

Summary
Somatic stem cells expand massively during tissue regeneration, which might require control of cell fitness, allowing elimination of non-competitive, potentially harmful cells. How or if such cells are removed to restore organ function is not fully understood. Here, we show that a substantial fraction of muscle stem cells (MuSCs) undergo necroptosis because of epigenetic rewiring during chronic skeletal muscle regeneration, which is required for efficient regeneration of dystrophic muscles. Inhibition of necroptosis strongly enhances suppression of MuSC expansion in a non-cell-autonomous manner. Prevention of necroptosis in MuSCs of healthy muscles is mediated by the chromatin remodeler CHD4, which directly represses the necroptotic effector Ripk3, while CHD4-dependent Ripk3 repression is dramatically attenuated in dystrophic muscles. Loss of Ripk3 repression by inactivation of Chd4 causes massive necroptosis of MuSCs, abolishing regeneration. Our study demonstrates how programmed cell death in MuSCs is tightly controlled to achieve optimal tissue regeneration.
Keywords: muscle stem cells, regeneration, necroptosis, muscle dystrophy, Chd4/NuRD, Ripk3
Graphical Abstract
Highlights
-
•
Necroptotic cell death of MuSCs is essential for efficient muscle regeneration
-
•
Inhibition of necroptosis exacerbates adverse crosstalk among mdx muscle stem cells
-
•
The CHD4/NuRD complex directly represses Ripk3-dependent necroptosis
-
•
Attenuated recruitment of CHD4 to Ripk3 locus lowers necroptosis threshold in dystrophy
Sreenivasan et al. describe the necessity to remove a subpopulation of muscle stem cells by necroptosis for efficient regeneration of chronically damaged skeletal muscles. Muscle stem cells acquire a lowered threshold for necroptosis by attenuating recruitment of the repressive CHD4/NuRD complex to the Ripk3 promotor.
Introduction
Skeletal muscle regeneration provides a paradigmatic example for the decisive role of tissue-resident stem cells and the necessity of cellular interactions to achieve organ restoration. Muscle stem cells (MuSCs; also known as satellite cells) are indispensable for muscle regeneration but require assistance and instructions from fibroblasts, endothelial cells, fibroadipogenic progenitor cells (FAPs), and immune cells, among others (Chargé and Rudnicki, 2004, Tidball, 2011, Relaix and Zammit, 2012). Such cells not only provide critical support for MuSCs, enabling their expansion, but might also promote secondary cell death (Saclier et al., 2013, Latroche et al., 2015, Tidball and Villalta, 2010, Joe et al., 2010, Forbes and Rosenthal, 2014). Secondary cell death is not necessarily harmful but might elicit beneficial effects. For example, programmed cell death in FAPs, induced by TNF-releasing inflammatory macrophages, limits fibrosis in acutely damaged skeletal muscles (Lemos et al., 2015). Moreover, MuSCs might engage in a battle of the “survival of the fittest” to ensure that damaged or less fit stem cells are eliminated (Bowling et al., 2019). It has been proposed that tissue stem cells are routinely lost and replaced in a process called neutral cell competition, but many questions related to such a machinery, including the mechanisms by which unfit cells are removed, have yet to be answered (Klein and Simons, 2011). At present, it is not known whether and to what extent MuSCs succumb to programmed cell death in acutely damaged and continuously regenerating dystrophic muscles. Moreover, very little attention has been paid to the specific mechanisms used for the elimination of parenchymal stem cells, although control of stem cell quantity and quality by programmed cell death during a phase of massive expansion might be as important to achieve proper tissue homeostasis and regeneration as the regulation of self-renewal and differentiation (Koren et al., 2018, Weinlich et al., 2017).
The pathology of Duchenne muscular dystrophy (DMD) is characterized by sustained activation of MuSCs and continuous inflammation, leading to pronounced cell death within the regenerating muscle tissue (Rosenberg et al., 2015). The efficiency of MuSC-dependent skeletal muscle regeneration depends strongly on the specific state and composition of inflammatory cells in the regenerating tissue, the release of inflammatory and anti-inflammatory cytokines, and the duration of inflammatory responses (Forbes and Rosenthal, 2014). Inflammatory conditions can induce various modes of programmed cell death, including apoptosis and different forms of regulated necrosis (Ashkenazi and Salvesen, 2014, Newton and Manning, 2016). TNF-α and IFN-γ, which are present in dystrophic muscles, trigger the recently identified prototypic form of regulated necrosis called necroptosis because of formation of the necrosome complex following association and auto- and transphosphorylation of the receptor-interacting serine/threonine-protein kinases RIPK1 and RIPK3. Phosphorylation of RIPK3 is essential to recruit and phosphorylate MLKL (mixed lineage kinase domain-like protein), which results in a late wave of JNK activation and production of reactive oxygen species (ROS), eventually causing oncotic cell death (Vanden Berghe et al., 2014, Grootjans et al., 2017). RIPK3-dependent necroptosis plays part in pathological processes such as heart muscle infarction (Zhang et al., 2016), inflammatory diseases (Welz et al., 2011), and tumor metastasis (Strilic et al., 2016), but a role in tissue regeneration or in the control of stem cells, including MuSCs, has not been investigated. Likewise, very little is known about a potential epigenetic regulation of necroptosis, when defining epigenetics as a stably heritable phenotype resulting from changes in a chromosome without alterations in the DNA sequence (Berger et al., 2009). So far, it has only been reported that inhibition of the epigenetic regulator UHRF1 (ubiquitin-like, containing PHD and RING finger domains 1) in Ripk3-null cancer cells reduces Ripk3 promoter methylation (Yang et al., 2017).
Here, we delineated the mode and role of MuSC death during skeletal muscle regeneration under acute and chronic disease conditions. We discovered that a subset of MuSCs undergoes either necroptotic or apoptotic cell death in dystrophic muscles, while acutely damaged or healthy muscles are devoid of necroptotic MuSCs. Unexpectedly, separate or combined inhibition of apoptosis and necroptosis in MuSCs impaired skeletal muscle regeneration and function in mdx mice. Co-culture experiments revealed that MuSCs from dystrophic muscles restricted expansion of healthy MuSCs, an effect that was strongly enhanced when necroptosis was blocked by Ripk3 inactivation in dystrophic MuSCs. To decipher the molecular basis for increased predisposition of dystrophic MuSCs for necroptosis, we conducted a short hairpin RNA (shRNA)-based screen. We found that CHD4, an essential component of the NuRD chromatin remodeling complex, completely suppresses expression of the necroptosis effector Ripk3 in healthy MuSCs. In contrast, CHD4-dependent repression of Ripk3 is partially alleviated in mdx MuSCs, allowing elimination of a subset of MuSCs by programmed cell death. Our data indicate that epigenetic regulation of necroptosis is critical for maintaining a healthy stem cell compartment in dystrophic muscles.
Results
Skeletal Muscle Dystrophy but Not Acute Muscle Injury Leads to Increased Necroptosis of MuSCs
To determine the mode and the extent of programmed cell death in MuSCs during acute muscle regeneration and in chronically regenerating dystrophic muscles, we performed immunofluorescence analysis of tibialis anterior (TA) muscles from wild-type (WT) mice injected with cardiotoxin (CTX) and from mdx mice. Apoptotic cells were detected by staining for cleaved CASP3. Necroptotic cells are unequivocally identified by staining for pMLKL (phosphorylated MLKL), a specific marker for necroptotic cell death (Hildebrand et al., 2014). However, because antibodies against pMLKL did not yield reliable results in our hands when using sections of dystrophic mouse tissues, we used antibodies against RIPK3 to identify cells that might potentially undergo programmed necrosis (Vanden Berghe et al., 2013). Induction of acute muscle injury by CTX injection into WT mice resulted in a major increase in apoptotic PAX7+ MuSCs, as expected (Tidball, 2011, Tidball and Villalta, 2010), whereas RIPK3+ MuSCs were essentially absent in both undamaged and acutely damaged muscles (Figures 1A–1C). Notably, however, and in stark contrast to WT muscles, mdx muscles harbored approximately equal numbers of both apoptotic and RIPK3+/PAX7+ MuSCs (Figures 1A–1C). Injection of CTX into TA muscles of mdx mice further enhanced the number of both apoptotic PAX7+/cleaved CASP3+ and PAX7+/RIPK3+ MuSCs (Figures S1A–S1C). Because RIPK3 expression alone is not an unequivocal marker for necroptosis, we performed electron microscopy (EM) analyses, allowing unbiased determination of cell death types in MuSCs via morphological criteria such as formation of pyknotic nuclei, fragmentation of nuclei, and intact or disrupted plasma membranes (Günther et al., 2013, Kostin et al., 2003, Shi and Garry, 2006, Boonsanay et al., 2016). Consistent with immunofluorescence staining, EM analysis confirmed that mdx muscles contain both apoptotic and necroptotic MuSCs, whereas undamaged WT muscles are devoid of dying MuSCs (Figures 1D and 1E).
Figure 1.

A Subpopulation of MuSCs Undergo Necroptotic or Apoptotic Cell Death in Chronic Muscle Disease
(A–C) Immunofluorescence staining of TA muscle cross-sections from control and mdx mice 2 weeks after CTX injury using antibodies against PAX7 and cleaved CASP3 to detect apoptosis (A) and PAX7 and RIPK3 to detect necroptosis (B) in MuSCs (n = 3 for each group). Scale bar, 25 μm.
(C) Quantification of cells identified in (A) and (B).
(D and E) EM images (MF, myofiber; # denotes chromatin condensation [apoptosis]; + denotes intact chromatin/nucleus [necrosis]; asterisk denotes MuSC disrupted membrane) (D) and quantification (E) of MuSCs from control (WT) and mdx mice undergoing apoptosis and necrosis (n = 3 for each group). Scale bar, 1 μm.
(F and G) Immunofluorescence staining for PAX7 and pMLKL to visualize (F) and quantify (G) MuSCs undergoing necroptosis in healthy (control) (n = 4) and Becker muscular dystrophy (BMD) patients (n = 4). Scale bar, 50 μm.
(H) Serum creatine kinase activity in control and BMD patients (n = 3 or 4 each).
Statistical analysis: ∗p < 0.05 and ∗∗p < 0.01, two-way ANOVA followed by Bonferroni post-test with alpha = 5%. All analyses indicated across the experiments were biological replicates unless otherwise stated.
To understand whether the occurrence of necroptotic MuSCs in dystrophic muscles is conserved between species, we analyzed human subjects with Becker muscular dystrophy (BMD) caused by in-frame deletions within the dystrophin gene (Emery, 2002). BMD patients exhibit a milder pathology in comparison with DMD patients, enabling better quantification of MuSCs, as muscle biopsies from human DMD patients often lack large areas of intact muscle tissue containing MuSCs (Dennett et al., 1988). Similar to mdx muscles in mice, we found increased numbers of PAX7+ MuSCs in human BMD muscles undergoing regeneration, as indicated by elevated serum muscle creatine kinase concentrations (Figures 1F–1H). More importantly, we detected several necroptotic PAX7+ MuSCs by co-staining with antibodies against pMLKL, a specific marker for necroptotic cell death (Hildebrand et al., 2014), in BMD muscle biopsies, whereas pMLKL+/PAX7+ MuSCs were never found in muscle biopsies of healthy individuals (Figures 1F and 1G).
In contrast to a previous study (Morgan et al., 2018), immunofluorescence analysis did not reveal the presence of any necroptotic myofibers in skeletal muscles of mdx mice and human BMD patients (Figures 1B and 1F). Consistent with this observation, RIPK3 protein was detected by western blot analysis only in isolated mdx MuSCs but neither in purified myofibers of WT and mdx mice nor in MuSCs from WT mice (Figures S1D and S1E). Taken together, the data demonstrate that a subset of MuSCs undergo apoptosis during acute injury-induced regeneration, whereas necroptotic cell death of MuSCs is an exclusive feature of dystrophic muscles, characterized by chronic inflammation and regeneration.
Inhibition of Necroptosis in MuSCs of mdx Mice Compromises Skeletal Muscle Regeneration and Muscle Function
The observation that a subset of MuSCs specifically undergoes necroptosis in dystrophic mdx muscles raised the question whether removal of MuSCs by programmed cell death is an unwanted byproduct of inflammatory processes in chronically regenerating muscles or serves a specific purpose. To answer this question, we generated Pax7CreERT2/Ripk3loxP/loxP and Pax7CreERT2/Ripk3loxP/loxP/mdx mice (hereafter Ripk3mKO and Ripk3mKO/mdx, respectively), which permit MuSC-specific deletion of Ripk3 in WT and mdx mice after administration of tamoxifen (TAM) (Figure 2A). Ripk3 is an essential component of the necroptosis pathway, and its deletion essentially abrogates necroptosis (Vanden Berghe et al., 2014, Grootjans et al., 2017). In agreement with previous studies that did not uncover a requirement of Ripk3 for normal development and tissue homeostasis (Zhao et al., 2017), we did not detect any discernable effects of Ripk3 inactivation on MuSC numbers, muscle morphology, and regeneration in mice with an intact dystrophin gene (Figures S1F–S1H).
Figure 2.

Necroptosis and Apoptosis of MuSCs Ameliorate Muscle Function in Chronic Muscle Disease
(A) Schematic representation of the tamoxifen regimen.
(B) Immunofluorescence staining of TA muscle cross-sections from mdx and mdx/Ripk3mKO mice 2 weeks after tamoxifen treatment using antibodies against PAX7 and cleaved CASP3 (n = 3 for each group). Scale bar. 25 μm.
(C) Quantification of apoptotic CASP3+/PAX7+MuSCs in TA muscle cross-sections.
(D) Quantification of total PAX7+ MuSCs in TA muscle cross-sections.
(E) Schematic representation of the tamoxifen regimen, z-VAD inhibitor treatment, muscle grip strength measurement, and time points of TA muscle collection.
(F–I) Immunofluorescence staining of TA muscle cross-sections from mdx and mdx/Ripk3mKO mice for PAX7 and cleaved CASP3 (F) and RIPK3 (H) 2 weeks after tamoxifen treatment and DMSO or z-VAD inhibitor treatment (n = 3 or 4 for each group) and quantified in (G) and (I), respectively. Scale bar, 25 μm.
(J) H&E staining of TA muscles sections from mdx and mdx/Ripk3mKO mice treated with DMSO or z-VAD inhibitor (n = 3 for each group). Scale bar, 100 μm.
(K) Quantification of muscle grip strength of mdx and mdx/Ripk3mKO mice treated with DMSO or z-VAD inhibitor (n = 3 or 4 for each group).
Statistical analysis: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.005, two-way ANOVA followed by Bonferroni post-test with alpha = 5%. All analyses indicated across the experiments were biological replicates unless otherwise stated.
We found that MuSC-specific inactivation of Ripk3 in mdx mice strongly increased the number of apoptotic MuSCs, indicating that inhibition of necroptosis facilitates enhanced activation of apoptosis in MuSCs of mdx mice (Figures 2B and 2C). Furthermore, the increased rate of apoptosis was associated with a reduction of PAX7+ MuSCs in Ripk3mKO/mdx compared with mdx mice (Figure 2D), indicating that prevention of necroptosis in mdx MuSCs blunts the increase of MuSC numbers. Notably, the number of CD45+ and CD68+ cells did not change in mdx muscles after inactivation of Ripk3, indicating that the inflammatory response is not altered after suppression of necroptosis in mdx MuSCs, which makes sense, as the major driver of inflammation in mdx muscles are damaged muscle fibers (Figures S1I–S1M). Consistently, CD45 and CD68 staining revealed that increased RIPK3 expression in dystrophic muscles is not limited to MuSCs but occurs in CD45+ and CD68+ cells as well (Figures S1I, S1K, and S1M).
To obtain additional insights into the physiological relevance of programmed cell death and to analyze whether inhibition of increased apoptosis prevents adverse effects of MuSC-specific Ripk3 inactivation on muscle morphology and function in mdx mice, we pharmacologically suppressed apoptosis in mdx mice by systemic administration of the cell-permeable, pan-caspase inhibitor z-VAD (Figure 2E). Inhibition of apoptosis with z-VAD significantly reduced the number of apoptotic PAX7+/cleaved CASP3+ MuSCs and moderately increased the number of PAX7+/RIPK3+ MuSCs in mdx muscles (Figures 2F–2I). In contrast, inactivation of Ripk3 essentially eliminated necroptotic MuSCs and increased the number of apoptotic PAX7+/cleaved CASP3+ in mdx muscles (Figures 2F–2I), indicating that removal of MuSCs in dystrophic muscles is secured by a reciprocal crosstalk between different programmed cell death pathways.
Interestingly, suppression of apoptosis did not improve muscle regeneration in Ripk3mKO/mdx compound mutants (Figure 2J), indicating that the impaired muscle morphology in Ripk3mKO/mdx is not caused by increased apoptosis but rather by the failure of MuSCs to restore damaged muscle fibers in continuously regenerating muscles. In contrast to the inhibition of necroptosis, suppression of apoptosis did not change the number of MuSCs in mdx mice, although we observed a further decline of MuSC numbers in compound Ripk3mKO/mdx mice after z-VAD treatment (Figure S1N). The data suggest that combined inhibition of necroptosis and apoptosis compromises the quality of the MuSC pool and thereby restricts its expansion. To determine whether the worsened muscle morphology in Ripk3mKO/mdx mice with and without z-VAD treatment has a functional impact on muscle strength, we performed grip strength tests. Consistent with the aggravated muscle pathology and attenuation of MuSC expansion, we found that inhibition of necroptosis by inactivation of Ripk3, suppression of apoptosis by z-VAD treatment, or a combination of both substantially reduced grip strength compared with untreated mdx mice (Figure 2K). The failure to improve muscle function by combined inhibition of cell death pathways corresponds to the further decline of MuSC numbers in compound Ripk3mKO/mdx mice treated with z-VAD. To further validate these results, we generated Casp8mKO/Ripk3mKO/mdx mice. TAM treatment of these mice led to genetic inactivation of both apoptosis and necroptosis specifically in MuSCs. Importantly, muscle grip strength tests of TAM-treated triple-mutant mice fully recapitulated the results obtained by treatment of Ripk3mKO/mdx mice with z-VAD (Figure S1O). Moreover, adult mdx/mlklKO mice lacking the necroptotic effector mlkl showed impaired tissue morphology and reduced muscle strength, confirming the importance of necroptosis for efficient regeneration in dystrophic muscles (Figures S1P and S1Q). Taken together, these data clearly demonstrate that failure to remove a potentially deleterious subpopulation of MuSCs through necroptosis and apoptosis reduces the fitness of the MuSC population, thereby compromising proper muscle regeneration and function.
Chronic Muscle Injury Predisposes Murine and Human MuSCs to Undergo Necroptosis
To gain further understanding into the cell autonomous role of cell death pathways, we next analyzed freshly isolated mouse and human MuSCs in culture. In comparison to healthy control cells, MuSCs from mdx mice and BMD patients showed a pronounced increase in the rate of necroptosis, as indicated by steady accumulation of EthD-III incorporating MuSCs (Krysko et al., 2008, Strilic et al., 2016) (Figures 3A and 3B; Videos S1 and S2). Treatment with the RIPK1 kinase inhibitor necrostatin-1 (Nec-1) (Degterev et al., 2005, Degterev et al., 2008) substantially reduced necroptotic cell death (Figures S2A and S2B; Videos S1 and S2), while inhibition of apoptosis with z-VAD increased necroptosis in MuSCs from human BMD patients, further confirming the crosstalk between necroptosis and apoptosis (Figure S2B; Videos S1 and S2). The dramatic increase in necroptotic mdx MuSCs in vitro demonstrates that necroptosis is not a rare event, although the rather small number of necroptotic (and apoptotic) cells detected in mdx muscles seems to indicate otherwise. We reason that phagocytic cells continuously remove dying cells in vivo, generating the false impression that necroptotic (and apoptotic) cell death is rare. The absence of phagocytes in cultures of MuSCs avoids this complication and allows accurate quantification of the full extent of necroptosis.
Figure 3.

Predisposition of MuSCs from Dystrophic Muscles for Necroptosis Reduces Adverse Crosstalk among mdx Muscle Stem Cells and Is Associated with Chromatin Reorganization
(A) EthD-III incorporation rates of MuSCs isolated from control and mdx mice in vitro.
(B) EthD-III incorporation rates of MuSCs isolated from healthy individuals and BMD patients in vitro (n = 3 for each group in both A and B).
(C) Schematic of co-culture setup with YFP+ WT MuSCs in direct monolayer co-culture with either YFP− WT or YFP−mdx MuSCs.
(D) Representative images of co-cultured MuSCs (insets are magnifications of indicated boxes). Scale bar, 100 μm.
(E) Quantification of YFP+ WT MuSCs incorporating EthD-III at endpoints as in (D) (n = 3 for each group).
(F) Schematic of transwell-assay setup. WT, mdx, and mdx/Ripk3mKO MuSCs are grown in the upper transwell; in the lower well, single WT MuSCs are grown in Matrigel at clonal density.
(G and H) Representative images of MuSC colony sizes after 4-day culture in Matrigel (G), quantified in (H). n = 3 for each group. Scale bar in (G): 100 μm.
(I) Volcano plot of ATAC-seq data showing increase of chromatin accessibility in mdx MuSCs (red) compared with control MuSCs (green). n = 2 or 3 for each group.
(J) Volcano plot of RNA-seq data showing increase of transcriptional upregulation in mdx MuSCs (red) compared with control MuSCs (green). n = 2 or 3 for each group.
(K) Correlation scores between chromatin accessibility from (I) and actively transcribed genes from (J).
(L) Heatmap representation of genes coding for chromatin remodelers with more (red) and less (blue) accessible chromatin in MuSCs derived from control and mdx mice (n = 2 for each group).
Statistical analysis: ∗∗p < 0.01 and ∗∗∗p < 0.005, two-way ANOVA followed by Bonferroni post-test with alpha = 5%. All analyses indicated across the experiments were biological replicates unless otherwise stated.
MuSC were isolated from wildtype mice and cultured in vitro in the presence or absence of different inhibitors for 58 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
MuSC were isolated from muscles of mdx mice and cultured in vitro in the presence or absence of different inhibitors for 70 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
The tightly regulated generation of necroptotic MuSCs in mdx muscles, together with the adverse effects of necroptosis inhibition on muscle regeneration, suggested that some MuSCs, which arise in the dystrophic environment, need to be removed to prevent undesirable effects on the remaining MuSC population. To test this hypothesis, we examined the effects of MuSCs isolated from mdx mice on YFP-labeled “healthy” MuSCs in co-culture experiments (Figures 3C–3E). Intriguingly, co-culture with mdx MuSCs induced pronounced EthD-III incorporation in YFP+ MuSCs, clearly indicating that dystrophic MuSCs are able to elicit deleterious non-autonomous effects on the MuSC population (Figures 3C–3E). We also found that direct physical contact is not required for dystrophic MuSCs to inhibit growth of MuSCs, as expansion of WT MuSC colonies in Matrigel was suppressed by mdx MuSCs, even when both cell types were separated by a membrane in transwell assays (Figures 3F–3H). Importantly, suppression of necroptosis in mdx MuSCs due to inactivation of Ripk3 exacerbated the deleterious non-cell-autonomous affects of mdx MuSCs and further reduced expansion of the MuSC population (Figures 3F–3H). RNA sequencing (RNA-seq) analysis of WT MuSCs co-cultured in transwell assays together with control, mdx, or mdx/Ripk3mKO MuSCs corroborated differential effects of mdx and mdx/Ripk3mKO MuSCs on the MuSC population (Figure S2D; Table S1). For example, we observed that signals derived from mdx/Ripk3mKO MuSCs reduced in WT MuSCs transcription of Egr3 and Malat1, which are involved in the proliferation and differentiation of muscle cells, respectively (Chen et al., 2017, Kurosaka et al., 2017).
In addition, we noted that the increased propensity of MuSCs from dystrophic muscles to undergo necroptosis was preserved for several days in vitro despite the absence of inflammatory or other signals (Figures 3A and 3B; Videos S1 and S2). This indicated an epigenetic control mechanism that forces MuSCs to “remember” previous pro-necroptotic conditions. To follow up this lead, we compared the chromatin state between WT and mdx MuSCs. Global assessment of chromatin organization by EM disclosed a massive relaxation of chromatin in MuSCs of mdx muscles, whereas WT muscles nearly exclusively contained MuSCs with highly condensed chromatin (Figures S2C and S2D). Furthermore, assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) revealed a dramatic increase of accessible open chromatin in mdx MuSCs compared with WT MuSCs, which went along with increased expression of corresponding genes (Figures 3I–3K; Table S1). Notably, numerous differentially accessible regions between mdx and WT MuSCs coded for chromatin remodelers and various cell death associated factors (Figure 3L; Table S2). We concluded that the continuous exposure to the regenerative, inflammatory microenvironment in mdx muscles results in stable opening of chromatin regions in MuSCs favoring necroptosis.
Chd4 Is Required for Muscle Regeneration and Suppresses Necroptosis of MuSCs
To pinpoint putative epigenetic factors that are responsible for the acquisition of a pro-necroptotic memory, we performed a lentiviral-based RNAi screen against a comprehensive array of known and putative chromatin modifiers targeting 634 genes in WT MuSCs (Fazzio et al., 2008) (Figure S3A; Table S3). As a readout, we used the self-renewal index (SI) of MuSCs, which already aided us in a previous screen (Zhang et al., 2015), assuming that any change in cellular survival due to programmed cell death will also impinge on the SI. We identified 58 genes that upon knockdown significantly changed the SI, 12 of which had previously been reported to regulate MuSC homeostasis, including Suv420h1 (Boonsanay et al., 2016), Ezh2 (Marchesi et al., 2012), Sirt1 (Ryall et al., 2015), Myocd (Long et al., 2007), and Sox15 (Lee et al., 2004) (Figure S3B). qRT-PCR and immunofluorescence staining confirmed the expression of potential candidate genes in MuSCs during myogenic differentiation (Figures S3C and S3D).
We became particularly attracted to Chd4, a core component of the NuRD deacetylation complex (Lai and Wade, 2011), as knockdown of Chd4 strongly reduced the number of MuSCs in vitro (Figure S3E). In addition, (1) knockdown of other components of the NuRD complex, including Hdac2, Mta1, and Rbbp4, yielded similar effects, and (2) expression of Chd4 was dramatically upregulated in activated, proliferating MuSCs (Figures S3B and S3C). To assess the effects of Chd4 inactivation in MuSCs in vivo, we generated Pax7CreERT/Chd4loxP/loxP mice (hereafter Chd4mKO) and verified loss of Chd4 expression after TAM administration (Figures S4A–S4C). Inactivation of the Chd4 gene in MuSCs under baseline conditions did not alter skeletal muscle morphology (Figures 4A and 4B; Figure S4D). However, we found that Chd4mKO skeletal muscles contained substantially smaller numbers of PAX7+ MuSCs in comparison with control littermates, which coincided with the emergence of necroptotic pMLKL+/PAX7+ MuSCs (Figures 4D–4F). Importantly, CTX-induced skeletal muscle regeneration was severely compromised in Chd4mKO mice (Figures 4A and 4C; Figure S4E), which was linked to a massive reduction of MuSCs and a reciprocal increase of necroptotic pMLKL+/PAX7+ MuSCs (Figures 4D–4F). We observed a striking 40% increase of pMLKL+/PAX7+ MuSCs in TA muscles of Chd4mKO mutant mice after CTX-induced injury, which is in contrast to the small numbers of necroptotic MuSCs in chronically regenerating mdx muscles (Figure 4G). This indicates that the massive surge of necroptotic cells in Chd4mKO-mutant mice overcomes the ability to efficiently clear dying necroptotic cells, whereas in dystrophic mdx muscles, gradually emerging necroptotic cells are swiftly removed, making their detection difficult.
Figure 4.

Chd4 Is Required for Muscle Regeneration and Suppresses Necroptosis of MuSCs
(A) Macroscopic images of uninjured and CTX-injured TA muscles from control and Chd4mKO mice.
(B and C) H&E staining of sections of uninjured muscle (B) and injured muscle (C) derived from muscles shown in (A). Scale bar (A–C), 100 μm.
(D) Quantification of PAX7+ cells from control and Chd4mKO muscles using PAX7 antibodies (n = 3 for each group).
(E–J) Immunofluorescence staining of TA muscle sections 2 weeks after CTX injury from control and mdx mice (n = 3 for each group) using antibodies against PAX7 and pMLKL to detect necroptosis (E–G) and PAX7 and cleaved CASP3 (H–J) to detect apoptosis (n = 5 for each group). Scale bar, 25 μm. (F, G, I, and J) Absolute and relative quantification of data represented in (E) and (H) (n = 3–5 for each group).
Statistical analysis: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗∗p < 0.001, two-way ANOVA followed by Bonferroni post-test with alpha = 5%. All analyses indicated across the experiments were biological replicates unless otherwise stated.
Interestingly, the number of apoptotic cleaved CASP3+/PAX7+ MuSCs was much smaller in acutely damaged Chd4mKO muscles compared with damaged WT control muscles, clearly indicating that inactivation of Chd4 shifts the mode of programmed cell death toward necroptosis (Figures 4H–4J). Consistent with the arrested proliferation after shRNA-mediated knockdown of Chd4 in MuSCs (Figure S3E), proliferation of MuSCs isolated from Chd4mKO mice was severely impaired (Figures S4G and S4H). Time-lapse imaging confirmed that MuSC-specific inactivation of Chd4 elicits necroptosis, as visualized by dramatically increased rates of EthD-III incorporation over time (Figures S4H and S4I; Video S3). In addition, EM analysis of TA sections confirmed the reduction of the total number of MuSCs and the increase of necroptotic MuSCs in Chd4mKO skeletal muscles (Figures S4K and S4L). We concluded that MuSC-specific loss of Chd4 triggers necroptosis of MuSCs and severely impairs muscle regeneration.
MuSC were isolated from muscles of Chd4mKO mutant mice and cultured in vitro in the presence or absence of different inhibitors for 58 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
CHD4/NuRD Directly Represses Expression of Ripk3 in MuSCs but Not in Embryonic Stem Cells
To gain a mechanistic understanding of how CHD4 regulates necroptosis of MuSCs, we characterized both the transcriptome and the proteome of cultured Chd4loxP/loxP MuSCs after adenovirus-mediated expression of GFP or Cre recombinase (Figures 5A and 5B; Figures S5A and S5B). Our analysis uncovered 178 and 325 differentially expressed genes (DEGs) after Chd4 inactivation on the basis of changes at the RNA and protein levels, respectively. The majority of genes was upregulated indicating that CHD4 acts primarily as a transcriptional repressor in MuSCs (Figures 5A and 5B; Table S4). Intriguingly, Ripk3 was among the top upregulated DEGs at both the transcript and protein levels (Figure S5B; Table S4), suggesting that CHD4 might regulate necroptosis via direct epigenetic repression of Ripk3. To test this hypothesis, we performed chromatin immunoprecipitation (ChIP) assays on different regions of the Ripk3 gene locus (Figure 5C). CHD4 binding was highly enriched at the promoter (prRipk3) but not at intragenic regions of the Ripk3 gene in MuSCs, which correlated with the absence of RNA polymerase II binding to the (repressed) Ripk3 promoter (Figures 5D and 5E). In contrast, ChIP assays revealed that CHD4 is not recruited to the Ripk3 promoter in mouse embryonic stem cells, despite the absence of polymerase II binding (Figures 5F and 5G). Furthermore, Ripk3 expression remained unchanged after inactivation of Chd4 in embryonic stem cells, indicating that repression of Ripk3 in embryonic stem cells and MuSCs is mediated by different mechanisms (Figure S5C).
Figure 5.

CHD4 Directly Suppresses Activation of Ripk3
(A and B) Volcano plots of RNA-seq (A) and proteomics data (B) visualizing significantly downregulated (red points) or upregulated (green points) transcripts and proteins after adeno-Cre-mediated inactivation of Chd4 in MuSCs from Chd4loxP/loxP mice (n = 4).
(C) Schematic representation of the locations of primers used for ChIP-qPCR to detect binding of Chd4 to the Ripk3 gene.
(D and E) qRT-PCR ChIP analyses of CHD4 binding (D) and POL II binding (E) in activated MuSCs (ASC).
(F and G) qRT-PCR ChIP analyses of CHD4 binding (F) and POL II binding (G) in embryonic stem cells (ESCs). IgG served as a negative control (n = 3 independent technical replicates for each group).
(H and I) qRT-PCR (H) and western blot (I) analyses of Ripk3 expression in control and mdx muscle tissues (n = 3).
(J and K) ChIP-qPCR analysis of CHD4 binding (J) and HDAC1 binding (K) to the promoter and intragenic regions of the Ripk3 gene in skeletal muscles from control and mdx mice (n = 3 independent technical replicates for each group).
(L) Heatmap representing gene expression of members of the NuRD complex (log2 expression) in quiescent MuSCs (QSC) from control (n = 2) and mdx (n = 3) mice on the basis of RNA-seq data.
Statiscal analysis: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.005, two-way ANOVA followed by Bonferroni post-test with alpha = 5%). All analyses indicated across the experiments were biological replicates unless otherwise stated.
Next, we investigated whether CHD4 regulates Ripk3 expression in MuSCs of dystrophic skeletal muscles. We detected dramatically elevated expression levels of Ripk3 mRNA and protein in muscles and MuSCs of mdx mice (Figures 5H and 5I; Figures S5D and S5E), which was matched by a massively decreased presence of CHD4 and HDAC1 at the Ripk3 promoter (Figures 5J and 5K). Interestingly, however, the expression of Chd4 and other components of the NuRD complex, including Hdac1, was not changed in mdx compared with WT muscles (Figure 5L; Figure S5F), indicating that diminished recruitment but not reduced expression of the CHD4/NuRD complex promotes Ripk3 expression and thereby elicits necroptosis of MuSCs in dystrophic muscles. Consistently, forced expression of Ripk3 in isolated WT MuSCs enhanced the number of MLKL+ necroptotic MuSCs even in the absence of any exogenous necroptosis-stimulating factors, demonstrating that elevated RIPK3 levels alone are sufficient to induce necroptosis of MuSCs (Figures S5G and S5H). Similar to the results obtained in murine MuSCs, knockdown of Chd4 in human myoblasts caused a dramatic upregulation of Ripk3 expression (Figure S5I). Moreover, CHD4 binding to the Ripk3 promoter was strongly reduced in human myoblasts derived from BMD patients compared with control human myoblasts, and we observed a corresponding upregulation of Ripk3 expression, exactly recapitulating the situation in mdx MuSCs (Figures S5J and S5K). Taken together, the data demonstrate that the CHD4/NuRD complex mediates direct epigenetic suppression of Ripk3 in murine and human MuSCs, which is alleviated in conditions of chronic muscle disease.
Inactivation of Ripk3 Restores the Impaired Regenerative Capacity of Chd4-Deficient MuSCs
To corroborate our finding that CHD4 controls necroptosis via regulation of Ripk3 expression, we generated Chd4mKO/Ripk3mKO mice allowing simultaneous and MuSC-specific inactivation of Chd4 and Ripk3. Strikingly, MuSC-specific inactivation of Ripk3 in Chd4mKO/Ripk3mKO compound mutants completely abrogated the occurrence of necroptotic PAX7+/pMLKL+ MuSCs observed in Chd4mKO mutants (Figures 6A and 6B; Figure S6A) and restored the total number of PAX7+ MuSCs under baseline conditions (Figure 6C). We then introduced a conditional Rosa26YFP allele into Chd4mKO and Chd4mKO/Ripk3mKO mutants and WT mice to trace the fate of recombined MuSCs after TAM treatment. Formation of expanding YFP+ MuSC colonies on ex vivo cultured single myofibers confirmed that loss of Ripk3 reestablishes cell proliferation and prevents cell death of Chd4mKO MuSCs. In contrast, MuSCs from Chd4mKO mutants with an intact Ripk3 allele uniformly incorporated EthD-III and underwent cell death via necroptosis (Figure S6B). Intriguingly, genetic inactivation of Ripk3 in Chd4mKO/Ripk3mKO double mutants markedly restored muscle regeneration, as indicated by formation of YFP+ myofibers after CTX injection, while virtually no YFP+ myofibers were detected in Chd4mKO single mutants (Figures 6D and 6E). Furthermore, Chd4mKO/Ripk3mKO compound mutants exhibited a substantial reduction of fibrosis after acute injury in comparison with Chd4mKO single mutants (Figure S6C). Although Chd4mKO/Ripk3mKO double mutants showed significantly improved regeneration, newly formed fibers were heterogeneously sized, and tissue morphology was not completely normal, indicating that CHD4 not only regulates Ripk3 but also plays an additional role at later stages of myogenic differentiation and muscle regeneration in vivo (Figure S6C). Treatment of Chd4mKO knockout mice with Nec-1s, a stable variant of Nec-1 (Takahashi et al., 2012), fully recapitulated the rescue of muscle regeneration in Chd4mKO/Ripk3mKO double mutants and increased the number of Pax7+ MuSCs in comparison with non-treated Chd4mKO controls (Figures 6D–6F). Consistently, Nec-1 but not the apoptosis inhibitor z-VAD prevented cell death in isolated Chd4mKO MuSCs almost completely and restored proliferation, adding further evidence that the primary reason for the failure of Chd4mKO MuSCs to proliferate is massively enhanced necroptotic cell death (Figures 6G and 6H; Video S3).
Figure 6.

Necrostatin Restores Proliferation of Chd4mKO MuSCs and Improves Skeletal Muscle Regeneration in Chd4mKO Mice
(A) Immunofluorescence staining for PAX7/pMLKL on TA muscle sections from Chd4mKO and Chd4mKO/Ripk3mKO mice. Scale bar, 25 μm.
(B and C) Quantification of PAX7+/pMLKL+ MuSCs (B) undergoing necroptosis and PAX7+ MuSCs (C) in control, Chd4mKO, and Chd4mKO/Ripk3mKO muscles (n = 3 for each group).
(D) Fluorescence images of uninjured (left) and injured (right) TA muscles from Chd4mKO/ROSA26YFP, Chd4mKO/Ripk3mKO/ROSA26YFP, and Chd4mKO/ROSA26YFP mice treated with Nec-1s 2 weeks after CTX-induced muscle injury (n = 3–5 for each group). Scale bar, 100 μm.
(E) Cross sections of injured TA muscles as in (D) indicating increased formation of YFP+ myofibers after Ripk3 inactivation. Scale bar, 25 μm.
(F) Quantification of MuSCs in Chd4mKO/ROSA26YFP TA muscles with and without Nec-1s treatment after CTX-induced muscle injury (n = 3–6 for each group.
(G) EthD-III incorporating Chd4mKO MuSCs after treatment with z-VAD, necrostatin-1, or DMSO (n = 3 for each group); scale bar, 100 μm.
(H) Quantification of EthD-III incorporating Chd4mKO MuSCs after 100 h of in vitro culture in a field of 1 mm2 following treatment with z-VAD, necrostatin-1, or DMSO (n = 3–6 for each group. Statisical analysis: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.005, two-way ANOVA followed by Bonferroni post-test with alpha = 5%).
All analyses indicated across the experiments were biological replicates unless otherwise stated.
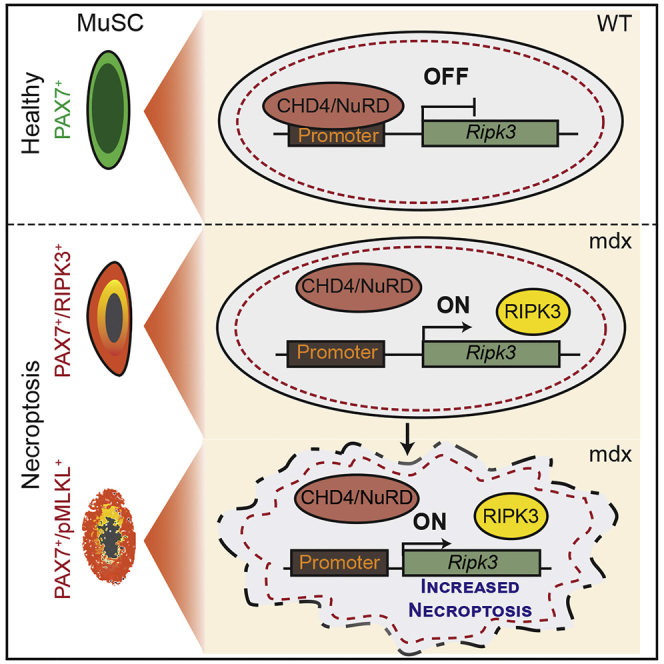
Taken together, these data demonstrate that the CHD4/NuRD complex is required for inhibition of necroptosis via active epigenetic suppression of Ripk3 in MuSCs under homeostatic conditions and during acute skeletal muscle regeneration. In contrast, CHD4/NuRD-mediated suppression of Ripk3 in MuSCs becomes attenuated in dystrophic muscles, which lowers the threshold for necroptosis and prevents exacerbation of dystrophic tissue remodeling.
Discussion
Here, we discovered that a subpopulation of MuSCs undergoes necroptotic cell death in chronically regenerating, dystrophic muscles in both mice and humans but not after acute muscle injury of healthy muscles. As inactivation of necroptosis compromised muscle regeneration in mdx mice, we postulate that controlled and limited removal of a subset of mdx MuSCs by programmed cell death serves an important role to assure the fitness of the MuSC compartment as a whole. In support of this, we demonstrated that mdx MuSCs exert adverse effects on the expansion of healthy MuSCs by a non-cell-autonomous mechanism, which is aggravated by inhibition of necroptosis in mdx MuSCs. Furthermore, we identified the molecular mechanism that prevents necroptosis of MuSCs in healthy and acutely damaged muscle but allows their partial elimination in chronically regenerating dystrophic muscles.
Programmed cell death plays a vital role in various developmental processes, for example, in the immune and the nervous systems, which are characterized by overproduction of cells. For example, superfluous cells that fail to acquire tolerated antigen specificities or synaptic connections need to be removed for proper organ function (Opferman et al., 2003, Nijhawan et al., 2000). Failure to remove dysfunctional or abnormal cells results in numerous diseases, including cancer and various immune disorders (Letai, 2017, Nagata and Tanaka, 2017). In addition, programmed cell is essential for fitness selection, allowing elimination of cells that are viable but “less fit” than surrounding cells. Most often, removal of so-called loser cells occurs by activation of apoptosis in the less fit cells (Hashimoto and Sasaki, 2019), by induction of a senescence-like program (Bondar and Medzhitov, 2010), or through engulfment by surrounding winner cells (Clavería et al., 2013). Our results seem to suggest that necroptosis is another mechanism by which “loser” cells are eliminated in an inflammatory environment.
Very little is known about a potential role and/or function of necroptosis during tissue regeneration, although increasing evidence suggests that removal of unwanted cells via necroptosis becomes particularly important under conditions of prolonged or extreme stress in which activation of apoptosis alone is not sufficiently effective to eliminate deleterious cells (Galluzzi et al., 2016). Only very recently it was shown that necroptotic death of proinflammatory microglia followed by repopulation to a pro-regenerative state is required to regenerate central nervous system myelin following injury (Lloyd et al., 2019). Our own study discloses a critical role of programmed cell death for the quality control of stem cells, which allows removal of functionally compromised stem cells, restraining expansion of other MuSCs during skeletal muscle repair by a non-cell-autonomous mechanism. The finding that inactivation of Ripk3 in MuSCs compromises regeneration and impairs muscle function in mdx mice was unexpected. One might assume that inhibition of programmed cell death increases the number of MuSCs in dystrophic muscles and improves morphology and function. However, we observed exactly the opposite effects, indicating that full expansion of MuSCs cannot take place before removal of potentially defective MuSCs. Pharmacological inhibition of apoptosis by z-VAD treatment or a combined suppression of necroptosis and apoptosis yielded similar results, further supporting our conclusion. However, inhibition of apoptosis by systemic z-VAD treatment not only affects MuSCs but will also enhance survival of other cell types, such as FAPs and inflammatory cells, which might also aggravate muscle pathology (Lemos et al., 2015).
Activation of either necroptosis or apoptosis might represent different sides of the same coin and serve the same purpose, as necroptosis is regarded as a “fail-safe” mechanism for cells unable to undergo apoptosis when required (Lu et al., 2014). However, in some cell types or conditions, necroptosis might be the primary mode of programmed cells death, as for example demonstrated for inflammatory microglia, which seem to lack any signs of apoptotic cell death (Lloyd et al., 2019). Furthermore, we disclosed the mechanism, which enables necroptotic cell death of MuSCs in dystrophic muscles. We found that MuSCs in mdx muscles express much higher levels of Ripk3 because of attenuated recruitment of the repressive CHD4/NuRD complex to the Ripk3 promoter. CHD4 and other components of the NuRD complex were initially identified in a screen to recognize factors regulating necroptosis in MuSCs. Complete genetic inactivation of Chd4 in MuSCs massively induced necroptosis, thus preventing expansion of MuSCs. Our experiments revealed that the mechanism to prevent untimely or disproportionate Ripk3 expression differs among cell types. Embryonic stem cells suppress Ripk3 expression, similar to MuSCs in healthy muscles, but without binding of CHD4 to the Ripk3 promoter, indicating that embryonic stem cells have evolved a different strategy to control Ripk3 expression, most likely due to different chromatin organization and a lowered requirement to initiate programmed cell death. In contrast, it has been described very recently that endothelial cells also rely on the NuRD complex to prevent excessive Ripk3 and subsequent vascular rupture under hypoxic conditions (Colijn et al., 2020). In more general terms, it seems likely that cells that readily initiate apoptosis benefit from lasting epigenetic suppression of Ripk3, whereas cells that are relatively resistant to apoptosis need a mechanism allowing rapid upregulation of Ripk3 expression, in case apoptosis fails. Treatment of Chd4-mutant MuSCs with necrostatin or inactivation of Ripk3 inhibited necroptosis and restored proliferation in vitro and partially in vivo, demonstrating that CHD4 is crucial to suppress aberrant upregulation of Ripk3 and necroptosis. Likewise, inactivation of Ripk3 significantly improved skeletal muscle regeneration defects in MuSC-specific Chd4 mutants, adding further evidence that Chd4 prevents aberrant induction of necroptosis in MuSCs. Notably, the Chd4mKO phenotype was not completely normalized by inactivation of Ripk3, which suggests additional functions of CHD4 during later stages of MuSC-dependent regeneration.
Tight control of Ripk3 expression is vital to restrict excessive initiation of necroptosis, as enhanced expression of Ripk3 alone is sufficient to induce necroptosis of MuSCs. In addition, our finding that reduction of MuSC numbers in uninjured Chd4 mutant muscles is completely reverted in Chd4mKO/Ripk3mKO mice confirms the decisive role of CHD4/NuRD in regulating RIPK3 levels for induction of necroptosis. We reason that upregulation of Ripk3 expression lowers the threshold for necroptosis induced by TNF superfamily members or other signals that are abundantly present in dystrophic muscles (Ashkenazi and Salvesen, 2014, De Paepe and De Bleecker, 2013). According to this model (Figure 7), high RIPK3 levels act as a permissive rather than an instructive signal for initiating necroptosis.
Figure 7.

Model
Attenuated epigenetic suppression of muscle stem cell necroptosis is required for efficient regeneration of dystrophic muscles. CHD4/NuRD suppresses Ripk3 expression and thereby necroptosis in MuSCs of healthy muscles (bottom). Chronic dystrophy continuously evocates a demand for regeneration and persistent activation of MuSCs. Necroptosis secures removal of defective MuSCs that otherwise inhibit MuSC expansion in a non-cell-autonomous manner if not eliminated. Necroptosis in MuSCs is mediated by attenuated recruitment of CHD4/NuRD to the Ripk3 promoter in dystrophic muscles and leads to a lasting increase in necroptosis even in the absence of pro-necroptotic signals (“memory” effect) (top).
In contrast to a recent study (Morgan et al., 2018), we did not obtain any evidence for necroptotic myofibers in mdx mice. Likewise, we did not observe beneficial but rather adverse effects of Ripk3 inactivation in MuSCs for muscle regeneration and function. Consistent with this observation, germline inactivation of Mlkl in mdx mice did not ameliorate but worsened the dystrophic phenotype. Hence, inactivation of programmed cell death pathways, including necroptosis, does not appear to be a viable option to treat dystrophic muscle disorders. Moreover, systemic inhibition of necroptosis by pharmacological intervention or in germline Ripk3- or Mlkl-mutant mice will inhibit necroptotic cell death of FAPs. This is important, as elimination of FAPs by programmed cells has been claimed to limit fibrosis in damaged skeletal muscles (Lemos et al., 2015). However, it remains to be seen whether necroptosis plays a similarly important role as apoptosis for elimination of FAPs in profibrotic conditions.
Interestingly, not the expression of Chd4 but its recruitment to the Ripk3 promoter was attenuated in both murine and human MuSCs from dystrophic muscles, indicating that either modifications of the CHD4/NuRD complex and/or absence of accessory proteins are responsible for relieved suppression of Ripk3 during chronic disease conditions. Compared with WT, cultured MuSCs from both murine and human dystrophic muscles showed a much higher rate of necroptosis even after several passages in vitro and despite the absence of cell death-inducing factors. This “memory” effect in MuSCs is particularly fascinating. Our data strongly indicate that the enhanced susceptibility of MuSCs from mdx muscle for necroptosis is based on reduced epigenetic repression of Ripk3 and not on the absence of dystrophin, as (1) we prevented necroptosis and restored proliferation of MuSCs by blocking the CHD4 target Ripk3; (2) expression of dystrophin is very low in MuSCs, although a function of dystrophin has been described for asymmetric division of MuSCs (Dumont et al., 2015); and (3) we observed the same increase in necroptosis in human BMD MuSCs, which do carry a partially functional in-frame mutation of the dystrophin gene (Emery, 2002).
Maintenance of the stem cell pool is particularly demanding in continuously regenerating dystrophic muscles. Our study uncovers that the MuSC population in dystrophic muscles needs to be purged by balanced and interconnected programmed cell death pathways to allow full stem cell expansion (Figure 7). Accordingly, we view necroptosis as a crucial process that facilitates optimized muscle repair by contributing to fitness selection but not as a mechanism that per se promotes tissue regeneration. Although we do not completely understand the necessity for enhanced MuSC quality control by programmed cell death in dystrophic muscles, potential explanations might include accumulation of malfunctioning MuSCs, which interferes with the expansion of a “healthy” stem cell pool or the ability to differentiate. Alternatively, some MuSCs might fail to respond to microenvironmental signals, are unable to re-enter the stem cell niche, or are on a potential path to malignant transformation. In support of this concept, mdx mice display strongly elevated rates of tumorigenic transformation in absence of p53, which not only safeguards genome stability but is also a strong cell death-inducing factor (Camboni et al., 2012, Preussner et al., 2018). Therefore, any attempt to increase the pool of stem cells for regenerative purposes needs to rule out that their physiological responsiveness and quality is jeopardized.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PAX7 mouse monoclonal antibody | DSHB | #MAB1675; RRID:AB_528428 |
| RIPK3 rabbit polyclonal antibody | Abcam | #ab56164; RRID:AB_2178667 |
| Cleaved Caspase-3 (Asp175) rabbit polyclonal antibody | Cell Signaling Technology | #966; RRID:AB_23411881 |
| Phospho-MLKL (Ser345) rabbit monoclonal antibody | Cell Signaling Technology | #62233; RRID:AB_2734703 |
| PAX7 mouse monoclonal antibody (Human) | Santa Cruz | #sc-81648; RRID:AB_2159836 |
| Phospho-MLKL(Ser358) rabbit monoclonal antibody (Human) | Cell Signaling Technology | #91689; RRID:AB_2732034 |
| SUMO1 rabbit monoclonal antibody | Abcam | #ab32058; RRID:AB_778173 |
| SENP5 rabbit polyclonal antibody | Sigma Aldrich | #SAB1300165; RRID:AB_10608890 |
| CHD4 (Mi-2 beta) rabbit polyclonal antibody | Active Motif | #39289; RRID:AB_261493 |
| CHD4 (D8B12) rabbit monoclonal antibody | Cell Signaling Technology | #11912; RRID:AB_2751014 |
| RIPK3 (G4G2A) rabbit monoclonal antibody | Cell Signaling Technology | #95702; RRID:AB_2721823 |
| GAPDH (14C10) rabbit monoclonal antibody | Cell Signaling Technology | #2118; RRID:AB_561053 |
| HDAC1 (10E2) Mouse monoclonal antibody | Cell Signaling Technology | #5356; RRID:AB_10612242 |
| Pol I/II/III RPB8 (B8-1) Mouse monoclonal antibody | Santa Cruz | #sc-21752; RRID:AB_675858 |
| Anti-Rabbit IgG | Diagenode | # C15410206; RRID:AB_2722554 |
| Anti-Mouse IgG | Diagenode | # C15400001-100; RRID: AB_2722553 |
| Plasmids | ||
| pMD2.G plasmid | pMD2.G plasmid was deposited by Didier Trono’s lab. | RRID:Addgene_12259 |
| psPAX2 plasmid | psPAX2 plasmid was deposited by Didier Trono’s lab. | RRID:Addgene # 12260 |
| Scrambled shrna: CCTAAGGTTAAGTCGCCCTCG CTCGAGCGAGGGCGACTTAACCTTAGG |
Sigma Aldrich | N/A |
| Chromatin modifier library shRNAs (Table S2) | Sigma Aldrich | N/A |
| Ripk3V5 plasmid | Sigma Aldrich | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tamoxifen | Sigma Aldrich | Cat# T5648 |
| Dispase | BD | Cat# 354235 |
| Collagenase, Type 2 | Worthington Biochemicals | Cat# CLS-2 |
| Percoll | Sigma Aldrich | Cat# P1644 |
| Matrigel Matrix | BD | Cat# 356234 |
| Trizol reagent | Invitrogen | Cat# 15596026 |
| Critical Commercial Assays | ||
| Click-iT EdU Kit | Invitrogen | Cat# C10337 |
| SuperScript II Reverse Transcriptase Kit | Invitrogen | Cat# 18091050 |
| DNeasy Blood & Tissue Kit | QIAGEN | Cat# 69504 |
| Nextera DNA Sample Preparation Kit | Illumina | Cat#FC-121-1030 |
| NextSeq500 platform using V2 chemistry | Illumina | Cat#FC-404-2004 |
| Deposited Data | ||
| RNaseq data (Figure S2; Table S1) | This paper | GEO: GSE134131 |
| ATACseq and RNaseq data (Figure 3; Table S2) | This paper | GEO: GSE117092; GEO: GSE134132 |
| RNaseq data (Figure 4; Table S4) | This paper | GEO: GSE117008 |
| Proteomics data (Figure 4; Table S4) | This paper | PRIDE: PXD010370 |
| Experimental Models: Cell Lines | ||
| HEK293FT | ATCC | Cat# PTA5077 |
| Experimental Models: Organisms/Strains | ||
| Chd4loxP/loxP mice | Dr. Pablo Gómez-del Arco | Stock No: 008462 |
| Ripk3loxP/loxP mice | Dr. Boris Strillic | N/A |
| MLKL knockout | Dr. Boris Strillic | N/A |
| Casp8loxP/loxP mice | Dr. Stephen Hedrick | N/A |
| Rosa26eYFP mice | Jackson Laboratory | Stock No: 006148 |
| mdx mice | Jackson Laboratory | Stock No: 001801 |
| Pax7::ZsGreen mice | Dr. Michael Kyba | Stock No: 029549 |
| Pax7CE mice | Dr. Chenming Fan | Stock No: 012476 |
| Software and Algorithms | ||
| R language (v3.4.1) | NA | www.r-project.org |
| GraphPad Prism 7 | GraphPad Software | N/A |
| STAR(v2.5.2b) | N/A | https://bioconda.github.io/ |
| Picard (v1.119) | N/A | https://bioconda.github.io/ |
| Gencode (version vM11) | N/A | https://www.gencodegenes.org/ |
| Oligonucleotides | ||
| Oligonucleotides used for genotyping and mRNA expression analysis are provided in Table S5 | N/A | |
Resource Availability
Lead Contact
Further information and reasonable requests for reagents may be directed to and will be fulfilled by Lead Contact, Thomas Braun (thomas.braun@mpi-bn.mpg.de).
Materials Availability
This study did not generate new unique reagents or new founder mice strains. All reagents are commercially available and listed in the key resource table.
Data availability
The accession number for the RNA-seq data related to Figure S2 and Table S1 reported in this paper is GEO: GSE134131. The accession number for the ATAC-seq data related to Figure 3 and Table S2 reported in this paper is GEO: GSE117092. The accession number for the RNA-seq data related to Figure 3 and Table S2 reported in this paper is GEO: GSE134132. The accession number for the RNA-seq data related to Figure 4 and Table S4 reported in this paper is GEO: GSE117008. The accession number for the Proteomics data related to Figure 4 and Table S4 reported in this paper is PRIDE: PXD010370.
Experimental Model and Subject Details
Mice
All animal experiments were performed in accordance with German animal protection laws and EU (Directive 2010/63/EU) ethical guidelines and were approved by the local governmental animal protection authority Regierungspräsidium Darmstadt.Animal experiments were performed in accordance with German animal protection laws and EU (Directive 2010/63/EU) ethical guidelines and were approved by the local governmental animal protection authority Regierungspräsidium Darmstadt. All mice used in this study were maintained on a C57BL/6 background, were drug and test naive, healthy prior to the studies, not used in previous procedures and kept in a barrier facility. Both female and male animals were analyzed in this study. None of the determined parameters in this study correlated with animal sex. Genotypes of all mice used in this study were determined using verified protocols with DNA isolated from tail or ear punch biopsies collected upon weaning. All mice were separated by sex and maintained in groups of 3-4 mice per cage in low-noise, temperature and humidity-controlled, individually ventilated cages. Generation of Chd4fl/fl (Gómez-Del Arco et al., 2016, Williams et al., 2004), Ripk3loxP/loxP, Mlkl−/− (Strilic et al., 2016) and Caspase8loxP/loxP mice (Beisner et al., 2005) have been described before. In addition, the following mouse lines were used: Pax7-CreERT2, R26RYFP, Pax7ZsGreen, Dmdmdx (mdx) (Lepper et al., 2009, Bosnakovski et al., 2008, Sicinski et al., 1989, Srinivas et al., 2001). Age-matched (8-12 week old) littermate controls were used to perform experiments unless otherwise stated. Treatment with tamoxifen was performed as indicated and previously reported (Günther et al., 2013). Controls throughout the study, unless stated otherwise, were tamoxifen treated littermates carrying the respective floxed alleles. Muscle injury was induced via cardiotoxin injection (50 μL of 0.06mg/ml solution in saline) to the tibialis anterior (T.A.) muscle. Contralateral uninjured T.A. muscles served as controls.
Necrostatin-1 treatment in mice
Nec-1s or 7-Cl−O-Nec-1 [5-((7-chloro-1H-indol-3-yl)methyl)-3-methylimidazolidine-2,4-dione], a stable variant of Necrostatin-1 was used to inhibit necroptosis in vivo (Takahashi et al., 2012). Intraperitoneal injections of Nec-1 s were performed at a concentration of 1.65 mg/kg of body weight (Strilic et al., 2016, Wang et al., 2017, Takahashi et al., 2012) 30 minutes prior to cardiotoxin (CTX) injury. Mice were sacrificed 14 days after muscle injury.
zVAD treatment in mice
Z-VAD(OMe)-FMK (zVAD), a pan-caspase inhibitor was used to specifically inhibit apoptosis in vivo (Iwata et al., 2003, Davis et al., 2013, Cohen, 1997). Intraperitoneal injections of zVAD were performed at a concentration of 2mg/kg of mice body weight along with tamoxifen treatment for 5 consecutive days prior to cardiotoxin (CTX) injury. In addition, mice were treated with zVAD inhibitor after CTX injury every second day until the mice were sacrificed as indicated.
Grip strength assays
Muscle grip strength was assessed based on the fore-limb hanging test as previously described (Aartsma-Rus and van Putten, 2014, Peled-Kamar et al., 1997). Essentially, performance is calculated as a ratio of [t/bw] where t = hanging time and bw = grams of body weight.
MuSC and myofiber culture
MuSC were isolated via FACS using the Pax7ZsGreen reporter allele as previously described (Kim and Braun, 2014, Bosnakovski et al., 2008) and cultured in DMEM GlutaMAX (GIBCO) containing 20% FCS 1%, Penicillin/Streptomycin, and 5ng/ml bFGF. HEK293FT cells (ATCC: # PTA5077) were used to produce lentiviral particles and were cultured in DMEM GlutaMAX (GIBCO) containing 10% FCS and 1% Penicillin/Streptomycin. Myofibers were isolated by enzymatic digestion of isolated FBD muscle with collagenase P (0.02%, Roche). Single myofibers were either fixed with 4% paraformaldehyde (PFA) immediately after digestion or after 3-day culturing in DMEM medium with 20% fetal calf serum (FCS) and basic fibroblast growth factor (bFGF) (5ng/ml). All cell lines, primary MuSCs, and myofibers were regularly analyzed for mycoplasma infections and grown in standard normoxic conditions (37°C and 5% CO2).
Co-culture experiments
MuSCs were purified from WT and mdx mutant mice by FACS purification as previously described (Kim and Braun, 2014). Co-cultures were performed on matrigel-coated 96-Greiner microclear plates in DMEM GlutaMAX (GIBCO) media containing 20% FCS 1%, Penicillin/Streptomycin, and 5ng/ml bFGF. To distinguish between WT and mdx MuSCs within the same well, WT cells were isolated from Tamoxifen-treated Pax7-CreERT2;R26RYFP. Cell death was measured by calculating the percentage of dying, EthD-III incorporating YFP+ MuSCs counter-stained with Hoechst dye to determine total number of cells. Each experiment was performed with three independent biological replicates.
Transwell assays
Transwell assays were performed on 96-transwell plates with polyester membranes of 8 μm pore size (Corning) wherein the matrigel-coated transwell insert was seeded with either either WT, mdx or Ripk3mKO/mdx MuSCS. In this setup, no transmigration of MuSCs from the upper transwell to the bottom well was observed. In the bottom well WT MuSCs were seeded at clonal density in undiluted matrigel. Culture was performed in DMEM GlutaMAX (GIBCO) containing 20% FCS 1%, Penicillin/Streptomycin, and 5ng/ml bFGF media. The number of MuSC colonies and cells per colony was counted after Pax7/DAPI staining. Each experiment was performed with three independent biological replicates.
Method Details
High-throughput screen and hit validation
Lentiviral-based RNAi screening was performed using a customized array of targeting shRNAs isolated from a genome-wide shRNA library (Sigma-Aldrich) as previously described (Sreenivasan et al., 2017, Zhang et al., 2015). All targeted genes are provided in (Table S3). Candidate genes were selected from a previously published RNAi screen for chromatin remodelers (Fazzio et al., 2008) and the gene list was further refined based on gene ontological analysis resulting in a total of 634 targeted genes. GenElute HP 96-Well Miniprep Kits (Sigma-Aldrich) was used to purify plasmids as described by the vendor.
RNA sequencing and Mass Spectrometry
RNaseq and mass spectrometry measurements were performed as previously described (Zhang et al., 2015). Briefly, FACS purified MuSC from Chd4loxP/loxP mice were expanded in vitro and subjected to adenoviral-mediated CRE recombination. Adenovirus encoding GFP served as a control. Samples were generated in biological triplicate and used for RNaseq and mass spectrometric measurements. Total RNA was isolated using commercial kits and according to the manufacturer’s protocol (RNAeasy Mini kit, QIAGEN). RNaseq libraries were constructed using Ion Total RNA-Seq Kit v2 (Life Technologies) and sequencing reactions were performed by Ion Torrent Proton platform with V3 chemistry (Ion PI Template OT2 200 Kit v3, Life Technologies) and PIV2 Chips (Ion PI Chip Kit v2, Life Technologies). For mass spectrometry measurements, whole cell lysates from MuSC obtained from Chd4loxP/loxP mice were subjected to adenoviral treatment (AdenoCRE/AdenoGFP), the samples were run on SDS-PAGE gels and stained with colloidal protein staining solution (Invitrogen). The gels were evenly sliced and subjected to in-gel digestion with trypsin. Released peptides were measured using a TQ-Orbitrap XL or a LTQ-Orbitrap Velos mass spectrometer (Thermo Fisher Scientific) equipped with a nanoelectrospray source (Proxeon). Raw data was analyzed using the MaxQuant software package (Cox et al., 2014).
ATACseq analysis
ATACseq analysis was performed as previously described (Buenrostro et al., 2013, Buenrostro et al., 2015). Briefly, 5000-25,000 FACS purified MuSC were subjected to ATAC Library preparation using Tn5 Transposase from Nextera DNA Sample Preparation Kit (Illumina). Cell pellet was resuspended in 50μl PBS and mixed with 25μl TD-Buffer, 2.5μl Tn5, 0.5μl 10% NP-40 and 22μl water. Cell/Tn5 mixture was incubated at 37°C for 30min with occasional snap mixing. Transposase treatment was followed by 30min incubation at 50°C together with 500mM EDTA pH8.0 for optimal recovery of digested DNA fragments. For neutralization of EDTA 100μl of 50mM MgCl2 was added followed by purification of the DNA fragments by MinElute PCR Purification Kit (QIAGEN). Amplification of Library together with Indexing was performed as previously described (Buenrostro et al., 2013). Libraries were mixed in equimolar ratios and sequenced on NextSeq500 platform using V2 chemistry. The resulting raw reads were assessed for quality, adaptor content and duplication rates with FastQC (Andrews, 2010). Trimmomatic version 0.36 was employed to trim reads after a quality drop below a mean of Q15 in a window of 5 nucleotides (Bolger et al., 2014). Only reads longer than 15 nucleotides were cleared for further analyses. Trimmed and filtered reads were aligned versus the Gencode mouse genome version mm10 (GRCm38, vM15) using STAR 2.5.3a with custom designed parameters (Dobin et al., 2013) and retaining only unique alignments to exclude reads with uncertain arrangement. Reads were further de-duplicated using Picard 2.9.0 (Picard: A set of tools for working with next generation sequencing data in the BAM format) to mitigate PCR artifacts leading to multiple copies of the same original fragment.
The Macs2 peak caller version 2.1.0 was employed to accommodate for the range of peak widths typically expected for ATACSeq (Zhang et al., 2008). The minimum q-value was set to −4 and FDR was changed to 0.0001. In order to determine thresholds for significant peaks, the data was manually inspected in IGV 2.3.52 browser (Robinson et al., 2011). Peaks overlapping ENCODE blacklisted regions (known mis-assemblies, satellite repeats) were excluded. In order to be able to compare peaks in different samples to assess reproducibility, the resulting lists of significant peaks were overlapped and unified to represent identical regions. Peak counts were recalculated using bigWigAverageOverBed (UCSC Tools) and normalized with DESeq2 (Anders and Huber, 2010). Peaks were annotated with the promoter (TSS +- 5000 nt) of the gene most closely located to the center of the peak based on reference data from GENCODE vM15.
ChIP and RT–qPCR
Chromatin Immunoprecipitation (ChIP) experiments were performed as described previously (Gómez-Del Arco et al., 2016). For muscle tissue, truChIP Tissue Chromatin Shearing Kit with SDS (Covaris) was used for chromatin sheraing. Cultured MuSC were fixed with 1% formaldehyde for 10 minutes and the reaction was quenched using 0.125M glycine for 5 minutes at room temperature. Cells were lysed and the enriched chromatin was sheared to 300-500bp using Bioruptor (Diagenode) and subjected to immunoprecipitation with indicated antibodies. Primers used for ChIP-qPCR are listed (Table S5). mRNA expression was verified via quantitative real-time PCR. RNA from MuSC and muscle tissue was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s protocol. Purified RNA was subjected to reverse transcriptase reaction in the presence of 25 ng/ml random primers and 2.5 mM dA/C/G/TTP with 10 U/ml SuperScript II Reverse Transcriptase (Invitrogen). Primers used for RT–qPCR are listed in (Table S5).
Immunofluorescence and morphological analysis
Immunofluorescence staining was performed in cultured MuSC and myofibers fixed in 4% PFA. Sections from snap frozen muscle were fixed in 4% PFA. Primary antibodies used for immunohistochemical staining are listed in the key resources table. Hematoxylin and Eosin staining were used to visualize muscle fibers. Masson’s trichrome staining was carried out using the ACCUSTAIN trichrome staining kit (Sigma-Aldrich) according to the manufacturer’s instructions. For EdU incorporation assays cultured MuSC were subjected to 10μM EdU for three hours prior to fixation. Click-iT EdU Imaging Kit (Invitrogen) was used to quantify EdU labeled cells according to the manufacturer’s protocol.
Cell death assay
Cell death assays were performed with MuSC isolated from control and mdx mice. z-VAD-fmk (20 μM, Cayman) and/or Necrostatin-1 (Nec-1; 70 μM, Enzo) were used to block apoptosis or Ripk1 activation and necroptosis, respectively. Stains of dying cells were visualized with EthD-III (1.6 μM, Biotium). For end-point experiments, Hoechst 33342 (2 μM, Thermo Scientific)-positive cells were used to count the total number of cells. Criteria for discriminating apoptotic and necrotic cells have been described previously (Strilic et al., 2016).
Western blot
Protein lysates from MuSC were obtained using protein lysis buffer I [66mM Tris-HCl and 2% SDS] supplemented with protease and phosphatase inhibitors (0.5 μg/ml benzamidine, 2 μg/ml aprotinin, 2 μg/ml leupetin, 2mM PMFS, 1mM Na3Vo4, 20mM NaF). Whole muscle cell lysates were generated by finely mincing isolated muscle tissue and resuspended in protein lysis buffer II (100 mM Tris-HCl, 10% SDS and 12.7 mM EDTA) supplemented with protease and phosphatase inhibitors as described above. Protein lysates were sonicated and clarified by centrifugation at 14000 rpm for 15 min. Protein concentrations were determined with DC Protein assay kit (Bio-Rad). Equal amounts of proteins were prepared in protein lysis buffer containing a final concentration of 15% glycerol, 50mM DTT and bromophenol blue. The samples were boiled at 95°C for 5 min and resolved by western blotting. Membranes were probed with the following antibodies: anti-Ripk3 (Cell signaling #95702), anti-CHD4 (Cell signaling #12011) and anti-GAPDH (Cell Signaling #2118). Quantification was performed by densitometric analysis using Image Lab 5.0 software (Bio-Rad).
Statistical Analysis
Animal studies were performed without blinding and no animals were excluded from the analysis. All experiments were performed in biological triplicates unless stated otherwise. Sample size for studies was chosen based on observed effect sizes and standard errors. Statistical analysis performed throughout the experiments either made use of one factorial ANOVA when multiple groups were compared or unpaired t test when two groups were compared. P values < 0.05 were considered statistically significant (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.005, ∗∗∗∗p < 0.0001). Mean values are depicted as ± s.e.m. and indicated in the figure legends. GraphPad Prism v5.03 or higher was used for data analysis.
Acknowledgments
We would like to thank the core facilities at Max Planck Institute Bad Nauheim (MPI-BN) for support with fluorescence-activated cell sorting (FACS) (A. Atzberger), EM (J. Pieske), and mass spectrometry (MS) services (A. Sokol and J. Graumann). We also thank A. Rothe, S.M. Liesener, and A. Marg for preparation of human patient samples and help with microscopy. We greatly appreciate the kind gift of caspase-8loxP/loxP mice from S.M. Hedrick (University of California, San Diego). This work was supported by the DFG (Cardiopulmonary Institute [CPI], SFB TRR81 TP A02 and SFB 1213, TP A02 and B02); the LOEWE Center for Cell and Gene Therapy; the German Center for Cardiovascular Research; the European Research Area Network on Cardiovascular Diseases (grant CLARIFY); and Foundation Leducq. P.G.A. is supported by MICINN (SAF2016-77816-P). P.M.-C. acknowledges funding from grants RTI2018-096068-B-I00, MDM-2014-0370, SEV-2015-0505, ERC-2016-AdG-741966, La Caixa-HEALTH-HR17-00040, MDA, UPGRADE-H2020-825825, AFM, and DPP-E. T.B. is a member of the German Center for Cardiovascular Research (DZHK) and of the German Center for Lung Research (DZL).
Author Contributions
K.S. performed most of the experiments and analyzed the data. A.I. performed western blot analyses. E.P. performed the adenoviral knockdown experiments for RNA-seq and liquid chromatography (LC)/MS measurements. C.K. performed bioinformatics analysis of ATAC-seq and RNA-seq analyses. M.K. generated the proteomics data. S.G. generated RNA-seq and ATAC-seq data. B.S. and S.O. provided the Ripk3flox, MlklKO mice and discussed data. P.M.-C. and E.P. provided the Chd4flox mice and discussed data. S.S. provided human patient samples. K.S., J.K., and T.B. designed the study, analyzed the data, and wrote the manuscript. All authors read and contributed to the final draft.
Declaration of Interests
The authors declare no competing interests.
Published: May 19, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107652.
Contributor Information
Johnny Kim, Email: johnny.kim@mpi-bn.mpg.de.
Thomas Braun, Email: thomas.braun@mpi-bn.mpg.de.
Supplemental Information
RNA analysis: Gene expression levels were considered significantly different when the following criteria were met: normalized read counts > 5, log2 fold change < −0.585 or > 0.585, and adjusted p value < 0.05 based on DESeq normalization. DESeq normalized read counts were used to identify significantly deregulated genes.
Normalized peaks from DESeq2 (Anders and Huber, 2010) were related to gene promoter regions (TSS +- 5000 nt) using reference data from GENCODE vM15. Peaks were classified as significantly different at a log2 fold change < −0.585 or > 0.585, and mean normalized read counts > 20 (WT versus mdx). Genes with differential peak inside the promoter region (ATACseq) or significantly differential, and normalized FPKM counts (RNaseq) were used for gene set enrichment analysis using KOBAS.
The table provides shRNA sequence for all known and putative chromatin modifiers in an array of 96-well plates (P1-38) (Zhang et al., 2015, Fazzio et al., 2008). Candidate hits were selected based on the self-renewal index (SI), which was calculated as described in the excel sheet.
RNA analysis: Gene expression levels were considered significantly different when the following criteria were met: normalized read counts > 5, log2 fold change < −0.585 or > 0.585, and adjusted p value < 0.05 based on DESeq normalization. Protein analysis: The MaxQuant software package (Version 1.6.1.0) was used to analyze raw data. Protein counts were classified as significantly different based on Student’s t test and p value < 0.05 comparing log2 LFQ intensities between CRE (Chd4 mutant) and GFP (Control). Calculations were done using the Perseus software (Version 1.6.0.8). DESeq normalized read counts and Log2 LFQ intensities were used to identify significantly deregulated genes/proteins.
References
- Aartsma-Rus A., van Putten M. Assessing functional performance in the mdx mouse model. J. Vis. Exp. 2014;(85) doi: 10.3791/51303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S., Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data.http://www.bioinformatics.babraham.ac.uk/projects/fastqc [Google Scholar]
- Ashkenazi A., Salvesen G. Regulated cell death: signaling and mechanisms. Annu. Rev. Cell Dev. Biol. 2014;30:337–356. doi: 10.1146/annurev-cellbio-100913-013226. [DOI] [PubMed] [Google Scholar]
- Beisner D.R., Ch’en I.L., Kolla R.V., Hoffmann A., Hedrick S.M. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J. Immunol. 2005;175:3469–3473. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- Berger S.L., Kouzarides T., Shiekhattar R., Shilatifard A. An operational definition of epigenetics. Genes Dev. 2009;23:781–783. doi: 10.1101/gad.1787609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondar T., Medzhitov R. p53-mediated hematopoietic stem and progenitor cell competition. Cell Stem Cell. 2010;6:309–322. doi: 10.1016/j.stem.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boonsanay V., Zhang T., Georgieva A., Kostin S., Qi H., Yuan X., Zhou Y., Braun T. Regulation of skeletal muscle stem cell quiescence by Suv4-20h1-dependent facultative heterochromatin formation. Cell Stem Cell. 2016;18:229–242. doi: 10.1016/j.stem.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Bosnakovski D., Lamb S., Simsek T., Xu Z., Belayew A., Perlingeiro R., Kyba M. DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp. Neurol. 2008;214:87–96. doi: 10.1016/j.expneurol.2008.07.022. [DOI] [PubMed] [Google Scholar]
- Bowling S., Lawlor K., Rodríguez T.A. Cell competition: the winners and losers of fitness selection. Development. 2019;146:dev167486. doi: 10.1242/dev.167486. [DOI] [PubMed] [Google Scholar]
- Buenrostro J.D., Giresi P.G., Zaba L.C., Chang H.Y., Greenleaf W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods. 2013;10:1213–1218. doi: 10.1038/nmeth.2688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buenrostro J.D., Wu B., Chang H.Y., Greenleaf W.J. ATAC-seq: a method for assaying chromatin accessibility genome-wide. Curr. Protoc. Mol. Biol. 2015;109:21.29.1–21.29.9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camboni M., Hammond S., Martin L.T., Martin P.T. Induction of a regenerative microenvironment in skeletal muscle is sufficient to induce embryonal rhabdomyosarcoma in p53-deficient mice. J. Pathol. 2012;226:40–49. doi: 10.1002/path.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chargé S.B., Rudnicki M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004;84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- Chen X., He L., Zhao Y., Li Y., Zhang S., Sun K., So K., Chen F., Zhou L., Lu L. Malat1 regulates myogenic differentiation and muscle regeneration through modulating MyoD transcriptional activity. Cell Discov. 2017;3:17002. doi: 10.1038/celldisc.2017.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavería C., Giovinazzo G., Sierra R., Torres M. Myc-driven endogenous cell competition in the early mammalian embryo. Nature. 2013;500:39–44. doi: 10.1038/nature12389. [DOI] [PubMed] [Google Scholar]
- Cohen G.M. Caspases: the executioners of apoptosis. Biochem. J. 1997;326:1–16. doi: 10.1042/bj3260001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colijn S., Gao S., Ingram K.G., Menendez M., Muthukumar V., Silasi-Mansat R., Chmielewska J.J., Hinsdale M., Lupu F., Griffin C.T. The NuRD chromatin-remodeling complex enzyme CHD4 prevents hypoxia-induced endothelial Ripk3 transcription and murine embryonic vascular rupture. Cell Death Differ. 2020;27:618–631. doi: 10.1038/s41418-019-0376-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J., Hein M.Y., Luber C.A., Paron I., Nagaraj N., Mann M. Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Mol. Cell. Proteomics. 2014;13:2513–2526. doi: 10.1074/mcp.M113.031591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis J., Kwong J.Q., Kitsis R.N., Molkentin J.D. Apoptosis repressor with a CARD domain (ARC) restrains Bax-mediated pathogenesis in dystrophic skeletal muscle. PLoS ONE. 2013;8:e82053. doi: 10.1371/journal.pone.0082053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Paepe B., De Bleecker J.L. Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediators Inflamm. 2013;2013:540370. doi: 10.1155/2013/540370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G.D., Mitchison T.J., Moskowitz M.A., Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- Degterev A., Hitomi J., Germscheid M., Ch’en I.L., Korkina O., Teng X., Abbott D., Cuny G.D., Yuan C., Wagner G. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennett X., Shield L.K., Clingan L.J., Woolley D.A. Becker and Duchenne muscular dystrophy: a comparative morphological study. Aust. Paediatr. J. 1988;24(Suppl 1):15–20. [PubMed] [Google Scholar]
- Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont N.A., Wang Y.X., von Maltzahn J., Pasut A., Bentzinger C.F., Brun C.E., Rudnicki M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015;21:1455–1463. doi: 10.1038/nm.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery A.E. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Fazzio T.G., Huff J.T., Panning B. An RNAi screen of chromatin proteins identifies Tip60-p400 as a regulator of embryonic stem cell identity. Cell. 2008;134:162–174. doi: 10.1016/j.cell.2008.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes S.J., Rosenthal N. Preparing the ground for tissue regeneration: from mechanism to therapy. Nat. Med. 2014;20:857–869. doi: 10.1038/nm.3653. [DOI] [PubMed] [Google Scholar]
- Galluzzi L., Bravo-San Pedro J.M., Kepp O., Kroemer G. Regulated cell death and adaptive stress responses. Cell. Mol. Life Sci. 2016;73:2405–2410. doi: 10.1007/s00018-016-2209-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Del Arco P., Perdiguero E., Yunes-Leites P.S., Acín-Pérez R., Zeini M., Garcia-Gomez A., Sreenivasan K., Jiménez-Alcázar M., Segalés J., López-Maderuelo D. The chromatin remodeling complex Chd4/NuRD controls striated muscle identity and metabolic homeostasis. Cell Metab. 2016;23:881–892. doi: 10.1016/j.cmet.2016.04.008. [DOI] [PubMed] [Google Scholar]
- Grootjans S., Vanden Berghe T., Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24:1184–1195. doi: 10.1038/cdd.2017.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Günther S., Kim J., Kostin S., Lepper C., Fan C.M., Braun T. Myf5-positive satellite cells contribute to Pax7-dependent long-term maintenance of adult muscle stem cells. Cell Stem Cell. 2013;13:590–601. doi: 10.1016/j.stem.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M., Sasaki H. Epiblast formation by TEAD-YAP-dependent expression of pluripotency factors and competitive elimination of unspecified cells. Dev. Cell. 2019;50:139–154.e5. doi: 10.1016/j.devcel.2019.05.024. [DOI] [PubMed] [Google Scholar]
- Hildebrand J.M., Tanzer M.C., Lucet I.S., Young S.N., Spall S.K., Sharma P., Pierotti C., Garnier J.M., Dobson R.C., Webb A.I. Activation of the pseudokinase MLKL unleashes the four-helix bundle domain to induce membrane localization and necroptotic cell death. Proc. Natl. Acad. Sci. USA. 2014;111:15072–15077. doi: 10.1073/pnas.1408987111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata A., Nishio K., Winn R.K., Chi E.Y., Henderson W.R., Jr., Harlan J.M. A broad-spectrum caspase inhibitor attenuates allergic airway inflammation in murine asthma model. J. Immunol. 2003;170:3386–3391. doi: 10.4049/jimmunol.170.6.3386. [DOI] [PubMed] [Google Scholar]
- Joe A.W., Yi L., Natarajan A., Le Grand F., So L., Wang J., Rudnicki M.A., Rossi F.M. Muscle injury activates resident fibro/adipogenic progenitors that facilitate myogenesis. Nat. Cell Biol. 2010;12:153–163. doi: 10.1038/ncb2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Braun T. Skeletal muscle stem cells for muscle regeneration. Methods Mol. Biol. 2014;1213:245–253. doi: 10.1007/978-1-4939-1453-1_20. [DOI] [PubMed] [Google Scholar]
- Klein A.M., Simons B.D. Universal patterns of stem cell fate in cycling adult tissues. Development. 2011;138:3103–3111. doi: 10.1242/dev.060103. [DOI] [PubMed] [Google Scholar]
- Koren E., Yosefzon Y., Ankawa R., Soteriou D., Jacob A., Nevelsky A., Ben-Yosef R., Bar-Sela G., Fuchs Y. ARTS mediates apoptosis and regeneration of the intestinal stem cell niche. Nat. Commun. 2018;9:4582. doi: 10.1038/s41467-018-06941-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostin S., Pool L., Elsässer A., Hein S., Drexler H.C., Arnon E., Hayakawa Y., Zimmermann R., Bauer E., Klövekorn W.P., Schaper J. Myocytes die by multiple mechanisms in failing human hearts. Circ. Res. 2003;92:715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- Krysko D.V., Vanden Berghe T., D’Herde K., Vandenabeele P. Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods. 2008;44:205–221. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- Kurosaka M., Ogura Y., Funabashi T., Akema T. Early growth response 3 (Egr3) contributes a maintenance of C2C12 myoblast proliferation. J. Cell. Physiol. 2017;232:1114–1122. doi: 10.1002/jcp.25574. [DOI] [PubMed] [Google Scholar]
- Lai A.Y., Wade P.A. Cancer biology and NuRD: a multifaceted chromatin remodelling complex. Nat. Rev. Cancer. 2011;11:588–596. doi: 10.1038/nrc3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latroche C., Gitiaux C., Chrétien F., Desguerre I., Mounier R., Chazaud B. Skeletal muscle microvasculature: a highly dynamic lifeline. Physiology (Bethesda) 2015;30:417–427. doi: 10.1152/physiol.00026.2015. [DOI] [PubMed] [Google Scholar]
- Lee H.J., Göring W., Ochs M., Mühlfeld C., Steding G., Paprotta I., Engel W., Adham I.M. Sox15 is required for skeletal muscle regeneration. Mol. Cell. Biol. 2004;24:8428–8436. doi: 10.1128/MCB.24.19.8428-8436.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos D.R., Babaeijandaghi F., Low M., Chang C.K., Lee S.T., Fiore D., Zhang R.H., Natarajan A., Nedospasov S.A., Rossi F.M. Nilotinib reduces muscle fibrosis in chronic muscle injury by promoting TNF-mediated apoptosis of fibro/adipogenic progenitors. Nat. Med. 2015;21:786–794. doi: 10.1038/nm.3869. [DOI] [PubMed] [Google Scholar]
- Lepper C., Conway S.J., Fan C.M. Adult satellite cells and embryonic muscle progenitors have distinct genetic requirements. Nature. 2009;460:627–631. doi: 10.1038/nature08209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A. Apoptosis and cancer. Annu. Rev. Cancer Biol. 2017;1:275–294. [Google Scholar]
- Lloyd A.F., Davies C.L., Holloway R.K., Labrak Y., Ireland G., Carradori D., Dillenburg A., Borger E., Soong D., Richardson J.C. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat. Neurosci. 2019;22:1046–1052. doi: 10.1038/s41593-019-0418-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long X., Creemers E.E., Wang D.Z., Olson E.N., Miano J.M. Myocardin is a bifunctional switch for smooth versus skeletal muscle differentiation. Proc. Natl. Acad. Sci. U S A. 2007;104:16570–16575. doi: 10.1073/pnas.0708253104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.V., Chen H.C., Walsh C.M. Necroptotic signaling in adaptive and innate immunity. Semin. Cell Dev. Biol. 2014;35:33–39. doi: 10.1016/j.semcdb.2014.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesi I., Fiorentino F.P., Rizzolio F., Giordano A., Bagella L. The ablation of EZH2 uncovers its crucial role in rhabdomyosarcoma formation. Cell Cycle. 2012;11:3828–3836. doi: 10.4161/cc.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan J.E., Prola A., Mariot V., Pini V., Meng J., Hourde C., Dumonceaux J., Conti F., Relaix F., Authier F.J. Necroptosis mediates myofibre death in dystrophin-deficient mice. Nat. Commun. 2018;9:3655. doi: 10.1038/s41467-018-06057-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata S., Tanaka M. Programmed cell death and the immune system. Nat. Rev. Immunol. 2017;17:333–340. doi: 10.1038/nri.2016.153. [DOI] [PubMed] [Google Scholar]
- Newton K., Manning G. Necroptosis and inflammation. Annu. Rev. Biochem. 2016;85:743–763. doi: 10.1146/annurev-biochem-060815-014830. [DOI] [PubMed] [Google Scholar]
- Nijhawan D., Honarpour N., Wang X. Apoptosis in neural development and disease. Annu. Rev. Neurosci. 2000;23:73–87. doi: 10.1146/annurev.neuro.23.1.73. [DOI] [PubMed] [Google Scholar]
- Opferman J.T., Letai A., Beard C., Sorcinelli M.D., Ong C.C., Korsmeyer S.J. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- Peled-Kamar M., Lotem J., Wirguin I., Weiner L., Hermalin A., Groner Y. Oxidative stress mediates impairment of muscle function in transgenic mice with elevated level of wild-type Cu/Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA. 1997;94:3883–3887. doi: 10.1073/pnas.94.8.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preussner J., Zhong J., Sreenivasan K., Gunther S., Engleitner T., Kunne C., Glatzel M., Rad R., Looso M., Braun T., Kim J. Oncogenic amplification of zygotic Dux factors in regenerating p53-deficient muscle stem cells defines a molecular cancer subtype. Cell Stem Cell. 2018;23:794–805.e4. doi: 10.1016/j.stem.2018.10.011. [DOI] [PubMed] [Google Scholar]
- Relaix F., Zammit P.S. Satellite cells are essential for skeletal muscle regeneration: the cell on the edge returns centre stage. Development. 2012;139:2845–2856. doi: 10.1242/dev.069088. [DOI] [PubMed] [Google Scholar]
- Robinson J.T., Thorvaldsdóttir H., Winckler W., Guttman M., Lander E.S., Getz G., Mesirov J.P. Integrative genomics viewer. Nat. Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A.S., Puig M., Nagaraju K., Hoffman E.P., Villalta S.A., Rao V.A., Wakefield L.M., Woodcock J. Immune-mediated pathology in Duchenne muscular dystrophy. Sci. Transl. Med. 2015;7:299rv4. doi: 10.1126/scitranslmed.aaa7322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryall J.G., Dell’Orso S., Derfoul A., Juan A., Zare H., Feng X., Clermont D., Koulnis M., Gutierrez-Cruz G., Fulco M., Sartorelli V. The NAD(+)-dependent SIRT1 deacetylase translates a metabolic switch into regulatory epigenetics in skeletal muscle stem cells. Cell Stem Cell. 2015;16:171–183. doi: 10.1016/j.stem.2014.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saclier M., Cuvellier S., Magnan M., Mounier R., Chazaud B. Monocyte/macrophage interactions with myogenic precursor cells during skeletal muscle regeneration. FEBS J. 2013;280:4118–4130. doi: 10.1111/febs.12166. [DOI] [PubMed] [Google Scholar]
- Shi X., Garry D.J. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692–1708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- Sicinski P., Geng Y., Ryder-Cook A.S., Barnard E.A., Darlison M.G., Barnard P.J. The molecular basis of muscular dystrophy in the mdx mouse: a point mutation. Science. 1989;244:1578–1580. doi: 10.1126/science.2662404. [DOI] [PubMed] [Google Scholar]
- Sreenivasan K., Braun T., Kim J. Systematic identification of genes regulating muscle stem cell self-renewal and differentiation. Methods Mol. Biol. 2017;1556:343–353. doi: 10.1007/978-1-4939-6771-1_19. [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C.S., William C.M., Tanabe Y., Jessell T.M., Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strilic B., Yang L., Albarrán-Juárez J., Wachsmuth L., Han K., Müller U.C., Pasparakis M., Offermanns S. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215–218. doi: 10.1038/nature19076. [DOI] [PubMed] [Google Scholar]
- Takahashi N., Duprez L., Grootjans S., Cauwels A., Nerinckx W., DuHadaway J.B., Goossens V., Roelandt R., Van Hauwermeiren F., Libert C. Necrostatin-1 analogues: critical issues on the specificity, activity and in vivo use in experimental disease models. Cell Death Dis. 2012;3:e437. doi: 10.1038/cddis.2012.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidball J.G. Mechanisms of muscle injury, repair, and regeneration. Compr. Physiol. 2011;1:2029–2062. doi: 10.1002/cphy.c100092. [DOI] [PubMed] [Google Scholar]
- Tidball J.G., Villalta S.A. Regulatory interactions between muscle and the immune system during muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2010;298:R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Berghe T., Grootjans S., Goossens V., Dondelinger Y., Krysko D.V., Takahashi N., Vandenabeele P. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods. 2013;61:117–129. doi: 10.1016/j.ymeth.2013.02.011. [DOI] [PubMed] [Google Scholar]
- Vanden Berghe T., Linkermann A., Jouan-Lanhouet S., Walczak H., Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat. Rev. Mol. Cell Biol. 2014;15:135–147. doi: 10.1038/nrm3737. [DOI] [PubMed] [Google Scholar]
- Wang Q., Zhou T., Liu Z., Ren J., Phan N., Gupta K., Stewart D.M., Morgan S., Assa C., Kent K.C., Liu B. Inhibition of receptor-interacting protein kinase 1 with necrostatin-1s ameliorates disease progression in elastase-induced mouse abdominal aortic aneurysm model. Sci. Rep. 2017;7:42159. doi: 10.1038/srep42159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinlich R., Oberst A., Beere H.M., Green D.R. Necroptosis in development, inflammation and disease. Nat. Rev. Mol. Cell Biol. 2017;18:127–136. doi: 10.1038/nrm.2016.149. [DOI] [PubMed] [Google Scholar]
- Welz P.S., Wullaert A., Vlantis K., Kondylis V., Fernández-Majada V., Ermolaeva M., Kirsch P., Sterner-Kock A., van Loo G., Pasparakis M. FADD prevents RIP3-mediated epithelial cell necrosis and chronic intestinal inflammation. Nature. 2011;477:330–334. doi: 10.1038/nature10273. [DOI] [PubMed] [Google Scholar]
- Williams C.J., Naito T., Arco P.G., Seavitt J.R., Cashman S.M., De Souza B., Qi X., Keables P., Von Andrian U.H., Georgopoulos K. The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity. 2004;20:719–733. doi: 10.1016/j.immuni.2004.05.005. [DOI] [PubMed] [Google Scholar]
- Yang Z., Jiang B., Wang Y., Ni H., Zhang J., Xia J., Shi M., Hung L.M., Ruan J., Mak T.W. 2-HG inhibits necroptosis by stimulating DNMT1-dependent hypermethylation of the RIP3 promoter. Cell Rep. 2017;19:1846–1857. doi: 10.1016/j.celrep.2017.05.012. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Günther S., Looso M., Künne C., Krüger M., Kim J., Zhou Y., Braun T. Prmt5 is a regulator of muscle stem cell expansion in adult mice. Nat. Commun. 2015;6:7140. doi: 10.1038/ncomms8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T., Zhang Y., Cui M., Jin L., Wang Y., Lv F., Liu Y., Zheng W., Shang H., Zhang J. CaMKII is a RIP3 substrate mediating ischemia- and oxidative stress-induced myocardial necroptosis. Nat. Med. 2016;22:175–182. doi: 10.1038/nm.4017. [DOI] [PubMed] [Google Scholar]
- Zhao Q., Yu X., Zhang H., Liu Y., Zhang X., Wu X., Xie Q., Li M., Ying H., Zhang H. RIPK3 mediates necroptosis during embryonic development and postnatal inflammation in Fadd-deficient mice. Cell Rep. 2017;19:798–808. doi: 10.1016/j.celrep.2017.04.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MuSC were isolated from wildtype mice and cultured in vitro in the presence or absence of different inhibitors for 58 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
MuSC were isolated from muscles of mdx mice and cultured in vitro in the presence or absence of different inhibitors for 70 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
MuSC were isolated from muscles of Chd4mKO mutant mice and cultured in vitro in the presence or absence of different inhibitors for 58 hr. Necroptotic cell death is indicated by EthD-III incorporation (red).
RNA analysis: Gene expression levels were considered significantly different when the following criteria were met: normalized read counts > 5, log2 fold change < −0.585 or > 0.585, and adjusted p value < 0.05 based on DESeq normalization. DESeq normalized read counts were used to identify significantly deregulated genes.
Normalized peaks from DESeq2 (Anders and Huber, 2010) were related to gene promoter regions (TSS +- 5000 nt) using reference data from GENCODE vM15. Peaks were classified as significantly different at a log2 fold change < −0.585 or > 0.585, and mean normalized read counts > 20 (WT versus mdx). Genes with differential peak inside the promoter region (ATACseq) or significantly differential, and normalized FPKM counts (RNaseq) were used for gene set enrichment analysis using KOBAS.
The table provides shRNA sequence for all known and putative chromatin modifiers in an array of 96-well plates (P1-38) (Zhang et al., 2015, Fazzio et al., 2008). Candidate hits were selected based on the self-renewal index (SI), which was calculated as described in the excel sheet.
RNA analysis: Gene expression levels were considered significantly different when the following criteria were met: normalized read counts > 5, log2 fold change < −0.585 or > 0.585, and adjusted p value < 0.05 based on DESeq normalization. Protein analysis: The MaxQuant software package (Version 1.6.1.0) was used to analyze raw data. Protein counts were classified as significantly different based on Student’s t test and p value < 0.05 comparing log2 LFQ intensities between CRE (Chd4 mutant) and GFP (Control). Calculations were done using the Perseus software (Version 1.6.0.8). DESeq normalized read counts and Log2 LFQ intensities were used to identify significantly deregulated genes/proteins.
Data Availability Statement
The accession number for the RNA-seq data related to Figure S2 and Table S1 reported in this paper is GEO: GSE134131. The accession number for the ATAC-seq data related to Figure 3 and Table S2 reported in this paper is GEO: GSE117092. The accession number for the RNA-seq data related to Figure 3 and Table S2 reported in this paper is GEO: GSE134132. The accession number for the RNA-seq data related to Figure 4 and Table S4 reported in this paper is GEO: GSE117008. The accession number for the Proteomics data related to Figure 4 and Table S4 reported in this paper is PRIDE: PXD010370.