

Abstract
Acute disseminated encephalomyelitis (ADEM) is a demyelinating, autoimmune disease of the central nervous system (CNS). It causes motor and sensory deficits, altered mental status and other neurological symptoms. Though rarely fatal, it has been associated with residual motor and neurocognitive deficits. Our case consisted of a 4-year-old girl who presented with fatigue and unsteady gait after a respiratory illness. During her hospital course, she became progressively weaker and experienced seizures. Imaging showed sections of demyelination in the CNS, and appropriate treatment was started. Additional labs resulted in positive Mycoplasma pneumoniae serum serology. Antimyelin oligodendrocyte glycoprotein (anti-MOG) antibodies were also found, which is a risk factor for relapsing, multiphasic ADEM. To our knowledge, this is the first case of anti-MOG antibody-associated ADEM due to M. pneumoniae infection. Our patient has made a complete recovery. The parents only report slightly increased fatigue and irritability.
Keywords: infectious diseases, infection (neurology), neurology
Background
Acute disseminated encephalomyelitis (ADEM) is an acute, autoimmune, demyelinating disease of the central nervous system (CNS) thought to be due to a preceding stimulus, such as a viral infection or immunisation. It is considered one of the most common demyelinating disorders of childhood, and has an annual incidence of approximately 0.3–0.6 per 100 000 children.1 The median age of onset is 5–8 years with a slight male predominance with studies reporting male:female ratios between 1.2:1 and 2.6:1.1 2 Mortality is low (1%–3%), but residual motor and neurocognitive deficits may persist.1 The presentation of ADEM can be diverse with various combinations of constitutional symptoms, altered mental status, motor and sensory deficits, and other neurological symptoms. Several studies demonstrated that despite resolution of lesions on MRI, some children had lower cognitive deficits in certain categories such as attention and visuospatial/visuomotor functioning.3 4 Although ADEM is typically monophasic, the presence of antibodies against myelin oligodendrocyte glycoprotein (anti-MOG antibodies) have been shown to have predictive value in relapsing, multiphasic ADEM.5 The current literature only reports anti-MOG antibody-associated ADEM after an influenza or Epstein-Barr virus (EBV) infection.6 To our knowledge, no cases of anti-MOG antibody-associated ADEM have been reported due to an Mycoplasma pneumoniae infection. Here we present one such case.
Case presentation
A 4-year-old girl presented to her primary care clinic with fever for 6 days, mild cough and congestion as well as weakness for the past day. Her highest fever of 102 °F (38.9°C) was 2 days prior to presentation and had been improving since. Though afebrile at presentation, her father reported increased fatigue and unsteady gait as well as mild cough and congestion, but no other symptoms. On physical exam, the patient was unsteady while standing and stumbled when trying to walk. No other abnormal findings were noted. The patient was sent to the emergency department (ED), where on repeat physical exam, the patient had mild dysmetria, and her gait tilted to the left side. Strength was unable to be assessed. The patient was admitted to the paediatric ward for further workup and imaging.
Six hours after admission, the patient began to experience non-bloody, non-bilious vomiting and worsening lethargy, decreased global tone, and truncal instability. During an episode of emesis, the patient had a 10 s episode of upper extremity stiffness, tachycardia and bruxism that was concerning for a seizure. A second episode of stiffness lasting approximately 1 min occurred 1 hour later. The next morning, she had two episodes of urinary incontinence. On repeat physical exam, she was unresponsive to tactile stimulus in all extremities, had bilateral ptosis, weakness of the left peripheral face, flattening of the left nasolabial fold, and was noted to have 3+ bilateral upper and lower extremity deep tendon reflexes (DTR) with bilateral clonus (2–3 beats), and an upgoing great toe on the right side (positive Babinski reflex).
Investigations
Laboratory tests in the ED were notable for a leucocyte count of 18.9×109/L (normal=5–15.5 ×109/L) with 86% neutrophil (normal=25%–71%). A urinalysis was unremarkable, and nasopharyngeal respiratory syncytial virus (RSV), and influenza PCR tests were negative. A CT scan of the head with intravenous contrast showed no mass, enhancement or abscess. The description of the study by radiology noted a ‘localised volume of loss of L anterior frontal lobe’ and a ‘focal hypodensity in L frontal periventricular white matter’. An MRI study of the head showed non-enhancing T2 hyperintensity abnormalities in ventral/dorsal pons, right dorsal medulla, left middle cerebellar peduncle and bilateral superior cerebellar peduncles, as well as grey matter abnormalities extending from the mid-cervical to mid-thoracic spinal cord (figures 1–4). An electroencephalography showed normal drowsiness and sleep evidenced by theta and delta waves.
Figure 1.
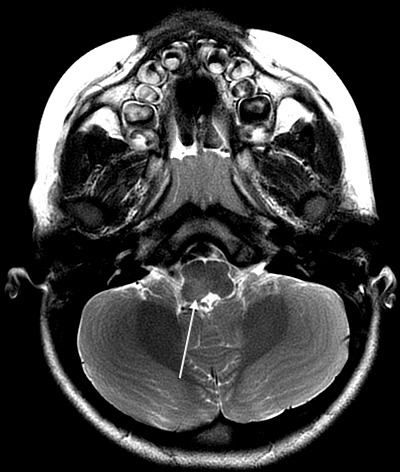
Axial T2 periodically rotated overlapping parallel lines with enhanced reconstruction (PROPELLER) image demonstrating hyperintensity in right dorsal medulla.
Figure 2.
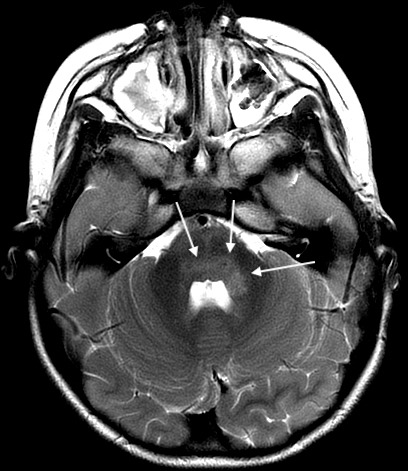
Axial T2 PROPELLER image demonstrating hyperintensities in bilateral dorsal pons and left middle cerebellar peduncle.
Figure 3.
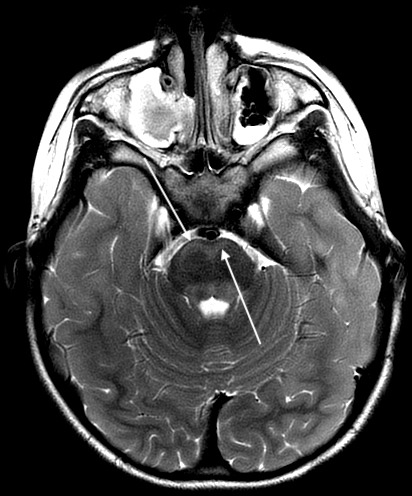
Axial T2 PROPELLER image demonstrating hyperintensity in bilateral ventral pons.
Figure 4.
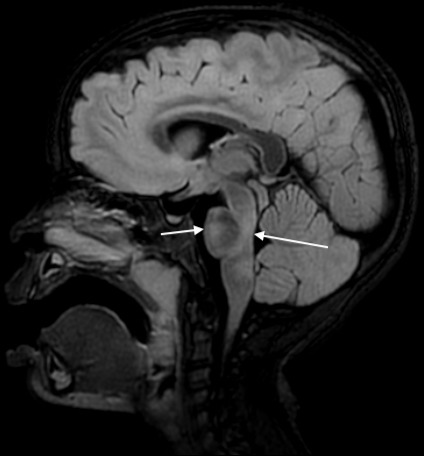
Sagittal fluid-attenuated inversion recovery (FLAIR) image demonstrating hyperintensities in ventral and dorsal pons.
Cerebrospinal fluid (CSF) showed lymphocytic predominant pleocytosis, with normal protein levels, negative bacterial cultures, negative PCR testing for enterovirus, herpes simplex virus (HSV) and varicella zoster virus (VZV). M. pneumoniae PCR from the CSF was negative. All studies performed on CSF were done at the same time. A qualitative serum M. pneumoniae IgM was positive and serum EBV serology was negative. On repeat MRI about a week later, the several hyperintensities, specifically in the medulla, dorsal pons, dorsal midbrain, left middle cerebellar peduncle and left superior cerebellar peduncle, previously reported, had become more hyperintense whereas those signal abnormalities in the cervical and mid-thoracic spinal cord had become less hyperintense. There were also new findings of non-enhancing T2 hyperintensities within the subcortical white matter in the frontal, parietal and temporal lobes (figures 5–11). At this time, serum antibodies for neuromyelitis optica and MOG were sent to an outside laboratory. Both were fluorescence-activated cell sorting (FACS) assays for IgG1 antibodies. The former was negative, but the latter was positive with a titre of 1:10 000 (normal <1:20).
Figure 5.
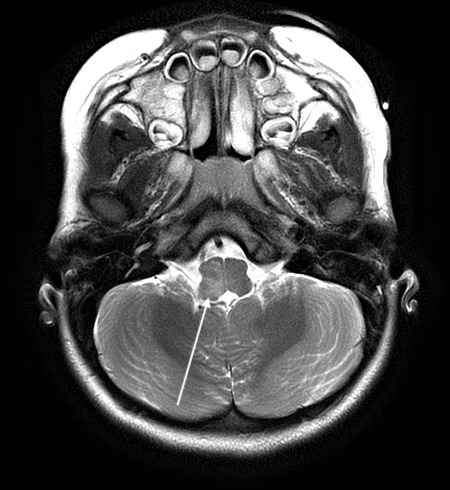
Axial T2 PROPELLER image demonstrating worsening of hyperintensity in right dorsal medulla.
Figure 6.
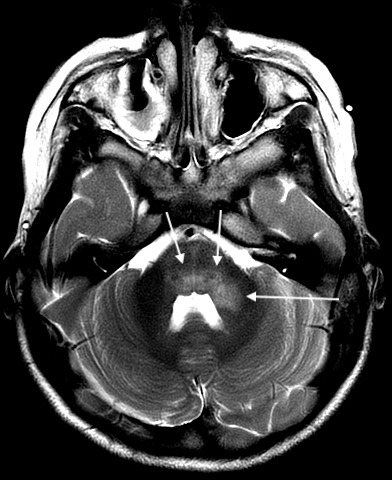
Axial T2 PROPELLER image demonstrating worsening of hyperintensities in bilateral dorsal pons and left middle cerebellar peduncle.
Figure 7.
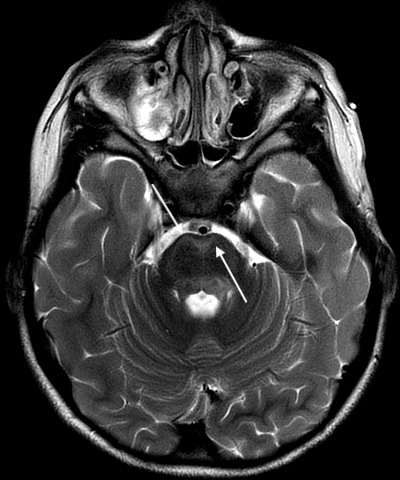
Axial T2 PROPELLER image demonstrating worsening of hyperintensities in bilateral ventral pons.
Figure 8.
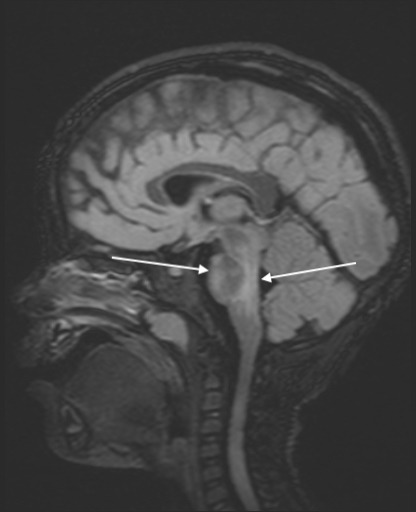
Sagittal FLAIR image demonstrating worsening hyperintensities in ventral and dorsal pons.
Figure 9.
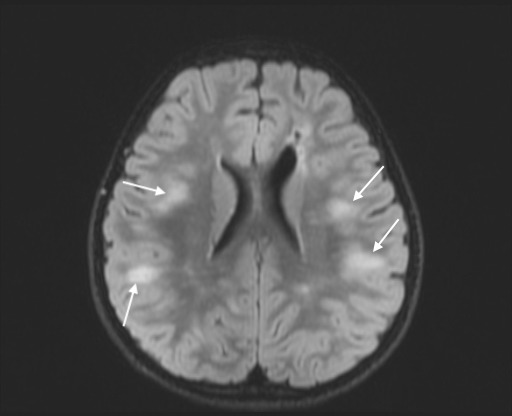
Axial FLAIR image showing multiple hyperintensities in subcortical white matter in parietal and temporal lobes.
Figure 10.
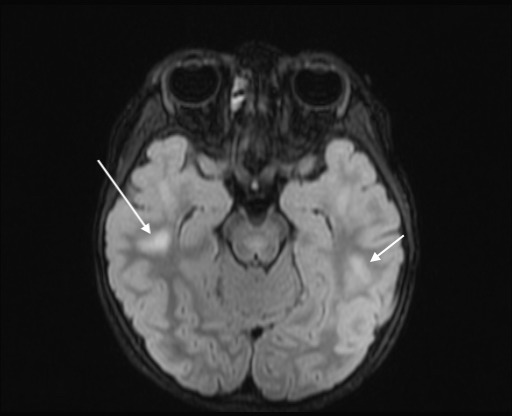
Axial FLAIR image showing multiple hyperintensities in subcortical white matter in bilateral temporal lobes.
Figure 11.
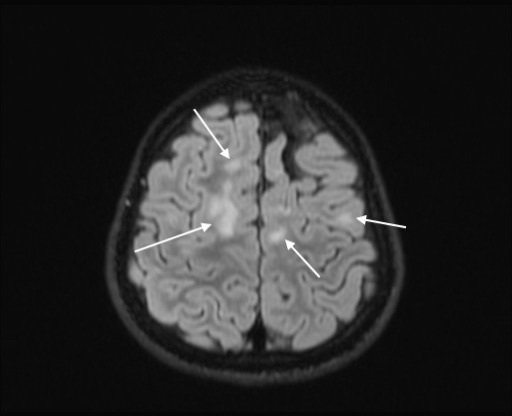
Axial FLAIR image showing multiple hyperintensities in subcortical white matter in frontal and temporal lobes.
Differential diagnosis
The differential diagnoses were very broad for this patient’s presentation. Acute cerebellar ataxia secondary to viral infection was initially considered due to the history of fever, respiratory symptoms and physical findings (mild dysmetria and gait ataxia) consistent with ataxia. Other diagnoses entertained during her admission included ADEM, acute flaccid myelitis (AFM), Guillain-Barre syndrome (GBS), encephalitis and toxin ingestion. Ultimately, acute cerebellar ataxia was discounted as the gait instability was thought to be due to weakness and not a primary cerebellar process. The presence of DTRs made GBS less probable. Infectious encephalitis secondary to several aetiologies such as M. pneumoniae, enterovirus, HSV, VZV was ruled out based on negative PCR testing from the CSF. Toxin ingestion was an unlikely aetiology as no substances in the patient’s household were identified which could have precipitated the symptoms. After the initial head MRI, AFM was the leading diagnosis based on the appearance and distribution of the grey matter lesions, but after the repeat MRI demonstrated hyperintensities within the white matter, ADEM was felt to be the most likely unifying diagnosis.
Treatment
Based on the MRI findings and concern for AFM, the patient was given intravenous immunoglobulin (IVIg) 1 g/kg per day for 2 days. Additionally, she received intravenous ceftriaxone and acyclovir, which were discontinued when the bacterial cultures and HSV and VZV PCR came back negative. After the positive serum M. pneumoniae IgM titre, the patient received a 5-day course of azithromycin. Additionally, based on the repeat MRI findings a week later, a 3-day course of intravenous methylprednisolone 20 mg/kg/day was given on hospital days 9–11.
Outcome and follow-up
After a 2-week hospitalisation, the patient was transferred to an outside facility for inpatient rehabilitation and was discharged 4 days later from the rehabilitation centre. On her initial follow-up 2 months later, she appeared to be at her baseline of normal health, with the exception of some ongoing difficulties with balance and a slight dysarthria. A repeat MRI performed 3 months after discharge reported significant improvement in both T2/FLAIR hyperintensities in subcortical white matter and T2 hyperintensities in pons, medulla and cerebellum. At a follow-up 6 months after initial presentation, her mother reported that the patient was at her neurological baseline except that she was more irritable and easily fatigued than prior to her illness. Because of positive anti-MOG antibody titre (and risk of multiphasic ADEM), the patient will be followed closely, but is generally doing well and has made a nearly complete recovery. A repeat anti-MOG titre was performed 6 months later. Though still positive, the titre was much lower (1:100). Based on the MRI findings and anti-MOG titre, the risk of multiphasic ADEM is considered low.
Discussion
The majority of ADEM cases have been associated with a preceding viral infection or an immunisation. Less commonly, ADEM occurs after a bacterial infection. Though rare, ADEM secondary to M. pneumoniae infection should be included in the differential for a patient with neurological symptoms following a respiratory disease given the ubiquity of M. pneumoniae infections. Of note, M. pneumoniae infection has been implicated in 5%–10% of patients with acute, febrile CNS disease.7
The pathogenesis of CNS disease due to M. pneumoniae infection is a topic of debate. On histopathological examination of white matter, ADEM is defined as ‘perivenular sleeves of demyelination associated with inflammatory infiltrates of myelin-laden macrophages, T and B lymphocytes, occasional plasma cells, and granulocytes’.1 M. pneumoniae has been postulated to contribute to the development of ADEM through both a direct neuroinvasion (M. pneumoniae DNA has been identified in CSF of patients via PCR) as well as by indirectly stimulating the production of steroid responsive autoantibodies directed against myelin proteins via molecular mimicry.7 A known example of this includes autoantibodies directed against a myelin glycoprotein, galactocerebroside, found in cases of M. pneumoniae-associated CNS disease.8 Direct neuroinvasion by M. pneumoniae may be associated with earlier onset of ADEM, while a delay in onset may be correlated with indirect disease caused by autoantibodies.8 Cerebrospinal fluid M. pneumoniae PCR failed to detect evidence of direct neuroinvasion in this patient. Instead our case may point towards an indirect mechanism because though some signal abnormalities were found on imaging early in the course of disease, MRI evidence of demyelination was not observed until later in the time course. One hypothesis of the indirect mechanism is molecular mimicry-induced production of antibodies against myelin proteins. This could lead to deposition of immune complexes in the small venules of the CNS leading to the perivenular inflammation seen in ADEM.7
An example of autoantibodies found in some cases of ADEM include anti-MOG antibodies (found on the surface of oligodendrocytes), most commonly associated with influenza and EBV infection. Similar to antibody production against galactrocerebroside, molecular mimicry caused by M. pneumoniae could induce production of antibodies against MOG. Interestingly, although anti-MOG antibodies in the setting of ADEM is typically transient, patients with persistent anti-MOG seropositivity are more likely to have a relapsing, multiphasic course of ADEM.5 9
As of 2013, MRI finding representative of demyelinating disease is required for ADEM diagnosis according to the International Pediatric Multiple Sclerosis Study Group.1 Findings can be varied, but are often diffuse, multiple, asymmetrical, T2 hyperintense lesions in both white and grey matter.10 As noted above, our patient showed both grey and white matter lesions throughout the CNS. Additionally, as in our patient, MRI findings may not be apparent on initial imaging.10 This could be due to a delayed indirect, autoimmune process that leads to ADEM.
Treatment for ADEM due to M. pneumoniae is based on empirical evidence from case reports. First-line treatment includes both immunosuppressive therapy such as an intravenous steroid plus an M. pneumoniae-specific antibiotic. In our case, this was methylprednisolone and azithromycin. Some patients have shown poor response to initial steroid therapy.11 Additionally, IVIg and plasmapheresis have been used if there is a poor response to intravenous steroids.11 Perhaps our patient showed excellent response to steroids because she was treated with two doses of IVIg previously when AFM was in the differential.
A brief note should be included about the serological testing used in this case. Cell-based assays such as FACS are considered the gold standard for the detection of anti-MOG-IgG1 antibody.12 Two studies provide good evidence about the sensitivity and specificity of the FACS. Using sera of 1109 patients, Waters et al found the assay to have a sensitivity of 24% and specificity of 100%.13 Similarly, a subsequent study by Waters et al compared three neuroimmunology laboratories and found all three had similar sensitivities (23.1%–27.5%) and specificities (98.1%–100%).14 The reported sensitivities and specificities for IgM serology for M. pnuemoniae infection vary between assays and studies. For example, Medjo et al found IgM serology to have a sensitivity of 81.82% and specificity of 100% when compared with IgG serology.15 On the contrary, Chang et al reported a lower sensitivity of 62.2% and specificity of 85.5%.16 To add to the discrepancies, Busson et al compared four IgM assays and found that although the sensitivities for all four were 100%, the specificities were lower (68.93%–80.58%).17 Clearly, the sensitivity and specificity of M. pneumoniae IgM serology are not uniformly reported. In this case, the combination of respiratory symptoms, positive M. pneumoniae serology and negative serology for other infectious causes points towards a true symptomatic M. pneumoniae infection as opposed to a false-positive or coincidental asymptomatic infection.
Though not a common presentation, secondary ADEM due to M. pneumoniae infection should be kept in the differential whenever a patient develops neurological symptoms following a respiratory illness. ADEM is not typically fatal but has been known to cause long-term disabilities. Treatment with intravenous steroids and an M. pneumoniae-specific antibody followed by IVIg and plasmapheresis if initial therapy fails should be implemented as soon as possible to prevent long-term consequences.
Learning points.
Though not common, acute disseminated encephalomyelitis (ADEM) should be on the differential for a patient with neurological signs after an infection.
Treatment for ADEM includes intravenous steroids and intravenous immunoglobulin as well as treatment of the underlying infection.
Follow-up with antimyelin oligodendrocyte glycoprotein antibody testing is necessary to test for relapsing, multiphasic ADEM.
Footnotes
Contributors: PB performed chart review and background research, drafted the manuscript and made edits. DP edited the manuscript and did background research. JI performed chart review and edited the document. LJC edited the document and approved the final version.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Parental/guardian consent obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1.Pohl D, Alper G, Van Haren K, et al. Acute disseminated encephalomyelitis. Neurology 2016;87:S38–45. 10.1212/WNL.0000000000002825 [DOI] [PubMed] [Google Scholar]
- 2.Cole J, Evans E, Mwangi M, et al. Acute disseminated encephalomyelitis in children: an updated review based on current diagnostic criteria. Pediatr Neurol 2019;100:26–34. 10.1016/j.pediatrneurol.2019.06.017 [DOI] [PubMed] [Google Scholar]
- 3.Gray MP, Gorelick MH, Encephalomyelitis AD. Acute disseminated encephalomyelitis.. Pediatr Emerg Care 2016;32:395–400. 10.1097/PEC.0000000000000825 [DOI] [PubMed] [Google Scholar]
- 4.Hahn CD, Miles BS, MacGregor DL, et al. Neurocognitive outcome after acute disseminated encephalomyelitis. Pediatr Neurol 2003;29:117–23. 10.1016/S0887-8994(03)00143-7 [DOI] [PubMed] [Google Scholar]
- 5.López-Chiriboga AS, Majed M, Fryer J, et al. Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-Associated disorders. JAMA Neurol 2018;75:1355–63. 10.1001/jamaneurol.2018.1814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakamura Y, Nakajima H, Tani H, et al. Anti-Mog antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: a case report. BMC Neurol 2017;17:76. 10.1186/s12883-017-0858-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.D’Alonzo R, Mencaroni E, Di Genova L, et al. Pathogenesis and treatment of neurologic diseases associated with Mycoplasma pneumoniae infection. Front Microbiol 2018;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer Sauteur PM, Jacobs BC, Spuesens EBM, et al. Antibody responses to Mycoplasma pneumoniae: role in pathogenesis and diagnosis of encephalitis? PLoS Pathog 2014;10:e1003983-e. 10.1371/journal.ppat.1003983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rossor T, Wright S, Duignan S, et al. Seizures at presentation are predictors of relapsing disease in children presenting with acute disseminated encephalomyelitis (S35.004). Neurology 2018;90:S35.004. [Google Scholar]
- 10.Kumar P, Kumar P, Sabharwal RK. Acute disseminated encephalomyelitis: case report and brief review. J Family Med Prim Care 2014;3:443–5. 10.4103/2249-4863.148145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Laila A, El-Lababidi RM, Hisham M, et al. A case of acute disseminated encephalomyelitis following Mycoplasma pneumoniae infection. IDCases 2018;12:41–3. 10.1016/j.idcr.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jarius S, Paul F, Aktas O, et al. Mog encephalomyelitis: international recommendations on diagnosis and antibody testing. J Neuroinflammation 2018;15:134. 10.1186/s12974-018-1144-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Waters P, Woodhall M, O'Connor KC, et al. Mog cell-based assay detects non-MS patients with inflammatory neurologic disease. Neurol Neuroimmunol Neuroinflamm 2015;2:e89. 10.1212/NXI.0000000000000089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Waters PJ, Komorowski L, Woodhall M, et al. A multicenter comparison of MOG-IgG cell-based assays. Neurology 2019;92:15. 10.1212/WNL.0000000000007096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Medjo B, Atanaskovic-Markovic M, Radic S, et al. Mycoplasma pneumoniae as a causative agent of community-acquired pneumonia in children: clinical features and laboratory diagnosis. Ital J Pediatr 2014;40:104. 10.1186/s13052-014-0104-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang H-Y, Chang L-Y, Shao P-L, et al. Comparison of real-time polymerase chain reaction and serological tests for the confirmation of Mycoplasma pneumoniae infection in children with clinical diagnosis of atypical pneumonia. J Microbiol Immunol Infect 2014;47:137–44. 10.1016/j.jmii.2013.03.015 [DOI] [PubMed] [Google Scholar]
- 17.Busson L, Van den Wijngaert S, Dahma H, et al. Evaluation of 10 serological assays for diagnosing Mycoplasma pneumoniae infection. Diagn Microbiol Infect Dis 2013;76:133–7. 10.1016/j.diagmicrobio.2013.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]