

Abstract
Viruses maximize their genetic coding capacity through a variety of biochemical mechanisms, including programmed ribosomal frameshifting (PRF), which facilitates the production of multiple proteins from a single mRNA transcript. PRF is typically stimulated by structural elements within the mRNA that generate mechanical tension between the transcript and ribosome. However, in this work, we show that the forces generated by the cotranslational folding of the nascent polypeptide chain can also enhance PRF. Using an array of biochemical, cellular, and computational techniques, we first demonstrate that the Sindbis virus structural polyprotein forms two competing topological isomers during its biosynthesis at the ribosome-translocon complex. We then show that the formation of one of these topological isomers is linked to PRF. Coarse-grained molecular dynamics simulations reveal that the translocon-mediated membrane integration of a transmembrane domain upstream from the ribosomal slip site generates a force on the nascent polypeptide chain that scales with observed frameshifting. Together, our results indicate that cotranslational folding of this viral protein generates a tension that stimulates PRF. To our knowledge, this constitutes the first example in which the conformational state of the nascent polypeptide chain has been linked to PRF. These findings raise the possibility that, in addition to RNA-mediated translational recoding, a variety of cotranslational folding or binding events may also stimulate PRF.
Keywords: protein folding, membrane protein, transmembrane domain, translocation, translation control, ribosome, alphavirus, programmed ribosomal frameshifting, topology, Sindbis virus, translocon, membrane protein folding, cotranslational folding
Introduction
Viruses have evolved numerous mechanisms to exploit the host machinery to increase the coding capacity of their highly constrained genomes. There are at least 27 viral genera that utilize programmed ribosomal frameshifting (PRF)4 to produce multiple proteins from a single transcript (RRID:SCR_006563). PRF is genetically encoded and minimally requires a portion of the transcript that contains a repetitive “slippery” heptanucleotide sequence (slip site) followed by a region that forms stimulatory RNA secondary structures (an ensemble of stem loops and/or pseudoknots) (1, 2). A collision between the translating ribosome and the stimulatory secondary structure increases the kinetic barrier to translocation, which causes the ribosome to dwell on the slip site (3–7). During this pause, the t-RNA that is annealed within the ribosomal P-site (and most often also the t-RNA in the A-site) (8) begins to sample alternative base-pairing interactions that shift the reading frame of the ribosome (9). Based on these mechanistic considerations, PRF is typically believed to be mediated at the level of RNA structure. Nevertheless, recent reports have also found that the efficiency of PRF can be tuned by a variety of regulatory proteins and/or miRNA (10–12).
PRF is utilized to temporally and stoichiometrically regulate protein production during viral replication and assembly. For instance, the alphavirus structural proteins are most often produced from a single polyprotein that is cleaved into the capsid (CP), E3, E2, 6K, and E1 proteins (Fig. 1A) (13). The E2 and E1 proteins are membrane glycoproteins that heterodimerize early in the assembly pathway. These dimeric units then form trimeric spike complexes, traffic to the plasma membrane, and initiate viral budding (14–16). A programmed ribosomal frameshift into the −1 reading frame (−1PRF) occurs with 10–15% efficiency during the translation of the 6K protein and gives rise to a secondary form of the polyprotein. This frameshifted polyprotein contains the TransFrame (TF) protein (13, 17), a known virulence factor (18–21), in place of the 6K and E1 proteins (Fig. 1B). Because −1PRF precludes the translation of E1, the efficiency of ribosomal frameshifting (1–48% in alphaviruses) (22) influences the stoichiometric ratio of the E1 and E2 glycoproteins and the net accumulation of spike complexes. Current evidence suggests that −1PRF is stimulated by a canonical poly-U slip site and a downstream RNA hairpin (23). However, an effort to map the stimulatory RNA structures within alphavirus polyproteins revealed that deletions within the predicted hairpin region are capable of reducing the efficiency of −1PRF but appear to be insufficient to knock out frameshifting completely (22). This observation suggests that there may be multiple regulatory mechanisms that mediate −1PRF within the alphavirus structural polyprotein.
Figure 1.

Structure and topological properties of the alphavirus polyprotein. A, a cartoon depicts the relative size and orientation of proteins within the major form of the alphavirus structural polyprotein. B, a cartoon depicts the relative size and orientation of proteins within the frameshifted form of the alphavirus structural polyprotein. C, a portion of the SINV structural polyprotein spanning the E2, 6K, and E1 proteins was scanned with the ΔG predictor using a 23-residue window (33). The predicted free energy difference associated with the cotranslational membrane integration of every possible 23-residue segment within the major form (black) and frameshifted form (orange) of the structural polyprotein is plotted as a function of the position of its central residue. The position of each predicted TM domain is indicated in blue. D, a cartoon depicts the manner in which the topological preferences of the guest TM domain (blue) influence the glycosylation state of the chimeric Lep protein. The glycosylation machinery is on the interior of the vesicle (the microsomal lumen). Membrane integration of the guest domain results in the production of a singly glycosylated product (bottom). The misintegration of the guest domain results in the production of a doubly glycosylated product (top). E, chimeric Lep constructs bearing putative TM domains from the SINV structural polyprotein were produced by in vitro translation in the presence of canine rough microsomes and analyzed by SDS-PAGE. A representative gel reveals the relative abundance of singly (G1) and doubly (G2) glycosylated translation products for each construct. Control reactions containing no RNA (no protein) and no rough microsomes (untargeted protein) are shown for the sake of comparison. These trends were consistently observed across five independent replicates.
−1PRF occurs during synthesis and processing of the nascent alphavirus structural polyprotein at the endoplasmic reticulum (ER) membrane. Following autoproteolytic cleavage of CP in the cytosol, a signal peptide at the N terminus of the E3 protein directs the nascent polyprotein to the ER lumen, where processing of the downstream proteins occurs. Localization of these segments within the lumen is essential to ensure that the E3, E2, and E1 ectodomains form their native disulfides and undergo glycosylation (15, 16, 24). Post-translational modifications are also critical for TF, which must be palmitoylated to reach the plasma membrane and incorporate into the viral envelope (25). The palmitoylated cysteines in TF are positioned near the edge of a putative transmembrane (TM) domain that is found in both TF and 6K (25, 26). Although these residues are present in both proteins, they are only palmitoylated in the context of the frameshifted polyprotein (25). Considering that palmitoylation only occurs on the cytosolic face of cellular membranes (27), the distinct modification state of the two forms of the polyprotein is therefore suggestive of an underlying difference in their topologies. In this study, we set out to gain insight into the interplay between −1PRF and the topology of the structural polyprotein. We first mapped the topology of the Sindbis virus (SINV) structural polyprotein. Our results demonstrate that the structural polyprotein forms two topological isomers. The predominant topology features two TM domains upstream of the −1PRF site, and its formation coincides with production of the 6K protein. Alternatively, the minor topology contains an additional TM domain upstream from the −1PRF site that is linked to the production of TF. Using protein engineering in conjunction with coarse-grained molecular dynamics (CGMD) simulations, we demonstrate that the efficiency of −1PRF depends upon the force generated by the translocon-mediated membrane integration of the extra TM domain within the minor topomer. Together, our observations highlight novel connections between the cotranslational folding, biosynthesis, and processing of the alphavirus structural polyprotein. Moreover, our findings reveal a novel mechanism that regulates the overall efficiency of −1PRF.
Results and discussion
Topological properties of the alphavirus structural polyprotein
The current model of the alphavirus structural polyprotein suggests that the E2 and 6K proteins each contain two TM domains (13, 28). However, there are two caveats to this model. First, cryo-EM structures reveal that the E2 protein only contains a single TM domain in the context of the viral envelope (29, 30). Although it has been speculated that a second TM domain within E2 is somehow extruded from the membrane during processing, the marginal hydrophobicity of this segment also raises the possibility that it may fail to undergo translocon-mediated membrane integration in the first place. Second, the hydrophobic portion of the SINV 6K protein is only 35 residues in length, which is quite short for a segment containing two putative TM domains and a loop. These ambiguous topological signals suggest that this portion of the polyprotein is frustrated and could potentially form multiple topological isomers (31), as has been suggested for the coronavirus E protein (32).
To survey the topological preferences of the E2-6K region, we scanned its sequence using a knowledge-based algorithm that predicts the energetics associated with the transfer of polypeptide segments from the translocon to the ER membrane (ΔG predictor) (33). Energetic predictions suggest that only the regions corresponding to the first hydrophobic segments within the E2 (TM1) and 6K (TM3) proteins are sufficiently hydrophobic to undergo robust membrane integration (ΔG < 0 kcal/mol; Fig. 1C). In contrast, the translocon-mediated membrane integration of the second hydrophobic segment within E2 (TM2) is predicted to be inefficient (Fig. 1C). To test these predictions, we measured the translocon-mediated membrane integration of each putative TM domain using a glycosylation-based translocation assay (34). Briefly, the sequences of each individual putative TM domain were cloned into a chimeric leader peptidase (Lep) reporter protein (34). Membrane integration of the putative TM segment (blue helix in Fig. 1D) results in the modification of only a single glycosylation site in Lep, whereas the passage of the segment into the lumen results in the modification of two glycosylation sites (Fig. 1D). Chimeric Lep proteins were produced by in vitro translation in the presence of canine rough microsomes, which contain native ER membranes and translocons. Consistent with predictions, Lep proteins containing TM1 and 3 acquire a single glycosyl modification, which suggests that these segments undergo robust translocon-mediated membrane integration (Fig. 1E). In contrast, the translocon-mediated membrane integration of the second putative TM domain of E2 (TM2) is significantly less efficient (Fig. 1E). These observations suggest that the E2 and 6K proteins are each likely to contain a single TM domain (TM1 and -3; Fig. 2A). It should also be noted that −1PRF only modifies the sequence of the loops downstream from these TM domains (Fig. 1C, orange line) and is therefore unlikely to impact their topological preferences.
Figure 2.
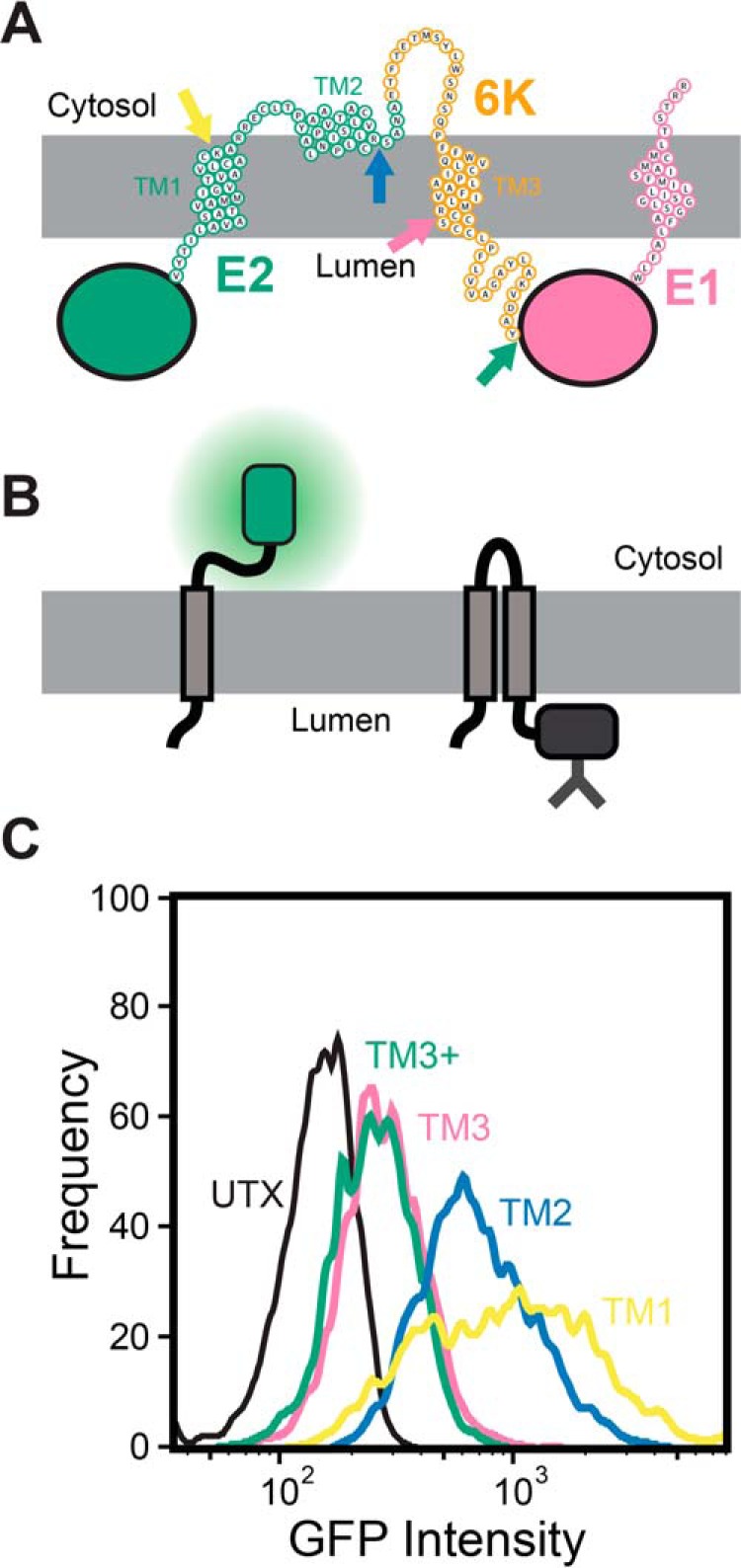
Topological properties of the major form of the SINV structural polyprotein. A, a cartoon depicts a putative topological model of the most abundant topology of the structural polyprotein that is consistent with computational and biochemical data. The positions at which gGFP reporter domains were fused to determine the cellular compartmentalization of the C-terminal portion of the segments corresponding to TM1 (yellow), TM2 (blue), TM3 (pink), and the C-terminal edge of the hydrophobic portion of 6K (TM3+, green) are indicated with arrows. B, a cartoon depicts the manner in which the cellular compartmentalization of the gGFP reporter domain alters its fluorescence. Topological signals that direct the gGFP domain into the cytosol will generate a fluorescent gGFP (left), whereas topological signals that direct the gGFP domain into the lumen (right) generate a glycosylated, nonfluorescent fusion domain. C, reporter constructs bearing a gGFP fusion downstream from TM1 (yellow), TM2 (blue), TM3 (pink), and the hydrophobic portion of the 6K protein (TM3+, green) were expressed in HEK293T cells, and cellular fluorescence intensities were analyzed by flow cytometry. A histogram from a representative trial depicts the gGFP intensities associated with 3,000 transfected cells expressing each reporter construct at a consistent expression level, as judged by the intensity of a bicistronic expression reporter.
Based on the computational and biochemical results in Fig. 1, we generated a topological model of the SINV structural polyprotein (Fig. 2A). This model correctly places the E2 and E1 ectodomains within the ER lumen and places the two palmitoylated cysteine residues in E2 (Cys-716 and Cys-718) within the cytosol (35, 36). To probe the topological preferences of the SINV polyprotein in the cell, we produced and characterized a series of reporter constructs that begin with the E3 protein and end at the C-terminal edge of each of the three putative TM domains within E2 and 6K (Fig. 2A and Fig. S1). Each of these fragments was genetically fused to a C-terminal cassette containing a short GS linker and glycosylatable GFP (gGFP) gene, which contains two glycosylation sites within the core of the enhanced GFP (eGFP) protein (37). Topological signals that direct the gGFP protein into the cytosol will produce a fluorescent gGFP, whereas the glycosylation of gGFP within the ER lumen renders the protein nonfluorescent (Fig. 2B). Each construct was then expressed in HEK293T cells, and flow cytometry was used to quantify the fluorescence intensity of the gGFP reporter at a consistent expression level, as judged by the intensity of a bicistronic reporter protein (Fig. S2). Expression of the reporter constructs containing gGFP downstream from TM1 and -2 (see Fig. 2A) generates fluorescent gGFP (Fig. 2C), which suggests that the C termini of these TM domains reside within the cytosol. In contrast, the reporter construct with gGFP downstream from TM3 (after Arg-785) exhibits an attenuated GFP signal at an equivalent expression level (Fig. 2C), which suggests that the gGFP fused to the C terminus of TM3 is projected into the ER lumen. Placement of the gGFP after the full stretch of hydrophobic amino acids in the 6K protein (after Tyr-807; see Fig. 2A) also results in an attenuated gGFP signal (TM3+ in Fig. 2C), which confirms that the full stretch of hydrophobic residues near TM3 only spans the membrane once. Thus, results from this cellular reporter assay (Fig. 2C) are consistent with predictions (Fig. 1C), in vitro translation data (Fig. 1E), and the model shown in Fig. 2A. These observations together confirm that the E1 and 6K proteins each contain a single TM domain in the most abundant form of the polyprotein.
Link between topology and −1 programmed ribosomal frameshifting
The topological properties of the structural polyprotein described above have implications for the manner in which it is processed at the ER membrane. Our model suggests the cluster of unmodified cysteines in the 6K protein (Cys-786, Cys-787, Cys-789, and Cys-790) reside at a C-terminal portion of TM3 that is projected into the ER lumen (Fig. 3A) and is therefore inaccessible to palmitoylating enzymes. However, these same residues are palmitoylated in the TF protein (25), which suggests that the orientation of TM3 must become inverted upon frameshifting to expose them to the cytosolic leaflet. Such an inversion could potentially occur as a consequence of the membrane integration of TM2 (Fig. 3B), which exhibits a weak propensity to undergo translocon-mediated membrane integration (Fig. 1E). Furthermore, the efficiency associated with the translocon-mediated membrane integration of TM2 (∼20%; Fig. 1E) is comparable with the frequency of −1PRF in the SINV polyprotein (∼16%) (22). Taken together, these observations potentially suggest a connection between the formation of a secondary topomer and −1PRF.
Figure 3.

Putative model for the interplay between translocon-mediated membrane integration and ribosomal frameshifting. A cartoon depicts the putative manner in which cotranslational folding of the nascent structural polyprotein is linked to −1PRF. A, a cartoon depicts the topological properties of the major form of the nascent structural polyprotein. TM2 is too polar to robustly partition into the membrane during translation (left). As a result, the E2 protein only contains a single TM domain in the context of the major form of the structural polyprotein (right). Cysteine residues that are conditionally palmitoylated in the frameshifted form of the polyprotein are shown in blue. B, a cartoon depicts the topological properties of the frameshifted form of the nascent structural polyprotein. TM2 is hydrophobic enough to occasionally partition into the membrane during translation (left), which imposes a tension on the ribosome that stimulates −1PRF. As a result, the E2 protein contains two TM domains in the context of the frameshifted form of the structural polyprotein (right). Cysteine residues that are conditionally palmitoylated in the frameshifted form of the polyprotein are shown in blue.
Based on these observations, we hypothesize that the translocon-mediated membrane integration of TM2 is mechanistically linked to −1PRF and the translation of the TF protein. Our model suggests mutations that alter the translocon-mediated membrane integration of TM2 should have a direct impact on −1PRF (Fig. 3). To test this hypothesis, we assessed whether mutations that alter the hydrophobicity of TM2 also influence −1PRF. We designed two double mutants that alter the hydrophobicity of TM2 by introducing hydrophobic residues at native polar residues, and vice versa. Both energetic predictions and in vitro translation measurements suggest the introduction of two nonnative leucine residues into TM2 (T738L/S739L, LL mutant) enhances the translocon-mediated membrane integration of TM2 (predicted ΔΔG = −1.7 kcal/mol; Fig. S3). In contrast, the introduction of two glutamate residues into TM2 (V735E/I736E, EE mutant) is predicted to increase its transfer free energy by 3.3 kcal/mol, which should significantly reduce its membrane integration efficiency (Fig. S3A). The effects of the EE substitutions appear to be subtle in the context of the Lep protein (Fig. S3B), although this likely reflects the limited dynamic range of this translocation assay (34, 38). Nevertheless, these results clearly show that the LL and EE mutations alter the translocon-mediated membrane integration of TM2.
To determine whether the cotranslational membrane integration of TM2 impacts translational recoding, we measured the effects of these substitutions on ribosomal frameshifting. PRF is most commonly measured using Dual-Luciferase reporters, which fuse luciferase domains to the 5′ (Renilla luciferase, 0-frame) and 3′ (firefly luciferase, −1 frame) of the gene of interest. The activity of firefly luciferase serves as a reporter for −1PRF and is normalized relative to translational efficiency based on the activity of Renilla luciferase. Current versions of these reporter constructs contain self-cleaving 2A segments that release these luciferase domains from the polypeptide of interest (39). Whereas the 2A elements are likely to somewhat efficiently release each fusion domain at some point during translation, the introduction of a soluble N-terminal domain could potentially compromise the fidelity of SRP-mediated targeting of the nascent chain to the translocon. To preserve the integrity of the signal peptide, we generated a series of reporter constructs in which translation begins at the native E3 signal peptide and continues until the ribosome reaches a fluorescent mKate fusion domain that is encoded in the −1 reading frame downstream from the PRF site (Fig. S4). To control for variations in transfection efficiency at the single-cell level, we included a downstream IRES cassette that drives the bicistronic expression of GFP from the reporter transcript. Reporter constructs encoding TM2 variants of the polyprotein were expressed in HEK293T cells, and cellular mKate intensities were quantitatively compared across cells within a discrete range of IRES-GFP intensities by flow cytometry (Fig. S5). The average mKate intensity among cells expressing a reporter construct bearing mutations that knock out the native ribosomal slip site (UUUUUUA → GUUCCUA, SSKO) is 79 ± 5% (n = 3, mean intensity change ± S.D.) lower than that among cells expressing the WT form of the reporter (Fig. 4A), which confirms that mKate intensities reflect the efficiency of −1PRF. The EE substitutions in TM2 decrease mKate intensity by 61 ± 16% relative to WT (n = 3, mean intensity change ± S.D.; Fig. 4A). In contrast, the LL substitutions increase the mKate intensity by 30 ± 11% (n = 3, mean intensity change ± S.D.; Fig. 4A). Differences in the distributions of cellular intensities were found to be both reproducible and statistically significant (n = 3, Mann–Whitney U test, α = 0.001). It should be emphasized that each of these reporters contains both the native slip site and stem loop regions and that these mutations do not alter their sequences. Thus, these findings demonstrate that −1PRF is sensitive to mutations that impact the membrane integration efficiency of TM2. Together, biochemical evidence suggests that TM2 is inefficiently recognized by the translocon (Fig. 1E), and cellular topology reporters suggest that this segment is most often localized within the cytosol (Fig. 2). Nevertheless, mutagenesis reveals that the propensity of the nascent chain to form a secondary topomer is positively correlated with −1PRF. These results are therefore suggestive of a mechanistic link between topogenesis and −1PRF.
Figure 4.
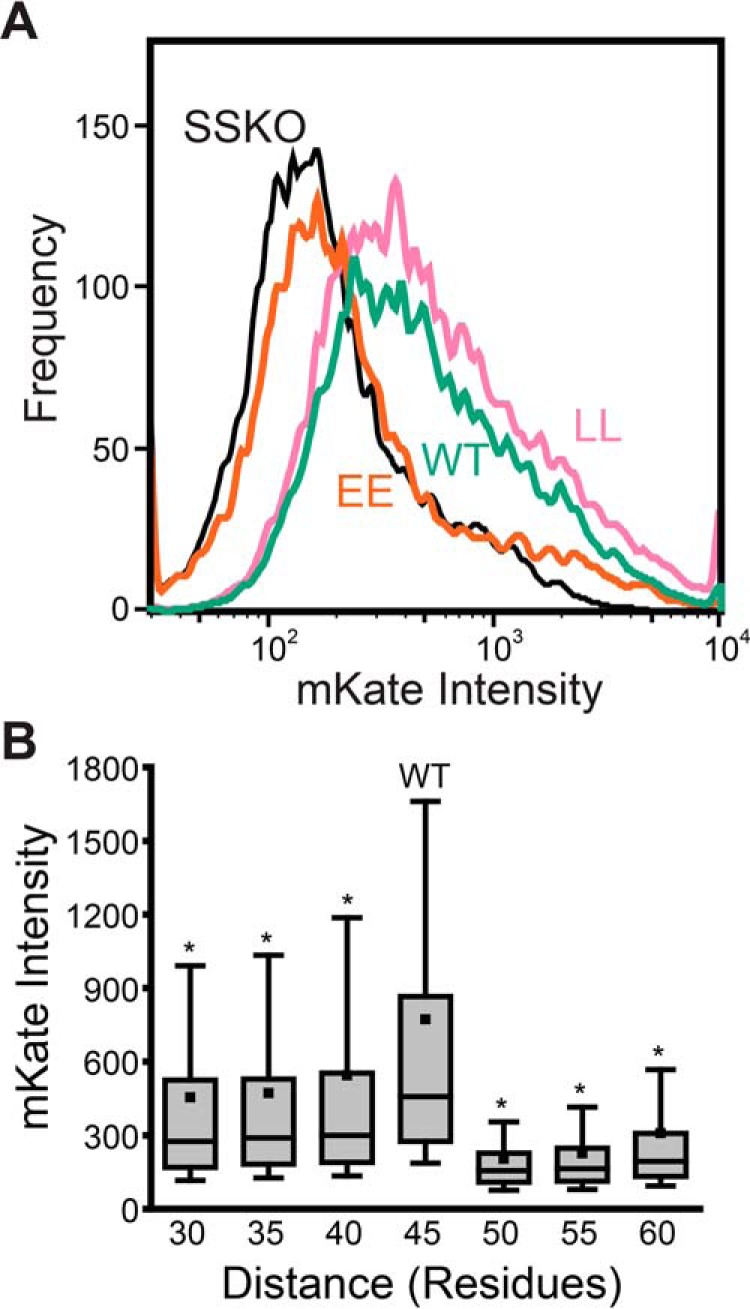
Influence of sequence modifications on −1 programmed ribosomal frameshifting. A fragment of the SINV structural polyprotein containing an mKate fused in the −1 reading frame downstream from the ribosomal slip site was used to compare the effects of sequence modifications on −1PRF levels in HEK293T cells. A, −1PRF reporter constructs containing the WT (green), the EE double mutant (orange), and the LL double mutant (pink) TM2 sequence were expressed in HEK293T cells, and cellular fluorescence intensities were analyzed by flow cytometry. A histogram depicts the mKate intensities associated with 3,000 cells expressing each reporter construct at a consistent expression level, as judged by the intensity of a bicistronic expression reporter. The intensity distribution of cells expressing a reporter construct containing the WT version of TM2 and a mutated slip site (UUUUUUA → GUUCCUA, SSKO, black) is also shown for reference. These trends were consistently observed across three independent biological replicates, and the distributions of cellular intensities for the EE and LL variant were found to be statistically distinct from those of WT at a significance level (α) of 0.001 according to a Mann–Whitney U test. B, −1PRF reporter constructs containing a series of deletions and G/S linker insertions within the loop between TM2 and -3 that alter the number of residues between the slip site and TM2 were expressed in HEK293T cells compared at a consistent expression level by flow cytometry. The distribution of fluorescent mKate reporter intensities from a representative experimental replicate is shown for cells expressing each reporter construct. The top and bottom edges of the box reflect the positions of the 75th and 25th percentile intensities of each distribution, respectively. The top and bottom whiskers reflect the positions of the 90th and 10th percentile intensities, respectively. The median intensities for each distribution are indicated by the horizontal line within the box, and the average intensities for each distribution are indicated with a square. Analysis of intensity distributions using a Mann–Whitney U test suggests that the distributions of each test construct are statistically distinct from that of the WT reporter (45 residues) at a significance level (α) of 0.001 (*). A similar trend and statistical confidence were observed in three independent biological replicates.
Impact of nascent chain forces on ribosomal frameshifting
The apparent link between cotranslational folding and ribosomal frameshifting has implications for the mechanism of −1PRF in the SINV structural polyprotein. The portion of the transcript containing the EE and LL mutations is over 100 nucleotides upstream from the ribosomal slip site and should therefore not perturb the stimulatory RNA structures that are currently believed to modulate −1PRF (2, 22, 23). These mutations instead alter the portion of the nascent chain that falls just outside of the ribosomal exit tunnel during −1PRF, which suggests that the nascent chain itself may stimulate frameshifting. Although it has yet to be implicated in ribosomal frameshifting, the cotranslational membrane integration and/or folding of the nascent chain is known to generate tension on the ribosome (41–44). Furthermore, the C-terminal residue of TM2 is positioned 45 residues upstream of the slip site, which corresponds to a distance that should maximize the tension on the nascent chain at the moment the slip site occupies the ribosomal active site (41, 42). Previous investigations have demonstrated that the force generated by the membrane integration of the nascent chain is sharply dependent upon this spacing (41, 42). Therefore, to assess the potential role of this force in ribosomal frameshifting, we generated a set of SINV −1PRF reporter constructs (used in Fig. 4A) containing a series of insertions and deletions that alter the distance between TM2 and the ribosomal slip site (see Table S1). Reporter constructs were then expressed in HEK293T cells, and −1PRF reporter intensities were quantitatively compared at a uniform expression level by flow cytometry (Fig. S5). A comparison of reporter intensities reveals that −1PRF is maximized at the WT distance of 45 residues (Fig. 4B). In all cases, deletions and insertions that change the distance between TM2 and the slip site result in large, statistically significant reductions in the relative intensity of the −1PRF reporter (Fig. 4B, n = 3, Mann–Whitney U test, α = 0.001). Moreover, the insertion of a 10-residue G/S linker decreases the intensity of the frameshift reporter by 76 ± 8% (n = 3, mean intensity change ± S.D.), which suggests that the membrane integration of TM2 is likely to be the primary driver of −1PRF within the SINV structural polyprotein. Nevertheless, the deletion of the region containing the stimulatory RNA hairpin downstream of the slip site abrogates PRF (Fig. S6), which suggests that both the hairpin and TM2 are needed for efficient PRF. Together, these findings suggest that topological signals within the SINV structural polyprotein generate a mechanical force that stimulates −1PRF.
To further explore the interplay between sequence, topology, and force, we carried out CGMD simulations of the translation and translocon-mediated membrane integration of the nascent structural polyprotein (44, 45). In these simulations, three-residue segments of the nascent chain are modeled as individual beads with physicochemical properties based on their constituent amino acids. New beads are translated at a rate of 5 amino acids/s and emerge from the ribosome-translocon complex into an environment with an implicit representation of the bilayer and cytosol supporting Movie (45). These simulations were previously found to sufficiently recapitulate several aspects of cotranslational membrane protein folding, including the formation of topological isomers and the generation of tension on the nascent chain (44, 46, 47). CGMD simulations of SINV polyprotein biosynthesis suggest that the nascent chain samples several different topological isomers (Fig. 5A), and that its topological heterogeneity persists after the polyprotein has cleared the translocon. TM2 undergoes translocon-mediated membrane integration (Fig. 5A, right) in only 44 ± 4% (mean ± S.D.) of the CGMD trajectories in which TM1 is correctly integrated into the membrane. Consistent with expectations, CGMD simulations suggest that the membrane integration efficiency of TM2 is enhanced by the LL mutations (51 ± 4%, mean ± S.D.) and reduced by the EE mutations (11 ± 3%). This finding provides additional evidence that the topological frustration within this domain (see Fig. 3) arises primarily from its marginal hydrophobicity.
Figure 5.

Coarse-grained molecular dynamics simulations of polyprotein biosynthesis. CGMD simulations were carried out to simulate biosynthesis of a series of SINV structural polyprotein variants, and the pulling force on the nascent chain was calculated at the point in which the ribosome occupies the slip site. A, representative snapshots from CGMD simulations are shown during translation at the slip site, which is the point during elongation at which pulling forces on the nascent chain are measured. The ribosomal exit tunnel is shown in brown. The translocon is shown in gray, and its lateral gate is highlighted in green. The nascent chain is shown in blue, except for the portions that correspond to TM1 and 2, which are highlighted in orange and red, respectively. The snapshot on the right depicts a representative trajectory in which TM2 passes through the lateral gate and into the membrane. The snapshot on the left depicts a representative trajectory in which TM2 fails to enter the translocon. B, −1PRF fluorescence reporter (mKate) intensity values for cells expressing a series of modified polyprotein variants were normalized relative to WT and plotted against the corresponding mean force measurements calculated from 560 CGMD trajectories. Error bars on the relative mKate intensity measurements reflect the S.D. from three independent biological replicates. Error bars are not shown for the mean force measurements due to the fact that the confidence intervals are smaller than the plotted date points. For reference, the 95% confidence intervals are shown in Table S1. The identity of each variant along with a linear fit of the data (dashes) are shown for reference (Pearson's r = 0.74, p = 0.036). C, pulling force measurements are compared among topological isomers for the WT (green), LL (pink), and EE (orange) polyprotein variants in which the number of TM2 residues (or beads) located within the translocon was found to vary. Values reflect the mean force, and error bars represent a 95% confidence interval. D, a histogram depicts the number of TM2 residues (or beads) within the translocon among the conformational trajectories sampled during biosynthesis of the WT (green), LL (pink), and EE (orange) variants of the SINV polyprotein.
To evaluate the connection between pulling forces on the nascent chain and frameshifting, we measured the tension on the nascent chain at the point of elongation when the slip site occupies the ribosomal active site supporting Movie (47). Pulling forces were highest for the LL variant, which averaged 4.2 pN higher than the WT. In contrast, the EE mutations reduce the pulling force on the nascent chain by an average of 2.1 pN relative to WT. These results are consistent with the hypothesis that differences in frameshifting arise from the effects of these mutations on the pulling force on the nascent chain. Simulations of polyprotein variants bearing insertions or deletions that alter the distance between TM2 and the ribosomal slip site indicate that the native distance (45 residues) is nearly optimal for the transmission of pulling force through the nascent chain (Table S1), which is consistent with the observed patterns in frameshifting (Fig. 4B). Overall, we find that the −1PRF efficiency associated with each polyprotein variant roughly scales with corresponding mean pulling force measurements from CGMD simulations (Pearson's r = 0.74, p = 0.036; Fig. 5B), which strongly suggests that the pulling forces generated by the translocon-mediated membrane integration of the nascent chain stimulate −1PRF.
An analysis of the spectrum of topological states sampled during translation reveals that the magnitude of the pulling force transmitted to the ribosome scales with the number of beads that occupy the translocon (Fig. 5C). This finding suggests that pulling forces are generated by the movement of hydrophobic transmembrane segments from the protein-conducting channel of the translocon to the hydrophobic membrane core, as has been established previously (40, 41, 47). The apparent variation in the conformation of TM2 at the translocon provides an explanation of the observed differences in pulling forces. Simulations suggest that differences in pulling forces arise from variations in the distribution of topological isomers that form during translation of these variants (Fig. 5D). The LL mutant predominately samples conformations where the majority of TM2 beads are in the translocon (Fig. 5A, right), whereas the EE mutant almost exclusively adopts conformations in which the majority of TM2 beads fall outside of the translocon and within the cytosol (Fig. 5A, left). As passage through the translocon is a prerequisite for membrane integration, the relationship between pulling forces and residence of the nascent chain within the translocon is consistent with our model for structural polyprotein biogenesis (Fig. 3) and confirms that the translocon-mediated membrane integration of TM2 stimulates −1PRF.
Survey of frameshifting elements among alphavirus structural polyproteins
Our model suggests that the hydrophobicity of TM2 and its distance from the slip site are the key determinants of the −1PRF efficiency within the SINV structural polyprotein. To assess whether this mechanism is likely to be operative within other alphaviruses, we surveyed six related structural polyproteins for similar sequence elements. Sequence scans carried out with the ΔG predictor reveal that each form of the alphavirus structural polyprotein contains a marginally hydrophobic TM domain upstream from the ribosomal slip site. Predicted transfer free energies associated with the translocon-mediated membrane integration of these putative TM domains range from +1.4 to +2.7 kcal/mol (Table 1), which suggests that the translocon-mediated membrane integration of these segments is likely to be inefficient. Consistent with this notion, CGMD simulations of the translation of these polyproteins indicate that the membrane integration efficiency of these segments ranges from 33 to 64% (Table 1). Furthermore, these marginally hydrophobic TM domains reside 44–52 residues upstream of their corresponding −1PRF sites (Table 1), which suggests that the tension generated by their translocon-mediated membrane integration is likely to be propagated back to the slip site (40, 41). Force measurements derived from CGMD simulations of polyprotein synthesis suggest that the tension in the nascent chain when the slip site occupies the ribosome is comparable with or greater than the tension generated during translation of the SINV variants characterized herein (Table 1). Taken together, these findings suggest that this −1PRF mechanism is likely to be conserved across the alphavirus genus. Additional investigations are needed to determine how nascent chain- and RNA-mediated −1PRF mechanisms are balanced against one another and how this mechanistic diversity ultimately influences viral evolution.
Table 1.
Comparison of the topological properties of alphavirus structural polyproteins
| Strain | Accession number | Predicted ΔGapp of TM2a | Membrane integration efficiency of TM2b | Distance from TM2 to slip site | Mean forcec | 95% confidence intervald |
|---|---|---|---|---|---|---|
| kcal/mol | residues | pN | ||||
| Sindbis virus | NC_001547 | 1.9 | 0.44 | 45 | 29.0 | 28.8–29.1 |
| Eastern equine encephalitis virus | NC_003899 | 2.4 | 0.51 | 45 | 31.1 | 31.0–31.2 |
| Middleburg virus | EF536323 | 1.6 | 0.33 | 52 | 34.0 | 33.9–34.1 |
| Sleeping disease virus | NC_003433 | 2.7 | 0.33 | 44 | 25.6 | 25.5–25.8 |
| Southern elephant seal virus | HM147990 | 2.1 | 0.34 | 47 | 25.2 | 25.0–25.3 |
| Semliki forest virus | NC_003215 | 2.1 | 0.64 | 49 | 33.2 | 33.1–33.4 |
| Venezuelan equine encephalitis virus | NC_001449 | 1.4 | 0.33 | 44 | 28.6 | 28.5–28.7 |
a Values reflect the minimum ΔGapp value as determined from a sequence scan of the full-length structural polyprotein using the ΔG predictor. (33)
b Values are derived from CGMD, and reflect the percentage of trajectories in which TM2 was found to adopt a transmembrane orientation.
c Values are derived from CGMD, and reflect the average force on the nascent chain while the ribosomal slip site occupies the ribosomal P-site.
d Confidence intervals are calculated by first assuming that errors on the mean force per trajectory are normally distributed, based on the large number of frames per trajectory and the long timescale in between frames. Then the overall confidence interval is obtained by bootstrapping the sampling error on the mean from the 560 trajectories, taking into account the errors on each trajectory.
Conclusions
Using an array of biochemical, cellular, and computational methods, we show that the nascent SINV structural polyprotein forms a spectrum of topological intermediates during biosynthesis and that −1PRF is primarily driven by the translocon-mediated membrane integration of a marginally hydrophobic TM domain within the E2 protein. We also provide evidence to suggest that this mechanism is likely to be conserved across the alphavirus genus. To date, the mechanistic basis of −1PRF has been generally attributed to the kinetic effects of mechanochemical forces that are generated by structural elements within the mRNA. Indeed, we do find that PRF in the SINV structural polyprotein depends upon the RNA stem loop downstream of the slip site (Fig. S6). Nevertheless, it is clear that the forces generated by the translocon-mediated membrane integration of TM2 dramatically enhance the frameshifting efficiency. To our knowledge, the findings reported herein constitute the first instance in which forces generated by conformational transitions in the nascent polypeptide chain have been implicated in the efficiency of PRF. Although additional investigations are needed to elucidate how pulling forces in the nascent chain physically stimulate −1PRF, a causative role for tension in both the transcript and nascent chain seems plausible, given that ribosomal frameshifting fundamentally arises from the movement of the tRNA with respect to the mRNA. It seems likely that cotranslational folding is one of many regulators, which include both host and viral effectors, that tune the net efficiency of PRF. This creates the potential for mechanistic diversity that could provide an evolutionary benefit for alphaviruses, as −1PRF is rendered tunable by either downstream or upstream mutations that impact the stability of the mRNA hairpin or the conformational ensemble of the nascent chain, respectively. This flexibility could also potentially provide the virus with a means of maintaining desired −1PRF levels in the presence of host factors that globally regulate −1PRF through mRNA interactions (12).
It should be noted that the implications of these findings potentially extend beyond the realm of viral proteins. A wide variety of molecular transitions have been found to generate tension within the nascent chain, including the folding of soluble domains near the ribosomal exit tunnel (43, 48) and the translocon-mediated membrane integration of nascent TM domains (40, 41). These observations suggest that the tension in the nascent chain should fluctuate as the structural features emerge from the ribosome (Fig. 6A), which may therefore provide the ribosome with a readout for the progress of cotranslational folding. In the case of the SINV structural polyprotein, the topological frustration within the nascent chain leads to the production of two competing topomers that generate distinct pulling forces on the ribosome in a manner that ultimately impacts the fidelity and processivity of translation (Fig. 6, B and C). This translational feedback constitutes a new form of mechanochemical allostery on the ribosome (48). Additional investigations are needed to explore the potential relevance of this type of cotranslational feedback to protein homeostasis. Indeed, interactions between the nascent chain and molecular chaperones are known to ratchet polypeptides across the membrane (49, 50) and may therefore contribute to pulling forces. This could potentially account for the fact that the deletion of components of the ribosome-associated chaperone complex has been found to attenuate −1PRF in yeast (51). Future investigations are needed to evaluate the full range of −1PRF effectors and how these are potentially exploited for regulatory purposes.
Figure 6.
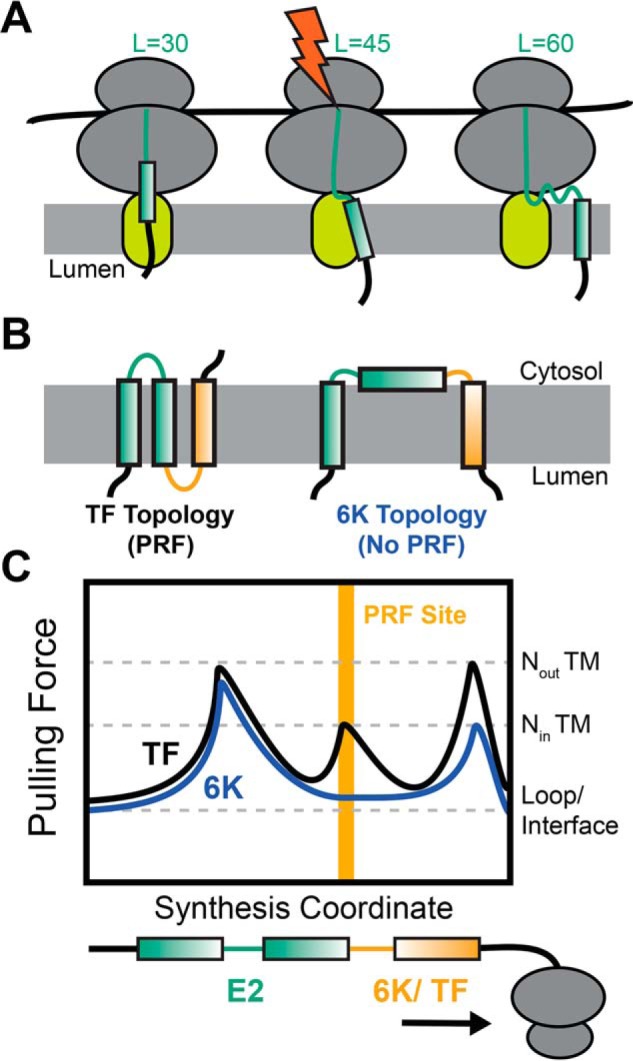
Interplay between topology, pulling force, and programmed ribosomal frameshifting. Cartoons depict the manner in which the translocon-mediated membrane integration of the nascent chain generates a fluctuation in pulling force that triggers PRF during synthesis of the SINV structural polyprotein. A, the pulling force generated by the translocon-mediated membrane integration of each TM domain generates a pulling force on the nascent chain that is maximized during the conjugation of the amino acid that lies ∼45 residues upstream of the C-terminal residue of the TM domain. B, the translocon-mediated membrane integration of TM2 is marginally efficient, which results in the formation of two topologies during translation of the SINV polyprotein. TM2 most often fails to undergo translocon-mediated membrane integration, which results in the formation of a topology featuring only two TM domains (TM1 and -3) in the form of the polyprotein containing the 6K protein. However, the translocon-mediated membrane integration of TM2 generates an alternative topology in the frameshifted form of the polyprotein containing the TF protein. C, a hypothetical plot of the fluctuations in the nascent chain pulling force during the translation of the two forms of the SINV structural polyprotein is shown. The translocon-mediated membrane integration of TM2 generates an extra pulling force on the nascent chain while the slip site occupies the ribosomal P-site, which stimulates −1PRF.
Experimental procedures
Computational predictions of topological energetics
The energetics associated with the translocon-mediated membrane integration of the nascent structural polyprotein were carried out using the ΔG predictor (RRID:SCR_018191) (34). These predictions are generated using a window scan function that sums depth-dependent free energies associated with the transfer of amino acids from the translocon to the ER membrane (33). Full sequence scans of varying window size were used to compare the predicted transfer free energies associated with each segment within the polyprotein to identify the segments that are most likely to undergo translocon-mediated membrane integration.
Plasmid preparation and mutagenesis
Chimeric Lep genes were generated in the context of a pGEM-based Lep expression vector (34) that was kindly provided by the laboratory of Gunnar von Heijne. Putative TM domains of interest were introduced into the H-segment position of this Lep construct using Gibson assembly. To probe the topology of and ribosomal frameshifting within the SINV structural polyprotein, a portion of the polyprotein containing the E3, E2, and 6K/TF proteins was first introduced downstream from the cytomegalovirus promoter within a pcDNA5 vector using Gibson assembly. To produce a series of reporter constructs for polyprotein topology, Gibson assembly was then used to replace the portion of the polyprotein gene downstream from each putative TM domain with a genetic cassette containing a 10-residue G/S linker, a glycosylatable eGFP gene (37), an internal ribosomal entry site (IRES), and an mKate gene, respectively (Fig. S1).
To produce a series of reporter constructs for ribosomal frameshifting, Gibson assembly was used to replace the portion of the polyprotein gene that falls 100 bp downstream from the ribosomal slip site in the 6K gene with a cassette containing an mKate gene in the −1 reading frame followed by an IRES and a GFP gene (Fig. S4). The frameshift reporter (mKate) was fused 100 nucleotides downstream from the slip site to avoid disrupting the stimulatory RNA hairpin downstream from the slip sites (22, 23). In addition to avoiding potential issues related to the impact of fusion domains on SRP-mediated targeting of the nascent chain, this design also avoids recently described artifacts associated with previous generations of the Dual-Luciferase reporter system in two ways (39). First, the transcript of our fluorescent expression reporter (eGFP) does not contain any cryptic splice sites. Second, the fluorescent −1PRF reporter protein (mKate) is liberated from the mutated portion of the nascent polypeptide through a native proteolytic cleavage site between the E2 and 6K/TF protein. Thus, mutations within TM2 should not impact the stability and/or turnover of mKate. An IRES-eGFP cassette was also incorporated into the downstream portion of the reporter transcript to facilitate comparisons of reporter intensities across cells with uniform expression level. Site-directed mutagenesis was used to introduce mutations into these constructs. Insertions and deletions were introduced using In-Fusion cloning (Takara, Mountain View, CA).
In vitro translation of chimeric Lep proteins
Chimeric Lep proteins were generated by in vitro translation as described previously (52). Briefly, mRNA for each chimeric Lep protein was produced from plasmids using the RiboMAX RNA production system in accordance with the manufacturer's instructions (Promega, Madison, WI). Lep proteins were then produced from mRNA by in vitro translation using rabbit reticulocyte lysate (Promega, Madison, WI) supplemented with canine pancreatic rough microsomes (tRNA Probes, College Station, TX) and EasyTag [35S]methionine (PerkinElmer Life Sciences). In vitro translation reactions were then diluted 1:4 into 1× SDS-PAGE sample buffer and separated using a 12% SDS-polyacrylamide gel. To image radioactive translation products, polyacrylamide gels were then dried, exposed to a phosphorimaging screen, and imaged using a Typhoon Imager (GE Healthcare). ImageJ software was then employed to process the data quantify the glycosylation state of each construct by densitometry.
Expression of fluorescent reporter constructs
Flow cytometry was used to compare the fluorescence intensity profiles of HEK293T cells expressing topology and frameshifting reporter constructs. Briefly, HEK293T cells were grown in Dulbecco's modified Eagle's medium (Gibco) containing 10% fetal bovine serum (Corning) and a penicillin/streptomycin antibiotic supplement (Gibco) in an incubator containing 5% CO2 at 37 °C. Reporter constructs were transiently expressed using Lipofectamine 3000 (Invitrogen) in accordance with the manufacturer's instructions. Cells were harvested 2 days post-transfection and analyzed using a BD LSRII flow cytometer (BD Biosciences). Cellular fluorescence profiles were analyzed using FlowJo software (Treestar, Ashland, OR). To compare cellular reporter intensities among cells with a uniform expression level, analysis of reporter intensity levels was restricted to cells that fell within a defined, uniform range of IRES-mKate or IRES-GFP expression reporter intensities. An example of the hierarchical gating strategy employed herein is shown in Figs. S2 and S5.
Coarse-grained simulations of polyprotein translation
CGMD simulations are based on a previously developed and tested approach (44, 45). Briefly, simulations are carried out in the context of a coarsened representation of the ribosome exit tunnel, Sec translocon, and nascent chain. The nascent chain is composed of beads that each represent three amino acids, and new beads are sequentially added to the nascent chain to explicitly simulate translation. Translation occurs at a rate of 5 aa/s, which mimics the rate of translation by eukaryotic ribosomes. Each bead interacts with the translocon, ribosome, and other beads in a manner that depends on the hydrophobicity and charge of its composite amino acids. Interactions with the solvent and lipid bilayer are modeled implicitly. The ribosome and Sec translocon are fixed in place, with the exception of the lateral gate of the translocon, which stochastically switches between the open and closed conformations in a manner dependent on the free energy difference between the two configurations.
The geometries of the ribosome and translocon are based on cryo-EM structures (53). Residue-specific interactions have been parameterized based on over 200 μs of simulations with the MARTINI forcefield. Fitting is performed using a Bayesian uncertainty quantification framework (54). This approach represents an update relative to previously published methodology, and the new parameters utilized herein are included in Table S2. All other parameters necessary to describe the system are available in previously published work (45). Integration is performed using overdamped Langevin dynamics with a diffusion coefficient of 253 nm2/s and a time step of 300 ns. Despite the significant simplifications involved in this model, the CGMD model has proven accurate in capturing the integration probabilities, topology distributions, and forces experienced in previous studies (44, 45, 47).
To obtain the distribution of topologies for various polyprotein mutants, the translation and integration of each sequence was simulated 560 times. To reduce computational cost, simulations only included the first three TMDs of the alphavirus polyprotein. To focus on the topological preferences of TM2, restraints were applied to enforce that TM1 adopts its native topological orientation. Pulling force measurements were performed by pausing translation when the −1PRF site resides within the ribosomal peptidyl transfer center. During this pause, the final bead was fixed in place, and the force on the bead exerted by the nascent chain was measured along the translocon channel axis. Due to the truncation of the exit tunnel in our model, the final bead corresponds to the amino acids 27 residues N-terminal of the −1PRF site. Translation is paused for 3 s, which is equivalent to the time it would take to translate five beads. This relatively short time window ensures that the distribution of polyprotein topologies is not affected by the pause. During this window, pulling forces were measured at a rate of 333 frames/s. To sample a wide range of topologies and conformations, each mutant was independently simulated 560 times. This protocol is analogous to force measurements performed in previous work, with the exception of a shortened pause duration (47).
Data availability
The cellular and biochemical data sets described herein will be made freely available by Jonathan Schlebach (Indiana University Department of Chemistry, jschleba@indiana.edu) upon request. All computational data and code relating to the CGMD simulations detailed herein will be made freely available by Thomas Miller III (California Institute of Technology Division of Chemistry and Chemical Engineering, tfm@caltech.edu) upon request. All remaining data are contained within the article. Viral sequences analyzed herein can be freely accessed through the National Center for Biotechnology Information (NCBI) using the following accession numbers: Sindbis virus (NC_001547), eastern equine encephalitis virus (NC_003899), Middleburg virus (EF536323), sleeping disease virus (NC_003433), southern elephant seal virus (HM147990), Semliki Forest virus (NC_003215), and Venezuelan equine encephalitis virus (NC_001449).
Author contributions
H. R. H., M. H. Z., V. N., W. D. P., T. F. M., S. M., and J. P. S. conceptualization; H. R. H., M. H. Z., L. M. C., V. N., W. D. P., T. F. M., S. M., and J. P. S. data curation; H. R. H., M. H. Z., V. N., W. D. P., S. M., and J. P. S. formal analysis; H. R. H., M. H. Z., L. M. C., V. N., W. D. P., and J. P. S. investigation; H. R. H., M. H. Z., and J. P. S. visualization; H. R. H., M. H. Z., L. M. C., V. N., W. D. P., and J. P. S. methodology; H. R. H., M. H. Z., L. M. C., W. D. P., T. F. M., and S. M. writing-review and editing; M. H. Z. and T. F. M. software; M. H. Z. validation; W. D. P., T. F. M., S. M., and J. P. S. supervision; W. D. P., T. F. M., S. M., and J. P. S. project administration; T. F. M. and J. P. S. resources; T. F. M., S. M., and J. P. S. funding acquisition; J. P. S. writing-original draft.
Supplementary Material
Acknowledgments
We thank Renuka Kudva, Gunnar von Heijne, and other members of the Schlebach and Mukhopadhyay laboratories for scientific input. We thank David Giedroc and Jonathan Dinman for editorial input. We thank Christiane Hassel and the Indiana University Bloomington Flow Cytometry Core Facility for experimental support. This work used the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges computer at the Pittsburgh Supercomputing Center through allocation TG-MCB160013 (55).
This research was supported in part by NIAID, National Institutes of Health, Grant R21AI142383 (to J. P. S. and S. M.) as well as NIGMS, National Institutes of Health, Grant R01GM125063 (to T. F. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article was selected as one of our Editors' Picks.
This article contains Tables S1 and S2, Figs. S1–S6, and supporting Movie.
- PRF
- programmed ribosomal frameshifting
- CGMD
- coarse-grained molecular dynamics
- eGFP
- enhanced green fluorescent protein
- ER
- endoplasmic reticulum
- gGFP
- glycosylatable green fluorescent protein
- HEK293T
- human embryonic kidney 293T cells
- IRES
- internal ribosome entry site
- Lep
- leader peptidase protein
- −1PRF
- −1 programmed ribosomal frameshifting
- SINV
- Sindbis virus
- TF
- TransFrame protein
- TM
- transmembrane
- SRP
- signal recognition particle
- CP
- capsid
- pN
- piconewtons.
References
- 1. Caliskan N., Peske F., and Rodnina M. V. (2015) Changed in translation: mRNA recoding by −1 programmed ribosomal frameshifting. Trends Biochem. Sci. 40, 265–274 10.1016/j.tibs.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Halma M. T. J., Ritchie D. B., Cappellano T. R., Neupane K., and Woodside M. T. (2019) Complex dynamics under tension in a high-efficiency frameshift stimulatory structure. Proc. Natl. Acad. Sci. U.S.A. 116, 19500–19505 10.1073/pnas.1905258116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Namy O., Moran S. J., Stuart D. I., Gilbert R. J., and Brierley I. (2006) A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature 441, 244–247 10.1038/nature04735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kim H. K., Liu F., Fei J., Bustamante C., Gonzalez R. L. Jr., and Tinoco I. (2014) A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U.S.A. 111, 5538–5543 10.1073/pnas.1403457111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caliskan N., Katunin V. I., Belardinelli R., Peske F., and Rodnina M. V. (2014) Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157, 1619–1631 10.1016/j.cell.2014.04.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chen J., Petrov A., Johansson M., Tsai A., O'Leary S. E., and Puglisi J. D. (2014) Dynamic pathways of −1 translational frameshifting. Nature 512, 328–332 10.1038/nature13428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi J., O'Loughlin S., Atkins J. F., and Puglisi J. D. (2020) The energy landscape of −1 ribosomal frameshifting. Sci. Adv. 6, eaax6969 10.1126/sciadv.aax6969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Korniy N., Samatova E., Anokhina M. M., Peske F., and Rodnina M. V. (2019) Mechanisms and biomedical implications of −1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 593, 1468–1482 10.1002/1873-3468.13478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bock L. V., Caliskan N., Korniy N., Peske F., Rodnina M. V., and Grubmüller H. (2019) Thermodynamic control of −1 programmed ribosomal frameshifting. Nat. Commun. 10, 4598 10.1038/s41467-019-12648-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belew A. T., Meskauskas A., Musalgaonkar S., Advani V. M., Sulima S. O., Kasprzak W. K., Shapiro B. A., and Dinman J. D. (2014) Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512, 265–269 10.1038/nature13429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Napthine S., Ling R., Finch L. K., Jones J. D., Bell S., Brierley I., and Firth A. E. (2017) Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun. 8, 15582 10.1038/ncomms15582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wang X., Xuan Y., Han Y., Ding X., Ye K., Yang F., Gao P., Goff S. P., and Gao G. (2019) Regulation of HIV-1 Gag-Pol expression by Shiftless, an inhibitor of programmed −1 ribosomal frameshifting. Cell 176, 625–635.e14 10.1016/j.cell.2018.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ramsey J., and Mukhopadhyay S. (2017) Disentangling the frames, the state of research on the alphavirus 6K and TF proteins. Viruses 9, E228 10.3390/v9080228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown R. S., Wan J. J., and Kielian M. (2018) The alphavirus exit pathway: what we know and what we wish we knew. Viruses 10, E89 10.3390/v10020089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lobigs M., Zhao H. X., and Garoff H. (1990) Function of Semliki Forest virus E3 peptide in virus assembly: replacement of E3 with an artificial signal peptide abolishes spike heterodimerization and surface expression of E1. J. Virol. 64, 4346–4355 10.1128/JVI.64.9.4346-4355.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mulvey M., and Brown D. T. (1996) Assembly of the Sindbis virus spike protein complex. Virology 219, 125–132 10.1006/viro.1996.0229 [DOI] [PubMed] [Google Scholar]
- 17. Firth A. E., Chung B. Y., Fleeton M. N., and Atkins J. F. (2008) Discovery of frameshifting in Alphavirus 6K resolves a 20-year enigma. Virol. J. 5, 108 10.1186/1743-422X-5-108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hallengärd D., Kakoulidou M., Lulla A., Kümmerer B. M., Johansson D. X., Mutso M., Lulla V., Fazakerley J. K., Roques P., Le Grand R., Merits A., and Liljeström P. (2014) Novel attenuated Chikungunya vaccine candidates elicit protective immunity in C57BL/6 mice. J. Virol. 88, 2858–2866 10.1128/JVI.03453-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Taylor A., Melton J. V., Herrero L. J., Thaa B., Karo-Astover L., Gage P. W., Nelson M. A., Sheng K. C., Lidbury B. A., Ewart G. D., McInerney G. M., Merits A., and Mahalingam S. (2016), Effects of an in-frame deletion of the 6k gene locus from the genome of Ross River virus. J. Virol. 90, 4150–4159 10.1128/JVI.03192-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Snyder J. E., Kulcsar K. A., Schultz K. L., Riley C. P., Neary J. T., Marr S., Jose J., Griffin D. E., and Kuhn R. J. (2013) Functional characterization of the alphavirus TF protein. J. Virol. 87, 8511–8523 10.1128/JVI.00449-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rogers K. J., Jones-Burrage S., Maury W., and Mukhopadhyay S. (2020) TF protein of Sindbis virus antagonizes host type I interferon responses in a palmitoylation-dependent manner. Virology 542, 63–70 10.1016/j.virol.2020.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chung B. Y., Firth A. E., and Atkins J. F. (2010) Frameshifting in alphaviruses: a diversity of 3′ stimulatory structures. J. Mol. Biol. 397, 448–456 10.1016/j.jmb.2010.01.044 [DOI] [PubMed] [Google Scholar]
- 23. Kendra J. A., Advani V. M., Chen B., Briggs J. W., Zhu J., Bress H. J., Pathy S. M., and Dinman J. D. (2018) Functional and structural characterization of the chickungunya virus translational recoding signals. J. Biol. Chem. 293, 17536–17545 10.1074/jbc.RA118.005606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Snyder A. J., Sokoloski K. J., and Mukhopadhyay S. (2012) Mutating conserved cysteines in the alphavirus e2 glycoprotein causes virus-specific assembly defects. J. Virol. 86, 3100–3111 10.1128/JVI.06615-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ramsey J., Renzi E. C., Arnold R. J., Trinidad J. C., and Mukhopadhyay S. (2017) Palmitoylation of Sindbis virus TF protein regulates its plasma membrane localization and subsequent incorporation into virions. J. Virol. 91, e02000–e2016 10.1128/JVI.02000-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gaedigk-Nitschko K., and Schlesinger M. J. (1990) The Sindbis virus 6K protein can be detected in virions and is acylated with fatty acids. Virology 175, 274–281 10.1016/0042-6822(90)90209-A [DOI] [PubMed] [Google Scholar]
- 27. Politis E. G., Roth A. F., and Davis N. G. (2005) Transmembrane topology of the protein palmitoyl transferase Akr1. J. Biol. Chem. 280, 10156–10163 10.1074/jbc.M411946200 [DOI] [PubMed] [Google Scholar]
- 28. Liljeström P., and Garoff H. (1991) Internally located cleavable signal sequences direct the formation of Semliki Forest virus membrane proteins from a polyprotein precursor. J. Virol. 65, 147–154 10.1128/JVI.65.1.147-154.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cheng R. H., Kuhn R. J., Olson N. H., Rossmann M. G., Choi H. K., Smith T. J., and Baker T. S. (1995) Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80, 621–630 10.1016/0092-8674(95)90516-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen L., Wang M., Zhu D., Sun Z., Ma J., Wang J., Kong L., Wang S., Liu Z., Wei L., He Y., Wang J., and Zhang X. (2018) Implication for alphavirus host-cell entry and assembly indicated by a 3.5 Å resolution cryo-EM structure. Nat. Commun. 9, 5326 10.1038/s41467-018-07704-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gafvelin G., and von Heijne G. (1994) Topological “frustration” in multispanning E. coli inner membrane proteins. Cell 77, 401–412 10.1016/0092-8674(94)90155-4 [DOI] [PubMed] [Google Scholar]
- 32. Ruch T. R., and Machamer C. E. (2012) The coronavirus E protein: assembly and beyond. Viruses 4, 363–382 10.3390/v4030363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hessa T., Meindl-Beinker N. M., Bernsel A., Kim H., Sato Y., Lerch-Bader M., Nilsson I., White S. H., and von Heijne G. (2007) Molecular code for transmembrane-helix recognition by the Sec61 translocon. Nature 450, 1026–1030 10.1038/nature06387 [DOI] [PubMed] [Google Scholar]
- 34. Hessa T., Kim H., Bihlmaier K., Lundin C., Boekel J., Andersson H., Nilsson I., White S. H., and von Heijne G. (2005) Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 433, 377–381 10.1038/nature03216 [DOI] [PubMed] [Google Scholar]
- 35. Ivanova L., and Schlesinger M. J. (1993) Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J. Virol. 67, 2546–2551 10.1128/JVI.67.5.2546-2551.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ryan C., Ivanova L., and Schlesinger M. J. (1998) Effects of site-directed mutations of transmembrane cysteines in Sindbis virus E1 and E2 glycoproteins on palmitylation and virus replication. Virology 249, 62–67 10.1006/viro.1998.9281 [DOI] [PubMed] [Google Scholar]
- 37. Lee H., Min J., von Heijne G., and Kim H. (2012) Glycosylatable GFP as a compartment-specific membrane topology reporter. Biochem. Biophys. Res. Commun. 427, 780–784 10.1016/j.bbrc.2012.09.138 [DOI] [PubMed] [Google Scholar]
- 38. Virkki M. T., Agrawal N., Edsbäcker E., Cristobal S., Elofsson A., and Kauko A. (2014) Folding of Aquaporin 1: multiple evidence that helix 3 can shift out of the membrane core. Protein Sci. 23, 981–992 10.1002/pro.2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Khan Y. A., Loughran G., and Atkins J. F. (2019) Contesting the evidence for −1 frameshifting in immune-functioning C-C chemokine receptor 5 (CCR5): the HIV co-receptor. bioRxiv 10.1101/513333 [DOI]
- 40. Ismail N., Hedman R., Schiller N., and von Heijne G. (2012) A biphasic pulling force acts on transmembrane helices during translocon-mediated membrane integration. Nat. Struct. Mol. Biol. 19, 1018–1022 10.1038/nsmb.2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cymer F., Ismail N., and von Heijne G. (2014) Weak pulling forces exerted on Nin-orientated transmembrane segments during co-translational insertion into the inner membrane of Escherichia coli. FEBS Lett. 588, 1930–1934 10.1016/j.febslet.2014.03.050 [DOI] [PubMed] [Google Scholar]
- 42. Cymer F., and von Heijne G. (2013) Cotranslational folding of membrane proteins probed by arrest-peptide-mediated force measurements. Proc. Natl. Acad. Sci. U.S.A. 110, 14640–14645 10.1073/pnas.1306787110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Goldman D. H., Kaiser C. M., Milin A., Righini M., Tinoco I. Jr., and Bustamante C. (2015) Mechanical force releases nascent chain-mediated ribosome arrest in vitro and in vivo. Science 348, 457–460 10.1126/science.1261909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhang B., and Miller T. F. 3rd. (2012) Long-timescale dynamics and regulation of Sec-facilitated protein translocation. Cell Rep. 2, 927–937 10.1016/j.celrep.2012.08.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Niesen M. J., Wang C. Y., Van Lehn R. C., and Miller T. F. 3rd (2017) Structurally detailed coarse-grained model for Sec-facilitated co-translational protein translocation and membrane integration. PLoS Comput. Biol. 13, e1005427 10.1371/journal.pcbi.1005427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Van Lehn R. C., Zhang B., and Miller T. F. 3rd. (2015) Regulation of multispanning membrane protein topology via post-translational annealing. Elife 4, e08697 10.7554/eLife.08697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Niesen M. J. M., Müller-Lucks A., Hedman R., von Heijne G., and Miller T. F. 3rd (2018) Forces on nascent polypeptides during membrane insertion and translocation via the Sec translocon. Biophys. J. 115, 1885–1894 10.1016/j.bpj.2018.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Leininger S. E., Narayan K., Deutsch C., and O'Brien E. P. (2019) Mechanochemistry in translation. Biochemistry 58, 4657–4666 10.1021/acs.biochem.9b00260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matlack K. E., Misselwitz B., Plath K., and Rapoport T. A. (1999) BiP acts as a molecular ratchet during posttranslational transport of prepro-α factor across the ER membrane. Cell 97, 553–564 10.1016/S0092-8674(00)80767-9 [DOI] [PubMed] [Google Scholar]
- 50. Brodsky J. L., Goeckeler J., and Schekman R. (1995) BiP and Sec63p are required for both co- and posttranslational protein translocation into the yeast endoplasmic reticulum. Proc. Natl. Acad. Sci. U.S.A. 92, 9643–9646 10.1073/pnas.92.21.9643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muldoon-Jacobs K. L., and Dinman J. D. (2006) Specific effects of ribosome-tethered molecular chaperones on programmed −1 ribosomal frameshifting. Eukaryot. Cell 5, 762–770 10.1128/EC.5.4.762-770.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roushar F. J., Gruenhagen T. C., Penn W. D., Li B., Meiler J., Jastrzebska B., and Schlebach J. P. (2019) Contribution of cotranslational folding defects to membrane protein homeostasis. J. Am. Chem. Soc. 141, 204–215 10.1021/jacs.8b08243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Voorhees R. M., Fernández I. S., Scheres S. H., and Hegde R. S. (2014) Structure of the mammalian ribosome-Sec61 complex to 3.4 Å resolution. Cell 157, 1632–1643 10.1016/j.cell.2014.05.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wu S., Angelikopoulos P., Tauriello G., Papadimitriou C., and Koumoutsakos P. (2016) Fusing heterogeneous data for the calibration of molecular dynamics force fields using hierarchical Bayesian models. J. Chem. Phys. 145, 244112 10.1063/1.4967956 [DOI] [PubMed] [Google Scholar]
- 55. Towns J., Cockerill T., Dahan M., Foster I., Gaither K., Grimshaw A., Hazlewood V., Lathrop S., Lifka D., Peterson G. D., Roskies R., Scott J. R., and Wilkins-Diehr N. (2014) XSEDE: accelerating scientific discovery. Comp. Sci. Eng. 16, 62–74 10.1109/MCSE.2014.80 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cellular and biochemical data sets described herein will be made freely available by Jonathan Schlebach (Indiana University Department of Chemistry, jschleba@indiana.edu) upon request. All computational data and code relating to the CGMD simulations detailed herein will be made freely available by Thomas Miller III (California Institute of Technology Division of Chemistry and Chemical Engineering, tfm@caltech.edu) upon request. All remaining data are contained within the article. Viral sequences analyzed herein can be freely accessed through the National Center for Biotechnology Information (NCBI) using the following accession numbers: Sindbis virus (NC_001547), eastern equine encephalitis virus (NC_003899), Middleburg virus (EF536323), sleeping disease virus (NC_003433), southern elephant seal virus (HM147990), Semliki Forest virus (NC_003215), and Venezuelan equine encephalitis virus (NC_001449).