

Abstract
N6-Methyladenosine (m6A) is the most prevalent modified base in eukaryotic mRNA and long noncoding RNA. Although candidate sites for the m6A modification are identified at the transcriptomic level, methods for site-specific quantification of absolute m6A modification levels are still limited. Herein, we present a facile method implementing a deoxyribozyme, VMC10, which preferentially cleaves the unmodified RNA. We leveraged reverse transcription and real-time quantitative PCR along with key control experiments to quantify the methylation fraction of specific m6A sites. We validated the accuracy of this method with synthetic RNA in which methylation fractions ranged from 0 to 100% and applied our method to several endogenous sites that were previously identified in sequencing-based studies. This method provides a time- and cost-effective approach for absolute quantification of the m6A fraction at specific loci, with the potential for multiplexed quantifications, expanding the current toolkit for studying RNA modifications.
Keywords: RNA methylation, RNA modification, DNA enzyme, reverse transcription, polymerase chain reaction (PCR), deoxyribozymes, N6-methyladenosine
Introduction
Over 100 types of RNA modifications have been identified to date. Among them, N6-methyladenosine (m6A)2 is most prevalent in mRNA and various long noncoding RNA (lncRNA) in higher eukaryotes (1). m6A modifications are widely involved in post-transcriptional gene regulation. The complex and dynamic nature of m6A-mediated regulation enables timely responses to signaling cues and large-scale modulation of gene expression. Therefore, m6A has been shown to be essential for development and associated with many human diseases (2, 3). The single methyl group is commonly deposited by either a methyltransferase writer complex composed of METTL3, METTL14, and WTAP (4) or by METTL16 methyltransferase (5) and is removed by either FTO (6) or ALKBH5 demethylase (7). Through its effects on RNA secondary structure and its interactions with m6A-binding proteins, m6A modifications affect essentially all known steps during an RNA's lifetime, including alternative splicing, polyadenylation, RNA export, translation, and degradation (8, 9). Despite m6A modification having a consensus DRACH motif (D = A, G, or U; R = G or A; and H = A, C, or U) (10, 11), the substoichiometric nature of m6A modification potentially creates large compositional heterogeneity in a single RNA species, i.e. each RNA of the same species may selectively carry m6A modification at one or a few DRACH motifs among all (12). Being able to quantify the extent of m6A modification at precise sites can greatly advance our current understanding of how changes in the m6A modification pattern (the site and fraction) are modulated by signaling cues and are then linked to various functional consequences.
Because of its important roles, techniques have been developed and applied to detect and quantify m6A modification. Detection of m6A modification is primarily facilitated by various high-throughput sequencing-based methods utilizing antibodies and chemical cross-linking (10, 11, 13, 14). Although these sequencing-based methods can map m6A candidate sites at the transcriptomic level, they cannot provide the fraction of modification at each site, because of factors such as antibody binding efficiency, specificity, and cross-linking reactivity (15). Real-time quantitative PCR (qPCR) was previously applied for locus-specific detection of pseudouridine (ψ) modification through chemical labeling of ψ residue, causing a shift in the melting peak of the resulting qPCR amplicons (16). Similar quantitative methods were recently developed for detection of m6A. These methods utilize enzymatic activities followed by qPCR, including differential ligation efficiency of T3 and T4 DNA ligases (17, 18), differential reverse transcription activity of Tth and BstI reverse transcriptases (19, 20), and a combination of selective elongation of DNA polymerase and ligation (21). Although these polymer elongation and ligation-based methods are successful at modification discrimination and can report the relative m6A abundance change, absolute quantification using these methods were only applied on MALAT1, an abundant lncRNA. In addition, the potential sequence dependence of these enzymatic activities requires caution for general applications to these methods (22, 23). Considering these potential pitfalls, absolute quantification using these methods would require calibration curves using fully modified and fully unmodified RNA for each target m6A site, which is expensive. The only available qPCR-independent method that can provide absolute quantification of m6A fraction site-specifically is site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and TLC (SCARLET) (24). However, the sophistication of the method and its requirement for radioactive labeling prevents its broad application. Very recently, endoribonuclease digestion-based sequencing methods have been developed that rely on selective cleavage of unmethylated A at the ACA motif (25, 26). These approaches provide single-base resolution for identification of modifications site with relative quantitative information but are limited to m6A sites carrying the ACA motif, as well as regions that contain relatively sparse ACA motifs (26).
To address these challenges, we present an easy-to-implement method for quantifying m6A fraction at specific loci from extracted total RNA. The method utilizes a deoxyribozyme (DR), VMC10, recently reported by Sednev et al. (27) to discriminate between A- and m6A-containing RNA (Fig. 1A). We chose VMC10 DR because it cleaves unmethylated A with reasonably high and robust efficiency, whereas its cleavage of m6A remains low even after a long incubation time (27). Sednev et al. (27) demonstrated that the remainder of intact RNA after VMC10 DR treatment is correlated with the known methylation level of specific sites of abundant endogenous RNAs. However, quantification of the m6A fraction requires additional characterization and correction of potential false positives and false negatives caused by various factors. Without these additional characterizations, modification levels of different m6A sites cannot be compared because of the sequence dependence of DR. In our method, we quantify and correct for the potential errors caused by sequence-dependent incomplete cleavage of the DR and the presence of nontarget RNAs from the total RNA. We show that our method can be used to robustly quantify the m6A fraction at specific loci on endogenous RNAs with a broad range of cellular abundance. We further show that the method can be adjusted for multiplexed quantification of m6A sites. Finally, we extensively discuss the limitations of the method and factors that need to be considered when applying this method.
Figure 1.

Workflow of the method. A, representative schematic of the active DR and the inactive DR (dDR). B, unmodified RNA is selectively cleaved by DR upstream of the target site, whereas m6A-modified RNA remains uncleaved. The remaining uncleaved RNAs are then quantified using RT with gene-specific reverse primer and qPCR. To control for variations in RNA input, an adjacent region on target RNA is also quantified with RT and qPCR as an internal reference. C, in the negative control sample, RNA is treated with a nonfunctional version of DR (dDR). Both m6A modified and unmodified RNA targets remain uncleaved and are subsequently quantified with RT and qPCR.
Results
Specificity and sequence dependence of DR cleavage efficiency
We first verified the cleavage efficiency of DR on a variety of fully modified or unmodified sites. For this purpose, we employed a 460-nt in vitro transcribed RNA from a gene block sequence with only one adenine in the sequence (referred to as “GB RNA” hereafter) and 35–41-mer synthetic RNA fragments with sequences around the MALAT1 2515, MALAT1 2577, and ACTB 1216 sites (Table S1). Each of these targets has either m6A or A at the respective m6A sites. The RNAs were treated with corresponding 40-mer VMC10 DR (referred to as DR for simplification hereafter) and subsequently analyzed by denaturing PAGE. For all the targets, the RNA fragments with unmethylated A were cleaved with high efficiency, and the cleavage efficiencies of modified RNAs were consistently below 5% (Fig. 2, A–D). In addition, the cleavage efficiencies on the unmodified RNAs were sequence-dependent (Fig. 2E), ranging from 50 to 82% for our tested cases.
Figure 2.
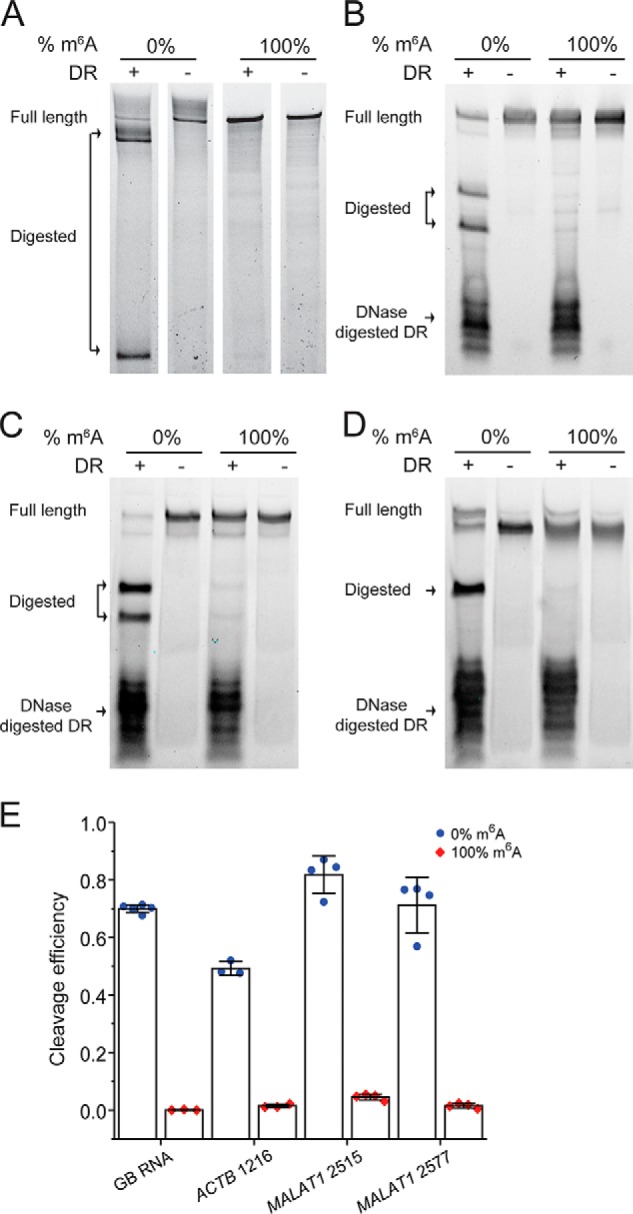
DR specifically cleaves unmodified RNAs, and its cleavage efficiency depends on sequence context around m6A sites. A–D, PAGE showing DR cleavage of 0 and 100% modified GB RNA (A) and RNA fragments containing modification site of ACTB 1216 (B), MALAT1 2515 (C), and MALAT1 2577 (D). E, bar plot of the cleavage efficiencies of m6A modified and unmodified target sites as quantified from PAGE. The error bars indicate means ± S.D. of three or four independent cleavage reactions. The DR is digested with DNase after DR cleavage reaction, because the DR may migrate to the same location as the input or cleaved RNA and affect data interpretation.
Method for absolute quantification of m6A fraction
We designed a quantification assay using RT-qPCR. As shown in Fig. 1, DR is designed for each modification site based on a VMC10 construct. The total RNA is subjected to DR treatment, during which only unmethylated RNAs upstream of the target site are cleaved. Thus, after the DR treatment, the amount of cleaved RNA should be inversely proportional to the methylation fraction (Fm) of RNA at the target site. The remaining RNA can be quantified using RT-qPCR. To control for any variations in the initial RNA input, we use RT-qPCR to also detect levels of adjacent uncleaved regions on the same target RNA as an internal reference.
Theoretically, the modified fraction calculated from qPCR can be written as,
| (Eq. 1) |
in which,
| (Eq. 2) |
where Ct+DR−m6A and Ct+DR−ref are the qPCR Ct values at the m6A site and a nearby reference site in the DR treated sample, whereas Ct−DR−m6A and Ct−DR−ref are the Ct values at the m6A and the reference site without DR digestion.
However, the measured ΔΔCt only reflects the digested fraction of the RNA substrate; i.e. Equation 1 only holds when digestion efficiency of the unmodified template is 100% and digestion efficiency of the modified template is 0%. Incomplete cleavage of unmodified A will lead to a false positive, and cleavage of the m6A will lead to a false negative. Based on the previous study and our tested cases (Fig. 2), the cleavage of VMC10 DR on m6A sequence is minimal, leading to insignificant error caused by false negative (Fig. S1). In addition, it is practically difficult and expensive to generate in vitro purified template containing 100% modified m6A to account for the exactly false-negative error at each m6A site of interest; we therefore left out the correction factor for false-negative error in our final calculation. On the other hand, the false-positive error can be significant because of the sequence-dependent incomplete cleavage of unmodified RNA by the DR and needs to be corrected for each m6A site of interest.
We therefore consider two major factors that may contribute to the false-positive error caused by incomplete digestion and quantify the effect of the two factors to extract the true modification fraction: the intrinsic sequence-dependent digestion efficiency and the presence of a large amount of nontarget RNAs from the total RNA extract. We define FDR as a correction factor to account for the incomplete DR digestion efficiency, which has to be determined for each m6A target (Fig. 2). We can determine FDR at each m6A site of interest by performing the DR digestion followed by RT-qPCR using the in vitro transcribed unmodified RNA,
| (Eq. 3) |
in which ΔΔCt is determined as in Equation 2. We define FN as the ratio of DR digestion efficiency of an RNA target in total RNA over digestion efficiency of a pure RNA target, to account for the potential drop of DR efficiency caused by the presence of nontarget RNAs. We can determine FN by performing DR digestion using the same in vitro transcribed unmodified RNA mixed with total RNA and compare with FDR from Equation 3,
| (Eq. 4) |
in which ΔΔCt is determined as in Equation 2. With the quantification of FDR and FN, the corrected modification fraction follows.
| (Eq. 5) |
We therefore can calculate Fm as follows.
| (Eq. 6) |
Validation of the absolute quantification of m6A fraction using pure RNA
To test the feasibility of the method to quantify the m6A methylation fraction, we used GB RNA with methylation fractions ranging from 0 to 100%. We performed DR treatment on GB RNA and estimated the cleaved fractions by denaturing PAGE. The cleaved fraction was linearly dependent on the methylation fraction of the input RNA (Fig. 3, A and B). Next, we tested whether we can use RT-qPCR to quantify the absolute methylation fraction. Because this quantification is performed on in vitro purified RNA, only FDR is needed to correct for Fm. Based on Equation 3, using the 100% unmodified GB RNA, we measured Ct+DR−m6A and Ct+DR−ref at the m6A site and a nearby reference site in the DR-treated sample and Ct−DR−m6A and Ct−DR−ref at the m6A and the reference site using a negative control containing identical amount of RNA but without the DR. We found that in addition to the expected larger Ct+DR−m6A compared with Ct−DR−m6A, there was a consistent difference between Ct+DR−ref and Ct−DR−ref. We speculated that this difference in Ct values at the reference site might be due to changes in the RNA secondary structure upon DR binding that can affect RT efficiency. To create a more accurate negative control, we designed a nonfunctional version of DR (“dead” DR or dDR) (Fig. 1, A and C), which has mutations in the AGC triplet, CG dinucleotide, and position 19 important for the catalytic activity of 8–17 family of enzymes (28). We tested the activity of dDR on multiple targets, for all of which digestion of RNA was undetectable (Figs. S2 and S3). Indeed, using the dDR-treated RNA as a negative control, the difference between Ct+DR−ref and Ct−DR−ref was eliminated (Fig. S4). Based on Equation 3, we determined FDR of the synthetic RNA to be 0.49 ± 0.08 (mean ± S.D.). With the FDR correction, we showed that the estimated Fm correlated well with the input m6A methylation fractions (Fig. 3C and Fig. S5).
Figure 3.

Validation of the method for absolute quantification of m6A fraction. GB RNA containing varied m6A fractions is used as a model system. A, PAGE showing DR cleavage fraction of the GB RNA. B, linear relationship between the input m6A fraction and the cleavage fraction of RNA by DR as quantified from the PAGE gel in A. The error bars indicate means ± S.D. for three biological replicates. C, estimated modification fraction as a function of input m6A fraction for the GB RNA. The error bars indicate means ± S.D. for at least three biological replicates.
DR cleavage efficiency in presence of nontarget RNAs
Next, we evaluated how the presence of total RNA affects the cleavage efficiency of the DR. The presence of the large amount of nontarget RNAs may compete for DR binding, consequently decreasing its cleavage efficiency at the target site in total RNA as opposed to purified RNA. We accounted for this potential decrease in efficiency with the FN correction factor, which we measured using three RNA transcripts: 1) the GB RNA used above, which is naturally missing in total RNA; 2) a PLAC2 RNA fragment containing two target sites, which is of low abundance in HeLa cell line; and 3) an unmethylated A site in the endogenous ACTB mRNA. The A1165 site on ACTB mRNA was chosen as the unmethylated A site because it was not detected in the sequencing-based studies (4, 29), nor does it contain the DRACH consensus motif. FDR of these three RNAs were measured with in vitro transcribed RNAs based on Equation 3 (Fig. 4, A and B). Then the in vitro transcribed GB RNA and the PLAC2 RNA fragment were spiked into the total RNA respectively to determine FN based on Equation 4. For the unmethylated A site in the ACTB mRNA, FN was determined by measuring the total RNA directly.
Figure 4.

The cleavage efficiency of DR is not compromised by the presence of total RNA. A and B, the cleavage efficiencies of the GB RNA, two m6A sites in PLAC2, and ACTB 1165 by 40-mer DR (A) and 60-mer DR (B) in the presence and absence of total RNA are determined by RT and qPCR. C, FN correction values for the GB RNA, two m6A sites in PLAC2, and ACTB 1165 for 40- and 60-mer DRs as determined from cleavage efficiencies in A and B. All error bars report means ± S.D. for three biological replicates.
To increase the binding specificity of DR, we compared a 60-mer DR and a 40-mer DR. We found that FDR values of 60-mer DR were higher than those of 40-mer DR (Fig. S2 and Fig. 4, A and B), likely because of a higher hybridization efficiency by 60-mer DR. FN values were consistently high for all tested RNAs, with the lowest FN values being 0.78 ± 0.02 for 40-mer DR and 0.93 ± 0.02 for 60-mer DR, demonstrating that the ability of our method to quantify m6A status should not be compromised by the presence of total RNA and that 60-mer can slightly outperform 40-mer DR (Fig. 4C). Overall, the average FN values were determined to be 0.94 ± 0.1 for 40-mer DR and 0.98 ± 0.05 for 60-mer DR. In addition, we compared the effect of DR concentration on FN values using PLAC2 m6A 2 DR as an example. We found that the cleavage efficiency is consistent at a wide range of DR concentrations as long as the molar concentration of DR is at least 10-fold higher than the molar concentration of RNA target (Fig. S6A). Likewise, the FN values stayed consistently at ∼1 for all tested concentrations of DR that were saturated compared with target RNA (Fig. S6B). Given these results, we can simplify Equation 6 to be as follows.
| (Eq. 7) |
Quantification of m6A fraction of endogenous sites
Having developed and validated our method, we applied it to determine the methylation fraction of several endogenous sites that were identified as potential m6A sites by RNA-Seq from more than one study: MALAT1 2515 (chr11 65500276), MALAT1 2577 (chr11 65500338), MALAT1 2611 (chr11 65500372), ACTB 1216 (chr7 5527743), LY6K 1171 (chr8 142703380), MCM5 2367 (chr22 35424323), SEC11A 1120 (chr15 84669674), INCENP 912 (chr11 62130275), INCENP 967 (chr11 62130330), INCENP 1060 (chr11 62130423), LMO7 2822 (chr13 75821377), and MRPL20 549 (chr1 1402080) (the genome position based on GRCh38.p13 primary assembly of m6A site is indicated in parentheses) (4, 14, 29). The selected RNAs vary from low to high abundance in HeLa cells, and some of them contain more than one modification site. Specifically, four of the targets (MALAT1 2515, MALAT1 2577, and MALAT1 2611, and ACTB 1216) were previously measured using the SCARLET assay (24), therefore serving as an additional validation of our method. To apply our method, a DR and a dDR were designed for each site. Because of the higher FDR and FN values with 60-mer DR, we chose to use the 60-mer DR for all endogenous RNAs. For each target site, we first generated in vitro transcribed RNAs containing the m6A sites of interest and performed DR digestion on these in vitro transcribed unmethylated RNAs to get FDR for each site. The FDR values were all greater than 0.49 and again varied among different RNAs (Fig. 5A and Fig. S3).
Figure 5.

Determination of m6A fraction of endogenous sites. A, the cleavage efficiencies (FDR) of the in vitro transcribed RNA by 60-mer DR as determined by RT and qPCR. B, determined m6A modification fractions of the 12 endogenous sites. C, determined m6A modification fractions for three endogenous targets using single and multiplexed measurements. All error bars report means ± S.D. for at least three biological replicates.
The methylation fractions of the endogenous sites were determined to range from 0.13 to 0.92 (Fig. 5B). Notably, our results show comparable methylation fractions for MALAT1 2515, MALAT1 2577, MALAT1 2611, and ACTB 1216 as in the SCARLET assay (24). Although the generally consistent results between our methods and SCARLET assay help validate our assay, we did notice that the values measured in our assay are slightly higher than those from SCARLET. One possible explanation for this slight variation can be the splint ligation step used in the SCARLET assay, in which the DNA oligonucleotide needs to be ligated to the RNase H cleaved RNA carrying either unmodified A or m6A at the 5′ end (24). It is possible that the splint ligation is less efficient for the m6A-containing RNA and therefore underestimates the m6A fraction in SCARLET assay.
Having validated that the method is able to quantify m6A fractions one site at a time, we investigated whether the method can be utilized in a multiplexed way. As a proof of concept, we chose to remeasure three m6A sites with varying levels of methylation: MALAT1 2611, ACTB 1216, and INCENP 912. For each biological replicate, the three corresponding active DRs were combined in one reaction, and the three dDRs were combined in another reaction. Similarly, RT reactions were also performed with combined RT primers for all three targets at either m6A site or internal control site, followed by separate qPCR for each target RNA. The multiplexed measurements of the methylation fractions were comparable with the ones we previously obtained in individual measurements (Fig. 5C). These results support that the method is accurate at measuring m6A fractions in a multiplexed fashion and can be further adapted for high-throughput measurements.
Effect of the nearby modifications on the DR cleavage efficiency
m6A modifications often exist in clusters (10). In addition, other types of RNA modifications are identified in mRNAs and lncRNAs (2). Therefore, the possible effect of the nearby modifications needs to be considered when applying this method. We designed synthetic RNA containing a nearby m6A, m1A, or ψ and measured their effects on cleavage efficiency of DR by PAGE analysis (Fig. 6 and Fig. S7). The cleavage efficiency was unaffected by the presence of m6A modification 2 and 4 nt upstream and downstream of the target site, suggesting that the method can be used to quantify the m6A fractions in RNA that contain m6A modifications in clusters. Furthermore, ψ modification caused a very minimal decrease in the cleavage efficiency of DR 2 nt away from target site and had no effect on the cleavage when present 4 nt away from the target site. Finally, m1A modification, which can affect the Watson–Crick base pairing, significantly decreased the cleavage efficiency at 2 nt away from the target site and moderately decreased the cleavage efficiency at 4 nt away from the target site. Overall, the results indicate that other nearby RNA modifications that do not affect the base pairing with the DR are not likely to affect the DR cleavage efficiency even when placed as close as only 2 nt away from the target site. However, nearby RNA modifications that weaken the base pairing with the DR will have a larger effect on the DR activity, but the effect decreases when the modification is more distal from the target site.
Figure 6.

The effects of nearby RNA modifications on cleavage efficiency (FDR) of DR. A, scheme of 35-nt synthetic RNA containing m6A, m1A, and ψ modifications. B, bar plot of the cleavage efficiencies of synthetic RNAs as quantified from PAGE. The error bars indicate means ± S.D. for three independent DR cleavage reactions.
Discussion
In summary, here we present a method for quantifying the absolute methylation fraction of potential m6A sites using a previously developed VMC10 DR (27), expanding the toolkit for site-specific quantification of m6A. In addition, the method can be adjusted for high-throughput quantification of m6A sites. Furthermore, as the VMC10 DR selectively cleaves the unmodified A, it can potentially be used to discriminate other modifications, such as m1A (30). We therefore expect that the DR-based quantification method can be easily applied to site-specific absolute quantifications of other RNA modifications.
Although this method is easy to implement, there are several limitations that need to be considered. First, the assay utilizes VMC10 DR, which has high cleavage efficiencies only on DGACH sequences, limiting its application on a subset of m6A sites with the DAACH sequences (27). Second, DR digestion efficiency varies among different sequences. Although low DR cleavage efficiency can be corrected by determining FDR for each modification site of interest using in vitro transcribed RNA, low DR efficiency can lead to less accurate quantification because of two reasons. First, a higher digestion efficiency leads to a larger ΔΔCt that reduces the measurement variation by qPCR. Conversely, low digestion efficiency will make the ΔΔCt too small to be accurately detected by qPCR. Second, the <5% cleavage efficiency on the modified RNA can lead to underestimation of the m6A fraction, and the percentage of underestimation depends on FDR (Fig. S1). A lower FDR will result in a larger underestimation. When FDR is 50%, a 5% cleavage of the modified RNA will result in a 10% underestimation of the m6A. Finally, the presence of a nearby modified nucleotide may affect the DR cleavage efficiency, depending on the type of the modification (Fig. 6).
To improve the accuracy of the measurement, there are also a few factors to note. First, for the synthetic RNA, we observed equal quality of Fm estimation using samples treated with dDR or samples lacking any DR as a negative control (Fig. 3C and Fig. S5). Nevertheless, we still recommend using dDR-treated sample as a negative control, because it corrects for potential changes in the RNA secondary structure caused by DR binding that can affect RT efficiency. Second, we recommend using 60-mer DR for quantification, because 60-mer DR overall has higher digestion efficiencies of unmethylated RNAs potentially because of a higher hybridization efficiency. Third, the quality of the primers used for RT and qPCR should be verified by performing calibration curves. Finally, we noticed that the largest source of technical variability in measurements originates from the RT step (compare error bars in Fig. 3, B and C). We therefore recommend performing multiple RT reactions for each DR-treated sample for a more accurate characterization.
Materials and methods
Cell culture and RNA extraction
HeLa cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% (v/v) fetal bovine serum (Thermo Fisher Scientific). The cells were grown at 37 °C under humidified conditions with 5% CO2. The total RNA was extracted using the RNeasy mini kit (Qiagen) according to the manufacturer's instructions.
In vitro transcription of endogenous RNA target fragments
The dsDNA templates for in vitro transcription were prepared by PCR with primers (Integrated DNA Technologies) that contain T7 promoter sequence and cDNA generated from total HeLa RNA. 1 μg of dsDNA templates were added to 100-μl reactions containing final concentrations of 2.5 mm each rNTP (New England Biolabs), 1× T7 polymerase reaction buffer (New England Biolabs), 4 mm MgCl2, 0.5 unit/μl SUPERase-In RNase inhibitor (Thermo Fisher Scientific), and 14 units/μl T7 polymerase (a kind gift from Dr. D. Bishop's Group). The reactions were incubated at 37 °C for 1 h. The transcript products were treated with DNase I recombinant (Roche) at 37 °C for 30 min and purified by phenol-chloroform extraction and ethanol precipitation. The primers are listed in Table S2.
In vitro transcription of 0 and 100% methylated GB RNA
A gene block containing a 460-nt random sequence with 51% GC content and one adenosine was purchased from Genewiz. The gene block sequence is listed in Table S1. The dsDNA template was amplified with primers containing an upstream T7 promoter sequence (Table S2). The in vitro reactions were carried out in the same conditions as for endogenous RNA targets, except that N6-methyladenosine-5′-triphosphate (Trilink Biotechnologies) was used instead of rATP for the generation of 100% methylated RNA. The transcript products were treated with DNase I recombinant (Roche) at 37 °C for 30 min and purified by phenol-chloroform extraction, 7% denaturing polyacrylamide gel, and ethanol precipitation.
Synthesis of RNA oligonucleotide
Unmodified phosphoramidites were purchased from Glen Research. Phosphoramidite of N6-methyladenosine was synthesized by following previously published procedure (17). RNA oligonucleotides were synthesized using Expedite DNA synthesizer at 1 μmol scale. After deprotection, RNA oligonucleotides were purified by PAGE.
Deoxyribozyme digestion
Total RNA (500 ng to 2 μg) or in vitro transcribed RNA fragments (50 nm) were mixed with 55.6 μm of either DR or dDR (Integrated DNA Technologies), 55.6 mm Tris-HCl (pH 7.5), and 166.7 mm NaCl in a final volume of 9 μl. The annealing of DR to the target site was facilitated by 5 min of incubation at 95 °C, followed by slow cooling to room temperature. After annealing, 1 μl of 200 mm MgCl2 was added to each reaction and incubated at 37 °C for 12 h. The final concentrations of the reagents in the incubation buffer are 50 μm of DR, 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, and 20 mm MgCl2 in a 10-μl reaction. The DR treatment of 35–40-nt MALAT1 2515, MALAT1 2577, and ACTB 1216 was carried out following the same protocol, except with stepwise cooling (95 °C for 5 min and 25 °C for 10 min) instead of slow cooling. To remove the DR after the digestion, 1.33 μl of 10× TURBO DNase buffer and 2 μl of TURBO DNase (Thermo Fisher Scientific) were added to the 10-μl DR reactions. The samples were incubated at 37 °C for 2 h. Subsequently, the DNase enzyme was inactivated by addition of EDTA (pH 7.5) to 15 mm final concentration and incubated at 75 °C for 10 min. The DR sequences are listed in the Table S3.
RNA digestion analyzed by PAGE
DR digestion reactions containing 50–100 ng of in vitro transcribed RNA or 35–40-nt synthetic RNAs were run on either 7% or 15% denaturing (7 m urea) PAGE, respectively. The gels were stained with SYBR green II RNA gel stain (Thermo Fisher Scientific) for 10 min and imaged with ChemiDocTM Imaging System (Bio-Rad). The cleavage efficiencies were analyzed with ImageJ using intensities of bands corresponding to the full-length RNA and the longer cleaved product.
Reverse transcription
For each DR-treated sample, separate RT reactions were performed with gene-specific reverse primers for the m6A region and internal reference site. Because of the presence of excess EDTA after DNase inactivation, either the reactions were significantly diluted or extra MgCl2 was added for maximum reverse transcriptase activity. The RNA was denatured at 70 °C for 5 min and then added to freshly prepared RT buffer with final concentration of 1 mm dNTPs (Thermo Fisher Scientific), 10% DMSO (Thermo Fisher Scientific), 10 mm DTT (Sigma–Aldrich), 250 nm of gene-specific reverse primer (Integrated DNA Technologies), and 20-fold dilution of reverse transcriptase from an iScript cDNA synthesis kit (Bio-Rad). The reactions were incubated at 25 °C for 5 min and at 46 °C for 20 min and heat-inactivated at 95 °C for 1 min. All primers are listed in Table S4.
qPCR
1 μl of cDNA was added into reaction mixture, containing 250 nm of each forward and reverse primers and 1× SsoAdvancedTM Universal SYBR® Green Supermix (Bio-Rad) in a final volume of 20 μl. The qPCRs were performed with CFX real-time PCR system (Bio-Rad), using preincubation of 95 °C for 30 s, followed by 40 cycles of 95 °C for 10 s and 60 °C for 30 s. The reactions were then subjected to melting curve analysis: 95 °C for 10 s and 65 °C for 5-s increments by 0.5 °C to 95 °C for 5 s. The data were analyzed with the supporting Bio-Rad CFX Maestro software. All primers are listed in Table S4. All error bars in the figures are means ± S.D. of multiple biological replicates. For in vitro prepared GB RNA with different input m6A fraction, biological replicates are defined as independently mixed GB RNA samples. For the cases of endogenous mRNAs, biological replicates are defined as independently extracted total RNA samples. The m6A fraction calculated for each biological replicate is from the average values of multiple technical replicates defined by independently performed RT reactions for each RNA sample.
Data availability
All data are contained within the manuscript and the supporting information file.
Author contributions
M. B., J. Z., and J. F. conceptualization; M. B., J. Z., Q. D., and E. M. H. data curation; M. B. formal analysis; M. B. validation; M. B. and J. F. writing-original draft; M. B., J. Z., Q. D., E. M. H., and J. F. writing-review and editing; J. Z., Q. D., E. M. H., and J. F. investigation; J. F. supervision; J. F. funding acquisition; J. F. project administration.
Supplementary Material
Acknowledgments
We thank Dr. C. He and Dr. T. Pan for useful discussion and Dr. A. Driouchi for comments on the manuscript.
This work was supported by funds from the Searle Scholars Program, National Institutes of Health Director's New Innovator Award 1DP2GM128185-01, and a pilot grant from the National Institutes of Health Center for the Dynamic RNA Epitranscriptomes at the University of Chicago (to J. F.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1–S4 and Figs. S1–S7.
- m6A
- N6-methyladenosine
- lncRNA
- long noncoding RNA
- qPCR
- quantitative PCR
- SCARLET
- site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and TLC
- DR
- deoxyribozyme
- GB RNA
- 460-nt in vitro transcribed RNA from a gene block sequence with only one adenine in the sequence
- dDR
- dead DR.
References
- 1. Boccaletto P., Machnicka M. A., Purta E., Piatkowski P., Baginski B., Wirecki T. K., de Crécy-Lagard V., Ross R., Limbach P. A., Kotter A., Helm M., and Bujnicki J. M. (2018) MODOMICS: a database of RNA modification pathways. 2017 update. Nucleic Acids Res. 46, D303–D307 10.1093/nar/gkx1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nachtergaele S., and He C. (2018) Chemical modifications in the life of an mRNA transcript. Annu. Rev. Genet. 52, 349–372 10.1146/annurev-genet-120417-031522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Maity A., and Das B. (2016) N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J. 283, 1607–1630 10.1111/febs.13614 [DOI] [PubMed] [Google Scholar]
- 4. Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., Dai Q., Chen W., and He C. (2014) A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10, 93–95 10.1038/nchembio.1432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Warda A. S., Kretschmer J., Hackert P., Lenz C., Urlaub H., Höbartner C., Sloan K. E., and Bohnsack M. T. (2017) Human METTL16 is a N6-methyladenosine (m6A) methyltransferase that targets pre-mRNAs and various non-coding RNAs. EMBO Rep. 18, 2004–2014 10.15252/embr.201744940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.-G., and He C. (2011) N6-Methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7, 885–887 10.1038/nchembio.687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zheng G., Dahl J. A., Niu Y., Fedorcsak P., Huang C.-M., Li C. J., Vågbø C. B., Shi Y., Wang W.-L., Song S.-H., Lu Z., Bosmans R. P., Dai Q., Hao Y.-J., Yang X., et al. (2013) ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49, 18–29 10.1016/j.molcel.2012.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kasowitz S. D., Ma J., Anderson S. J., Leu N. A., Xu Y., Gregory B. D., Schultz R. M., and Wang P. J. (2018) Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet. 14, e1007412 10.1371/journal.pgen.1007412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shi H., Wei J., and He C. (2019) Where, when, and how: context-dependent functions of RNA methylation writers, readers, and erasers. Mol. Cell 74, 640–650 10.1016/j.molcel.2019.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Meyer K. D., Saletore Y., Zumbo P., Elemento O., Mason C. E., and Jaffrey S. R. (2012) Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149, 1635–1646 10.1016/j.cell.2012.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., and Rechavi G. (2012) Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485, 201–206 10.1038/nature11112 [DOI] [PubMed] [Google Scholar]
- 12. Liu N., Dai Q., Zheng G., He C., Parisien M., and Pan T. (2015) N6-Methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518, 560–564 10.1038/nature14234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chen K., Lu Z., Wang X., Fu Y., Luo G.-Z., Liu N., Han D., Dominissini D., Dai Q., Pan T., and He C. (2015) High-resolution N6-methyladenosine (m6A) map using photo-crosslinking-assisted m6A sequencing. Angew. Chem. Int. Ed. Engl. 54, 1587–1590 10.1002/anie.201410647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Linder B., Grozhik A. V., Olarerin-George A. O., Meydan C., Mason C. E., and Jaffrey S. R. (2015) Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat. Methods 12, 767–772 10.1038/nmeth.3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helm M., and Motorin Y. (2017) Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet. 18, 275–291 10.1038/nrg.2016.169 [DOI] [PubMed] [Google Scholar]
- 16. Lei Z., and Yi C. (2017) A radiolabeling-free, qPCR-based method for locus-specific pseudouridine detection. Angew. Chem. Int. Ed. Engl. 56, 14878–14882 10.1002/anie.201708276 [DOI] [PubMed] [Google Scholar]
- 17. Dai Q., Fong R., Saikia M., Stephenson D., Yu Y. T., Pan T., and Piccirilli J. A. (2007) Identification of recognition residues for ligation-based detection and quantitation of pseudouridine and N6-methyladenosine. Nucleic Acids Res. 35, 6322–6329 10.1093/nar/gkm657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Liu W., Yan J., Zhang Z., Pian H., Liu C., and Li Z. (2018) Identification of a selective DNA ligase for accurate recognition and ultrasensitive quantification of N6-methyladenosine in RNA at one-nucleotide resolution. Chem. Sci. 9, 3354–3359 10.1039/C7SC05233B [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Harcourt E. M., Ehrenschwender T., Batista P. J., Chang H. Y., and Kool E. T. (2013) Identification of a selective polymerase enables detection of N6-methyladenosine in RNA. J. Am. Chem. Soc. 135, 19079–19082 10.1021/ja4105792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang S., Wang J., Zhang X., Fu B., Song Y., Ma P., Gu K., Zhou X., Zhang X., Tian T., and Zhou X. (2016) N6-Methyladenine hinders RNA- and DNA-directed DNA synthesis: application in human rRNA methylation analysis of clinical specimens. Chem. Sci. 7, 1440–1446 10.1039/C5SC02902C [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao Y., Wang Y., Tang Q., Wei L., Zhang X., and Jia G. (2018) An elongation- and ligation-Based qPCR amplification method for the radiolabeling-free detection of locus-specific N6-methyladenosine modification. Angew. Chem. Int. Ed. Engl. 57, 15995–16000 10.1002/anie.201807942 [DOI] [PubMed] [Google Scholar]
- 22. Potapov V., Ong J. L., Langhorst B. W., Bilotti K., Cahoon D., Canton B., Knight T. F., Evans T. C. Jr., and Lohman G. J. S. (2018) A single-molecule sequencing assay for the comprehensive profiling of T4 DNA ligase fidelity and bias during DNA end-joining. Nucleic Acids Res. 46, e79 10.1093/nar/gky303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harada K., and Orgel L. E. (1993) Unexpected substrate specificity of T4 DNA ligase revealed by in vitro selection. Nucleic Acids Res. 21, 2287–2291 10.1093/nar/21.10.2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu N., Parisien M., Dai Q., Zheng G., He C., and Pan T. (2013) Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19, 1848–1856 10.1261/rna.041178.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang Z., Chen L.-Q., Zhao Y.-L., Yang C.-G., Roundtree I. A., Zhang Z., Ren J., Xie W., He C., and Luo G.-Z. (2019) Single-base mapping of m6 A by an antibody-independent method. Sci. Adv. 5, eaax0250 10.1126/sciadv.aax0250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garcia-Campos M. A., Edelheit S., Toth U., Safra M., Shachar R., Viukov S., Winkler R., Nir R., Lasman L., Brandis A., Hanna J. H., Rossmanith W., and Schwartz S. (2019) Deciphering the “m6A code” via antibody-independent quantitative profiling. Cell 178, 731–747.e16 10.1016/j.cell.2019.06.013 [DOI] [PubMed] [Google Scholar]
- 27. Sednev M. V., Mykhailiuk V., Choudhury P., Halang J., Sloan K. E., Bohnsack M. T., and Höbartner C. (2018) N6-Methyladenosine-sensitive RNA-cleaving deoxyribozymes. Angew. Chem. Int. Ed. Engl. 57, 15117–15121 10.1002/anie.201808745 [DOI] [PubMed] [Google Scholar]
- 28. Santoro S. W., and Joyce G. F. (1997) A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. U.S.A. 94, 4262–4266 10.1073/pnas.94.9.4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ke S., Pandya-Jones A., Saito Y., Fak J. J., Vågbø C. B., Geula S., Hanna J. H., Black D. L., Darnell J. E. Jr., and Darnell R. B. (2017) m6A mRNA modifications are deposited in nascent pre-mRNA and are not required for splicing but do specify cytoplasmic turnover. Genes Dev. 31, 990–1006 10.1101/gad.301036.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tserovski L., Marchand V., Hauenschild R., Blanloeil-Oillo F., Helm M., and Motorin Y. (2016) High-throughput sequencing for 1-methyladenosine (m1A) mapping in RNA. Methods 107, 110–121 10.1016/j.ymeth.2016.02.012 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript and the supporting information file.