

The LysRS-Ap4A axis impairs cGAS-STING pathway activation, delaying nucleic acid-driven innate immune responses.
Abstract
Inflammation is an essential part of immunity against pathogens and tumors but can promote disease if not tightly regulated. Self and non-self-nucleic acids can trigger inflammation, through recognition by the cyclic GMP-AMP (cGAMP) synthetase (cGAS) and subsequent activation of the stimulator of interferon genes (STING) protein. Here, we show that RNA:DNA hybrids can be detected by cGAS and that the Lysyl-tRNA synthetase (LysRS) inhibits STING activation through two complementary mechanisms. First, LysRS interacts with RNA:DNA hybrids, delaying recognition by cGAS and impeding cGAMP production. Second, RNA:DNA hybrids stimulate LysRS-dependent production of diadenosine tetraphosphate (Ap4A) that in turn attenuates STING-dependent signaling. We propose a model whereby these mechanisms cooperate to buffer STING activation. Consequently, modulation of the LysRS-Ap4A axis in vitro or in vivo interferes with inflammatory responses. Thus, altogether, we establish LysRS and Ap4A as pharmacological targets to control STING signaling and treat inflammatory diseases.
INTRODUCTION
Dinucleotides are bioactive molecules for which a signaling role in mammalian cells has emerged in recent years. In particular, cyclic dinucleotides, such as cyclic guanosine monophosphate–adenosine monophosphate (cGAMP), have been described as activators of the inflammatory response (1). In the presence of self- or non-self–nucleic acid species, the cyclic GMP-AMP synthase (cGAS) DNA sensor produces cGAMP (2), activating inflammatory responses (3). A pivotal step in the mounting of this response to immune-stimulatory nucleic acids is the interaction of cGAMP with the stimulator of interferon genes (STING) (4, 5), which leads to the recruitment of the tank binding kinase 1 (TBK1) and transcription factors, including interferon response factor 3 (IRF3) (6). The assembly of this STING signalosome leads to the phosphorylation of IRF3 that translocates into the nucleus to orchestrate the transcription of a set of genes that includes proinflammatory cytokines and type I interferon (IFN) (6). Dysregulation of the cGAS-STING pathway fuels several inflammatory human pathologies, including autoimmune, autoinflammatory, and malignant disorders (7). The development of means to act on the cGAS-STING signaling pathway is therefore paramount to current therapies aiming to curb disease-associated inflammation (8). Small molecules that modulate cGAS-STING pathway activation have been identified (9), opening novel investigation avenues. Yet, in certain pathologies, what determines the outcome of STING activation or inhibition remains unknown (10). This underscores the pressing need for a better understanding of the mechanisms regulating the cGAS-STING pathway in different cellular contexts.
Cytosolic double-stranded DNA (dsDNA) species are the main activators of the cGAS-STING pathway, but recently, RNA:DNA hybrids have also been described as possible ligands for cGAS. However, the molecular mechanisms involved in their recognition and in the regulation of ensuing inflammatory cytokines production are poorly described. RNA:DNA hybrids can arise as by-products of several physiological processes, including DNA replication, immunoglobulin class switching and hypermutation, or double-strand DNA break repair (11). Endogenous and exogenous reverse transcriptases also contribute to generating RNA:DNA hybrids (12, 13). Regardless of their origin, these moieties can threaten cellular integrity, by promoting unwanted triggering of inflammatory responses (12–14). However, while RNA:DNA hybrids can stimulate cGAS-dependent cGAMP production in vitro (14), it is still unknown whether such interaction can occur in cells.
To identify proteins involved in the detection of RNA:DNA hybrids and in the regulation of associated inflammatory responses, we used a biochemistry approach. We thereby identified the Lysyl tRNA synthetase (LysRS), which is a component of the cytosolic multi-tRNA synthetase complex (MSC), as a direct binding partner of RNA:DNA hybrids. While LysRS plays major roles in translation through aminoacylation of the cognate tRNA, noncanonical functions have been described (15, 16), including regulation of immune response (17–19). This prompted us to investigate the role of LysRS in detection and regulation of RNA:DNA hybrid–induced inflammatory responses. We thus demonstrate that RNA:DNA hybrids can be detected by cGAS and that LysRS can regulate subsequent STING activation through two complementary mechanisms. On one hand, LysRS interacts with cytosolic RNA:DNA hybrids, delaying recognition by cGAS. On the other hand, stimulation of cells with RNA:DNA hybrids leads to LysRS-dependent diadenosine tetraphosphate (Ap4A) production. This second messenger can associate with STING, delaying its activation. We therefore show that the LysRS-Ap4A pathway contributes to regulate nucleic acid-dependent cGAS-STING activation.
RESULTS
The MSC interacts with RNA:DNA hybrids
To identify RNA-DNA interacting proteins, we used a blunt synthetic 40-nt-long RNA:DNA hybrid bearing a 5′ biotin (BRNA:DNA; fig. S1A). We first verified the stability of synthetic RNA:DNA hybrids by immunofluorescence (fig. S1B) in mouse embryonic fibroblasts (MEFs), together with their ability to elicit a proinflammatory response in MEF, MEF knocked out for Sting (MEFSting−/−) or cGas (MEFcGas−/−). The latter was assessed by measuring Ifnβ mRNA levels by real-time quantitative polymerase chain reaction (RT-qPCR) (Fig. 1A) and IRF3 phosphorylation (pIRF3) by Western blot (WB; Fig. 1B). As expected (14), synthetic RNA:DNA hybrids induced IFN production in a cGAS-STING–dependent manner. We next performed in vitro pull-down assays by incubating cytosolic extracts (S100) from HeLa-S3 cells (fig. S1C) with BRNA:DNA hybrids before pull-down using streptavidin-affinity magnetic beads and elution (Fig. 1C, left). Biotinylated single-stranded RNA (ssBRNA) was included as a control. Eluted material was resolved by SDS–polyacrylamide gel electrophoresis (PAGE) and either silver-stained (Fig. 1C, right) or processed by mass spectrometry. Protein enrichment data revealed substantial interaction with all members of the MSC that is a central actor in protein synthesis, composed by eight tRNA synthetases: GluRSEPRS, IleRSIARS, LeuRSLARS, GlnRSQARS, MetRSMARS, ArgRSRARS, LysRSKARS, AspRSDARS, and three scaffold proteins: p43AIMP1, p38AIMP2, and p18AIMP3 (fig. S1D) (20). Using WB, we confirmed that proteins of the MSC interact with BRNA:DNA hybrids, but not with ssBRNA or ssBDNA, in vitro (Fig. 1D). We recapitulated the specific interaction of the MSC with RNA:DNA hybrids in human (Fig. 1E) and murine cells (Fig. 1F) upon transfection of BRNA:DNA hybrids and streptavidin pull-down. We therefore show that the MSC is recruited to RNA:DNA hybrids both in vitro and in cells.
Fig. 1. MSC is recruited to RNA:DNA hybrids.
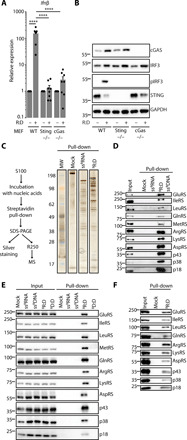
(A) Mean (±SEM), Ifnβ mRNA levels in WT-MEF, MEFSting−/−, and MEFcGAS−/− transfected or not with RNA:DNA hybrids (R:D); n = 8. t test, ****P < 0.0001. (B) WB analysis of whole-cell extracts of cells treated as in (A). Membranes were probed with the indicated antibodies. (C) Left: Experimental scheme. MS, mass spectrometry; ssBRNA, biotinylated ssRNA; BR:D, biotinylated RNA:DNA hybrids. Right: Silver staining of samples obtained following the experimental scheme. Numbers indicate molecular weight (MW) in kDa. (D) WB analysis of pull-down performed as in (C), except that biotinylated ssDNA (ssBDNA) was included as a control. (E) HeLa cells were transfected or not with biotinylated BR:D, ssBRNA, or biotinylated dsDNA (BD:D) before whole-cell extract preparation and pull-down using streptavidin affinity beads. Input and eluates were analyzed by WB using the indicated antibodies. (F) As in (E), except that WT-MEF were transfected with BR:D before pull-down. Input and eluates were analyzed by WB using the indicated antibodies. All immunoblots show representative experiments.
The Lyslyl tRNA synthetase interacts directly with RNA:DNA hybrids
We next investigated which member of the MSC is responsible for its interaction with RNA:DNA hybrids. Three members of the MSC complex, namely, LysRS, AspRS, and p43, comprise oligonucleotide/oligosaccharide binding (OB)–fold domains that are predicted to bind nucleic acids (21). Thus, we tested whether these proteins can directly interact with RNA:DNA hybrids using recombinant glutathione S-transferase (GST)–tagged proteins (fig. S2A). GST was used as negative control. We incubated purified proteins with BRNA:DNA hybrids before pull-down using streptavidin affinity beads and analysis by WB using an anti-GST antibody. We thereby show that LysRS displays strong binding to RNA:DNA hybrids (Fig. 2A) and that this interaction is Ribonuclease H (RNaseH)–sensitive (Fig. 2B). We further characterized the binding of LysRS to RNA:DNA hybrids, using full-length LysRS or LysRS constructs harboring either a N-terminal deletion of the putative OB-fold nucleic acid binding domain (LysRS∆N) or a C-terminal deletion of the catalytic aminoacyl transferase domain (LysRS∆C) (22). Recombinant GST-LysRS, GST-LysRS∆N, GST-LysRS∆C, and GST (fig. S2B) were used in in vitro pull-down experiments using BRNA:DNA hybrids immobilized on streptavidin affinity beads. This showed that LysRS interacts with RNA:DNA hybrids through its N-terminal domain (Fig. 2C). This binding is sensitive to RNaseH treatment (Fig. 2D). We additionally confirmed that murine LysRS (fig. S2B) binds RNA:DNA hybrids (fig. S2C) and that this binding is RNaseH sensitive (fig. S2D) as observed for human LysRS (Fig. 2, A to C). Thus, our in vitro experiments shown that LysRS can directly bind RNA:DNA hybrids.
Fig. 2. The Lyslyl tRNA synthetase interacts directly with RNA:DNA hybrids.
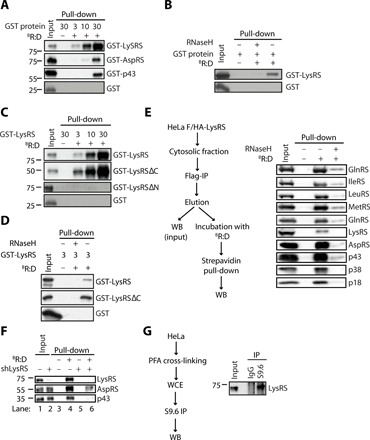
(A) In vitro interaction assay. Three, 10, and 30 pmol of recombinant LysRS, AspRS, or p43 were incubated with 20 pmol of BR:D before pull-down using streptavidin affinity beads. Input and eluates were analyzed by WB using anti-GST antibody. (B) As in (A), except that 3 pmol of recombinant LysRS was incubated with 20 pmol of BR:D before pull-down on streptavidin affinity beads. Where indicated, nucleic acids were treated with RNaseH before pull-down. Input and eluates were analyzed by WB using anti-GST antibody. (C) As in (A), except that 3, 10, and 30 pmol of recombinant LysRS, LysRSΔN, or LysRSΔC were incubated with 20 pmol of BR:D before pull-down using streptavidin affinity beads. Input and eluates were analyzed by WB using anti-GST antibody. (D) Three pmol of recombinant LysRS, LysRS∆C, or GST were incubated with 20 pmol of BR:D before pull-down using streptavidin affinity beads. Where indicated, nucleic acids were treated with RNaseH before pull-down. Input and eluates were analyzed by WB using anti-GST antibody. (E) Reciprocal immunoprecipitation. F/HA-LysRS was Flag-purified from the cytosolic fraction of HeLa cells and used as input material for pull-down using 20 pmol of BR:D and streptavidin affinity beads. Where indicated, nucleic acids were treated with RNaseH before pull-down. Input and eluates were analyzed by WB using the indicated antibodies. (F) Whole-cell extracts from A549 cells stably expressing shLuc or shLysRS were used in streptavidin pull-downs using BR:D. Input and eluates were analyzed by WB using the indicated antibodies. (G) Whole cell extracts (WCEs) from paraformaldehyde (PFA) cross-linked HeLa cells were immunoprecipitated with the S9.6 anti–RNA:DNA hybrids antibody or mock immunoglobulin G (IgG) (left). Input and eluates were analyzed by WB using an anti-LysRS antibody (right). All immunoblots show representative experiments. IP, immunoprecipitation.
Our in vitro binding data further suggest that LysRS may be the key to the recruitment of MSC proteins to RNA:DNA hybrids. To test this, we first FLAG-purified overexpressed FLAG-hemagglutinin (HA)–tagged LysRS (F/HA-LysRS) from the cytosolic fraction of HeLa cells. Eluted material was used as input for pull-down using streptavidin-immobilized BRNA:DNA hybrids (Fig. 2E, left). WB analysis using antibodies targeting proteins of the MSC complex revealed that all tested members coprecipitated with RNA:DNA hybrids (Fig. 2E, right). We confirmed the specificity of this interaction by RNaseH treatment of the BRNA:DNA hybrids before binding to the streptavidin affinity beads (Fig. 2E, right). We next knocked down LysRS in cells before pull-down assays using BRNA:DNA hybrids. This showed that decreased levels of LysRS (Fig. 2F, compare lanes 1 and 2) are accompanied by decreased association of other tested components of the MSC (Fig. 2F, compare lanes 4 and 6). This indicates that the presence of LysRS is the key to the recruitment of the MSC to RNA:DNA hybrids in cells. Last, we questioned whether LysRS interacts with endogenous RNA:DNA hybrids. To this aim, we adapted the S9.6 antibody–based DNA:RNA hybrid immunoprecipitation protocol [adapted from (13)]. Using the HeLa cell line that presents endogenous RNA:DNA hybrids (fig. S2E), we confirmed that endogenous RNA:DNA hybrids immunoprecipitated LysRS (Fig. 2G). This was further confirmed by experiments where endogenous GlnRS was immunopurified, before assessment of the presence of other components of the MSC and extraction of GlnRS-associated DNA (fig. S2F). WB analysis of the eluates confirmed the presence of LyRS (fig. S2F, middle), and quantitative PCR analysis showed an enrichment of sequences belonging to the high-copy satellite II region of the human genome (fig. S2F, right) that is reported to produce RNA:DNA hybrids (23). Thus, together, our data show that within the MSC complex LysRS is the main mediator of the interaction with RNA:DNA hybrids.
The Lysyl tRNA synthetase inhibits RNA:DNA hybrid-induced IFN production
While the main reported function of LysRS within the MSC complex is to aminoacylate its cognate tRNA, there are reports of additional noncanonical functions, including negative regulation of immune responses (15, 16). Thus, we interrogated whether LysRS can modulate RNA:DNA hybrid–associated inflammation. To this aim, we transduced WT-MEF with a LysRS targeting short hairpin RNA (shRNA) (shLysRS) or with a control Luciferase targeting shRNA (shLuc), before transfection or not with RNA:DNA hybrids for 3, 6, and 12 hours. Cells were harvested and analyzed for Ifnβ mRNA levels by RT-qPCR (Fig. 3A), as well as by WB against phosphorylated IRF3 (pIRF3) (Fig. 3B). This showed that the absence of LysRS leads to up-regulated basal Ifnβ mRNA levels, which are further increased upon transfection of RNA:DNA hybrids. The kinetics of Ifnβ expression were faster in LysRS–knocked-down cells, suggesting that LysRS delays activation of the IFN response. These results were confirmed using the three prime repair exonuclease 1 (Trex1)–deficient MEF (MEFTrex1−/−), which is a well-characterized model of chronic inflammation that presents high levels of RNA:DNA hybrids together with elevated STING-dependent Ifnβ and interferon stimulated genes (ISGs) mRNA levels (fig. S3, A and B) (24). Following knockdown of LysRS in MEFTrex1−/−, we observed increased Ifnβ mRNA levels (Fig. 3C and fig. S3C) and increased levels of pIRF3 both at steady state and upon RNA:DNA hybrid stimulation (Fig. 3D and fig. S3D). Conversely, overexpression of F/HA-LysRS, followed by RT-qPCR analysis showed decreased steady-state and RNA:DNA hybrid–induced Ifnβ mRNA levels (Fig. 3E), accompanied by decreased levels of pIRF3 (Fig. 3F). In further support of the role of LysRS in controlling innate immune responses, primary mouse neonatal fibroblasts heterozygous for LysRS (MNFsLysRS+/−; fig. S3E) showed higher basal Ifnβ mRNA levels (Fig. 3G) than their wild-type counterparts (MNFsLysRS+/+), together with elevated ISG mRNA levels, including Ifit1, Ifit2, Isg15, and Cxcl10 (fig. S3F). Consistent with their ISG expression profile, MNFsLysRS+/− poorly support herpes simplex virus (HSV) type 1 and 2 replication (Fig. 3, H and I). Thus, our data demonstrate that LysRS negatively regulates Ifnβ expression in a model of chronic inflammation and that the absence of LysRS leads to the establishment of an antiviral state.
Fig. 3. The Lysyl tRNA synthetase inhibits RNA:DNA hybrid–induced IFN production.
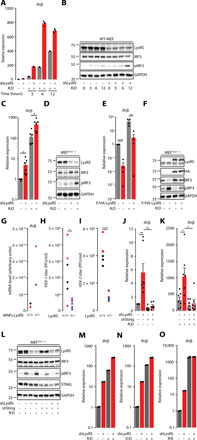
(A) Mean (±SEM) Ifnβ mRNA levels in shLysRS-expressing versus shLuc-expressing WT-MEF transfected or not with R:D for 3, 6, 9, and 12 hours. Representative graph (n = 4). (B) Whole-cell extracts from cells treated as in (A) were analyzed by WB using indicated antibodies. (C) Mean (±SEM) Ifnβ mRNA levels in shLysRS-expressing versus shLuc-expressing MEFTrex1−/− transfected or not with R:D for 6 hours (n = 7). (D) Whole-cell extracts from cells treated as in (C) were analyzed by WB using indicated antibodies.(E) Mean (±SEM) Ifnβ mRNA levels in MEFTrex1−/− overexpressing F/HA-LysRS following transfection or not with R:D for 6 hours (n = 4). (F) Whole-cell extracts from cells treated as in (E) were analyzed by WB using indicated antibodies. (G) Ifnβ mRNA levels in WT-MNF (mouse neonatal fibroblast) and MNFLysR +/− (cells derived from two independent 1-day-old mice). Different colors indicate different mice. (H and I) WT-MNF and MNFLysR +/− were infected with HSV-1 (H) or HSV-2 (I) at multiplicity of infection (MOI) =1, and viral titers were measured 24 hours later. Data represent biological triplicates of cells derived from two independent 1-day-old mice. Different colors indicate different mice. (J and K) Mean (±SEM) Ifnβ mRNA levels in MEFTrex1−/− transduced with shLuc, shLysRS, shSting, or shLysRS and shSting prior (J) or after (K) transfection with R:D for 6 hours. Data are expressed relative to shLuc-expressing cells (n = 7). (L) WB analysis of whole-cell extracts from experiment performed as in (J) and (K). (M) Ifnβ mRNA levels in shLuc versus shLysRS-treated MEFTrex1−/− after transfection with R:D for 6 hours. (N) Ifnβ mRNA levels in shLuc versus shLysRS-treated MEFTrex1−/− after transfection with dsDNA (D:D) for 6 hours. (O) Ifnβ mRNA levels in shLuc versus shLysRS-treated MEFTrex1−/− after transfection with dsRNA (R:R) for 6 hours. Results in (M) to (O) are presented as mean Ifnβ mRNA levels from technical duplicates (representative experiments; n = 3). (C, E, I, J, and K) Unpaired t test, *P < 0.05, **P < 0.01, and ****P < 0.0001. All immunoblots show representative experiments. PFU, plaque-forming units.
Detection of RNA:DNA hybrids has been previously reported to lead to the activation of STING (Fig. 1A) (14). Thus, we next assessed whether LysRS-mediated inhibition of Ifnβ production may rely on inhibition of STING activation. To this aim, we knocked down both LysRS and Sting in MEFTrex1−/− and measured Ifnβ mRNA and pIRF3 levels by RT-qPCR and WB, respectively. This showed that while the absence of LysRS leads to increased Ifnβ mRNA levels at both steady state (Fig. 3J) and upon stimulation with RNA:DNA hybrids (Fig. 3K), such increase is lost in the absence of STING (Fig. 3, J and K). Consistently, knockdown of LysRS led to increased pIRF3 levels (Fig. 3L) that are lost in the double knockdown. We confirmed the dependency on STING by knocking down LysRS in WT-MEF before transfection with either RNA:DNA hybrids (Fig. 3M) and dsDNA (Fig. 3N), which are known to trigger STING-dependent signaling, or dsRNA (Fig. 3O), which signals through a different pathway (25). The absence of LysRS led to increased Ifnβ mRNA levels in response to RNA:DNA hybrids and dsDNA but not in response to dsRNA (Fig. 3, M to O). This is supported by increased levels of pIRF3 (fig. S3G). Thus, together, our data demonstrate that LysRS delays STING-dependent Ifnβ expression.
LysRS delays the detection of RNA:DNA hybrids by cGAS
We next questioned the molecular mechanism through which LysRS inhibits STING activation. It was previously suggested that cGAS may bind to RNA:DNA hybrids because (i) in vitro RNA:DNA hybrids can stimulate cGAS-dependent cGAMP production (14) and (ii) RNA:DNA-dependent Ifnβ expression requires cGAS (Fig 1, A and B) (14) . Given that we show that LysRS interacts with RNA:DNA hybrids (Fig. 2A), we hypothesize that LysRS may inhibit STING activation by preventing the association of cGAS with RNA:DNA hybrids. To test this hypothesis, we first monitored whether cGAS interacts with the RNA:DNA hybrids used in our assays. To this aim, T98G cells, which are cGAS deficient, were engineered to stably express FLAG-HA–tagged cGAS (F/HA-cGAS). The functionality of overexpressed cGAS was assesses by monitoring the phosphorylation status of IRF3 after transfection with RNA:DNA hybrids and dsDNA (fig. S4A). We prepared cytosolic and nuclear extracts (fig. S4B) according to (26) from cGAS overexpressing cells, and the cytosolic fraction was used for in vitro pull-down experiments using BRNA:DNA hybrids, dsBDNA, and ssBDNA immobilized on streptavidin affinity beads. Pulled-down material was assessed by WB, showing that cGAS binds RNA:DNA hybrids, albeit at lower levels than dsDNA. Next, we transfected F/HA-cGAS–expressing T98G cells with BRNA:DNA hybrids and dsBDNA before pull-down using streptavidin affinity beads. WB analysis of the pulled-down material showed that in cells, cGAS interacts both with dsDNA and with RNA:DNA hybrids (Fig. 4B). Thus, together, our data demonstrate that cGAS interacts with RNA:DNA hybrids.
Fig. 4. LysRS delays the detection of RNA:DNA hybrids by cGAS.
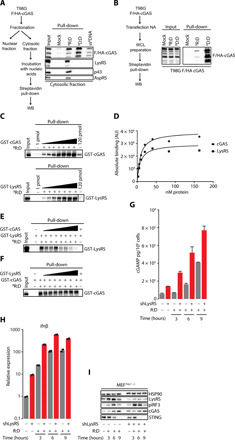
(A) In vitro interaction. Left: Experimental scheme. T98G cells stably expressing F/HA-cGAS (T98GF/HA-cGAS) were fractionated. The obtained cytosolic fraction was incubated with either 20 pmol of BR:D, BD:D, or ssBDNA before streptavidin affinity pull-down. Right: Input and eluates were immunoblotted using anti-HA antibody to detect F/HA-cGAS and the indicated antibodies. (B) In cell interaction. Left: Experimental scheme. T98GF/HA-cGAS cells were transfected or not with BR:D, and BD:D. Six hours after transfection, cells were harvested, and whole-cell extracts were prepared and used for streptavidin affinity pull-down. Right: Inputs and eluates were immunoblotted using anti-HA antibody. (C) In vitro streptavidin pull-downs were performed using 20 pmol of BR:D and increasing amounts of recombinant proteins (2.5, 5, 10, 20, 40, 80, and 160 nM). Biotin (20 pmol) was used as mock ligand at the lowest and highest doses of recombinant proteins. Input and eluates were immunoblotted with anti-GST antibody. (D) Signals from WB in (C) were quantified by ImageJ and plotted in Prism, and data were fitted by the nonlinear regression method for one-site specific binding. x axis: amounts (nM) of recombinant protein and y axis: absolute binding expressed as arbitrary units (AU). Representative graph (n = 4). (E and F) Competition experiments. Streptavidin-immobilized BR:D were incubated with 10 nM LysRS and increasing doses of cGAS (2.5, 5, 10, 20, 40, 80, and 160 nM) (E) or with 10 nM cGAS and increasing doses of LysRS (2.5, 5, 10, 20, 40, 80, and 160 nM) (F). Input and eluates were immunoblotted with either anti-LysRS or anti-cGAS antibodies as indicated. (G) LysRS was knocked down in MEFTrex1−/− before transfection with R:D for 3, 6, and 9 hours. Cells were harvested, and cell pellets were processed for intracellular cGAMP measurements using the cGAMP ELISA Kit (CISBIO). Graph represents mean ± SD from technical duplicates of pg cGAMP per 106 cells. Representative graph (n = 3). (H) Ifnβ mRNA levels, measured in cells treated as in (G), are expressed as mean (±SEM) from technical duplicates. Data are expressed relative to shLuc-expressing cells. Representative graph (n = 3). (I) Whole-cell extracts from experiment presented in (G) were analyzed by WB using the indicated antibodies. All immunoblots show representative experiments.
Having established that both LysRS and cGAS can interact with RNA:DNA hybrids, we next wished to investigate the affinity of these interactions by calculating the dissociation constant (Kd) of the interaction of cGAS or LysRS with RNA:DNA hybrids. To this end, we performed in vitro streptavidin pull-down assays using increasing amounts of recombinant proteins (fig. S4C) and a fixed amount of BRNA:DNA hybrids. Biotin was used as a mock ligand in this assay, at the lowest and highest doses of tested proteins. WB analysis (Fig. 4C) and quantification of the interaction (Fig. 4D) show a KD of ~8.05 nM for cGAS and of ~11.3 nM for LysRS. This indicates that LysRS and cGAS have similar affinity for RNA:DNA hybrids in vitro. Competition experiments showed that in vitro cGAS can compete with LysRS for association to RNA:DNA hybrids (Fig. 4E), while LysRS cannot replace cGAS (Fig. 4F).
Knowing that LysRS is an abundant protein and that cGAS is an ISG, their relative abundance in cells may be the key in their ability to compete. Therefore, to assess whether LysRS can interfere with the recruitment of cGAS to RNA:DNA hybrids, we measured intracellular cGAMP levels in the absence of LysRS, at steady state and upon RNA:DNA hybrid stimulation (Fig. 4G). This showed that knockdown of LysRS leads to higher levels of intracellular cGAMP both at steady state and upon stimulation with RNA:DNA hybrids, which correlate with higher levels of Ifnβ mRNA (Fig. 4H) and pIRF3 (Fig. 4I). Thus, our data indicate that LysRS interferes with cGAMP production in response to RNA:DNA hybrids, further implying that LysRS likely delays recognition of RNA:DNA hybrids by cGAS.
Ap4A prevents STING-dependent Ifnβ expression
A role for LysRS in regulation of immune responses was previously suggested (15). In particular, under immunological stress, LysRS produces Ap4A, an evolutionarily conserved second messenger (27), that regulates both gene expression and immune responses (17, 18, 28). This led us to question whether the presence of RNA:DNA hybrids could be sensed by LysRS as a form of immunological stress and consequently stimulate the production of Ap4A, the latter potentially contributing to the inhibition of STING-mediated immune responses. To test this hypothesis, we transfected WT-MEF with RNA:DNA hybrids and measured intracellular Ap4A levels at 0, 3, 6, 9, and 12 hours after transfection (Fig. 5A). Activation of the cGAS-STING pathway was controlled by WB analysis of pIRF3 levels (Fig. 5B). This showed that RNA:DNA hybrids stimulate Ap4A production. Next, to assess whether RNA:DNA hybrid–dependent Ap4A production requires LysRS, we knocked down LysRS before transfection with RNA:DNA hybrids for 12 hours (Fig. 5, C and D). LysRS knockdown led to the expected decrease of intracellular Ap4A levels. In addition, the ability of RNA:DNA hybrids to stimulate Ap4A production is lost in LysRS knock-down cells, establishing RNA:DNA hybrids as trigger for LysRS-dependent Ap4A production.
Fig. 5. Ap4A inhibits STING-dependent Ifnβ expression.
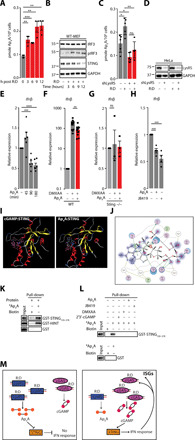
(A) Mean (±SEM) Ap4A levels in WT-MEFs transfected or not with R:D for 3, 6, 9, or 12 hours are expressed as pmol of Ap4A per 106 cells (n = 3). (B) Whole-cell extracts from cells treated as in (A) were analyzed by WB using the indicated antibodies. (C) Mean (±SEM) Ap4A levels in HeLa cells stably expressing shLuc or shLysRS transfected or not with R:D hybrids for 12 hours are expressed as pmol of Ap4A per 106 cells (n = 5). (D) Whole-cell extracts from cells treated as (C) were analyzed by WB using the indicated antibodies. (E) Mean (±SEM) Ifnβ mRNA levels in untreated MEFTrex1−/− versus MEFTrex1−/− treated with 500 μM Ap4A for 45, 90, or 180 min (n = 6). (F) Mean (±SEM) Ifnβ mRNA levels in WT-MEF stimulated with 200 μM DMXAA and treated, or not, with 500 μM Ap4A for 2 hours (n = 13). Data are normalized to nontreated control. (G) Mean (±SEM) Ifnβ mRNA levels in MEFSting−/− stimulated with 200 μM DMXAA and treated, or not, with 500 μM Ap4A for 2 hours (n = 3). Data are normalized to nontreated control. (H) Mean (±SEM) Ifnβ mRNA levels in MEFTrex1−/− treated with 500 μM Ap4A or 500 μM JB419 for 3 hours versus untreated MEFTrex1−/− (n = 4). (I) The 3D modeling of docked cGAMP (left) or Ap4A (right) in the cleft of STING obtained using Amber force field and the crystal structure of human STING complexed to cGAMP (PDB: 4KSY). Ap4A has adopted a horseshoe-like conformation. (J) Predicted molecular interactions that stabilize Ap4A in the active site of STING. Atoms are pink spheres (acidic, red outline; basic, blue outline). Interactions are depicted by arrows showing the direction of the electron exchange (donor or acceptor), with backbone interactions in blue and sidechain interactions in green. The dotted contour defines the proximity of the available conformational space for the depicted compound. (K) WB analysis of in vitro binding experiment using 100 μg of GST, GST-HINT or GST-STING139–378, and 10 μM streptavidin-immobilized biotinylated Ap4A. (L) WB analysis of in vitro binding experiment performed using 100 μg of GST or GST-STING139–378 and 10 μM streptavidin-immobilized biotinylated Ap4A. Competition was performed with 100 μM cGAMP, DMXAA, JB419, or Ap4A as indicated. (M) Proposed model. At steady state (left), LysRS interacts with R:D, delaying the recruitment of cGAS to R:D. LysRS interaction with R:D also stimulates Ap4A production, which buffers STING activation. In the absence of LysRS (right), cGAS detects R:D, producing cGAMP and resulting in STING activation. IFN is produced, inducing the establishment of a positive feedback loop, which increases the total amount of cGAS and additional ISGs. (A, C, E, F, G, and H) Unpaired t test: ns, nonsignificant; *P < 0.05, **P < 0.01, and ****P < 0.0001. All immunoblots show representative experiments.
To investigate whether Ap4A could have an impact on STING-dependent inflammation, we treated MEFTrex1−/− with Ap4A for 45, 90, and 180 min. We observed that Ap4A decreased Ifnβ mRNA levels over the three-hour time course (Fig. 5E). In addition, simultaneously treating WT (WT-MEF) and STING knockout MEFs (MEFSting−/−) with DMXAA and Ap4A showed that, while cotreatment reduced the ability of DMXAA to induce Ifnβ expression in WT-MEF by half (Fig. 5F), no decrease was observed in MEFSting−/− (Fig. 5G). Because it is reported that Ap4A has poor capacity to cross the plasma membrane (29) and is quickly catabolized in cells (30), we next tested the ability of JB419 [nonhydrolyzable analog of Ap4A (31)] to modulate Ifnβ expression. We observed that treatment of MEFTrex1−/− with JB419 leads to decreased Ifnβ mRNA levels, comparable to those observed upon treatment with Ap4A (Fig. 5H). These experiments suggest that Ap4A and its stable JB419 nonhydrolyzable analog can decrease Ifnβ mRNA levels in a model of STING-dependent inflammation. In addition, Fig. 5G suggests that Ap4A acts through STING, opening the possibility that Ap4A may interact with STING. Therefore, we next questioned whether Ap4A acts through STING to delay the type I IFN response by binding to the cGAMP binding pocket of STING. To this aim, we used Amber force field (32) and the previously published cocrystal of STING with cGAMP [Protein Data Bank (PDB): 4KSY] (4) to compare the binding of Ap4A and cGAMP to STING. This predicted that cGAMP and Ap4A adopt a similar three-dimensional (3D) conformation upon docking to STING (Fig. 5I and fig. S5A). Comparison of the interactions between the in silico–docked Ap4A to STING (Fig. 5J) to those in the crystal structure of STING and cGAMP (fig. S5B) indicates that similar interactions are established. Comparison of the potential energies indicates that the Ap4A:STING complex is more dynamic than the cGAMP:STING complex and that the interaction of Ap4A with STING is thermodynamically less stable (table S1). This was confirmed by molecular dynamics simulations showing that the Ap4A:STING complex is highly unstable compared to cGAMP:STING (fig. S5C).
On the basis of these predictions, we performed in vitro binding assays using recombinant GST-tagged C-terminal binding domain of mouse STING (GST-STING139–378; fig. S5D) and streptavidin-immobilized biotinylated Ap4A. GST and GST-tagged mouse Histidine triad nucleotide-binding protein 1 (Hint1) (fig. S5D), reported to bind Ap4A (33), were included as negative and positive controls, respectively. We observed that, as predicted in silico, STING139–378 interacts with Ap4A (Fig. 5K). Also in agreement with in silico predictions (table S1), competition experiments using unlabeled Ap4A, cGAMP, or DMXAA as competitors showed that binding of biotinylated Ap4A to STING can be easily displaced (Fig. 5L). The JB419 nonhydrolyzable analog of Ap4A also competed for the binding of STING to Ap4A (Fig. 5L), further suggesting that JB419 also interacts with STING. Thus, these data indicate that Ap4A can bind to STING. Our observations also confirm the in silico predictions that Ap4A and cGAMP share the same binding pocket on STING and that the binding of Ap4A to STING is less stable as compared to that of cGAMP. We therefore propose a model where LysRS and Ap4A regulate the activation of STING by two complementary mechanisms. First, LysRS recognizes cytosolic RNA:DNA hybrids, delaying their detection by cGAS. Second, binding of LysRS to RNA:DNA hybrids stimulates the production of Ap4A, which delays the binding of cGAMP to STING, resulting in delayed IFN response (Fig. 5M, left). In the absence of LysRS, cGAS recognizes RNA:DNA hybrids leading to a higher cGAMP production and subsequent type I IFN response (Fig. 5M, right).
LysRS-mediated negative regulation of Ifnβ expression is conserved in zebrafish
Last, we wished to challenge our finding in vivo in zebrafish embryos, taking advantage of the close evolutionary resemblance of their innate antiviral pathways to that of humans (34). We first verified that recombinant zebrafish LysRS (zLysRS) interacts with RNA:DNA hybrids in vitro (Fig. 6A). We then performed whole-body knockdown of the unique zebrafish ortholog of LysRS (zLysRS) by injecting wild-type one-cell zebrafish embryos with a splice-blocking morpholino targeting zlysrs (Fig. 6B) or with a control morpholino. Microscopy analysis of fish morphology showed no developmental defect (Fig. 6C). Quantification of the expression levels of type I IFN (ifnφ1) and the IFN-response gene isg15 at 72 hours after injection showed that knocking down lysrs led to a global up-regulation of the IFN response (Fig. 6D). This establishes that negative regulation of type I IFN by LysRS is conserved in fish and mammals.
Fig. 6. LysRS-mediated negative regulation of Ifnβ expression is conserved in zebrafish.
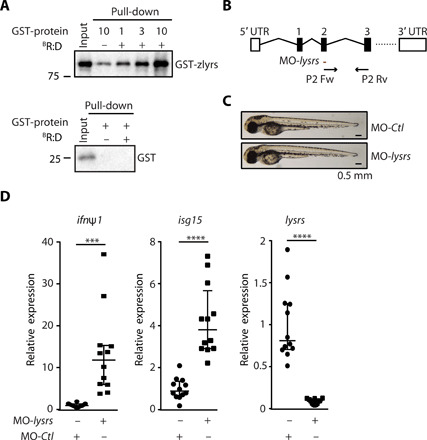
(A) WB analysis of pull-down experiments performed as in Fig. 2D, except that 1, 3, and 10 pmol of recombinant zebrafish LysRS (zlysrs) protein were used; (n = 3). (B) Schematic representation of the zebrafish lysrs locus. The region targeted by lysrs-targeting morpholino oligo (MO-lysrs) and primers used for amplification of lysrs are indicated by a red line or black arrows, respectively. Boxes, exons; straight lines, introns. (C) Larvae injected with MO-lysrs or with control MO (MO-Ctl). (D) lysrs, ifnφ1, and isg15 mRNA levels were quantified by RT-qPCR in larvae injected with MO-lysrs or with MO-Ctl. Median (±SEM) of 12 pools of three independent larvae per condition. Unpaired t test. ***P < 0.001, ****P < 0.01. All blots show representative experiments.
DISCUSSION
In summary, we show that RNA:DNA hybrids are bona fide ligands of cGAS and that LysRS represses RNA:DNA hybrid–dependent cGAS-STING activation through two complementary mechanisms. First, LysRS interacts with RNA:DNA hybrids, preventing, or delaying, their recognition by cGAS. Consistently, knocking down LysRS leads to enhanced cGAMP production, attesting to increased cGAS activation. This infers that in the absence of LysRS the bioavailability of RNA:DNA hybrids for recognition by cGAS is enhanced. Considering that cGAS is an ISG and that LysRS is highly expressed in cells, their relative abundance is likely to be a key determinant of STING activation. In pathological situations where acute nucleic acid–induced STING stimulation occurs, such as upon chemotherapy or radiotherapy regimens or viral infections (35), it is likely that masking of RNA:DNA hybrids by LysRS is critical. Second, we identify here that simulation with RNA:DNA hybrids leads to LysRS-dependent Ap4A production that, in turn, delays the activation of type I IFN responses by binding to the cGAMP binding pocket of STING, confirming its signaling function in mammalian cells (17, 18, 28).
LysRS-dependent inhibition of STING is likely complementary to existing cellular processes that regulate cytosolic levels of nucleic acids to avoid unwanted triggering of the cGAS-STING pathway (7). Enzymes such as deoxyribonuclease II (DNase II), TREX 1, RNaseH 1, or RNaseH 2 are in charge of degrading cytosolic nucleic acids, preventing their detection. Thus, at resting state, despite the potential generation of RNA:DNA hybrids through processes such as transcription-replication collision or immunoglobulin class switching (11), it is likely that LysRS is mostly engaged in translation. Hence, in the absence of such nucleases, the LysRS-Ap4A pathway may play a pivotal role. For example, it is reported that the absence of RNaseH2 leads to up-regulation of STING-dependent type I IFN responses through the appearance of dsDNA species containing ribonucleotides that induce DNA damage rather than through an increase of total RNA:DNA hybrids (36). While it can be expected that, in this model, LysRS would not have its inhibitory effect, due to the lack of RNA:DNA hybrids, it would be important to assess the impact of treatment with Ap4A or JB419. In addition, it has been shown that members of the MSC can interfere with retroviral life cycles. In particular, LysRS interferes with the HIV-1 replication cycle (15) and that HIV-1 infection promotes nuclear translocation of LysRS (37), a process that may lead to the production of Ap4A (18), favoring the viral life cycle by decreasing the antiviral response. It would be therefore important to test whether LysRS is able to bind HIV-1–derived RNA:DNA hybrids, whether this leads to Ap4A production, and the impact of the latter on the viral life cycle.
While our work reveals that the LysRS-Ap4A axis is a key pathway involved in physiological regulation of STING activation, it also raises questions regarding the involvement of the MSC in regulating the innate immune responses. Other members of the MSC, such as bifunctional glutamate/proline-tRNA ligase (EPRS) (38), have already been shown to play roles in the regulation of immune responses. Thus, it would be important to examine whether knocking down additional members of the MSC, together with LysRS, can enhance the effect of LysRS on the cGAS-STING pathway. In addition, we also show that LysRS-mediated inhibition of type I IFN response is a conserved mechanism in mammals and fish. However, it is likely that the overall involved molecular mechanisms differ. While it has been described that in vitro, the zebrafish orthologs of cGAS can produce cGAMP (39), it has been previously reported that cGAS is dispensable for innate immune detection, to the contrary of probable ATP-dependent RNA helicase DDX41 and ATP-dependent RNA helicase A (DHX9), in zebrafish (40). It would therefore be important to test whether LysRS-mediated inhibition of IFN responses may be mediated by sequestration of RNA:DNA hybrids from other sensors than cGAS.
Last, our data also show that the JB419 cell-permeant nonhydrolyzable analog of Ap4A (31) antagonizes STING to levels similar to those observed for Ap4A. Thus, this compound bears the potential for translation to the clinic and opens the way to the design of derived compounds with higher affinity for STING or increased retention time in the cGAMP-binding pocket. While such compounds would have to be carefully assessed for side effects and cytotoxicity before testing in animal models of chronic inflammation, our study provides basis for the design of a novel class of pharmaceutical inhibitors of STING. In addition, activating endogenous LysRS to produce Ap4A may also be a relevant approach to study the effect of long-term exposure to these compounds on global immune responses. Our work thus warrants further studies to better characterize the role of the LysRS-Ap4A axis in human pathologies where inflammatory responses are reported.
MATERIALS AND METHODS
Study design
The present study aimed at identifying the molecular mechanisms governing regulation of STING activation. Experiments performed in vitro, in cells, and in vivo were carefully controlled. In vitro experiments performed using recombinant proteins or cell lysates were performed independently using different protein preparations. Experiments performed in cells were performed in technical duplicates or triplicates, and biological repetitions are indicated in the figure legends. In vivo experiments in zebrafish were repeated twice on pools of three individual larvae. Morpholino injections were blinded. No outlier was excluded in the course of the analyses.
Cells and cell cultures
293T, A549, HeLa, HeLa-S3, WT-MEF, MEFSting−/−, MEFcGas−/−, MEFTrex1−/−, T98G, and T98GF/H-cGAS were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% penicillin/streptomycin, and 1% glutamine. T98G overexpressing F/HA-cGAS were generated by transducing parental T98G with viral particles produced by using the pOZ-F/HAcGAS construct and selected with puromycin. Cells were maintained in DMEM supplemented with 10% FBS, 1% penicillin/streptomycin, and 1% glutamine. A549 were obtained from S. Goodborn. WT-MEF, MEFSting−/−, and MEFcGas−/− were a gift of S. R. Paludan, MEFTrex1−/− were obtained from J. Rehwinkel, and parental T98G were obtained from C. Goujon.
Mouse neonatal fibroblast
MNFs were purified as described by the Krishma Halai laboratory. Briefly, skin was isolated from newborns at day 1, incubated in Krebs-Ringer Hepes (KRH) buffer containing 10 mM Hepes and 1% penicillin/streptomycin and minced in pieces with a scalpel before digestion in KRH/ATB buffer supplemented with collagenase (1 mg/ml) at 37°C. After centrifugation at 1000 rpm for 5 min, the supernatant was discarded, and the isolated cells put in culture in DMEM, 20% FBS, and 1% penicillin/streptomycin.
Plasmids
shRNAs targeting LysRS and STING were cloned into the pSUPERIOR.retro.puro vector from Oligoengine, according to the manufacturer’s instructions. N-terminally Flag- and HA-tagged codon-optimized mouse LysRS (MWG Eurofins) was cloned into the pOZ retroviral vector. For bacterial expression of recombinant proteins, the human LysRS gene was amplified by PCR from complementary DNA (cDNA) of HeLa cells. The cDNA and codon-optimized human cGAS (MWG Eurofins) were cloned into the pGEX-4T1 plasmid. Codon-optimized mouse LysRS was cloned into the pGEX-4T1 plasmid. Danio rerio LysRS (zLysRS) was amplified by PCR from the cDNA of zebrafish larvae and cloned in pGEX-4T1.
Viral particle production and infection
shRNA-expressing retroviral particles were produced by cotransfection of 2 × 106 293T cells with 5 μg of shRNA-containing pSUPERIOR, 2.5 μg of murine leukemia virus (MLV) GagPol, and 2.5 μg of A-MLV envelope, using the standard calcium phosphate transfection protocol. Retroviral particles containing the transgene encoding Flag- and HA-tagged LysRS (F/HA-LysRS) were produced following the same procedure except that cells were cotransfected with 5 μg of pOZ-F/HALysRS, 2.5 μg of MLV GagPol, and 2.5 μg of A-MLV envelope. Viral particles were harvested 48 hours after transfection, filtered with 0.45 μM filters, and used for transduction.
For knockdown of LysRS, 105 cells were seeded 24 hours before transduction. Medium was replaced 10 hours after transduction, and transfection was performed 72 hours later. A similar procedure was used for knockdown of LysRS and STING, except that retroviral particles containing shRNAs targeting LysRS and STING were added at the same time. For expression of F/HA-LysRS, 5 × 105 cells were seeded 24 hours before transduction. Twenty-four hours later, viral particles were added, and medium was replaced after 10 hours. RNA:DNA hybrids were transfected 96 hours later.
HSV plaque assay
MNFs were seed at a density of 0.5 to 1 × 105 cells per well in a 24-well plate (Nunc) in 500 μl of DMEM + 1% FBS 24 hours before infection with HSV-1 KOS or HSV-2 333 at multiplicity of infection (MOI) of 0.1. Supernatants were harvested 24 hours later, and virus yield was quantified using the plaque titration assay on Vero cells.
Protein purification
Recombinant GST-HINT, GST-LysRS, GST-cGAS, GST-STING (amino acids 139 to 378), and GST-ZfLysRS were produced in Escherichia coli BL21 cells. E. coli transformed with GST or GST expression constructs were grown in LB medium at 37°C to an absorbance at 600 nm of 0.6 before induction with 0.25 mM isopropyl-β-d-1-thiogalactopytanoside overnight at 16°C. Bacteria were harvested by centrifugation, resuspended in TETN-100 buffer [50 mM tris-HCl (pH 8), 100 mM NaCl, 1 mM EDTA, and 0.1% Triton X-100, supplemented with lysozyme (2 mg/ml; Sigma), 10 mM β-mercaptoethanol, and 0.5 mM phenylmethylsulfonyl fluoride (PMSF)], and incubated on ice for 30 min. Salt and detergent concentrations were increased, respectively, to 400 mM and 0.5% before sonication. Lysates were resuspended in TETN-400 buffer [50 mM tris-HCl (pH 8), 400 mM NaCl, 1 mM EDTA, and 0.5% Triton X-100, supplemented with 10 mM β-mercaptoethanol and 0.5 mM PMSF] and were clarified by centrifugation at 13,000 rpm for 30 min at 4°C before incubation with the appropriate volume of glutathione-Sepharose beads for 4 hours at 4°C. Sepharose beads were washed three times with ice-cold TETN-400, and recombinant proteins were eluted with elution buffer [150 mM NaCl and 50 mM tris-HCl (pH 8)] supplemented with 30 mM reduced l-glutathione. Eluates were quantified by Coomassie staining.
Nuclear cytoplasmic fractionation
T98GF/H-cGAS cells were fractionated according to Gentili et al. (26) Briefly, cells were harvested and cell pellet was washed once in 400 μl of cold cytoplasmic lysis (CL) buffer [10 mM Hepes (pH 7.9), 10 mM KCl, and 1.5 mM MgCl2] in the presence of 0.5 mM PMSF and centrifuged at 300g for 4 min. The supernatant was discarded, and cell pellet was lysed by adding three cell pellet volumes of cold CL for 10 min on ice, with gentle flicking. Cytosolic extract was obtained by adding 0.625% final of NP-40 for an additional of 5 min on ice, with gentle flicking. Nuclei were pelleted at 16,000g for 5 min, and cytosolic extracts were collected in a fresh tube. Nuclei were then lysed by adding three cell pellet volumes of cold nuclear lysis buffer [420 mM NaCl, 20 mM Hepes (pH 7.9), 1.5 mM MgCl2, 0.2 mM EDTA, 25% glycerol, and 0.5 mM PMSF] for 15 min on ice, with gentle flicking. Nuclear lysates were vortexed and sonicated for 20 min at 4°C in a Bioruptor ultrasonic bath. Nuclear lysates were cleared by centrifugation at 16000g for 5 min.
Biotinylated nucleic acid pull-down using recombinant protein
In vitro interaction between RNA:DNA hybrids and recombinant proteins was performed using Dynabeads M280. Beads were blocked overnight in blocking buffer [20 mM Hepes (pH 7.9), 15% glycerol, 0.05% NP-40, and 50 mM NaCl, supplemented with 2 mM dithiothreitol (DTT), 100 mM NaCl, and bovine serum albumin (BSA; 10 mg/ml)]. After three washes in 1× wash buffer [5 mM tris-HCl (pH 7.5), 1 mM EDTA, and 1 M NaCl], M280 beads were coupled to nucleic acids according to the manufacturer’s instructions. Briefly, beads were incubated in 30 μl of 2× wash buffer [10 mM tris-HCl (pH 7.5), 2 mM EDTA, and 2 M NaCl] containing 20 pmol of BRNA:DNA hybrids at 25°C for 15 min. Protein binding was performed at 37°C for 30 min with increasing amounts of recombinant proteins in 20 mM Hepes (pH 7.9), 15% glycerol, 0.05% NP-40, and 150 mM NaCl, supplemented with 2 mM DTT, 2 mM PMSF, and RNase inhibitor. When indicated, hybrids were treated with 30 U of RNaseH (Ambion) in RNaseH buffer [50 mM tris-HCl (pH 8), BSA (50 ng/ml), 1 mM DTT, and 4 mM MgCl2] for 1 hour at 30°C, before binding to the beads. After three washes, bound material was eluted in 30 μl of Laemmli buffer. Pull-down in competition between hcGAS and hLysRS was performed as described above except that the two proteins were added at the same time to the beads coupled to the BRNA:DNA hybrids.
Biotinylated nucleic acid pull-down using cell extracts
Interaction of endogenous protein and transfected biotinylated nucleic acids was tested by transfecting HeLa cells with nucleic acids (2 μg/ml) using JetPrime according to the manufacturer’s protocol. Six hours after transfection, cells were harvested and lysed in TENTG-150 [20 mM tris-HCl (pH 7.4), 0.5 mM EDTA, 150 mM NaCl, 10 mM KCl, 0.5% Triton X-100, 1.5 mM MgCl2, and 10% glycerol, supplemented with 10 mM β-mercaptoethanol and 0.5 mM PMSF] on ice for 30 min. Lysates were centrifuged at 13,000 rpm for 30 min at 4°C. Equal amounts of whole-cell lysates were incubated for 3 hours at 4°C on a wheel with 3 μg (30 μl) Dynabeads M280 blocked overnight at 4°C as described above. After three washes in 20 mM tris-HCl (pH 7.4), 0.5 mM EDTA, 0.05% Triton, 0.1% Tween, 150 mM NaCl, 10% glycerol, and 5 mM MgCl2, bound material was eluted in Laemmli buffer.
In vitro biotinylated nucleic acid pull-down
Pull-down was carried out using 3 μg (30 μl) of Dynabeads M280 per condition. Beads were blocked overnight as described above. After three washes in 1× wash buffer, 100 pmol of nucleic acid was coupled to M280 beads according to the manufacturer’s instructions before equilibration in dialysis buffer [20 mM tris-HCl (pH 7.4), 0.5 mM EDTA, 150 mM NaCl, 10% glycerol, and 1.5 mM MgCl2]. Cytosolic extracts from HeLa S3 cells, prepared using the Dignam protocol, were diluted in dialysis buffer supplemented with 2% Tween, 1% Triton, 10 mM β-mercaptoethanol, and 0.5 mM PMSF and centrifuged at 13,000 rpm for 30 min at 4°C. One milliliter of diluted lysate was added to the beads and incubated at 4°C on a rocker for 4 hours in low-binding tubes (Axygen). Three consecutive washes were performed in 20 mM tris-HCl (pH 7.4), 0.5 mM EDTA, 0.05% Triton, 0.1% Tween, 150 mM NaCl, 10% glycerol, and 5 mM MgCl2. Tubes were changed at first and last washes. Bound material was eluted in 30 μl of Laemmli buffer.
Pull-down using biotinylated Ap4A and recombinant proteins
In vitro interaction between STING and biotinylated Ap4A was tested using Dynabeads M280, blocked as described above. Ten micromolar of biotinylated Ap4A (BioLog) or biotin was bound to the beads at 25°C for 30 min in 2× wash buffer. Thirty, 50, or 100 μg of recombinant proteins (mSTING, mHINT, and GST) was incubated with the beads for 1 hour at 4°C on the wheel in a final volume of 200 μl in 150 mM NaCl, 50 mM tris-HCl (pH 8.0), 10 mM MgCl2, BSA (0.1 μg/μl), 10 mM β-mercaptoethanol, and 0.5 mM PMSF. Competition was performed after protein binding using a 10-fold excess of the competitor (DMXAA, 2′3′-cGAMP, Ap4A, or JB419) for 1 hour at 4°C on wheel in a final volume of 200 μl in the buffer described above. After three washes in 150 mM NaCl, 50 mM tris-HCl (pH 8.0), 10 mM MgCl2, 0.1% Tween, BSA (0.1 μg/μl), 10 mM β-mercaptoethanol, and 0.5 mM PMSF, bound material was eluted in 30 μl of Laemmli buffer.
S9.6 immunoprecipitation
Endogenous RNA:DNA hybrids were immunoprecipitated using the S9.6 antibody. Briefly, HeLa cells were crosslinked with 1% paraformaldehyde (PFA) for 20 min at room temperature. PFA was quenched by incubation with 125 mM glycine for 5 min at room temperature. Cells were harvested and lysed in TENTG-150 on ice for 30 min. Lysates were centrifuged at 13,000 rpm at 4°C for 30 min. The soluble fraction was precleared for 20 min at 4°C with 15 μl of agarose protein G beads, followed by incubation overnight at 4°C with 30 μl of agarose protein G beads coupled to 10 μg/ml of either irrelevant mouse immunoglobulin G (IgG) or S9.6 antibody. Beads were washed five times in TENTG-150 buffer, and immunoprecipitated material was eluted in Laemmli buffer.
RNA extraction and RT-qPCR
RNA was extracted using TRIzol (Invitrogen) and treated with TURBO DNase (Ambion) according to manufacturer’s protocols. Reverse transcription (SuperScript IV reverse transcriptase, Invitrogen) and qPCR using specific primers were performed using SYBR Green Master Mix (Takara) and LightCycler 480 cycler (Roche). mRNA levels were normalized to Gapdh mRNA levels except in zebrafish experiments where mRNA levels were normalized to zebrafish actin (zactin).
Whole-cell extract preparation and immunoblot
Cells were either lysed in 3 packed cell volume (PCV) of radioimmunoprecipitation assay buffer [150 mM NaCl, 50 mM tris-HCl (pH 8), 0.5% sodium deoxycholate, 0.1% SDS, and 1% NP-40] supplemented with 0.5 mM PMSF for 15 min at 4°C or 5 PCV of TENTG-150 for 30 min at 4°C. Lysates were centrifuged 30 min at 13,000 rpm, and supernatants were collected for immunoblot. For phosphorylated protein analysis, buffer was supplemented with PhosphoSTOP (Roche) before whole-cell extraction. Protein quantification was performed using Bradford assay. Samples were resolved on SDS-PAGE, and proteins were transferred onto nitrocellulose membranes. Primary antibodies used include anti-phospho IRF3 (1:500; Cell Signaling 4D4G; 1:500; Abcam Ab76493), anti-IRF3 (1:1000; Cell Signaling D6I4C), anti-phospho TBK1 (1:1000; Cell Signaling D52C2), anti-TBK1 (1:1000; Cell Signaling D1B4), anti-STING (1:1000; Cell Signaling D2P2F), anti-HA (1:10000; Roche), anti–glyceraldehyde-phosphate dehydrogenase (GAPDH; 1:5000; Proteintech Europe 800004-1-Ig), anti-AMP1 (1:1000; Bethyl Laboratories A304896A-M), anti-DARS (1:1000; Bethyl Laboratories A304799A-M), anti-human LysRS (1:1000; Bethyl Laboratories A300630A-M), anti-mouse LysRS (1:2000; Proteintech Europe 14951-1AP), anti-GST (1:10000; Bethyl Laboratories A190122A), anti PARP-1 (1:500; Santa Cruz Biotechnology F-2 sc8007), anti-tubulin (1:2000; Proteintech Europe 66031-1-Ig), anti–TREX-1 (1:250; Santa Cruz Biotechnology C-11 sc133112), and anti-cGAS mouse specific (1:1000; Cell Signaling D3080). The S9.6 antibody was a gift from S. Leppla. All secondary antibodies (Cell Signaling) were used at 1:2000 dilution. Signal was visualized with SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific), and images were acquired on a ChemiDoc (Bio-Rad).
Morpholino knock-down experiments
Eggs of wild-type AB zebrafish (one-cell stage) were microinjected with 4 ng of the KARS- specific antisense morpholino (MO3i3, TCCATATTCGCTACTCATCGTACA) or with a control morpholino (MOctl, GAAAGCATGGCATCTGGATCATCGA). The KARS-specific MO targets the splice donor site between exon 3 and intron 4. Efficiency of knockdown was assessed by RT-qPCR with splice-sensitive primers [TGGACCCCAATCAATACTTCAAG (forward) and GGTCTCCAGGCTGAAGGTGGTTAT (reverse)]. Embryos then developed with no obvious morphological defects at 28°C. At 72 hours after injection, embryos were collected for gene expression analysis by RT-qPCR.
Molecular modeling
Molecular docking of Ap4A on STING was performed using ZDOCK version 3.0 using the STING crystal structure (PDB: 4KSY). RDOCK was used to refine and quickly evaluate the results obtained by ZDOCK. Energy minimization was performed to remove the geometrical strain from the top-ranking poses of Ap4A during the docking experiments. Top hit complexes of Ap4A and STING were subjected to extensive energy minimization run using the Amber99 force field as it is implemented into Gromacs, version 4.5.5, via the Gromita graphical interface, version 1.07. An implicit generalized born solvation was chosen at this stage, in an attempt to speed up the energy minimization process.
The interaction space and binding potential of each docking conformation were further explored by subjecting the molecular complexes to unrestrained molecular dynamics simulations using the Gromacs suite, version 4.5.5. Molecular dynamics took place in a periodic environment, which was subsequently solvated with sphingosylphosphoryl choline water using the truncated octahedron box extending to 7 Å from each molecule. Partial charges were applied, and the molecular systems were neutralized with counter ions as required. The temperature was set to 300 K, and the step size was set to 2 fs. The total run of each molecular complex was 100 ns, using the NVT ensemble in a canonical environment. NVT stands for number of atoms, volume, and temperature that remain constant throughout the calculation. The results of the molecular dynamics simulations were collected into a molecular trajectory database for further analysis.
Determination of intracellular Ap4A levels in MEF and HeLa
Cellular nucleotide extraction and Ap4A determination were performed using a sensitive luminescence-based assay. Briefly, nucleotides were extracted from cells by adding 5 ml of ice-cold trichloroacetic acid and subsequent neutralization with an equal volume of 0.6 M tri-octylamine in 1,1,2-trichlorotrifluoroethane. Following centrifugation at 500g for 5 min, the top aqueous layer was removed, and 10 U of thermo-sensitive alkaline phosphatase (Thermo Fisher Scientific) was added to degrade adenosine triphosphate (ATP). Remaining nucleotides were concentrated using DEAE-Sephacel beads. Following mixing and centrifugation at 12,000g for 1 min, beads were washed with water. Nucleotides bound to DEAE-Sephacel were eluted twice with 1 M triethylammonium bicarbonate (pH 8.5), and eluates were vacuum-dried. Nucleotides from the two eluates were combined and dissolved in 120 μl of Ap4A assay buffer [25 mM Hepes-NaOH (pH 7.8) and 5 mM magnesium acetate]. To remove any remaining ATP, a further 10 U of alkaline phosphatase was added, incubated at 37°C, and subsequently incubated at 95°C to denature alkaline phosphatase. Ap4A levels in 10 μl of sample were determined by adding 50 μl of Bactiter GLO (Promega). Background ATP levels were first measured on a Berthold Lumat 9507, and once stabilized, recombinant Ap4A hydrolase was added to cleave Ap4A (ATP + AMP), and levels of ATP were measured by the increase in luminescence. Levels of Ap4A in samples were determined by comparison to standards and expressed as pmol per 106 cells.
cGAMP ELISA
cGAMP enzyme-linked immunosorbent assay (ELISA) was performed according to the manufacturer’s protocol using the Cayman Chemical 2′3′-cGAMP ELISA Kit (Bertin Bioreagents). MEFTrex−/− were seeded at 3 × 106 in 150-cm2 plates 24 hours before transduction. Forty-eight hours later, cells were seeded at 3 × 106, and 24 hours later, cells were transfected with RNA:DNA hybrids (2 µg/ml). For cGAMP quantification, cells were harvested, counted, washed in phosphate-buffered saline (PBS), pelleted, and frozen in 500 μl of 80% MeOH/H2O (80/20, v/v) at −80°C until extraction. Samples were thawed and subjected to five freeze/thaw cycle in liquid nitrogen. Lysates were centrifuged at 16,000 rcf at 4°C for 20 min. The recovered supernatants were subjected to speed vacuum drying in SAVAN 110 SpeedVac Concentrator at 65°C for 2 hours. The pellets were resuspended in 50 μl of RNase/DNase-free water.
Radiolabeling
For radiolabeling experiments, HeLa cells were lysed in TENTG-150 buffer, and DNA was extracted with phenol/chloroform/isoamyl (pH 8) (12/12/1). DNA was subsequently dephosphorylated using Shrimp Alkaline Phosphatase (rSAP) New England Biolabs (NEB) before labeling with γ32P ATP for 30 min at 37°C using the T4 polynucleotide kinase (NEB). Subsequent RNaseH treatment was performed with 10, 20, or 40 U following the manufacturer’s protocol. Unbound radiolabeled nucleotides were removed using Illustra Microspin G-50 Columns before resolution on 5% acrylamide, 0.5% tris borate ethylamide gel, and autoradiography. Images were acquired using Thyphoon FLA 7000.
Cell treatment and transfection
Cells were transfected with JetPrime transfection reagent (Polyplus) at 1:2 ratio with various nucleic acids at 2 μg/ml. DMXAA (Invivogen) was used at 200 μM in Opti-MEM (Gibco). Ap4A (Sigma-Aldrich) or JB419 was used at 500 μM in Opti-MEM.
Immunofluorescence and microscopy analysis
WT-MEF were plated 24 hours before transfection with Cyanin3-labeled RNA:DNA hybrids (2 μg/ml) in Opti-MEM using the JetPrime reagent, according to the manufacturer’s instructions. Two hours after transfection, cells were detached with 0.5 mM EDTA in PBS and plated on fibronectin-treated coverslips. Four hours later, cells were fixed with 4% PFA in PBS, and permeabilization was performed with 0.1% Triton X-100 in PBS for 5 min at room temperature (RT). After blocking in PBS containing 0.1% Tween (PBS-T) and 5% BSA for 30 min at RT, cells were incubated overnight with the S9.6 antibody at 1/200 dilution in PBS-T, 5% BSA.RNaseH treatment before primary antibody incubation was performed, when indicated, with 60 U of RNaseH (Ambion) in RNaseH buffer in a humid chamber overnight at 30°C. Secondary antibody incubation was performed for 1 hour at RT. Cytoskeleton was stained with Actin Green 488 ReadyProbes reagent (Molecular Probes by Life Technologies), and the nucleus with 4′,6-diamidino-2-phenylindole. Vectashield reagent was used to mount the coverslips. Z-stacks were acquired by a Apotome Z2 microscope by Zeiss with ZEN (blue edition) software, and images were processed with Fiji.
Statistical analysis
Statistical analysis was performed using GraphPad Prism version 7. Unpaired or one-column t test was performed as indicated in figure legends.
Compounds (PubChem CID)
DMXAA (Vadimezan): 123964
Ap4A (Diadenosine tetraphosphate): 21706
2′3′ cGAMP (cyclic guanosine monophosphate-adenosine monophosphate): 135564529
Oligonucleotide sequences
mIFNβ fw 5′-CTGCGTTCCTGCTGTGCT TCTCCA-3′
mIFNβ rv 5′-TTCTCCGTCATCTCCATA GGGATC-3′
mSTING fw 5′-TTGGGTACTTGGGGTTGATCTT-3′
mSTING rv 5′-GCACCACTGAGCATGTT GTTATG-3′
mLysRS fw 5′-GTACTGCCCTGGAATAC GGG-3′
mLysRS rv 5′-TTTCAGTGGTCGCTGCAG TT-3′
mGAPDH fw 5′-TTCACCACCATGGAGAAGGC-3′
mGAPDH rv 5′-GGCATCGACTGTGGTCATGA-3′
zfACTIN fw 5′ CGAGCTGTCTTCCCATCCA 3′
zfACTIN rv 5′-TCACCAACGTAGCTGTCTTTCTG-3′
zfIFNϕ fw 5′-GAATGGCTTGGCCGATACAGGATA-3′
zfIFNϕ rv 5′-TCCTCCACCTTTGACTTGTCCATC-3′
zfLysRS fw P2 5′-TGGACCCCAATCAATACTTCAAG-3′
zfLysRS rv P2 5′-GGTCTCCAGGCTGAAGGTGGTTAT 3′
Morpholino sequences
MO3i3 5′-TCCATATTCGCTACTCATCGTACAT-3′
Control MO 5′-TACCAAAAGCTCTCTTATCGAGGGA-3′
Oligo sequences for annealing
RNA 2 or BRNA 2 were annealed with RNA 5 to generate non biotinylated or biotinylated dsRNA; RNA 2 or BRNA 2 were annealed with DNA 9 to generate nonbiotinylated or biotinylated RNA:DNA hybrids; DNA 9 or BDNA 9 were annealed with DNA 6 to obtain nonbiotinylated or biotinylated dsDNA. Annealing was performed in a MasterCycler nexus gradient (Eppendorf) in annealing buffer [60 mM NaCl, 5 mM tris-HCl (pH 7.5), and 0.2 mM EDTA] using the following program: 4 min at 95°, 85°, 82°, 80°, 78°, 75°, and 72°C, followed by 10 min at 70°C and slow cooling down to 10°C over a period of 2 hours. The quality of annealed material was controlled by running 500 ng of annealed material on a 10% nondenaturing acrylamide gel using ssRNA and ssDNA as controls.
RNA 2: UUU CAA UUC CUU UUA GGA UUA AUC UUG AAG AUA GAG UUA A
RNA 5: UUA ACU CUA UCU UCA AGA UUA AUC CUA AAA GGA AUU GAA A
DNA 9: TTA ACT CTA TCT TCA AGA TTA ATC CTA AAA GGA ATT GAA A
DNA 6: TTT CAA TTC CTT TTA GGA TTA ATC TTG AAG ATA GAG TTA A
BRNA 2: BIO- UUU CAA UUC CUU UUA GGA UUA AUC UUG AAG AUA GAG UUA A
BDNA 9: BIO- TTA ACT CTA TCT TCA AGA TTA ATC CTA AAA GGA ATT GAA A
shRNA sequences
shSTING fw (mouse)
5′-GATCCCCTGATTCTACTATCGTCTTATTCAAGAGATAAGACGATAGTAGAATCATTTTTC-3′
shSTING rv (mouse)
5′-TCGAGAAAAA TGATTCTACTATCGTCTT ATCTCTTGAATAAGACGA TAGTAGAATCAGGG-3′
shLysRS fw (mouse) 1
5′-GATCCCCGAATCAGCATGGTAGAAGATTCAAGAGATCTTCTACCATGCTGATTCTTTTTC-3′
shLysRS rv (mouse) 1
5′-TCGAGAAAAAGAATCAGCATGGTAGAAGATCTCTTGAATCTTCTACCATGCTGATTCGGG-3′
shLysRS fw (mouse) 2
5′-GATCCCCGAAATTTATCGTCCGCTCTTTCAAGAGAAGAGCGGACGATAAATTTCTTTTTC-3′
shLysRS rv (mouse) 2
5′-TCGAGAAAAAGAAATTTATCGTCCGCTCTTCTCTTGAAAGAGCGGACGATAAATTTCGGG-3′.
shLysRS fw (mouse) 3
5′-GATCCCCGCCTTTCATCACCTATCACTTCAAGAGAGTGATAGGTGATGAAAGGCTTTTTC-3′
shLysRS rv (mouse) 3
5′-TCGAGAAAAAGCCTTTCATCACCTATCACTCTCTTGAAGTGATAGGTGATGAAAGGCGGG-3′
shLysRS fw (human)
5′-GATCCCCCAAGGTATCGCCAGAGATATTCAAGAGATATCTCTGGCGATACCTTGTTTTTC-3′
shLysRS rv (human)
5′-TCGAGAAAAACAAGGTATCGCCAGAGATATCTCTTGAATATCTCTGGCGATACCTTGGGG 3′
Supplementary Material
Acknowledgments
We thank J. Rehwinkel for the gift of immortalized MEF and MEFTrex1−/− generated from the mice of T. Lindhal and S. Leppla for the S9.6 antibody. We acknowledge the imaging facility MRI, member of the national infrastructure France-BioImaging infrastructure supported by the French National Research Agency (ANR-10-INBS-04, “Investments for the future”) and the IERP unit of INRA. We thank J. Déjardin, M. Benkirane, and S. McLennan for reading the manuscript. We also thank S. Chang and L. Balcon for technical support. We thank J. Venables for professional editing of the manuscript. Funding: Work in N.L.’s laboratory was supported by grants from the European Research Council (ERC STG 637763), the ANRS (109560), the “fondation ARC pour la recherche sur le cancer” (PJA20141201605), the Labex EpiGenMed, “Investissements d’avenir” program, (ANR-10-LABX-12-01), and Merck Sharp and Dohme Avenir (M.S.D. – GnoSTic). J.G. was supported by the ERC (637763) followed by an ANRS fellowship. K.P. was supported by the ERC (637763) followed by the Labex EpiGenMed (ANR-10-LABX-12-01). A.-L.V. was supported by M.S.D. and ERC (637763), B.A. and S.D. were supported by the ERC (637763), and C.T. was supported by M.S.D. I.K.V. was supported by the ERC (637763) and the « Prix Roger PROPICE pour la recherche sur le cancer du pancréas » of the Fondation pour la Recherche Médicale (FRM). We acknowledge the SIRIC Montpellier Cancer Grant INCa_Inserm_DGOS_12553 for support. Work in S.K.’s laboratories was funded by M.S.D. Work in N.J.J.’s and A.S.M.’s laboratories were funded by grants CR863 and CR968 and a Research Development Fund grant from North West Cancer Research (UK). Work in P.B.’s laboratory benefited from institutional support from “Institut National de la Recherche Agronomique” (INRA) and from funding and facilities from the TEFOR project–Investissement d’avenir–ANR-II-INBS-0014. S.R.P. is funded by the European Research Council (ERC-AdG ENVISION; 786602), the Novo Nordisk Foundation (NNF18OC0030274), and the Lundbeck Foundation (R198-2015-171 and R268-2016-3927). Work in B.N.’s laboratory was, in part, financially supported by statutory funds of the Centre of Molecular and Macromolecular Studies, Polish Academy of Sciences. Author contributions: Conceptualization: J.G. and N.L. Methodology: J.G, P.B., B.N., N.J.J., S.R.P., S.K., and C.L. Investigation: J.G., K.P., I.K.V., D.V., T.P., A.-L.V., A.S.M., R.K., N.J.J., A.H., B.A., S.D., and C.T. Writing: N.L., D.V., S.R.P., C.L., and J.G. Visualization: D.V. and J.G. Funding acquisition: N.L. Competing interests: B.N., R.K., N.L., and J.G. are inventors on a patent (STING inhibitors and their therapeutic uses) related to this work filed with European Patent Office (no. 19306072.0, 9 September 2019). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaax3333/DC1
REFERENCES AND NOTES
- 1.Danilchanka O., Mekalanos J. J., Cyclic dinucleotides and the innate immune response. Cell 154, 962–970 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gao P., Ascano M., Wu Y., Barchet W., Gaffney B. L., Zillinger T., Serganov A. A., Liu Y., Jones R. A., Hartmann G., Tuschl T., Patel D. J., Cyclic [G(2′,5′)pA(3′,5′)p] is the metazoan second messenger produced by DNA-activated cyclic GMP-AMP synthase. Cell 153, 1094–1107 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun L., Wu J., Du F., Chen X., Chen Z. J., Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang X., Shi H., Wu J., Zhang X., Sun L., Chen C., Chen Z. J., Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol. Cell 51, 226–235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ablasser A., Goldeck M., Cavlar T., Deimling T., Witte G., Röhl I., Hopfner K.-P., Ludwig J., Hornung V., cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 498, 380–384 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X.-D., Wu J., Gao D., Wang H., Sun L., Chen Z. J., Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341, 1390–1394 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartmann G., Nucleic acid immunity. Adv. Immunol. 133, 121–169 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patel S., Jin L., TMEM173 variants and potential importance to human biology and disease. Genes Immun. 20, 82–89 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu J.-J., Zhao L., Hu H.-G., Li W.-H., Li Y.-M., Agonists and inhibitors of the STING pathway: Potential agents for immunotherapy. Med. Res. Rev. 40, 1117–1141 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Khoo L. T., Chen L.-Y., Role of the cGAS–STING pathway in cancer development and oncotherapeutic approaches. EMBO Rep. 19, e46935 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Santos-Pereira J. M., Aguilera A., R loops: New modulators of genome dynamics and function. Nat. Rev. Genet. 16, 583–597 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Brégnard C., Guerra J., Déjardin S., Passalacqua F., Benkirane M., Laguette N., Upregulated LINE-1 activity in the fanconi anemia cancer susceptibility syndrome leads to spontaneous pro-inflammatory cytokine production. EBioMedicine 8, 184–194 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rigby R. E., Webb L. M., Mackenzie K. J., Li Y., Leitch A., Reijns M. A. M., Lundie R. J., Revuelta A., Davidson D. J., Diebold S., Modis Y., MacDonald A. S., Jackson A. P., RNA:DNA hybrids are a novel molecular pattern sensed by TLR9. EMBO J. 33, 542–558 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mankan A. K., Schmidt T., Chauhan D., Goldeck M., Höning K., Gaidt M., Kubarenko A. V., Andreeva L., Hopfner K.-P., Hornung V., Cytosolic RNA:DNA hybrids activate the cGAS–STING axis. EMBO J. 33, 2937–2946 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Motzik A., Nechushtan H., Foo S. Y., Razin E., Non-canonical roles of lysyl-tRNA synthetase in health and disease. Trends Mol. Med. 19, 726–731 (2013). [DOI] [PubMed] [Google Scholar]
- 16.Park S. G., Schimmel P., Kim S., Aminoacyl tRNA synthetases and their connections to disease. Proc. Natl. Acad. Sci. U.S.A. 105, 11043–11049 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee Y. N., Nechushtan H., Figov N., Razin E., The function of lysyl-tRNA synthetase and Ap4A as signaling regulators of MITF activity in FcepsilonRI-activated mast cells. Immunity 20, 145–151 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Yannay-Cohen N., Carmi-Levy I., Kay G., Yang C. M., Han J. M., Kemeny D. M., Kim S., Nechushtan H., Razin E., LysRS serves as a key signaling molecule in the immune response by regulating gene expression. Mol. Cell 34, 603–611 (2009). [DOI] [PubMed] [Google Scholar]
- 19.Park S. G., Kim H. J., Min Y. H., Choi E.-C., Shin Y. K., Park B.-J., Lee S. W., Kim S., Human lysyl-tRNA synthetase is secreted to trigger proinflammatory response. Proc. Natl. Acad. Sci. U.S.A. 102, 6356–6361 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deutscher M. P., The eucaryotic aminoacyl-tRNA synthetase complex: Suggestions for its structure and function. J. Cell Biol. 99, 373–377 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Flynn R. L., Zou L., Oligonucleotide/oligosaccharide-binding fold proteins: A growing family of genome guardians. Crit. Rev. Biochem. Mol. Biol. 45, 266–275 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwon S. B., Yu J. E., Park C., Lee J., Seong B. L., Nucleic acid-dependent structural transition of the intrinsically disordered N-terminal appended domain of human Lysyl-tRNA synthetase. Int. J. Mol. Sci. 19, E3016 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bersani F., Lee E., Kharchenko P. V., Xu A. W., Liu M., Xega K., MacKenzie O. C., Brannigan B. W., Wittner B. S., Jung H., Ramaswamy S., Park P. J., Maheswaran S., Ting D. T., Haber D. A., Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. U.S.A. 112, 15148–15153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim Y. W., Sanz L. A., Xu X., Hartono S. R., Chedin F., Genome-wide DNA hypomethylation and RNA:DNA hybrid accumulation in Aicardi–Goutières syndrome. eLife 4, 626 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tatematsu M., Funami K., Seya T., Matsumoto M., Extracellular RNA sensing by pattern recognition receptors. J. Innate Immun. 10, 398–406 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gentili M., Lahaye X., Nadalin F., Nader G. P. F., Lombardi E. P., Herve S., De Silva N. S., Rookhuizen D. C., Zueva E., Goudot C., Maurin M., Bochnakian A., Amigorena S., Piel M., Fachinetti D., Londoño-Vallejo A., Manel N., The N-terminal domain of cGAS determines preferential association with centromeric DNA and Innate immune activation in the nucleus. Cell Rep. 26, 3798 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Despotović D., Brandis A., Savidor A., Levin Y., Fumagalli L., Tawfik D. S., Diadenosine tetraphosphate (Ap4A) - an E. coli alarmone or a damage metabolite? FEBS J. 284, 2194–2215 (2017). [DOI] [PubMed] [Google Scholar]
- 28.Marriott A. S., Vasieva O., Fang Y., Copeland N. A., McLennan A. G., Jones N. J., NUDT2 disruption elevates diadenosine tetraphosphate (Ap4A) and down-regulates immune response and cancer promotion genes. PLOS ONE 11, e0154674 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vartanian A., Alexandrov I., Prudowski I., McLennan A., Kisselev L., Ap4A induces apoptosis in human cultured cells. FEBS Lett. 456, 175–180 (1999). [DOI] [PubMed] [Google Scholar]
- 30.Carmi-Levy I., Yannay-Cohen N., Kay G., Razin E., Nechushtan H., Diadenosine tetraphosphate hydrolase is part of the transcriptional regulation network in immunologically activated mast cells. Mol. Cell. Biol. 28, 5777–5784 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Krakowiak A., Pęcherzewska R., Kaczmarek R., Tomaszewska A., Nawrot B., Stec W. J., Evaluation of influence of Ap4A analogues on Fhit-positive HEK293T cells; cytotoxicity and ability to induce apoptosis. Bioorg. Med. Chem. 19, 5053–5060 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Duan Y., Wu C., Chowdhury S., Lee M. C., Xiong G., Zhang W., Yang R., Cieplak P., Luo R., Lee T., Caldwell J., Wang J., Kollman P., A point-charge force field for molecular mechanics simulations of proteins based on condensed-phase quantum mechanical calculations. J. Comput. Chem. 24, 1999–2012 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Lee Y.-N., Razin E., Nonconventional involvement of LysRS in the molecular mechanism of USF2 transcriptional activity in FcεRI-activated mast cells. Mol. Cell. Biol. 25, 8904–8912 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Langevin C., Aleksejeva E., Passoni G., Palha N., Levraud J.-P., Boudinot P., The antiviral innate immune response in fish: Evolution and conservation of the IFN system. J. Mol. Biol. 425, 4904–4920 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Motwani M., Pesiridis S., Fitzgerald K. A., DNA sensing by the cGAS–STING pathway in health and disease. Nat. Rev. Genet. 20, 657–674 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Mackenzie K. J., Carroll P., Lettice L., Tarnauskaitė Ž., Reddy K., Dix F., Revuelta A., Abbondati E., Rigby R. E., Rabe B., Kilanowski F., Grimes G., Fluteau A., Devenney P. S., Hill R. E., Reijns M. A. M., Jackson A. P., Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Duchon A. A., St. Gelais C., Titkemeier N., Hatterschide J., Wu L., Musier-Forsyth K., HIV-1 exploits a dynamic multi-aminoacyl-tRNA synthetase complex to enhance viral replication. J. Virol. 91, e01240-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee E.-Y., Lee H.-C., Kim H.-K., Jang S. Y., Park S.-J., Kim Y.-H., Kim J. H., Hwang J., Kim J.-H., Kim T.-H., Arif A., Kim S.-Y., Choi Y.-K., Lee C., Lee C.-H., Jung J. U., Fox P. L., Kim S., Lee J.-S., Kim M. H., Infection-specific phosphorylation of glutamyl-prolyl tRNA synthetase induces antiviral immunity. Nat. Immunol. 17, 1252–1262 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rolf J., Siedentop R., Lütz S., Rosenthal K., Screening and identification of novel cGAS homologues using a combination of in vitro and in vivo protein synthesis. Int. J. Mol. Sci. 21, E105 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ge R., Zhou Y., Peng R., Wang R., Li M., Zhang Y., Zheng C., Wang C., Conservation of the STING-mediated cytosolic DNA sensing pathway in zebrafish. J. Virol. 89, 7696–7706 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaax3333/DC1