

By Max Bingham, PhD
Hyperuricemia Linked to Diabetes Development in Uricase Knockout Mice
Hyperuricemia likely accelerates diabetes development by inhibiting β-cell survival, but it is unlikely to be the cause of diabetes, according to Lu et al. (p. 1149). Moreover, therapy to reduce plasma urate levels had only limited effect on diabetes incidence or β-cell death in the mouse model used in the study. The authors suggest that the research might help explain part of the apparent relationship between hyperuricemia and diabetes. The study focuses on a mouse model with a knockout of the gene for uricase, which is the enzyme that degrades uric acid. Experiments compared the effects (against wild-type controls) of a high-fat diet on parameters such as glucose and insulin tolerance and urate and creatinine levels. They found that mice with the knockout had significantly raised levels of serum urate compared with controls over 56 weeks. Many other parameters remained the same as controls with the exception of the knockout mice having significantly reduced triglyceride levels. Glucose and insulin tolerance tests, meanwhile, showed that the knockout mice had impaired glucose tolerance but not insulin resistance, indicating they did not spontaneously develop diabetes. Lu et al. found that, following a high-fat diet, the knockout mice had similar insulin sensitivity compared with controls but eventually developed severe glucose intolerance. They also report that the treatment of hyperuricemia with a urate-lowering drug did not fully reverse the development of diabetes or β-cell apoptosis. Commenting more widely, author Jie Lu told Diabetes: “These results demonstrate that hyperuricemia per se does not cause diabetes and that urate-lowering treatment cannot reverse β-cell apoptosis or reduce diabetes incidence. However, we stress the benefits of treating hyperuricemia in nonworsening β-cell death, ameliorating tubulointerstitial damage and postponing the development of diabetes. These data provide new evidence in the remaining controversy over whether asymptomatic hyperuricemia should be treated and might also offer novel insights toward a therapeutic regime for diabetes.”
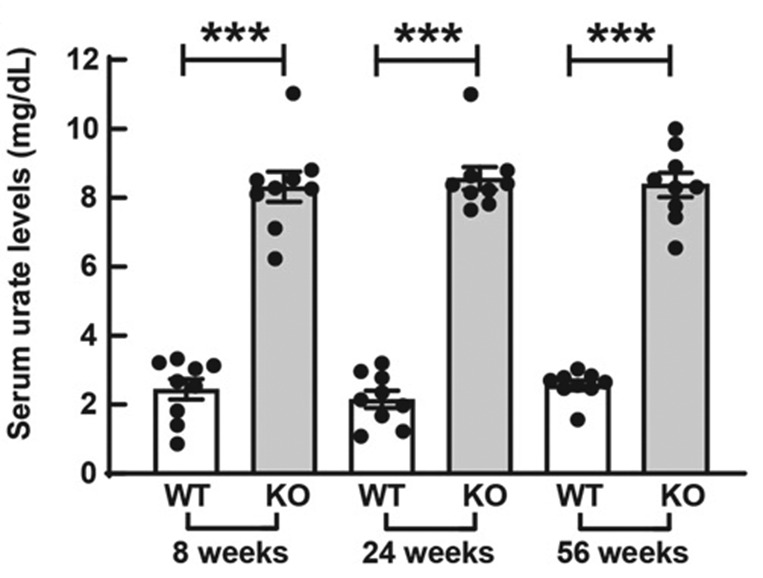
Raised serum urate levels in Uox knockout (KO) mice compared with wild-type (WT) control mice over 56 weeks.
Myo-inositol Oxygenase Key to Tubulointerstitial Injury in Diabetes
The key to understanding renal tubulointerstitial injury in diabetes may lie with the extent of expression of myo-inositol oxygenase (MIOX), according to Sharma et al. (p. 1248). Specifically, they suggest that MIOX overexpression accentuates such injury while its deficiency shields kidneys from injury by dampening oxidative and endoplasmic reticulum stress. They go on to explain many of the mechanisms and pathways that might be involved; however, with the investigation focused on mouse models and cell-based experiments, they refrain from suggesting that any targets for treatment or protection exist yet. The findings come from a series of experiments with transgenic mice that overexpress MIOX (MIOX-TG), mice with a knockout for MIOX (MIOX-KO), and wild-type mice as controls. The authors also included a crossbred mouse, comprising Ins2 Akita and MIOX-KO, as a kidney injury rescue model. They report that MIOX-TG mice (i.e., with MIOX overexpression) had worse renal function—in terms of kidneys that had increased oxidant and endoplasmic reticulum stress—than the other mouse models. They also had perturbed redox metabolism in the form of altered ratios of NAD/NADH and GSH/GSSG and, in addition, increased expression of NOX4. There was also evidence of increased apoptosis and related molecules, including increased TGF-b signaling and related signaling transducers and altered expression of markers of endoplasmic reticulum stress. Imaging also uncovered evidence of accentuated tubulointerstitial fibrosis in mice overexpressing MIOX. In comparison, the MIOX-KO and Ins2Akita/KO mice had minimal or reduced changes in terms of fibrosis. The authors suggest it is conceivable that the various processes may increase the actions of others, which would in turn accelerate the progression of injury. Commenting further, author Yashpal S. Kanwar told us: “Collectively, these studies revealed an uncharacterized role for MIOX in accentuating oxidant and endoplasmic reticulum stress–mediated tubulointerstitial injury. Our investigation hypothesizes that MIOX may serve as a potential therapeutic target that could possibly ameliorate progression of diabetic nephropathy.”
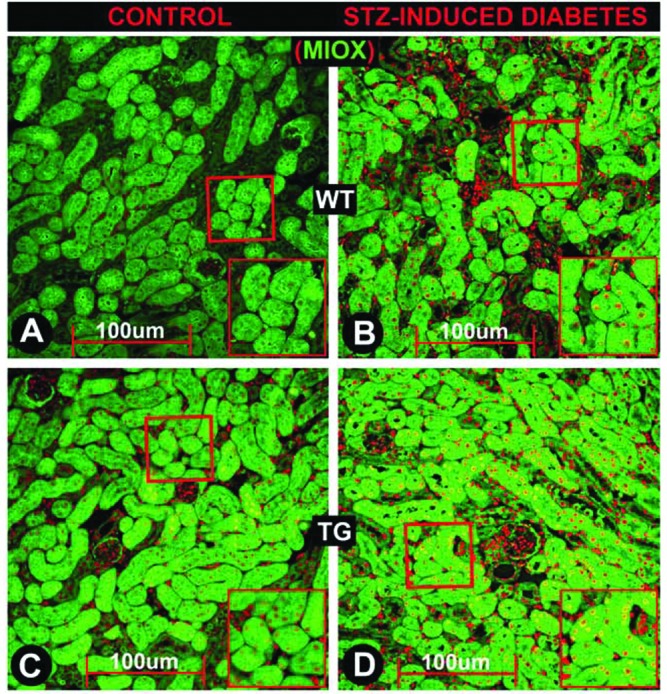
Expression of MIOX in wild-type (WT) and transgenic (TG) mice under control (left) and diabetes (right) conditions.
Adipose Triglyceride Lipase Identified as Key Factor in β-Cell Dysfunction in Diabetes
Adipose triglyceride lipase (ATGL) is a key lipase involved in the mobilization of lipid droplets in human β-cells, according to Liu et al. (p. 1178). Specifically, it seems its disruption results in lipid droplet accumulation in β-cells and impaired insulin secretion, potentially explaining β-cell dysfunction in type 2 diabetes. The findings come from an analysis of various forms of islets and human pseudoislet cell models and looked at the effects of silencing ATGL on lipid droplet mobilization, or lipolysis, under various scenarios including that of diabetes. They found that exposing human islets from donors without diabetes to glucose increased rates of lipolysis. In comparison, however, islets from donors with diabetes exposed to high levels of glucose showed no increase in lipolysis, suggesting lipolysis dysregulation and triglyceride accumulation. After initially identifying ATGL as responsible for glucose-induced lipolysis in INS1 cells, the authors found that silencing ATGL in human pseudoislets impairs glucose-induced lipolysis and increased the number and size of lipid droplets. To explain the effects, they went on to look at SNARE complex proteins in ATGL-deficient INS1 cells and human pseudoislets and found that one particular protein, syntaxin 1a or STX1A, was significantly reduced when ATGL was missing. Further experiments suggested that the reduced levels of STX1A were likely the result of reduced palmitoylation. Commenting more widely, author Yumi Imai stated: “Lipid droplets in β-cells have been underappreciated, as they are difficult to find in mouse islets. We focused on a human islet cell model and identified a previously unrecognized molecular link between the impairment in lipid droplet mobilization and β-cell dysfunction in type 2 diabetes. Our study raises new questions about how lipolysis is dysregulated in diabetes and whether the restoration of lipolysis serves as a new strategy to improve β-cell function in type 2 diabetes, which we hope to address in the future.”
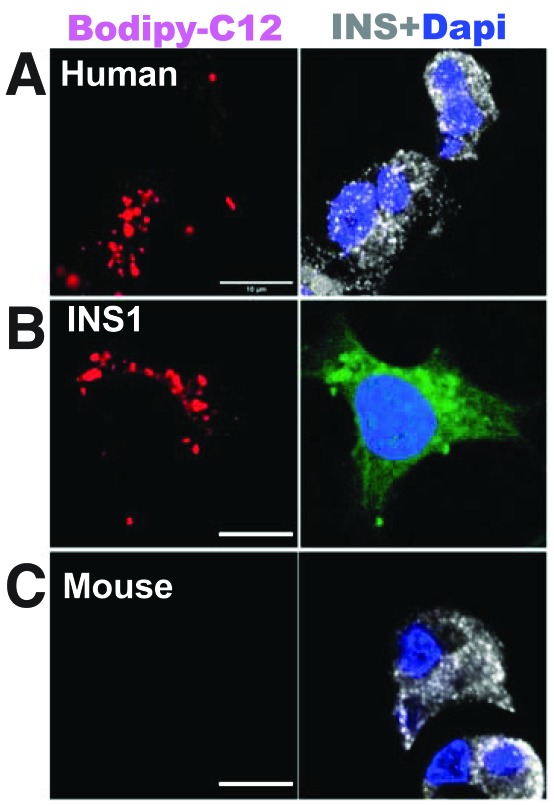
Human (A) and INS1 (B) cells, but not mouse cells (C), show lipid droplets (left) and increased lipolysis (right) in response to glucose.
Central KATP Channels Modulate the Suppression of Endogenous Glucose Production by Hyperglycemia
About half of endogenous glucose production is suppressed by hyperglycemia, and this is likely due to mediation by central KATP channels, according to Carey et al. (p. 1140). They suggest that the findings and the mechanisms surrounding the idea of “glucose effectiveness” might point toward novel therapeutic targets for improving glycemic control in type 2 diabetes. Previous studies suggest that regulation of endogenous glucose production via central KATP channels in type 2 diabetes appears to be impaired. The findings come from a study involving nine humans without diabetes who were subjected to a pair of 7-h euglycemic-hyperglycemic clamp tests and involved the differential administration of either oral glyburide (a KATP antagonist) or saline as a placebo. Rats, as a model, were then used to explore more of the mechanisms. The authors found that under various fixed conditions (e.g., for insulin, glucose, and many other factors) and with administration of the saline control, hyperglycemia suppressed endogenous glucose production by 59% in the final hour of the procedure. In comparison, administration of glyburide resulted in hyperglycemia suppressing endogenous glucose production by only 32%. The authors then used a combination of antagonist (glyburide) and agonist (diazoxide) in normal healthy male rats and found that antagonism with glyburide significantly reduced the effects of hyperglycemia on endogenous glucose production. Administration of diazoxide, the agonist, meanwhile abolished all of the positive effects of the antagonist glyburide. Author Meredith Hawkins commented: “With growing recognition that many neurological and psychiatric conditions are linked to altered glucose metabolism, these parallel studies in rodents and humans demonstrate the importance of central signaling pathways in restraining glucose production. Specifically, these studies build on previous literature by showing that these pathways contribute to normal suppression of glucose production by hyperglycemia. Additional studies are warranted to further delineate this regulation, how it is affected by neurologic conditions, and how it might be restored in individuals with diabetes.”
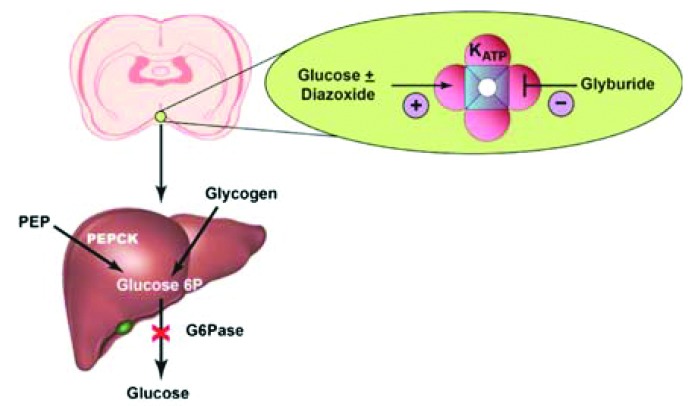
Schematic of the opposing actions of glyburide and diazoxide at different sites on the same KATP channel.