

Abstract
Warburg Micro syndrome is a rare autosomal recessive disease due to mutation in the RAB3GAP1, RAB3GAP2, RAB18 and TBC1D20 genes. It is commonly seen in consanguineous marriages, characterized by optic (microcornea, microphthalmia, congenital cataracts), neurologic )microcephaly, corpus callosum hypoplasia, severe mental retardation( and hypogonadism; some non-typical findings could be present (cardiomyopathy, peripheral neuropathy). We report a novel homozygous mutation in the RAB3GAP1 gene in a 7-month-old boy from healthy nonconsanguineous parents from the same village in Syria, with bilateral congenital cataracts, hypogonadism, muscular hypotonia and severe developmental delay. Whole exome sequencing (WES) showed a homozygous mutation in the c.2195del p.(Pro732Glnfs*6) in exon 19 of the RAB3GAP1 gene, which is likely pathogenic and correlates with Warburg Micro syndrome type 1.
Keywords: Warburg syndrome, whole-exome sequencing, RAB3GAP1, Warburg Micro syndrome type 1
INTRODUCTION
Warburg Micro syndrome (WARBM) is a rare autosomal recessive disorder characterized by postnatal growth retardation, hypoplasia of the corpus callosum, microcephalus, delayed motor development, severe intellectual disability, microphthalmia, microcornea, congenital cataracts, optic atrophy and hypogonadism [1].
This syndrome that consists of four subtypes (1, 2, 3 and 4) recognized till date are caused by mutations in RAB3GAP1 on chromosome 2q21.3, RAB3GAP2 on chromosome 1q41, RAB18 on chromosome 10p21.1 and TBC1D2 on chromosome 20p13, respectively, and are clinically indistinguishable [2, 3].
Mutations of RAB3GAP1 are most prevalent and represent 40% of WARBM patients [2]. RAB3GAP1 comprises 24 exons and codes the catalytic subunit of a Rab GTPase activating protein having Rab3 subfamily specificity (RAB3A, RAB3B, RAB3C and RAB3D) [4, 5]. Rab3 proteins are crucial for ordinary growth of the eye and brain [4]. Here, we report a novel RAB3GAP1 homozygous mutation in a child with WARBM.
CASE REPORT
A 7-month-old boy presented to Children’s University Hospital—Damascus with Ambiguous genitalia, muscular hypotonia, developmental delay and a history of corrected congenital bilateral cataract.
Antenatal period and delivery were uneventful, and the child was born at 38 weeks gestation by vaginal delivery with 3000 g birth weight, 49 cm length and 35 cm occipital-frontal circumference.
The boy was born to clinically healthy non-consanguineous parents from the same village, and he is the youngest of his two unaffected siblings, and the father and mother aged 37 and 33 years old, respectively, when our patient was born.
Physical examination showed bilateral medial squint, microphthalmia, muscular hypotonia, ambiguous genitalia (small penis and empty scrotum) (Fig. 1). Facial features include a prominent nasal root, relatively short nose and large ears. His weight was normal 8.4 kg. Furthermore, he had motor milestones and language delay, optic atrophy and visual impairment with no seizures.
Figure 1.
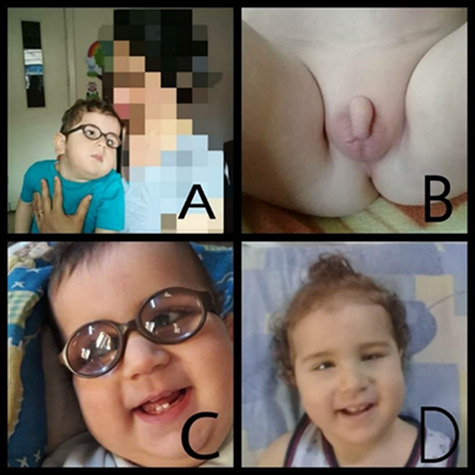
Pictures of the patient illustrating A. Severe hypotonia, B. hypogonadism, C. corrected cataract, D. bilateral medial squint.
Laboratory evaluation revealed decreased circulating cortisol level 5.1 mg/dl at 8 a.m., other pituitary hormones were normal, high lactate levels turned out to be normal afterwards, abdominal ultrasonography detected bilateral undescended testes and magnetic resonance imaging of brain (Fig. 2) revealed diffuse brain atrophy and periventricular demyelinating lesions. Electrocardiogram, bone scan, neonatal TORCH and basic metabolic panel were normal.
Figure 2.
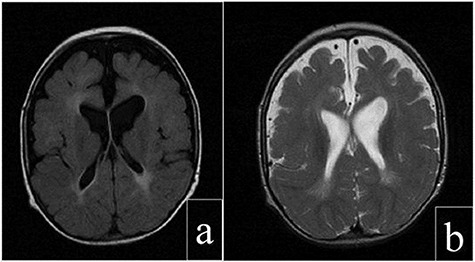
Cerebral magnetic resonance images of the patient: (a) axial T2 FLAIR view showing periventricular increased signal. (b) Axial FRFSE T2 view showing enlargement of the subarachnoid space, ventricles and sylvian fissures.
G-banded chromosome analysis revealed a normal male karyotype. Whole-exome sequencing (CentoXome GOLD®) supplementary revealed novel RAB3GAP1 gene mutation associated with autosomal recessive WARBM 1.
In his every 3-month follow-ups, his neurodevelopment and muscular tone somewhat improved as a result of the physical therapy, but he showed intellectual disabilities.
DISCUSSION
We report a 7-month-old boy with bilateral congenital cataract, ambiguous genitalia and developmental delay associated with novel RAB3GAP1 homozygous mutation. Pathogenic variants in RAB3GAP1 gene are associated with WARBM type 1, an autosomal recessive disease, characterized by ocular (congenital cataract, microphthalmia), brain anomalies (agenesis/hypoplasia of the corpus callosum, abnormal myelination), severe mental retardation and microgenitalia [1, 2].
Reviewing the literature, WARBM cases resulted from consanguineous marriage [4, 6–8]. However, the parents of our patient are non-consanguineous, with unaffected siblings. This finding is contrary to previous studies that have suggested that consanguineous marriages increase the risk for congenital malformations and autosomal recessive disorders.
Usually, bilateral congenital cataracts are associated with TORCH infections, which tested negative in our patient.
Diagnosis of WABM is based on clinical features and molecular genetics [6]; our patient had visual impairment, optic atrophy, severe developmental delay and genital abnormalities. These clinical findings along with low cortisol and high lactate suggested metabolic or mitochondrial diseases but were ruled out because of the negative basic metabolic panel and the spontaneous improvement of the neurodevelopment of the child. So a genetic disease was suspected to be the cause, subsequently identified and confirmed by a whole-exome sequencing, revealing a homozygous likely pathogenic variant c.2195del p.(Pro732Glnfs*6) in exon 19 in RAB3GAP1 gene associated with autosomal recessive WARBM 1.
The treatment of WABM is directed toward the present symptoms, there is no specific treatment, the cataracts of the patient were surgically removed, he had an orchidopexy for his undescended testes and he is undergoing the physical therapy.
WABM is a genetic disease with huge medical and social costs; many individuals with WABM will require assistance as both their physical and mental delays deteriorate as they grow.
As an avoidance measure, consanguineous marriage should be avoided because of the increased risk between it and autosomal recessive diseases [9].
This case presents a novel RAB3GAP1 homozygous mutation in a child with WARBM type 1 to nonconsanguineous parents, which suggest more research to give information about this rare autosomal recessive disease. Its treatment and protection would prove be a fruitful area for further work in future.
ACKNOWLEDGEMENTS
We would like to thank Prof. André Megarbane for his efforts in the acquisition of the genetic approach to this case and the parents for helping us.
CONFLICT OF INTEREST
There is no conflict of interest.
FUNDING
There is no funding.
CONSENT
Informed patient consent was obtained.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not required.
GUARANTOR
Prof. Diana Alasmar.
REFERENCES
- 1. Warburg M, Sjö O, Fledelius HC, Pedersen SA. Autosomal recesssive microcephaly, microcornea, congenital cataract, mental retardation, optic atrophy, and Hypogenitalism: micro syndrome. JAMA Pediatr 1993;147:1309–12. doi: 10.1001/archpedi.1993.02160360051017. [DOI] [PubMed] [Google Scholar]
- 2. Handley MT, Morris-Rosendahl DJ, Brown S, Macdonald F, Hardy C, Bem D et al. Mutation Spectrum in RAB3GAP1, RAB3GAP2, and RAB18 and genotype–phenotype correlations in Warburg micro syndrome and Martsolf syndrome. Hum Mutat 2013;34:686–96. doi: 10.1002/humu.22296. [DOI] [PubMed] [Google Scholar]
- 3. Imagawa E, Fukai R, Behnam M, Goyal M, Nouri N, Nakashima M et al. Two novel homozygous RAB3GAP1 mutations cause Warburg micro syndrome. Human Genome Variation 2015;2:15034. doi: 10.1038/hgv.2015.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aligianis IA, Johnson CA, Gissen P, Chen D, Hampshire D, Hoffmann K et al. Mutations of the catalytic subunit of RAB3GAP cause Warburg micro syndrome. Nat Genet 2005;37:221–3. doi: 10.1038/ng1517. [DOI] [PubMed] [Google Scholar]
- 5. Nagano F, Sasaki T, Fukui K, Asakura T, Imazumi K, Takai Y. Molecular cloning and characterization of the noncatalytic subunit of the Rab3 subfamily-specific GTPase-activating protein. J Biol Chem 1998;273:24781–5. doi: 10.1074/jbc.273.38.24781. [DOI] [PubMed] [Google Scholar]
- 6. Dursun F, Guven A, Morris-Rosendahl D. Warburg micro syndrome. J Pediatr Endocrinol Metab: JPEM 2012;25:379–82. doi: 10.1515/jpem-2011-0459. [DOI] [PubMed] [Google Scholar]
- 7. Abdel-Salam GM, Hassan NA, Kayed HF, Aligianis IA. Phenotypic variability in micro syndrome: report of new cases. Genetic Counseling (Geneva, Switzerland) 2007;18:423–35. [PubMed] [Google Scholar]
- 8. Graham JM Jr, Hennekam R, Dobyns WB, Roeder E, Busch D. MICRO syndrome: an entity distinct from COFS syndrome. Am J Med Genet A 2004;128a:235–45. doi: 10.1002/ajmg.a.30060. [DOI] [PubMed] [Google Scholar]
- 9. Salway S, Ali P, Ratcliffe G, Such E, Khan N, Kingston H et al. Responding to the increased genetic risk associated with customary consanguineous marriage among minority ethnic populations: lessons from local innovations in England. J Community Genet 2016;7:215–28. doi: 10.1007/s12687-016-0269-1. [DOI] [PMC free article] [PubMed] [Google Scholar]