

Key Points
Question
Does programmed cell death 1 immune checkpoint inhibition with nivolumab improve overall survival compared with bevacizumab treatment for patients with recurrent glioblastoma?
Findings
In this randomized phase 3 clinical trial of 369 patients diagnosed with recurrent glioblastoma treated with nivolumab, an improved survival benefit was not observed in patients who received nivolumab compared with bevacizumab-treated control patients.
Meaning
Additional research is needed; nivolumab monotherapy did not improve overall survival compared with bevacizumab in the treatment of recurrent glioblastoma. A study of nivolumab in combination with radiotherapy and temozolomide in patients with newly diagnosed glioblastoma with methylated MGMT promoter is ongoing.
This randomized clinical trial examines the effect of programmed cell death 1 immune checkpoint inhibition with nivolumab on overall survival for patients with recurrent glioblastoma.
Abstract
Importance
Clinical outcomes for glioblastoma remain poor. Treatment with immune checkpoint blockade has shown benefits in many cancer types. To our knowledge, data from a randomized phase 3 clinical trial evaluating a programmed death-1 (PD-1) inhibitor therapy for glioblastoma have not been reported.
Objective
To determine whether single-agent PD-1 blockade with nivolumab improves survival in patients with recurrent glioblastoma compared with bevacizumab.
Design, Setting, and Participants
In this open-label, randomized, phase 3 clinical trial, 439 patients with glioblastoma at first recurrence following standard radiation and temozolomide therapy were enrolled, and 369 were randomized. Patients were enrolled between September 2014 and May 2015. The median follow-up was 9.5 months at data cutoff of January 20, 2017. The study included 57 multicenter, multinational clinical sites.
Interventions
Patients were randomized 1:1 to nivolumab 3 mg/kg or bevacizumab 10 mg/kg every 2 weeks until confirmed disease progression, unacceptable toxic effects, or death.
Main Outcomes and Measures
The primary end point was overall survival (OS).
Results
A total of 369 patients were randomized to nivolumab (n = 184) or bevacizumab (n = 185). The MGMT promoter was methylated in 23.4% (43/184; nivolumab) and 22.7% (42/185; bevacizumab), unmethylated in 32.1% (59/184; nivolumab) and 36.2% (67/185; bevacizumab), and not reported in remaining patients. At median follow-up of 9.5 months, median OS (mOS) was comparable between groups: nivolumab, 9.8 months (95% CI, 8.2-11.8); bevacizumab, 10.0 months (95% CI, 9.0-11.8); HR, 1.04 (95% CI, 0.83-1.30); P = .76. The 12-month OS was 42% in both groups. The objective response rate was higher with bevacizumab (23.1%; 95% CI, 16.7%-30.5%) vs nivolumab (7.8%; 95% CI, 4.1%-13.3%). Grade 3/4 treatment-related adverse events (TRAEs) were similar between groups (nivolumab, 33/182 [18.1%]; bevacizumab, 25/165 [15.2%]), with no unexpected neurological TRAEs or deaths due to TRAEs.
Conclusions and Relevance
Although the primary end point was not met in this randomized clinical trial, mOS was comparable between nivolumab and bevacizumab in the overall patient population with recurrent glioblastoma. The safety profile of nivolumab in patients with glioblastoma was consistent with that in other tumor types.
Trial Registration
ClinicalTrials.gov Identifier: NCT02017717
Introduction
Glioblastoma has a poor prognosis, with a 5-year survival rate of less than 10%.1,2 Nearly all patients experience recurrence following standard-of-care surgical resection, radiotherapy, and temozolomide.2,3,4 Treatment options at recurrence are limited, and no therapy has prolonged overall survival (OS) in this setting, which underscores the need for novel therapeutic interventions in this patient population.4
Use of immunotherapy to promote antitumor immune response is an area of active research in the treatment of glioblastoma. Accumulating evidence suggests that immune cells are able to enter, proliferate, and function in the central nervous system (CNS), and resident macrophages can express major histocompatibility complex II antigens and T-cell costimulatory cytokines on activation.5 These data and results from murine glioma models showing improved survival with checkpoint inhibitors6 suggest that immune checkpoint blockade may be a potential treatment option for glioblastoma.
Nivolumab is a fully human immunoglobulin G4 monoclonal antibody targeting the programmed death-1 (PD-1) immune checkpoint receptor. The safety of nivolumab in recurrent glioblastoma was demonstrated in the phase 1 safety lead-in cohorts of the CheckMate 143 randomized clinical trial (NCT02017717).7 On the basis of these safety results,7 a randomized, open-label, phase 3 cohort was initiated to compare the efficacy and safety of nivolumab vs bevacizumab in patients with first recurrence of glioblastoma.
Methods
Study Design and Patients
The trial protocol is available in Supplement 1, and the statistical analysis plan is included in Supplement 2. Cohort 2 of the CheckMate 143 trial was a randomized, open-label, phase 3 trial conducted at 57 clinical sites in 12 countries. Eligible patients had histologically confirmed World Health Organization grade IV recurrent glioblastoma or gliosarcoma (as defined by Response Assessment in Neuro-Oncology criteria8) after first-line treatment with radiotherapy and temozolomide, were 18 years or older, had a Karnofsky performance status of 70 or higher, and were 28 days or longer from prior surgery and 12 weeks or more from prior radiation. Patients who had more than 1 recurrence of glioblastoma, had a diagnosis of secondary glioblastoma, or required escalating or chronic supraphysiological doses of corticosteroids (>10 mg/d prednisone equivalents [dexamethasone equivalents]; determined by the investigator) to treat symptomatic cerebral edema were ineligible. Additional inclusion and exclusion criteria are listed in the eMethods in Supplement 3. Enrollment was increased by 120 patients to compensate for patient voluntary withdrawal in the bevacizumab arm.
The study protocol was approved by the institutional review board or independent ethics committee of each participating institution. The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice, as defined by the International Conference on Harmonisation. All patients provided written informed consent prior to enrollment. Randomization and masking methods are described in the eMethods in Supplement 3.
Study Procedures
Patients received 3 mg/kg of nivolumab or 10 mg/kg of bevacizumab intravenously every 2 weeks. Study treatment continued until investigator-assessed progressive disease or onset of toxic effects requiring permanent discontinuation of study treatment. Patients could continue study treatment following first evidence of progression until confirmed by follow-up magnetic resonance imaging (MRI) within 12 weeks if there was evidence of investigator-assessed clinical benefit and adequate tolerability.
Tumor assessments were performed by the investigator using contrast-enhanced MRI at baseline, day 1 of weeks 7 and 13, and every 8 weeks thereafter.8 Follow-up for survival occurred every 3 months. Adverse events (AEs) were assessed continuously during treatment and for 100 days or more after the end of treatment according to National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0). At the time of enrollment, data on MGMT promoter methylation status (as locally assessed) were collected without information on method of assessment; testing was not required for enrollment. PD-L1 testing methods are described in the eMethods in Supplement 3.
Outcomes
The primary end point was OS, defined as the time from randomization to death from any cause, assessed for each group using the Kaplan-Meier method. Secondary end points were OS rate at 12 months, investigator-assessed progression-free survival (PFS; defined as time from randomization to disease progression or death from any cause), and investigator-assessed objective response rate (ORR; defined as confirmed complete response [CR] or partial response [PR]). Exploratory end points included safety and OS in prespecified patient subgroups, including MGMT promoter methylation status (methylated vs unmethylated) and baseline corticosteroid use (yes [within 5 days of first dose] vs no). Because corticosteroids suppress the immune response,9 additional analyses were performed to explore whether no baseline corticosteroid use had a survival benefit based on patients’ MGMT promoter methylation status.
Statistical Analysis
The final analysis of OS was planned for when 300 or more deaths were reported among 369 randomized patients, providing approximately 92% power with an overall type I error of 0.05. Overall survival was compared between treatment groups using a 2-sided log-rank test stratified by the presence or absence of measurable disease at baseline. Kaplan-Meier methodology was used to estimate survival in each group, including medians (95% CI) and OS rates, and the hazard ratios (HRs [95% CIs]) were estimated using a Cox proportional hazards model adjusted for measurable disease. Additional statistical methods are described in the eMethods in Supplement 3. The software used for statistical analyses was SAS statistical software (version 9.2; SAS Institute, Inc), and the data cutoff date for the analysis was January 20, 2017.
Results
Patients and Treatments
From September 2014 through May 2015, 369 patients were randomized to nivolumab (n = 184) or bevacizumab (n = 185) (Figure 1). Demographics and baseline clinical characteristics were relatively well balanced between treatment groups. Patients in the nivolumab group had a numerically longer median time interval from diagnosis to recurrence (10.1 months [range, 3.4-49.6 months] vs 8.5 months [range, 0-38.2 months]) (Table 1). No patients used the NovoTTF-100L system during the study.
Figure 1. Study Profile.

CONSORT diagram showing the number of patients in CheckMate 143 cohort 2 who were enrolled, treated with nivolumab or bevacizumab, discontinued treatment, and analyzed for efficacy and safety. Q2W indicates every 2 weeks.
Table 1. Patient Demographics and Baseline Characteristics.
| Characteristic | No. (%) | |
|---|---|---|
| Nivolumab (n = 184) | Bevacizumab (n = 185) | |
| Age, median (range), y | 55.5 (22-77) | 55.0 (22-76) |
| <65 y | 142 (77.2) | 156 (84.3) |
| Male | 116 (63.0) | 119 (64.3) |
| Histopathologic diagnosis | ||
| Glioblastoma | 183 (99.5) | 184 (99.5) |
| Gliosarcoma | 1 (0.5) | 1 (0.5) |
| Radiotherapy completed | 184 (100.0) | 185 (100.0) |
| Temozolomide received | 183 (99.5) | 185 (100.0) |
| Median No. of prior temozolomide cycles (range) | 6.0 (0-42) | 5.0 (1-26) |
| Time from last RT dose to first dose of study drug | ||
| No. of patients | 182 | 163 |
| Median (range), mo | 8.8 (1.8-47.5) | 6.9 (1.1-36.9) |
| Time from initial diagnosis to recurrence | ||
| Median (range), mo | 10.1 (3.4-49.6) | 8.5 (0-38.2) |
| <1 y | 108 (58.6) | 139 (75.1) |
| ≥1 y | 76 (41.3) | 46 (24.9) |
| Karnofsky performance status at study entry | ||
| 100 | 42 (22.8) | 25 (13.5) |
| 90 | 71 (38.6) | 78 (42.2) |
| 80 | 50 (27.2) | 57 (30.8) |
| 70 | 19 (10.3) | 24 (13.0) |
| <70 | 2 (1.1) | 0 |
| Not reported | 0 | 1 (0.5) |
| Measurable target lesion(s) | 153 (83.2) | 156 (84.3) |
| Target lesion size, median (range), mm2 | 859.0 (100-5278) | 854.0 (110-4030) |
| Site of target lesion(s) | ||
| Temporal lobe | 64 (34.8) | 54 (29.2) |
| Frontal lobe | 49 (26.6) | 53 (28.6) |
| Parietal lobe | 23 (12.5) | 27 (14.6) |
| Occipital lobe | 12 (6.5) | 11 (5.9) |
| Cerebellum | 0 | 2 (1.1) |
| Brain stem | 1 (0.5) | 0 |
| Insula | 0 | 1 (0.5) |
| Other | 20 (10.9) | 28 (15.1) |
| MGMT promoter methylation status | ||
| Methylated | 43 (23.4) | 42 (22.7) |
| Unmethylated | 59 (32.1) | 67 (36.2) |
| Not reported | 82 (44.6) | 76 (41.1) |
| PD-L1 expression level | ||
| <1% | 107 (58.2) | 114 (61.6) |
| ≥1% | 48 (26.1) | 35 (18.9) |
| Not quantifiable | 29 (15.8) | 36 (19.5) |
| Corticosteroid usea | ||
| Yes | 73 (39.7) | 79 (42.7) |
| <2 mg/d | 20 (10.9) | 25 (13.5) |
| ≥2 to <4 mg/d | 27 (14.7) | 26 (14.1) |
| ≥4 mg/d | 26 (14.1) | 28 (15.1) |
| No | 111 (60.3) | 106 (57.3) |
Abbreviations: PD-L1, programmed death ligand 1; RT, radiotherapy.
Dexamethasone equivalents.
Of 369 randomized patients, 347 received study treatment with nivolumab (n = 182 [52.4%]) or bevacizumab (n = 165 [47.6%]). Final analysis was performed when 301 patients had died. At data cutoff (January 20, 2017), median (range) follow-up was 9.8 (1.3-26.3) months in the nivolumab group and 9.4 (0-26.8) months in the bevacizumab group, and 175 of 184 patients (95%) in the nivolumab group and 158 of 185 patients (85%) in the bevacizumab group had permanently discontinued study treatment; the most common reasons were disease progression (nivolumab, n = 162 [89.0%]; bevacizumab, n = 132 [80.0%]) and study drug-associated toxic effects (nivolumab, n = 6 [3.3%]; bevacizumab, n = 11 [6.7%]) (Figure 1). Duration of study treatment and number of doses are described in the eResults in Supplement 3.
Efficacy
No statistical difference was observed in the risk of death between groups (HR, 1.04; 95% CI, 0.83-1.30; P = .76); 154 of 184 patients (83.7%) in the nivolumab group died vs 147 of 185 patients (79.5%) in the bevacizumab group. Median OS (mOS) was similar: 9.8 months (95% CI, 8.2-11.8 months) with nivolumab vs 10.0 months (95% CI, 9.0-11.8 months) with bevacizumab (HR, 1.04; 95% CI, 0.83-1.30; P = .76) (Figure 2A). Median PFS was 1.5 months (95% CI, 1.5-1.6 months) with nivolumab and 3.5 months (95% CI, 2.9-4.6 months) with bevacizumab (HR, 1.97; 95% CI, 1.57-2.48; P < .001) (Figure 2B).
Figure 2. Overall Survival (OS) and Progression-Free Survival (PFS) in All Patients.
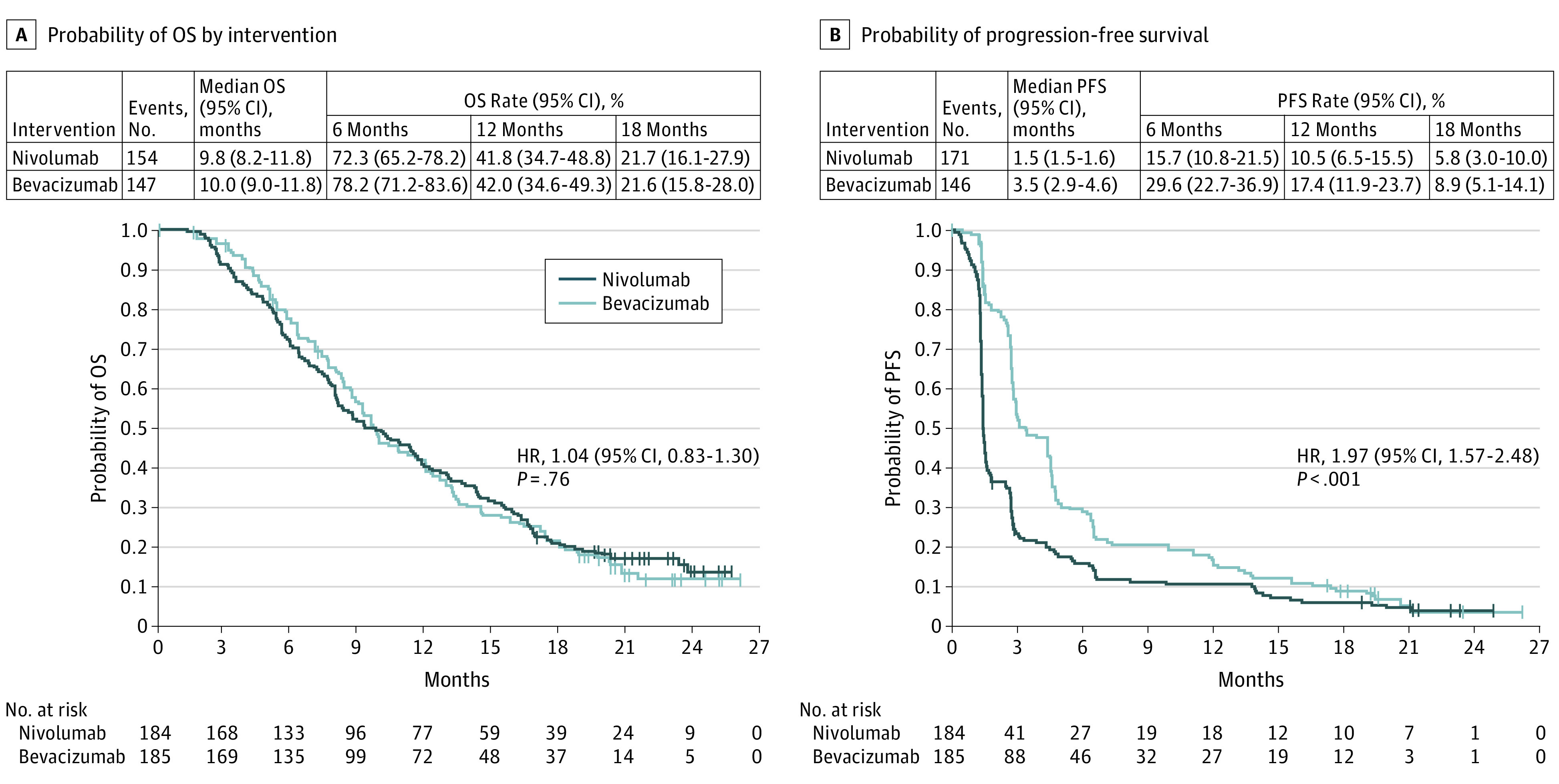
A, The number of events; median OS; OS rates at 6, 12, and 18 months; and the Kaplan-Meier curve for OS in all patients treated with nivolumab or bevacizumab. B, The number of events; median PFS; PFS rates at 6, 12, and 18 months; and the Kaplan-Meier curve for PFS per investigator assessment in patients treated with nivolumab or bevacizumab. Symbols indicate censored observations. Hazard ratios (HRs) and CIs were estimated using a Cox proportional hazards model.
The ORR in patients evaluable for response in the nivolumab (n = 153) and bevacizumab (n = 156) groups was 7.8% (95% CI, 4.1%-13.3%) and 23.1% (95% CI, 16.7%-30.5%) (eTable 1 in Supplement 3). Additional ORR data are presented in the eResults in Supplement 3. Responses were numerically more durable with nivolumab than with bevacizumab, with respective duration-of-response median (range) of 11.1 (0.6-18.7) months and 5.3 (3.1-24.9) months.
Exploratory Analyses
Overall, OS was generally similar between prespecified patient subgroups (Figure 3A). Yet, among patients with no baseline corticosteroid use, the HR for nivolumab vs bevacizumab was 0.84 (95% CI, 0.62-1.15), and among patients with baseline corticosteroid use, the HR for nivolumab vs bevacizumab was 1.41 (95% CI, 1.01-1.97) (Figure 3A). The difference in mOS between patients with baseline corticosteroid use and those without was thus greater with nivolumab (7.0 vs 12.6 months) than with bevacizumab (8.9 vs 11.8 months) (eFigure 1 in Supplement 3).
Figure 3. Overall Survival (OS) in Prespecified Patient Subgroups Defined by Baseline Clinical Characteristics.
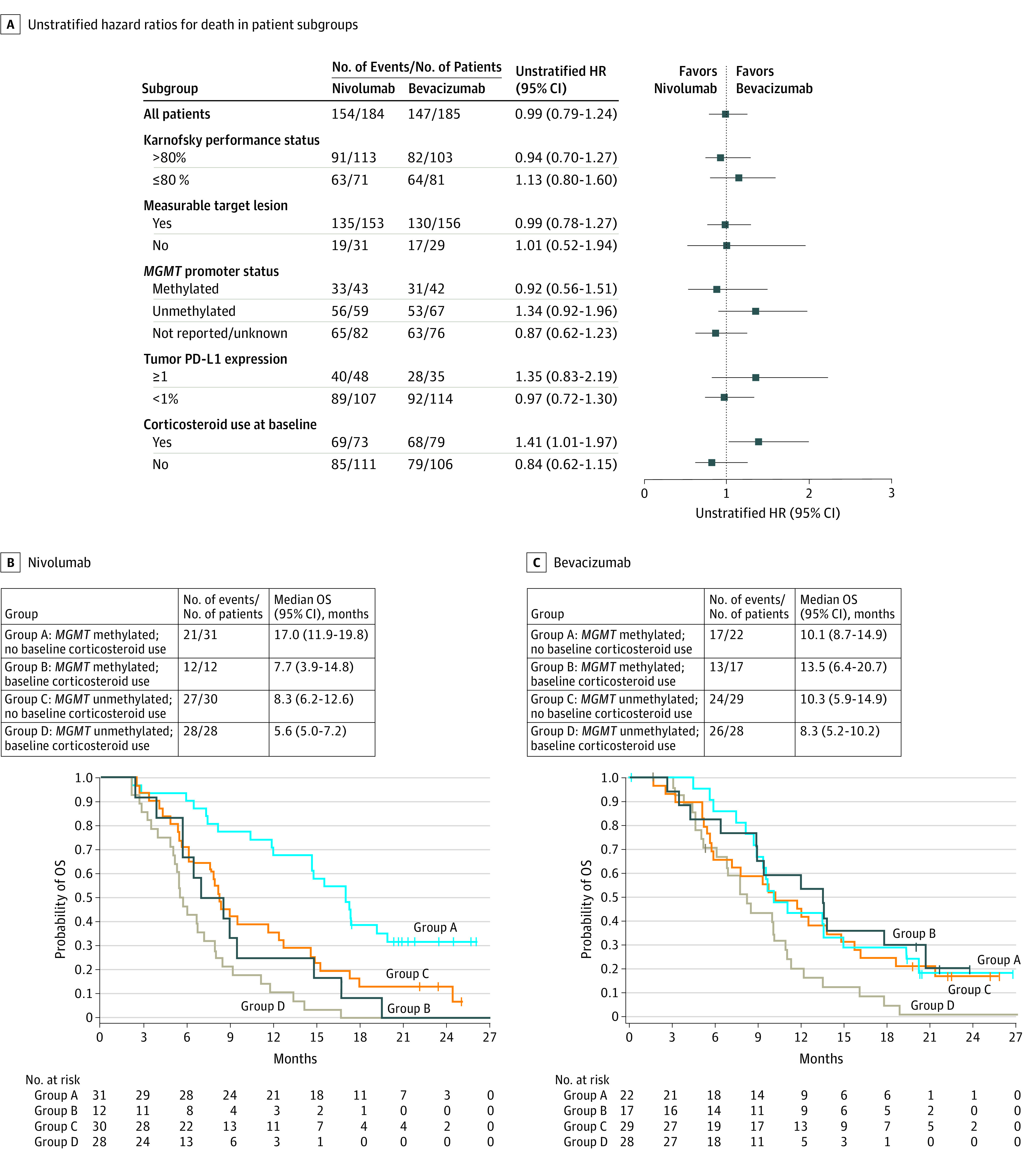
A, Forest plot of unstratified hazard ratios (HRs) for death in the analysis of treatment effect in prespecified patient subgroups according to baseline characteristics. B, Exploratory post hoc analyses of the number of events, median OS, and Kaplan-Meier curves for OS in prespecified patient subgroups treated with nivolumab. C, Exploratory post hoc analyses for bevacizumab. Subgroups include patients with methylated tumors who did not receive corticosteroids at baseline, patients with methylated tumors who received corticosteroids at baseline, patients with unmethylated tumors who did not receive corticosteroids at baseline, and patients with unmethylated tumors who received corticosteroids at baseline. Symbols indicate censored observations; HRs and CIs were estimated using a Cox proportional hazards model.
The mOS was longer in patients with tumors with a methylated MGMT promoter in both treatment groups (eFigure 2 in Supplement 3). There was a trend for inferior mOS with nivolumab in patients with unmethylated MGMT promoter tumors (HR, 1.34; 95% CI, 0.92-1.96) but not in patients with methylated MGMT promoter tumors (HR, 0.92; 95% CI, 0.56-1.51) (Figure 3A) (eResults in Supplement 3). Other disease characteristics, such as performance status (Figure 3A) or size of residual tumor, were not associated with OS.
Hypothesis-generating subgroup analyses were conducted to evaluate OS in prespecified subgroups. In a multivariable Cox proportional hazards model analysis, no baseline corticosteroid use (HR, 0.59; 95% CI, 0.36-0.95) and methylated MGMT promoter status (HR, 0.47; 95% CI, 0.29-0.78) were each associated with longer OS in the nivolumab group (eTable 2 in Supplement 3). With bevacizumab, methylated MGMT promoter status was associated with longer OS (HR, 0.54; 95% CI, 0.32-0.89) (eTable 2 in Supplement 3), but no baseline corticosteroid use was not. On the basis of these results, the combined association of baseline MGMT promoter methylation status and corticosteroid use with OS was evaluated. Among patients with methylated MGMT promoter and no baseline corticosteroid use, a trend toward longer mOS was observed in nivolumab-treated patients than in bevacizumab-treated patients (17.0 vs 10.1 months; HR, 0.58; 95% CI, 0.30-1.11) (Figure 3, B and C) (eResults in Supplement 3).
Safety
Any-grade TRAEs occurred at similar rates in the nivolumab (103/182; 56.6%) and bevacizumab (95/165; 57.6%) groups, with the most common being fatigue in the nivolumab group and hypertension in the bevacizumab group (Table 2). Similar rates of grade 3/4 TRAEs were reported with nivolumab (33/182; 18.1%) and bevacizumab (25/165; 15.2%). Neurological TRAEs were reported in 25 of 182 (13.7%) nivolumab-treated patients (grade 3/4, 8 [4.4%]) and 16 of 165 (9.7%) bevacizumab-treated patients (grade 3/4, 2 [1.2%]); no individual neurological TRAEs were reported in 5% or more of patients. Serious TRAEs are described in the eResults in Supplement 3.
Table 2. Treatment-Related Adverse Events of Any Grade Reported in 5% or More Patients and Grade 3/4 Reported in 2 or More Patients in Either Treatment Group.
| Patients | No. (%) | |||
|---|---|---|---|---|
| Nivolumab (n = 182)a | Bevacizumab (n = 165)a | |||
| Any Grade | Grade 3/4 | Any Grade | Grade 3/4 | |
| Any treatment-related adverse event | 103 (56.6) | 33 (18.1) | 95 (57.6) | 25 (15.2) |
| Treatment-related adverse events occurring in ≥5% of patients in either group | ||||
| Fatigue | 38 (20.9) | 6 (3.3) | 23 (13.9) | 0 |
| Hypertension | 1 (0.5) | 0 | 37 (22.4) | 13 (7.9) |
| Nausea | 7 (3.8) | 0 | 9 (5.5) | 0 |
| Rash | 12 (6.6) | 0 | 4 (2.4) | 0 |
| Pruritus | 11 (6.0) | 0 | 2 (1.2) | 0 |
| Diarrhea | 10 (5.5) | 0 | 2 (1.2) | 0 |
| Increased alanine aminotransferase | 10 (5.5) | 4 (2.2) | 0 | 0 |
| Other grade 3/4 treatment-related adverse events occurring in ≥2 patients in either group | ||||
| Headache | 8 (4.4) | 2 (1.1) | 8 (4.8) | 0 |
| Increased lipase | 7 (3.8) | 4 (2.2) | 1 (0.6) | 0 |
| Pulmonary embolism | 1 (0.5) | 1 (0.5) | 6 (3.6) | 5 (3.0) |
| Seizure | 5 (2.7) | 2 (1.1) | 0 | 0 |
| Cerebrovascular accident | 0 | 0 | 4 (2.4) | 2 (1.2) |
| Pneumonitis | 4 (2.2) | 2 (1.1) | 0 | 0 |
| Increased amylase | 3 (1.6) | 2 (1.1) | 1 (0.6) | 0 |
| Hyperglycemia | 2 (1.1) | 2 (1.1) | 0 | 0 |
Patients who received study treatment.
Immune-mediated AEs (IMAEs) reported in 2% or more of patients are shown in eTable 4 in Supplement 3; the most common were diarrhea (nivolumab, 27 [14.8%]; bevacizumab, 13 [7.9%]), increased alanine aminotransferase (15 [8.2%]; 9 [5.5%], respectively), and rash (17 [9.3%]; 7 [4.2%], respectively). No treatment-related deaths were reported.
Discussion
The CheckMate 143 trial was the first randomized phase 3 study to investigate an immune checkpoint inhibitor in patients with a primary brain tumor. The study did not meet the primary end point of improved OS with nivolumab vs bevacizumab; OS was comparable between treatment groups. The PFS and ORR were numerically better in the bevacizumab group. Durations of response were numerically longer in the nivolumab group. Toxic effects were consistent with the known safety profiles of nivolumab and bevacizumab.10,11 No new safety signals were observed, including no apparent increase in the incidence of neurological TRAEs.
Hypothesis-generating data from subgroup analyses indicated that corticosteroid use at baseline, a known prognostic factor for patients with glioblastoma,12 seemed to be disproportionally and unfavorably associated with outcomes in the nivolumab group. Patients requiring corticosteroids to treat symptomatic cerebral edema may have more rapidly progressive disease and may not have sufficient time to derive benefit from immunotherapy. Furthermore, direct effects of corticosteroids on T-cell function might abrogate activation or priming of the immune system.13
The association of MGMT promoter methylation, a well-known prognostic factor for patients with glioblastoma,14 with survival was also analyzed. Longer mOS was observed in patients with methylated tumors than in patients with unmethylated tumors in both treatment groups. The difference in mOS between patients with vs without methylated MGMT promoter tumors was numerically greater in the nivolumab group than in the bevacizumab group. The post hoc subgroup analyses indicated that the subgroup of patients with glioblastoma with methylated MGMT promoter and no baseline corticosteroid dependence may be most likely to derive benefit from immune checkpoint inhibition.
Limitations
Study limitations include the small number of patients in the subgroup analyses, lack of standardized MGMT promoter methylation status assessment, insufficient data on quality of life assessments, and use of archival tissue collected at the time of initial diagnosis for biomarker analyses.
Conclusions
To our knowledge, the CheckMate 143 randomized clinical trial is the first phase 3 study investigating the use of a PD-1 inhibitor in patients with recurrent glioblastoma. The study did not meet the primary end point of OS. The safety profile of nivolumab in patients with glioblastoma was consistent with that in other tumor types. Patients with methylated MGMT promoter glioblastoma and no baseline corticosteroid use may potentially derive benefit from treatment with immune checkpoint inhibition.
Trial Protocol
Statistical Analysis Plan
eMethods
eResults
eTable 1. Response Rates, Time to Response, and Duration of Response per Investigator Assessment
eTable 2. Multivariable Analysis of Baseline Factors Associated With Overall Survival
eTable 3. Subsequent Therapies
eTable 4. Immune-Mediated Adverse Events Reported in ≥ 2% of Patients in Either Group
eFigure 1. OS based on Baseline Corticosteroid Use
eFigure 2. OS based on MGMT Methylation Status in Either Group
eFigure 3. Sensitivity Analysis of the Association of MGMT Promoter Methylation Status With OS
eFigure 4. Sensitivity Analysis of the Association of MGMT Promoter Methylation Status and Baseline Corticosteroid Use With OS
eFigure 5. Corticosteroid Use in Dexamethasone Equivalents in the Nivolumab and Bevacizumab Treatment Groups
References
- 1.Stupp R, Hegi ME, Mason WP, et al. ; European Organisation for Research and Treatment of Cancer Brain Tumour and Radiation Oncology Groups; National Cancer Institute of Canada Clinical Trials Group . Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009;10(5):459-466. doi: 10.1016/S1470-2045(09)70025-7 [DOI] [PubMed] [Google Scholar]
- 2.Weller M, van den Bent M, Tonn JC, et al. ; European Association for Neuro-Oncology (EANO) Task Force on Gliomas . European Association for Neuro-Oncology (EANO) guideline on the diagnosis and treatment of adult astrocytic and oligodendroglial gliomas. Lancet Oncol. 2017;18(6):e315-e329. doi: 10.1016/S1470-2045(17)30194-8 [DOI] [PubMed] [Google Scholar]
- 3.Omuro A, DeAngelis LM. Glioblastoma and other malignant gliomas: a clinical review. JAMA. 2013;310(17):1842-1850. doi: 10.1001/jama.2013.280319 [DOI] [PubMed] [Google Scholar]
- 4.Weller M, Cloughesy T, Perry JR, Wick W. Standards of care for treatment of recurrent glioblastoma—are we there yet? Neuro Oncol. 2013;15(1):4-27. doi: 10.1093/neuonc/nos273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lim M, Xia Y, Bettegowda C, Weller M. Current state of immunotherapy for glioblastoma. Nat Rev Clin Oncol. 2018;15(7):422-442. doi: 10.1038/s41571-018-0003-5 [DOI] [PubMed] [Google Scholar]
- 6.Reardon DA, Gokhale PC, Klein SR, et al. Glioblastoma eradication following immune checkpoint blockade in an orthotopic, immunocompetent model. Cancer Immunol Res. 2016;4(2):124-135. doi: 10.1158/2326-6066.CIR-15-0151 [DOI] [PubMed] [Google Scholar]
- 7.Omuro A, Vlahovic G, Lim M, et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20(5):674-686. doi: 10.1093/neuonc/nox208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963-1972. doi: 10.1200/JCO.2009.26.3541 [DOI] [PubMed] [Google Scholar]
- 9.Gustafson MP, Lin Y, New KC, et al. Systemic immune suppression in glioblastoma: the interplay between CD14+HLA-DRlo/neg monocytes, tumor factors, and dexamethasone. Neuro Oncol. 2010;12(7):631-644. doi: 10.1093/neuonc/noq001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ferris RL, Blumenschein G Jr, Fayette J, et al. Nivolumab for recurrent squamous-cell carcinoma of the head and neck. N Engl J Med. 2016;375(19):1856-1867. doi: 10.1056/NEJMoa1602252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedman HS, Prados MD, Wen PY, et al. Bevacizumab alone and in combination with irinotecan in recurrent glioblastoma. J Clin Oncol. 2009;27(28):4733-4740. doi: 10.1200/JCO.2008.19.8721 [DOI] [PubMed] [Google Scholar]
- 12.Pitter KL, Tamagno I, Alikhanyan K, et al. Corticosteroids compromise survival in glioblastoma. Brain. 2016;139(Pt 5):1458-1471. doi: 10.1093/brain/aww046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Min L, Hodi FS, Kaiser UB. Corticosteroids and immune checkpoint blockade. Aging (Albany NY). 2015;7(8):521-522. doi: 10.18632/aging.100797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hegi ME, Diserens AC, Gorlia T, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352(10):997-1003. doi: 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial Protocol
Statistical Analysis Plan
eMethods
eResults
eTable 1. Response Rates, Time to Response, and Duration of Response per Investigator Assessment
eTable 2. Multivariable Analysis of Baseline Factors Associated With Overall Survival
eTable 3. Subsequent Therapies
eTable 4. Immune-Mediated Adverse Events Reported in ≥ 2% of Patients in Either Group
eFigure 1. OS based on Baseline Corticosteroid Use
eFigure 2. OS based on MGMT Methylation Status in Either Group
eFigure 3. Sensitivity Analysis of the Association of MGMT Promoter Methylation Status With OS
eFigure 4. Sensitivity Analysis of the Association of MGMT Promoter Methylation Status and Baseline Corticosteroid Use With OS
eFigure 5. Corticosteroid Use in Dexamethasone Equivalents in the Nivolumab and Bevacizumab Treatment Groups