with sildenafil in patients with chronic heart failure: a double-blind, placebo-controlled clinical trial. J Card Fail. 2008;14(3):189–197. doi: 10.1016/j.cardfail.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 210.Lewis GD, Shah R, Shahzad K, et al. Sildenafil improves exercise capacity and quality of life in patients with systolic heart failure and secondary pulmonary hypertension. Circulation. 2007;116(14):1555–1562. doi: 10.1161/CIRCULATIONAHA.107.716373. [DOI] [PubMed] [Google Scholar]
- 211.Wilkins MR, Paul GA, Strange JW, et al. Sildenafil versus Endothelin Receptor Antagonist for Pulmonary Hypertension (SERAPH) study. Am J Respir Crit Care Med. 2005;171(11):1292–1297. doi: 10.1164/rccm.200410-1411OC. [DOI] [PubMed] [Google Scholar]
- 212.Guazzi M, Vicenzi M, Arena R, Guazzi MD. PDE5 inhibition with sildenafil improves left ventricular diastolic function, cardiac geometry, and clinical status in patients with stable systolic heart failure: results of a 1-year, prospective, randomized, placebo-controlled study. Circ Heart Fail. 2011;4(1):8–17. doi: 10.1161/CIRCHEARTFAILURE.110.944694. [DOI] [PubMed] [Google Scholar]
- 213.Kim KH, Kim HK, Hwang IC, et al. PDE 5 inhibition with udenafil improves left ventricular systolic/diastolic functions and exercise capacity in patients with chronic heart failure with reduced ejection fraction; a 12-week, randomized, double-blind, placebo-controlled trial. Am Heart J. 2015;169(6):813–822. doi: 10.1016/j.ahj.2015.03.018. [DOI] [PubMed] [Google Scholar]
- 214.Fernandes AMS, Andrade AC, Barroso ND, et al. The immediate effect of sildenafil on right ventricular function in patients with heart failure measured by cardiac magnetic resonance: a randomized control trial. PLoS One. 2015;10(3):1–10. doi: 10.1371/journal.pone.0119623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Guazzi M, Vicenzi M, Arena R, Guazzi MD. Pulmonary hypertension in heart failure with preserved ejection fraction: a target of phosphodiesterase-5 inhibition in a 1-year study. Circulation. 2011;124(2):164–174. doi: 10.1161/CIRCULATIONAHA.110.983866. [DOI] [PubMed] [Google Scholar]
- 216.Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309(12):1268–1277. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Andersen M, Ersbøll M, Axelsson A, et al. Sildenafil and diastolic dysfunction after acute myocardial infarction in patients with preserved ejection fraction: the Sildenafil and Diastolic Dysfunction after Acute Myocardial Infarction (SIDAMI) trial. Circulation. 2013;127(11):1200–1208. doi: 10.1161/CIRCULATIONAHA.112.000056. [DOI] [PubMed] [Google Scholar]
- 218.Hoendermis ES, Liu LCY, Hummel YM, et al. Effects of sildenafil on invasive haemodynamics and exercise capacity in heart failure patients with preserved ejection fraction and pulmonary hypertension: a randomized controlled trial. Eur Heart J. 2015;36(38):2565–2573. doi: 10.1093/eurheartj/ehv336. [DOI] [PubMed] [Google Scholar]
- 219.Liu L, Hummel Y, van der Meer P, et al. Effects of sildenafil on cardiac structure and function, cardiopulmonary exercise testing and health-related quality of life measures in heart failure patients with preserved ejection fraction and pulmonary hypertension. Eur J Heart Fail. 2017;19(1):116–125. doi: 10.1002/ejhf.662. [DOI] [PubMed] [Google Scholar]
- 220.Taylor RE, Shows KH, Zhao Y, Pittler SJ. A PDE6A promoter fragment directs transcription predominantly in the photoreceptor. Biochem Biophys Res Commun. 2001;282(2):543–547. doi: 10.1006/bbrc.2001.4605. [DOI] [PubMed] [Google Scholar]
- 221.Fawcett L, Baxendale R, Stacey P, et al. Molecular cloning and characterization of a distinct human phosphodiesterase gene family: PDE11A. Proc Natl Acad Sci U S A. 2000;97(7):3702–3707. doi: 10.1073/pnas.050585197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Hetman JM, Soderling SH, Glavas NA, Beavo JA. Cloning and characterization of PDE7B, a cAMP-specific phosphodiesterase. Proc Natl Acad Sci U S A. 2000;97(1):472–476. doi: 10.1073/pnas.97.1.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Patrucco E, Albergine MS, Santana LF, Beavo JA. Phosphodiesterase 8A (PDE8A) regulates excitation-contraction coupling in ventricular myocytes. J Mol Cell Cardiol. 2010;49(2):330–333. doi: 10.1016/j.yjmcc.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Tian X, Vroom C, Ghofrani HA, et al. Phosphodiesterase 10A upregulation contributes to pulmonary vascular remodeling. PLoS One. 2011;6(4):1–12. doi: 10.1371/journal.pone.0018136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Lee DI, Zhu G, Sasaki T, et al. Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature. 2015;519(7544):472–476. doi: 10.1038/nature14332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Zhang L, Bouadjel K, Manoury B, Vandecasteele G, Fischmeister R, Leblais V. Cyclic nucleotide signalling compartmentation by PDEs in cultured vascular smooth muscle cells. Br J Pharmacol. 2019;176(11):1780–1792. doi: 10.1111/bph.14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Scott NJA, Rademaker MT, Charles CJ, Espiner EA, Richards AM. Hemodynamic, hormonal, and renal actions of phosphodiesterase-9 inhibition in experimental heart failure. J Am Coll Cardiol. 2019;74(7):889–901. doi: 10.1016/j.jacc.2019.05.067. [DOI] [PubMed] [Google Scholar]
- 228.Oliver PM, Fox JE, Kim R, et al. Hypertension, cardiac hypertrophy, and sudden death in mice lacking natriuretic peptide receptor A. Proc Natl Acad Sci U S A. 1997;94(26):14730–14735. doi: 10.1073/pnas.94.26.14730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Kuhn M, Holtwick R, Baba HA, Perriard JC, Schmitz W. Progressive cardiac hypertrophy and dysfunction in atrial natriuretic peptide receptor (GC-A) deficient mice. Heart. 2002;87(4):368–374. doi: 10.1136/heart.87.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Tamura N, Ogawa Y, Chusho H, et al. Cardiac fibrosis in mice lacking brain natriuretic peptide. Proc Natl Acad Sci U S A. 2000;97(8):4239–4244. doi: 10.1073/pnas.070371497. [DOI] [PMC free article] [PubMed] [Google Scholar]