

Combining multicellular signaling profiles with in vivo imaging reveals how nanotherapy can block local resistance pathways.
Abstract
Interpreting how multicellular interactions in the tumor affect resistance pathways to BRAF and MEK1/2 MAPK inhibitors (MAPKi) remains a challenge. To investigate this, we profiled global ligand-receptor interactions among tumor and stromal/immune cells from biopsies of MAPK-driven disease. MAPKi increased tumor-associated macrophages (TAMs) in some patients, which correlated with poor clinical response, and MAPKi coamplified bidirectional tumor-TAM signaling via receptor tyrosine kinases (RTKs) including AXL, MERTK, and their ligand GAS6. In xenograft tumors, intravital microscopy simultaneously monitored in situ single-cell activities of multiple kinases downstream of RTKs, revealing MAPKi increased TAMs and enhanced bypass signaling in TAM-proximal tumor cells. As a proof-of-principle strategy to block this signaling, we developed a multi-RTK kinase inhibitor nanoformulation that accumulated in TAMs and delayed disease progression. Thus, bypass signaling can reciprocally amplify across nearby cell types, offering new opportunities for therapeutic design.
INTRODUCTION
The mitogen-activated protein kinase (MAPK)/extracellular signal–regulated kinase (ERK) pathway plays a vital role in the regulation of cellular growth and survival. Aberrant MAPK signaling drives cancer progression in many malignancies and often arises due to activating alterations in the pathway’s key components including the small GTPase KRas (KRAS) and the serine/threonine-protein kinase that it activates, BRAF (v-Raf murine sarcoma viral oncogene homolog B). BRAF mutations are especially common in melanoma and papillary thyroid cancer, while KRAS mutations occur most frequently in pancreatic and colorectal cancers. In addition, BRAF and KRAS gene expression can be up-regulated, and this is especially the case for ovarian cancer (OVCA), which exhibits among the highest rates of RAS or RAF copy number amplification [CNA; 20 to 27% based on The Cancer Genome Atlas (TCGA) datasets] (1) and up-regulated gene expression (2–5) compared to other cancer types.
The frequency of aberrant MAPK/ERK activation across diverse cancer types has spurred extensive development of MAPK inhibitors (MAPKi). BRAF and MEK1/2 kinase inhibitors (BRAFi and MEKi, respectively) have been proven effective in treating patients with BRAFV600E-mutant melanoma, anaplastic thyroid, and non–small cell lung cancer, yet disease progression or recurrence often occurs within 12 months in patients with advanced disease. In other cancers, clinical trials of MEKi such as trametinib and selumetinib are ongoing, but results have been mixed, especially in disseminated malignancies of the gastrointestinal tract and peritoneal cavity. For example, a phase 2 trial of selumetinib in low-grade serous OVCA (LGSOC) demonstrated a modest response rate uncorrelated with the presence of BRAF or KRAS mutation (6), and mixed results have been seen with MEKi in BRAF-mutant metastatic colorectal carcinoma (7). Thus, a need exists to understand why durable response to MAPKi has been so difficult to achieve in patients and how responses could be improved.
Strides have been made in understanding mechanisms of resistance to MAPKi in BRAF-mutant disease. Both ERK pathway reactivation and bypass signaling downstream of receptor tyrosine kinases (RTKs) have been implicated and can arise through mechanisms involving genetic mutation (8, 9), genomic or transcriptional amplification (10, 11), posttranslational up-regulation (12), and engagement with cells and extracellular matrix of the tumor microenvironment (13–15). Uncovered mechanisms of MAPKi resistance have translated into improved clinical outcomes: ERK pathway reactivation following BRAFi motivated the strategy to combine BRAFi with MEKi, and combination BRAFi + MEKi safely outperforms single-agent therapy in melanoma (16). In other cases, putative resistance mechanisms have less efficiently translated to the clinic. For instance, bypass signaling through the RTK epidermal growth factor receptor (EGFR) has been observed as a possible explanation of MAPKi resistance in BRAF-mutant colorectal cancer, yet clinical trials combining BRAFi, MEKi, and EGFR inhibition have shown variable efficacy (7, 17). Currently, basket or bucket trials are increasingly used to target cancers based on markers of pathway dysregulation or genetic mutation rather than by cancer type, for instance, with the BRAFi vemurafenib in multiple nonmelanoma BRAFV600-mutant cancers, including glioma, sarcoma, pancreatic, and ovarian cancers (18). Emerging response data from these basket trials may help shed light on which MAPKi resistance mechanisms are most generalizable. For now, despite the growing catalog of resistance pathways, it can be unclear which are most important across cancer types and from patient to patient. Most work to date has examined MAPKi resistance from a cancer cell–autonomous perspective or has been very targeted in examining intercellular interactions between specific cell types within tumor tissue. It is increasingly apparent that bypass signaling in MAPKi resistance may be dependent on multiple diverse ligands in the tumor microenvironment, yet dissecting these intercellular signaling networks remains a challenge.
To overcome these issues, we pursued a multistep approach to elucidate new drug-resistance mechanisms arising from the tumor microenvironment. First, we applied systems-level analyses of bulk and single-cell patient gene expression data to parse the cellular and molecular contributions of the tumor microenvironment to MAPKi response. We focused on BRAF-mutant malignant melanoma treated with BRAFi + MEKi and MAPK-dysregulated OVCA, treated with MEKi to reflect current standard-of-care therapy and ongoing clinical trial activity, respectively. This analysis highlighted the enrichment of tumor-associated macrophages (TAMs) in MAPK-dysregulated disease and their correlation with poor treatment outcome. It also implicated reciprocated growth factor/RTK signaling between cancer cells and TAMs in MAPKi resistance. In the second step, targeted protein profiling of human cancer cell lines cocultured with macrophages (MΦ) confirmed these hypotheses and demonstrated that MΦ-supplied growth factors can promote prosurvival bypass signaling in cancer cells associated with elevated c-Jun N-terminal kinase (JNK) activity. Then, to visualize TAM-dependent bypass signaling in situ within the tumor, we developed an intravital microscopy approach to simultaneously visualize the real-time activities of ERK and JNK in cancer cells as they respond to MAPKi. This in vivo imaging revealed enhanced bypass signaling especially in TAM-neighboring cancer cells across tumor types. In a final step, for a proof-of-principle strategy to exploit this cross-talk, we developed a nanotherapy aimed at targeting the tumor microenvironment, with a formulation similar to those of clinical-stage nanomedicines (19, 20). Nanoparticle-foretinib (NanoFore) efficiently delivered high levels of the multi-RTK inhibitor (RTKi) foretinib—which targets the implicated RTKs AXL (tyrosine-protein kinase receptor UFO), MERTK (proto-oncogene tyrosine-protein kinase MER), and MET [hepatocyte growth factor (HGF) receptor, c-MET]—to TAMs and TAM-neighboring tumor cells. NanoFore combined with MAPKi to extend survival, while mere TAM depletion did not. Overall, this work demonstrates how global ligand/receptor coexpression analysis and in vivo microscopy can inform new translational strategies to overcome drug resistance in metastatic disease.
RESULTS
TAMs correlate with MAPKi resistance in patients, and TAMs coexpress, with cancer cells, ligands and receptors of the AXL/MERTK/TYRO3 family
Although numerous cancer cell–autonomous mechanisms of MAPKi resistance have been reported (8–12, 21–23), a systematic perspective of how intercellular dynamics in the tumor may contribute to overall clinical response is still lacking. We hypothesized that changes in the cellular composition of the tumor microenvironment could suggest putative cancer cell–nonautonomous mechanisms of MAPKi resistance. The bioinformatic tool CIBERSORT quantified leukocytes from bulk gene expression data of 19 BRAFV600-mutant melanoma biopsies derived from nine patients, taken before treatment and at disease progression (fig. S1A) (24). Patients received combined dabrafenib (BRAFi) and trametinib (MEKi) (n = 7) or monotherapy (n = 1 for each drug). We correlated changes in relative cell type abundance before and after treatment with the best response in tumor burden in those patients (Fig. 1A). CIBERSORT infers individual immune cell populations based on gene signatures from isolated cell populations, including “M2” [interleukin-4 (IL-4)–treated], “M1” [lipopolysaccharide (LPS)/interferon-γ (IFN-γ)–treated], and “M0” (untreated) MΦ populations. While increases in individual signatures for M0 and M2-like MΦ only moderately correlated with worse clinical response, the linear combinations of all MΦ subsets [M0 + M1 + M2] and especially [M0 + M2] were significantly correlative (Fig. 1, B and C, and fig. S1B). Poor responders did not have lower pretreatment MΦ, demonstrating that dynamic changes in TAM abundance and relative polarization contributions, as opposed to the initial levels, were more strongly associated with clinical outcome (fig. S1A). Thus, these pilot clinical data suggest that TAM behavior may be influencing response to MAPKi in patients with BRAF-mutant melanoma.
Fig. 1. Resistance-associated MΦ signaling networks in MAPK-mutant tumors.
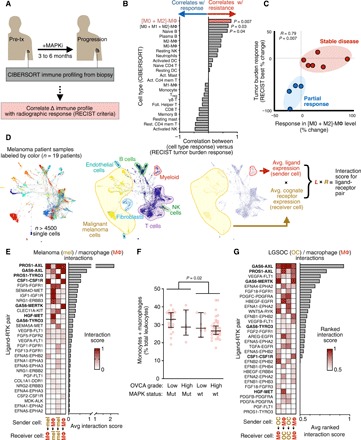
(A) Schematic depicting correlation analysis of patient biopsy immune profiling with radiographic response, used to generate data in (B) and (C). (B and C) From matched pre-MAPKi and at-progression biopsies, leukocyte change was correlated with best change in tumor burden following MAPKi in patients with melanoma (n = 9), shown across all CIBERSORT-quantified cell types (B) and with individual patient data points for the most significant immune correlate (C) (Spearman exact test with false discovery rate correction). Treg, regulatory T cells; NK, natural killer; wt, wild type; DC, dendritic cells. (D) SPRING visualization of single-cell RNA-sequencing (scRNA-seq) data from patients with melanoma, shown with individual cells pseudocolored according to the patient from which they were isolated (left) or to their annotated cell type (center). For global ligand-receptor coexpression analysis, average ligand expression levels of sender cells were multiplied with average cognate receptor expression levels of receiver cells (right). (E) Top growth factor/RTK coexpression tabulated from data in (D) and ranked according to scores between melanoma cells and MΦ (n = 19 patients). FGF, fibroblast growth factor; FGFR, fibroblast growth factor receptor. (F) Monocyte and MΦ abundance was quantified from OVCA biopsies using CIBERSORT and compared across tumors with or without RAS-MAPK–associated mutations (n = 69, medians ± interquartile range, two-tailed Mann-Whitney U test). (G) Top growth factor/RTK coexpression tabulated from LGSOC cancer cells (n = 3 patients) and ascites MΦ (n = 5 patients).
We next examined which molecular pathways TAMs may be communicating through to influence MAPKi response in tumor cells. We performed a systematic analysis of global ligand and matched receptor coexpression on a single-cell RNA sequencing (scRNA-seq) dataset consisting of over 4500 immune (CD45+) and nonimmune (CD45−, including malignant and stromal) cells from 19 patients with malignant melanoma (Fig. 1D) (25). By cross-referencing scRNA-seq with a database of >1100 high-confidence, literature-supported interactions between ligands and receptors (26–28), we systematically estimated the magnitude of cell communication among and across cell types within tumor tissue (fig. S1C). This analysis was largely dominated by highly expressed adhesion receptors (integrins), extracellular matrix components (collagen and laminin), and components of the human leukocyte antigen system and therefore made comparisons within classes of receptors difficult (fig. S1C). Consequently, guided by the initial TAM associations we identified (Fig. 1, B and C), along with literature implicating RTKs (10–12, 21–23), we quantified the roughly 200 known ligand-RTK interactions selectively between cancer cells and TAMs. Autocrine (homotypic) and paracrine (heterotypic) interactions were ranked between “sender cells” acting as ligand sources and “receiver cells” expressing cognate receptors (Fig. 1D). This analysis revealed ligand-mediated signaling along pathways already associated with MAPKi resistance, including colony-stimulating factor 1 (CSF1)/CSF1 receptor (CSF1R), HGF/MET, and fibroblast growth factor/fibroblast growth factor receptor (Fig. 1E). Signaling in these pathways was largely asymmetric, such that cancer cells served as either the sender or receiver but not both. In contrast, high levels of joint ligand-receptor expression were observed among the AXL/MERTK/TYRO3 family of RTKs, and the analysis indicated mutual, reciprocal communication such that cancer cells could serve as both senders and receivers. Growth arrest-specific 6 (GAS6) and protein S (PROS1) are both ligands of the AXL/MERTK/TYRO3 family shown to be relevant in this analysis, with GAS6 more selectively expressed in TAMs compared to tumor cells (Fig. 1E). These signaling pathways are not restricted solely to TAMs, as other leukocyte and stromal populations—including myeloid subpopulations of monocytes, dendritic cells, neutrophils, and multiple MΦ polarization subsets across the spectrum of polarization phenotypes (29, 30)—can express their components. Nonetheless, MΦ express relatively high levels in both patients and mice compared to other cell types (fig. S1, D to F). Overall, this analysis suggests that TAMs can supply growth factors to amplify signaling of RTKs previously implicated in cancer-intrinsic kinase inhibitor resistance and cancer cells can, in turn, amplify TAM signaling along AXL, MERTK, and CSF1R pathways traditionally associated with M2-like tumor-promoting phenotypes (31, 32).
Because basket trials are designed around similar genomic alterations, some emergent mechanisms of resistance may also be similar, even across different cancer types. We therefore studied the extent to which our findings in melanoma might extend to OVCA, since KRASG12D and BRAFV600E mutations are prevalent in certain OVCA subtypes (for instance, >50% prevalence in some LGSOC and serous borderline populations) (33), RAS or RAF expression can be up-regulated in OVCA compared to other cancer types (see Materials and Methods for statistical details), and OVCA is less studied in the context of MAPKi, shows poor prognosis, and has been poorly responsive to MAPKi therapy in clinical trials (6). Although no publicly available datasets currently exist for OVCA biopsies before and after MAPKi, we were able to profile the immune composition of 69 OVCA patient biopsies acquired from primary debulking surgery (34). CIBERSORT analysis of this dataset indicated that monocytes and MΦ were at baseline highly abundant in OVCA, especially in MAPK-mutant tumors primarily of the LGSOC subtype, suggesting that they may contribute in MAPKi resistance as was implicated in melanoma (Fig. 1F). To assess signaling pathways expression data from purified CD16+ MΦ, we tabulated joint expression of ligands and cognate RTKs using bulk RNA expression data from purified CD16+ BDCA1− MΦ from the ascites of five patients with OVCA (35), along with EpCAM+ (epithelial cell adhesion molecule–positive) epithelial cells from the primary tumors, metastases, and ascites of three patients with LGSOC (Fig. 1G) (36). As found in patients with melanoma, this analysis similarly showed the potential for cross-talk between cancer cells and TAMs—occurring not only unidirectionally via CSF1 and its receptor (CSF1-CSF1R), along with HGF and its receptor (HGF-MET), but also in a mutually reciprocated manner via AXL and its ligand GAS6 (Fig. 1G).
MAPKi elicits TAM accumulation in patient-derived and mouse models of BRAFV600E cancer
To better understand how TAMs may affect MAPKi responses, we quantified TAMs across multiple BRAFV600-mutant models of cancer, before and after MAPKi treatment. Nu/nu mice containing functional TAMs but deficient in adaptive immune repertoires were used, in support of immunogenic mouse and human cancer models. Flow cytometry revealed that TAMs, defined as GFP− (green fluorescent protein–negative) CD45+ CD11b+ F4/80+, significantly increased following MAPKi treatment with dabrafenib and trametinib in subcutaneously implanted mouse melanoma tumors derived from ultraviolet B (UVB)–irradiated genetically engineered BrafV600E/wt Pten−/− Cdkn2−/− YUMMER1.7 cells (Fig. 2A) (37). This model was used because it mimics clinical responses of human melanoma to BRAFi, where initial sensitivity is followed by emergent resistance and its mutational burden from UVB exposure supports tumor-immune examination. We confirmed the flow cytometry results by confocal imaging of tumors using Macrin, a fluorescent dextran nanoparticle (dextran-NP) characterized to be selective for MΦ (38), which, in this work, was labeled as TAMs with >90% selectivity (Fig. 2B and fig. S2A). Basket trials notwithstanding, MAPKi trials focused on OVCA have used single-agent MEKi (NCT00551070 and NCT02101788), as opposed to BRAFi or BRAFi + MEKi. Guided by this treatment design, similar TAM accumulation was seen in intraperitoneally disseminated tumor models generated using BRAFV600E ES2 human OVCA cells or BRAFV600E PtD patient-derived OVCA cells following MEKi treatment with trametinib (Fig. 2, C and D). Longitudinal intravital microscopy of ES2 xenografts growing within a dorsal skin-fold window chamber revealed that MEKi elicited sustained TAM accumulation over 96 hours, especially at the tumor periphery (Fig. 2, E and F), which corresponded with a slight decrease in cross-sectional tumor area (Fig. 2G). GFP signal from tumor cells was better preserved over time at the tumor margin compared to the tumor core (Fig. 2H), suggesting a combination of treatment-independent central necrosis and a preservation of tumor viability within the TAM-rich tumor periphery. Overall, these results demonstrate that multiple BRAF-mutant mouse models of cancer display MAPKi-induced TAM accumulation over 1 to 4 days after treatment.
Fig. 2. MAPKi increases TAMs in BRAF-mutant melanoma and OVCA mouse models.
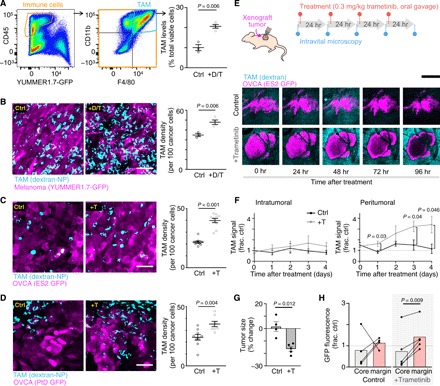
(A) YUMMER1.7 melanoma tumors were excised and analyzed by flow cytometry for TAMs (GFP− CD45+ CD11b+ F4/80+), 24 hours after three daily doses of dabrafenib (30 mg/kg) and trametinib (0.3 mg/kg) (+D/T; n ≥ 3). (B to D) Representative images (left) and quantification (right) of dextran-NP+ TAMs (cyan) within GFP-labeled (magenta) (B) YUMMER1.7, (C) intraperitoneally disseminated ES2 OVCA, or (D) intraperitoneally disseminated PtD OVCA tumors. At least one tumor each from n = 3 nu/nu mice per group was excised 24 hours after three daily doses of MAPKi (+T trametinib alone; two-tailed t test). Ctrl, control. Scale bars, 50 μm. (E to H) Schematic depicting daily imaging and trametinib treatment schedule for mice bearing ES2 xenograft tumors implanted in dorsal skin-fold window chambers (top). Representative intravital microscopy of OVCA and TAMs (bottom) (E) and their quantification (F) to (H) are shown (n ≥ 4 nu/nu mice per group, two-tailed t test). Scale bar, 250 μm. Tumor size and fluorescence were compared at 0 and 96 hours. Data are means ± SEM for all.
To investigate whether tumor cell–secreted factors could be influencing TAM accumulation upon MAPKi, we cultured MAPK-dysregulated OVCA cell lines—BRAF-mutant ES2, KRAS-mutant OVCAR8, and EGFR-overexpressing OVCA429 exhibiting elevated p-ERK (phosphorylated ERK) signaling (39)—with or without trametinib and then measured supernatant levels of 27 cytokines using a multiplexed bead-based immunoassay (Bio-Rad/Luminex). RANTES [also known as chemokine (C-C motif) ligand 5 (CCL5)]—a chemokine known to attract leukocytes via its cognate receptor, CCR5 (40)—was most up-regulated following MAPKi (fig. S2B). We then tested whether perturbing CCR5 signaling could reduce TAMs in MAPKi-treated mice bearing OVCA tumors. Subjects treated with both trametinib and maraviroc, a CCR5 antagonist U.S. Food and Drug Administration (FDA)–approved for HIV treatment, showed less TAM accumulation compared to subjects treated with trametinib alone (fig. S2C). Overall, these data suggest that TAM accumulation following MAPKi at least, in part, depends on RANTES/CCL5 signaling in the ES2 OVCA model.
MΦ-derived GAS6 and HGF sensitize cancer cells to inhibition of bypass signaling following MAPKi
We next tested whether the presence of TAMs could affect cytotoxicity of MAPKi on cancer cells. Conditioned medium was generated from M2-MΦ (defined hereafter as cells polarized in vitro by IL-4; see fig. S3A for characterization of MΦ polarization markers after IL-4 treatment) derived from U937 cells treated with dose responses of the MEKi trametinib for 24 hours. Incubation with this conditioned medium yielded substantial increases in the concentrations at which trametinib killed 50% of ES2 OVCA cells (Fig. 3A). Similar protective effects were observed with trametinib in the KRAS-mutant CT26 murine colorectal cancer cell line, as well as the BRAFi vemurafenib in the ES2 cell line (fig. S3B). Transwell coculture experiments using M2-MΦ derived from primary peripheral blood mononuclear cells (PBMCs), as well as the MAPK-dysregulated OVCA cell lines ES2, OVCA429, and OVCAR8, also reduced the cytotoxic impact of trametinib (Fig. 3B). Transwell coculture with M1-MΦ (defined hereafter as cells polarized by LPS/IFN-γ in vitro) derived from PBMCs showed less impact, such that two of three cell lines exhibited no change in trametinib-induced cell killing with coculture (fig. S3C). Overall, these results suggest that TAM impacts on MAPKi cytotoxicity are heterogeneous across MΦ culture models and polarization states but, in general, promote resistance.
Fig. 3. MAPKi polarizes MΦs toward an alternatively activated, HGF- and GAS6-producing phenotype.
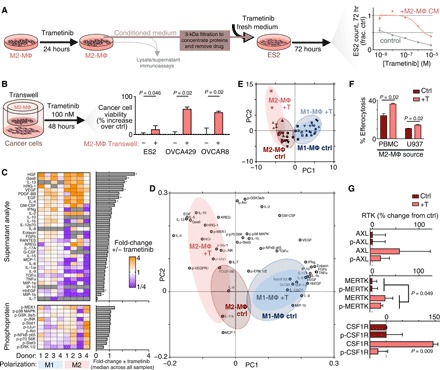
(A) Schematic of M2-MΦ conditioned medium experiments (left) and data showing ES2 cell count following 72-hour trametinib ± conditioned medium from M2-MΦ that were pretreated with 24-hour trametinib (right) (n = 3). (B) OVCA viability was measured by propidium iodide and annexin V staining for three cell lines treated for 48 hours with 100 nM trametinib ± transwell coculture with M2-MΦ (averaged across five donors with n = 3 reps per donor, two-tailed t test). (C) Heat map of supernatant and lysate proteins from PBMC-derived M1-MΦ and M2-MΦ. Analyte levels of MΦs treated with 24-hour trametinib are plotted relative to their respective no-treatment controls (n = 3, *P < 0.05, two-tailed Mann-Whitney U test with false discovery rate correction). GM-CSF, granulocyte-MΦ CSF; TGFα, transforming growth factor–α; TNFα, tumor necrosis factor–α; p-NFκB, phosphorylation of nuclear factor κB. (D and E) Loading (D) and score (E) plots generated from principal components analysis (PCA) of data from (C). Red shadings represent M2-MΦ, blue shadings represent M1-MΦ, and gray shading superimposed on each represents control conditions. (F) MΦ uptake of trametinib-treated ES2 cell debris following pretreatment of M2-MΦ with (+T) trametinib (n = 3, two-tailed t test). (G) RTK levels of U937-derived M2-MΦ, 24 hours after trametinib (n = 3 and n = 6 for pooled MERTK two-tailed t test). Data are means ± SEM for all.
We next investigated potential mechanisms of ligand-RTK cross-talk explaining MΦ-promoted MAPKi resistance. We anticipated that cancer-MΦ cross-talk would be dependent on multiple interrelated signaling pathways and therefore used a systems-level strategy based on multiplexed protein profiling combined with principal components analysis (PCA). We measured how 11 phosphoproteins, as well as supernatant accumulation of 33 growth factors and cytokines, changed with trametinib treatment across a panel of PBMC-derived M1-MΦ and M2-MΦ (Fig. 3C). This study revealed (i) that HGF and GAS6 were among the most highly up-regulated factors produced by trametinib-treated MΦ, (ii) that M2-MΦ, in general, produced more growth factors than M1-MΦ, and (iii) that MΦ phosphosignaling response was mixed across PBMC donors. To better interpret these heterogeneous and yet intercorrelated data, we performed PCA to distill key axes of covariation across measurements among these four groups: M1-MΦ ± trametinib and M2-MΦ ± trametinib (Fig. 3, D and E). In an unsupervised statistical process, the PCA scores revealed distinct separation between the two MΦ control groups (Fig. 3E), and variable loadings indicated which groups of measurements were driving these separations across the samples (Fig. 3D). PCA suggested that trametinib elicited a greater effect on M2-MΦ, since there was little separation with trametinib treatment in M1-MΦ. The upper-left quadrant of the scores and loadings plot was most associated with trametinib-induced effects from M2-MΦ and notably included growth factors (GAS6, HGF, and neuregulin 1/heregulin), anti-inflammatory cytokines such as IL-10, and signaling response in MΦ via up-regulated p-JNK, p-cJUN, and p-MEK1 (Fig. 3D). Trametinib-treated M2-MΦ (data associated with Fig. 3C) produced 76% the absolute level of GAS6 and 665% of HGF relative to trametinib-treated ES2 OVCA cells (data associated with fig. S2B), suggesting that both cell types can bidirectionally supply GAS6, whereas M2-MΦ serves as the primary HGF source upon MEKi (fig. S3D). In addition, experiments in MΦ differentiated from U937 demonstrated similarly up-regulated GAS6 and HGF with trametinib (fig. S3E).
In MΦ, AXL and MERTK are known to mediate engulfment of apoptotic cell material in a GAS6-dependent manner (efferocytosis) (28, 31, 41), and we observed enhanced efferocytosis across two MΦ models following trametinib (Fig. 3F). In these experiments, joint MΦ levels of total and phosphorylated MERTK mildly increased with trametinib (Fig. 3G), although the effects on GAS6 production were greater (Fig. 3C). Total CSF1R levels also increased with trametinib (Fig. 3G and fig. S3F). Overall, these results collectively indicate that trametinib affects MΦ to enhance their M2-like behaviors, including clearance of cell debris and growth factor production.
From the above data, we hypothesized that MΦ-derived growth factor production was amplifying RTK-mediated bypass signaling in cancer cells including especially the JNK pathway, along with reactivation of the ERK pathway. In ES2 cells, MAPKi increased AXL and MET levels on the cell surface while decreasing their accumulation in the supernatant (fig. S4A). This finding was consistent with prior reports of reduced proteolytic shedding as a posttranslational resistance mechanism to MAPKi (12, 31) and was associated here with cancer cell–autonomous induction of p-MET (fig. S4B). With evidence that MAPKi could enhance AXL and MET levels on cancer cells, we next tested whether trametinib-treated MΦ could influence the effectiveness of drugs blocking AXL and MET signaling. The addition of an AXL-Fc “ligand trap” or an HGF-neutralizing antibody enhanced trametinib cytotoxicity in the presence of conditioned medium generated from M2-MΦ treated with trametinib for 24 hours (Fig. 4A). In contrast, neither treatment affected trametinib response in cells grown without the same conditioned medium (Fig. 4A). In the presence of conditioned medium from M1-MΦ treated with trametinib for 24 hours, only the HGF-neutralizing antibody was sensitizing, which is consistent with previous observations that GAS6 and HGF up-regulation was stronger in M2-MΦ rather than in M1-MΦ (fig. S4C). Overall, these results suggest that M2-MΦ amplifies the dependence of cancer cells on ligand-mediated AXL and MET signaling during MAPKi.
Fig. 4. MΦs enhance cancer cell sensitivity to RTKi following MAPKi.
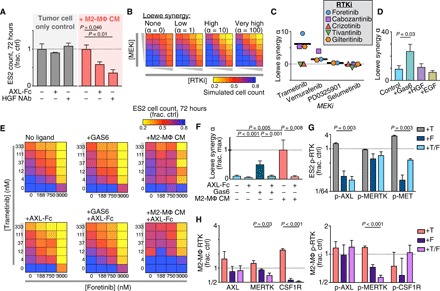
(A) ES2 cell count, 72 hours after treatment with 100 nM trametinib ± AXL-Fc ligand trap, HGF-neutralizing antibody (HGF NAb), and conditioned medium from M2-MΦ treated with 24-hour trametinib (M2-MΦ CM) (n = 3, two-tailed t test). (B) Illustrative dose-response surfaces for two model drugs with increasing degrees of the Loewe synergy parameter α. (C) α was calculated from ES2 cells treated for 72 hours with MEKi + RTKi across n = 24 combinations of concentrations and n = 2 replicates (thick line, median). (D to F) α was calculated as in (C) for trametinib + foretinib, but with ES2 cultured in recombinant growth factor (100 ng/ml) (D) or AXL-Fc (1 μg/ml), GAS6 (400 ng/ml), and/or M2-MΦ-CM (E and F) (n = 2, two-tailed t test). (G and H) p-RTK levels were measured in ES2 (G) and RTK and p-RTK levels in U937-derived M2-MΦ (H) 24 hours after treatment with trametinib (T), foretinib (F), or the combination (n = 3, two-tailed t test). For all except (C), data are means ± SEM.
With evidence that cancer cells and MΦ signal through multiple related RTKs, we hypothesized that an effective therapeutic strategy would require simultaneous multikinase inhibition. Several multi-RTKi have reached the clinic, and several (especially those targeting MERTK and CSF1R) are associated with promoting an antitumor polarization phenotype in MΦ (40). We screened a small panel of five RTKi—specifically, those with high-affinity targets including AXL (gilteritinib), MET (crizotinib and tivantinib), and both (foretinib and cabozantinib)—for their ability to combine with four different MEKi to kill cancer cells. ES2 cells were treated with varying combined dose responses of MEKi and RTKi, and the resulting cell viability measurements were then fit to a mathematical model of Loewe synergy (Fig. 4B). This model infers an interaction parameter α that reflects the degree of antagonistic or synergistic effects between two drugs. Of all 15 tested combinations, cotreatment with trametinib and foretinib yielded the highest α (Fig. 4C and fig. S4D). Foretinib is a multikinase inhibitor with high affinity toward AXL, MERTK, MET, vascular endothelial growth factor (VEGF) receptor, and CSF1R and has been tested in phase 1/2 trials. In efficiently inhibiting all of these RTKs, foretinib may be considered less specific than the other RTKi examined, therefore suggesting that its superior activity is due to simultaneously targeting multiple compensating RTKs (not just AXL, MERTK, or MET individually). Consistent with the model that MΦ amplify RTK-mediated bypass signaling, trametinib/foretinib synergy was enhanced with both MΦ conditioned medium and recombinant GAS6 (Fig. 4, D to F). In contrast, synergy was reduced with an AXL-Fc ligand trap, indicating that MΦ-enhanced drug synergy was dependent on GAS6 (Fig. 4, E and F). Foretinib cotreatment reduced trametinib-induced AXL and MET phosphoprotein levels in ES2 (Fig. 4G). Further, protein levels of CSF1R, MERTK, and p-MERTK were all decreased with foretinib in MΦ, indicating that foretinib affected RTK in both cancer cells and MΦ (Fig. 4H and fig. S4E).
In vivo microscopy reveals that TAM-proximal cancer cells exhibit distinct bypass signaling in a localized manner
Although the above in vitro and clinical evidence suggest that TAMs may enhance bypass signaling in cancer cells following MAPKi, it has traditionally been difficult to visualize this signaling as it occurs in situ within tumor tissue. To accomplish this, we developed a new intravital microscopy strategy that enables simultaneous quantification of multiple phosphosignaling activities in vivo and at a single-cell level. We hypothesized that imaging downstream signaling pathway activities would be better suited to capture the integrated impact of multiple converging RTKs. To identify the most relevant downstream signaling pathway to image, we measured phosphorylated levels of 3 implicated RTKs (AXL, MERTK, and MET) and 11 key intracellular signaling nodes, across three OVCA cell lines, as they responded to trametinib in the presence or absence of M1-MΦ or M2-MΦ coculture in vitro (figs. S4B and S5A). Correlation analysis using these 756 measurements revealed that phosphorylation of JNK and its substrate cJUN were together most correlative with phosphorylation of AXL, MERTK, and MET (Fig. 5A), similarly as we found earlier in M2-MΦ (Fig. 3D). In ES2 cells treated with trametinib, in vitro coculture with M2-MΦ resulted in an attenuated decrease in p-JNK levels, while also protecting against inhibition of p-ERK downstream of MEK (Fig. 5B). Thus, this analysis suggested that MΦ-supplied factors could elevate ERK and JNK signaling activities in neighboring cancer cells.
Fig. 5. Intravital microscopy reveals altered bypass signaling in TAM-proximal cancer cells.
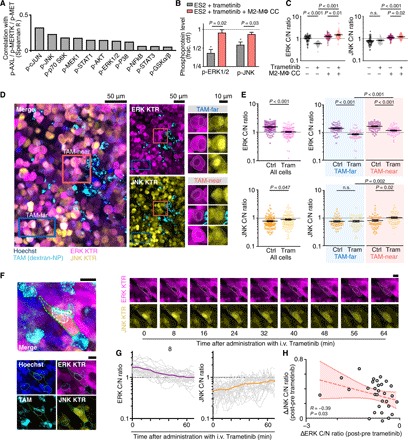
(A) Levels of 11 intracellular phosphoproteins were correlated with three p-RTK (p-AXL/p-MERTK/p-MET) levels across three OVCA cell lines, three MΦ coculture conditions, and ±24-hour 100 nM trametinib. A total of n = 756 total Luminex measurements were considered (see figs. S4B and S5A). (B) Phosphoproteins were measured in ES2 cells, 24 hours after trametinib ± transwell M2-MΦ coculture (CC) (n = 3, two-tailed t test). (C) Cytoplasmic-to-nuclear (C/N) ratios from ERK and JNK kinase translocation reporter (KTRs) were measured in ES2 cells 24 hours after treatment with trametinib ± transwell coculture with M2-MΦ (n = 60 cells across six wells, *P < 0.05, two-tailed t test). (D and E) Representative confocal microscopy (D) of disseminated intraperitoneal tumors, showing ES2 cells expressing ERK and JNK KTRs near and far from TAMs, from nu/nu xenografts 24 hours after treatment (trametinib, 0.3 mg/kg, by mouth), quantified in (E) (n = 120 cells across n = 12 tumors and three mice per group, two-tailed t test). Scale bars, 50 μm or 10 μm as indicated). (F to H) Time-lapse intravital microscopy of ES2 xenografts imaged following intravenous (i.v.) trametinib treatment. Representative images (F) and quantification (G and H) are shown. Scale bar, 10 μm. In (G), thin and thick lines denote single cells and their mean, and in (H), data points denote single cells at 60 min after treatment (n = 20 cells across two tumors, Spearman’s exact test). Data are means ± SEM for (B), (C), and (E). n.s., not significant.
To directly examine these findings in vivo, we used a fluorescent kinase translocation reporter (KTR) system for visualizing ERK and JNK activity. Transgenic kinase substrates based on canonical endogenous substrates shuttle in and out of the nucleus depending on their phosphorylation (42). KTRs are fused to fluorescent proteins, allowing single-cell kinase activity to be quantified by measuring the ratio of cytoplasmic-to-nuclear (C/N) fluorescence (Fig. 5C and fig. S5B). In vitro, KTRs showed decreased ERK and increased JNK activities with trametinib treatment, albeit with narrow dynamic range (Fig. 5C). In vivo, using both ES2 and patient-derived PtD orthotopic xenografts of disseminated OVCA, KTR imaging revealed that tumor cells exhibited distinct signaling behavior when in close proximity (<40 μm) to TAMs (Fig. 5, D and E, and fig. S5, C and D). More specifically, both models showed that 24 hours following treatment with trametinib, TAM-proximal cancer cells exhibited higher ERK and JNK signaling activities compared to tumor cells far (>40 μm) from TAMs (Fig. 5, D and E, and fig. S5, C and D).
We next performed time-lapse intravital microscopy of KTR-expressing ES2 xenograft tumors to understand the in vivo dynamics of TAM-influenced bypass signaling. Tumor-bearing mice were anesthetized on a heated stage and were imaged before and for roughly 1 hour following intravenous trametinib administration. Time-dependent increases in JNK activity of single ES2 cells near TAMs were significantly correlated with decreases in ERK activity, suggesting that JNK activation was not due to trametinib inaccessibility within the tumor (Fig. 5, F to H). While photobleaching occurred due to repeated imaging, this did not affect quantification of KTR C/N ratios, and C/N ratios in untreated tumors remained relatively constant over the same time frame (fig. S5, E and F). In summary, in vivo microscopy in multiple models revealed that TAMs correlate with altered JNK bypass signaling in cancer cells in a spatially localized fashion.
Nanoformulated foretinib accumulates in TAM-rich tumors following MAPKi and combines with trametinib to block disease progression
Given evidence that MAPKi induces local signaling between TAMs and cancer cells, we hypothesized that formulation of foretinib within TAM-accumulating nanoparticles could efficiently target TAM-amplified bypass signaling (43). We based the foretinib nanoformulation (NanoFore) on polymeric micelles composed of biodegradable and clinically used materials poly(lactic-co-glycolic) acid (PLGA) and the copolymer poly(ε-caprolactone)–block–methoxy poly(ethylene glycol) (PCL-b-mPEG) (Fig. 6A and fig. S6). This formulation is similar to those used in recent reports describing high TAM accumulation of nanoparticles and gradual release of the drug payload to neighboring cancer cells (44). NanoFore exhibited physicochemical properties (hydrodynamic size, 58 nm), slow drug release rate (1.6% over 72 hours), and systemic pharmacokinetics, all consistent with behavior seen in clinical formulations (fig. S6). To understand how MAPKi pretreatment might affect NanoFore uptake by TAM, we performed gene set enrichment analysis (GSEA) on a recently published screen that measured the effect of CRISPR gene silencing on nanoparticle uptake by U937 cells. Genes were associated with higher phagocytosis score ranking if their silencing was associated with up-regulated phagocytosis, and GSEA was used to determine whether more highly ranked genes were enriched for members of an a priori defined gene set. Encouragingly, we found that silencing members of the RAS/RAF/MEK/ERK pathway was significantly associated with enhanced nanoparticle phagocytosis (Fig. 6B), suggesting that NanoFore delivery could be a viable strategy for targeting TAMs that have increased in number, and potentially in phagocytic activity, as a result of MAPKi.
Fig. 6. Nanoformulated multikinase inhibitor efficiently accumulates in TAMs of MAPKi-treated tumors to extend survival and reduce bypass signaling.
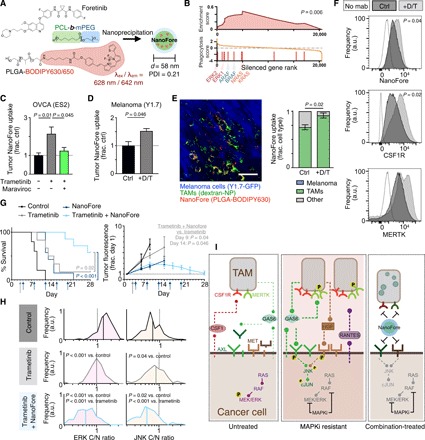
(A) Schematic of foretinib nanoparticle encapsulation (NanoFore). PDI, polydispersity index. (B) GSEA of a genome-wide CRISPR screen, showing that RAS/RAF/MEK/ERK pathway silencing is enriched for enhanced phagocytic activity in U937 cells (GSEA permutation test). (C and D) NanoFore accumulation was measured 24 hours after treatment with trametinib (T) or dabrafenib + trametinib (D/T) in ES2 (C) and YUMMER1.7 (“Y1.7”) (D) tumors by fluorescence reflectance imaging (FRI), 24 hours after MAPKi and maraviroc (n ≥ 3). (E) Corresponding with (D), representative confocal microscopy (left) and quantification (right) measured colocalization between NanoFore and TAMs in YUMMER1.7 tumors (n ≥ 3). Scale bar, 50 μm. a.u., arbitrary units. (F) Flow cytometry of YUMMER1.7 tumors quantified NanoFore uptake, CSF1R, and MERTK levels in dextran-NP+ TAMs (n ≥ 3). (G) Nu/nu mice bearing intraperitoneal ES2 tumors were treated with trametinib (daily starting on day 3; gray arrow), NanoFore (every 4 days; blue arrows), or the combination and were monitored for reaching the experimental end point (top; n ≥ 5, two-tailed log-rank test) and imaged by FRI (bottom; n ≥ 3, two-tailed t test). (H) Corresponding to (G), single-cell ERK and JNK activities were measured at time of experimental end point for individual subjects (n = 120 cells across 12 tumors, two-tailed t test). For (C) to (E) and G, data are means ± SEM (two-tailed t test). (I) Summary of schematic. Untreated tumor cells with constitutive RAS/RAF/MEK/ERK signaling produce soluble AXL and MET RTK ectodomains; TAMs express MERTK and CSF1R and accumulate in MAPK-driven tumors (left). MAPKi using dabrafenib and/or trametinib increases TAMs in resistant tumors, reduces AXL and MET ectodomain shedding in cancer cells, leads to AXL and MET accumulation on the cancer cell surface, enhances production of GAS6 by TAMs and cancer cells, enhances HGF production by TAMs, and stimulates cancer cell production of the TAM-recruiting cytokine RANTES/CCL5. TAM-proximal cancer cells exhibit elevated JNK/cJUN phosphosignaling (center), but this can be inhibited by NanoFore, which is efficiently taken up by TAMs (right).
Fluorescently labeled NanoFore accumulated efficiently and selectively in the tumors of mice bearing either disseminated intraperitoneal OVCA or melanoma xenografts, as measured by fluorescence reflectance imaging (FRI) of excised tumors and healthy organs 24 hours following NanoFore administration (fig. S6, B and C). FRI also revealed that bulk NanoFore accumulation was increased in both models by MAPKi treatment (Fig. 6, C and D). Treatment with the CCR5 antagonist maraviroc, which we described above as reducing TAM accumulation following MAPKi (fig. S2C), reduced the accumulation of NanoFore in MAPKi-treated tumors (Fig. 6C). Confocal microscopy of tumors demonstrated that fluorescent NanoFore accumulated preferentially in dextran-NP+ TAMs, as opposed to GFP-labeled cancer cells and other stromal cells (Fig. 6E). Consequently, MAPKi-induced improvement in NanoFore accumulation can be at least, in part, attributed to the higher number of TAMs in treated tumors. Moreover, TAMs in MAPKi-treated tumors accumulated more NanoFore on a per-cell basis (Fig. 6F), which was fitting with the CRISPR screen analysis (Fig. 6B). Consistent with in vitro studies, MAPKi led to increased levels of CSF1R and MERTK on TAMs (although the latter was not statistically significant), therefore confirming in vivo expression of NanoFore RTK targets (Fig. 6F). Thus, in both models, MAPKi improved NanoFore accumulation in tumors via increased TAM levels and enhanced TAM uptake on a per-cell basis.
We next examined the ability of NanoFore to block TAM-mediated bypass signaling and disease progression in tumor-bearing mice treated with MAPKi. As expected with similar nanoformulations, NanoFore retained cytotoxicity in vitro compared to solvent-based administration in cell culture assays (fig. S6F). In vivo, both trametinib and NanoFore monotherapies extended median survival by only 4 days in the aggressive ES2 model. However, the combination of NanoFore and trametinib delayed disease progression such that the median survival end point was not reached in this study; survival was therefore extended by at least 19 days, conservatively (Fig. 6G). ES2 cells in this model expressed both ERK and JNK KTRs, and therefore, additional reporters for bioluminescence were not used to minimize the number of transgenic constructs. Nonetheless, the ERK KTR was bright enough to be detectable by FRI, and the extended animal survival correlated with decreased tumor fluorescence in live subjects (Fig. 6G and fig. S6G). Tumors were also analyzed for ERK and JNK activity at the experimental end point, showing that NanoFore blocked JNK signaling that was elevated in tumors exposed to trametinib monotherapy (Fig. 6H). Because foretinib inhibits CSF1R in addition to AXL, MET, and others, we also examined the extent to which TAM accumulation was affected by repeated NanoFore. Combination-treated subjects euthanized at their survival end points had fewer TAMs than their trametinib-treated counterparts, but TAM levels still remained comparable to levels seen in untreated tumors (fig. S6H). Further, combination therapy was also significantly more effective than either monotherapy in the CT26 colorectal cancer allograft model (fig. S6I).
To investigate whether the efficacy of NanoFore could simply be explained through its activity toward CSF1R and its ability to reduce TAM accumulation, we tested how TAM depletion strategies affected MAPKi action in the same model of intraperitoneal ES2 OVCA as used above. In contrast to the improvements seen with NanoFore, MΦ depletion via intraperitoneal injections of α-CSF1R monoclonal antibody (mAb) or clodronate liposomes did not significantly enhance the ability of trametinib to extend survival in subjects with disseminated ES2 tumors (fig. S7, A to C). Nonetheless, cotreatment with α-CSF1R mAb somewhat diminished tumor burden in the ovary (fig. S7, D to E). These results suggest that blocking the overall accumulation of TAMs does not itself markedly improve MAPKi efficacy in this model. Therefore, the efficacy of NanoFore at least involves more nuanced aspects of TAM behavior such as polarization toward different cytokine and growth factor communication profiles.
DISCUSSION
This work describes a new targetable, multicellular mechanism of how bypass signaling in cancer cells can be reciprocally amplified via cross-talk with TAMs in the tumor microenvironment (Fig. 6I). Our findings in multiple MAPK-dysregulated melanoma and OVCA models suggest that, in some cases, resistance mechanisms to therapies in basket trials may be generalizable and therefore targetable with the same combination therapies across cancers with the same driver alterations but different organ sites of origin. We build on extensive prior studies that describe how signaling through RTKs in cancer cells can promote alternative modes of phosphosignaling that circumvent the intended cytotoxic effects of targeted kinase inhibitors, including through RTKs such as AXL, MERTK, and MET that are implicated here. However, past reports have largely focused on cancer cell–autonomous signaling, or targeted, unidirectional signaling from one cell type to another. Although well-known asymmetric signaling pathways were identified in this study, including CSF1-CSF1R and RANTES/CCL5-CCR5 signaling from cancer cells to TAMs and HGF-MET signaling from TAMs to cancer cells, we also found mutually reciprocated signaling among the RTKs AXL and MERTK to be especially important. AXL and MERTK exhibit distinct functions, yet they are structurally similar, bind the ligand GAS6 to participate in efferocytosis, and are inhibited by many of the same drugs (45). In cancer cells, AXL has been associated with invasive, mesenchymal, chemoresistant, and even dormancy phenotypes, and MERTK overexpression drives proliferation in cancer types, including melanoma (46). In leukocytes, AXL and MERTK can promote immunosuppressive signaling that is enhanced by the efferocytosis of apoptotic material (28, 31, 41). Furthermore, ligand-dependent AXL activity can be amplified by the spatial clustering of phosphatidylserine-bound GAS6 on apoptotic bodies (47) and inhibited by GAS6 binding to proteolytically shed soluble receptor (31). In the context of this prior understanding, our data suggest a model that MAPKi-induced tumor cell killing, combined with an accumulation of GAS6-producing TAMs, a reduction in soluble AXL receptor shed from cancer cells, and an elevation of AXL on the cell surface of cancer cells, collectively leads to enhanced GAS6-mediated signaling in close spatial proximity to TAM. On the basis of the initial analysis of clinical data, in this work, we focused on cross-talk between the innate immune system and cancer cells. However, AXL and MERTK inhibition are reported to improve response to immune checkpoint blockade in mice, they can be expressed by other stromal populations, and it is likely that joint GAS6-mediated communication between cancer cells and TAMs will have important implications for T cell function and immunotherapy response.
Real-time in vivo microscopy of signaling pathway activities, as well as imaging of patient-derived and orthotopic cancer models, revealed that bypass signaling was spatially biased on a microscopic scale within the tumor and amplified in cancer cells near TAMs. While our in vitro data translated well to our in vivo findings, other cell types within the tumor microenvironment may also influence drug response and can be explored in future work using the framework presented here. For example, melanoma-associated fibroblasts have been shown to be paradoxically activated by MAPKi and can promote drug resistance via extracellular matrix remodeling, and cancer cell signaling, for instance, via focal adhesion kinase, within three-dimensional contexts can lead to distinct drug responses compared to what is seen on tissue culture plastic (15). Further, patients with OVCA often present with ascites, which contains a suspension of cell types including tumor cells, fibroblasts, mesothelial cells, and neutrophils, and therefore may also play a contributory role in clinical response (48). Although no substantial differences have been observed in hazard ratios between males versus females in pivotal MAPKi clinical trials for BRAF-mutant melanoma or colorectal cancer, it is still possible that competing resistance mechanisms are more or less prevalent depending on gender. Future work could investigate whether patient gender may influence the relationship between TAMs and MAPKi resistance. As motivation, data from humans, rodents, and/or primary cell cultures have shown that MΦ from females can exhibit greater phagocytic capacity and IL-10 production in some circumstances (49). Androgens, progesterone, and/or estradiol can affect MΦ polarization state and recruitment, for instance, via decreased expression of chemotactic factors such as CCL2 and inducible nitric oxide synthase (49).
Here, our intravital microscopy results raise the possibility of using TAM-directed drug delivery strategies to improve the efficiency with which therapeutics reach their targets, while minimizing potentially toxic systemic exposure. Although precedent exists for triple-combination kinase inhibitor regimens, toxicity can be especially problematic for promiscuous drugs that simultaneously inhibit multiple pathways. We identified bypass signaling through multiple related RTK pathways, and the most synergistic drug combination we tested used foretinib, which potently inhibits multiple RTKs and has known toxicity issues when administered in traditional oral formulations (50). In a recent phase 1 study of foretinib combined with the EGFR inhibitor erlotinib in non–small cell lung cancer, dose reductions occurred in the majority of patients. The only patients with AXL+ cancers by histology whose tumors shrunk on treatment also expressed MET, therefore highlighting the potential importance of assessing and targeting multiple signaling pathways simultaneously (50). To efficiently deliver foretinib to TAMs and nearby cancer cells, we used a nanoformulation strategy similar to what is being clinically developed for other toxic anticancer drugs (19, 20). In prior studies where both the nanoparticle and its drug payload were coimaged in vivo, drug-loaded nanoparticles were observed to highly accumulate in TAMs and gradually release their drug payload to neighboring cancer cells (44, 51). Although we did not directly visualize the drug payload in this study, we nonetheless found that the nanoparticle vehicle efficiently accumulated in TAMs, and cancer cells showed a reduction in JNK bypass signaling that was known to be elevated in TAM-proximal cells. Overall, these results collectively suggest that NanoFore is able to efficiently be delivered to disseminated OVCA, inhibit signaling in cancer cells, and reduce disease progression. We anticipate that these results will motivate further investigation into distinct toxicity and efficacy profiles of NanoFore compared to its traditional orally delivered counterpart, including those related to myelotoxicities and immune-related adverse events.
Companion biomarkers are likely to be necessary for successful translation of treatments that target the bypass-signaling mechanisms described in this work. Here, we identify multiple relevant signaling pathways (including via AXL and MET) involved in MAPKi resistance and directly observe their heterogeneous behavior across the tumor microenvironment, therefore complicating their quantification and interpretation. In addition to identifying biomarkers for certain signaling pathways, therapeutic nanoparticles are increasingly recognized as heterogeneously accumulating in solid cancers due to variable microenvironmental features related to the enhanced permeability and retention (EPR) effect, and biomarkers are likewise being proposed and tested to stratify patients based on EPR predictors (20). One promising strategy has been to noninvasively quantify TAMs by magnetic resonance imaging or positron emission tomography/computed tomography (PET/CT) using nanomaterials (52). We demonstrate here that TAMs correlate both with bypass signaling and with nanomaterial delivery in orthotopic tumor models, and we image TAMs using the dextran-NP Macrin, which is clinically translatable and can be imaged by PET/CT (38). Other possible strategies include using the magnetic nanoparticle ferumoxytol (Feraheme) to image TAMs in patients, since ferumoxytol is FDA approved for the treatment of anemia and already has been successfully tested in preclinical and clinical studies as a predictive marker of nanotherapeutic delivery (53). Together, this study combines scRNA-seq, systems-level analysis of coculture experiments, and intravital microscopy to shed new perspective on how signaling pathways adapt across cell populations in response to MAPKi, and we offer translationally relevant imaging and therapeutic strategies to target this resistance pathway in patients.
MATERIALS AND METHODS
Study design
The objective of this study was to understand the influence of TAMs on MAPKi response in various MAPK-driven cancers. Computational analysis using patient gene expression data was performed to generate testable hypotheses on the association of specific immune cell subsets and ligand-receptor interactions with therapeutic resistance. Subsequently, coculture and conditioned medium experiments were designed with proteomic and phenotypic end points to investigate TAM-tumor cross-talk in vitro. Last, animal imaging and efficacy studies were designed to visualize localized, reciprocal signaling and assess the impact of our TAM-avid multikinase inhibitor in combination with MAPKi. Experiments were performed with ≥3 independent replicates or as described in the figure captions; data collection and treatment group assignment were predetermined; no outliers were excluded. Previous experiments and their corresponding power analyses informed the group sizes of the present study (12, 51, 54, 55). Where possible, computational and statistical analysis was performed in a manner blinded to the treatment group identities, and image acquisition, postprocessing, and algorithms were applied with unbiased parameters across treatment groups.
Meta-analysis of RAS and RAF genetics across cancer types
Rates of CNA were assessed using cBioPortal (1). Querying for RAS and RAF genes (ARAF, BRAF, CRAF, KRAS, NRAS, and HRAS), serous OVCA exhibited the highest rate of amplification (20%, 117 of 584) in the 2018 TCGA pan-cancer analysis compared to any other of the 70 cancer types that were quantified across 10,953 patients and 32 TCGA pan-cancer studies (1). Among 579 patients in the provisional ovarian serous cystadenocarcinoma TCGA dataset with CNA reported, 27% (158) exhibited RAS or RAF amplification (1). Gene expression of KRAS was analyzed across multiple datasets comparing tumor versus normal tissue, using Oncomine (oncomine.org) (2). In this analysis, 190 studies across all cancer types were ranked according to KRAS up-regulation in cancer versus corresponding normal tissue. On the basis of gene ranking, the second and sixth most highly up-regulated datasets were for OVCA: Hendrix et al. (103 patients) (56) and TCGA (586 patients), respectively.
CIBERSORT analysis of patient biopsies
CIBERSORT uses a reference gene signature of roughly 550 differentially expressed genes from 22 leukocyte types to estimate the relative fraction of each within bulk transcriptional profiles of samples (57). We applied this deconvolution algorithm to datasets accessed on the National Center for Biotechnology Information Gene Expression Omnibus. We used bulk gene expression data of pretreatment and progression BRAFV600-mutant melanoma biopsies from 10 patients (GSE61992) (24) and of 69 primary high- or low-grade OVCA biopsies with or without MAPK (BRAF/KRAS/ERBB2) mutations (GSE12172) (34). ERBB2 was included in this analysis as it is known in LGSOC to be associated with constitutive RAS-MAPK signaling in LGSOC (34).
Ligand-receptor interaction analysis
Ligand-receptor interaction analysis of the melanoma patient scRNA-seq data from GSE72056 (25) was performed, as previously described (26). Briefly, each ligand-receptor interaction between cell type A and cell type B was scored as the product of average receptor expression across all type A cells and the average ligand expression across all type B cells. The reference list of known and literature-supported interactions was obtained from published datasets and then annotated for growth factor—RTK interactions. Ligand-receptor interaction analysis of the bulk RNA expression data from GSE40484 (35) and GSE73091 (36) was performed similarly, with the exception that the ranks of ligand and receptor expression were used in place of absolute values. Expression data from GSE40484 for CD16+ BDCA1− MΦs were averaged across five patients with OVCA, and expression data from GSE73091 for EpCAM+ epithelial cells were averaged across the primary tumors, metastases, and ascites of three patients with LGSOC.
Gene set enrichment analysis
GSEA using a custom RAS/RAF/MEK/ERK pathway gene set was performed on a published magnetic CRISPR screen originally aimed at identifying regulators of phagocytosis using negatively charged 300-nm-diameter particles in U937 cells (58, 59). The custom gene set consisted of the canonical pathway members KRAS, NRAS, HRAS, ARAF, BRAF, RAF1, MAP2K1, MAP2K2, MAPK1, and MAPK3. MAP3K8 was also included, as it is a (albeit less well-known) MAPK pathway agonist that activates ERK primarily through MEK-dependent mechanisms without a requirement for RAF signaling (60, 61). Enrichment score and nominal P values were calculated from the GSEA Java Applet (Broad Institute).
Cell culture and materials
ES2, CT26, and U937 cells were obtained from American Type Culture Collection; PtD and OVCA429 cells were provided by D. Pepin [Massachusetts General Hospital (MGH)] and M. Birrer (MGH/University of Alabama), respectively. OVCAR8 cells were provided by S. Bhatia (Massachusetts Institute of Technology) and YUMMER1.7 cells by M. Bosenberg (Yale). Cell lines were obtained through material transfer agreements and grown according to vendor guidelines. Where relevant, cells were then seeded into tissue culture plates such that they were ~80% confluent by the time of experiment end point. Under conditions where cells were treated with trametinib (Selleck Chem), a 100 nM dose was used, unless otherwise specified. Recombinant human AXL-Fc chimera protein (R&D Systems) or human HGF-neutralizing antibody (AB-294-NA, R&D Systems) was used where indicated at concentrations of 1 and 10 μg/ml, respectively. Recombinant GAS6, HGF, or EGF protein (PeproTech) was used at 100 ng/ml. For the in vitro synergy screen, drugs were dosed up to the following maximum concentrations: 333 nM trametinib, 5 μM vemurafenib, 5 μM PD0325901, 5 μM selumetinib, 3 μM foretinib, 6 μM cabozantinib, 3 μM crizotinib, 3 μM tivantinib, and 6 μM gilteritinib. MEKi and RTKi were diluted three- and fourfold, respectively. All inhibitors were purchased from Selleck Chem. PrestoBlue (Invitrogen) was used as a resazurin-based assay for proliferation/cytotoxicity throughout, following the manufacturer’s guidelines in a standard 96-well microtiter plate format. Five thousand cells per well were plated overnight before drug treatment the following day, and PrestoBlue was then used to measure viable cell count at 72 hours after treatment, unless otherwise stated.
Monocyte isolation from healthy female donors
PBMCs were isolated from peripheral blood leukapheresis samples from healthy female donors (STEMCELL Technologies) via density gradient centrifugation (Lymphoprep, STEMCELL Technologies). PBMCs were washed 2× with 2% heat-inactivated fetal bovine serum (FBS) and 1 mM EDTA in phosphate-buffered saline (PBS) to remove contaminating platelets and then frozen down in cryopreservation medium [90% heat-inactivated FBS and 10% dimethyl sulfoxide (DMSO)] until further use. Immediately before differentiation into MΦs, PBMCs were thawed for monocyte isolation via immunomagnetic negative selection (EasySep human monocyte isolation kit, STEMCELL Technologies).
MΦ differentiation
MΦ differentiated from PBMCs were used whenever possible, but for experiments limited by cell numbers, U937-derived MΦ were used instead. For monocytes isolated from healthy donors, monocytes were plated at a density of 2 × 105 cells/ml and granulocyte-MΦ CSF (50 ng/ml) was added for M1-MΦ or MΦ CSF (50 ng/ml) for M2-MΦ predifferentiation. On day 4, an additional 50% of the same cytokine-containing medium was added. On day 7, predifferentiation medium was removed and replaced with activation medium containing IFN-γ (20 ng/ml) + LPS (50 ng/ml; Sigma-Aldrich) for M1-MΦ or IL-4 (20 ng/ml) for M2-MΦ. Twenty-four hours later, MΦs were detached with ice-cold 10 mM EDTA in PBS for seeding into experimental culture plates. For differentiation of U937 monocytes into MΦs, a similar protocol was performed, except monocytes were plated at a density of 5 × 105 cells/ml and incubated with medium containing phorbol 12-myristate 13-acetate (50 ng/ml; Sigma-Aldrich) for 1 day in place of the 6-day predifferentiation step. Growth factors and cytokines were from PeproTech. MΦ differentiation was validated by flow cytometric staining. M1-MΦ from PBMCs expressed increased CD68 and CD80, while M2-MΦ from both cell sources expressed increased CD68 and CD163 relative to their respective monocyte precursors (fig. S3A).
Conditioned medium and coculture assays
Conditioned medium was generated by treating MΦ with trametinib, concentrated using Amicon spin filters (3 kDa; EMD Millipore) to remove any remaining free drug, and then resuspended in fresh medium, along with the appropriate concentration of fresh drug. For transwell coculture assays, we used 24-well plates containing polycarbonate transwell inserts with 0.4-μm pores (Corning). MΦ (5 × 104) were seeded in the inner insert, while 1 × 105 or 5 × 104 OVCA cells were seeded in the well bottom for end point signaling measurements (24-hour incubation) or phenotypic measurements (48-hour incubation), respectively.
Luminex assays
The Bio-Plex Pro Human Cytokine 27-plex Assay (#M500KCAF0Y, Bio-Rad) was used to measure levels of cytokines associated with varied types of immune response, while individually coupled magnetic beads were used to detect growth factors associated with implicated ligand-RTK interactions from our computational coexpression analysis (Fig. 1, E and G). To generate our own immunoassays, we used capture and detection antibodies against human neuregulin 1 beta 1, GAS6, HGF, EGF, amphiregulin, heparin-binding EGF, VEGF, and transforming growth factor–α purchased as DuoSets from R&D Systems. For antibody-bead coupling, Bio-Plex Pro magnetic COOH beads (Bio-Rad) were activated with EDC [N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide; Sigma-Aldrich] and sulfo-NHS (N-hydroxysulfosuccinide; Pierce) in 100 mM phosphate buffer (pH 6.3) for 20-min shaking at room temperature and then incubated with capture antibody (0.1 mg/ml) in 50 mM Hepes buffer (pH 7.4) overnight shaking at 4°C. The following day, beads were washed 3× in 1% bovine serum albumin (BSA) in PBS and then stored at 4°C. Immunoassays were adapted to a 384-well plate format, performed on 1× or 1/10× dilutions of clarified supernatants according to the manufacturers’ protocols, and read on a FLEXMAP 3D system (Luminex Corp). For each analyte, five-parameter logistic regression was used to fit standard curves for absolute quantification. Measured levels of analytes were normalized on the basis of the micro bicinchoninic acid protein assay (Pierce), and the Benjamini-Hochberg procedure was used to correct for multiple comparisons between treatment conditions.
A custom Bio-Plex phosphoprotein panel was used to measure levels of intracellular kinases and consisted of immunoassays detecting phosphorylated versions of 11 key signaling nodes: cJun (pSer63), JNK (pThr183/pTyr185), p70 S6K (pThr421/pSer424), MEK1 (pSer217/pSer221), signal transducers and activators of transcription 1 (Stat1) (pTyr701), Akt (pSer473), ERK1/2 (pThr202/pTyr204 and Thr185/pTyr187), p38 MAPK (pThr180/pTyr182), nuclear factor κB p65 (pSer536), Stat3 (pTyr705), and glycogen synthase kinase 3a/b (pSer21/pSer9). R&D DuoSets against human AXL, MERTK, MET, and CSF1R were used to generate immunoassays against these RTKs. For RTK pTyr measurements, an anti–pan-pTyr antibody (4G10, Sigma-Aldrich) was used in place of the RTKs’ corresponding detection antibodies. Bio-Plex cell lysis buffer (Bio-Rad) was used to lyse cells for intracellular kinase measurements, while NP40 cell buffer [20 mM tris-HCl, 150 mM NaCl, 2 mM EDTA, 1% NP-40, and 10% glycerol (pH 7.4)] was used for RTK measurements. Lysis buffers contained protease and phosphatase inhibitors per manufacturer guidelines (Bio-Rad). In both cases, immunoassays were performed on clarified lysates diluted twofold in assay buffer consisting of 1% BSA in PBS.
Efferocytosis assay
We generated fluorescent apoptotic bodies by treating CellTracker Deep Red–labeled (Invitrogen) ES2 cells with 10 mM trametinib for 48 hours. Supernatants were harvested and apoptotic bodies were purified by sequential centrifugation for 10 min at 300g and then 10 min at 2000g (62). Apoptotic bodies were resuspended in fresh medium and incubated with pretreated M2-MΦ (100 nM trametinib or DMSO control for 24 hours) for 4 hours. MΦs were detached with 10 mM EDTA, stained with propidium iodide for viability, and then analyzed with an Accuri C6 flow cytometer to assess uptake of CellTracker-labeled apoptotic bodies.
Generation and characterization of KTR-expressing cell lines
KTR-expressing ES2 or PtD cell lines were established by lentiviral transfection with the ERK KTR mClover and JNK KTR mRuby2 constructs, gifted from M. Covert (Addgene plasmids #59150 and #59154) (42). For in vitro characterization, reporter cell lines were seeded 40,000 cells per well into a 24-well plate (Ibidi) and then treated the following day with EGF (PeproTech), anisomycin (Sigma-Aldrich), or JNK-IN-7 (MedChemExpress). After a 24-hour incubation period, cells were fixed with warm 4% paraformaldehyde in PBS for 15 min, stained with Hoechst 33342 (1 μg/ml; Invitrogen) for 10 min, and imaged with a Leica DMI 6000 microscope and Oasis Surveyor software. ImageJ [National Institutes of Health (NIH)] was used to quantify the C/N intensity ratios of mClover and mRuby2 fluorescence. Similar experiments were also performed ±trametinib and ±transwell coculture with M2-MΦ.
Nanoparticle-foretinib synthesis and characterization
NanoFore was synthesized via nanoprecipitation by first dissolving 5 mg of PCL5kDa-b-mPEG5kDa block copolymer (Advanced Polymer Materials Inc.; manufacturer provided Mn, 10 kDa; polydispersity index, 1.19, lot 01-07-160-1), 1 mg of PLGA (50:50 PLGA, 30 to 60 kDa; Sigma-Aldrich), and 1 mg of foretinib (LC Labs) in 300 μl of DMF (dimethylformamide). An equal volume of acetonitrile was added, and a nanoprecipitation procedure was performed by adding the solution dropwise to 20 ml of deionized H2O (dI-H2O) under stirring. After overnight stirring, the solution was filtered through 0.45-μm syringe filters (Whatman) and then concentrated using 100-kDa molecular weight cutoff (MWCO) spin filters (Amicon, Millipore). For characterization, hydrodynamic radius and zeta-potential measurements were performed via dynamic light scattering (Malvern Zetasizer) in dI-H2O at a concentration of approximately 0.1 mg/ml (drug + polymer mass). For quantification of nanoparticle drug loading, nanoparticles were dissolved with the addition of four-part DMF and then analyzed on a high-performance liquid chromatography (HPLC) system with in-line photodiode array detector (Waters). Foretinib concentration within nanoparticle samples was quantified via integration of absorbance peaks at 332 nm relative to a standard curve of known foretinib concentrations. Drug release was measured by diluting NanoFore 1:20 in PBS at 37°C for 72 hours and by filtering the sample using 100-kDa MWCO spin filters (Amicon, Millipore). Flow-through and concentrated nanoparticle were both analyzed by HPLC for relative foretinib concentration. For imaging of NanoFore pharmacokinetics and cellular uptake, fluorescently labeled PLGA-BODIPY630 was used during the nanoformulation reaction, synthesized exactly as previously described by EDC/NHS coupling of PLGA30–60kDa to BODIPY-630/650-NH2 (λex/λem = 628/642 nm) (38).
Animal studies
All animal research was performed in accordance with guidelines from the Institutional Subcommittee on Research Animal Care and with approval of the Institutional Animal Care and Use Committee. Experimental models of intraperitoneally disseminated OVCA were established by injecting 5 × 106 KTR-expressing ES2 or PtD cells suspended in 200 μl of PBS into the peritoneal cavity of 6- to 10-week-old female nu/nu mice (MGH Cox 7). Tumors were allowed to grow for roughly 2 weeks before treatment for imaging studies and for roughly 1 week before treatment for efficacy studies. CT26 (1 × 106) or YUMMER1.7 cells were implanted in the flanks of 6- to 10-week-old female BALB/c (JAX) or nu/nu mice, respectively, and treatments in these studies began upon palpable tumor formation (average diameter, 7 mm ± 0.5 mm in the CT26 model).
Mice were randomly assigned into treatment groups, and no significant difference in body weight was observed across groups upon assignment. Where indicated, mice were treated with trametinib (0.3 mg/kg; 1% DMSO, 0.5% methylcellulose, and 0.2% Tween-80 in water; Selleck Chem), dabrafenib (30 mg/kg; 1% DMSO, 0.5% methylcellulose, and 0.2% Tween-80 in water; Selleck Chem), and/or maraviroc (30 mg/kg; 5% DMSO, 0.5% 0.1 N HCl in water; Selleck Chem) via oral gavage (63, 64). For clodronate liposome treatment, 150 μl of either clodronate-loaded (5 mg/ml) or PBS-loaded liposomes (Liposoma BV) were injected intraperitoneally on day 1 of the study, followed by 50 μl every 3 days thereafter (until subject reached experimental end point). For α-CSF1R antibody treatment, 200 μg of either InVivoMAb anti-mouse CSF1R (AFS98, BioXCell) or rat immunoglobulin G2a isotype control (2A3¸BioXCell) in 200 μl of PBS was injected intraperitoneally beginning on day 1 of the study, followed by every 2 to 3 days thereafter (until subject reached experimental end point). NanoFore treatments were given by tail vein or intraperitoneal injection upon immediate dilution into final 50 or 200 μl of 1× PBS, respectively. Foretinib (10 mg/kg) was used for the NanoFore efficacy study in the intraperitoneal ES2 model, while 30 mg/kg was used for all other animal studies. Subcutaneous tumor growth was monitored daily by caliper, and tumor volume was calculated using the formula V = 4/3πr3, where r = 1/4 × (length + width). In all studies, subjects were monitored daily, and a body condition score of ≤2, ascites or tumor burden affecting mobility or causing signs of pain or distress, or a tumor size exceeding 1 cm in diameter triggered the experimental end point for euthanasia and calculating animal survival. Intraperitoneal tumor burden was longitudinally tracked via whole-body fluorescence imaging using the Ami HT imaging system (Spectral Instruments Imaging). Aura software (Spectral Instruments Imaging) was used to quantify total emission (photons per second) associated with the ES2 ERK KTR (λex/λem = 465/510 nm) within equally sized regions of interest drawn around each mouse.
Flow cytometry
OVCA cells were gently detached with Versene (Gibco) before staining. For phenotypic measurements, cells were stained with annexin V fluorescein isothiocyanate and propidium iodide (Dead Cell Apoptosis Kit, Invitrogen). For RTK expression measurements, cells were stained with anti-human AXL-AF700 (108724, R&D Systems) and anti-human MET-PE (95106, R&D Systems), as well as a LIVE/DEAD fixable stain (Invitrogen) to gate out dead cells. After washing and resuspension, samples were run on an Accuri C6 flow cytometer (BD Biosciences), and data were analyzed using FlowJo software (FlowJo LLC).
For flow cytometry measurements of harvested animal tumors, tumors were digested for 30 min at 37°C in a solution containing two RPMIs: one 3× digestion medium [collagenase (10 U/ml), collagenase IV (400 U/ml), and deoxyribonuclease I (30 U/ml) in Hank’s balanced salt solution (HBSS)]. Digested tumors were filtered through 70-μm strainers, centrifuged for 5 min at 450g and 7°C. Following cell counting, pellets were resuspended in staining buffer (2% heat-inactivated FBS in HBSS), filtered again through 40-μm strainers, and incubated with a cocktail composed of DRAQ7 (BioLegend, for dead cell exclusion) and the following antibodies: anti-CD45 (30-F11), anti-CD11b (M1/70), anti-F4/80 (T45-2342), anti-CSF1R (AFS98), and anti-Mer (DS5MMER, Thermo Fisher Scientific). All antibodies except anti-Mer were purchased from BioLegend. After washing and resuspension, samples were run on a LSR II flow cytometer (BD Biosciences).
Tissue microscopy
Dextran-NP was administered intravenously, and 24 hours later, mice were euthanized and tumor-bearing organs (ovaries, uterine fat pads, liver, and omentum) were harvested for immediate imaging on a FluoView FV1000MPE confocal imaging system (Olympus America). Images were acquired with a XLFluor 2× air objective [numerical aperture (NA), 0.14; Olympus] or a XLUMPLFLN 20× water immersion objective (NA, 1.0; Olympus). Fluorescence channels were sequentially excited with 405-, 473-, 559-, and 635-nm diode lasers, combined with DM405/473/559/635-nm dichroic beam splitters. SDM473/560/640-nm beam splitters were used for emitted light, detected using BA 430- to 455-nm, BA 490- to 540-nm, BA 575- to 620-nm, and BA 655- to 755-nm emission filters (Olympus). Numbers of fluorescently labeled MΦs and ERK KTR-expressing tumor cells were quantified using maximum intensity projections across an equal number of 20× 3-zoom z-stack slices in ImageJ (NIH). Intensity measurements of ERK and JNK KTRs from images acquired with the same acquisition settings were background-corrected before calculating C/N ratios for single-tumor cells. For time-lapse intravital microscopy, 2 × 106 ES2-ECJR cells in 50 μl of PBS were injected under the fascia following implantation of titanium dorsal skin-fold window chambers (APJ Trading) in nu/nu mice. Analgesic buprenorphine was used before and for 3 days following implantation, and surgeries were performed under 2% isoflurane supplied with O2 (2 liters/min). Chambers were sealed with sterile coverslips, and mice were prophylactically supplied with antibiotic in drinking water. Xenograft tumors were allowed to grow for approximately 2 weeks before treatment and imaging on the FV1000MPE system. On the day of the imaging experiment, the mice were anesthetized with isoflurane, tail-vein catheters were placed, and subjects were immobilized onto a heated microscope stage (37°C) and monitored under anesthesia for the experiment duration. Ex vivo FRI was performed using an OV110 imaging system (Olympus). For microscopy analysis, fluorescence intensities were quantified following background subtraction. Dextran-NPs and fluorescent derivatives were prepared, as previously described, using Pacific Blue–NHS ester (38). Briefly, carboxymethyl dextran was activated with EDC and NHS, cross-linked with l-lysine in 2-(N-morpholino)ethanesulfonic acid buffer, and purified by dialysis, following previous publication (38). This product (hydrodynamic size, ~18 nm) (38), or 500-kDa amino-dextran (Thermo Fisher Scientific; hydrodynamic radius, ~16 nm) (65) in application to time-lapse intravital microscopy, was reacted with Pacific Blue–NHS ester or VT680XL-NHS ester (e.g., Fig. 5F) at room temperature and purified again by a PD-10 column (GE Healthcare) or 30-kDa Amicon spin filter (Millipore) (38). Approximately 5 nmol dye per mouse was used for imaging, measured by absorbance (NanoDrop, Thermo Fisher Scientific).
Statistical analysis
PCA was performed on mean-centered, variance-normalized data using PLS Toolbox (Eigenvector Research) for MATLAB (MathWorks). Simulation and inference of parameters for the Loewe model of drug-drug synergy were implemented, as described previously, using MATLAB (12), and SE in this analysis was quantified via jackknife leave-one-out cross-validation. Throughout the report, results were displayed as means ± SEM, unless otherwise stated, with α = 0.05 level of significance. Statistical analyses were performed with GraphPad Prism 7.0 or MATLAB.
Supplementary Material
Acknowledgments
We thank R. J. Sullivan (MGH) for helpful guidance and S. Schmidt (MGH) for assistance with mouse model imaging. Funding: This work was supported, in part, by NIH/NCI grants R00CA207744, R00CA207866, U01CA206997, R01CA206890, T32CA079443, R01GM069668, R01CA96504, U54CA112967, and U54CA217377, the NSF Graduate Research Fellowship Program (S.J.W. and G.L.), and an American Thyroid Association/Thyroid Cancer Survivors’ Association Research Grant. Author contributions: S.J.W., M.A.M., D.A.L., and R.W. developed the concept. M.A.M., S.J.W., D.A.L., and R.W. wrote the paper. All authors designed the experiments, analyzed the results, and edited the manuscript. Competing interests: R.W. is a cofounder of T2Biosystems and Lumicell, serves as a scientific advisor for ModeRNA Therapeutics, Tarveda Therapeutics, and Alivio Therapeutics. None of these activities are related to the manuscript. M.A.M., D.A.L., and M.J.O. are co-inventors of a patent related to the described research. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding author (M.A.M.). Requests for collaboration involving materials used in this research will be fulfilled provided that a written agreement is executed in advance between MGH and the requesting parties.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz8521/DC1
REFERENCES AND NOTES
- 1.Gao J., Aksoy B. A., Dogrusoz U., Dresdner G., Gross B., Sumer S. O., Sun Y., Jacobsen A., Sinha R., Larsson E., Cerami E., Sander C., Schultz N., Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhodes D. R., Kalyana-Sundaram S., Mahavisno V., Varambally R., Yu J., Briggs B. B., Barrette T. R., Anstet M. J., Kincead-Beal C., Kulkarni P., Varambally S., Ghosh D., Chinnaiyan A. M., Oncomine 3.0: Genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia 9, 166–180 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ramaswamy S., Tamayo P., Rifkin R., Mukherjee S., Yeang C.-H., Angelo M., Ladd C., Reich M., Latulippe E., Mesirov J. P., Multiclass cancer diagnosis using tumor gene expression signatures. Proc. Natl. Acad. Sci. 98, 15149–15154 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Su A. I., Welsh J. B., Sapinoso L. M., Kern S. G., Dimitrov P., Lapp H., Schultz P. G., Powell S. M., Moskaluk C. A., Frierson H. F. Jr., Hampton G. M., Molecular classification of human carcinomas by use of gene expression signatures. Cancer Res. 61, 7388–7393 (2001). [PubMed] [Google Scholar]
- 5.Ramaswamy S., Ross K. N., Lander E. S., Golub T. R., A molecular signature of metastasis in primary solid tumors. Nat. Genet. 33, 49–54 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Farley J., Brady W. E., Vathipadiekal V., Lankes H. A., Coleman R., Morgan M. A., Mannel R., Yamada S. D., Mutch D., Rodgers W. H., Birrer M., Gershenson D. M., Selumetinib in women with recurrent low-grade serous carcinoma of the ovary or peritoneum: An open-label, single-arm, phase 2 study. Lancet Oncol. 14, 134–140 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corcoran R. B., André T., Atreya C. E., Schellens J. H. M., Yoshino T., Bendell J. C., Hollebecque A., McRee A. J., Siena S., Middleton G., Muro K., Gordon M. S., Tabernero J., Yaeger R., O’Dwyer P. J., Humblet Y., De Vos F., Jung A. S., Brase J. C., Jaeger S., Bettinger S., Mookerjee B., Rangwala F., Van Cutsem E., Combined BRAF, EGFR, and MEK inhibition in patients with BRAFV600E-mutant colorectal cancer. Cancer Discov. 8, 428–443 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Whittaker S., Kirk R., Hayward R., Zambon A., Viros A., Cantarino N., Affolter A., Nourry A., Niculescu-Duvaz D., Springer C., Marais R., Gatekeeper mutations mediate resistance to BRAF-targeted therapies. Sci. Transl. Med. 2, 35ra41 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Wagle N., Van Allen E. M., Treacy D. J., Frederick D. T., Cooper Z. A., Taylor-Weiner A., Rosenberg M., Goetz E. M., Sullivan R. J., Farlow D. N., Friedrich D. C., Anderka K., Perrin D., Johannessen C. M., Kenna A. M., Cibulskis K., Kryukov G., Hodis E., Lawrence D. P., Fisher S., Getz G., Gabriel S. B., Carter S. L., Flaherty K. T., Wargo J. A., Garraway L. A., MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 4, 61–68 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan J. S., Whittle M. C., Nakamura K., Abell A. N., Midland A. A., Zawistowski J. S., Johnson N. L., Granger D. A., Jordan N. V., Darr D. B., Usary J., Kuan P.-F., Smalley D. M., Major B., He X., Hoadley K. A., Zhou B., Sharpless N. E., Perou C. M., Kim W. Y., Gomez S. M., Chen X., Jin J., Frye S. V., Earp H. S., Graves L. M., Johnson G. L., Dynamic reprogramming of the kinome in response to targeted MEK inhibition in triple-negative breast cancer. Cell 149, 307–321 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Villanueva J., Vultur A., Lee J. T., Somasundaram R., Fukunaga-Kalabis M., Cipolla A. K., Wubbenhorst B., Xu X., Gimotty P. A., Kee D., Santiago-Walker A. E., Letrero R., D’Andrea K., Pushparajan A., Hayden J. E., Brown K. D., Laquerre S., McArthur G. A., Sosman J. A., Nathanson K. L., Herlyn M., Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by cotargeting MEK and IGF-1R/PI3K. Cancer Cell 18, 683–695 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller M. A., Oudin M. J., Sullivan R. J., Wang S. J., Meyer A. S., Im H., Frederick D. T., Tadros J., Griffith L. G., Lee H., Weissleder R., Flaherty K. T., Gertler F. B., Lauffenburger D. A., Reduced proteolytic shedding of receptor tyrosine kinases is a post-translational mechanism of kinase inhibitor resistance. Cancer Discov. 6, 382–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T., Xiao M., Ge Y., Krepler C., Belser E., Lopez-Coral A., Xu X., Zhang G., Azuma R., Liu Q., Liu R., Li L., Amaravadi R. K., Xu W., Karakousis G., Gangadhar T. C., Schuchter L. M., Lieu M., Khare S., Halloran M. B., Herlyn M., Kaufman R. E., BRAF inhibition stimulates melanoma-associated macrophages to drive tumor growth. Clin. Cancer Res. 21, 1652–1664 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith M. P., Sanchez-Laorden B., O’Brien K., Brunton H., Ferguson J., Young H., Dhomen N., Flaherty K. T., Frederick D. T., Cooper Z. A., Wargo J. A., Marais R., Wellbrock C., The immune microenvironment confers resistance to MAPK pathway inhibitors through macrophage-derived TNFα. Cancer Discov. 4, 1214–1229 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hirata E., Girotti M. R., Viros A., Hooper S., Spencer-Dene B., Matsuda M., Larkin J., Marais R., Sahai E., Intravital imaging reveals how BRAF inhibition generates drug-tolerant microenvironments with high integrin β1/FAK signaling. Cancer Cell 27, 574–588 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dummer R., Ascierto P. A., Gogas H. J., Arance A., Mandala M., Liszkay G., Garbe C., Schadendorf D., Krajsova I., Gutzmer R., Chiarion-Sileni V., Dutriaux C., de Groot J. W. B., Yamazaki N., Loquai C., Moutouh-de Parseval L. A., Pickard M. D., Sandor V., Robert C., Flaherty K. T., Encorafenib plus binimetinib versus vemurafenib or encorafenib in patients with BRAF-mutant melanoma (COLUMBUS): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 19, 603–615 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Van Cutsem E., Huijberts S., Grothey A., Yaeger R., Cuyle P.-J., Elez E., Fakih M., Montagut C., Peeters M., Yoshino T., Wasan H., Desai J., Ciardiello F., Gollerkeri A., Christy-Bittel J., Maharry K., Sandor V., Schellens J. H. M., Kopetz S., Tabernero J., Binimetinib, encorafenib, and cetuximab triplet therapy for patients with BRAFV600E-mutant metastatic colorectal cancer: Safety lead-in results from the phase III BEACON colorectal cancer study. J. Clin. Oncol. 37, 1460–1469 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hyman D. M., Puzanov I., Subbiah V., Faris J. E., Chau I., Blay J.-Y., Wolf J., Raje N. S., Diamond E. L., Hollebecque A., Gervais R., Elez-Fernandez M. E., Italiano A., Hofheinz R. D., Hidalgo M., Chan E., Schuler M., Lasserre S. F., Makrutzki M., Sirzen F., Veronese M. L., Tabernero J., Baselga J., Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N. Engl. J. Med. 373, 726–736 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ashton S., Song Y. H., Nolan J., Cadogan E., Murray J., Odedra R., Foster J., Hall P. A., Low S., Taylor P., Ellston R., Polanska U. M., Wilson J., Howes C., Smith A., Goodwin R. J., Swales J. G., Strittmatter N., Takáts Z., Nilsson A., Andren P., Trueman D., Walker M., Reimer C. L., Troiano G., Parsons D., De Witt D., Ashford M., Hrkach J., Zale S., Jewsbury P. J., Barry S. T., Aurora kinase inhibitor nanoparticles target tumors with favorable therapeutic index in vivo. Sci. Transl. Med. 8, 325ra17 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Shi J., Kantoff P. W., Wooster R., Farokhzad O. C., Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 17, 20–37 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Paraiso K. H., Xiang Y., Rebecca V. W., Abel E. V., Chen Y. A., Munko A. C., Wood E., Fedorenko I. V., Sondak V. K., Anderson A. R., Ribas A., Palma M. D., Nathanson K. L., Koomen J. M., Messina J. L., Smalley K. S., PTEN loss confers BRAF inhibitor resistance to melanoma cells through the suppression of BIM expression. Cancer Res. 71, 2750–2760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun C., Wang L., Huang S., Heynen G. J., Prahallad A., Robert C., Haanen J., Blank C., Wesseling J., Willems S. M., Zecchin D., Hobor S., Bajpe P. K., Lieftink C., Mateus C., Vagner S., Grernrum W., Hofland I., Schlicker A., Wessels L. F., Beijersbergen R. L., Bardelli A., Di Nicolantonio F., Eggermont A. M., Bernards R., Reversible and adaptive resistance to BRAF(V600E) inhibition in melanoma. Nature 508, 118–122 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Prahallad A., Sun C., Huang S., Di Nicolantonio F., Salazar R., Zecchin D., Beijersbergen R. L., Bardelli A., Bernards R., Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483, 100–103 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Long G. V., Fung C., Menzies A. M., Pupo G. M., Carlino M. S., Hyman J., Shahheydari H., Tembe V., Thompson J. F., Saw R. P., Howle J., Hayward N. K., Johansson P., Scolyer R. A., Kefford R. F., Rizos H., Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat. Commun. 5, 5694 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Tirosh I., Izar B., Prakadan S. M., Wadsworth M. H., Treacy D., Trombetta J. J., Rotem A., Rodman C., Lian C., Murphy G., Fallahi-Sichani M., Dutton-Regester K., Lin J.-R., Cohen O., Shah P., Lu D., Genshaft A. S., Hughes T. K., Ziegler C. G., Kazer S. W., Gaillard A., Kolb K. E., Villani A.-C., Johannessen C. M., Andreev A. Y., Van Allen E. M., Bertagnolli M., Sorger P. K., Sullivan R. J., Flaherty K. T., Frederick D. T., Jané-Valbuena J., Yoon C. H., Rozenblatt-Rosen O., Shalek A. K., Regev A., Garraway L. A., Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kumar M. P., Du J., Lagoudas G., Jiao Y., Sawyer A., Drummond D. C., Lauffenburger D. A., Raue A., Analysis of single-cell RNA-seq identifies cell-cell communication associated with tumor characteristics. Cell Rep. 25, 1458–1468.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ramilowski J. A., Goldberg T., Harshbarger J., Kloppmann E., Lizio M., Satagopam V. P., Itoh M., Kawaji H., Carninci P., Rost B., Forrest A. R., A draft network of ligand–receptor-mediated multicellular signalling in human. Nat. Commun. 6, 7866 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stanford J. C., Young C., Hicks D., Owens P., Williams A., Vaught D. B., Morrison M. M., Lim J., Williams M., Brantley-Sieders D. M., Balko J. M., Tonetti D., Earp H. S. III, Cook R. S., Efferocytosis produces a prometastatic landscape during postpartum mammary gland involution. J. Clin. Invest. 124, 4737–4752 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arlauckas S. P., Garren S. B., Garris C. S., Kohler R. H., Oh J., Pittet M. J., Weissleder R., Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 8, 5842–5854 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zilionis R., Engblom C., Pfirschke C., Savova V., Zemmour D., Saatcioglu H. D., Krishnan I., Maroni G., Meyerovitz C. V., Kerwin C. M., Choi S., Richards W. G., De Rienzo A., Tenen D. G., Bueno R., Levantini E., Pittet M. J., Klein A. M., Single-cell transcriptomics of human and mouse lung cancers reveals conserved myeloid populations across individuals and species. Immunity 50, 1317–1334.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller M. A., Sullivan R. J., Lauffenburger D. A., Molecular pathways: Receptor ectodomain shedding in treatment, resistance, and monitoring of cancer. Clin. Cancer Res. 23, 623–629 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engblom C., Pfirschke C., Pittet M. J., The role of myeloid cells in cancer therapies. Nat. Rev. Cancer 16, 447–462 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Grisham R. N., Garg K., Zhou Q., Iasonos A., Berger M. F., Dao F., Hyman D. M., Levine D. A., Solit D. B., Aghajanian C., A comprehensive analysis of BRAF and KRAS mutation status in low-grade serous (LGS) and serous borderline (SB) ovarian cancer (OC). J. Clin. Oncol. 30, 5030 (2012). [Google Scholar]
- 34.Anglesio M. S., Arnold J. M., George J., Tinker A. V., Tothill R., Waddell N., Simms L., Locandro B., Fereday S., Traficante N., Russell P., Sharma R., Birrer M. J.; AOCS Study Group, deFazio A., Chenevix-Trench G., Bowtell D. D., Mutation of ERBB2 provides a novel alternative mechanism for the ubiquitous activation of RAS-MAPK in ovarian serous low malignant potential tumors. Mol. Cancer Res. 6, 1678–1690 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Segura E., Touzot M., Bohineust A., Cappuccio A., Chiocchia G., Hosmalin A., Dalod M., Soumelis V., Amigorena S., Human inflammatory dendritic cells induce Th17 cell differentiation. Immunity 38, 336–348 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Gao Q., Yang Z., Xu S., Li X., Yang X., Jin P., Liu Y., Zhou X., Zhang T., Gong C., Wei X., Liu D., Sun C., Chen G., Hu J., Meng L., Zhou J., Sawada K., Fruscio R., Grunt T. W., Wischhusen J., Vargas-Hernández V. M., Pothuri B., Coleman R. L., Heterotypic CAF-tumor spheroids promote early peritoneal metastatis of ovarian cancer. J. Exp. Med. 216, 688–703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J., Perry C. J., Meeth K., Thakral D., Damsky W., Micevic G., Kaech S., Blenman K., Bosenberg M., UV-induced somatic mutations elicit a functional T cell response in the YUMMER1.7 mouse melanoma model. Pigment Cell Melanoma Res. 30, 428–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim H. Y., Li R., Ng T. S. C., Courties G., Rodell C. B., Prytyskach M., Kohler R. H., Pittet M. J., Nahrendorf M., Weissleder R., Miller M. A., Quantitative imaging of tumor-associated macrophages and their response to therapy using 64Cu-labeled macrin. ACS Nano 12, 12015–12029 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lok G. T., Chan D. W., Liu V. W., Hui W. W., Leung T. H., Yao K. M., Ngan H. Y., Aberrant activation of ERK/FOXM1 signaling cascade triggers the cell migration/invasion in ovarian cancer cells. PLOS ONE 6, e23790 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P., Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 14, 399–416 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seitz H. M., Camenisch T. D., Lemke G., Earp H. S., Matsushima G. K., Macrophages and dendritic cells use different Axl/Mertk/Tyro3 receptors in clearance of apoptotic cells. J. Immunol. 178, 5635–5642 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Regot S., Hughey J. J., Bajar B. T., Carrasco S., Covert M. W., High-sensitivity measurements of multiple kinase activities in live single cells. Cell 157, 1724–1734 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodell C. B., Arlauckas S. P., Cuccarese M. F., Garris C. S., Li R., Ahmed M. S., Kohler R. H., Pittet M. J., Weissleder R., TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng. 2, 578–588 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miller M. A., Zheng Y.-R., Gadde S., Pfirschke C., Zope H., Engblom C., Kohler R. H., Iwamoto Y., Yang K. S., Askevold B., Kolishetti N., Pittet M., Lippard S. J., Farokhzad O. C., Weissleder R., Tumour-associated macrophages act as a slow-release reservoir of nano-therapeutic Pt(IV) pro-drug. Nat. Commun. 6, 8692 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Myers S. H., Brunton V. G., Unciti-Broceta A., AXL inhibitors in cancer: A medicinal chemistry perspective. J. Med. Chem. 59, 3593–3608 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Graham D. K., DeRyckere D., Davies K. D., Earp H. S., The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 14, 769–785 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Zweemer A. J. M., French C. B., Mesfin J., Gordonov S., Meyer A. S., Lauffenburger D. A., Apoptotic bodies elicit Gas6-mediated migration of AXL-expressing tumor cells. Mol. Cancer Res. 15, 1656–1666 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kipps E., Tan D. S., Kaye S. B., Meeting the challenge of ascites in ovarian cancer: New avenues for therapy and research. Nat. Rev. Cancer 13, 273–282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Klein S. L., Flanagan K. L., Sex differences in immune responses. Nat. Rev. Immunol. 16, 626–638 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Leighl N. B., Tsao M. S., Liu G., Tu D., Ho C., Shepherd F. A., Murray N., Goffin J. R., Nicholas G., Sakashita S., Chen Z., Kim L., Powers J., Seymour L., Goss G., Bradbury P. A., A phase I study of foretinib plus erlotinib in patients with previously treated advanced non-small cell lung cancer: Canadian cancer trials group IND.196. Oncotarget 8, 69651–69662 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller M. A., Chandra R., Cuccarese M. F., Pfirschke C., Engblom C., Stapleton S., Adhikary U., Kohler R. H., Mohan J. F., Pittet M. J., Weissleder R., Radiation therapy primes tumors for nanotherapeutic delivery via macrophage-mediated vascular bursts. Sci. Transl. Med. 9, eaal0225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weissleder R., Nahrendorf M., Pittet M. J., Imaging macrophages with nanoparticles. Nat. Mater. 13, 125–138 (2014). [DOI] [PubMed] [Google Scholar]
- 53.Miller M. A., Arlauckas S., Weissleder R., Prediction of anti-cancer nanotherapy efficacy by imaging. Nanotheranostics 1, 296–312 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Arlauckas S. P., Garris C. S., Kohler R. H., Kitaoka M., Cuccarese M. F., Yang K. S., Miller M. A., Carlson J. C., Freeman G. J., Anthony R. M., Weissleder R., Pittet M. J., In vivo imaging reveals a tumor-associated macrophage–mediated resistance pathway in anti–PD-1 therapy. Sci. Transl. Med. 9, eaal3604 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Miller M. A., Gadde S., Pfirschke C., Engblom C., Sprachman M. M., Kohler R. H., Yang K. S., Laughney A. M., Wojtkiewicz G., Kamaly N., Bhonagiri S., Pittet M. J., Farokhzad O. C., Weissleder R., Predicting therapeutic nanomedicine efficacy using a companion magnetic resonance imaging nanoparticle. Sci. Transl. Med. 7, 314ra183 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hendrix N. D., Wu R., Kuick R., Schwartz D. R., Fearon E. R., Cho K. R., Fibroblast growth factor 9 has oncogenic activity and is a downstream target of Wnt signaling in ovarian endometrioid adenocarcinomas. Cancer Res. 66, 1354–1362 (2006). [DOI] [PubMed] [Google Scholar]
- 57.Newman A. M., Liu C. L., Green M. R., Gentles A. J., Feng W., Xu Y., Hoang C. D., Diehn M., Alizadeh A. A., Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haney M. S., Bohlen C. J., Morgens D. W., Ousey J. A., Barkal A. A., Tsui C. K., Ego B. K., Levin R., Kamber R. A., Collins H., Tucker A., Li A., Vorselen D., Labitigan L., Crane E., Boyle E., Jiang L., Chan J., Rincón E., Greenleaf W. J., Li B., Snyder M. P., Weissman I. L., Theriot J. A., Collins S. R., Barres B. A., Bassik M. C., Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nat. Genet. 50, 1716–1727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Johannessen C. M., Boehm J. S., Kim S. Y., Thomas S. R., Wardwell L., Johnson L. A., Emery C. M., Stransky N., Cogdill A. P., Barretina J., Caponigro G., Hieronymus H., Murray R. R., Salehi-Ashtiani K., Hill D. E., Vidal M., Zhao J. J., Yang X., Alkan O., Kim S., Harris J. L., Wilson C. J., Myer V. E., Finan P. M., Root D. E., Roberts T. M., Golub T., Flaherty K. T., Dummer R., Weber B. L., Sellers W. R., Schlegel R., Wargo J. A., Hahn W. C., Garraway L. A., COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gruosso T., Garnier C., Abelanet S., Kieffer Y., Lemesre V., Bellanger D., Bieche I., Marangoni E., Sastre-Garau X., Mieulet V., Mechta-Grigoriou F., MAP3K8/TPL-2/COT is a potential predictive marker for MEK inhibitor treatment in high-grade serous ovarian carcinomas. Nat. Commun. 6, 8583 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Théry C., Amigorena S., Raposo G., Clayton A., Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 30, 3.22. 1–3.22. 29 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Jing Y., Zhang Z., Ma P., An S., Shen Y., Zhu L., Zhuang G., Concomitant BET and MAPK blockade for effective treatment of ovarian cancer. Oncotarget 7, 2545–2554 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Walker D. K., Abel S., Comby P., Muirhead G. J., Nedderman A. N. R., Smith D. A., Species differences in the disposition of the CCR5 antagonist, UK-427,857, a new potential treatment for HIV. Drug Metab. Dispos. 33, 587–595 (2005). [DOI] [PubMed] [Google Scholar]
- 65.Armstrong J. K., Wenby R. B., Meiselman H. J., Fisher T. C., The hydrodynamic radii of macromolecules and their effect on red blood cell aggregation. Biophys. J. 87, 4259–4270 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/21/eaaz8521/DC1