

A microcapsule platform was constructed for modulating immunization microenvironments and potentiating vaccination against cancer.
Abstract
Therapeutic cancer vaccines that harness the immune system to reject cancer cells have shown great promise for cancer treatment. Although a wave of efforts have spurred to improve the therapeutic effect, unfavorable immunization microenvironment along with a complicated preparation process and frequent vaccinations substantially compromise the performance. Here, we report a novel microcapsule-based formulation for high-performance cancer vaccinations. The special self-healing feature provides a mild and efficient paradigm for antigen microencapsulation. After vaccination, these microcapsules create a favorable immunization microenvironment in situ, wherein antigen release kinetics, recruited cell behavior, and acid surrounding work in a synergetic manner. In this case, we can effectively increase the antigen utilization, improve the antigen presentation, and activate antigen presenting cells. As a result, effective T cell response, potent tumor inhibition, antimetastatic effects, and prevention of postsurgical recurrence are achieved with various types of antigens, while neoantigen was encapsuled and evaluated in different tumor models.
INTRODUCTION
As a new therapeutic modality, immunotherapy has elicited much interest and shown potential for treating cancers (1–3). One of the most attractive immunotherapy strategies is cancer vaccination, which depends on the uptake of antigens, and the activation and lymph node homing of professional antigen-presenting cells (APCs) to elicit T cell responses (4, 5). The final stage is recognition and elimination of tumor cells by effector T cells (6). However, the direct use of tumor antigens is ineffective by several factors that are attributable to unfavorable microenvironments in vivo (7). For example, if free antigens were quickly cleared by metabolic processes, then APCs would fail to capture enough cargo to process for presentation (8). Although antigen uptake can be improved by increasing the antigen dose, in this case, APCs are still unlikely to be activated enough to provide sufficient costimulatory signals to T cells (9). Internalized exogenous antigens are usually processed and then presented by major histocompatibility complex II (MHC-II) molecules, which are not responsible for inducing cellular responses to therapeutic tumor vaccines (10, 11).
The abovementioned limitations have led to the need for strategies to modulate immunization microenvironments for tumor vaccination. One popular approach is the development of Toll-like receptor agonists, such as CpG oligodeoxynucleotides (12, 13), monophosphoryl lipid A (MPLA) (14, 15), and flagellin peptides (16, 17). According to several reports, the cellular response is indeed ameliorated through the coadministration of these agonists (18, 19). An alternative is the development of nanoparticle delivery system, which has also attracted increasing interest (20, 21). With surface functionalization, nanoparticles can efficiently enhance cellular uptake, APC activation, and cross-presentation (22, 23). Unfortunately, these strategies remain unsatisfactory, since they only focus on one part of the immunization microenvironment. Furthermore, the U.S. Food and Drug Administration (FDA)–unapproved adjuvant components, complicated preparation processes, and frequent vaccination requirements also detract from successfully moving these strategies from the bench to the clinic (24, 25). Therefore, developing a facile approach for constructing a versatile platform that can synergistically modulate more aspects of immunization microenvironments (such as antigen release kinetics and APC recruitment features) remains necessary but challenging (26).
In light of these considerations, we herein constructed self-healing microcapsules to modulate the immunization microenvironments that are more ideal for anticancer vaccination. Briefly, antigen molecules were efficiently loaded into polylactic acid (PLA)–based gigaporous microspheres in a postdiffusion manner, and through a mild sealing process, the superficial pores were able to heal, yielding antigen-loaded microcapsules. After vaccination, these microcapsules remained at the injection site and formed an antigen depot. Accompanying microcapsule degradation, antigen internalization was consistently increased because of coordinated sustained antigen release and APC recruitment. During this process, lactic acid sourced from the degraded products created a favorable acidic environment, which facilitated antigen uptake, cross-presentation, APC recruitment, and APC activation. These elaborately modulated microenvironments synergistically induced a flow of activated APCs that homed to the lymph nodes and continuously induced T cells to attack tumor cells. Using various types of antigens [ovalbumin (OVA) protein, mucin 1 (MUC1) peptide, and neoantigen], tumors (lymphoma, melanoma, and breast cancer), and models (primary growth, metastasis, and postsurgical recurrence), we systematically verified the superior therapeutic effects and safety of using these self-healing microcapsules for potent cancer vaccinations.
RESULTS
Self-healing microcapsules facilitated antigen loading
Reducing rapid antigen clearance in vivo is a prerequisite for potent vaccination (27, 28). Although alum fails to induce cellular response (29, 30), its distinctive capacity for detaining antigen at the vaccination site provide a good hint (31). One promising alternative adjuvant is microcapsule, wherein the antigen can be loaded and protected from degradation (32). In a typical microencapsulation process, the antigen should be dissolved with a polymer in an organic solvent, which is followed by the emulsification through mechanical stirring or homogenization. Such a complicated and harsh process can significantly compromise antigen stability and loading content, which is unfavorable for vaccination (33, 34).
To address this problem, we developed a mild and efficient method for antigen microencapsulation (Fig. 1A). Inspired by our previous work (35), we first used a double-emulsion method to prepare gigaporous microspheres, wherein PLA (an FDA-approved polymer) was used as the main matrix, and poly(ethylene glycol)-b-poly-dl-lactide (PELA; an amphiphilic polymer) was used as the emulsifier. Using this method, the structural properties could be well controlled by tuning the emulsification power (fig. S1A), the osmotic gradient (fig. S1B), and the pore evolution time (fig. S1C). After optimization, the resulting microspheres exhibited numerous open pores (diameter ≈ 2 μm) on their surface (Fig. 1, A and B) with an interconnected porous network (pore size ≈ 5 μm) inside (Fig. 1, C and D), and the porosity reached 82% (fig. S1D). During the adsorption process, the gigaporous structure significantly facilitated massive antigens to penetrate the microspheres (fig. S1H). As “self-healing” is a unique phenomenon of PLA that occurs at temperatures close to PLA’s glass transition temperature (36, 37) (Tg = 41.42°C; fig. S1E), spontaneous rearrangement of the polymer chains was triggered by gentle infrared (IR) irradiation (fig. S1F). As a result, the pores on the surface healed enough to form microcapsules (Fig. 1, A′ and B′), and large amounts of antigens were encapsulated (Fig. 1, C′ and D′), with a loading content of up to 20% (fig. S1G). Unlike traditional preencapsulation methods, the key features of our new postencapsulation approach were a simple mixing process, a lack of antigen exposure to organic solvent, and mild processing conditions, which resulted in large improvements in the antigen stability and loading content.
Fig. 1. Strategy of using self-healing microcapsules to modulate immunization microenvironments for cancer vaccination.
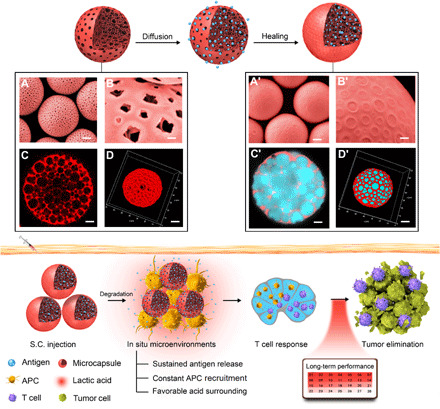
Through a diffusion manner and a healing process, the antigen could be efficiently loaded in the microcapsules. The corresponding characterizations of gigaporous microspheres and antigen-loaded microcapsules are displayed below: (A and A′) Scanning electron microscopy (SEM) images in a magnified view. Scale bars, 10 μm. (B and B′) SEM images in a local feature. Scale bars, 1 μm. (C and C′) Confocal laser scanning microscopy (CLSM) images in two-dimensional (2D) cut view. Scale bars, 5 μm. (D and D′) 3D reconstruction. Scale bars, 10 μm. Although without traditional molecular adjuvant, these microcapsules could still create in situ beneficial immunization microenvironments at the vaccination site, wherein sustained antigen release, constant APC recruitment, and favorable acidic surrounding collaborated effectively. As a result, a potent T cell response and tumor elimination could be achieved. S.C., subcutaneous.
Spatiotemporal synergy of antigen release and APC recruitment maximized antigen utilization
After successful microencapsulation, we next assessed the antigen release kinetics, which have been demonstrated to be highly correlated with the efficiency of generating immunity. As shown in Fig. 2A, the fluorescence of free Cy5-OVA quickly disappeared, within 1 day, due to rapid antigen clearance. Gigaporous microspheres with open pores prolonged the metabolization period of this mixed antigen to approximately 1 week, which was still unsatisfactory. In contrast, healed microcapsules formed an antigen depot (fig. S2A) and a sustained antigen release microenvironment, persisting over 3 weeks at the injection site. Initially, an antigen release of ~50% within 5 days provided an adequate supply of antigens for priming the immune system. With further degradation (fig. S2, B to G), most of the remaining antigens gradually released till 21 days, which afforded a sustained and sufficient antigen supply and thereby maintained the immune response.
Fig. 2. Spatiotemporal confluence of antigen release and APC recruitment maximized antigen utilization.
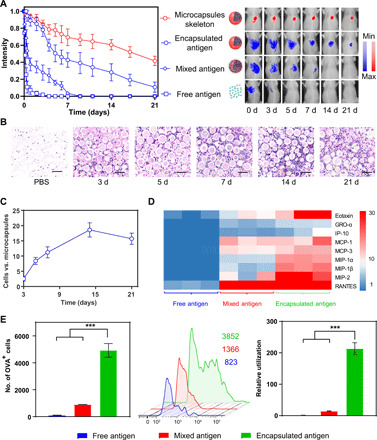
(A) Quantitative fluorescence intensity (left) and corresponding representative images (right) of antigen (blue)/microcapsules (red) at different time points. (B) Representative histological images of local tissues trapping microcapsules at different time points. PBS, phosphate-buffered saline. Scale bars, 50 μm. (C) The ratio of recruited cells versus local microcapsules. (D) Heat map representation of typical chemokine secretion at local injection site after 5 days of injection of different treatments. The color of the respective box in one row represents the value of the chemokine secretion in one sample compared with the normal expression level in untreated mice tissues. GRO-α, growth-regulated oncogene-alpha; IP-10, IFN-gamma-Inducible protein 10. (E) Comparison of number of OVA+ cells (left), intracellular OVA-Cy5 fluorescence intensity (middle; the number of mean fluorescence intensity was showed in corresponding colors), and utilization of OVA (right) after 5 days of injection of different formulations. The utilization of OVA at different formulations was calculated by formula: number of OVA+ cells × fluorescence intensity and normalized by utilization of free OVA. All bars represent means ± SD (n = 3).
In addition to the sustained antigen release, our microcapsules with suitable size (~50 μm) (fig. S3) also demonstrated their superior capacity to continuously attract APCs with great vigor (fig. S3, A and B). As shown in hematoxylin and eosin (H&E) images and the corresponding quantitative calculation, one microcapsule could attract an average of three cells by day 3 and up to 20 cells by day 14 (Fig. 2, B and C), which could be attributed to the up-regulation of chemokines (38, 39) [such as monocyte chemoattractant protein-1 (MCP-1), MCP-3, macrophage inflammatory protein-1α (MIP-1α), MIP-1β, MIP-2, eotaxin, and RANTES] (Fig. 2D and fig. S3, C, D, and H). Both the antigen release behavior and the chemotactic gradient clearly worked in synchrony: Sustained antigen release was accompanied by continuous APC recruitment. Such a spatiotemporal confluence could enhance the probability of APCs encountering the released antigen from the “microcapsule arsenal.” Compared to the scattered and starving APCs in the free antigen and mixed antigen groups at the vaccination site, the number of OVA+ APCs in the encapsulation group jumped to 5000 (Fig. 2E), and the corresponding number of internalized OVA molecules was also much higher. In this case, the relative antigen utilization of the encapsulation group was calculated to be 200-fold higher than that of the free antigen group (Fig. 2E). For dendritic cells (DCs) alone, this value was further amplified 800-fold because of their superior performance over macrophages at antigen internalization (fig. S4). Considering the function of DCs, this result may also benefit subsequent antigen presentation (40).
Acidic surroundings favored APC recruitment, antigen uptake, activation, and cross-presentation
Considering the long-term retention of microcapsules at the vaccination site, we next assessed local pH change during microcapsule degradation, as PLA, the main microcapsule matrix, was degraded into lactic acid (41, 42). To monitor this in situ, we administered microcapsules loaded with pH-sensitive dye and observed changes in their fluorescence with two-photon confocal laser scanning microscopy (CLSM) (Fig. 3A and fig. S2G). At the beginning of vaccination (day 0), the microcapsules were yellow, indicating a neutral microenvironment (NM) (pH 7.2), and they became green 3 days later (pH 6.5), suggesting that microcapsule degradation indeed created an acidic environment. The pH value was maintained over the following 2 weeks due to a balance between microcapsule degradation and in vivo metabolism.
Fig. 3. Local AM improved antigen uptake, cross-presentation, and APC activation.
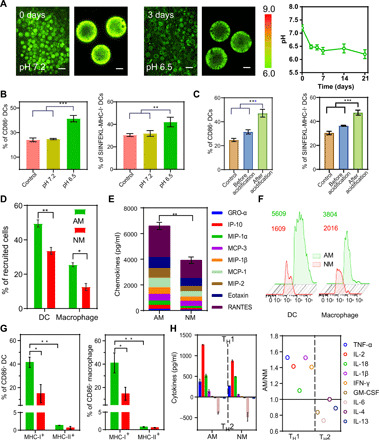
(A) Two-photon fluorescent images of pH-sensitive dye loaded microcapsules in vivo (left) and pH value quantification during microcapsule degradation (right). (B) Activation of DCs induced by acidic environment in vitro. (C) Activation of DCs before/after acidification of NM microcapsules in vitro. (D) Comparison of recruited DCs and macrophages number at day 5 under different microenvironments [AM represents an acidic microenvironment with pH 6.5, and NM represents a neutral microenvironment with pH 7.2)]. (E) In vivo concentrations of the typical chemokines under different microenvironments. (F) Comparison of antigen uptake amounts in DCs and macrophages after 5 days of injection (mean fluorescence intensity was shown in corresponding colors). (G) Comparative evaluation of cross-presentation (MHC-1) in DCs and macrophages. (H) In vivo concentration of the indicated cytokines from explanted microcapsule tissues at AM and NM (left) and their one-on-one ratios (right, value > 1, represented up-regulated cytokines; value < 1, represented down-regulated cytokines). GM-CSF, granulocyte-macrophage colony-stimulating factor. All bars represent means ± SD (n = 3).
Because lactic acid plays roles in many physiological activities (43–46), we investigated the effect on APCs of the aforementioned special acidic microenvironment (AM) derived from lactic acid. To this end, we first tuned the cell culture medium with lactic acid and evaluated the effect on DC activation and presentation in vitro. As shown in Fig. 3B, DCs cultured in pH 7.2 showed almost no change on their activation (indicated by CD86 expression) or presentation (indicated by MHC-I expression). On the contrary, these two indicators could be significantly improved upon pH 6.5, indicating lactic acid as a stimulant for DCs. Similar results were also observed, when we incubated DCs with the degradation product of microcapsules (fig. S5, A to D). For further verification, NM microcapsules were established as the control group by coencapsulating NaHCO3, which can neutralize lactic acid during degradation. Incubation with NM microcapsules had almost no effect on the DC activation and presentation (Fig. 3C). However, acidification with lactic acid again elevated the expression level of CD86 and MHC-I, thus confirming the stimulative effect sourced from the microcapsule degradation product (lactic acid) rather than the microcapsule skeleton.
The above results prompted us to evaluate the effect of acidic surroundings in vivo. As shown in Fig. 3D, both DCs and macrophages were recruited more efficiently in the AM group, since the AM outperformed the NM at chemokine induction at the vaccination site (Fig. 3E). Meanwhile, the special AM promoted antigen uptake. Compared to the NM group, the intracellular amounts of fluorescent antigen [detected by flow cytometry (FC)] in the AM group were 3.5-fold higher for DCs and 1.9-fold higher for macrophages (Fig. 3F), which could be attributed to enhanced macropinocytosis via the stimulation of acid-sensing ion channel actin polymerization and subsequent micropinocytosis (47, 48). The mild extracellular acidosis further promoted APC maturation. In the CD86+ APC population, the expression of MHC-I was more than 40-folds of MHC-II in the AM group, indicating the significant promotion of antigen cross-presentation by microcapsules. However, the MHC-I expression decreased more than half in the NM group, in turn, indicating the important role of AM for antigen cross-presentation (Fig. 3G and fig. S5E). In addition, the superiority of the AM was also reflected in T helper cell (TH1) preference, as higher concentrations of TH1-type cytokines and lower concentrations of TH2-type cytokines were present at the injection site in the AM group than in the NM group. Correspondingly, the AM/NM ratio surpassed one for each TH1-type cytokine while falling below one for most TH2-type cytokines (Fig. 3H). Such a TH1 inclination would facilitate subsequent cellular response (49, 50).
Potent cellular immune response
Having demonstrated the favorable immunization microenvironments created by our self-healing microcapsules, we next evaluated the subsequent T cell proliferation and activation after a single vaccination. As shown in Fig. 4A, a very distinguishing proliferation of OVA-specific CD8+ T cell proliferation could be observed after administration of the various vaccines. Compared with pure antigen (G2), an equivalent dose of antigen mixed with gigaporous microspheres (G3) resulted in a further improvement. Once OVA was encapsulated in microcapsules (G4), a large number of APCs would be effectively activated and then home to the lymph nodes (fig. S6, A and B). Correspondingly, we observed the most robust proliferation, with 93.8% of OVA-specific CD8+ T cells undergoing division in this scenario. Moreover, we also detected granzyme B, an important effector of cytotoxic T lymphocyte (CTL) cytotoxicity, in the splenocytes of immunized mice. Again, the G4 group had the highest percentage of granzyme B–secreting CD8+ cells (Fig. 4B), reflecting good activation.
Fig. 4. Potent cellular immune response against OVA model antigen.
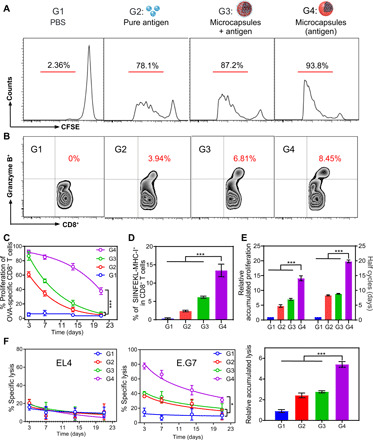
(A) Flow cytometric analysis of OVA-specific CD 8+ T cell (from OT-1 mice) proliferation in lymph nodes after different treatments [G1, PBS control; G2, pure antigen group (200 μg); G3, antigen mixed with gigaporous microspheres; G4, antigen encapsulated in healed microcapsules; the total antigen dose in G2 equaled one single administration in G3/G4]. (B) Comparison of CD8+ granzyme B+ T cells in splenocyte population analyzed by the FC. (C) Proliferation of OVA-specific CD8+ T cells (from OT-1 mice) at different time points (the curve was analyzed using a nonlinear regression). (D) Comparison of OVA-specific CD8+ T cell amounts using pentamer at day 14 after vaccination. (E) Calculation of accumulated proliferation performance (left) and corresponding half cycles (right; the half-cycles for G1 was not applicable). (F) In vitro killing assay showing the percentage of specific lysis using EL4 cells (mock control) or E.G7 cells (specific target) at different time points and the comparison of corresponding accumulated lysis performance. All bars represent means ± SD (n = 3).
A longer duration yielded even better results. The capacity of OVA-specific CD8+ T cell expansion in the G2 and G3 groups fell to mediocre immediately due to rapid clearance of the administered antigen, while the sustained antigen release in the G4 group significantly prevented such a quick decrease from occurring (Fig. 4C). Taking the 14th day for an example, G4 group increased the portion of OVA-specific T cells up to 13.5%, while this value in G2 and G3 groups was only 2.4 and 6.2%, respectively (Fig. 4D). On the basis of the nonlinear regression of these groups in Fig. 4C, we further obtained the half cycle of the reduced T cell expansion trend (Fig. 4E), which quantitatively reflected the decay rate. Compared with the short half cycles of G2 and G3 groups, the period was extended to ≈20 days in the G4 group. Correspondingly, the cumulative performance of OVA-specific CD8+ T cell proliferation in the G4 group was improved to 15-fold. These distinctive proliferation dynamics led to significant differences in the capabilities of the groups to lyse target cells. The G4 group exhibited the best cytotoxicity toward E.G7 lymphoma cells (a derivative of OVA-expressing EL4 cells), whereas no damage to EL4 cells was detected (Fig. 4F), indicating effective and specific clearance by OVA-specific CD8+ T cells. Moreover, the lysis rate in the G4 group remained above 30% after 3 weeks, once again demonstrating the superior long-term effects in the G4 group. As a result, the cumulative target cell lysis performance of the G4 group was far superior to that of the other groups (Fig. 4F), indicating the great promise that this formulation held for inducing continuous and effective therapeutic effects in vivo.
Safe and effective therapeutic
The abovementioned results prompted us to evaluate the therapeutic effect in an established E.G7-OVA tumor model. The mice were challenged with E.G7-OVA cells at the axillary and subsequently received single vaccination with different formulations (Fig. 5A). As shown in Fig. 5B, administration of antigen alone at a general dose (60 μg) in G2 group resulted in almost no inhibition of tumor growth, because of rapid antigen clearance. Although the therapeutic effect could be slightly ameliorated in the G3 group (equivalent dose), the survival time was extended only for 1 week. With the help of microcapsule in G4 group, the tumor development could be significantly delayed, and the survival rate after 30 days jumped to 100% (Fig. 5C). Nevertheless, this performance, in our opinion, was compromised by the general dose, since the amount of released antigen at each time point was diluted. In this aspect, we raised the dose to 200 μg (G4+ group) and further gained a great improvement (Fig. 5B) mainly due to the increased antigen cross-presentation (figs. S2E, S3, E and F, and S5E). Specifically, most mice remained tumor free, and only one death occurred during observation time. Similar satisfactory results were also observed in B16-MUC1 primary tumor model (fig. S7, A to C).
Fig. 5. Antitumor efficacy and safe evaluation in an E.G7-OVA tumor model.
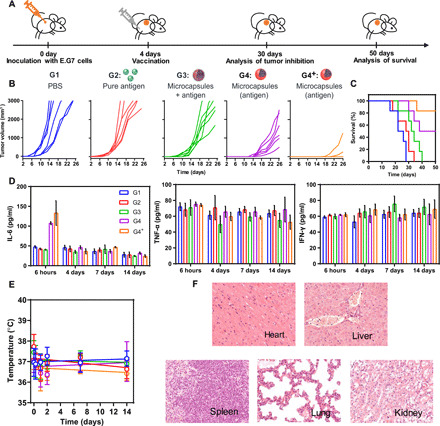
(A) The scheme of E.G7 tumor inhibition. (B) E.G7-OVA tumor volume development after different vaccinations [G1, PBS control; G2, pure peptides group (60 μg); G3, free antigen mixed with gigaporous microspheres group (60 μg); G4, antigen encapsulated in healed microcapsules group (60 μg); G4+, antigen encapsulated in healed microcapsules with an increased dose group (200 μg)]. Each line represents one animal. Mice were euthanized as their tumor volumes had reached 3000 mm3. (C) The survival time of immunized mice in E.G7-bearing mouse model. (D) Measurement of cytokine storm–related cytokines, interleukin-6 (IL-6), interferon-γ (IFN-γ), and tumor necrosis factor–α (TNF-α). (E) Body temperature changes of mice in different groups. (F) Representative H&E images of tissue slices in G4+. All bars represent means ± SD (n = 6).
We also performed a safety evaluation of the aforementioned vaccines. Considering the potential risk of cytokine storms during traditional immunotherapy, we first evaluated three typical indicators, tumor necrosis factor–α (TNF-α), interferon-γ (IFN-γ), and interleukin-6 (IL-6) (Fig. 5D). The TNF-α and IFN-γ values always kept very similarly to the untreated group after vaccination. Although IL-6 values in the microcapsule groups increased at the beginning, they remained in a safe range and eventually returned to baseline. Furthermore, biochemical markers, including aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN), lactic dehydrogenase (LDH), and alkaline phosphatase (ALP), all remained at normal levels (table S1). In addition, no obvious effects on body temperature were observed (Fig. 5E), and H&E staining of the main organs showed no obvious inflammatory infiltrates or toxicity (Fig. 5F). Together, these data demonstrated the in vivo biosafety of using our self-healing microcapsules for cancer vaccination.
Considering the universality and severity of cancer metastasis in the clinic, we then evaluated the antimetastatic effect of our vaccination platform. To this end, a malignant metastatic model was established through intravenous inoculation of B16-MUC1 cells, and the mice then received single vaccination of different formulations with equivalent dose (200 μg of antigen) (Fig. 6A). Eighteen days later, lung and kidney metastases had developed spontaneously and were clearly observed in tissue samples and histological sections (G1 group). Compared with the unsatisfactory antimetastatic effect in the G2 group, the G4 group exhibited a great inhibition of lung and kidney metastases (Fig. 6, B and C). With the combination of small dose of MPLA doped in PLA framework (G5), almost no metastases could be detected (Fig. 6, B and C), which was mainly due to the enhancement on DCs recruitment, activation, and cross-presentation (figs. S2F, S3F, and S5E). Correspondingly, we observed a stable body weight (Fig. 6D) and a prolonged survival time (Fig. 6E), which significantly outperform other counterparts.
Fig. 6. Antimetastasis performance in malignant B16-MUC1 mouse models.
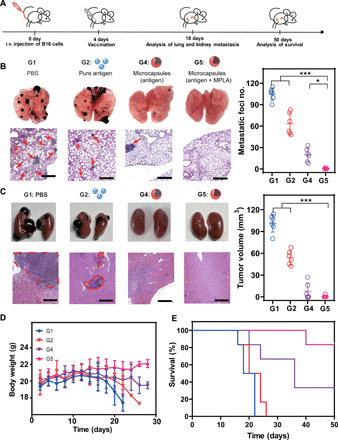
(A) Scheme of B16 antimetastasis model (several mice per group were dissected, photographed and sliced at day 18). G1, PBS control; G2, pure antigen group (200 μg); G4, antigen (200 μg) encapsulated in healed microcapsules; G5: antigen (200 μg) encapsulated in healed microcapsules with 3 μg of MPLA. (B) Representative lung photographs, H&E-stained lung slices, and corresponding quantification of lung metastasis nodules after different treatments. Scale bars, 500 μm. (C) Representative kidney photographs, H&E-stained kidney slices. Scale bars, 500 μm. Corresponding tumor volumes of metastasis nodules (right) after different treatments. (D) Body weight changes after different treatments. (E) The survival time of mice with different treatments. All bars represent means ± SD (n = 6). i.v., intravenous.
As an emerging precision medicine technology, neoantigen-based vaccination has shown bright prospects to fight against cancers, especially those highly malignant tumors (such as melanoma, glioblastoma, and triple-negative breast cancer) (51, 52). Encouraged by this, we also testified the feasibility of using our microcapsule platform-based neoantigen formulation for precision medicine against triple-negative breast cancer (Fig. 7A). For the prediction of neoantigen peptides, triple-negative breast tumor (4T1) tissue and normal tissue were sequenced and compared. After synthesizing and verifying these peptides (fig. S7, D, E, H, and I), different formulations were prepared for the single vaccination to 4T1 tumor-bearing mice with equivalent dose (200 μg). As shown in Fig. 7B, the proportion of polyfunctional CD8+ T cells in splenocytes increased in the sequence of G1, G2, AS04 (commercialized adjuvant mixed with neoantigen peptides), G4, and G5 groups and their specific response to the neoantigen peptides enhanced with a same order (fig. S7, F and G). The immunosuppressive tumor microenvironments in the above groups thus could be gradually ameliorated, which was verified by the increased CD8+ T cells with superior activity in tumor (Fig. 7C), while decreased regulatory T cells in tumor (Fig. 7D). Compared with the AS04 group, our microcapsule formulation without MPLA (G4 group) still showed a further improvement on tumor inhibition. Once low dose of MPLA was loaded in our microcapsule (G5 group), we achieved the most potent tumor inhibition effect (Fig. 7E).
Fig. 7. Evaluation of vaccination performance with neoantigen peptides.
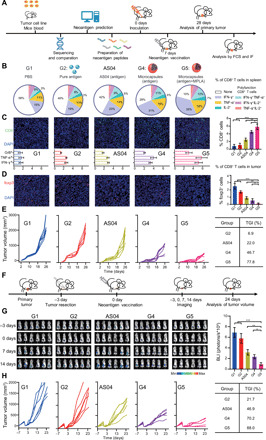
(A) Scheme of neoantigen production and vaccination in primary tumor model. G1, PBS control; G2, pure antigen group (200 μg); AS04, antigen (200 μg) mixed with AS04 adjuvant group. G4, antigen (200 μg) encapsulated in healed microcapsules; G5, antigen (200 μg) encapsulated in healed microcapsules with 3 μg of MPLA. IF, immunofluorescence. (B) Enumeration of the numbers of polyfunctional CD8+ T cells in splenocytes from mice immunized with various vaccine formulations. Polyfunctional: secreted two or more effector cytokines for killing tumor cells. (C) Representative immunofluorescence image of tumor slices, corresponding portion of CD8+ T cells, and phenotyping of infiltrated CD8+ T cells (by FCS analysis) in 4T1 tumor after different treatments. GrB+, granzyme B+; DAPI, 4′,6-diamidino-2-phenylindole. (D) Representative immunofluorescence image of tumor slices, corresponding portion of foxp3 T cells in 4T1 tumor after different treatments. (E) Individual tumor growth kinetics of primary tumors and tumor growth inhibition rates of different treatments. (F) The scheme vaccination in postsurgical recurrence model. (G) All bioluminescence images (BLI) of 4T1-luc tumors before/after surgical resection and quantitative statistics of BLI tumor burden at day 14. BLI values are represented as photons per second per square centimeter per steradian in regions of recurrent tumors (n = 6). (H) Individual tumor growth kinetics of postsurgical recurrence models and tumor growth inhibition (TGI) rates of different treatments (n = 6). All bars represent means ± SD.
As the neoantigen should be predicted from the patient tumor tissue, this vaccination modality may be preferred for the prevention of postsurgical recurrence. To simulate the practical application of neoantigen vaccine in clinical situation, we lastly established a tumor recurrence model by surgically resecting most of the primary 4T1 tumor in the mammary fat pad (Fig. 7F). As the 4T1 cells had been modified with luciferase expression, we therefore could monitor the therapeutic effect via the bioluminescence signal after single vaccination. Compared with the phosphate-buffered saline (PBS) group that relapsed with aggressive signal spreading over abdomen area, vaccination with neoantigen alone also failed to induce a potent inhibition on the tumor recurrence (Fig. 7G). Although this could be gradually ameliorated in AS04 and G4 groups, all mice still suffered from an obvious recurrence after 2 weeks and irrepressible tumor development (Fig. 7, G and H). On the contrary, very weak sign of Luc-4T1 and significant inhibition of recurrent tumor were observed in G5 group, indicating the most efficient inhibition of residual tumor cells compared with other counterparts.
DISCUSSION
Most local delivery systems shared a common feature that a complex combination of antigen/adjuvants/cytokines was loaded in the systems (53, 54). To enhance the final immune response, excess components and repeat immunizations were always applied, which not only caused the waste of vaccine components but also resulted in more concerns about the safety and clinical applicability. Meanwhile, an important reason why many vaccines failed to maximize their treatment capacity was the ignorance of complicated immunization microenvironments in situ. The importance of synergistically modulating immunization microenvironments revealed herein can supply a comprehensive and systematic view for most vaccine formulations as well as other local delivery systems in muscles and even tumors. As a result, our system was ensured to be fully used to exhibit potent immune performance with a single immunization.
Another feature of most local delivery carriers, such as cryogel and poly(lactide-co-glycolide) (PLG) matrices (55, 56), mainly only served as antigen deposit for sustained antigen/adjuvant/cytokines release in situ. In addition to this well-known capacity, our PLA microcapsules also found a new potentiality, which has not been reported yet. The AM, created by PLA degradation product, owned the superiority to significantly enhance cellular immune response via improved cell recruitment, antigen uptake, cross-presentation, and TH1-type cytokines secretion, which further explained the aforementioned good performance by the microcapsules. This finding not only offers us more inspirations to enhance the immune response but also provides more opportunities for PLA-based materials in immunology.
Compared with those fashionable particles synthesized with new materials (57, 58), our microcapsules might seem mediocre at the first glance. However, we achieved the aforementioned superiorities with such an FDA-approved material and rational and ingenious design, which could definitely pave the way from bench to clinic. Meanwhile, the proposed unique postencapsulation and self-healing features of porous microcapsules also exhibited an easy-manipulative and efficient paradigm for constructing a versatile vaccine platform. In this aspect, various cargos, including different types of tumor antigens, exosomes, chemical drugs, nanoparticles, and even their combinations, can also be loaded to extend the applications to precise medicine, tissue engineering, and drug delivery. In addition, loading additional hydrophilic or hydrophobic adjuvants is also a simple task for our encapsulation system, the flexibility and efficiency mentioned above not only enables us to achieve desired immune intensity and duration for diverse immunotherapy applications but also guarantees convenience and safety.
In summary, we herein developed a novel and facile PLA microcapsule–based platform for high-performance cancer vaccination. The special self-healing feature provided a mild and efficient paradigm for antigen microencapsulation. After vaccination, the gradual degradation of microcapsules resulted in sustained release of encapsulated antigen, while the microcapsules with suitable size (~50 μm) could efficiently recruit APCs. Such a spatiotemporal synergy maximized antigen utilization. Notably, we also demonstrated that the acidic surrounding sourced from the degradation components played important roles on the APC recruitment, antigen uptake, APC activation, and cross-presentation. With the cooperation of these advantages, effective T cell response, tumor inhibition, antimetastasis, and postsurgical recurrence prevention were achieved after a single immunization, while side effects were not notable. Together, these results strongly demonstrated that our microcapsule system could effectively serve as a safe, high-performance platform for cancer therapeutic vaccines.
Although promising, several works remain before translating our self-healing microcapsule for vaccination from bench to clinic. Considering the robust antibody and immune memory responses that were observed (fig. S8, A and B), the possibility of developing prophylactic tumor vaccines should be evaluated. Meanwhile, given that this microcapsule-based vaccine has shown potent T cell response, combination with immune checkpoint inhibitors, such as programed death 1 (PD-1) antibody, is worthy exploring to avoid T cell exhaustion and further improve the therapeutic performance (fig. S8, C and D). In addition, the efficacy of our vaccine formulation should be further assessed in the humanized patient-derived tumor xenograft model, and related work with patient-derived tumor neoantigens is in progress.
MATERIALS AND METHODS
Experimental design
The objective of this study was to improve subcutaneous immune environment and enhance T cell response. To this end, we developed self-healing microcapsules as vaccine carrier, verified the synergistic effect of microcapsules in T cell response enhancement, and evaluated tumor inhibition loading with various antigens in three different tumor cells and five mouse tumor models, respectively. For each animal experiment, mice were randomized on the basis of tumor volumes. Investigators were blinded to treatment groups when monitoring tumor volumes and health status of mice. All tumor inhibition experiments were repeated at least twice. Statistical analyses were conducted when applicable and were included in the figure legends. All animal experiments were performed in compliance with guide of care and use of laboratory animals.
Materials and reagents
PLA [relative molecular mass (Mw), 12 kDa] and PELA (Mw 40 kDa) were purchased from Dai Gang Company (Shandong, China). Poly(vinyl alcohol) (PVA-217) was ordered from Kuraray (Tokyo, Japan). OVA, MPLA, and lactic acid were supplied by Sigma-Aldrich (St. Louis, MO, USA). SAPDRTRPAP (MUC1-specific MHC-I) peptide and SIINFEKL (OVA-specific MHC-I) peptide were synthesized by Ji Er Biochemical Company. Carboxyfluorescein diacetate succinimidyl ester (CFSE) dye, Micro BCA (bicinchoninic acid) protein assay kit, Tissue Protein Extraction Reagent (T-Per), and Halt protease inhibitor cocktail were purchased from Thermo Fisher Scientific (Waltham, MA, USA). ToxinSensor Chromogenic LAL (limulus amebocyte lysate) Endotoxin Assay Kit was purchased from GenScript (Beijing, China). The medium for DCs, splenocyte cells, and tumor cells culture were RPMI 1640 (Gibco, Carlsbad, CA, USA) with 10% (v/v) fetal bovine serum (FBS) (Gibco, Carlsbad, CA, USA). ProcartaPlex Mouse TH1/TH2 and Chemokine Panel 1 (20 plex) and all mouse antibodies enzyme-linked immunosorbent assay (ELISA) and fluorochrome-conjugated anti-mouse antibodies were obtained from eBioscience (San Diego, CA, USA), unless otherwise indicated. Recombinant mouse granulocyte-macrophage colony-stimulating factor and IL-4 were obtained from PeproTech (Rocky Hill, NJ, USA). Cy5, Cy7, Nilered, and seminaphthorhodafluor-1 (SNARF-1) were purchased from Fanbo Biochemicals Company (Beijing, China). Collagenase D and deoxyribonuclease (DNase) were purchased from Roche (Basel, Switzerland). IFN-γ enzyme-linked immunospot (ELISpot) was purchased from Mabtech. Mouse lymphocyte separation medium, concanavalin A, and brefeldin A were purchased from Solarbio (Solarbio Science & Technology Co. Ltd., Beijing, China). All other reagents were of analytical grade.
Cell line and animal
EL4 and E.G7-OVA (derivative of EL4) cells were provided by the State Key Laboratory of Biochemical Engineering (Beijing, China), and MS+B16 cell expressing MUC1 protein was provided by the National Engineering Laboratory for AIDS Vaccine and the Key Laboratory for Molecular Enzymology and Engineering (Changchun, China) (59, 60). OT-1 mice used were provided by the State Key Laboratory of Biochemical Engineering (Beijing, China).
C57BL/6 mice and Balb/c mice, 6 to 8 weeks of age, were obtained from Vital River Laboratories (Beijing, China). This study was performed in strict accordance with the Regulations for the Care and Use of Laboratory Animals and Guideline for Ethical Review of Animal (China, GB/T 35892-2018). All animal experiments were reviewed and approved by the Animal Ethics Committee of the Institute of Process Engineering (approval ID: IPEAECA 2016139).
Fabrication and characterization of microcapsule vaccination
The double emulsion and solvent extraction methods were used to prepare gigaporous PLA microspheres, as previously described with some modification. Briefly, 0.5 ml of 0.5% (w/t) NaCl was added into 2 ml of ethyl acetate containing 100 mg of compound (PLA and PELA). The primary emulsion was prepared by sonication (120 W; Digital Sonifier 450, Branson Ultrasonics Corp., Danbury, CT, USA) for 20 s in an ice bath, and then the prepared primary emulsion was added into 15 ml of 3% (w/t) PVA (external water phase), following homogenizing at 3000 rpm for 90 s (ULTRA-TURRAX, T18 basic, IKA, Germany). The obtained emulsion was poured into 30 ml of deionized water following magnetically stirred to evolution and presolidification at 100 rpm for 30 min, and then 500 ml of deionized water was added to solidify the microspheres. The solidified microcapsules were collected by centrifugation, washed three times with deionized water, and stored at 4°C for later use. In addition, MPLA-loaded microspheres were prepared by adding MPLA (10 μl of chloroform solution) into the oil phase before emulsion.
To prepare the closed microcapsules loading protein (OVA)/peptide (SAPDRTRPAP), protein/peptide was mixed with microspheres at 4°C overnight, followed by healing for 2 hours at ≈40°C using IR irradiation. The shape and surface morphology of gigaporous microspheres and healed microcapsules were observed using a JSM-6700F scanning electron microscopy (SEM) (JEOL, Japan). The inner morphology of microspheres before and after healing was characterized by CLSM TCS SP8 (Leica, Germany). The size distribution of microspheres and microcapsules was measured by laser diffraction using Mastersizer 2000 (Malvern, UK).
Microcapsules without traditional molecular adjuvant were used in the evaluation of in situ APC recruitment (Fig. 2), acid microenvironment (Fig. 3), cellular immune response (Fig. 4), and E.G7 antitumor inhibition (Fig. 5). Microcapsules with low dose of MPLA were prepared to testify the therapy effect of our vaccine platform in B16-MUC1 metastasis model (Fig. 6) and 4T1 primary tumor and recurrence model (Fig. 7).
Quality control of our vaccine formulations
The detection of residual solvents (ethyl acetate and chloroform)
For ethyl acetate, 30 m by 0.53 mm inner diameter (i.d.) column (DB-624) was used for gas chromatography (GC). The column oven temperature program was as follows: 50°C (5 min) to 10°C/min to 180°C (5 min). The injection temperature was 200°C, and the detector temperature was 250°C [flame ionization detector (FID)]. The head space was sampled as follows: The vial was kept at 80°C for 30 min, pressurized for 3 min, and injected for 1 min into the GC column. For chloroform, 60 m by 0.32 mm i.d. column (HP-1) was used for GC. The column oven temperature program was as follows: 70°C (10 min) to 20°C/min to 200°C (15 min). The injection temperature was 150°C, and the detector temperature was 250°C (FID). The head space was sampled as follows: The vial was kept at 80°C for 20 min, pressurized for 3 min, and injected for 1 min into the GC column. The residual of ethyl acetate was only 0.004%, which is much lower than the limit in United States Pharmacopeia 36 (USP36) (0.5%). Notably, not even a signal of chloroform was detected by GC, indicating no chloroform residue in our vaccine formulation.
The detection of endotoxin contamination
This assay was executed with the GenScript ToxinSensor Chromogenic LAL Endotoxin Assay Kit by following the corresponding protocols. The contents in G2, G3, and G4 groups were separately detected as ~0.13, ~0.19, and ~0.20 endotoxin units (EU), which were lower than the endotoxin limit (0.25 EU/ml in USP36).
Regent purity
The purity of PLA, OVA, and antigen peptide was more than 97%, and the regents used were analytically pure (analytical research grade).
Characterization of gigaporous microspheres and healed microcapsules
The inner pore size distribution and porosity of microspheres were determined by Mercury Injection Apparatus (AutoPore IV 9500, Micromeritics). For the glass transition temperature of mixture (PLA and PELA) measurement, lyophilized mixture was analyzed by differential scanning calorimetry (DSC Q2000). The process of antigen penetrating into microcapsule inner structures was observed by CLSM TCS SP8 (Leica, Germany).
Evaluation of loading efficiency for OVA
The microcapsules-encapsulated OVA content was determined in triplicate by incubating 3 mg of lyophilized microcapsules in 1 ml of 0.1 M NaOH solution under gentle shaking overnight. Protein concentration in the solution was determined using a Micro BCA protein assay kit (Pierce). The loading efficiency (LE) of the microspheres was calculated by the following equations
In vivo antigen release and polymer degradation behavior
To test the duration of OVA at different vaccination formulations and degradation period of polymer particles in vivo, OVA was labeled with water-soluble fluorescence probe Cy5-NHS ester (SE), and microcapsule was stained with hydrophobic near-IR fluorescence probe Cy7 during the process of preparing microspheres. Mice were injected subcutaneously with free Cy5-OVA (60 μg), Cy7-labeled microcapsules mixed with Cy5-OVA (60 μg) and Cy5-OVA (200 μg), Cy7-labeled microcapsules encapsulated Cy5-OVA (60 μg) and Cy5-OVA (200 μg), and Cy7-labeled microcapsules encapsulated Cy5-OVA (200 μg). At different time intervals, the mice were scanned using an in vivo imaging system FX Pro (Kodak) and euthanized to extract the tissue at injection site following to weight.
In vitro antigen release and polymer degradation behavior
To evaluate the influence of addition of adjuvant and different pH solution encapsulated in microcapsules on degradation and antigen release, OVA protein was prepared to 10 mg/ml and encapsulated into microcapsules. Then, the microcapsules were washed with deionized water and placed in 37°C. Microcapsules morphology was observed using SEM, and antigen release was determined using a Micro BCA protein assay kit at days 0, 0.5, 1, 3, 5, 7, 14, and 21.
Analysis of release mechanism in vivo
For the observation of antigen distribution in microcapsules in vivo during the period of metabolism, mice were euthanized at different time points after injecting microcapsules encapsulated Cy5-OVA. The implanted microcapsules were removed to view the changed morphology using SEM, and the resident antigen in microcapsules was visualized with CLSM.
Evaluation of degradability and cellular recruitment of microcapsules
Animals were injected with 3 mg of blank microcapsules, and subcutaneous nodules were explanted at various time points. The extracted tissues were weighted and extracted to test the protein concentration, according to the instruction mentioned in Materials and Methods (see the “In vivo chemokine analysis” section).
Biocompatibility experiment in vivo
To evaluate the fitness of recruited cells at injection site, microcapsules were injected into animals and extracted along with partial tissues at various time points. The tissues were cut into pieces, stained with a live-dead dye, and visualized using CLSM.
Identification of cellular recruitment at injection site
Animals were injected with 3 mg of blank microcapsules and subcutaneous-formed nodules were explanted at various time points. Then, nodules were paraffin-embedded, sectioned, and stained with H&E. The recruited cells and microcapsules at the injection site were observed using Vectra platform (Caliper Life Sciences, Hopkinton, USA) and quantified using Inform software (Caliper Life Sciences, Hopkinton, USA).
Activation of DCs in vitro
To evaluate activation of primary DCs resulting from microcapsules, bone marrow–derived cells were derived using standard techniques. Briefly, bone marrow cells were isolated from male C57BL/6J mice and cultured in RPMI 1640–based medium supplemented with 10% FBS, and 500 μg of microcapsules were added. At day 5, DCs were harvested and used for experiment. DC differentiation and activation were confirmed using the phycoerythrin (PE)–CD11c, PE-Cy7-CD80, APC-CD40, fluorescein isothiocyanate (FITC)–CD86, eFlour450–MHC-I, and BV605–MHC-II antibodies.
To investigate activation of primary DCs due to degradation components source from microcapsules, the microcapsules were incubated in cell culture medium at 37°C with 0.02% NaN3. Every week, we collected all supernatant by centrifugation and supplemented the same volume of cell culture medium. The supernatant together with antigen was then added into the DC culture medium for 24 hours, and the CD86 and MHC-I expressions were detected.
To evaluate the role of lactic acid and oligomers in primary DC activation, the concentration of lactic acid in each week and supernatant collected as above was determined using the Lactate Assay Kit. Standard procedure following the instructions was executed. Subsequently, equivalent lactic acid was added into culture medium according to different concentrations of supernatant. After 24 hours of coincubation with DCs and antigen, the CD86 and MHC-I expressions were detected.
To clarify the important role of acid environment in primary DC activation, pH values of DC culture medium were tuned to 7.2 and 6.5 by lactic acid. After 24 hours of coincubation with DCs and antigen, the CD86 and MHC-I expressions were detected. Similarly, primary DCs were cocultured with NM microcapsules for 24 hours, and then the pH was tuned to 6.5. After 24 hours, the CD86 and MHC-I expressions were detected.
DCs and macrophages recruitment in lymph nodes
The draining lymph nodes (dLNs) were harvested and measured from the immunized mice at different time points, and single-cell suspensions from dLNs were prepared by mechanical disruption and isolated from red cell using red blood cell (RBC) lysis buffer. Cells were collected by centrifugation and counted using a handheld automated cell counter (Millipore). Statistic cells were stained with FITC-CD11c and APC-F4/80 antibodies for analyzing the number of DCs and macrophages, and PE-CD40, APC-Cy7-CD80, eFlour 450–MHC-II, and BV605–MHC-I antibodies were used for analyzing mature extent of DCs and macrophages in dLNs. The photograph in fig. S6 was shot by X.X. (Institute of Process Engineering, Chinese Academy of Sciences).
Antibody activation
To evaluate the serum antibody level, blood samples were harvested on days 14, 28, 35, and 42 after immunization with different vaccination formulations (G1, PBS; G2, 200 μg of OVA; G4, 200 μg of OVA encapsulated within 3 mg of microspheres; G5, 200 μg of OVA encapsulated 3 mg of microcapsules loading 3 μg of MPLA, n = 6), and anti-OVA immunoglobulin G levels were determined by ELISA analysis.
Evaluation of antigen utilization in vivo
Sixty micrograms of free antigen Cy5-OVA or mixed with microspheres or encapsulated into microcapsules were injected into mice, respectively. Tissues warping microcapsules were extracted at day 5. Tissues were processed through mechanical disruption, washed, and suspended in cold PBS; the resulting single-cell suspension was centrifuged to remove supernatant. The bottom cells were resuspended in 1 ml of an enzyme cocktail composed of collagenase D (1 ml/ml) and recombinant DNase I (100 U/ml) in RPMI 1640 for 30 min at 37°C, following wash with PBS and filtered through a 20-μm cell strainer. Cells were then stained with FITC-conjugated CD11c and eFlour 450–conjugated F4/80 to specifically identify DCs and macrophages analysis. The percentages of OVA+ cells, CD11c+OVA+ cells, and F4/80+OVA+ cells were measured using a BD LSR Fortessa FC and analyzed using FlowJo software (version 7.6).
Measurement of pH change in situ
To monitor the pH changes inside microcapsules during the degradation in vivo, microcapsules loading probe SNARF-1 were injected subcutaneously into mice. The probe was excited at 488 nm and emitted at 580- and 640-nm wavelengths. The ratio (I 640/I 580) variations with time at injection site were accessed using two-photon confocal microscope at days 0, 3, 5, 7, 14, and 21. To evaluate the real pH change by the ratio variations, a standard curve was made between ratio variations and assigned pH medium (6.0, 6.5, 7.0, 7.5, 8.0, 8.5, and 9.0) with the same detected conditions.
Activation of DCs and macrophages in vivo
To verify the cell recruitment and activation ability of AM created by microcapsules, microcapsules encapsulating 20 mM NaHCO3 group were evaluated as a control experiment to establish a neutral microenvironment in vivo, and then microcapsules loaded with PBS and NaHCO3 were subcutaneously administrated into animals, respectively, and extracted at day 5. The subcutaneous tissues were cut off extra parts to ensure same weight of tissues. The single-cell suspension was prepared following the method described before. Cells were pelleted and counted using a handheld automated cell counter (Millipore), then FITC-conjugated CD11c and APC-conjugated F4/80 antibodies were managed to analyze DCs and macrophages recruitment, and APC-Cy7-conjugated CD86, eFlour 450–conjugated MHC-I, and Percp-Cy5.5-conjugated MHC-II stains were conducted for DCs and macrophages maturation analysis.
To investigate the influence of antigen dose, adjuvant and particle size on the APC recruitment, activation, and cross-presentation, different vaccine formulations were prepared and injected into C57BL/6J mice. Then, animals were euthanized, and recruited cells were harvested for staining and analyzed by FC at days 7, 14, and 21. Accordingly, subcutaneous nodules were sliced and stained by H&E for histological observation, counted by Inform software, and analyzed.
Comparison of cytokine and chemokine secretion at AM and NM in vivo
To determine concentration of cytokines and chemokines in vaccination site, tissue samples were extracted from the animals implanted with microcapsules loading PBS and NaHCO3 after 5 days, respectively, and followed the method described before to extract the tissue protein. Then, protein was analyzed using ProcartaPlex Mouse TH1/TH2 and Chemokine Panel 1, according to the manufacturer’s instructions.
In vivo chemokine analysis
Six- to eight-weeks-old male C57BL/6 mice were injected with 60 μg of free OVA or mixed with microspheres or encapsulated into microcapsules, respectively. All vaccine formulation was antigen-dose equivalent. At day 5, the tissues at the injection site were extracted from the animals and sheared to fragment between 1 and 3 mm, followed by digesting using T-Per reagent and homogenized to debris with sonication. Protease inhibitor was added to protect the protein from endogenous proteases during the digest. Then, the supernatant of tissue debris solution was analyzed using ProcartaPlex Mouse Chemokine Panel 1, according to the manufacturer’s instructions.
Comparative antigen uptake between AM and NM in vivo
To determine the utilization of antigen in different pH microenvironments, 60 μg of Cy5-OVA in PBS or 20 mM NaHCO3 were encapsulated into microcapsules, respectively, following injection into animals. Tissues warping microcapsules with similar weight were extracted at day 5 to prepare single-cell suspension using the method before described. Cells were then counted and stained with FITC-conjugated CD11c and eFlour 450–conjugated F4/80 to specifically identify DCs and macrophages analysis. The percentage of OVA+ cells, CD11c+OVA+ cells, and F4/80+OVA+ cells were measured using a BD LSRFortessa FC and analyzed using FlowJo software.
Cytotoxicity activity of CTL
To evaluate antigen-specific CTL activity at various time points, spleens (n = 3) of immunized mice were isolated to prepare single-cell suspension. The splenocytes were stimulated with the SIINFEKL (OVA-specific MHC-I) peptide for 3 days in RPMI 1640 medium containing recombinant IL-2 (20 U/ml). Subsequently, these activated effector cells were incubated with mitomycin-treated E.G7 cells or EL4 target cells. The CTL activity was evaluated at 50:1 ratios of effector cells to target cells (E/T ratios) in an LDH cytotoxicity detection assay.
OVA-specific CTL cells proliferation induced by microcapsule vaccines
Separated OT-1 T cells were used to evaluate the proliferation of OVA-specific CD8 T cells in vivo. CFSE-labeled OT-1 T cells (2 × 106) were intravenously administrated after immunization. Then, animals were euthanized, and LN cells were harvested and stained with PE-CD3 and e-Flour–CD8 antibodies. The division of OVA-specific CD8 T cells was assessed by FC.
Quantification analysis of OVA-specific CD8 T cells using pentamer
Splenocytes of mice, which were immunized with OVA protein, were harvested and stained at day 14 for FC analysis. CD3 (PerCP-Cy5.5), CD8 (PE-Cy7), and SIINFEKL-pentamer (PE) were used to mark OVA-specific CD8 T cells.
Evaluation of effector T cells response
To assess the proportion of granzyme B+ CD8 T cells, the splenocytes of immunized mice were stimulated ex vivo with SIINFEKL (5 μg/ml) for 6 hours. The cells were then fixed, permeabilized, stained with anti-granzyme and anti-CD8α antibodies, and analyzed by FC. To evaluate effector memory response, these cells were stained with PerCP-Cy5.5-CD3, PE-Cy7-CD8α, APC-Cy7-CD44, and Pacific-Blue-CD62L antibodies and analyzed by FC.
Evaluation of health condition
The temperature of mice treated with different vaccination formulations were monitored continually using a microprobe thermometer (RET-3, Physitemp Instruments Inc.). The blood of mice was collected through the retro-orbital route at various time points (6 hours, 3, 7, and 14 days) for monitoring of early and delayed cytokine storm responses. TNF-α, IL-6, and IFN-γ were quantified using the respective ELISA kits, as per the manufacturer’s instructions. To determine the serum biochemical parameters, at days 28 and 56, blood samples were collected to detect the levels of AST, ALT, BUN, ALP, and LDH using an automated analyzer (Hitachi 917, Hitachi Ltd., Tokyo, Japan). For a histological analysis of the effect on organs, the spleens, livers, kidneys, and lungs were collected and fixed in 4% (v/v) formaldehyde. These tissues were processed for H&E staining and histological examination using Vectra platform.
Evaluation of tumor growth inhibition at an E.G7-bearing mice model
Female C57BL/6 mice were randomly assigned to different groups (G1: PBS, 100 μl; G2: pure antigen, 60 μg of OVA; G3: 60 μg of OVA mixed with 3 mg of microspheres; G4: 60 μg of OVA encapsulated within 3 mg of microcapsules; G4+: 200 μg of OVA encapsulated within 3 mg of microcapsules; n = 6). To establish tumor models, E.G7 cells (1 × 106) were injected into the left axillary region of the C57BL/6 mice on day 0. Vaccines were subcutaneously injected into the lower right flank at day 4 when the tumor was palpable. After the vaccination for 14 days, several mice were euthanized, and their spleens were harvested for FCS analysis. To monitor tumor progression, tumor sizes were measured continually and represented as 1/2 × L × W2 (cubic millimeters), where L is the longest and W is the shortest tumor diameter.
Evaluation of antimetastasis at B16-MUC1 mice models
For antimetastasis evaluation, 2 × 105 MS+B16 cells were intravenously inoculated into animals ahead of the same vaccination procedure that was adopted. Vaccines were subcutaneously injected on day 4. After the vaccination for 14 days, mice were euthanized, and their lungs and kidneys were harvested. Tumor nodules on lungs and kidneys exceeding 2 mm in diameter were counted manually, and tumor sizes on kidneys were measured using a slide caliper, and calculation as that of B16 tumor-bearing mice was adopted. The rest of the mice in groups were continuously monitored to the end points. All photographs in Fig. 6 were shot by T.Y. (Institute of Process Engineering, Chinese Academy of Sciences).
Neoantigen prediction
Human lymphocyte antigen (HLA) typing of the paired normal and tumor samples were performed from whole-exome sequencing using POLYSOLVER (v1.0) and Bwakit (v0.7.11), and the comprehensive HLA types were used for further neoantigen prediction. All nonsilent mutations were translated into 21-mer peptide sequences using in-house software centered on mutated amino acid. Then, the 21-mer peptide was used to create 8- to 11-mer peptide via a sliding window approach for prediction of MHC-I binding affinity. NetMHCpan (v3.0) was used to determine the binding strength of mutated peptides to patient-specific HLA alleles. The predicted peptides were scored and ranked according to the multiple criteria using in-house software. Peptides with scores higher than 0 were selected. On the basis of the prediction score, eight peptides were selected from the prediction library of 19,197 MHC-I key neoantigen peptides and prepared as an immunization pool against 4T1 tumor model. The sequence, HLA subtype, and corresponding gene of neoantigen peptides have been summarized in fig. S7E. The neoantigen raw sequence data were deposited at National Center for Biotechnology Information, National Institutes of Health under BioProject: PRJNA600867, available at http://ncbi.nlm.nih.gov/bioproject/600867.
Evaluation of tumor growth inhibition and postsurgical recurrence in 4T1 breast cancer mice models
Female Balb/c mice were randomly assigned to different groups (G1: PBS, 100 μl; G2: pure antigen, 200 μg of neoantigen peptides; G3: 200 μg of neoantigen peptides encapsulated with 3 mg of microspheres; AS04: 200 μg of neoantigen peptides mixed with 100 μl of AS04 adjuvant; G4: 200 μg of neoantigen peptides encapsulated within 3 mg of microcapsules; G5: 200 μg of neoantigen peptides and 3 μg of MPLA encapsulated within 3 mg of microcapsules; n = 6). To establish tumor models, 4T1 cells (1.5 × 106) were injected into the mammary fat pad region of the Balb/c mice on day 0. Vaccines were subcutaneously injected into the lower right flank at day 7 when the tumor was palpable. After the vaccination for 14 days, several mice were euthanized, and their tumor tissues were harvested for FCS analysis. To monitor tumor progression, tumor sizes were measured continually and represented as 1/2 × L × W2 (cubic millimeters), where L is the longest and W is the shortest tumor diameter.
For postsurgical recurrence, surgical section was executed after luciferase-transferred 4T1 tumor volume reached 200 mm3. The similar intensity of the bioluminescence in each group after the surgical operation was required, and then the same vaccination procedure was adopted. Vaccines were subcutaneously injected on day 4. Mice were imaged at days −3, 0, 7, and 14 with the same dose of d-luciferin by an In Vivo Imaging System (IVIS) optical imaging system (Caliper Life Sciences Inc., PerkinElmer). The rest of the mice in groups were continuously monitored to the end points.
IFN-γ ELISpot assay
The assay was performed according to the manufacturer’s instructions (Mabtech). Briefly, standard 96-well plates (Millipore) were coated with anti-mouse IFN-γ antibody diluted 1:200 in sterile PBS (final concentration, 15 μg/ml). Splenocytes from 4T1 tumor-bearing mice were plated at 4 × 105 cells per well, in duplicate, with 4T1 neoantigen peptides at 10 nM. After overnight restimulation at 37°C, plates were washed and incubated with biotinylated anti-mouse IFN-γ antibody, washed, and incubated for 2 hours at room temperature with streptavidin-ALP conjugated antibody. After extensive washing, 100 μl per well of the substrate (bromochloroindolyl phosphate-1–nitro blue tetrazolium step solution, Pierce) was added to measure spot development. The washing plates were thoroughly washed with distilled water to stop the reaction. Plates were allowed to air-dry completely, and spots were counted using an automated ELISpot reader (AT-Spot 2100, China).
T cell peptide-specific polyfunctional T cells analysis of splenocyte
Polyfunctionality T cell responses were detected by intracellular cytokine staining using FC. At the end of survival, splenocytes were harvested and incubated with RBC Lysing Buffer (Solarbio, China) for 10 min at room temperature and then were washed and diluted with RPMI 1640 medium (Gibco-BRL) with 10% FBS into 96-well plates at 1 × 106 per well. Peptides and brefeldin A (Sigma-Aldrich, USA) were added, respectively, at 1 nM and 1 μg/ml. After incubating for 16 hours at 37°C, splenocytes were surface-stained with anti-CD3 (EF506), anti-CD4 (EF450), and anti-CD8 (BV605) and intracellular-stained with IFN-γ (PE), TNF-α (FITC), and IL-2 (APC) after fixation/permeabilization. The stained samples were acquired through a CytoFLEX flow cytometer, and the data were analyzed by CytExpert software.
Evaluation of the combined therapy effect with PD-1 antibody
C57BL/6 J mice were randomly assigned to different groups (G5 group, vaccination of microcapsule loading with OVA protein (200 μg) and MPLA (3 μg); G5 + aPD-1 group, G5 vaccination combined with twice intraperitoneal injections of PD-1 antibody (100 μg) at days 4 and 7). Subsequent detections were performed at days 3 and 21. Corresponding OVA-specific CD8+ T cell proliferation assay seen above, female Balb/c mice were randomly assigned to different groups [G1: PBS, 100 μl; G5: vaccination of microcapsule (3 mg) loading with neoantigen peptides (200 μg) and MPLA (3 μg); G5 + aPD-1 group: G5 vaccination combined with twice intraperitoneal injections of PD-1 antibody (100 μg)] at days 4 and 7. To establish tumor models, 4T1 cells (1 × 106) were injected into the mammary fat pad region of the Balb/c mice on day 0. Vaccines were subcutaneously injected into the lower right flank at day 4 when the tumor was palpable. For other detailed experiment descriptions, see above.
Statistics guidelines
All results were expressed as means ± SD unless otherwise noted. Differences between two groups were performed using an unpaired, two-tailed Student’s t test. Differences among more than two groups were evaluated by one-way analysis of variance (ANOVA) with the significance determined by Tukey-adjusted t tests. *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We appreciated the National Engineering Laboratory for AIDS Vaccine and Key Laboratory for Molecular Enzymology and Engineering (Changchun, China) for supplying the MS+B16 expressing MUC1 cells. Funding: This work was supported by the National Key R&D Program of China (2017YFA0207900), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29040303), and the National Natural Science Foundation of China (nos. 21622608 and 21821005). Author contributions: G.M. and W.W. conceived and designed experiments. X.X., T.Y., and S.W. performed immune effects evaluation. X.N. contributed to microcapsule construction. X.X. and T.Y. performed tumor experiments. J.W., S.Q., X.G., F.L., W.W., and G.M. assisted in data analysis. X.X., T.Y., W.W., and G.M wrote the paper. All authors edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay7735/DC1
REFERENCES AND NOTES
- 1.Liu S. Y., Wei W., Yue H., Ni D. Z., Yue Z. G., Wang S., Fu Q., Wang Y. Q., Ma G. H., Su Z. G., Nanoparticles-based multi-adjuvant whole cell tumor vaccine for cancer immunotherapy. Biomaterials 34, 8291–8300 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Yue H., Wei W., Gu Z., Ni D., Luo N., Yang Z., Zhao L., Garate J. A., Zhou R., Su Z., Ma G., Exploration of graphene oxide as an intelligent platform for cancer vaccines. Nanoscale 7, 19949–19957 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Wang C., Ye Y., Hu Q., Bellotti A., Gu Z., Tailoring biomaterials for cancer immunotherapy: Emerging trends and future outlook. Adv. Mater. 29, 1606036 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Liang F., Lindgren G., Sandgren K. J., Thompson E. A., Francica J. R., Seubert A., De Gregorio E., Barnett S., O’Hagan D. T., Sullivan N. J., Koup R. A., Seder R. A., Loré K., Vaccine priming is restricted to draining lymph nodes and controlled by adjuvant-mediated antigen uptake. Sci. Transl. Med. 9, eaal2094 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Mellman I., Coukos G., Dranoff G., Cancer immunotherapy comes of age. Nature 480, 480–489 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran E., Turcotte S., Gros A., Robbins P. F., Lu Y.-C., Dudley M. E., Wunderlich J. R., Somerville R. P., Hogan K., Hinrichs C. S., Parkhurst M. R., Yang J. C., Rosenberg S. A., Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer. Science 344, 641–645 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaiko G. E., Horvat J. C., Beagley K. W., Hansbro P. M., Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 123, 326–338 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amidi M., Mastrobattista E., Jiskoot W., Hennink W. E., Chitosan-based delivery systems for protein therapeutics and antigens. Adv. Drug Deliv. Rev. 62, 59–82 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Disis M. L., Schiffman K., Guthrie K., Salazar L. G., Knutson K. L., Goodell V., dela Rosa C., Cheever M. A., Effect of dose on immune response in patients vaccinated with an HER-2/neu intracellular domain protein–Based vaccine. J. Clin. Oncol. 22, 1916–1925 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Watts C., Capture and processing of exogenous antigens for presentation on MHC molecules. Annu. Rev. Immunol. 15, 821–850 (1997). [DOI] [PubMed] [Google Scholar]
- 11.Mellman I., Steinman R. M., Dendritic cells: Specialized and regulated antigen processing machines. Cell 106, 255–258 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Klinman D. M., Immunotherapeutic uses of CpG oligodeoxynucleotides. Nat. Rev. Immunol. 4, 249–259 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Zhang Y., Cui Z., Kong H., Xia K., Pan L., Li J., Sun Y., Shi J., Wang L., Zhu Y., Fan C., One-shot immunomodulatory nanodiamond agents for cancer immunotherapy. Adv. Mater. 28, 2699–2708 (2016). [DOI] [PubMed] [Google Scholar]
- 14.Mata-Haro V., Cekic C., Martin M., Chilton P. M., Casella C. R., Mitchell T. C., The vaccine adjuvant monophosphoryl lipid A as a TRIF-biased agonist of TLR4. Science 316, 1628–1632 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Bryant C. E., Spring D. R., Gangloff M., Gay N. J., The molecular basis of the host response to lipopolysaccharide. Nat. Rev. Microbiol. 8, 8–14 (2010). [DOI] [PubMed] [Google Scholar]
- 16.Burdelya L. G., Krivokrysenko V. I., Tallant T. C., Strom E., Gleiberman A. S., Gupta D., Kurnasov O. V., Fort F. L., Osterman A. L., DiDonato J. A., Feinstein E., Gudkov A. V., An agonist of Toll-like receptor 5 has radioprotective activity in mouse and primate models. Science 320, 226–230 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sfondrini L., Rossini A., Besusso D., Merlo A., Tagliabue E., Mènard S., Balsari A., Antitumor activity of the TLR-5 ligand flagellin in mouse models of cancer. J. Immunol. 176, 6624–6630 (2006). [DOI] [PubMed] [Google Scholar]
- 18.Kasturi S. P., Skountzou I., Albrecht R. A., Koutsonanos D., Hua T., Nakaya H. I., Ravindran R., Stewart S., Alam M., Kwissa M., Villinger F., Murthy N., Steel J., Jacob J., Hogan R. J., García-Sastre A., Compans R., Pulendran B., Programming the magnitude and persistence of antibody responses with innate immunity. Nature 470, 543–547 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni D., Qing S., Ding H., Yue H., Yu D., Wang S., Luo N., Su Z., Wei W., Ma G., Biomimetically engineered demi-bacteria potentiate vaccination against cancer. Adv. Sci. 4, 1700083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bachmann M. F., Jennings G. T., Vaccine delivery: A matter of size, geometry, kinetics and molecular patterns. Nat. Rev. Immunol. 10, 787–796 (2010). [DOI] [PubMed] [Google Scholar]
- 21.Yue H., Ma G., Polymeric micro/nanoparticles: Particle design and potential vaccine delivery applications. Vaccine 33, 5927–5936 (2015). [DOI] [PubMed] [Google Scholar]
- 22.Lynn G. M., Laga R., Darrah P. A., Ishizuka A. S., Balaci A. J., Dulcey A. E., Pechar M., Pola R., Gerner M. Y., Yamamoto A., Buechler C. R., Quinn K. M., Smelkinson M. G., Vanek O., Cawood R., Hills T., Vasalatiy O., Kastenmüller K., Francica J. R., Stutts L., Tom J. K., Ryu K. A., Esser-Kahn A. P., Etrych T., Fisher K. D., Seymour L. W., Seder R. A., In vivo characterization of the physicochemical properties of polymer-linked TLR agonists that enhance vaccine immunogenicity. Nat. Biotechnol. 33, 1201–1210 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.den Brok M. H., Büll C., Wassink M., de Graaf A. M., Wagenaars J. A., Minderman M., Thakur M., Amigorena S., Rijke E. O., Schrier C. C., Adema G. J., Saponin-based adjuvants induce cross-presentation in dendritic cells by intracellular lipid body formation. Nat. Commun. 7, 13324 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sangha R., Butts C., L-BLP25: A peptide vaccine strategy in non–small cell lung cancer. Clin. Cancer Res. 13, s4652–s4654 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Zhao L., Seth A., Wibowo N., Zhao C.-X., Mitter N., Yu C., Middelberg A. P., Nanoparticle vaccines. Vaccine 32, 327–337 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Irvine D. J., Swartz M. A., Szeto G. L., Engineering synthetic vaccines using cues from natural immunity. Nat. Mater. 12, 978–990 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Danhier F., Ansorena E., Silva J. M., Coco R., Le Breton A., Préat V., PLGA-based nanoparticles: An overview of biomedical applications. J. Control. Release 161, 505–522 (2012). [DOI] [PubMed] [Google Scholar]
- 28.Talmadge J. E., The pharmaceutics and delivery of therapeutic polypeptides and proteins. Adv. Drug Deliv. Rev. 10, 247–299 (1993). [Google Scholar]
- 29.Petrovsky N., Novel human polysaccharide adjuvants with dual Th1 And Th2 potentiating activity. Vaccine 24, S26–S29 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schnare M., Barton G. M., Holt A. C., Takeda K., Akira S., Medzhitov R., Toll-like receptors control activation of adaptive immune responses. Nat. Immunol. 2, 947–950 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Coffman R. L., Sher A., Seder R. A., Vaccine adjuvants: Putting innate immunity to work. Immunity 33, 492–503 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris W., Steinhoff M. C., Russell P. K., Potential of polymer microencapsulation technology for vaccine innovation. Vaccine 12, 5–11 (1994). [DOI] [PubMed] [Google Scholar]
- 33.Mak W. C., Cheung K. Y., Trau D., Influence of different polyelectrolytes on layer-by-layer microcapsule properties: Encapsulation efficiency and colloidal and temperature stability. Chem. Mater. 20, 5475–5484 (2008). [Google Scholar]
- 34.Wei Y., Wang Y. X., Wang W., Ho S. V., Qi F., Ma G. H., Su Z. G., Microcosmic mechanisms for protein incomplete release and stability of various amphiphilic mPEG-PLA microspheres. Langmuir 28, 13984–13992 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Na X.-M., Gao F., Zhang L.-Y., Su Z.-G., Ma G.-H., Biodegradable microcapsules prepared by self-healing of porous microspheres. ACS Macro Lett. 1, 697–700 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Desai K.-G. H., Schwendeman S. P., Active self-healing encapsulation of vaccine antigens in PLGA microspheres. J. Control. Release 165, 62–74 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reinhold S. E., Desai K. G. H., Zhang L., Olsen K. F., Schwendeman S. P., Self-healing microencapsulation of biomacromolecules without organic solvents. Angew. Chem. Int. Ed. Engl. 51, 10800–10803 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engelhardt E., Toksoy A., Goebeler M., Debus S., Bröcker E. B., Gillitzer R., Chemokines IL-8, GROα, MCP-1, IP-10, and Mig are sequentially and differentially expressed during phase-specific infiltration of leukocyte subsets in human wound healing. Am. J. Pathol. 153, 1849–1860 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Melgarejo E., Medina M. Á., Sánchez-Jiménez F., Urdiales J. L., Monocyte chemoattractant protein-1: A key mediator in inflammatory processes. Int. J. Biochem. Cell Biol. 41, 998–1001 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Figdor C. G., de Vries I. J. M., Lesterhuis W. J., Melief C. J., Dendritic cell immunotherapy: Mapping the way. Nat. Med. 10, 475–480 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Grizzi I., Garreau H., Li S., Vert M., Hydrolytic degradation of devices based on poly (dl-lactic acid) size-dependence. Biomaterials 16, 305–311 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Zhu Y., Romain C., Williams C. K., Sustainable polymers from renewable resources. Nature 540, 354–362 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Pedersen T. H., Nielsen O. B., Lamb G. D., Stephenson D. G., Intracellular acidosis enhances the excitability of working muscle. Science 305, 1144–1147 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Sahlin K., Edström L., Sjöholm H., Hultman E., Effects of lactic acid accumulation and ATP decrease on muscle tension and relaxation. Am. J. Physiol. 240, C121–C126 (1981). [DOI] [PubMed] [Google Scholar]
- 45.Immke D. C., McCleskey E. W., Lactate enhances the acid-sensing Na+ channel on ischemia-sensing neurons. Nat. Neurosci. 4, 869–870 (2001). [DOI] [PubMed] [Google Scholar]
- 46.Kraut J. A., Madias N. E., Metabolic acidosis: Pathophysiology, diagnosis and management. Nat. Rev. Nephrol. 6, 274–285 (2010). [DOI] [PubMed] [Google Scholar]
- 47.Tong J., Wu W.-N., Kong X., Wu P.-F., Tian L., Du W., Fang M., Zheng F., Chen J.-G., Tan Z., Gong F., Acid-sensing ion channels contribute to the effect of acidosis on the function of dendritic cells. J. Immunol. 186, 3686–3692 (2011). [DOI] [PubMed] [Google Scholar]
- 48.Vermeulen M., Giordano M., Trevani A. S., Sedlik C., Gamberale R., Fernández-Calotti P., Salamone G., Raiden S., Sanjurjo J., Geffner J. R., Acidosis improves uptake of antigens and MHC class I-restricted presentation by dendritic cells. J. Immunol. 172, 3196–3204 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Chen Q., Ghilardi N., Wang H., Baker T., Xie M. H., Gurney A., Grewal I. S., de Sauvage F. J., Development of Th1-type immune responses requires the type I cytokine receptor TCCR. Nature 407, 916–920 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Luo Z., Wu Q., Yang C., Wang H., He T., Wang Y., Wang Z., Chen H., Li X., Gong C., Yang Z., A powerful CD8+ T-cell stimulating D-tetra-peptide hydrogel as a very promising vaccine adjuvant. Adv. Mater. 29, 1601776 (2017). [DOI] [PubMed] [Google Scholar]
- 51.Verdegaal E. M. E., de Miranda N. F. C. C., Visser M., Harryvan T., van Buuren M. M., Andersen R. S., Hadrup S. R., van der Minne C. E., Schotte R., Spits H., Haanen J. B. A. G., Kapiteijn E. H. W., Schumacher T. N., van der Burg S. H., Neoantigen landscape dynamics during human melanoma-T cell interactions. Nature 536, 91–95 (2016). [DOI] [PubMed] [Google Scholar]
- 52.Yadav M., Jhunjhunwala S., Phung Q. T., Lupardus P., Tanguay J., Bumbaca S., Franci C., Cheung T. K., Fritsche J., Weinschenk T., Modrusan Z., Mellman I., Lill J. R., Delamarre L., Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature 515, 572–576 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Ali O. A., Emerich D., Dranoff G., Mooney D. J., In situ regulation of DC subsets and T cells mediates tumor regression in mice. Sci. Transl. Med. 1, 8ra19 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J., Li W. A., Choi Y., Lewin S. A., Verbeke C. S., Dranoff G., Mooney D. J., Injectable, spontaneously assembling, inorganic scaffolds modulate immune cells in vivo and increase vaccine efficacy. Nat. Biotechnol. 33, 64–72 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bencherif S. A., Warren Sands R., Ali O. A., Li W. A., Lewin S. A., Braschler T. M., Shih T.-Y., Verbeke C. S., Bhatta D., Dranoff G., Mooney D. J., Injectable cryogel-based whole-cell cancer vaccines. Nat. Commun. 6, 7556 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ali O. A., Huebsch N., Cao L., Dranoff G., Mooney D. J., Infection-mimicking materials to program dendritic cells in situ. Nat. Mater. 8, 151–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moon J. J., Suh H., Bershteyn A., Stephan M. T., Liu H., Huang B., Sohail M., Luo S., Um S. H., Khant H., Goodwin J. T., Ramos J. T., Chiu W., Irvine D. J., Interbilayer-crosslinked multilamellar vesicles as synthetic vaccines for potent humoral and cellular immune responses. Nat. Mater. 10, 243–251 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nochi T., Yuki Y., Takahashi H., Sawada S., Mejima M., Kohda T., Harada N., Kong I. G., Sato A., Kataoka N., Tokuhara D., Kurokawa S., Takahashi Y., Tsukada H., Kozaki S., Akiyoshi K., Kiyono H., Nanogel antigenic protein-delivery system for adjuvant-free intranasal vaccines. Nat. Mater. 9, 572–578 (2010). [DOI] [PubMed] [Google Scholar]
- 59.Zhang H., Liu C., Zhang F., Geng F., Xia Q., Lu Z., Xu P., Xie Y., Wu H., Yu B., Wu J., Yu X., Kong W., MUC1 and survivin combination tumor gene vaccine generates specific immune responses and anti-tumor effects in a murine melanoma model. Vaccine 34, 2648–2655 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Posey A. D. Jr., Schwab R. D., Boesteanu A. C., Steentoft C., Mandel U., Engels B., Stone J. D., Madsen T. D., Schreiber K., Haines K. M., Cogdill A. P., Chen T. J., Song D., Scholler J., Kranz D. M., Feldman M. D., Young R., Keith B., Schreiber H., Clausen H., Johnson L. A., June C. H., Engineered CAR T cells targeting the cancer-associated Tn-glycoform of the membrane Mucin MUC1 control adenocarcinoma. Immunity 44, 1444–1454 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaay7735/DC1