

Sequence function relationships help to understand how imine reductase enzymes perform reductive amination in water.
Abstract
Imine reductases (IREDs) have shown great potential as catalysts for the asymmetric synthesis of industrially relevant chiral amines, but a limited understanding of sequence activity relationships makes rational engineering challenging. Here, we describe the characterization of 80 putative and 15 previously described IREDs across 10 different transformations and confirm that reductive amination catalysis is not limited to any particular subgroup or sequence motif. Furthermore, we have identified another dehydrogenase subgroup with chemoselectivity for imine reduction. Enantioselectivities were determined for the reduction of the model substrate 2-phenylpiperideine, and the effect of changing the reaction conditions was also studied for the reductive aminations of 1-indanone, acetophenone, and 4-methoxyphenylacetone. We have performed sequence-structure analysis to help explain clusters in activity across a phylogenetic tree and to inform rational engineering, which, in one case, has conferred a change in chemoselectivity that had not been previously observed.
INTRODUCTION
Imine reductases (IREDs) are nicotinamide adenine dinucleotide phosphate (NADPH)–dependent enzymes that have been applied to the stereoselective synthesis of chiral amines through asymmetric imine reduction and reductive amination (1, 2). A number of pharmaceuticals, agrochemicals, and natural products contain one or more chiral amine functionalities, with examples including the anti-Parkinson’s agent pramipexole (3), the tuberculosis treatment ethambutol (4), and the natural product histrionicotoxin (5). While amine dehydrogenases and transaminases have previously been exploited in the direct conversion of carbonyl-containing compounds to the corresponding chiral primary amines (6, 7), only IREDs have been shown to catalyze reductive amination with multiple different amine nucleophiles, including the secondary amine pyrrolidine (8), at concentrations within an order of magnitude of that of the cosubstrate ketone or aldehyde (9). Asymmetric reductive amination requires high chemoselectivity to avoid direct reduction of the carbonyl starting material, and existing chemical catalyst systems that fulfill this requirement tend to be limited to a particular class of amine nucleophile, such as anilines (10) or ammonia (11).
The first IREDs were identified by Mitsukura et al. (12, 13) on the basis of their ability to catalyze the enantioselective reduction of the synthetic imine 2-methylpyrroline. Subsequently, a much broader range of wild-type IREDs have been found and applied to the synthesis of a range of cyclic amines, including tetrahydroisoquinolines and piperidines (14), 3-thiazolines (15), bulky α,β-unsaturated imines (16), and 2H-1,4-benzoxazines (17). IREDs have also been applied in cascades for the deracemization of racemic amines (18) and for the direct synthesis of chiral amines from simple precursors (Fig. 1) (19, 20), in some cases with closed-loop recycling of the nicotinamide cofactor (21, 22). Recently, they have been used in new chemoenzymatic methods for the synthesis of pharmaceutically relevant dibenz[c,e]azepines (23) and 2,2-disubstituted azepanes (24). However, despite promising results in the reductive aminations of cyclohexanone and a range of aldehydes, for ketones such as 1-indanone and 4-fluorophenylacetone, more than 20 equivalents of amine nucleophile were required to achieve only moderate conversions (2, 9, 25). Likewise, although good conversions were observed in the reductive amination of simple cyclic ketones such as cyclohexanone with ammonia, no conversion has yet been observed using ammonia for the amination of aldehydes. The scope of reactions performed by IREDs in nature is generally unknown, although the ArpDHII enzyme from Streptomyces argillaceus, which shares 36% sequence identity with the reductive aminase from Aspergillus oryzae (AspRedAm) (2), has been assigned a role in argimycin biosynthesis (26).
Fig. 1. Synthetic applications of IREDs.
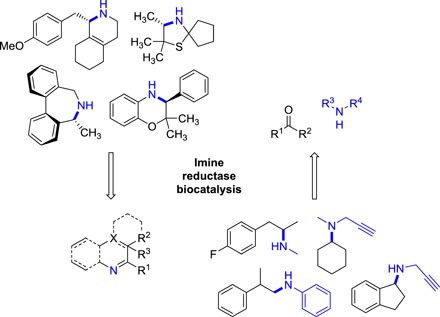
Representative examples of biotransformations performed with IRED to date (2, 9, 15, 16, 23, 25).
In the course of their biochemical characterization, several specific residues have been proposed to help control IRED selectivity and substrate scope. Hussain et al. (14) had observed that the D172A and D172L variants of R-IRED from Streptomyces sp. GF3587 both showed decreases in conversion for more bulky substrates, typically also with a reduction in ee (enantiomeric excess), and Aleku et al. (2) then showed that a Q240A mutant of AspRedAm gives improved conversions with certain ketones. However, France et al. (8) noted that only one of the six “key residues” that had been proposed to confer activity in fungal reductive aminases was conserved among several bacterial IREDs, which gave high conversions in reductive amination. Nonetheless, the importance of these residues in reductive aminase catalysis is supported by crystallographic data and engineering studies (27). The diversity of active site motifs in characterized IREDs indicates that this activity may not be confined to a narrow subset of dehydrogenases and could be less unusual than first anticipated.
With a view to applying IREDs on scale, Roiban and co-workers (9) studied the reductive amination of a variety of carbonyl substrates with aniline and related nucleophiles. Conversions of up to 99% were achieved with 1.1 equivalents of aniline in the reductive amination of cyclohexanone (0.4 g), and a more recent study by Bornadel et al. (28) demonstrated intensification of an IRED-catalyzed reductive amination reaction to reach a space time yield of 12.9 g liter−1 hour−1. Most recently, Roiban and co-workers have described the directed evolution of an IRED for the synthesis of a lysine specific demethylase 1 inhibitor on multikilogram scale, with the transformation run under acidic conditions. The most improved variant had significantly higher activity and stability under the desired reaction conditions compared to the wild-type enzyme, but because of the lack of an effective colorimetric assay, the work required a sophisticated automated platform. Of 296 amino acids, 256 were varied by single-site saturation mutagenesis, and in total, more than 9000 variants were screened by ultraperformance liquid chromatography across three rounds of mutagenesis (29).
To explore structure-activity relationships across the IRED family more deeply, we prepared a panel of 95 enzymes, containing 80 putative IREDs from fungi, bacteria, and plants alongside 13 previously described IREDs (8, 9, 13, 14, 24, 30) and two RedAms (2, 27). Here, we describe their biochemical characterization in five different reductive aminations and the reduction of 2-phenylpiperideine (1), to establish the substrate scope of wild-type IREDs for chemical synthesis and the further characterization of a subset of these in seven more reductive aminations (Fig. 2). For a reductive amination with aniline that typically gave low conversion, the use of cosolvents has been explored as a way of alleviating substrate inhibition and increasing conversion without mutagenesis. An alternative structural analysis approach was then explored, whereby contacts were determined between amino acid residues, considering all rotameric states, and the docked substrate, which was given a certain degree of conformational freedom. These hypothetical contact points were compared across the sequences to determine potential structure-activity relationships. On the basis of this analysis, an enzyme with negligible activity was mutated to establish the validity of the structure-activity model. Structural differences between fungal RedAms and newly characterized IREDs from plants were also examined, to explore whether domain swapping is necessary for IRED chemoselectivity. Last, we tested a new hypothesis about the ubiquity of reductive amination activity by comparing the conversions obtained with the biosynthetic bacterial IRED ArpDHII with those previously reported with the reductive aminase AspRedAm.
Fig. 2. IRED-catalyzed reductive aminations described in this work.
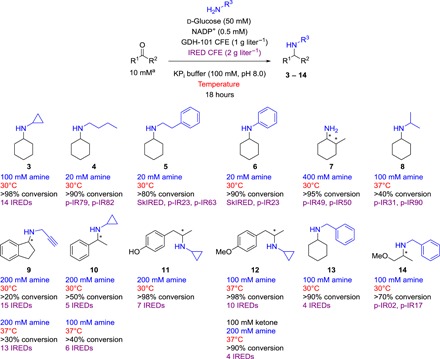
Amine nucleophile concentrations (blue), reaction temperatures (red), maximum conversions (black), and the IREDs with which the reported maximum conversions were achieved (purple) are shown for each product. Conversions were determined by gas chromatography–flame-ionization detection (GC-FID) with comparison to chemically synthesized standards. Enantioselectivities were not determined. Superscripted a indicates that ketone concentration was 10 mM [5 μl from a 1 M stock solution in dimethyl sulfoxide (DMSO)] except where otherwise stated, in which case, it was added neat. CFE, cell-free extract.
RESULTS
Sequence selection and initial characterization
For the purposes of this study, we identified a diverse selection of genomic putative IREDs, which were then cloned into Escherichia coli. Putative IREDs were initially selected from bacterial and fungal sources on the basis of searches of the UniProt database (31) and the laboratory culture collection for putative proteins with sequence identity of >30% with AspRedAm or a bit score higher than 50, but without bias toward particular active site motifs. Subsequently, the search was narrowed to proteins identified within plant genomes, and the presence of an nicotinamide adenine dinucleotide (NAD)–binding domain (PF03446) was used to select 26 putative dehydrogenases from a group with no more than 27% sequence identity with the fungal RedAm.
The resulting enzyme panel contains previously uncharacterized enzymes from a range of prokaryotes and eukaryotes, ranging from 17 to 62% sequence identity to AspRedAm. The diversity of this panel is demonstrated with different putative IREDs across the whole tree of life (fig. S1). Rather than falling into a single clade, the fungal enzymes are divided into two groups, with two further outliers in the putative IREDs p-IR36 and p-IR62 from Fonsecaea multimorphosa and Pochonia chlamydosporia, respectively. The putative actinobacterial IREDs are also split across the tree. The putative dehydrogenases that are of plant origin fall into a distinct clade and match two different Pfam signatures, PF03446 (the NAD-binding domain of 6-phosphogluconate dehydrogenases) and PF14833. The former spans approximately 160 residues from the N terminus and is detected in all the sequences considered here. For the putative IREDs from plants, the first 120 residues from the N terminus in the same NAD-binding domain also match the signature for the NAD-binding domain of 3-hydroxyisobutyrate dehydrogenases, which have been compared to IREDs in sequence studies by Fademrecht et al. (32).
The putative IREDs were initially screened for conversion in the asymmetric reduction of 1 to 2-phenylpiperidine 2. Soluble expression was interpreted as good, moderate, or poor, and the observed conversions and enantioselectivities were compared to those of R-IRED and S-IRED from Streptomyces sp. (12, 13) that were used as positive controls (fig. S2). A clarified lysate of cells containing empty pET-28b(+) vector was used as a negative control. Of 90 cell-free extracts, 57 putative IREDs showed moderate or good expression based on SDS–polyacrylamide gel electrophoresis. Sixty percent of the enzymes (54 putative IREDs) gave at least 10% conversion in the reduction of 1 after 18 hours, including 10 of those that did not express well, and 30% of the panel gave >98% conversion. Of the enzymes that gave at least 10% conversion to 2, 21 putative IREDs gave >90% ee, although only one of these enzymes was (R)-selective. While S-IRED and p-IR49 were (R)-selective for 2, the closely related p-IR23, p-IR24, p-IR48, and p-IR68 were all highly (S)-selective. Having collated a diverse panel of enzymes, we then sought to explore their synthetic potential in a range of reductive amination reactions, with a view to comparing reaction outcomes across the sequence space.
Nucleophile scope in biocatalytic reductive amination
To explore conversions with a lower (2:1) stoichiometry of amine nucleophile to cyclohexanone, we characterized the enzyme panel with respect to conversion in the reductive aminations of cyclohexanone with butylamine, phenethylamine, and aniline. The previously characterized R-IRED and S-IRED from Streptomyces sp. (12, 13), SkIRED from Streptomyces kanamyceticus (30), AspRedAm, and its homolog AtRedAm (2) were also included as positive controls. Butylamine and aniline were chosen for being chemically distinct, and since phenethylamine shares properties with both, the resulting set of conversions would illustrate whether the aryl group or the lone pair stabilization of aniline have a more significant effect on conversion. While the products of these transformations are not known to be of practical interest, aside from any value added through amination, they allowed us to map variations in conversion and substrate preference across the panel.
In the reductive amination of cyclohexanone with cyclopropylamine to yield N-cyclopropylcyclohexylamine 3, 57 of the putative IREDs gave at least 10% conversion (Fig. 3), and the activities largely correlate with those seen in the reduction of 1. However, while p-IR05 gave no measurable conversion in imine reduction, it gave 38% conversion to 3. This effect is even more marked for p-IR14, p-IR19, and p-IR83, all of which gave more than 80% higher conversion in this reductive amination reaction compared to imine reduction. The opposite substrate preference is also noticeable in some cases, particularly with p-IR11 (81% conversion to 2 and 21% conversion to 3).
Fig. 3. Reductive amination with primary amines and ammonia.
(A) Heterologous expression in E. coli; (B) conversion to 3 with cyclohexanone and 100 mM cyclopropylamine (up to >99%); (C) conversion to 4 with cyclohexanone and 20 mM butylamine (up to 93%); (D) conversion to 5 with cyclohexanone and 20 mM phenethylamine (up to 88%); and (E) conversion to 6 with cyclohexanone and 20 mM aniline (up to 96%). Cyan and indigo bars illustrate conversions to two diastereomers of 7 with 2-methylcyclohexanone and 400 mM ammonium chloride (up to 98% combined). Previously described IREDs are labeled in blue. All transformations were performed with 10 mM ketone at 30°C. Conversions were determined by GC-FID with comparison to chemically synthesized standards, with darker colors and larger bars corresponding to higher conversions.
In general, full conversion was not observed with butylamine, phenethylamine, or aniline, with only 13 of 90 enzymes appearing to give more than 50% conversion to N-butylcyclohexylamine 4 after 18 hours, compared to 52 of 90 for 3. Conversions in the reductive amination of cyclohexanone with phenethylamine to yield N-cyclohexylphenethylamine 5 and aniline to yield N-cyclohexylaniline 6 were somewhat correlated, although only seven enzymes gave >30% conversion to 6. SkIRED that was first described in 2013 (30) proved to be one of the most active biocatalysts for the reductive amination of cyclohexanone with aniline, giving 92% conversion. Of the enzymes that gave less than 20% conversion to 3, only p-IR65 gave any significant conversion in reductive aminations at low stoichiometries. This IRED was also the only enzyme in the panel which gave >99% conversion in cyclic imine reduction without concomitant high conversion in the reductive amination of cyclohexanone.
Direct reductive amination with ammonia continues to be of interest for the synthesis of chiral primary amines and has advantages over enzymatic transamination in terms of its atom economy. The reductive amination of 2-methylcyclohexanone with ammonia to give 2-methylcyclohexylamine 7 was therefore investigated with the same enzyme panel. Ammonia was applied at a higher concentration of 400 mM (40 equivalents with respect to 2-methylcyclohexanone), with the ammonium chloride solution also acting as the reaction buffer. Although the conversions to 7 correlated well with the total conversions to 3, the reactions with ammonia typically did not go to completion. The syn/anti-selectivity was also quite poor, which suggests that the methyl group has a limited influence on the mode of binding, although there are some general trends where enzymes in the same clade largely favor the same diastereomer. For example, AspRedAm, AtRedAm, p-IR9, and p-IR16 all favor one product, whereas p-IR58, p-IR88, p-IR78, p-IR50, p-IR89, and p-IR90 favor the other.
On the basis of the initial results, 35 of the least active IREDs were excluded from further screening with more challenging substrates. In experiments to date, IREDs have been found to give lower conversions with more sterically demanding amines. For example, with purified AspRedAm, 20 equivalents of isopropylamine were required to obtain 94% conversion in the reductive amination of cyclohexanone to yield N-isopropylcyclohexylamine 8 (2). Forty-five novel and 13 previously characterized IREDs were screened in the same reaction with 10 equivalents isopropylamine (fig. S3), and the highest conversions to product were obtained with p-IR31 (44%) and p-IR90 (49%). However, 53% of the panel gave no conversion, indicating that screening of multiple wild-type enzymes remains necessary to find hits for more challenging nucleophile substrates.
Biocatalytic reductive amination with novel ketone substrates
The smaller panel of IREDs was then tested in the reductive amination of a number of prochiral ketones (Fig. 4). The reaction of 1-indanone with propargylamine was studied at both 30° and 37°C, with greater conversions to N-propargyl-1-aminoindan 9 being observed across the panel at the higher temperature. Acetophenone, a precursor to active pharmaceutical ingredients such as rivastigmine (33), was then used as a substrate with either 10 equivalents of cyclopropylamine at 37°C or 20 equivalents at 30°C. While none of the enzymes gave full conversion to N-(1-phenylethyl)cyclopropanamine 10, the mean conversion to product at 30°C was higher than for that of 1-indanone (19% versus 10%), as was the highest conversion obtained (59% versus 28%). When mapped phylogenetically, the enzymes’ conversion to 10 correlates well with that observed for the reductive amination of 1-indanone, although some show a preference for one over the other. Again, the IREDs gave higher conversions at 37°C than at 30°C, although the highest conversions (59% with p-IR58 and 55% with p-IR13) were obtained at 30°C with the higher concentration of cyclopropylamine. Some groups of related enzymes give similar conversions, with the fungal enzymes generally giving better conversions than R-IRED, SkIRED, and their bacterial homologs.
Fig. 4. Reductive amination of prochiral ketones.
(A) Heterologous expression in E. coli; (B) conversion to 9 with 10 mM 1-indanone and 200 mM propargylamine at 30°C (up to 28%); (C) conversion to 9 with 10 mM 1-indanone and 200 mM propargylamine at 37°C (up to 37%); (D) conversion to 10 with 10 mM acetophenone and 200 mM cyclopropylamine at 30°C (up to 59%); (E) conversion to 10 with 10 mM acetophenone and 100 mM cyclopropylamine at 37°C (up to 50%); (F) conversion to 11 with 10 mM 4-hydroxyphenylacetone and 100 mM cyclopropylamine at 30°C (up to >99%); (G) conversion to 12 with 10 mM 4-methoxyphenylacetone and 100 mM cyclopropylamine at 37°C (up to >99%); and (H) conversion to 12 with 100 mM 4-methoxyphenylacetone and 200 mM cyclopropylamine at 37°C (up to 96%). Previously described IREDs are labeled in blue. Conversions were determined by GC-FID with comparison to chemically synthesized standards, with darker colors corresponding to higher conversions.
The stability and substrate scope of the novel IREDs was further investigated through the reductive amination of two phenylacetone derivatives under different conditions. The mean conversions in the reductive aminations of 4-hydroxyphenylacetone with cyclopropylamine to give 4-[2-(cyclopropylamino)propyl]phenol 11 at 30°C and those of 4-methoxyphenylacetone with cyclopropylamine to give N-[1-(4-methoxyphenyl)-2-propanyl]cyclopropanamine 12 at 37°C were 24 and 35%, respectively, with 10 mM ketone, although both reactions went to completion with at least seven IREDs. For the reductive amination of 4-methoxyphenylacetone, with 10 mM ketone and 100 mM cyclopropylamine, 10 enzymes gave >90% conversion. When the transformation was repeated with 100 mM ketone and 200 mM amine nucleophile, only p-IR06, p-IR13, p-IR16, and p-IR23 gave >90% conversion, indicating that these enzymes have greater stability under increased substrate loading. Substrate inhibition is a limiting factor in the application of enzymes on a larger scale, the ability of these enzymes to tolerate high substrate loading demonstrates these enzymes are good tools for process chemistry. Note that the increased ketone concentration without a proportional increase in the amount of cyclopropylamine leads to an improved reaction stoichiometry of 2:1, which has not previously been demonstrated for IRED-catalyzed reductive amination of a phenylacetone derivative.
Last, the enzyme panel was characterized in terms of its activity in the reductive amination of methoxyacetone to yield N-benzyl-1-methoxy-2-propylamine 13 (fig. S4), to explore the possibility of accessing members of the chloroacetanilide family of herbicides. The panel was also screened in the reductive amination of cyclohexanone with benzylamine to yield N-benzylcyclohexylamine 14, with the conversion data serving as a control for variations in amine substrate scope. Three enzymes, p-IR06, p-IR17, and p-IR79, gave greater than 30% conversion with methoxyacetone, and these IREDs also gave >60% conversion in the reductive amination of cyclohexanone with benzylamine. Several typically productive enzymes including p-IR16, p-IR23, p-IR63, and SkIRED gave no conversion with methoxyacetone.
Reaction intensification with aniline
With a large set of sequence and conversion data in hand, we elected to explore the use of cosolvents as a way of increasing conversion in reductive amination without resorting to directed evolution. The reductive amination of cyclohexanone with aniline catalyzed by p-IR89 was selected as a model transformation in which moderate conversion had been obtained, but the reaction did not go to completion within 24 hours. To profile the effect of solvents with different polarities, we investigated different percentages of cyclohexane, methanol, dimethyl sulfoxide (DMSO), and tert-butyl methyl ether (MTBE) (table S1). Of the four tested cosolvents, cyclohexane was the best tolerated, with 10, 20, and 50% by volume all giving higher conversions than a purely aqueous transformation [up to 41% with 10% (v/v) cyclohexane]. MTBE and ethanol were inhibitory, while up to 20% (v/v) DMSO was well tolerated (31% conversion after 18 hours). A complete lack of DMSO appeared to have a deleterious effect on conversion compared to the screening conditions, where the addition of substrates from 1 M stock solutions gave a final composition of 3% (v/v) DMSO. In further experiments, conversion to 6 reached 81% when the reaction was run with 50 mM cyclohexanone and 100 mM aniline with a 200-μl layer of cyclohexane cosolvent (Table 1). Pushpanath et al. (7) have previously demonstrated that a biphasic reaction system can lead to improved conversions with amine dehydrogenases, an effect that was attributed to alleviation of product inhibition, and this would explain the improvement in conversion observed here, since aniline has a relatively low solubility in water compared to cyclohexane.
Table 1. Reaction intensification using cyclohexane cosolvent.
Conversions to 6 obtained in the reductive amination of cyclohexanone with aniline at different substrate and cosolvent loadings, as determined by GC-FID. All reactions contained 500 μl of aqueous components and variable total volumes from 700 to 900 μl.
| Entry | [Cyclohexanone] (mM) | [Aniline] (mM) | Cyclohexane (μl) | Conversion to 6 (%) |
| 1 | 50 | 100 | 200 | 81 |
| 2 | 50 | 100 | 250 | 75 |
| 3 | 50 | 100 | 300 | 76 |
| 4 | 75 | 100 | 300 | 74 |
| 5 | 100 | 100 | 300 | 59 |
| 6 | 50 | 100 | 350 | 75 |
| 7 | 50 | 100 | 400 | 71 |
Structure-activity relationships
In analyzing the results of screening across a variety of ketone and amine substrate combinations, we observed markedly different substrate specificities between several enzymes with high sequence identity and similar expression levels. We also observed some grouping of enantioselectivities in imine reduction based on sequence similarity. To shed light on some of the observed trends, we performed a sequence-structure analysis using the SmartScaffold approach. This analysis involves advanced, multitemplate homology modeling, flexible docking, and a custom functionality Structure and Ligand-based and Active site Prediction (SLAP) that defines the extended active site through the study of steric clashes and contacts between the protein model and a conformational ensemble of the best scoring docked poses.
On the basis of structure-guided sequence alignment of active site residues for six enzymes from the original panel, we determined that the two strongly (R)-selective enzymes (p-IR49 and S-IRED) have the residues Pro121, Phe176, Met/Ile210, and Ser243 based on AspRedAm numbering, while several of the strongly (S)-selective enzymes including R-IRED, p-IR64, and p-IR48 have Thr121, Met/Leu176, Phe/Trp210, and Ala243. The Pro121/Phe176 combination corresponds to the SkIRED 139/194 motif used by Fademrecht et al. (32) in their selectivity classification of putative IREDs, although this categorization does not hold true for all IREDs described to date (9, 34).
We also observed that several bacterial IREDs, such as SkIRED and p-IR23, gave higher conversions in the reductive amination reactions with phenethylamine and aniline than the fungal reductive aminases AspRedAm and AtRedAm. A comparison of the active site residues highlighted in mechanistic studies of fungal RedAms (27) across eight of the most active novel IREDs, as well as AspRedAm, SkIRED, p-IR23, and p-IR48, shows that only Asp169 is fully conserved, although the bacterial IREDs do have several residues in common (table S2). Because of the diversity of sequences within the panel, a more detailed comparison focused on groups of well-expressed IREDs with high sequence identity that gave markedly different conversions.
p-IR23 and p-IR48 were both well expressed and gave >99% conversion to product in the reductive amination of cyclohexanone with cyclopropylamine, but p-IR23 gave much higher conversion in the reductive amination with aniline (96% versus 5%). Homology models of p-IR23 and p-IR48 were generated using the “closed” form of AspRedAm [Protein Data Bank (PDB) code: 5G6S] as a template, and the substrates cyclohexanone and aniline were docked to determine which residues they would be likely to interact with. The result was a list of 23 residues that could come into contact with one or both of the substrates (Fig. 5A). By limiting the analysis to this subset of residues, it becomes clearer how the activity of p-IR48 in the reductive amination of cyclohexanone with aniline might be improved through rational mutagenesis, especially since 19 are conserved between p-IR23 and p-IR48, with four amino acids potentially having a greater effect on substrate binding. The amino acids in AspRedAm-equivalent positions 170 (Ser/Thr), 176 (Leu/Met), and 180 (Met/Leu) are the most similar in terms of their chemical properties, making 244 (Leu/Phe) a clear candidate for mutagenesis. Two mutants of p-IR48 were generated, in which either AspRedAm-equivalent residue 244 (p-IR48 F246L) or residues 170, 176, 180, and 244 (p-IR48 T172S/M178L/L182M/F246L) were switched with those in p-IR23. These enzymes were expressed, purified, and tested for conversion in the reductive amination of cyclohexanone with two equivalents of aniline at several time points alongside purified wild-type p-IR23 and p-IR48 (Fig. 5B). The quadruple mutant of p-IR48 was unstable and gave negligible, but the F246L mutant gave increased conversion compared to the wild-type (6% versus 2% over 24 hours). This mutation also gave rise to an increase in the amount of alcohol by-product formed due to direct reduction of the ketone. Since the conversion to both possible products showed a threefold increase from the wild-type to the mutant, it is not clear whether the mutation has increased alcohol dehydrogenase activity at the expense of IRED activity or whether opening of the active site has resulted in higher activity in all capacities.
Fig. 5. Rational mutagenesis for reductive amination with aniline.
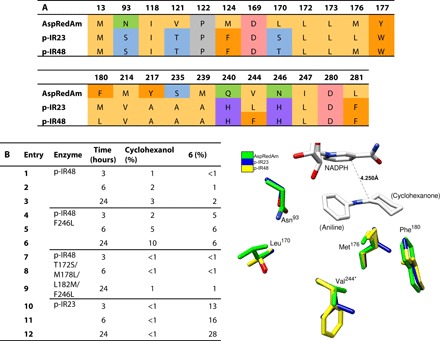
(A) Comparisons of theoretical extended active site residues for the reductive amination of cyclohexanone with aniline in AspRedAm to the analogous residues in the novel IREDs p-IR23 and p-IR48. (B) Conversions in the reductive amination of cyclohexanone with aniline with purified wild-type p-IR23, wild-type p-IR48, p-IR48 F246L, and p-IR48 T172S/M178L/L182M/F246L at different time points. Conversions were determined by GC-FID with comparison to a chemically synthesized standard. The adjacent image shows the positions of nonconserved residues with 6 docked in the active site, based on homology modeling. Residue 244* is attached to the second protein chain in the dimer.
We also considered whether the different residues at AspRedAm-equivalent position 210 (Tyr in p-IR23 versus Phe in p-IR48), which were not picked up using the analysis platform but had been shown to confer changes in activity for AspRedAm (2), might explain the difference in conversion with aniline. To explore this, we tested the corresponding F209Y mutant of p-IR48 for conversion to 6 under the same conditions. However, no conversion to the secondary amine was observed. Although this does not confirm that the list of contact points identified by the SmartScaffold tool is exhaustive, in this case, it appears that residue 210 is also not solely responsible for differences in conversion between the two wild-type enzymes.
The same sequence-structure analysis can be used to make similar comparisons with the other IRED and substrate combinations. For example, p-IR08 and p-IR13 both gave full conversion in the reductive amination of 4-hydroxyphenylacetone with cyclopropylamine to yield 11, yet p-IR07 and p-IR28 gave only 30 and 11% conversion respectively. Here, the results of homology modeling with AspRedAm and docking with 11 indicated 41 residues that could potentially be in contact with the product, of which 23 are conserved across all five enzymes (table S3). Similarly, no single sequence motif appears to preclude activity toward methoxyacetone, although 10 of 27 identified contact point residues are not shared by any other active enzymes in the panel.
Last, noting that the novel IREDs from plants have low sequence identity with AspRedAm and fall into a distinct clade on the phylogenetic tree, we used homology modeling to investigate their structure. p-IR14 from Arabidopsis thaliana shares 26% sequence identity to the β-hydroxyacid dehydrogenase from the same organism (β-HADAt; PDB code: 3DOJ) but only 18% sequence identity with AspRedAm. Although these enzymes showed markedly lower soluble expression in E. coli, several including p-IR14, p-IR-31, and p-IR56 gave >90% conversion in the reductive amination of cyclohexanone with cyclopropylamine. Crucially, this transformation requires that the enzyme be chemoselective for the imine over the cyclohexanone, direct reduction of which would yield cyclohexanol. Not only do these enzymes have different amino acids in all areas of their structure proposed to be important for catalysis, but on the basis of homology modeling with β-HADAt, they also appear to have a different quaternary structure to previously characterized IREDs such as AspRedAm (fig. S5). Rather than interlocking, the two subunits sit parallel, a structural difference that had previously been proposed to distinguish β-hydroxyacid dehydrogenases from IREDs (32). These results, along with recent mutagenesis studies performed by Nestl and co-workers (35), indicate that IREDs may not require domain swapping to exhibit their unique catalytic properties.
Reductive amination with ArpDHII from S. argillaceus
In the course of this study, an IRED (ArpDHII) was identified in the biosynthesis of polyketide alkaloids in S. argillaceus (26). This enzyme has 36% identity with AspRedAm, more than p-IR23, p-IR48, and many of the other active biocatalysts for reductive amination discussed herein (fig. S6). On the basis of the data obtained through screening, we came to believe that reductive amination catalysis is a promiscuous activity common to almost all IREDs and felt that similar conversions being obtained using an IRED with a known cyclic imine natural substrate would support this conclusion. Purified ArpDHII was therefore tested for conversion in several biotransformations that had previously been reported with purified AspRedAm, to compare its activity and substrate scope (table S4) (2). ArpDHII gave lower conversion than AspRedAm in 7 of 10 reactions, and no conversion was observed in the reductive amination of 1-indanone with 250 mM propargylamine. However, the S. argillaceus enzyme did give good conversion to 3 (93% after 18 hours), and the reductive amination of hydrocinnamaldehyde was successful with three different amine nucleophiles, giving >89% conversion to product in all cases.
DISCUSSION
In this study, we have provided a more complete description of the IREDs and RedAms as a diverse family of promiscuous dehydrogenases and improved tools to map IRED sequence-activity relationships. We identified 45 novel IREDs, 33 of which gave >10% conversion in reductive amination at a 2:1 amine:ketone stoichiometry, from a panel of 80 putative dehydrogenase enzymes including 25 from plant origin in the “twilight zone” of sequence identity with AspRedAm. Of these, 12 enzymes were found to catalyze the reductive amination of cyclohexanone with cyclopropylamine (>10% conversion), including one with just 17.1% sequence identity with AspRedAm (p-IR56 from Nicotiana attenuata).
Empirical characterization of the putative IREDs alongside 15 previously described IREDs across 12 substrate combinations in reductive amination led to the identification of biocatalysts for six new substrate combinations, including six enzymes that effectively (>70% conversion) catalyze the reductive amination of 4-methoxyphenylacetone at 100 mM ketone concentration and a 1:2 stoichiometry with cyclopropylamine. However, the reductive aminations of both 1-indanone and acetophenone appear to always require >10 equivalents of amine nucleophile to achieve good conversion to product. Of the novel IREDs, p-IR09 and p-IR16 have been identified as good scaffolds for engineering, since they are active, tolerate increased substrate concentrations, express well, and their close homology with AspRedAm means that their active site residues are more likely to be mapped accurately. Nonetheless, no single enzyme gave the best conversion for every substrate combination, and no single clade contains all the most active enzymes for a given transformation. For example, p-IR57 shares only 23.5% sequence identity with SkIRED, yet they gave comparable conversions to 3 and 11. For the reductive amination of cyclohexanone with aniline, where substrate inhibition limits turnover, a biphasic reaction system with cyclohexane has been shown to significantly improve total conversion to product. This highlights the significant role of reaction conditions in biocatalysis, a factor that is often not explored in initial screening of wild-type enzymes or engineered variants.
Despite their diversity, the active novel IREDs herein all catalyze reductive amination of the model substrate cyclohexanone, yet no single enzyme was found to give the highest conversion across all the tested reaction conditions and substrate combinations. Nonetheless, where resources are limited, we conclude that a lack of conversion in both the reduction of 1 and the reductive amination of cyclohexanone with cyclopropylamine might be a sufficient basis to exclude an enzyme from further screening.
On the basis of the comparison of conversions to product with positioning on a phylogenetic tree, it appears that the substrate scope of an IRED in terms of carbonyl compounds and amine nucleophiles might be controlled by different regions in the protein structure. A sequence-structure analysis has been used to map possible contact points for a subset of enzymes and substrate pairs, and interchange of residues between p-IR23 and p-IR48 highlighted a possible role of Leu/Phe244 (AspRedAm numbering) in catalysis and selectivity. This residue is also the only identified contact point for the reductive amination of 4-hydroxyphenylacetone with cyclopropylamine, which varies between IREDs p-IR07 and p-IR08, despite the considerable difference in conversion obtained with cell-free extracts containing the two enzymes (30 and 99%, respectively). Both were well expressed in E. coli, so it seems unlikely that the discrepancy would be due to a difference in biocatalyst concentration alone, although stability could play a role. However, while for 6, the higher-converting p-IR23 does not have Phe at this position, for 12, the presence of Phe at position 244 is associated with the higher conversion.
The approach described herein would be bolstered by an improved active site model based on crystal structures with complementary molecular dynamics simulations, to account for domain movements during catalysis (2, 27), and the use of this method to perform semirational engineering would most likely expedite the development of new IRED-catalyzed processes compared to a very broad directed evolution approach such as that described by Schober et al. (29). Where the goal is to improve an enzyme’s stability under process parameters, it can be necessary to target residues away from the active site, and tools to predict the results of these changes are still in their infancy. Even with our systematic approach, we have demonstrated that it is difficult to effect a change in activity of an IRED, especially with the concomitant effects of sequence changes on heterologous expression and stability.
Last, the study of the biosynthetic enzyme ArpDHII showed that an IRED that evolved to reduce cyclic imines can still effectively perform intermolecular reductive amination with certain substrates and suggests that “reductive aminase” activity observed to date in enzymes of fungal is likely to be a result of enzyme promiscuity. It appears that the majority of IREDs can usefully be applied in intermolecular reductive amination and this activity is not limited to a single set of active site residues. For the reductive amination of cyclohexanone with aniline, the highest conversions were obtained with enzymes from bacteria. The very high conversions to 3, and in all three reductive aminations of hydrocinnamaldehyde, suggest that ArpDHII shares the excellent chemoselectivity of AspRedAm. We believe that this is because ArpDHII evolved to only reduce the cyclic imine nigrifactin without also reducing the carbonyl in the hydrolyzed aminoketone form or the polyketide intermediates in argimycin biosynthesis (26). The occurrence of IRED-catalyzed reductive amination as a promiscuous activity could explain why it is a relatively slow process compared to other biocatalytic aminations and also why, in our case, attempts to engineer for an increase in reductive amination activity have also lead to increased ketoreduction.
In summary, the data presented herein demonstrate that IRED sequence-activity relationships are highly substrate dependent, although many residues close to and within the active site are highly conserved between enzymes. The knowledge that an enzyme with cyclic imine natural substrates can also catalyze intermolecular reductive amination provides a new avenue for biocatalyst discovery, and sequence-structure analysis offers a way to limit the scope of saturation mutagenesis experiments, which is of benefit in directed evolution experiments where chromatographic analysis can become cost-limiting. There remains a strong need for reliable high-throughput screening methods, with the potential benefits from IRED engineering having been robustly demonstrated (29). In addition, the fact that direct ketoreduction activity can be instilled in IREDs through point mutations supports earlier work, indicating that the chemoselectivity of dehydrogenases is tunable through mutagenesis (35). However, this type of promiscuity has implications in terms of other assays, since the activity of wild-type and mutant IREDs in reductive amination has frequently been measured using NADPH oxidation assays, in which imine reduction and ketoreduction are indistinguishable (2, 27, 36).
MATERIALS AND METHODS
Chemicals, media, and reagents
Commercially available chemicals and reagents were purchased from Sigma-Aldrich (Poole, Dorset, UK), Alfa Aesar (Karlsruhe, Germany), or Fluorochem (Hadfield, Derbyshire, UK) unless otherwise stated and used without further purification. High-performance liquid chromatography solvents were obtained from Sigma-Aldrich (Poole, Dorset, UK) or ROMIL (Waterbeach, Cambridge, UK), and gas chromatography (GC) gases were obtained from BOC Gas (Guildford, UK). Ammonium hydroxide (23 to 30 weight % in water) and benzylamine were purchased from Acros Organics (Geel, Belgium), and NADP+ was purchased from Prozomix (Haltwhistle, Northumberland, UK). Tris base, potassium dihydrogen phosphate, dipotassium phosphate, glucose, magnesium sulfate, sodium hydroxide, hydrochloric acid, and agarose were purchased from Thermo Fisher Scientific (Loughborough, Leicestershire, UK). Isopropyl-β-d-thiogalactopyranoside (IPTG), kanamycin sulfate, LB agar, LB broth, 2xYT broth, and autoinduction media were purchased from Formedium (Hunstanton, Norfolk, UK). Compounds 1 and 2 were prepared as described by Hussain et al. (14); compounds 3, 4, 8, and 13 were prepared as described by Aleku et al. (2); and compound 6 was prepared as described by France et al. (8). Where necessary, primers were purchased from Eurofins Analytik (Hamburg, Germany). Nucleotides, polymerases, phosphatases, restriction enzymes, and protein ladders were purchased from New England Biolabs (Ipswich, MA, USA). Miniprep and gel extraction kits were purchased from QIAGEN (Hilden, Germany). SYBR Safe dye and chemically competent E. coli BL21 (DE3) and DH5α cells were purchased from Invitrogen (Carlsbad, CA, USA). CDX-901 glucose dehydrogenase (GDH) was purchased from Codexis (Redwood City, CA, USA), and GDH-101 was supplied by Johnson Matthey (Cambridge, UK).
Software and sequence-structure analysis
Protein and DNA sequences were selected and downloaded from the UniProt (31) and National Center for Biotechnology Information online databases (37). Plasmid and primer design were performed using SnapGene software (GSL Biotech, Chicago, USA). Phylogenetic trees were produced using the Clustal Omega online tool (38), and dendrograms for the presentation of screening data were produced using the iToL (Interactive Tree of Life) online tool (European Molecular Biology Laboratory, Heidelberg, Germany) (39). The proprietary SmartScaffold approach from Johnson Matthey was used for the sequence and structure analysis. The approach involves numerous steps, including advanced, multitemplate homology modeling, flexible docking, and a custom functionality (SLAP) that defines the extended active site through the study of steric clashes and contacts between the protein model and a conformational ensemble of the best scoring docked poses. The homology modeling and docking use the Prime and Glide functionalities of Maestro 11.9 software (Schrödinger, Cambridge, MA, USA) with default parameters.
Selection and cloning of putative IRED sequences
Putative IRED sequences were selected either by a BLAST with the sequence of AspRedAm (2) as a query, across the full database, or limited to mammalian, cyanobacterial, plant, or extremophile genomes or by a search of the Kyoto Encyclopaedia of Genes and Genomes (40) for proteins with >30% sequence identity to AspRedAm, or a bit-score of greater than 50, within the genomes of organisms in the laboratory culture collection. Where possible, these enzymes were cloned from live cultures, and where this was unsuccessful, the gene was instead purchased as a synthetic construct in a generic vector. Two more were cloned from purchased genomic DNA, but the rest of the putative IREDs for the panel were purchased from GeneArt (Regensburg, Germany) as DNA strings that were codon-optimized for E. coli using their GeneOptimizer online tool (41). These were subcloned directly into a cut pET28b(+) vector purchased from Novagen (Darmstadt, Germany), using NdeI and XhoI restriction sites by means of an In-Fusion HD Cloning kit (Takara Bio, Kusatsu, Japan) according to the manufacturer’s protocol. The constructs were then transformed into chemically competent E. coli BL21 (DE3) and DH5α and were also transferred to Prozomix Ltd. for production according to their internal protocols (24). Sequencing of the colonies resulting from subcloning into pET-28b(+) revealed several deviations from the expected sequences (see section S2.4), and in the case of one enzyme from our in-house culture collection, the resulting protein sequence showed significant divergence from that listed in the UniProt database (31), most likely due to cloning from a different Streptomyces rimosus strain. The enzymes that were successfully cloned were assigned codes from p-IR01 to p-IR90. Of the 80 previously uncharacterized putative IREDs, 21 are of fungal origin. In addition to these, a further 22 IREDs from plants and 1 enzyme of mammalian origin were selected. The rest of the panel was made up of IREDs from a range of bacterial origins.
Expression and purification of IREDs
Single colonies of E. coli BL21 (DE3) cells transformed with the required plasmid were used to inoculate 5-ml precultures in LB medium supplemented with kanamycin (30 mg liter−1), which were grown overnight at 37°C and 250 rpm. For all IREDs except AspRedAm, which was produced as previously described (2), 2-liter baffled flasks containing 400 ml of LB medium were autoclaved and then supplemented with kanamycin (30 mg liter−1), before being inoculated with the overnight cultures. The cells were grown in a shaking incubator at 37°C and 250 rpm to an optical density at 600 nm of 0.6 to 0.8, whereby protein expression was induced by addition of IPTG (0.1 mM). Cell growth was continued at 20°C and 250 rpm overnight. The cells were then harvested by centrifugation [4°C and 2831 relative centrifugal field (rcf) for 20 min] and washed with potassium phosphate buffer [100 mM (pH 7.0)]. Wet cells were resuspended in 500% (v/w) Tris (hydroxymethyl) aminomethane hydrochloride (Tris-HCl) buffer [100 mM (pH 8.0)], and lysis was performed by ultrasonication (4°C for 20 min, 30-s pulse, 30-s off, repeat 20 times). The resulting suspension was centrifuged (39,191 rcf for 40 min), and the supernatant was used directly or flash-frozen in liquid nitrogen and either stored at −80°C or lyophilized and stored at −20°C until use.
Biocatalytic reduction of 1 with cell-free extract
To a pH-adjusted solution of d-glucose (10 μmol, 20 mM) and NADP+ (0.25 μmol, 0.5 mM) in tris-HCl buffer [100 mM (pH 8.0)] were added B. subtilis GDH cell-free extract (50 μl) and IRED cell-free extract (2.0 mg, 4 g liter−1) from solutions (10 g liter−1) in tris-HCl buffer [100 mM (pH 8.0)]. Imine 1 (5 μmol, 10 mM) was added from a 1 M stock solution in DMSO to give a total reaction volume of 500 μl. Reactions were incubated at 30°C and 250 rpm for 24 hours, after which they were quenched by addition of 5 M NaOH (50 μl) and extracted into tert-butyl methyl ether (2 × 500 μl). The organic fractions were combined, dried over anhydrous MgSO4, and analyzed for conversion by GC–flame-ionization detection (FID). The samples were subsequently derivatized by addition of triethylamine (5 μl) and acetic anhydride (5 μl), followed by incubation at 30°C and 250 rpm for 5 min. The reactions were quenched with 5 M NaOH (50 μl), and the phases were separated. The organic phase was dried over anhydrous MgSO4 and analyzed for enantiomeric excess by GC-FID. We estimate the limit of detection to be approximately 0.05 mM, or 0.5% conversion/1% ee in 10 mM transformation, and the limit of quantification to be closer to 1 mM or 10% conversion/20% ee for a typical transformation, based on an SD of 5%.
Typical procedure for screening in biocatalytic reductive amination with cell-free extract
To pH-adjusted solutions of amine nucleophile (10 μmol, 20 mM) in tris-HCl buffer [100 mM (pH 8.0)] in a 96-well plate were added d-glucose (50 μmol, 50 mM) and NADP+ (0.25 μmol, 0.5 mM) followed by GDH-101 cell-free extract (0.5 mg, 1.0 g liter−1) and IRED cell-free extract (2.0 mg, 4 g liter−1) from solutions (10 g liter−1) in tris-HCl buffer [100 mM (pH 8.0)]. The ketone substrate (5 μmol, 10 mM) was added from a 1 M stock solution in DMSO to give a total reaction volume of 500 μl. Reactions were incubated at 30°C and 300 rpm for 24 hours, after which they were quenched by addition of 1 M NaOH (300 μl) and extracted into tert-butyl methyl ether (2 × 500 μl) using a Hamilton MicroLab STAR liquid handling system. The combined organic phases were analyzed by GC-FID. Where derivatization was required, triethylamine (5 μl) and acetic anhydride (5 μl) were added, and the samples were incubated at 30°C and 250 rpm for 5 min. The reactions were quenched with 1 M NaOH (300 μl), and the phases were separated. Where cosolvents or substrate concentrations above 10 mM were investigated, all substrates were added neat or from a pH-adjusted aqueous solution. For transformations with 100 mM ketone, the substrate was added neat to avoid increasing the final percentage of DMSO.
Biocatalytic reductive aminations with ArpDHII
To a pH-adjusted solution of amine nucleophile in tris-HCl buffer [100 mM (pH 9.0)] were added d-glucose (12.5 μmol, 25 mM) and NADP+ (0.25 μmol, 0.5 mM) followed by B. subtilis GDH cell-free extract (50 μl) and purified ArpDHII (0.5 mg, 1 g liter−1) from solutions (10 g liter−1) in tris-HCl buffer [100 mM (pH 9.0)]. The ketone or aldehyde substrate (2.5 μmol, 5 mM) was added from a 1 M stock solution in DMSO to give a total reaction volume of 500 μl. Reactions were incubated at 30°C and 250 rpm for 24 hours, after which they were quenched by addition of 5 M NaOH (50 μl) and extracted into tert-butyl methyl ether (2 × 500 μl). The combined organic phases were dried over anhydrous MgSO4 and analyzed by GC-FID.
Biocatalytic reductive aminations with other purified IREDs and variants
To a solution of d-glucose (20 μmol, 20 mM) and NADP+ (0.5 μmol, 0.5 mM) in tris-HCl buffer [100 mM (pH 8.0)] were added B. subtilis GDH cell-free extract (100 μl) and purified IRED (1 mg, 1 g liter−1) from a solution (10 g liter−1) in potassium phosphate (KPi) buffer [100 mM (pH 8.0)]. Cyclohexanone (10 μmol, 10 mM) and aniline (20 μmol, 20 mM) were added from 1 M stock solutions in DMSO to give a total reaction volume of 1 ml. Reactions were incubated at 30°C and 250 rpm, with 200-μl aliquots taken at defined time intervals, quenched by addition of 5 M NaOH (50 μl), and extracted into tert-butyl methyl ether (2 × 200 μl). The combined organic phases were dried over anhydrous MgSO4 and analyzed by GC-FID.
Supplementary Material
Acknowledgments
We acknowledge G. Aleku for insights regarding enzyme discovery. Funding: S.L.M. thanks Johnson Matthey both for funding through an industrial CASE studentship and for additional contributions of resources and expertise. R.S.H. acknowledges the European Research Council for funding through the H2020 program. J.R.M. acknowledges the Industrial Biotechnology Innovation Centre (IBioIC) and Biotechnology and Biological Sciences Research Council (BBSRC) funding through a CASE studentship with Prozomix Ltd. J.I.R. acknowledges AstraZeneca for funding through an industrial CASE studentship. N.J.T. is grateful to the European Research Council for the award of an advanced grant (742987). Author contributions: S.L.M. designed the studies, selected and cloned enzymes, synthesized and characterized chemical standards, performed screening, and prepared the manuscript. A.P. helped guide the project and performed sequence-structure analysis. R.S.H. performed mutagenesis, expression, and purification and assisted with enzyme cloning. J.R.M. performed enzyme screening and sequence analysis. U.K., J.L.G., J.M.-S., and J.I.R. provided advice and assistance with selection and cloning of enzymes. D.W. performed bioinformatic analysis. S.B. assisted with enzyme screening. C.J.T. performed reaction optimization studies. B.D. and N.J.T. supervised and guided the project. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. All IREDs are commercially available through Prozomix Ltd. as lyophilized cell-free extracts. Plasmids encoding IRED of interest may also be obtained from Prozomix Ltd. or the University of Manchester. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay9320/DC1
REFERENCES AND NOTES
- 1.Mangas-Sanchez J., France S. P., Montgomery S. L., Aleku G. A., Man H., Sharma M., Ramsden J. I., Grogan G., Turner N. J., Imine reductases (IREDs). Curr. Opin. Chem. Biol. 37, 19–25 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Aleku G. A., France S. P., Man H., Mangas-Sanchez J., Montgomery S. L., Sharma M., Leipold F., Hussain S., Grogan G., Turner N. J., A reductive aminase from Aspergillus oryzae. Nat. Chem. 9, 961–969 (2017). [DOI] [PubMed] [Google Scholar]
- 3.Parkinson Study Group , Pramipexole vs. levodopa as initial treatment for Parkinson disease: A randomized controlled trial. JAMA 284, 1931–1938 (2000). [DOI] [PubMed] [Google Scholar]
- 4.Conde M. B., Efron A., Loredo C., De Souza G. R., Graça N. P., Cezar M. C., Ram M., Chaudhary M. A., Bishai W. R., Kritski A. L., Chaisson R. E., Moxifloxacin versus ethambutol in the initial treatment of tuberculosis: A double-blind, randomised, controlled phase II trial. Lancet 373, 1183–1189 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mortari M. R., Schwartz E. N., Schwartz C. A., Pires O. R. Jr., Santos M. M., Bloch C. Jr., Sebben A., Main alkaloids from the Brazilian dendrobatidae frog Epipedobates flavopictus: Pumiliotoxin 251D, histrionicotoxin and decahydroquinolines. Toxicon 43, 303–310 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Savile C. K., Janey J. M., Mundorff E. C., Moore J. C., Tam S., Jarvis W. R., Colbeck J. C., Krebber A., Fleitz F. J., Brands J., Devine P. N., Huisman G. W., Hughes G. J., Biocatalytic asymmetric synthesis of chiral amines from ketones applied to sitagliptin manufacture. Science 329, 305–309 (2010). [DOI] [PubMed] [Google Scholar]
- 7.Pushpanath A., Siirola E., Bornadel A., Woodlock D., Schell U., Understanding and overcoming the limitations of Bacillus badius and Caldalkalibacillus thermarum amine dehydrogenases for biocatalytic reductive amination. ACS Catal. 7, 3204–3209 (2017). [Google Scholar]
- 8.France S. P., Howard R. M., Steflik J., Weise N. J., Mangas-Sanchez J., Montgomery S. L., Crook R., Kumar R., Turner N. J., Identification of novel bacterial members of the imine reductase enzyme family that perform reductive amination. ChemCatChem 10, 510 (2018). [Google Scholar]
- 9.Roiban G.-D., Kern M., Liu Z., Hyslop J., Tey P. L., Levine M. S., Jordan L. S., Brown K. K., Hadi T., Ihnken L. A. F., Brown M. J. B., Efficient biocatalytic reductive aminations by extending the imine reductase toolbox. ChemCatChem 9, 4475 (2017). [Google Scholar]
- 10.Storer R. I., Carrera D. E., Ni Y., MacMillan D. W. C., Enantioselective organocatalytic reductive amination. J. Am. Chem. Soc. 128, 84–86 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Gallardo-donaire J., Hermsen M., Wysocki J., Ernst M., Rominger F., Trapp O., Hashmi A. S. K., Schäfer A., Comba P., Schaub T., Direct asymmetric ruthenium-catalyzed reductive amination of alkyl–Aryl ketones with ammonia and hydrogen. J. Am. Chem. Soc. 140, 355–361 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Mitsukura K., Suzuki M., Shinoda S., Kuramoto T., Yoshida T., Nagasawa T., Purification and characterization of a novel (R)-imine reductase from Streptomyces sp. GF3587. Biosci. Biotechnol. Biochem. 75, 1778–1782 (2011). [DOI] [PubMed] [Google Scholar]
- 13.Mitsukura K., Kuramoto T., Yoshida T., Kimoto N., Yamamoto H., Nagasawa T., A NADPH-dependent (S)-imine reductase (SIR) from Streptomyces sp. GF3546 for asymmetric synthesis of optically active amines: Purification, characterization, gene cloning, and expression. Appl. Microbiol. Biotechnol. 97, 8079–8086 (2013). [DOI] [PubMed] [Google Scholar]
- 14.Hussain S., Leipold F., Man H., Wells E., France S. P., Mulholland K. R., Grogan G., Turner N. J., An (R)-imine reductase biocatalyst for the asymmetric reduction of cyclic imines. ChemCatChem 7, 579–583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zumbrägel N., Merten C., Huber S. M., Gröger H., Enantioselective reduction of sulfur-containing cyclic imines through biocatalysis. Nat. Commun. 9, 1949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yao P., Xu Z., Yu S., Wu Q., Zhu D., Imine reductase-catalyzed enantioselective reduction of bulky α,β-unsaturated imines en route to a pharmaceutically important morphinan skeleton. Adv. Synth. Catal. 361, 556 (2019). [Google Scholar]
- 17.Zumbrägel N., Machui P., Nonnhoff J., Gröger H., Enantioselective biocatalytic reduction of 2H-1,4-benzoxazines using imine reductases. J. Org. Chem. 84, 1440–1447 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Heath R. S., Pontini M., Hussain S., Turner N. J., Combined imine reductase and amine oxidase catalyzed deracemization of nitrogen heterocycles. ChemCatChem 8, 117 (2016). [Google Scholar]
- 19.France S. P., Hussain S., Hill A. M., Hepworth L. J., Howard R. M., Mulholland K. R., Flitsch S. L., Turner N. J., One-pot cascade synthesis of mono- and disubstituted piperidines and pyrrolidines using carboxylic acid reductase (CAR), ω-transaminase (ω-TA), and imine reductase (IRED) biocatalysts. ACS Catal. 6, 3753–3759 (2016). [Google Scholar]
- 20.Ramsden J. I., Heath R. S., Derrington S. R., Montgomery S. L., Mangas-Sanchez J., Mulholland K. R., Turner N. J., Biocatalytic N-alkylation of amines using either primary alcohols or carboxylic acids via reductive aminase cascades. J. Am. Chem. Soc. 141, 1201–1206 (2019). [DOI] [PubMed] [Google Scholar]
- 21.Montgomery S. L., Mangas-Sanchez J., Thompson M. P., Aleku G. A., Dominguez B., Turner N. J., Direct alkylation of amines with primary and secondary alcohols through biocatalytic hydrogen borrowing. Angew. Chem. Int. Ed. 56, 10491–10494 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Tavanti M., Mangas-Sanchez J., Montgomery S. L., Thompson M. P., Turner N. J., A biocatalytic cascade for the amination of unfunctionalised cycloalkanes. Org. Biomol. Chem. 15, 9790–9793 (2017). [DOI] [PubMed] [Google Scholar]
- 23.France S. P., Aleku G. A., Sharma M., Mangas-Sanchez J., Howard R. M., Steflik J., Kumar R., Adams R. W., Slabu I., Crook R., Grogan G., Wallace T. W., Turner N. J., Biocatalytic routes to enantiomerically enriched dibenz[c, e]azepines. Angew. Chem. Int. Ed. 56, 15589–15593 (2017). [DOI] [PubMed] [Google Scholar]
- 24.Zawodny W., Montgomery S. L., Marshall J. R., Finnigan J. D., Turner N. J., Clayden J., Chemoenzymatic synthesis of substituted azepanes by sequential biocatalytic reduction and organolithium-mediated rearrangement. J. Am. Chem. Soc. 140, 17872–17877 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Matzel P., Gand M., Höhne M., One-step asymmetric synthesis of (R)- and (S)-rasagiline by reductive amination applying imine reductases. Green Chem. 19, 385–389 (2017). [Google Scholar]
- 26.Ye S., Braña A. F., González-Sabín J., Morís F., Olano C., Salas J. A., Méndez C., New insights into the biosynthesis pathway of polyketide alkaloid argimycins P in Streptomyces argillaceus. Front. Microbiol. 9, 252 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharma M., Mangas-Sanchez J., France S. P., Aleku G. A., Montgomery S. L., Ramsden J. I., Turner N. J., Grogan G., A mechanism for reductive amination catalyzed by fungal reductive aminases. ACS Catal. 8, 11534–11541 (2018). [Google Scholar]
- 28.Bornadel A., Bisagni S., Pushpanath A., Montgomery S. L., Turner N. J., Dominguez B., Technical considerations for scale-up of imine-reductase-catalyzed reductive amination: A case study. Org. Process Res. Dev. 23, 1262–1268 (2019). [Google Scholar]
- 29.Schober M., MacDermaid C., Ollis A. A., Chang S., Khan D., Hosford J., Latham J., Ihnken L. A. F., Brown M. J. B., Fuerst D., Sanganee M. J., Roiban G.-D., Chiral synthesis of LSD1 inhibitor GSK2879552 enabled by directed evolution of an imine reductase. Nat. Catal. 2, 909–915 (2019). [Google Scholar]
- 30.Rodríguez-Mata M., Frank A., Wells E., Leipold F., Turner N. J., Hart S., Turkenburg J. P., Grogan G., Structure and activity of NADPH-dependent reductase Q1EQE0 from Streptomyces kanamyceticus, which catalyses the R-selective reduction of an imine substrate. ChemBioChem 14, 1372–1379 (2013). [DOI] [PubMed] [Google Scholar]
- 31.The UniProt Consortium , UniProt: The universal protein knowledgebase. Nucleic Acids Res. 45, D158–D169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fademrecht S., Scheller P. N., Nestl B. M., Hauer B., Pleiss J., Identification of imine reductase-specific sequence motifs. Proteins 84, 600–610 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Emre M., Aarsland D., Albanese A., Byrne E. J., Deuschl G., De Deyn P. P., Durif F., Kulisevsky J., van Laar T., Lees A., Poewe W., Robillard A., Rosa M. M., Wolters E., Quarg P., Tekin S., Lane R., Rivastigmine for dementia associated with Parkinson’s disease. N. Engl. J. Med. 351, 2509–2518 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Velikogne S., Resch V., Dertnig C., Schrittwieser J. H., Kroutil W., Sequence-based in -silico discovery, characterisation, and biocatalytic application of a set of imine reductases. ChemCatChem 10, 3236–3246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lenz M., Fademrecht S., Sharma M., Pleiss J., Grogan G., Nestl B. M., New imine-reducing enzymes from β-hydroxyacid dehydrogenases by single amino acid substitutions. Protein Eng. Des. Sel. 31, 109–120 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Matzel P., Krautschick L., Höhne M., Photometric characterization of the reductive amination scope of the imine reductases from Streptomyces tsukubaensis and Streptomyces ipomoeae. ChemBioChem 18, 2022–2027 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Sayers E. W., Agarwala R., Bolton E. E., Brister J. R., Canese K., Clark K., Connor R., Fiorini N., Funk K., Hefferon T., Holmes J. B., Kim S., Kimchi A., Kitts P. A., Lathrop S., Lu Z., Madden T. L., Marchler-Bauer A., Phan L., Schneider V. A., Schoch C. L., Pruitt K. D., Ostell J., Database resources of the national center for biotechnology information. Nucleic Acids Res. 47, D23–D28 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., Thompson J. D., Higgins D. G., Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Mol. Syst. Biol. 7, 539 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Letunic I., Bork P., Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ogata H., Goto S., Sato K., Fujibuchi W., Bono H., Kanehisa M., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Raab D., Graf M., Notka F., Schödl T., Wagner R., The GeneOptimizer algorithm: Using a sliding window approach to cope with the vast sequence space in multiparameter DNA sequence optimization. Syst. Synth. Biol. 4, 215–225 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay9320/DC1