

Abstract
Nitric oxide/cyclic guanosine monophosphate (cGMP) signaling is compromised in Alzheimer’s disease (AD), and phosphodiesterase 5 (PDE5), which degrades cGMP, is upregulated. Sildenafil inhibits PDE5 and increases cGMP levels. Integrating previous findings, we determine that most doses of sildenafil (especially low doses) likely activate peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α) via protein kinase G-mediated cyclic adenosine monophosphate (cAMP) response element binding protein (CREB) phosphorylation and/or Sirtuin-1 activation and PGC1α deacetylation. Via PGC1α signaling, low-dose sildenafil likely suppresses β-secretase 1 expression and amyloid-β (Aβ) generation, upregulates antioxidant enzymes, and induces mitochondrial biogenesis. Plus, sildenafil should increase brain perfusion, insulin sensitivity, long-term potentiation, and neurogenesis while suppressing neural apoptosis and inflammation. A systematic review of sildenafil in AD was undertaken. In vitro, sildenafil protected neural mitochondria from Aβ and advanced glycation end products. In transgenic AD mice, sildenafil was found to rescue deficits in CREB phosphorylation and memory, upregulate brain-derived neurotrophic factor, reduce reactive astrocytes and microglia, decrease interleukin-1β, interleukin-6, and tumor necrosis factor-α, decrease neural apoptosis, increase neurogenesis, and reduce tau hyperphosphorylation. All studies that tested Aβ levels reported significant improvements except the two that used the highest dosage, consistent with the dose-limiting effect of cGMP-induced phosphodiesterase 2 (PDE2) activation and cAMP depletion on PGC1α signaling. In AD patients, a single dose of sildenafil decreased spontaneous neural activity, increased cerebral blood flow, and increased the cerebral metabolic rate of oxygen. A randomized control trial of sildenafil (ideally with a PDE2 inhibitor) in AD patients is warranted.
Keywords: Alzheimer’s disease, cyclic GMP, mitochondrial biogenesis, peroxisome proliferator-activated receptor gamma coactivator 1-alpha, sildenafil citrate
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia worldwide, and AD patients and their families urgently require novel therapeutics to prevent and slow the progression of this devastating disorder. Hallmarks of AD include amyloid-β (Aβ) peptide secretion and deposition into neuritic plaques, tau protein hyperphosphorylation and neurofibrillary tangle formation, metal ion dyshomeostasis [1–9], oxidative stress and lipid, nucleic acid, and protein damage [10–13], abortive cell cycle reentry [14–26], neuroinflammation and microbial dysbiosis [27–33], insulin resistance [34, 35], cerebrovascular dysfunction [36–38], synaptic dysfunction [39, 40], neuronal loss, endoplasmic reticulum stress [41–44], and mitochondrial dysfunction [45–48].
Sildenafil (Viagra) is a drug used to treat erectile dysfunction and pulmonary arterial hypertension that inhibits phosphodiesterase 5 (PDE5) (Fig. 1). PDE5 degrades cyclic guanosine monophosphate (cGMP). Upstream of cGMP, normally, the amino acid L-arginine is converted by three varieties of the enzyme nitric oxide synthase (NOS) into nitric oxide (NO). NO is a small cell-permeable gas molecule that diffuses across the plasma membrane and activates soluble guanylyl cyclase (sGC). sGC converts guanosine triphosphate (GTP) into cGMP [49]. However, in AD, the activity of the NOS/NO/cGMP pathway is severely impaired. NOS activity is significantly decreased in AD patients’ superior frontal gyri and hippocampi compared to age-matched controls [50], even though aberrant neuronal NOS (nNOS) protein expression has been observed in a subpopulation of isocortical pyramidal neurons in AD patients’ brains [51, 52] and the intensity of astrocyte endothelial NOS (eNOS) and inducible NOS (iNOS) expression had increased in AD patients’ deep cortical layers [52, 53]. NO-induced soluble sGC (but not basal sGC or particulate guanylyl cyclase) activity was found to be decreased by 50% in AD patients’ superior temporal cortices compared to controls [54]. cGMP levels were found to be significantly lower in AD patients’ cerebrospinal fluid (CSF) compared to controls, and decreases in levels of cGMP correlated with CSF Aβ42 levels [55], comorbid depression [56], and cognitive decline as measured by Mini-Mental State Examination (MMSE) [55, 57].
Fig. 1.
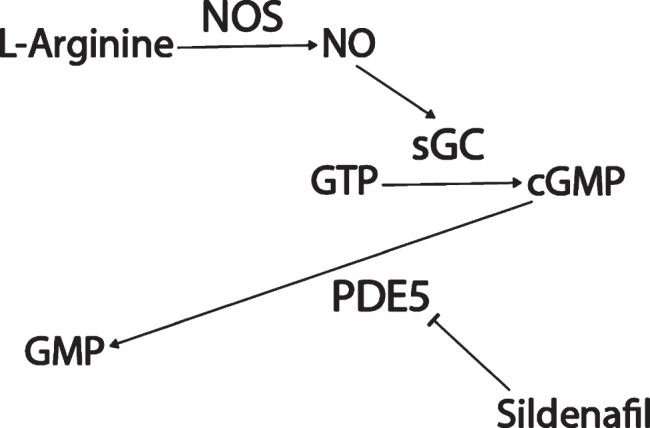
Sildenafil mechanism of action.
Potentially contributing to this cGMP depletion, PDE5 protein levels have been found to be significantly upregulated in AD patients’ temporal cortices [55], and PDE5 mRNA levels are significantly elevated in AD patients’ entorhinal cortices compared to controls according to a meta-analysis of mRNA datasets (p = 0.001, FDR = 0.018; Fig. 2) [58]. Granted, low PDE5 mRNA expression in the human CNS has been reported relative to peripheral tissue [59–61], and no specific hybridization signal was observed for PDE5 mRNA in the brains of aged and AD patients using radioactive in situ hybridization histochemistry in one study, drawing skepticism as to whether PDE5 inhibition has relevance to AD [62–64]. Regardless though, the weight of the evidence suggests that PDE5 is expressed (albeit relatively less than in peripheral tissues) in the normal human brain [58–61] and that, moreover, it is upregulated in the AD entorhinal and temporal cortices [55, 58], supporting the notion that PDE5 may be an important therapeutic target in AD.
Fig. 2.

PDE5A mRNA levels upregulated in the AD entorhinal cortex.
cGMP/PGC1α signaling
This multi-mechanism NOS/NO/cGMP signaling dysfunction is an important therapeutic target in AD for multiple reasons. One reason is that cGMP is responsible for increasing the expression and activity of peroxisome proliferator-activated receptor-γ coactivator 1α (PGC1α). PGC1α overexpression (or low-dose NO) suppresses the expression of β-secretase 1 (BACE1) [65], the rate-limiting enzyme in Aβ generation, suggesting that PGC1α activity suppresses Aβ generation. In addition, PGC1α is a master transcriptional regulator of mitochondrial biogenesis, oxidative respiration [66, 67], fatty acid β-oxidation [68], and antioxidant defense [69]. PGC1α upregulates multiple antioxidant genes, including mitochondrial manganese superoxide dismutase (MnSOD), when bound to Forkhead box O3a (Foxo3a) and deacetylated by Sirtuin-1 (SIRT1) [69]. However, PGC1α protein levels are significantly lower in AD patients’ hippocampi compared to controls [48], and mitochondrial biogenesis and MnSOD expression are impaired there [45, 48]. Sildenafil has the potential to reverse the hippocampal PGC1α suppression in AD. 10μM sildenafil treatment for 24 hours in vitro and 0.3 mg/kg sildenafil in vivo have been shown to induce PGC1α expression and mitochondrial biogenesis by increasing cGMP in renal proximal tubular cells and mouse renal cortex, respectively [70]. In addition, sildenafil has been shown to upregulate SOD and catalase activities in rat liver (1.48 mg/kg sildenafil) and human blood (100 mg sildenafil one time dosage) [71, 72].
Multiphasic regulation of PGC1α by sildenafil and cGMP
NOS/NO/sGC/cGMP signaling upregulates PGC1α in diverse cell types, including not only renal proximal tubular cells [70], but also brown adipocytes, U937 monocytic cells, HeLa cervical cancer-derived cells, white 3T3-L1 adipocytes [73, 74], and probably neurons [75]. In mice, subcortical brain tissue responded to hypoxia with PGC1α upregulation (as well as with mitochondrial biogenesis) in a manner that depended on nNOS expression [75], so the nNOS/NO/cGMP/PGC1α pathway does appear to be active in neurons.
An important caveat to the claim that sildenafil and cGMP activate PGC1α though is that NOS/NO/cGMP appears to be a multiphasic rheostat of PGC1α signaling whose outcome depends on cGMP concentration, duration, and crosstalk with cAMP signaling mediated by phosphodiesterase 3 (PDE3) and phosphodiesterase 2 (PDE2) activity. Low concentrations of cGMP compete for the active site of PDE3 [76], thereby impeding PDE3 from degrading cAMP and increasing cAMP levels. By contrast, high cGMP concentrations allosterically activate PDE2, thereby promoting cAMP degradation [76, 77]. Mirroring this, low-dose sildenafil (up to 1μM) inhibits PDE3, resulting in cAMP accumulation [76], whereas high-dose sildenafil (10 and 100μM) activates PDE2, resulting in cAMP degradation [76] and suppression of the cAMP/EPAC/adenosine monophosphate-activated protein kinase (AMPK) pathway [76, 78, 79]. For this reason, excessively high doses of sildenafil resulted in inferior therapeutic responses compared to low doses in preclinical models of non-alcoholic hepatic steatosis and obesity [76]. This caveat may be relevant to AD as well. The dose-dependent effect of sildenafil and cGMP signaling on cAMP levels should affect PGC1α regulation since cAMP is required for robust PGC1α expression and mitochondrial biogenesis in AD [48, 67]. On top of this, although NO/sGC/cGMP signaling typically promotes PGC1α activity, it also simultaneously activates the protein kinase G (PKG)/phosphoinositide 3-kinase (PI3K)/Akt signaling cascade, which decreases PGC1α activity [80–83]; opposing this negative regulation of PGC1α by cGMP, cAMP inhibits Akt via PKA and Rap1b (Fig. 4) [84]. In other words, cGMP per se simultaneously increases and decreases PGC1α signaling by distinct mechanisms, with crosstalk with cAMP pathways probably determining whether PGC1α is activated or inhibited overall. Therefore, since high doses of sildenafil activate PDE2 and decrease cAMP levels, high-dose sildenafil would be expected to be less effective than low-dose sildenafil at activating PGC1α, inducing mitochondrial biogenesis, upregulating antioxidant genes, and downregulating BACE1 [76].
Fig. 4.

How sildenafil and cGMP may regulate PGC1α.
Low and high but not moderate cGMP levels activate PGC1α?
Consistent with the dose-dependent effect of sildenafil and cGMP on PGC1α, 10μM of 8-Br-cGMP treatment for 24 hours upregulated PGC1α mRNA expression and mitochondrial biogenesis in renal proximal tubular cells [70]. By contrast, in endothelial cells, 100μM 8-Br-cGMP treatment for 6–24 hours downregulated PGC1α [80, 83]. In addition, treatment for less than 12 hours with NO donors decreased PGC1α expression, whereas treatment for 24 hours or more increased PGC1α expression [83]. NO-triggered PGC1α downregulation and consequent reactive oxygen species (ROS) production were required for endothelial cell migration, an effect that was mediated by NO-activated PI3K/Akt signaling and Akt phosphor-inhibition of Foxo3a [80].
Surprisingly though, much higher dosages of cGMP than 100μM upregulate PGC1α like lower doses do: for example, PGC1α was upregulated in brown adipocytes treated with 1 mM 8-Br-cGMP for 4 days [73]. In human monocytic U937 cells, rat L6 myotubes, and rat PC12 neurosecretory cells, 3 mM 8-Br-cGMP treatment every day for 6 days upregulated PGC1α, as well as Nrf1 and Tfam, mitochondrial proteins Cox IV and Cytochrome C, and mitochondrial DNA content [66]. Furthermore, 6 days of 3 mM 8-Br-cGMP in these three cell types resulted in increased oxidative phosphorylation-coupled oxygen consumption [66].
Therefore, there appears to be a U-shaped dose-response curve between cGMP concentrations and PGC1α expression, with PGC1α being downregulated by 100μM cGMP for 6–24 hours [80, 83] but upregulated by 10μM cGMP or 1–3 mM cGMP for 24 hours or longer (Fig. 3) [66, 70, 73]. This might be because 100μM cGMP corresponds to the 10–100μM dosage of sildenafil that is sufficient to activate PDE2 and lower cAMP [76], whereas much higher dosages of cGMP and sildenafil nevertheless stimulate mitochondrial biogenesis and PGC1α transcription [66, 73] because supraphysiological cGMP signaling is sufficient to overcome the cAMP depletion to induce robust PGC1α activity independently of cAMP, perhaps via cAMP response element binding protein (CREB) phosphorylation and SIRT1 activation (see next subsection). If so, this would not be unprecedented: in fibroblasts, PDE2 overexpression was sufficient to lower cAMP levels and induce a transition into a pro-fibrotic myofibroblast phenotype, an alternation that cGMP elevating agents reversed, suggesting that sufficiently high cGMP levels can overcome the deficits to cellular signaling induced by PDE2-mediated cAMP depletion [77]. Therefore, it appears that, for optimal PGC1α expression, mitochondrial biogenesis, and BACE1 downregulation, either sufficiently low or high sildenafil dosages must be used, or sildenafil must be co-administered with a PDE2 inhibitor.
Fig. 3.

Multiphasic dose response of PGC1α expression to cGMP concentration.
In addition to this complex but clinically relevant dose-specific effect, the mechanism by which NO/cGMP typically upregulates PGC1α remains unresolved [67]. Two potential mechanisms involving either CREB or SIRT1 will now be described.
How sildenafil and cGMP activate PGC1α
In the hippocampus during long-term potentiation and in other neural tissues, both the cGMP/PKG and the cAMP/PKA pathways contribute to CREB phosphorylation [85–90], and PKA-mediated CREB phosphorylation promotes PGC1α transcription [67], so cGMP/PKG/CREB signaling might promote PGC1α expression as well. In addition, cGMP may contribute to the post-translational regulation of PGC1α. Once transcribed and translated, the stability, subcellular localization, and co-activator activity of PGC1α proteins are regulated by multiple post-translational modifications. For example, PGC1α can be inhibited via phosphorylation by Akt [81, 82], S6 Kinase [91], or glycogen synthase kinase 3β (GSK3β) [74, 92], or via acetylation by general control of amino acid synthesis 5 (GCN5) [81, 93–95] or p300 [96]. Conversely, PGC1α can be activated via phosphorylation by adenosine monophosphate activated protein kinase (AMPK) [97–101] or p38 mitogen activated protein kinase (MAPK) [101, 102], via methylation by protein arginine methyltransferase 1 (PRMT1) [103], or via deacetylation by SIRT1 (Fig. 5) [69, 93, 94, 96, 101, 104–107]. Interestingly given the SIRT1-mediated activation of PGC1α, sildenafil has been shown to upregulate SIRT1 in heart, cardiomyocytes [108], serum, and subcutaneous adipose tissue [109]. cGMP analogues upregulated SIRT1 expression in white adipose tissue [110, 111] and in osteoblasts [112]. Mice with osteoblast-specific PKGII overexpression exhibited increased SIRT1 expression [112]. The PKG inhibitor KT-5823 blocked the relief of spinal allodynia induced by resveratrol, a SIRT1 activator [113, 114]. In hypoxic myocardial cells, 1μM sildenafil treatment decreased PGC1α protein acetylation [115, 116]. Therefore, it is possible that sildenafil may promote PGC1α deacetylation in some contexts via cGMP/PKG/SIRT1 signaling (Fig. 5).
Fig. 5.
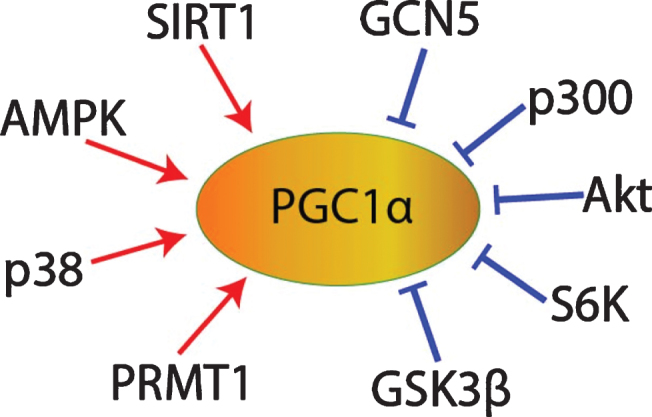
PGC1α post-translational regulation.
Benefits of sildenafil in AD
Since most doses of sildenafil/cGMP activate PGC1α and PGC1α signaling induces mitochondrial biogenesis [67, 70], upregulates antioxidant enzymes [69], and suppresses BACE1 expression [65], sildenafil should provide significant benefits to patients with AD. In addition to its PGC1α-specific benefits, sildenafil promotes smooth muscle relaxation and vasodilation via cGMP [117], which might provide additional benefit to patients with AD since hypoperfusion is also a significant impairment in AD patients’ brains [37, 38, 118]. Sildenafil suppresses apoptosis in hypoxic neurons [117, 119] and promotes neurogenesis [120–123], so it might slow the loss of AD neurons and promote the replenishment of new ones. Furthermore, sildenafil improves insulin sensitivity and endothelial inflammation in patients [124–126], so sildenafil might also promote insulin sensitivity and suppress inflammation in AD brains. Since cGMP/PKG signaling mediates long-term potentiation via CREB phosphorylation [85–90, 117], sildenafil should improve the learning and memory impairments associated with AD. Therefore, in theory, most doses of sildenafil should improve multiple hallmarks of AD, including excessive Aβ generation, impaired mitochondrial biogenesis, oxidative stress, neuroinflammation, hypoperfusion, insulin resistance, neuron loss, insufficient neurogenesis, and memory deficits.
Sildenafil and AD comorbidities and risk factors
It is also important to consider the effects of sildenafil on common AD comorbidities and/or risk factor conditions, such as type II diabetes, cardiovascular diseases, and depression, since many AD patients suffer from one or more of these conditions.
Regarding the effect of sildenafil in depression, NOS/NO/sGC/cGMP and serotonin signaling tend to oppose each other. cGMP triggers cerebral vasodilation [127], whereas serotonin induces cerebral vasoconstriction [128, 129]. NOS/NO/sGC/cGMP/PKG signaling activates the serotonin transporter (SERT), inducing serotonin reuptake [130–132]. For this reason, sildenafil might be expected to make selective serotonin reuptake inhibitor (SSRI) antidepressants less effective. However, there do not appear to be any reports of this being the case, and sildenafil has been used safely and successfully to treat erective dysfunction in patients taking SSRIs [133]. Moreover, sildenafil itself has been shown to exert an antidepressant effect in mice [134].
Regarding the effects of sildenafil in type II diabetes, it has been found in a randomized, double-blind, placebo-controlled study that 25 mg thrice daily for 3 months sildenafil increases insulin sensitivity in patients with pre-diabetes, indicating that sildenafil might be beneficial for patients with AD and type II diabetes [125]. Regarding the effects of sildenafil and cardiovascular diseases, despite early concerns [135], sildenafil usage does not appear to contribute to myocardial infarction or sudden cardiac death risk [136]. In fact, treatment of erectile dysfunction in patients who had had a myocardial infarction with PDE5 inhibitors (but not alprostadil) correlated with a reduced risk of mortality and hospitalization for heart failure (n = 43,145) [137]. Preclinically, in a mouse model of hypercholesterolemia, sildenafil decreased aortic atherosclerotic plaques by 40% [138]. Furthermore, sildenafil decreases cardiac hypertrophy [139]. Therefore, sildenafil treatment in AD patients with comorbid cardiovascular diseases would be expected to be safe and potentially beneficial.
MATERIALS AND METHODS
To determine the current progress in studying sildenafil and AD, we searched PubMed for “sildenafil Alzheimer’s.” Both preclinical and clinical studies were reviewed. Results that were not about the effect of sildenafil on patients or preclinical models with AD (e.g., studies about the interaction between cGMP and Aβ in long-term potentiation) were discarded. To ensure clinical relevancy, studies about derivates of sildenafil were also discarded.
RESULTS
As per the methods section, two in vitro studies, ten rodent studies, one systematic review, and two pilot studies in patients were included. Overall, all the studies supported the use of sildenafil in AD (see Table 1 for a summary of results).
Table 1.
Literature review results
| Dosage | Population | Results | Study |
| 10–100μM sildenafil | HT-22 hippocampal neuronal cells exposed to Aβ 25 - 35 | Sildenafil rescued mitochondrial Ca2 + overload and dysfunction due to Aβ 25 - 35 by opening ATP-sensitive K + channels | [140] |
| 20μM sildenafil | HT-22 hippocampal neuronal cells exposed to advanced glycation end products | Sildenafil decreased mitochondrial permeability transition pore opening and apoptosis via HO1 upregulation | [141] |
| Variable, 3 mg/kg | Scopolamine-induced cholinergic dysfunction mice | Sildenafil rescued memory | [143] |
| Variable, primarily 3 mg/kg/day intraperitoneal sildenafil | APP/PS1 mice | Sildenafil rescued long-term potentiation, CREB phosphorylation, memory, and decreased Aβ levels | [144] |
| 6 mg/kg, intraperitoneal daily for 3 months | APP/PS1 mice | Sildenafil improved memory, amyloid plaque load, inflammation, and neurogenesis | [145] |
| 2 mg/kg sildenafil twice daily for 4 months | APP/PS1 mice | Sildenafil rescued memory and amyloid pathology, downregulated PDE5, and increased NOS, NO, and cGMP levels | [146] |
| Sildenafil was dissolved in 0.9% saline at a concentration of 1.0 mg/ml. 10.0 mg/kg of this solution was administered intraperitoneally with an injection volume of 0.1 ml/10 g. | APP/PS1 mice | Sildenafil improved memory, decreased levels of Aβ, IL-1β, IL-6 and TNF-α, and increased p-CREB | [147] |
| 15 mg/kg sildenafil daily for 10 weeks in water | J20 mice | Sildenafil improved memory, tau hyperphosphorylation, and Akt and GSK3β phosphorylation, but not prefrontal cortex Aβ 42 levels | [61] |
| 15 mg/kg daily sildenafil, intraperitoneal | Tg2576 AD mice | Sildenafil improved memory, tau but not frontal cortex amyloid pathology, inhibited GSK3β, decreased CDK5 p25/35 ratio, upregulated BDNF and Arc | [148] |
| Variable | Aged mice | Sildenafil improved spatial memory retention but not acquisition | [64] |
| 3 mg/kg intraperitoneal sildenafil daily for 3 weeks | Aged mice | Sildenafil decreased double-stranded DNA breaks and pro-apoptotic caspase-3 and Bax and upregulated antiapoptotic Bcl2 and BDNF | [149] |
| 7.5 mg/kg sildenafil intraperitoneally once daily for 4 weeks | SAMP8 mice | Sildenafil improved amyloid and tau pathology, memory, and gliosis | [145, 150] |
| 7.5 mg/kg sildenafil intraperitoneally once daily for 4 weeks | SAMP8 mice | Sildenafil decreased JNK phosphor-activation in the hippocampus, tau phosphorylation, and memory deficits | [151] |
| 50 mg sildenafil, single dosage | 10 AD patients | Sildenafil decreased spontaneous neural activity in right hippocampus | [152] |
| 50 mg sildenafil, single dosage | 14 AD patients | Sildenafil increased cerebral metabolic rate of oxygen and cerebral blood flow in 12 patients, decreased cerebral vascular reactivity in 8 patients | [127] |
HT-22 hippocampal neuronal cells treated with Aβ 25 - 35 exhibited mitochondrial calcium overload, which was associated with ATP depletion, ROS generation, permeability transition pore opening, caspase-9 activation, and cell death. Sildenafil prevented these effects by promoting the opening of ATP-sensitive K + channels [140]. In cultured HT-22 hippocampal neuronal cells, exposure to advanced glycation end products [141] (a risk factor for AD [142]) induced mitochondrial ROS generation, depleted intracellular ATP, opened the mitochondrial permeability transition pore, released cytochrome C, activated caspase-3, and initiated apoptosis. Treatment with sildenafil upregulated heme oxygenase 1 (HO1), protected mitochondria from permeability transition pore opening and cytochrome C release, and decreased caspase-3 activation and apoptosis [141]. HO1 expression was required for sildenafil-induced protection of mitochondrial reductive capacity and permeability transition pore opening [141], suggesting that sildenafil protected mitochondria via HO1 upregulation.
In mice with cholinergic dysfunction mimicking AD due to scopolamine injection, sildenafil rescued maze performance, with 3 mg/kg appearing to be more efficacious than the 1.5 or 4.5 mg/kg dosages [143].
In hippocampal slices from transgenic amyloid precursor protein (APP)/presenilin 1 (PS1) AD mice, 50 nM sildenafil rescued the deficits in tetanus-induced long-term potentiation in the Schaffer collateral pathway caused by the APP/PS1 genotype [144]. In vivo sildenafil treatment produced similar results. In APP/PS1 mice, a one-time dosage of 3 mg/kg sildenafil rescued contextual fear memory. Daily intraperitoneal dosages for 2-3 weeks of 3 mg/kg sildenafil partially rescued spatial working memory deficits on the radial arm water maze test. Similar benefits were found for daily dosages of 3 mg/kg sildenafil 9–12 weeks after treatment cessation, indicating a long-term benefit that persists even after treatment cessation. Sildenafil also rescued long-term spatial reference memory on the Morris water maze and the probe trial. Sildenafil treatment rescued tetanus-induced CREB phosphorylation in the CA1 hippocampus to normal levels. 3 weeks of daily 3 mg/kg sildenafil was sufficient to reduce Aβ 40 and Aβ 42 levels in cerebral cortex samples [144].
In APP/PS1 mice administered sildenafil 6 mg/kg intraperitoneally daily for 3 months, significant improvements were documented in behavioral tests (nesting behavior, arm entries in the Y maze, Morris water maze escape latency and path length), as well as immunoreactivity of inflammatory microglial and astrocytic markers ionized calcium binding adaptor molecule 1 (Iba1) and glial fibrillary acidic protein (GFAP), neurogenesis as shown by NeuN-positive neurons and doublecortin (DCX)-positive cells in dentate gyrus, and amyloid plaque burden [145].
In APP/PS1 AD mice, 2 mg/kg sildenafil twice daily for 4 months rescued cognition as shown by spontaneous alternation and escape from electrical stimulation in the Y-maze test, decreased amyloid pathology as shown by decreased cortical and hippocampal AβPP, Aβ 40, and Aβ 42 levels, decreased PDE5 expression, and increased nNOS, eNOS, iNOS, NO, and cGMP levels [146].
Sub-chronic intraperitoneal sildenafil treatment in APP/PS1 mice was found to improve memory as shown by novel object recognition preference, downregulated proinflammatory cytokines interleukin-1β (IL-1β), intereukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) in the hippocampus, decreased hippocampal soluble Aβ 40 and Aβ 42 expression, and increased CREB phosphorylation [147].
A recent systematic review found that, on the Morris water maze and the T-maze, sildenafil improved spatial memory retention but not acquisition in aged mice [64]. Interestingly, this review reported that PDE5 is not located in brain structures critical for AD based on the lack of specific hybridization of a PDE5 mRNA probe [62–64], but as noted, other groups have found increased PDE5 mRNA or protein expression in AD patients’ entorhinal [58] or temporal cortices [55], respectively, and PDE5 mRNA expression has been found by multiple groups in the normal human brain, albeit at lower levels than in peripheral tissues [58–61].
In J20 AD mice, 15 mg/kg sildenafil daily for 10 weeks in drinking water resulted in improved performance on the Morris water maze, decreased tau hyperphosphorylation, and increased Akt and GSK3β phosphorylation, but it did not alter prefrontal cortex Aβ 42 levels [61].
In Tg2576 AD mice, 15 mg/kg/day intraperitoneal sildenafil significantly rescued learning and memory deficits as shown by the Morris water maze and fear conditioning tasks, reduced hippocampal tau hyperphosphorylation, GSK3β activity, and the CDK5 p25/p35 ratio, upregulated hippocampal p-CREB and c-Fos following fear conditioning training, and increased hippocampal expression of brain-derived neurotrophic factor (BDNF) and activity-regulated cytoskeletal-associated protein (Arc) (an immediate early response gene involved in memory encoding) [148]. However, sildenafil did not affect total Aβ 42 levels in the frontal cortex [148].
In aged mice, 3 mg/kg intraperitoneal sildenafil daily for 3 weeks decreased double-stranded DNA breaks and apoptotic cells as visualized by the terminal deoxyuridine triphosphate nick end labeling (TUNEL) assay in the CA1 hippocampus, downregulated proapoptotic proteins caspase-3 and B-cell lymphoma 2-associated X (Bax), upregulated antiapoptotic B-cell lymphoma protein-2 (Bcl2) and BDNF, downregulated AβPP expression, and suppressed the age-associated shift in the Aβ 42/Aβ 40 ratio [149].
In the senescence accelerated mouse-prone 8 (SAMP8) mouse model of accelerated aging and sporadic AD, 7.5 mg/kg sildenafil for 4 weeks improved cognitive performance as shown by the Morris water maze and the passive avoidance test [145, 150, 151], tau hyperphosphorylation [145, 151], inflammation as shown by GFAP downregulation, and amyloid pathology as shown by downregulation of BACE1, cathepsin B, and hippocampal Aβ 42 [150]. Sildenafil also activated Akt and inhibited GSK3β, calpain, cyclin-dependent kinase 5 (CDK5) [145, 150], and c-Jun N-terminal kinase (JNK) [151].
Sildenafil in AD patients
In 10 AD patients, a single 50 mg dose of sildenafil significantly decreased spontaneous neural activity in the right hippocampus as shown by the fractional amplitude of low-frequency fluctuations recorded on functional magnetic resonance imaging of the blood oxygen level-dependent signal, a parameter that had been shown to be aberrantly increased in AD patients’ hippocampi and parahippocampal gyri [152]. In 12 elderly patients with AD, a single dosage of 50 mg of sildenafil significantly increased the cerebral metabolic rate of oxygen and cerebral blood flow [127]. In 8 AD patients, it decreased cerebrovascular reactivity [127].
DISCUSSION
As predicted in the introduction, preclinical studies that tested these parameters have found that sildenafil rescued CREB phosphorylation, long-term potentiation, and learning and memory [61, 143–148, 150, 151], increased neurogenesis [145], and decreased neuroinflammation [145, 147, 150]. In addition, these studies consistently found that sildenafil decreased tau hyperphosphorylation and related parameters [61, 145, 148, 150, 151]. This might be in part because the MnSOD downregulation in AD hippocampal neurons contributes to tau hyperphosphorylation [45,153], and low-dose sildenafil appears to upregulate MnSOD via PGC1α activation [69–72]. The relatively high 15 mg/kg dosages appear to have resulted in decreased tau hyperphosphorylation predominantly because high-dose sildenafil activated the cGMP/PKG/PI3K/Akt pathway, leading to increased inhibitory Ser9 phosphorylation of tau kinase GSK3β [61, 80, 148].
Discrepancies have been reported, however, regarding the effect of sildenafil on Aβ levels: most of the studies showed that sildenafil decreases Aβ levels [144–147, 150], but the two studies using the highest dosages (15 mg/kg) reported no effect on frontal cortex Aβ levels [61, 148]. This can be understood through the lens of the U-shaped dose-response curve of sildenafil and cGMP on PGC1α signaling documented in the introduction: low-dose sildenafil and cGMP appear to activate PGC1α and suppress BACE1 expression [65, 70, 76], whereas high-dose sildenafil and 100μM cGMP appear to inhibit PGC1α due to crosstalk with cAMP and PDE2 signaling, failing to suppress BACE1 expression. In other words, it is possible that the 15 mg/kg sildenafil studies may not have decreased Aβ levels significantly [61, 148] because that dosage activates PDE2, depletes cAMP [76], and inhibits cAMP-signaled PGC1α activation and BACE1 repression [65, 67]. Another intriguing observation is that only the 15 mg/kg studies reported Akt activation and/or GSK3β inhibition [61, 148], suggesting the possibility that the 15 mg/kg dosages activated Akt [61, 148] and may have consequently repressed PGC1α expression and its anti-amyloidogenic properties (Fig. 4) [80–82].
Interestingly, in vitro studies found that sildenafil protected mitochondria from Aβ or AGEs via ATP-sensitive K + channels or HO1 upregulation, respectively [140, 141], suggesting that sildenafil may promote mitochondrial function via multiple mechanisms, some of which may be independent of PGC1α.
In patients, an especially promising finding is that 50 mg sildenafil increased the cerebral metabolic rate of oxygen [127]. This effect might be accounted for totally by the increases in the cerebral blood flow [127], or it may have been partially mediated as well by PGC1α-regulated mitochondrial biogenesis. However, none of the studies reviewed measured PGC1α mRNA, protein, or acetylation levels, nor other markers of mitochondrial biogenesis, making it impossible to evaluate this possibility. Nor was the effect of sildenafil on SIRT1 activity or the SIRT1/PGC1α pathway explored in any of these studies. Nor did these studies examine the effect of sildenafil on markers of insulin resistance or antioxidant enzyme expression. Future preclinical studies in transgenic AD mice should address these points directly to assess the possible role of SIRT1 and PGC1α activity in sildenafil-induced Aβ suppression, mitochondrial biogenesis, and antioxidant enzyme expression in AD, as well as the putative effect of sildenafil on insulin resistance. Future studies should also assess the potential of combining sildenafil with a PDE2 inhibitor to increase sildenafil’s maximal effective dosage continuously throughout the treatment duration. This would bypass the dose-limiting effect of sildenafil on PDE2 activation and cAMP depletion [76], allowing for robust simultaneous cAMP and cGMP signaling and therefore maximal PGC1α activity, mitochondrial biogenesis, antioxidant enzyme expression, and BACE1 repression [48, 65–67, 69, 70, 73–75, 96, 108, 109, 112, 115, 116, 154–156]. The best candidate for this role would be propentofylline, a potent and broad-spectrum PDE inhibitor and methyl xanthine derivate like caffeine that is particularly effective at inhibiting cGMP-stimulated PDE2 activity and PDE4 [157]. Propentofylline would be superior to other PDE2 inhibitors primarily because 300 mg of it taken thrice daily one hour before meals has been tested and found to be safe and effective in mild to moderate AD patients in 5 phase III clinical trials [158–164]. Intriguingly, a recent review by Heckman and colleagues opined that, based on the preclinical evidence, inhibition of PDE2, PDE4, and PDE5 seemed to hold the most promise for the treatment of AD [165], and sildenafil and propentofylline administered together would potently inhibit these three therapeutic targets simultaneously [157].
Ultimately, a randomized control trial of sildenafil should be undertaken in AD patients to assess the clinical benefits of long-term sildenafil administration in this population compared to elderly controls. This RCT should use the 50 mg/day dosage [127]. As outcome measures, the RCT should test cognition on the MMSE and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale, comorbid depression on the Geriatric Depression Scale [56], amyloid and tau pathology binding with 2-(1-6-[(2-[F- 18]fluoroethyl)(methyl)amino]-2-naphthylethylidene)malononitrile positron emission tomography (FDDNP-PET) [166, 167], cerebral blood flow with MRI [127], the cerebral metabolic rate of oxygen with MRI [127, 168, 169], the cerebral metabolic rate of glucose with 18F-fluoro-deoxyglucose positron emission tomography (FDG-PET) [34, 35, 170–174], inflammation with CSF IL-1β, IL-6, and TNF-α [124–126, 147, 175–182], NOS/NO/sGC/cGMP/PGC1α pathway dysfunction with CSF cGMP [55, 56], and antioxidant enzyme activity with urine 8-oxo-2’-deoxyguanosine as an indirect biomarker [45, 69, 71, 72, 183].
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
- [1]. Li X, Jiang LH (2018) Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis 9, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2]. Suh SW, Jensen KB, Jensen MS, Silva DS, Kesslak PJ, Danscher G, Frederickson CJ (2000) Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer’s diseased brains. Brain Res 852, 274–278. [DOI] [PubMed] [Google Scholar]
- [3]. Yumoto S, Kakimi S, Ishikawa A (2018) Colocalization of aluminum and iron in nuclei of nerve cells in brains of patients with Alzheimer’s disease. J Alzheimers Dis 65, 1267–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4]. Smith MA, Zhu X, Tabaton M, Liu G, McKeel DW, Cohen ML, Wang X, Siedlak SL, Dwyer BE, Hayashi T, Nakamura M, Nunomura A, Perry G (2010) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19, 353–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5]. Forbes WF, McLachlan DR (1996) Further thoughts on the aluminum-Alzheimer’s disease link. J Epidemiol Community Health 50, 401–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6]. Xu L, Zhang W, Liu X, Zhang C, Wang P, Zhao X (2018) Circulatory levels of toxic metals (aluminum, cadmium, mercury, lead) in patients with Alzheimer’s disease: A quantitative meta-analysis and systematic review. J Alzheimers Dis 62, 361–372. [DOI] [PubMed] [Google Scholar]
- [7]. McLachlan DR, Smith WL, Kruck TP (1993) Desferrioxamine and Alzheimer’s disease: Video home behavior assessment of clinical course and measures of brain aluminum. Ther Drug Monit 15, 602–607. [PubMed] [Google Scholar]
- [8]. Perl DP, Good PF (1987) The association of aluminum Alzheimer’s disease, and neurofibrillary tangles. J Neural Transm Suppl 24, 205–211. [PubMed] [Google Scholar]
- [9]. Rui D, Yongjian Y (2010) Aluminum chloride induced oxidative damage on cells derived from hippocampus and cortex of ICR mice. Brain Res 1324, 96–102. [DOI] [PubMed] [Google Scholar]
- [10]. Nunomura A, Perry G, Aliev G, Hirai K, Takeda A, Balraj EK, Jones PK, Ghanbari H, Wataya T, Shimohama S, Chiba S, Atwood CS, Petersen RB, Smith MA (2001) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60, 759–767. [DOI] [PubMed] [Google Scholar]
- [11]. Butterfield DA, Sultana R (2007) Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: Insights into the progression of this dementing disorder. J Alzheimers Dis 12, 61–72. [DOI] [PubMed] [Google Scholar]
- [12]. Bradley MAA, Markesbery WRR, Lovell MAA (2010) Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med 48, 1570–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13]. Lovell MA, Soman S, Bradley MA (2011) Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev 132, 443–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Vincent I, Jicha G, Rosado M, Dickson DW (1997) Aberrant expression of mitotic Cdc2 / Cyclin B1 kinase in degenerating neurons of Alzheimer’s disease brain. J Neurosci 17, 3588–3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Nagy Z, Esiri MM, Cato AM, Smith AD (1997) Cell cycle markers in the hippocampus in Alzheimer’s disease. Acta Neuropathol 94, 6–15. [DOI] [PubMed] [Google Scholar]
- [16]. Frade JM, López-Sánchez N (2010) neuronal tetraploidy induced by p75 A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75 NTR. Cell Cycle 9, 1934–1941. [DOI] [PubMed] [Google Scholar]
- [17]. Westra JW, Barral S, Chun J (2009) A reevaluation of tetraploidy in the Alzheimer’s disease brain. Neurodegener Dis 6, 221–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18]. Walton CC, Perea G, Frade JM, Barrio-Alonso E, Hernández-Vivanco A, Walton CC, Perea G, Frade JM (2018) Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19]. Fuchsberger T, Lloret A, Viña J, Fuchsberger T, Lloret A, Viña J (2017) New functions of APC/C ubiquitin ligase in the nervous system and its role in Alzheimer’s disease. Int J Mol Sci 18, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20]. Bonda DJ, Evans TA, Santocanale C, Llosá JC, Viña J, Bajic VP, Castellani RJ, Siedlak SL, Perry G, Smith MA, Lee HG (2009) Evidence for the progression through S-phase in the ectopic cell cycle re-entry of neurons in Alzheimer disease. Aging (Albany NY) 1, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Zhu X, Lee H, Perry G, Smith MA (2007) Alzheimer disease, the two-hit hypothesis: An update. Biochim Biophys Acta 1772, 494–502. [DOI] [PubMed] [Google Scholar]
- [22]. Zhu X, Siedlak SL, Wang Y, Perry G, Castellani RJ, Cohen ML, Smith MA (2008) Neuronal binucleation in Alzheimer disease hippocampus. Neuropathol Appl Neurobiol 34, 457–465. [DOI] [PubMed] [Google Scholar]
- [23]. Bushman DM, Kaeser GE, Siddoway B, Westra JW, Rivera RR, Rehen SK, Yung YC, Chun J (2015) Genomic mosaicism with increased amyloid precursor protein (APP) gene copy number in single neurons from sporadic Alzheimer’s disease brains. Elife 4, doi: 10.7554/eLife.05116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24]. Yang Y, Geldmacher DS, Herrup K (2001) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 21, 2661–2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. Iourov IY, Vorsanova SG, Liehr T, Yurov YB (2009) Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: Differential expression and pathological meaning. Neurobiol Dis 34, 212–220. [DOI] [PubMed] [Google Scholar]
- [26]. Mosch B, Morawski M, Mittag A, Lenz D, Tarnok A, Arendt T (2007) Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J Neurosci 27, 6859–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27]. Miklossy J (2008) Chronic inflammation and amyloidogenesis in Alzheimer’s disease –role of spirochetes. J Alzheimers Dis 13, 381–391. [DOI] [PubMed] [Google Scholar]
- [28]. Alonso R, Pisa D, Fernández-Fernández AM, Carrasco L (2018) Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front Aging Neurosci 10, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29]. Piacentini R, De Chiara G, Li Puma DD, Ripoli C, Marcocci ME, Garaci E, Palamara AT, Grassi C (2014) HSV-1 and Alzheimer’s disease: More than a hypothesis. Front Pharmacol 5, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30]. Fülöp T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE (2018) Role of microbes in the development of Alzheimer’s disease: State of the Art - An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front Genet 9, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31]. Devanand DP (2018) Viral hypothesis and antiviral treatment in Alzheimer’s disease. Curr Neurol Neurosci Rep 18, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32]. Osorio C, Kanukuntla T, Diaz E, Jafri N, Cummings M, Sfera A (2019) The post-amyloid era in Alzheimer’s disease: Trust your gut feeling. Front Aging Neurosci 11, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33]. Moir RD, Lathe R, Tanzi RE (2018) The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14, 1602–1614. [DOI] [PubMed] [Google Scholar]
- [34]. Talbot K, Wang H, Kazi H, Han L-Y, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE (2012) Demonstrated brain insulin resistance in Alzheimer ’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122, 1316–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35]. Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, Xu XJ, Wands JR, de la Monte SM (2005) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - is this type 3 diabetes? J Alzheimers Dis 7, 63–80. [DOI] [PubMed] [Google Scholar]
- [36]. Ramos-Cejudo J, Wisniewski T, Marmar C, Zetterberg H, Blennow K, de Leon MJ, Fossati S (2018) Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EBioMedicine 28, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37]. Ishii K, Sasaki M, Yamaji S, Sakamoto S, Kitagaki H, Mori E (1998) Paradoxical hippocampus perfusion in mild-to-moderate Alzheimer’s disease. J Nucl Med 39, 293–298. [PubMed] [Google Scholar]
- [38]. Ishii K, Kitagaki H, Kono M, Mori E (1996) Decreased medial temporal oxygen metabolism in Alzheimer’s disease shown by PET. J Nucl Med 37, 1159–1165. [PubMed] [Google Scholar]
- [39]. Overk CR, Masliah E (2014) Pathogenesis of synaptic degeneration in Alzheimer’s disease and Lewy body disease. Biochem Pharmacol 88, 508–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40]. Selkoe DJ (2002) Alzheimer’s disease is a synaptic failure. Science 298, 789–791. [DOI] [PubMed] [Google Scholar]
- [41]. Ohno M (2018) PERK as a hub of multiple pathogenic pathways leading to memory deficits and neurodegeneration in Alzheimer’s disease. Brain Res Bull 141, 72–78. [DOI] [PubMed] [Google Scholar]
- [42]. Ohno M (2014) Roles of eIF2alpha kinases in the pathogenesis of Alzheimer’s disease. Front Mol Neurosci 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43]. de la Monte SM1, Re E, Longato L, Tong M (2012) Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease.S217-S. J Alzheimers Dis 30(Suppl 2), 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44]. de la Monte SM (2012) Triangulated mal-signaling in Alzheimer’s disease: Roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J Alzheimers Dis 30(Suppl 2), S231–S249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45]. Majd S, Power JHT (2018) Oxidative stress and decreased mitochondrial superoxide dismutase 2 and peroxiredoxins 1 and 4 based mechanism of concurrent activation of AMPK and mTOR in Alzheimer’s disease. Curr Alzheimer Res 15, 1–13. [DOI] [PubMed] [Google Scholar]
- [46]. Hirai K, Aliev G, Nunomura A, Fujioka H, Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters H V, Tabaton M, Shimohama S, Cash AD, Siedlak SL, Harris PL, Jones PK, Petersen RB, Perry G, Smith MA (2001) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21, 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47]. Moreira PI, Siedlak SL, Wang X, Santos MS, Oliveira CR, Tabaton M, Nunomura A, Szweda LI, Aliev G, Smith MA, Zhu X, Perry G (2007) Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 66, 525–532. [DOI] [PubMed] [Google Scholar]
- [48]. Sheng B, Wang X, Su B, Lee HG, Casadesus G, Perry G, Zhu X (2012) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120, 419–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49]. Fiscus RR (2002) Involvement of cyclic GMP and protein kinase G in the regulation of apoptosis and survival in neural cells. Neurosignals 11, 175–190. [DOI] [PubMed] [Google Scholar]
- [50]. Liu P, Fleete MS, Jing Y, Collie ND, Curtis MA, Waldvogel HJ, Faull RLM, Abraham WC, Zhang H (2014) Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol Aging 35, 1992–2003. [DOI] [PubMed] [Google Scholar]
- [51]. Lüth HJ, Holzer M, Gertz H-J, Arendt T (2000) Aberrant expression of nNOS in pyramidal neurons in Alzheimer’s disease is highly co-localized with p21 ras and p16 INK4a. Brain Res 852, 45–55. [DOI] [PubMed] [Google Scholar]
- [52]. Lüth H-JJ, Münch G, Arendt T (2002) Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res 953, 135–143. [DOI] [PubMed] [Google Scholar]
- [53]. Lüth HJ, Holzer M, Gärtner U, Staufenbiel M, Arendt T (2001) Expression of endothelial and inducible NOS-isoforms is increased in Alzheimer’s disease, in APP23 transgenic mice and after experimental brain lesion in rat: Evidence for an induction by amyloid pathology. Brain Res 913, 57–67. [DOI] [PubMed] [Google Scholar]
- [54]. Bonkale WL, Winblad B, Ravid R, Cowburn RF (1995) Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci Lett 187, 5–8. [DOI] [PubMed] [Google Scholar]
- [55]. Ugarte A, Gil-Bea F, García-Barroso C, Cedazo-Minguez Á, Ramírez MJ, Franco R, García-Osta A, Oyarzabal J, Cuadrado-Tejedor M (2015) Decreased levels of guanosine 3’, 5’-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol Appl Neurobiol 41, 471–482. [DOI] [PubMed] [Google Scholar]
- [56]. Hesse R, Lausser L, Gummert P, Schmid F, Wahler A, Schnack C, Kroker KS, Otto M, Tumani H, Kestler HA, Rosenbrock H, von Arnim CAF (2017) Reduced cGMP levels in CSF of AD patients correlate with severity of dementia and current depression. Alzheimers Res Ther 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57]. Ahmed NS (2019) Tadalafil: 15 years’ journey in male erectile dysfunction and beyond. Drug Dev Res 80, 683–701. [DOI] [PubMed] [Google Scholar]
- [58]. Xu M, Zhang DF, Luo R, Wu Y, Zhou H, Kong LL, Bi R, Yao YG (2018) A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement 14, 215–229. [DOI] [PubMed] [Google Scholar]
- [59]. Lakics V, Karran EH, Boess FG (2010) Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 59, 367–374. [DOI] [PubMed] [Google Scholar]
- [60]. Loughney K, Hill TR, Florio VA, Uher L, Rosman GJ, Wolda SL, Jones BA, Howard ML, McAllister-Lucas LM, Sonnenburg WK, Francis SH, Corbin JD, Beavo JA, Ferguson K (1998) Isolation and characterization of cDNAs encoding PDE5A, a human cGMP-binding, cGMP-specific 3’,5’-cyclic nucleotide phosphodiesterase. Gene 216, 139–147. [DOI] [PubMed] [Google Scholar]
- [61]. García-Barroso C, Ricobaraza A, Pascual-Lucas M, Unceta N, Rico AJ, Goicolea MA, Sallés J, Lanciego JL, Oyarzabal J, Franco R, Cuadrado-Tejedor M, García-Osta A (2013) Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 64, 114–123. [DOI] [PubMed] [Google Scholar]
- [62]. Reyes-Irisarri E, Markerink-Van Ittersum M, Mengod G, De Vente J (2007) Expression of the cGMP-specific phosphodiesterases 2 and 9 in normal and Alzheimer’s disease human brains. Eur J Neurosci 25, 3332–3338. [DOI] [PubMed] [Google Scholar]
- [63]. Blokland A, Menniti FS, Prickaerts J (2012) PDE Inhibition and cognition enhancement. Expert Opin Ther Pat 22, 349–354. [DOI] [PubMed] [Google Scholar]
- [64]. Devan BD, Pistell PJ, Duffy KB, Kelley-Bell B, Spangler EL, Ingram DK (2014) Phosphodiesterase inhibition facilitates cognitive restoration in rodent models of age-related memory decline. Neurorehabilitation 34, 101–111. [DOI] [PubMed] [Google Scholar]
- [65]. Kwak Y-D, Wang R, Li J, Zhang Y-W, Xu H, Liao F-F (2011) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener 6, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66]. Nisoli E, Falcone S, Tonello C, Cozzi V, Palomba L, Fiorani M, Pisconti A, Brunelli S, Cardile A, Francolini M, Cantoni O, Carruba MO, Moncada S, Clementi E (2004) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A 101, 16507–16512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67]. Fernandez-Marcos PJ, Auwerx J (2011) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93, 884S–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68]. Wang H, Peiris TH, Mowery A, Lay J Le, Gao Y, Greenbaum LE (2008) CCAAT/enhancer binding protein-β is a transcriptional regulator of peroxisome-proliferator-activated receptor-γ coactivator-1α in the regenerating liver. Mol Endocrinol 22, 1596–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69]. Olmos Y, Sánchez-Gómez FJ, Wild B, García-Quintans N, Cabezudo S, Lamas S, Monsalve M (2013) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxidants Redox Signal 19, 1507–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70]. Whitaker RM, Wills LP, Stallons LJ, Schnellmann RG (2013) CGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharmacol Exp Ther 347, 626–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71]. Perk H, Armagan A, Nazıroğlu M, Soyupek S, Hoscan MB, Sütcü R, Ozorak A, Delibas N (2008) Sildenafil citrate as a phosphodiesterase inhibitor has an antioxidant effect in the blood of men. J Clin Pharm Ther 33, 635–640. [DOI] [PubMed] [Google Scholar]
- [72]. Sheweita S, Salama B, Hassan M (2015) Erectile dysfunction drugs and oxidative stress in the liver of male rats. Toxicol Rep 2, 933–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73]. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO (2003) Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 299, 896–899. [DOI] [PubMed] [Google Scholar]
- [74]. Gureev AP, Shaforostova EA, Popov VN (2019) Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front Genet 10, 435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75]. Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H, Piantadosi CA (2008) Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 28, 2015–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76]. Banerjee J, Bruckbauer A, Thorpe T, Zemel MB (2019) Biphasic effect of sildenafil on energy sensing is mediated by phosphodiesterases 2 and 3 in adipocytes and hepatocytes. Int J Mol Sci 20, E2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77]. Vettel C, Lämmle S, Ewens S, Cervirgen C, Emons J, Ongherth A, Dewenter M, Lindner D, Westermann D, Nikolaev VO, Lutz S, Zimmermann WH, El-Armouche A (2014) PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am J Physiol Heart Circ Physiol 306, H1246–H1252. [DOI] [PubMed] [Google Scholar]
- [78]. Laurent A-C, Bisserier M, Lucas A, Tortosa F, Roumieux M, De Régibus A, Swiader A, Sainte-Marie Y, Heymes C, Vindis C, Lezoualc’h F (2015) Exchange protein directly activated by cAMP 1 promotes autophagy during cardiomyocyte hypertrophy. Cardiovasc Res 105, 55–64. [DOI] [PubMed] [Google Scholar]
- [79]. Omar B, Zmuda-Trzebiatowska E, Manganiello V, Göransson O, Degerman E (2009) Regulation of AMP-activated protein kinase by cAMP in adipocytes: Roles for phosphodiesterases, protein kinase B, protein kinase A, Epac and lipolysis. Cell Signal 21, 760–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80]. Borniquel S, García-Quintáns N, Valle I, Olmos Y, Wild B, Martínez-Granero F, Soria E, Lamas S, Monsalve M (2010) Inactivation of Foxo3a and subsequent downregulation of PGC-1 alpha mediate nitric oxide-induced endothelial cell migration. Mol Cell Biol 30, 4035–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81]. Xiong S, Salazar G, San Martin A, Ahmad M, Patrushev N, Hilenski L, Nazarewicz RR, Ma M, Ushio-Fukai M, Alexander RW (2010) PGC-1αserine 570 phosphorylation and GCN5-mediated acetylation by angiotensin II drive catalase down-regulation and vascular hypertrophy. J Biol Chem 285, 2474–2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82]. Li X, Monks B, Ge Q, Birnbaum MJ (2007) Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1α transcription coactivator. Nature 447, 1012–1016. [DOI] [PubMed] [Google Scholar]
- [83]. Borniquel S, Valle I, Cadenas S, Lamas S, Monsalve M (2006) Nitric oxide regulates mitochondrial oxidative stress protection via the transcriptional coactivator PGC-1α . FASEB J 20, 1889–1891. [DOI] [PubMed] [Google Scholar]
- [84]. Lou L, Urbani J, Ribeiro-Neto F, Altschuler DL (2002) cAMP inhibition of Akt is mediated by activated and phosphorylated Rap1b. J Biol Chem 277, 32799–32806. [DOI] [PubMed] [Google Scholar]
- [85]. Lu Y-F, Hawkins RD (2002) Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J Neurophysiol 88, 1270–1278. [DOI] [PubMed] [Google Scholar]
- [86]. Chen Y, Zhuang S, Cassenaer S, Casteel DE, Gudi T, Boss GR, Pilz RB (2003) Synergism between calcium and cyclic GMP in cyclic AMP response element-dependent transcriptional regulation requires cooperation between CREB and C/EBP-beta. Mol Cell Biol 23, 4066–4082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87]. Fiorito J, Deng S, Landry DW, Arancio O (2018) Targeting the NO/cGMP/CREB phosphorylation signaling pathway in Alzheimer’s disease. In Neurochemical Basis of Brain Function and Dysfunction, Heinbockel T, Csoka AB, eds. IntechOpen, doi: 10.5772/intechopen.81029 [DOI]
- [88]. Ciani E, Guidi S, Bartesaghi R, Contestabile A (2002) Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: Implication for a survival role of nitric oxide. J Neurochem 82, 1282–1289. [DOI] [PubMed] [Google Scholar]
- [89]. Nagai-Kusuhara A, Nakamura M, Mukuno H, Kanamori A, Negi A, Seigel GM (2007) cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp Eye Res 84, 152–162. [DOI] [PubMed] [Google Scholar]
- [90]. Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O (2005) Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci 25, 6887–6897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91]. Lustig Y, Ruas JL, Estall JL, Lo JC, Devarakonda S, Laznik D, Choi JH, Ono H, Olsen JV, Spiegelman BM (2011) Separation of the gluconeogenic and mitochondrial functions of pgc-1α through s6 kinase. Genes Dev 25, 1232–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92]. Anderson RM, Barger JL, Edwards MG, Braun KH, O’connor CE, Prolla TA, Weindruch R (2008) Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7, 101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93]. Dominy JE, Lee Y, Gerhart-Hines Z, Puigserver P (2010) Nutrient-dependent regulation of PGC-1α’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta 1804, 1676–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94]. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P (2007) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α . EMBO J 26, 1913–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95]. Lerin C, Rodgers JT, Kalume DE, Kim SH, Pandey A, Puigserver P (2006) GCN5 acetyltransferase complex controls glucose metabolism through transcriptional repression of PGC-1α. Cell Metab 3, 429–438. [DOI] [PubMed] [Google Scholar]
- [96]. Nemoto S, Fergusson MM, Finkel T (2005) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem 280, 16456–16460. [DOI] [PubMed] [Google Scholar]
- [97]. Jäer S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A 104, 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98]. Garcia D, Shaw RJ (2017) AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol Cell 66, 789–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99]. Wan Z, Root-Mccaig J, Castellani L, Kemp BE, Steinberg GR, Wright DC (2014) Evidence for the role of AMPK in regulating PGC-1 alpha expression and mitochondrial proteins in mouse epididymal adipose tissue. Obesity 22, 730–738. [DOI] [PubMed] [Google Scholar]
- [100]. Leick L, Fentz J, Biensø RS, Knudsen JG, Jeppesen J, Kiens B, Wojtaszewski JFP, Pilegaard H (2010) PGC-1α is required for AICAR-induced expression of GLUT4 and mitochondrial proteins in mouse skeletal muscle.E456-E. Am J Physiol Endocrinol Metab 299, 465. [DOI] [PubMed] [Google Scholar]
- [101]. Tang BL (2016) Sirt1 and the mitochondria. Mol Cells 39, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102]. Barger PM, Browning AC, Garner AN, Kelly DP (2001) p38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor α: A potential role in the cardiac metabolic stress response. J Biol Chem 276, 44495–44501. [DOI] [PubMed] [Google Scholar]
- [103]. Teyssier C (2005) Activation of nuclear receptor coactivator PGC-1 by arginine methylation. Genes Dev 19, 1466–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104]. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P (2005) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434, 113–118. [DOI] [PubMed] [Google Scholar]
- [105]. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F, Messadeq N, Milne J, Lambert P, Elliott P, Geny B, Laakso M, Puigserver P, Auwerx J (2006) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell 127, 1109–1122. [DOI] [PubMed] [Google Scholar]
- [106]. Gurd BJ (2011) Deacetylation of PGC-1a by SIRT1: Importance for skeletal muscle function andexercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab 36, 589–597. [DOI] [PubMed] [Google Scholar]
- [107]. Amat R, Planavila A, Chen SL, Iglesias R, Giralt M, Villarroya F (2009) SIRT1 controls the transcription of the PGC-1a gene in skeletal muscle through PGC-1a auto-regulatory loop and interaction with MyoD. J Biol Chem 284, 21872–21880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108]. Shalwala M, Zhu S-G, Das A, Salloum FN, Xi L, Kukreja RC (2014) Sirtuin 1 (SIRT1) activation mediates sildenafil induced delayed cardioprotection against ischemia-reperfusion injury in mice. PLoS One 9, e86977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109]. Fiore D, Gianfrilli D, Giannetta E, Galea N, Panio G, di Dato C, Pofi R, Pozza C, Sbardella E, Carbone I, Naro F, Lenzi A, Venneri MA, Isidori AM (2016) PDE5 inhibition ameliorates visceral adiposity targeting the miR-22/SIRT1 pathway: Evidence from the CECSID trial. J Clin Endocrinol Metab 101, 1525–1534. [DOI] [PubMed] [Google Scholar]
- [110]. Nisoli E, Tonello C, Cardile A, Cozzi V, Bracale R, Tedesco L, Falcone S, Valerio A, Cantoni O, Clementi E, Moncada S, Carruba MO (2005) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310, 314–317. [DOI] [PubMed] [Google Scholar]
- [111]. Kitada M, Ogura Y, Koya D (2016) The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging (Albany NY) 8, 2290–2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112]. Ayrapetyan T (2018) Defective NO/cGMP/PKG signaling downregulates Sirt1 expression in aged osteoblasts. UC San Diego. ProQuest ID: AYRAPETYAN_ucsd_0033M_17722. Merritt ID: ark:/13030/m5f819f3.
- [113]. Shao H, Xue Q, Zhang F, Luo Y, Zhu H, Zhang X, Zhang H, Ding W, Yu B (2014) Spinal SIRT1 activation attenuates neuropathic pain in mice. PLoS One 9, e100938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114]. Bermúdez-Ocaña DY, Ambriz-Tututi M, Pérez-Severiano F, Granados-Soto V (2006) Pharmacological evidence for the participation of NO-cyclic GMP-PKG-K+ channel pathway in the antiallodynic action of resveratrol. Pharmacol Biochem Behav 84, 535–542. [DOI] [PubMed] [Google Scholar]
- [115]. Jia H, Guo Z, Yao Y (2019) PDE5 inhibitor protects the mitochondrial function of hypoxic myocardial cells. Exp Ther Med 17, 199–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116]. Li N, Yuan Y, Li S, Zeng C, Yu W, Shen M, Zhang R, Li C, Zhang Y, Wang H (2016) Pde5 inhibitors protect against post-infarction heart failure. Front Biosci (Landmark Ed) 21, 1194–1210. [DOI] [PubMed] [Google Scholar]
- [117]. Domek-łopacińska KU, Strosznajder JB (2010) Cyclic GMP and nitric oxide synthase in aging and Alzheimer’s disease. Mol Neurobiol 41, 129–137. [DOI] [PubMed] [Google Scholar]
- [118]. Alsop DC, Casement M, De Bazelaire C, Fong T, Press DZ (2008) Hippocampal hyperperfusion in Alzheimer’s disease. Neuroimage 42, 1267–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119]. Uazzi MAG, Amaja MIS, Caretti A, Bianciardi P, Ronchi R, Fantacci M, Guazzi M, Samaja M (2008) Phosphodiesterase-5 inhibition abolishes neuron apoptosis induced by chronic hypoxia independently of hypoxia-inducible factor-1α signaling. Exp Biol Med 233, 1222–1230. [DOI] [PubMed] [Google Scholar]
- [120]. Wang L, Zhang ZG, Zhang RL, Chopp M (2005) Activation of the PI3-K/Akt pathway mediates cGMP enhanced-neurogenesis in the adult progenitor cells derived from the subventricular zone. J Cereb Blood Flow Metab 25, 1150–1158. [DOI] [PubMed] [Google Scholar]
- [121]. Dias Fiuza Ferreira E, Valério Romanini C, Cypriano PE, Weffort de Oliveira RM, Milani H (2013) Sildenafil provides sustained neuroprotection in the absence of learning recovery following the 4-vessel occlusion/internal carotid artery model of chronic cerebral hypoperfusion in middle-aged rats. Brain Res Bull 90, 58–65. [DOI] [PubMed] [Google Scholar]
- [122]. Zhang R, Wang Y, Zhang L, Zhang Z, Tsang W, Lu M, Zhang L, Chopp M (2002) Sildenafil (Viagra) induces neurogenesis and promotes functional recovery after stroke in rats. Stroke 33, 2675–2680. [DOI] [PubMed] [Google Scholar]
- [123]. Zhang RL, Chopp M, Roberts C, Wei M, Wang X, Liu X, Lu M, Zhang ZG (2012) Sildenafil enhances neurogenesis and oligodendrogenesis in ischemic brain of middle-aged mouse. PLoS One 7, e48141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124]. Zemel MB, Kolterman O, Rinella M, Vuppalanchi R, Flores O, Barritt AS, Siddiqui M, Chalasani N (2019) Randomized controlled trial of a leucine-metformin-sildenafil combination (NS-0200) on weight and metabolic parameters. Obesity 27, 59–67. [DOI] [PubMed] [Google Scholar]
- [125]. Ramirez CE, Nian H, Yu C, Gamboa JL, Luther JM, Brown NJ, Shibao CA (2015) Treatment with sildenafil improves insulin sensitivity in prediabetes: A randomized, controlled trial. J Clin Endocrinol Metab 100, 4533–4540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126]. Aversa A, Vitale C, Volterrani M, Fabbri A, Spera G, Fini M, Rosano GMC (2008) Chronic administration of Sildenafil improves markers of endothelial function in men with Type 2 diabetes. Diabet Med 25, 37–44. [DOI] [PubMed] [Google Scholar]
- [127]. Sheng M, Lu H, Liu P, Li Y, Ravi H, Peng SL, Diaz-Arrastia R, Devous MD, Womack KB (2017) Sildenafil improves vascular and metabolic function in patients with Alzheimer’s disease. J Alzheimers Dis 60, 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128]. Grome JJ, Harper AM (1983) The effects of serotonin on local cerebral blood flow. J Cereb Blood Flow Metab 3, 71–77. [DOI] [PubMed] [Google Scholar]
- [129]. Geday J, Hermansen F, Rosenberg R, Smith DF (2005) Serotonin modulation of cerebral blood flow measured with positron emission tomography (PET) in humans. Synapse 55, 224–229. [DOI] [PubMed] [Google Scholar]
- [130]. Steiner JA, Carneiro AMD, Wright J, Matthies HJG, Prasad HC, Nicki CK, Dostmann WR, Buchanan CC, Corbin JD, Francis SH, Blakely RD (2009) cGMP-dependent protein kinase iα associates with the antidepressant-sensitive serotonin transporter and dictates rapid modulation of serotonin uptake. Mol Brain 2, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131]. Wong A, Zhang YW, Jeschke GR, Turk BE, Rudnick G (2012) Cyclic GMP-dependent stimulation of serotonin transport does not involve direct transporter phosphorylation by cGMP-dependent protein kinase. J Biol Chem 287, 36051–36058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132]. Zhang YW, Gesmonde J, Ramamoorthy S, Rudnick G (2007) Serotonin transporter phosphorylation by cGMP-dependent protein kinase is altered by a mutation associated with obsessive-compulsive disorder. J Neurosci 27, 10878–10886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133]. Nurnberg HG, Seidman SN, Gelenberg AJ, Fava M, Rosen R, Shabsigh R (2002) Depression, antidepressant therapies, and erectile dysfunction: Clinical trials of sildenafil citrate (Viagra ®) in treated and untreated patients with depression. Urology 60, 58–66. [DOI] [PubMed] [Google Scholar]
- [134]. Socała K, Nieoczym D, Pieróg M, Szuster-Ciesielska A, Wyska E, Wlaź P (2016) Antidepressant-like activity of sildenafil following acute and subchronic treatment in the forced swim test in mice: Effects of restraint stress and monoamine depletion. Metab Brain Dis 31, 1095–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135]. Cheitlin MD, Hutter AM, Brindis RG, Ganz P, Kaul S, Russell RO, Zusman RM, Forrester JS, Douglas PS, Faxon DP, Fisher JD, Gibbons RJ, Halperin JL, Hochman JS, Kaul S, Weintraub WS, Winters WL, Wolk MJ (1999) Use of sildenafil (Viagra) in patients with cardiovascular disease. Circulation 99, 168–177. [DOI] [PubMed] [Google Scholar]
- [136]. Kontaras K, Varnavas V, Kyriakides ZS (2008) Does sildenafil cause myocardial infarction or sudden cardiac death? Am J Cardiovasc Drugs 8, 1–7. [DOI] [PubMed] [Google Scholar]
- [137]. Andersson DP, Trolle Lagerros Y, Grotta A, Bellocco R, Lehtihet M, Holzmann MJ (2017) Association between treatment for erectile dysfunction and death or cardiovascular outcomes after myocardial infarction. Heart 103, 1264–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138]. Balarini CM, Leal MA, Gomes IBS, Pereira TMC, Gava AL, Meyrelles SS, Vasquez EC (2013) Sildenafil restores endothelial function in the apolipoprotein E knockout mouse. J Transl Med 11, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139]. Yang HM, Jin S, Jang H, Kim JY, Lee JE, Kim J, Kim HS (2019) Sildenafil reduces neointimal hyperplasia after angioplasty and inhibits platelet aggregation via activation of cGMP-dependent protein kinase. Sci Rep 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140]. Son Y, Kim K, Cho HR (2018) Sildenafil protects neuronal cells from mitochondrial toxicity induced by β-amyloid peptide via ATP-sensitive K + channels. Biochem Biophys Res Commun 500, 504–510. [DOI] [PubMed] [Google Scholar]
- [141]. Sung SK, Woo JS, Kim YH, Son DW, Lee SW, Song GS (2016) Sildenafil ameliorates advanced glycation end products-induced mitochondrial dysfunction in HT-22 hippocampal neuronal cells. J Korean Neurosurg Soc 59, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142]. Perrone L, Grant WB (2015) Observational and ecological studies of dietary advanced glycation end products in national diets and Alzheimer’s disease incidence and prevalence. J Alzheimers Dis 45, 965–979. [DOI] [PubMed] [Google Scholar]
- [143]. Devan BD, Sierra-Mercado D, Jimenez M, Bowker JL, Duffy KB, Spangler EL, Ingram DK (2004) Phosphodiesterase inhibition by sildenafil citrate attenuates the learning impairment induced by blockade of cholinergic muscarinic receptors in rats. Pharmacol Biochem Behav 79, 691–699. [DOI] [PubMed] [Google Scholar]
- [144]. Puzzo D, Staniszewski A, XianDeng S, Privitera L, Leznik E, Liu S, Zhang H, Feng Y, Palmeri A, Landry DW, Arancio O (2009) Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J Neurosci 29, 8075–8086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145]. Zhu L, Yang JY, Xue X, Dong YX, Miao FR, Wang YF, Xue H, Wu CF (2015) A novel phosphodiesterase-5 Inhibitor: Yonkenafil modulates neurogenesis, gliosis to improve cognitive function and ameliorates amyloid burden in an APP/PS1 transgenic mice model. Mech Ageing Dev 150, 34–45. [DOI] [PubMed] [Google Scholar]
- [146]. Jin F, Gong Q-HH, Xu Y-SS, Wang L-NN, Jin H, Li F, Li L-SS, Ma Y-MM, Shi J-SS (2014) Icariin, a phoshphodiesterase-5 inhibitor, improves learning and memory in APP/PS1 transgenic mice by stimulation of NO/cGMP signalling. Int J Neuropsychopharmacol 17, 871–881. [DOI] [PubMed] [Google Scholar]
- [147]. Zhang J, Guo J, Zhao X, Chen Z, Wang G, Liu A, Wang Q, Zhou W, Xu Y, Wang C (2013) Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav Brain Res 250, 230–237. [DOI] [PubMed] [Google Scholar]
- [148]. Cuadrado-Tejedor M, Hervias I, Ricobaraza A, Puerta E, Pérez-Roldán JM, García-Barroso C, Franco R, Aguirre N, García-Osta A (2011) Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br J Pharmacol 164, 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149]. Puzzo D, Loreto C, Giunta S, Musumeci G, Frasca G, Podda MV, Arancio O, Palmeri A (2014) Effect of phosphodiesterase-5 inhibition on apoptosis and beta amyloid load in aged mice. Neurobiol Aging 35, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150]. Orejana L, Barros-Miñones L, Jordan J, Cedazo-Minguez A, Tordera RM, Aguirre N, Puerta E (2015) Sildenafil decreases BACE1 and cathepsin B levels and reduces APP amyloidogenic processing in the SAMP8 mouse. J Gerontol Ser A Biol Sci Med Sci 70, 675–685. [DOI] [PubMed] [Google Scholar]
- [151]. Orejana L, Barros-Miñones L, Aguirre N, Puerta E (2013) Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp Gerontol 48, 565–571. [DOI] [PubMed] [Google Scholar]
- [152]. Samudra N, Motes M, Lu H, Sheng M, Diaz-Arrastia R, Devous M, Hart J, Womack KB (2019) A pilot study of changes in medial temporal lobe fractional amplitude of low frequency fluctuations after sildenafil administration in patients with Alzheimer’s disease. J Alzheimers Dis 70, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153]. Melov S, Adlard PA, Morten K, Johnson F, Golden TR, Hinerfeld D, Schilling B, Mavros C, Masters CL, Volitakis I, Li Q-X, Laughton K, Hubbard A, Cherny RA, Gibson B, Bush AI (2007) Mitochondrial oxidative stress causes hyperphosphorylation of tau. PLoS One 2, e536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154]. Gerhart-Hines Z, Dominy JE, Blättler SM, Jedrychowski MP, Banks AS, Lim JH, Chim H, Gygi SP, Puigserver P (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD +. Mol Cell 44, 851–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155]. Wang R, Li JJ, Diao S, Kwak YD, Liu L, Zhi L, Büeler H, Bhat NR, Williams RW, Park EA, Liao FF (2013) Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab 17, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156]. Han B, Jiang W, Liu H, Wang J, Zheng K, Cui P, Feng Y, Dang C, Bu Y, Wang QM, Ju Z, Hao J (2020) Upregulation of neuronal PGC-1α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 10, 2832–2848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157]. Meskini N, Némoz G, Okyayuz-Baklouti I, Lagarde M, Prigent AF (1994) Phosphodiesterase inhibitory profile of some related xanthine derivatives pharmacologically active on the peripheral microcirculation. Biochem Pharmacol 47, 781–788. [DOI] [PubMed] [Google Scholar]
- [158]. Rother M, Erkinjuntti T, Roessner M, Kittner B, Marcusson J, Karlsson I (1998) Propentofylline in the treatment of Alzheimer’s disease and vascular dementia: A review of phase III trials. Dement Geriatr Cogn Disord 9, 36–43. [DOI] [PubMed] [Google Scholar]
- [159]. Frampton MA, Harvey RJ, Kirchner V (2003) Propentofylline for dementia. Cochrane Database Syst Rev, CD002853. [DOI] [PubMed]
- [160]. Rother M (1999) A 72-week, placebo-controlled study assessing propentofylline’s safety, efficacy, and impact on disease progression in patients with Alzheimer’s disease. Eur Neuropsychopharmacol 9, 319. [Google Scholar]
- [161]. Kittner B, De Deyn PP, Erkinjuntti T (2000) Investigating the natural course and treatment of vascular dementia and Alzheimer’s disease. Parallel study populations in two randomized, placebo-controlled trials. Ann N Y Acad Sci 903, 535–541. [DOI] [PubMed] [Google Scholar]
- [162]. Kittner B (1999) Using a combined randomized start/withdrawal design to assess propentofylline’s effects on disease progression in Alzheimer’s disease and vascular dementia: Results of clinical studies. Eur Neuropsychopharmacol 9, 320. [Google Scholar]
- [163]. Rother M (1999) Long-term effects of propentofylline in patients with Alzheimer’s disease: Safety, efficacy, and impact on disease progession. J Am Geriatr Soc 47, s2. [Google Scholar]
- [164]. Rother M (1999) Propentofylline versus placebo in patients with Alzheimer’s disease: A 72-week study examining safety, efficacy, and impact on disease progression. Neurology 52, 172.9921868 [Google Scholar]
- [165]. Heckman PRA, Blokland A, Prickaerts J (2017) From age-related cognitive decline to Alzheimer’s disease: A translational overview of the potential role for phosphodiesterases. Adv Neurobiol 17, 135–168. [DOI] [PubMed] [Google Scholar]
- [166]. Small GW, Siddarth P, Li Z, Miller KJ, Ercoli L, Emerson ND, Martinez J, Wong KP, Liu J, Merrill DA, Chen ST, Henning SM, Satyamurthy N, Huang SC, Heber D, Barrio JR (2018) Memory and brain amyloid and tau effects of a bioavailable form of curcumin in non-demented adults: A double-blind, placebo-controlled 18-month trial. Am J Geriatr Psychiatry 26, 266–277. [DOI] [PubMed] [Google Scholar]
- [167]. Shoghi-Jadid K, Small GW, Agdeppa ED, Kepe V, Ercoli LM, Siddarth P, Read S, Satyamurthy N, Petric A, Huang SC, Barrio JR (2002) Localization of neurofibrillary tangles and beta-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry 10, 24–35. [PubMed] [Google Scholar]
- [168]. Xu F, Ge Y, Lu H (2009) Noninvasive quantification of whole-brain cerebral metabolic rate of oxygen (CMRO2) by MRI. Magn Reson Med 62, 141–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169]. Liu P, Xu F, Lu H (2013) Test-retest reproducibility of a rapid method to measure brain oxygen metabolism. Magn Reson Med 69, 675–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170]. de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, Tsui W, Kandil E, Scherer AJ, Roche A, Imossi A, Thorn E, Bobinski M, Caraos C, Lesbre P, Schlyer D, Poirier J, Reisberg B, Fowler J (2001) Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET). Proc Natl Acad Sci U S A 98, 10966–10971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171]. De Leon MJ, Mosconi L, Blennow K, DeSanti S, Zinkowski R, Mehta PD, Pratico D, Tsui W, Saint Louis LA, Sobanska L, Brys M, Li Y, Rich K, Rinne J, Rusinek H (2007) Imaging and CSF studies in the preclinical diagnosis of Alzheimer’s disease. Ann N Y Acad Sci 1097, 114–145. [DOI] [PubMed] [Google Scholar]
- [172]. Mosconi L, De Santi S, Li J, Tsui WH, Li Y, Boppana M, Laska E, Rusinek H, de Leon MJ (2008) Hippocampal hypometabolism predicts cognitive decline from normal aging. Neurobiol Aging 29, 676–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173]. Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, De Leon MJ (2009) FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging 36, 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174]. Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147, 180–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175]. Cojocaru IM, Cojocaru M, Miu G, Sapira V (2011) Study of interleukin-6 production in Alzheimer’s disease. Rom J Intern Med 49, 55–58. [PubMed] [Google Scholar]
- [176]. Cacabelos R, Alvarez XA, Fernández-Novoa L, Franco A, Mangues R, Pellicer A, Nishimura T (1994) Brain interleukin-1 beta in Alzheimer’s disease and vascular dementia. Methods Find Exp Clin Pharmacol 16, 141–151. [PubMed] [Google Scholar]
- [177]. Cheng X, He P, Lee T, Yao H, Li R, Shen Y (2014) High activities of BACE1 in brains with mild cognitive impairment. Am J Pathol 184, 141–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178]. Chang R, Yee K-L, Sumbria RK (2017) Tumor necrosis factor α Inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis 9, 1179573517709278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179]. Diniz BS, Teixeira AL, Ojopi EB, Talib LL, Mendonça VA, Gattaz WF, Forlenza OV (2011) Higher serum sTNFR1 level predicts conversion from mild cognitive impairment to Alzheimer’s disease. J Alzheimers Dis 22, 1305–1311. [DOI] [PubMed] [Google Scholar]
- [180]. Lourenco M V, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espírito-Santo S, Campello-Costa P, Houzel J-C, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG (2013) TNF-α mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s β-amyloid oligomers in mice and monkeys. Cell Metab 18, 831–843. [DOI] [PubMed] [Google Scholar]
- [181]. Montgomery SL, Mastrangelo MA, Habib D, Narrow WC, Knowlden SA, Wright TW, Bowers WJ (2011) Ablation of TNF-RI/RII expression in Alzheimer’s disease mice leads to an unexpected enhancement of pathology. Am J Pathol 179, 2053–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182]. Wu Y-Y, Hsu J-L, Wang H-C, Wu S-J, Hong C-J, Cheng IH-J (2015) Alterations of the neuroinflammatory markers IL-6 and TRAIL in Alzheimer’s disease. Dement Geriatr Cogn Dis Extra 5, 424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183]. Zengi O, Karakas A, Ergun U, Senes M, Inan L, Yucel D (2011) Urinary 8-hydroxy-2’-deoxyguanosine level and plasma paraoxonase 1 activity with Alzheimer’s disease. Clin Chem Lab Med 50, 529–534. [DOI] [PubMed] [Google Scholar]