

CONSPECTUS.
From structure elucidation and biogenesis to synthetic methodology and total synthesis, terpene natural products have profoundly influenced the development of organic chemistry. Moreover, their myriad functional attributes range from fragrance to pharmaceuticals and have had great societal impact. Ruzicka’s formulation of the “biogenetic isoprene rule,” a Nobel Prize winning discovery now over 80 years old, allowed for identification of higher order terpene (aka “isoprenoid”) structures from simple five-carbon isoprene fragments. Notably, the isoprene rule still holds pedagogical value to students of organic chemistry today. Our laboratory has completed syntheses of over two dozen terpene and meroterpene structures to date, and the isoprene rule has served as a key pattern recognition tool for our synthetic planning purposes. At the strategic level, great opportunity exists in finding unique and synthetically simplifying ways to connect the formal C5 isoprene fragments embedded in terpenes. Biomimetic cationic polyene cyclizations represent the earliest incarnation of this idea, which has facilitated expedient routes to certain terpene polycycle classes. Nonetheless, a large swath of terpene chemical space remains inaccessible using this approach.
In this Account, we describe strategic insight into our endeavors in terpene synthesis published over the last five years. We show how biosynthetic understanding, combined with a desire to utilize abundant and inexpensive [C5]n building blocks, has led to efficient, abiotic syntheses of multiple complex terpenes with disparate ring systems. Informed by nature, but unconstrained by its processes, our synthetic assembly exploits chemical reactivity across diverse reaction types—including radical, anionic, pericyclic, and metal-mediated transformations.
First, we detail an 8-step synthesis of the cembrane diterpene chatancin from dihydrofarnesal using a bio-inspired–but not -mimetic–cycloaddition. Next, we describe the assembly of the antimalarial cardamom peroxide using a polyoxygenation cascade to fuse multiple units of molecular oxygen onto a dimeric skeleton. This 3–4 step synthesis arises from (–)-myrtenal, an inexpensive pinene oxidation product. We then show how a radical cyclization cascade can forge the hallmark cyclooctane ring system of the complex sesterterpene 6-epi-ophioblin N from two simple polyprenyl precursors, (–)-linalool and farnesol. To access the related, more complex metabolite 6-epi-ophiobolin A, we exploited the plasticity of our synthetic route and found that use of geraniol (C10) rather than farnesol (C15) gave us the flexibility needed to address the additional oxidation found in this congener. Following this work, we describe two strategies to access several guaianolide sesquiterpenes. Retrosynthetic disconnection to monoterpenes, carvone or (–)-linalool, coupled with a powerful allylation strategy allowed us to address guaianolides with disparate stereochemical motifs. Finally, we examine a semisynthetic approach to the illicium sesquiterpenes from the abundant 15-carbon feedstock terpene (+)-cedrol using an abiotic ring shift and multiple C–H oxidation reactions inspired by a postulated biosynthesis of this natural product class.
Graphical Abstract
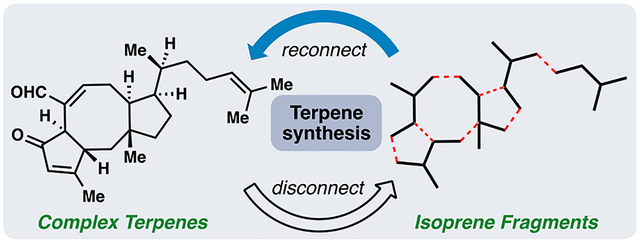
INTRODUCTION
Despite originating from a single biosynthetic building block, the incredibly vast terpene chemical space contains well over 50,000 discrete molecules and hundreds of unique polycyclic ring systems.1 Terpene synthases are responsible for the oligomerization of five-carbon dimethylallyl pyrophosphate (DMAPP) units and their further cyclization to (poly)cyclic mono- (C10), sesqui- (C15), di- (C20), sester- (C25), and triterpenes (C30).2 Additional enzymatic tailoring, typically via hydroxylation and subsequent esterification, converts these simple, hydrocarbon materials into impressive chemical libraries with rich three-dimensional complexity and widely varying oxidation patterns. Additionally, meroterpenes, which are produced by transferase-mediated attachment of prenyl, geranyl, or farnesyl groups to natural products otherwise arising from polyketide, amino acid (i.e. shikimate), and alkaloid biosynthetic pathways, further serve to amplify the number and structural diversity of terpene-like small molecules.
Given the immense heterogeneity of terpenoids, there remains no unifying strategy for their synthetic construction. Approaches are often determined on a case-by-case basis guided by the precise structure of the carbocyclic ring system found in the terpene target.3 A large body of synthetic work has sought to emulate aspects of nature’s cationic cyclization and rearrangement pathways for stereocontrolled synthesis.4 However, a large number of exotic terpene rings systems including many with medium or large ring sizes are not efficiently prepared in the laboratory at the present time with these strategies.
Our laboratory has reported the syntheses of an assortment of biologically active terpene (see 1–4, 10-16)5 and meroterpene (see 5–9)6 targets during the past five years (Figure 1). While these structures are largely unrelated, we approached all of our syntheses by considering both their proposed biosynthesis (if known) and Ruzicka’s isoprene rule.7 We devised synthetic plans which maximized the number of isoprene (or more generally [C5]n) fragments in potential starting materials, since these units are conserved throughout all chiral pool and achiral terpene building blocks.8 The synthetic processes capable of connecting the fragments were considered only after identifying potential terpene starting materials. Herein, we highlight five distinct terpene projects in our laboratory guided by these ideas; each established concise routes to the target structures from simple [C5]n building blocks.
Figure 1.
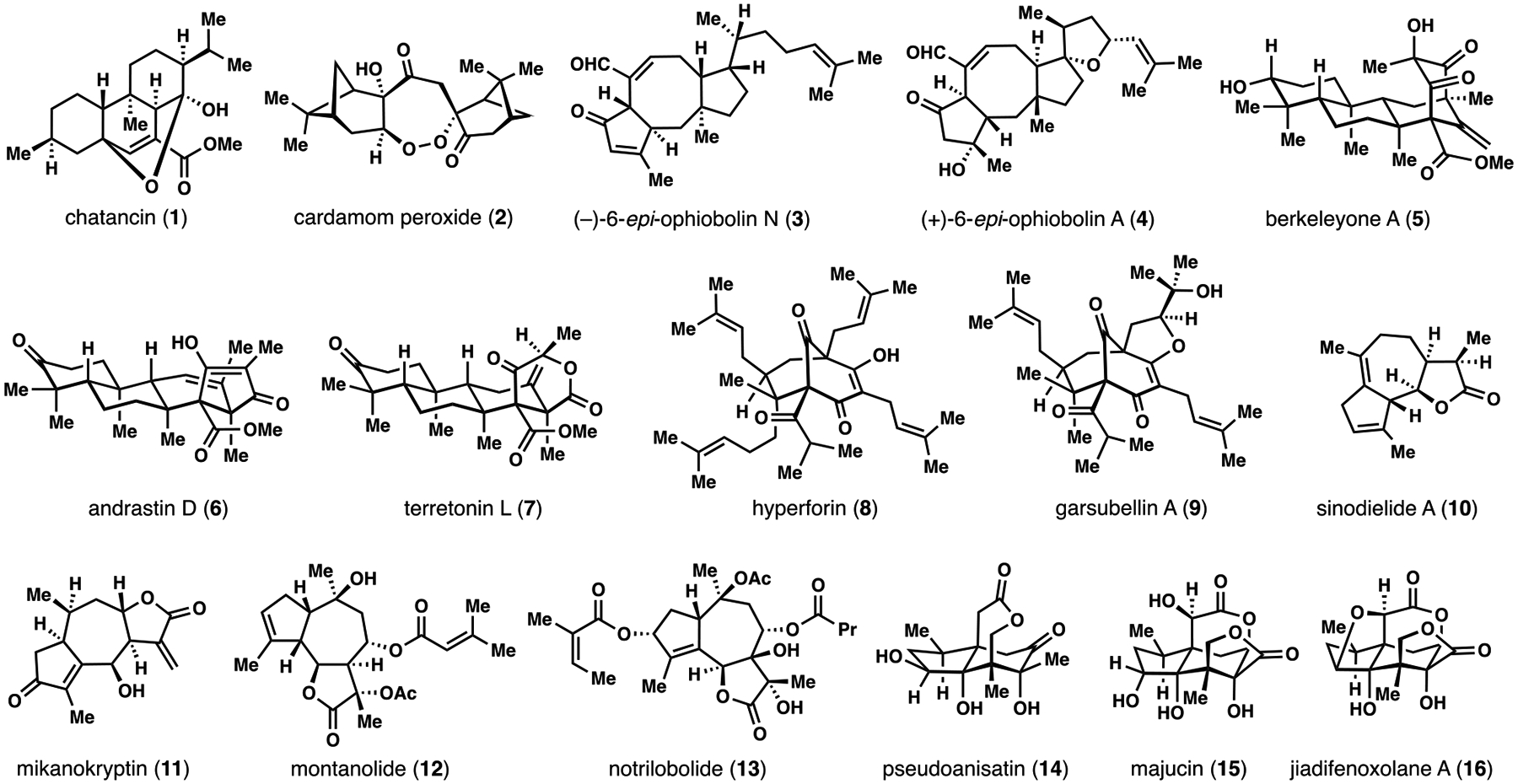
Selected terpene and meroterpene natural products synthesized in the Maimone laboratory (2014–2019).
TOTAL SYNTHESIS OF CHATANCIN
In search of naturally occurring platelet activating factor (PAF) antagonists, Sato and co-workers isolated the polycyclic diterpene chatancin (1) from Sarcophyton sp., a soft coral found off the coast of Japan.9 Chatancin (1) was found to both inhibit PAF-induced platelet aggregation and PAF receptor binding at low to submicromolar concentrations (Figure 2A). The intriguing terpene skeleton of 1 is suggestive of potential cembranoid biosynthetic origins (see 18), and its complex polycyclic ring system is proposed to arise from a transannular [4+2] cycloaddition between an alkene and a pyran, furan, or pyrylium-type (see 17) diene.10 The latter, pyrylium-based pathway is favored energetically according to calculations by Tantillo.11,12 Inspired by the unique polycyclic framework and bioactivity of chatancin (1), the groups of Gössinger,13 Deslongchamps,10 Maimone,5b and Ding14 have each reported distinct total syntheses.
Figure 2.
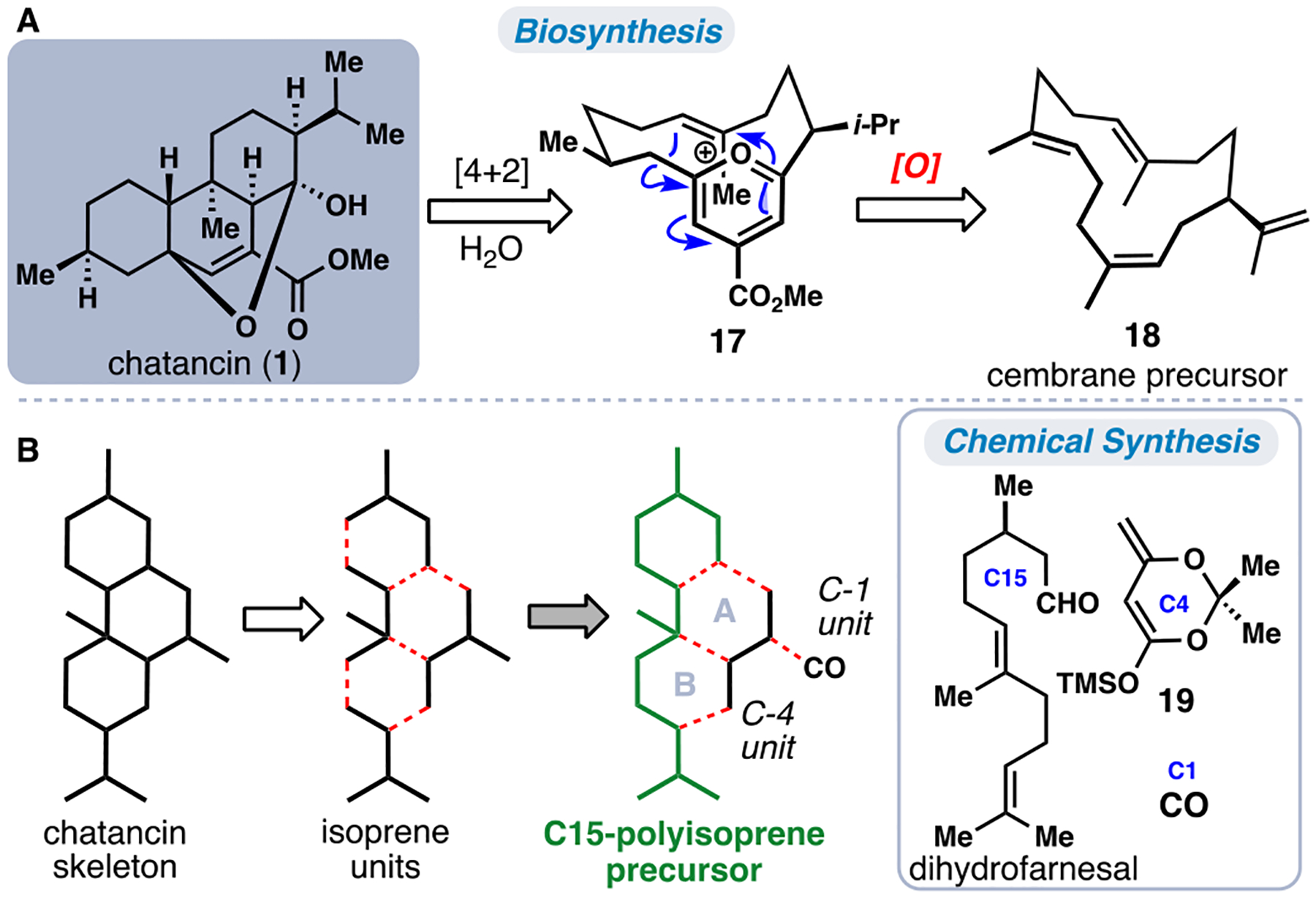
The marine cembranoid chatancin (1). (A) Proposed biosynthesis. (B) Isoprenoid building block-based disconnection.
We analyzed the chatancin carbocyclic skeleton tracing it back to its constituent formal isoprene units (Figure 2B). Reconnecting three of these fragments gave rise to a C15-polyprenyl precursor (shown in green), representative of a farnesyl-type chain. From this fragment, five additional carbon atoms and two six-membered rings (see rings A and B) comprise the core of 1. Diverging from the biosynthesis of 1, which presumably arises from a macrocyclic precursor, we envisioned that entropic considerations would make initial formation of ring A easier. In the forward sense, dihydrofarnesal, silylketene acetal 19,15 and carbon monoxide were utilized as discrete chemical building blocks (Figure 2A).
We began our synthetic studies by forging an aldol adduct from 19 and (S)-2,3-dihydrofarnesal (available in one-step) through a BF3•OEt2-mediated Mukaiyama aldol process,15 which was oxidized in-situ with Dess-Martin Periodinane to afford compound 20 (Scheme 1). Heating 20 in refluxing toluene cleanly elicited thermal acetone extrusion and subsequent cyclization to pyrone 21, which was triflated in-situ (Tf2O, Et3N) to yield vinyl triflate 22.16 Attachment of the final carbon atom was achieved via a Pd-catalyzed methoxycarbonylation (Pd(OAc)2/DPEPhos/CO/MeOH) to generate a methyl ester intermediate, which underwent a pyrone-alkene [4+2] cycloaddition with careful heating (PhMe, 100 °C). Noatbly, more forcing conditions caused loss of CO2 and generation of a diene. While this cycloaddition proceeded in high yield (90%), two diastereomers (see 23 and 24) out of a possible 4, were formed in equal quantities. To construct the final ring of chatancin, the terminal prenyl group was chlorinated (SO2Cl2), and intermediate 25 was subjected to a zinc-mediated addition to the neighboring lactone. Finally, hydrogenation of the isopropenyl group (H2, Pd/C) in 26 afforded chatancin (1) in 8 steps from commercially available materials (13% overall yield) and without the need for protecting groups.
Scheme 1.
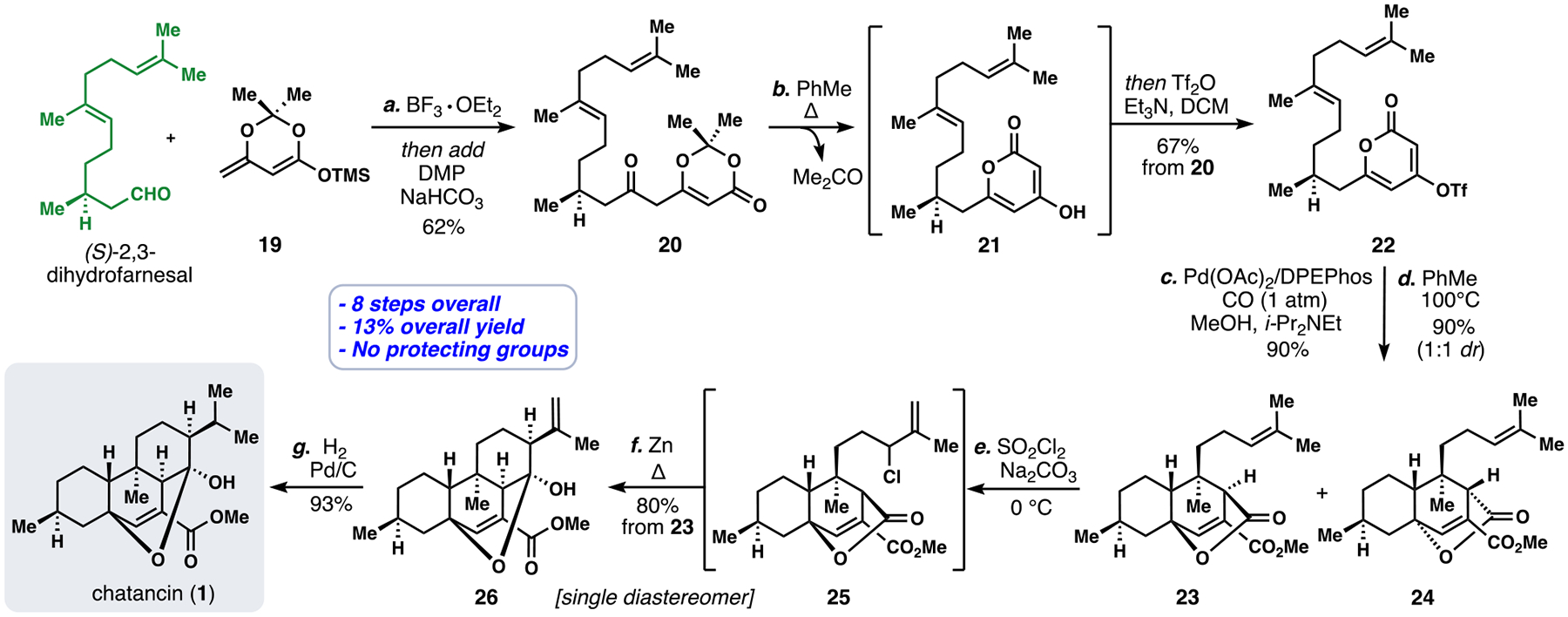
Total Synthesis of chatancin (1).
TOTAL SYNTHESIS OF CARDAMOM PEROXIDE
The cardamom peroxide (2), isolated from Amomum krervanh Pierre (Siam cardamom), is a 1,2-dioxepane-containing terpenoid, which possesses strong inhibitory activity against P. falciparum malaria (Figure 3A).17 We became interested in this natural product to investigate its possible biosynthetic origins and antimalarial mechanism of action. We speculated that the 7-membered endoperoxide might be formed in nature via an air oxidation process, in analogy to the biosynthesis of the flagship antimalarial endoperoxide natural product artemisinin.18 In applying this hypothesis to 2, we envisioned that peroxyradical 27 might serve as a key biosynthetic intermediate capable of undergoing diastereoselective 7-endo radical addition to forge the endoperoxide core and ultimately provide 2 after further oxygenation and hydroperoxide reduction (Figure 3A).19 With this sequence in mind, generating 27 became our primary synthetic objective.
Figure 3.
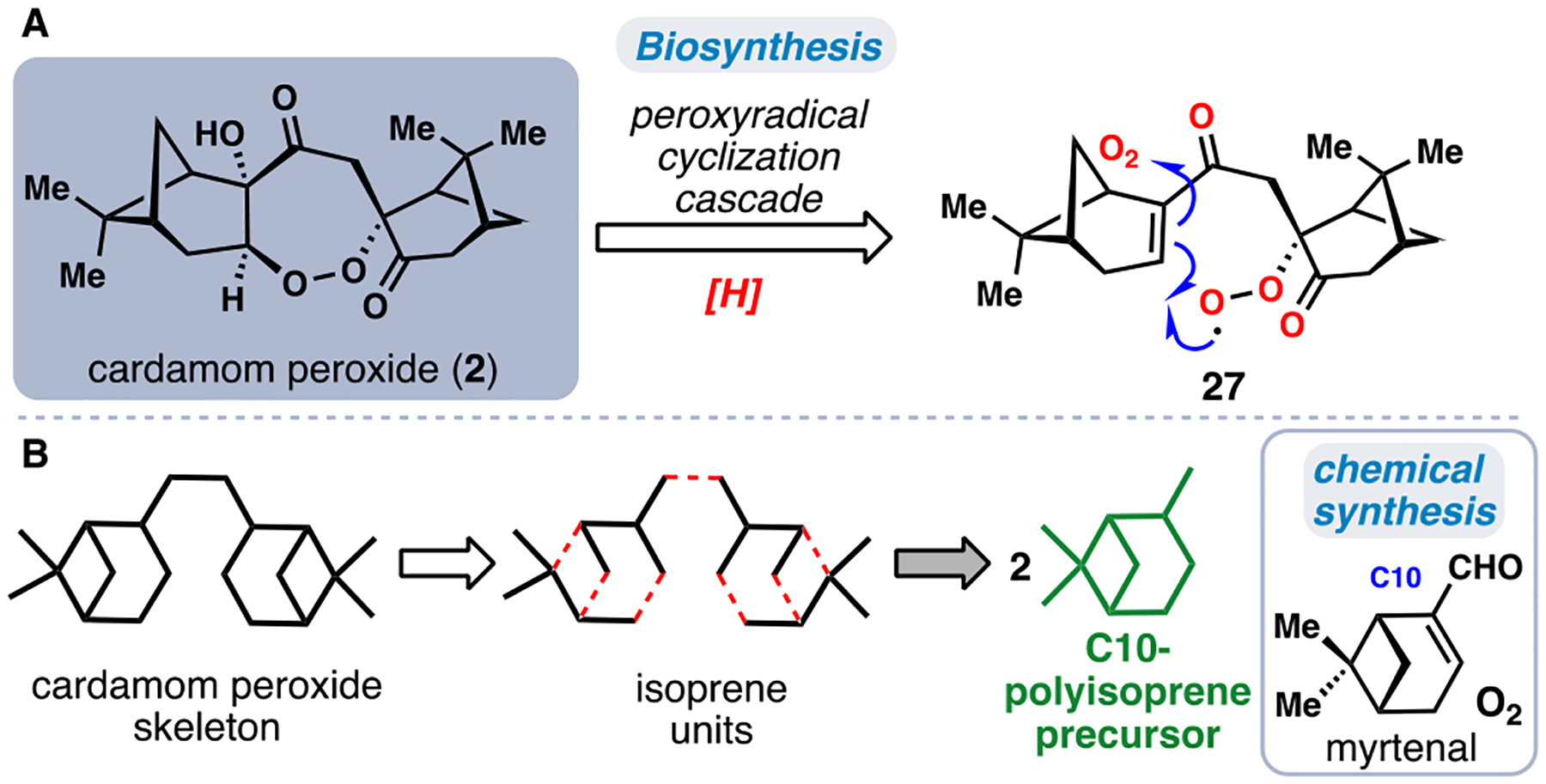
The antimalarial cardamom peroxide (2). (A) Proposed biosynthetic endoperoxidation cascade. (B) Isoprenoid building block-based disconnection.
Although the carbon skeleton of the cardamom peroxide can be disconnected into its constituent isoprene fragments (Figure 3B), the two intact pinene-type bicyclo[3.1.1]heptane units found in 2 suggest that dimerization of an bicyclic monoterpene would be a prudent disconnection. Furthermore, this synthetic strategy might illuminate how nature constructs the core of this C20 terpenoid.
We initially evaluated several paths to a dimeric monoterpene intermediate with adequate handles for exploring the polyoxygenation cascade.5a Ultimately, a McMurry reductive coupling of (–)-myrtenal, an oxidation product of pinene, proved capable of generating C2-symmetric triene 28 on a gram scale (Scheme 2). Subjecting this material to singlet oxygen (3O2, methylene blue) resulted in a desymmetrizing [4+2] cycloaddition, which was followed by a one-pot Kornblum–DeLaMare fragmentation with DBU to give intermediate 29. Next, oxidation of 29 with DMP provided bis-enone 30. In subsequent efforts to streamline the synthesis, we found that 30 could be prepared in one-pot from compound 28 through a cobalt(II)/salen-catalyzed formal radical fragmentation of the initial [4+2] cycloadduct (see 31 for possible mechanism) followed by in-situ oxidation with DMP.20
Scheme 2.
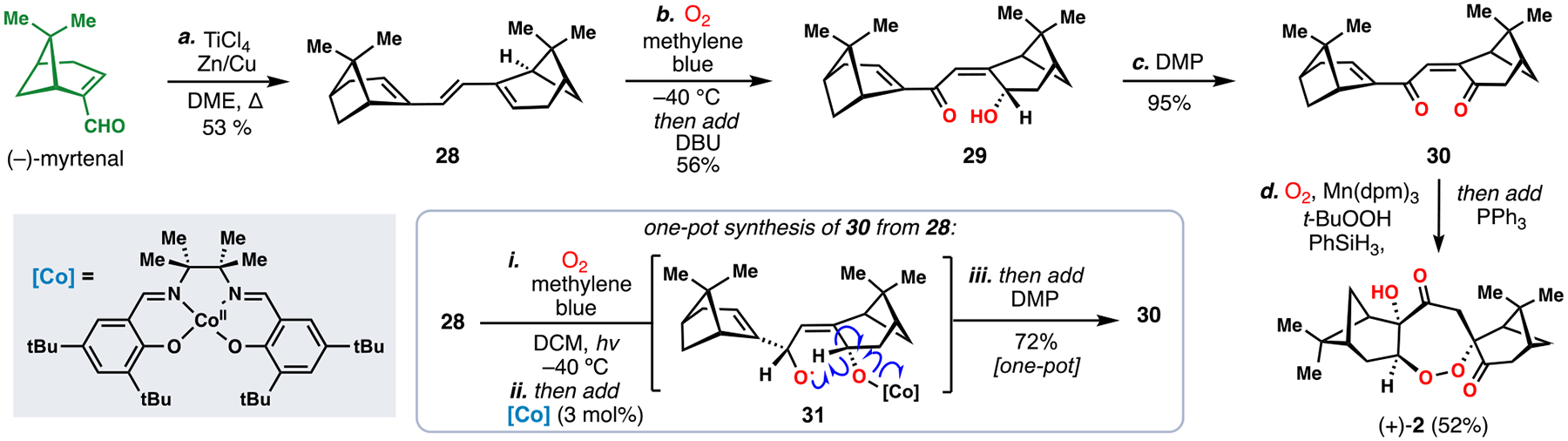
Total synthesis of the cardamom peroxide in 3 or 4 steps.
With 30 in hand, we sought conditions to generate peroxyradical 27–and ultimately 2–a task in which we faced several challenges in regio-, chemo-, and diastereocontrol. Owing to their mild conditions, the Co-catalyzed Mukaiyama radical hydroperoxidation,21 and subsequent Mn-catalyzed variant of Magnus,22 offered ideal solutions to this problem. Given the electron-deficient enone system, Mn-based catalysts were superior, and a protocol employing Mn(dpm)3 and tBuOOH as oxidant proved optimal for generating the cardamom peroxide (2) – a 52% isolated yield was obtained after in-situ reduction with triphenylphosphine. Notably, 2 was formed as a single isomer during this process with the two chiral pinene unit guiding all aspects of stereocontrol. Additionally, the quantity of material produced in this 3–4 step route, over half a gram of natural product, enabled further antimalarial evaluation.20
Our work on the synthesis of the cardamom peroxide via a polyoxygenation cascade provided insight into how unfunctionalized terpene fragments can be rapidly modified with molecular oxygen to yield complex natural products. Further applications of this concept are described in our synthetic work on guaianolide sesquiterpenes (Scheme 7).
Scheme 7.
Total synthesis of complex slovanolide-type guaianolides from carvone.
TOTAL SYNTHESIS OF OPHIOBOLIN SESTERTERPENES
The ophiobolin sesterterpenes, which possess a complex 5,8,5-fused ring system, are historically significant phytotoxins which have recently stimulated significant biological attention due to their potent and diverse anticancer activities and the presence of reactive functional groups.23 While cyclooctane-containing natural products have received much interest from the total synthesis community for decades,24 only the groups of Kishi,25 Nakada,26 and Maimone5c,,5i have realized full solutions to members of this growing terpene class.
The biosynthesis of the ophiobolin ring system is proposed to occur from geranylfarnesyl pyrophosphate through 5,11-fused macrocyclic carbocation 32 and a subsequent transannular cyclization (Figure 4A).27 While this process initially forges a cis-5,8-fused ring system, subsequent epimerization adjacent to the ketone group in the ophiobolins is facile; thus, nearly all members have isolated counterparts with epimeric C-6 stereochemistry (see 3 for example). Inspired by this impressive cascade, we sought to develop our own polycyclization process, which led to the realization of a short total synthesis of 3 in 2016.5c Reducing the ophiobolin skeleton to its constituent isoprene units and strategic fragment re-assembly revealed the possibility of utilizing an uninterrupted 15-carbon polyprenyl chain in our synthetic plan (Figure 4B). Thus, the ophiobolin skeleton could be disconnected to a [C5]3 building block and C6, C3, and C1 fragments. In the forward direction, (–)-linalool (of which only the 6 carbons shown in blue contribute to the target), trimethylsulfonium iodide (C1), trichloroacetyl chloride (C2), diiodomethane (C1), and farnesol (C15) were utilized to construct the carbon scaffold of this intricate C25 terpene.
Figure 4.
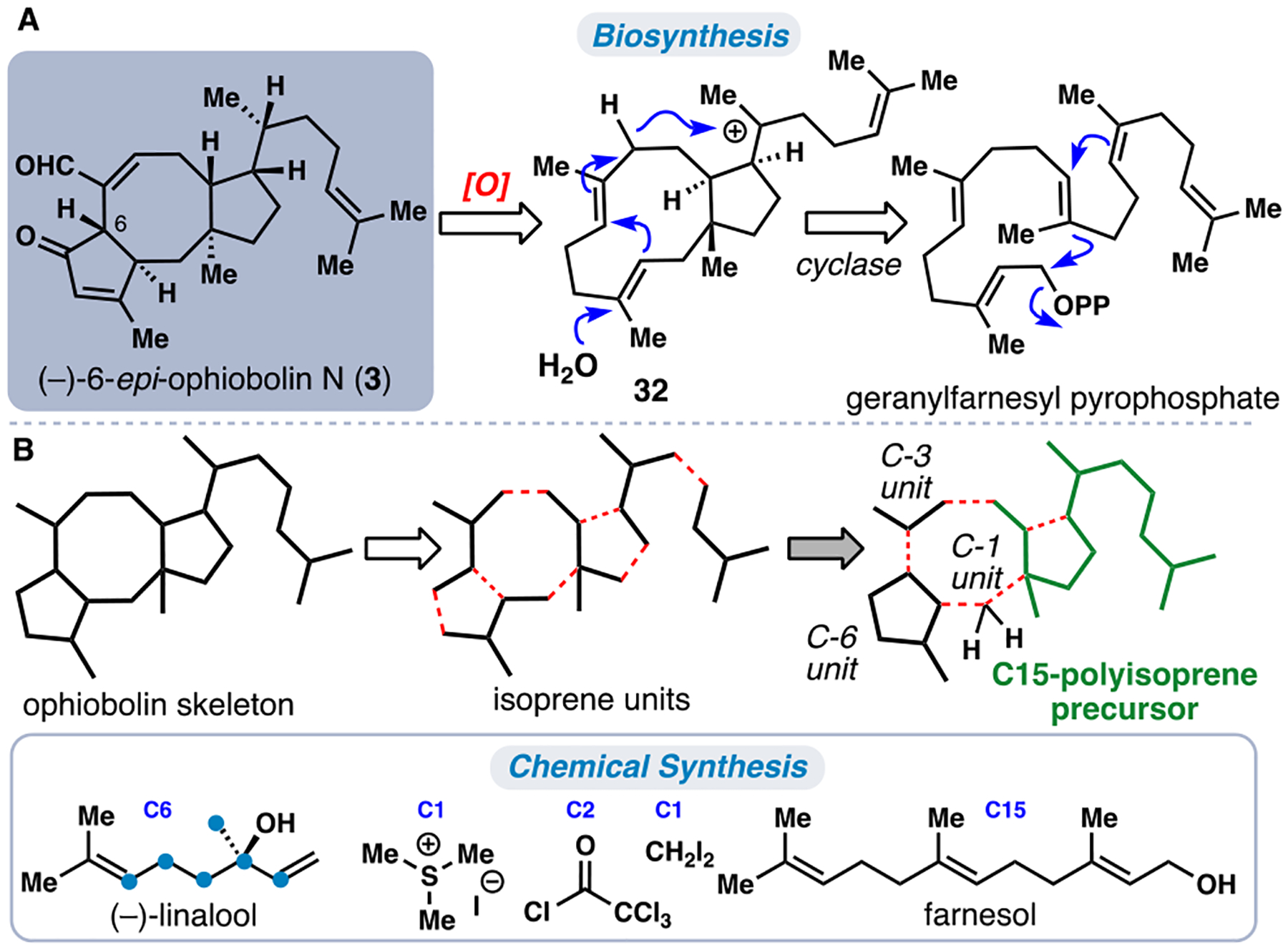
Representative ophiobolin sesterterpene (–)-6-epi-ophiobolin N (3). (A) Cyclase-mediated construction of the ophiobolin 5,8,5-fused ring system (B) Isoprenoid building block-based disconnection.
We began our synthetic work with chiral iodide 33, available via the asymmetric cyclopropanation of farnesol with diiodomethane and a subsequent Appel reaction according to the method of Charette (Scheme 3).28 Notably, this fragment represents a merge of the C15 and C1 units from our retrosynthetic plan (Figure 4B). Treating 33 with t-BuLi triggered lithium-halogen exchange and cyclopropane fragmentation generating an organolithium species that underwent transmetallation with CuI and subsequent 1,4-addition to cyclopentenone 34 (prepared in two-steps from (–)-linalool via ring closing metathesis, silylation, and allylic oxidation).5c The conjugate addition occurred with moderate facial selectivity (3:1 dr) favoring approach of the nucleophile opposite to the OTBS group, and the resulting enolate formed could be acylated with trichloroacetyl chloride. After cyclopentanone reduction (DIBAL/n-BuLi), 35 was isolated. In the key step of the synthesis, 35 was exposed to radical-generating conditions (Et3B/O2, (TMS)3SiH, thiol), which initiated a reductive 8-endo/5-exo cascade cyclization generating polycycle 37. The structure of the thiol catalyst employed affected the stereochemistry of the C15 stereocenter; using bulky TADDOL-based thiol 36,5c, 29 the correct configuration was favored (dr = 3.4:1).30 The final carbon of the target was appended via the Corey-Chaykovsky reaction, generating a spiro-epoxide intermediate that was exposed to the powerful reducing agent lithium naphthalenide inducing a dehalogenative epoxide opening. Presumably, this process generated intermediate trianion 38 leading to diol 39 after work-up. From 39, double Swern oxidation and subsequent treatment with TsOH to induce dehydration generated 3 in only 9 steps.
Scheme 3.
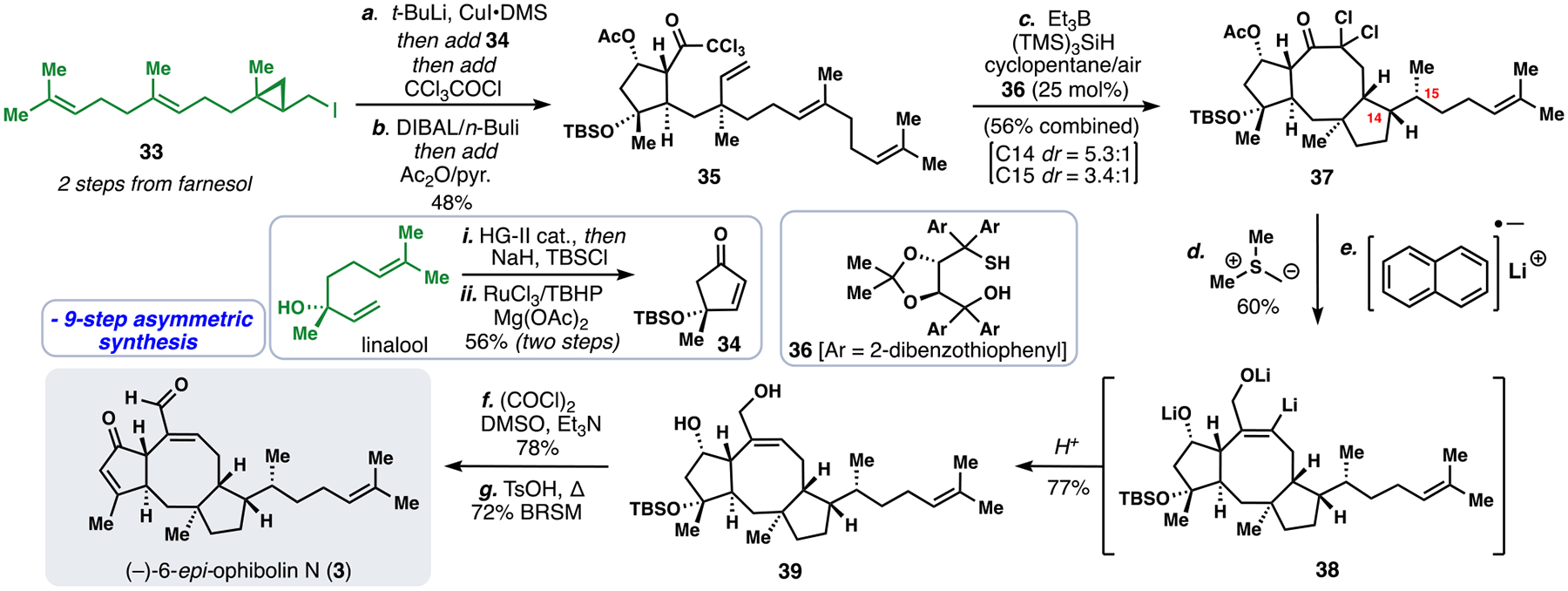
9-Step total synthesis of (–)-6-epi-ophiobolin N (3).
While the synthesis of 3 laid the groundwork for concise synthetic entry into the ophiobolins, several challenges remained to construct more complex and diverse members. First, the unfunctionalized nature of 3 presents hurdles when contemplating formation of the hallmark spirocyclic THF ring motif found in many ophiobolins such as 4. Second, as a result of the facial selectivity of the initial conjugate addition, our synthesis of 3 constructed the natural product in its unnatural, levorotary, enantiomeric form. Lastly, the final elimination of H2O in the synthesis of 3 was required to address the incorrect relative stereochemical relationship between the tertiary hydroxyl stereocenter and the rest of the ophiobolin scaffold. To address the first problem, we returned to our isoprene-based analysis of ophiobolins (Figure 5). Rather than utilizing a C15 farnesyl chain, we elected to employ the shorter C10 polyisoprene building block geraniol; it was envisioned that this would allow greater flexibility in installing the additional carbons and oxygenation needed for the spirocyclic THF ring. Thus, a revised blueprint for (+)-6-epi-ophiobolin A (4) was devised (Figure 5).
Figure 5.
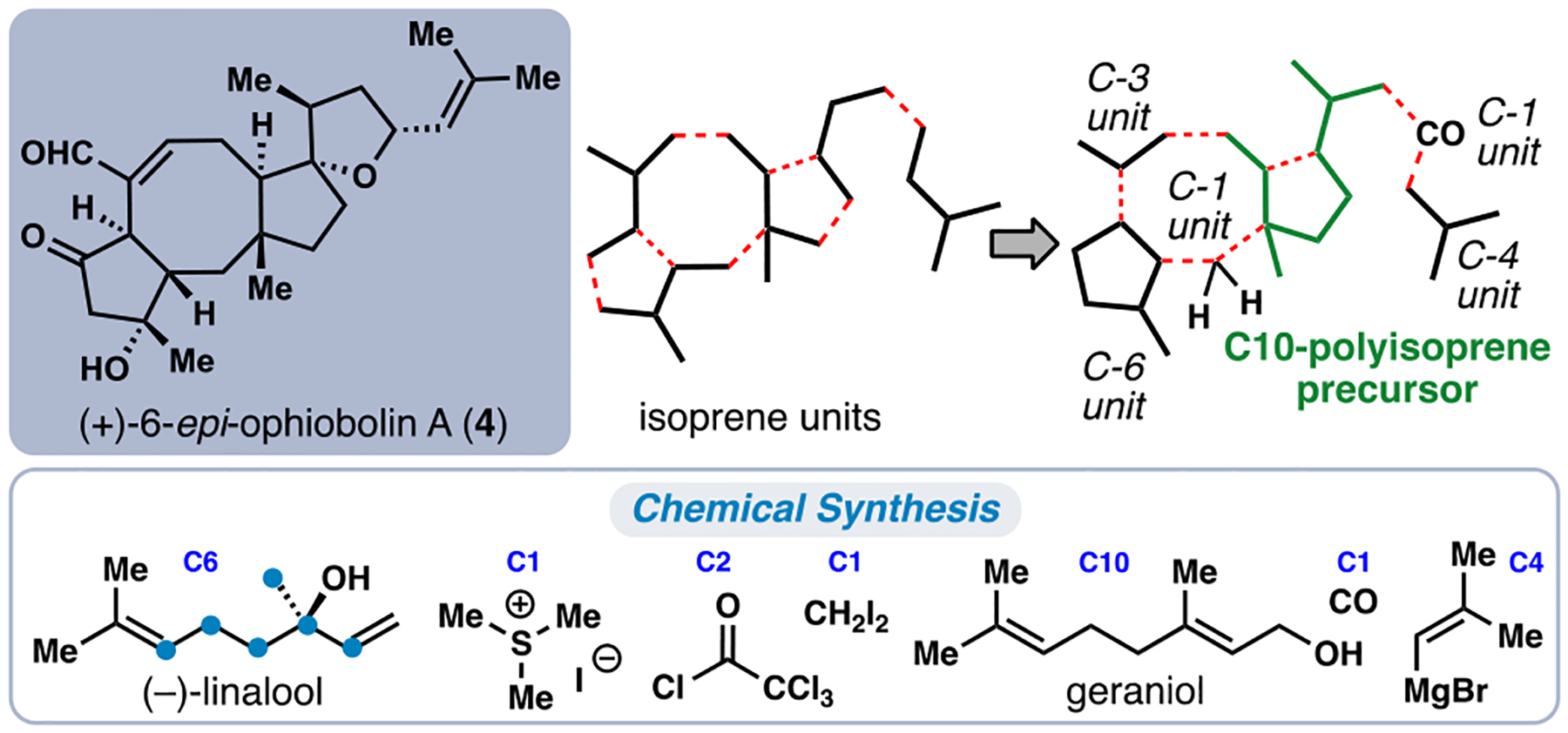
Revised isoprenoid building block-based disconnection for construction of complex ophiobolin members such as 4.
We began our synthesis of 4 with iodide 40 which was prepared from geraniol in an analogous fashion to 33 albeit with the opposite absolute configuration (Scheme 4).28, 5i Akin to 33, this material took part in an anionic ring opening/conjugate addition sequence with enone 34. In a divergence from past work, the resulting enolate was quenched with Mukaiyama’s dehydrogenation reagent (N-tert-butylbenzenesulfinimidoyl chloride) delivering enone 41.31 Conjugate reduction of this material with PhMe2SiCu–H (generated in-situ from DIBAL and PhMe2SiCu) and enolate trapping with trichloroacetyl chloride furnished 42, thus serving to formally “correct” the facial selectivity of the initial conjugate addition reaction.32 Subjecting 42 to Cu-mediated atom transfer radical cyclization conditions brought about an 8-endo/5-exo radical cascade, but unlike the conversion of 35 to 37 a non-reductive pathway ensued, and thus the final product (see 43) possessed a key alkene handle poised for further functionalization. Following oxidation state manipulations, compound 44 was generated and reacted productively with singlet oxygen in a diastereoselective ene reaction (3:1 dr) producing tertiary alcohol 45 in 42% isolated yield.
Scheme 4.
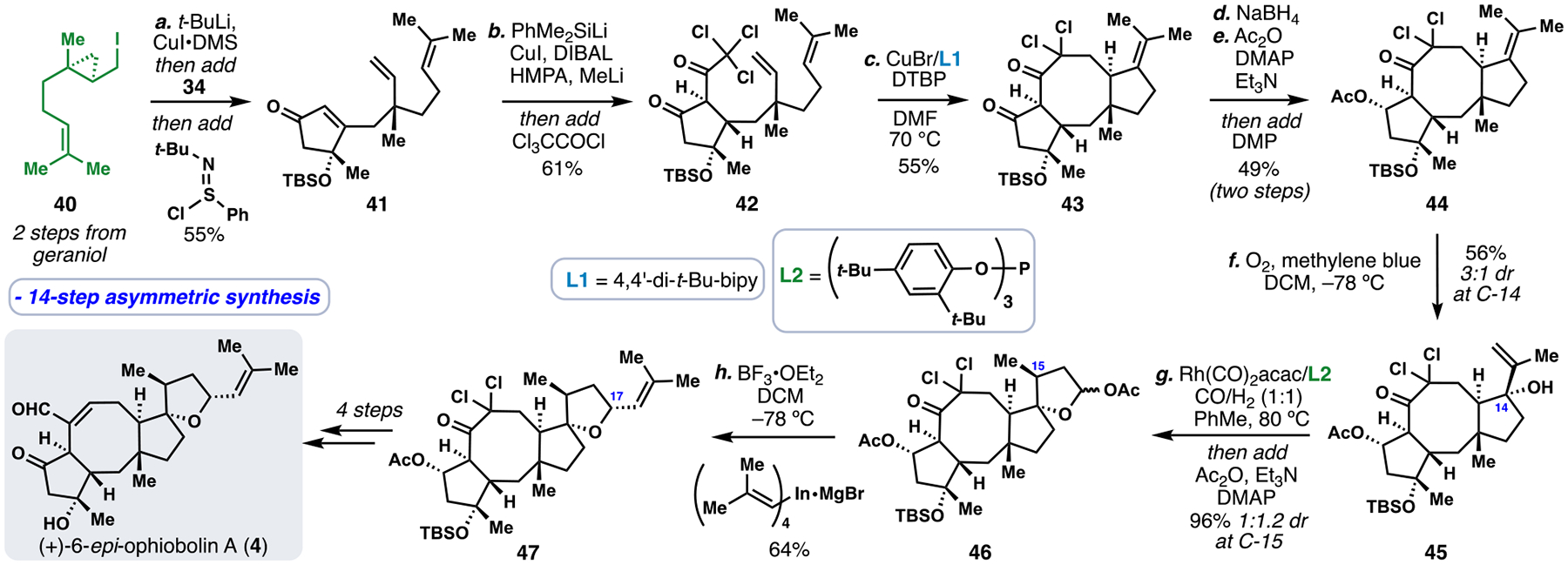
Total synthesis of (+)-6-epi-ophiobolin A (4).
To complete assembly the THF ring, 45 was first subjected to rhodium-catalyzed hydroformylation conditions,33 producing cyclic lactol acetate 46 after in-situ acetalization (Scheme 4). While the yield of this transformation was nearly quantitative, a lack of diastereoselectivity at the newly-formed methyl stereocenter led to diminished overall efficiency (44% yield of isomer 46). In a second key step, we successfully utilized the tetraorganoindate nucleophile shown in a diastereoselective alkenylation of an in-situ generated oxocarbenium ion thus delivering 47 with excellent diastereocontrol. Finally, a four-step sequence consisting of Corey-Chaykovsky epoxidation, lithium naphthalenide-mediated reductive dehalogenative epoxide opening, fluoride-mediated desilylation, and Swern oxidation delivered (+)-6-epi-ophiobolin in 14-steps from geraniol.
The total synthesis of ophiobolins 3 and 4 showcase how strategic, polyprenyl-based cyclizations can be used to generate synthetically challenging polycycles in an efficient manner rivaling the intricacies of Nature’s cyclase-mediated solutions. Moreover, these works once again give testament to the power of radical cascades in complex terpene synthesis.34
TOTAL SYNTHESIS OF GUAIANOLIDE SESQUITERPENES
The plant families Asteraceae and Apiaceae are major producers of bioactive sesquiterpene metabolites including the large family of 5,7-fused guaianolide lactones.35 Many of these natural products possess cytotoxic properties and have been popular synthetic targets for decades (Figure 6).35,5h Although full biosynthetic roadmaps to guaianolide members from Asteraceae and Apiaceae are incomplete, a germacranolide-type macrocycle is generally accepted as a precursor to this family of natural products (Figure 6A).36 Nevertheless, the enzymatic machinery within Asteraceae and Apiaceae differ, and each family forms products with distinct stereochemical and oxidation patterns (see 10 and 11 for example). These structural nuances pose a challenge for a unifying synthetic strategy toward guaianolides and can confound chiral-pool-based strategies wherein starting materials may be available in only one enantiomeric series.8
Figure 6.
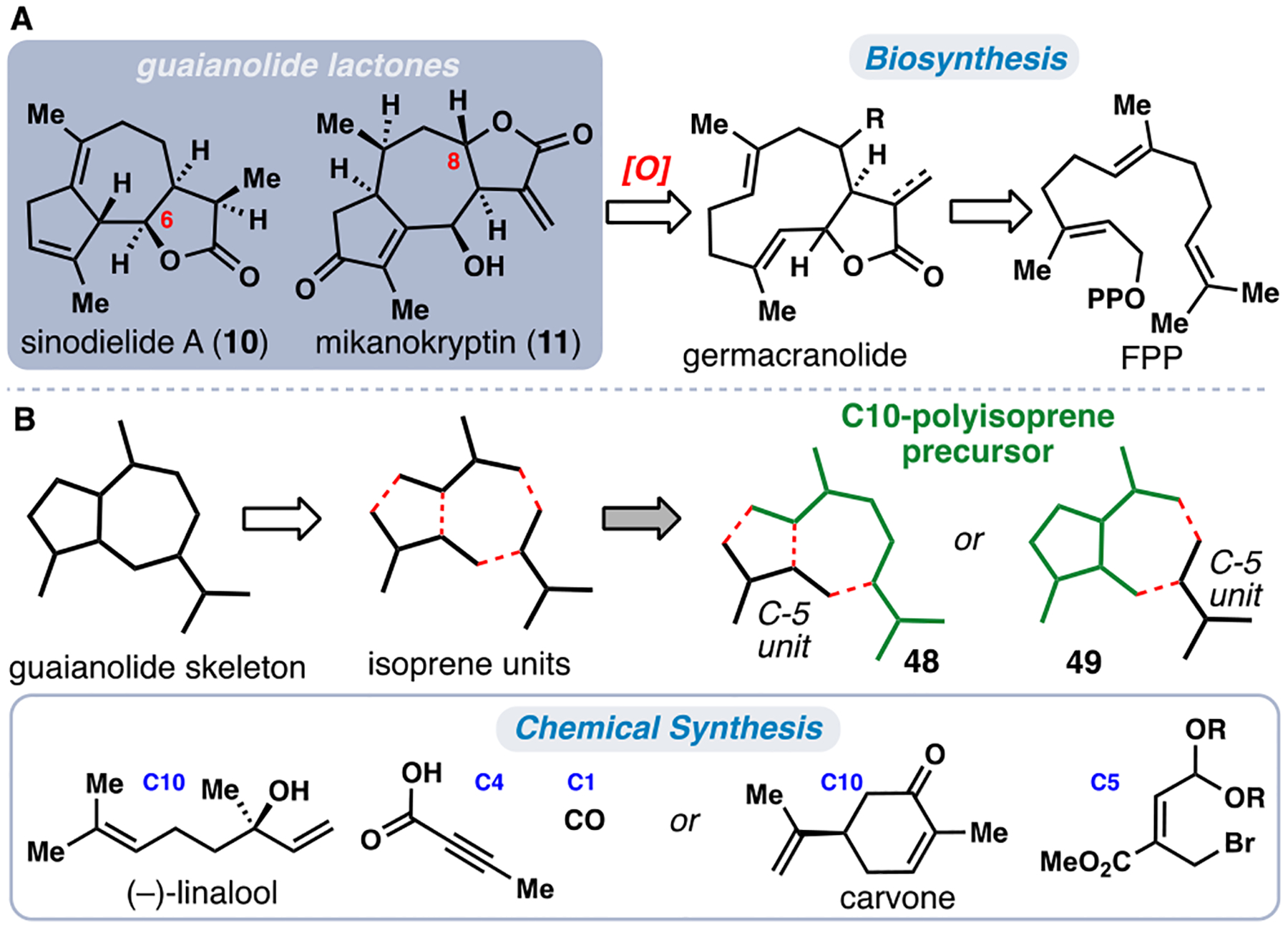
Representative guaianolide sesquiterpenes. A) Biosynthesis occurs from a germacranolide precursor. B) Two distinct isoprene-based disconnections.
We identified two reasonable [C5]n-based disconnections (Figure 6B) during our isoprene-based analysis of the guaianolide core. In the first approach, a linear, 10-carbon monoterpene was selected (see 48 in green) and merged with a C5 fragment. The second disconnection, which has been employed previously, uses a ten-carbon cyclopentane building block (see 49 in green), which is typically prepared by ring contraction of a cyclic monoterpene (Figure 6B).5h,8 The availability of many cyclic monoterpenes in either enantiomeric form lent greater strength to the latter strategy for its capability to access a wider array of guaianolides. In each case, unification of a C10 chiral-pool building block with a C5 or C4 + C1 fragment was key to the overall strategy. In the forward sense, (–)-linalool and carvone were utilized as the [C5]2 components (Figure 6B).
Employing the first strategy, we initially designed a route to the simple, Apiaceae-derived member sinodielide A (10), which leverages the chiral tertiary alcohol of (–)-linalool to direct formation of the guaianolide five-membered ring (Scheme 5).5h To this end, (–)-linalool was transformed into ester 50 (NaHMDS, but-2-ynoic pivalic anhydride) which underwent a moderately diastereoselective intramolecular Pauson-Khand cyclization to provide bicyclic lactone 51. DIBAL reduction and protection (TESOTf, collidine) of the major diastereomer followed by chlorination of the prenyl group with sulfuryl chloride yielded 52. The protected primary allylic alcohol was selectively oxidized to the corresponding aldehyde (CrO3/pyridine) to provide a key intermediate for the 7-membered ring-forming reaction. Gratifyingly, this material underwent NHK-type coupling (NiCl/CrCl3) to provide the guaianolide skeleton-containing product 53 with 9:1 dr and establishing cis stereochemistry at C-6 and C-7. A hydroboration-oxidation process gave lactone 54 and deprotonation followed by a kinetic proton quench gave isomerically pure lactone 55. Finally, conversion to 10 was achieved via a one-pot reductive allylic transposition and subsequent dehydration of the remaining tertiary alcohol.
Scheme 5.
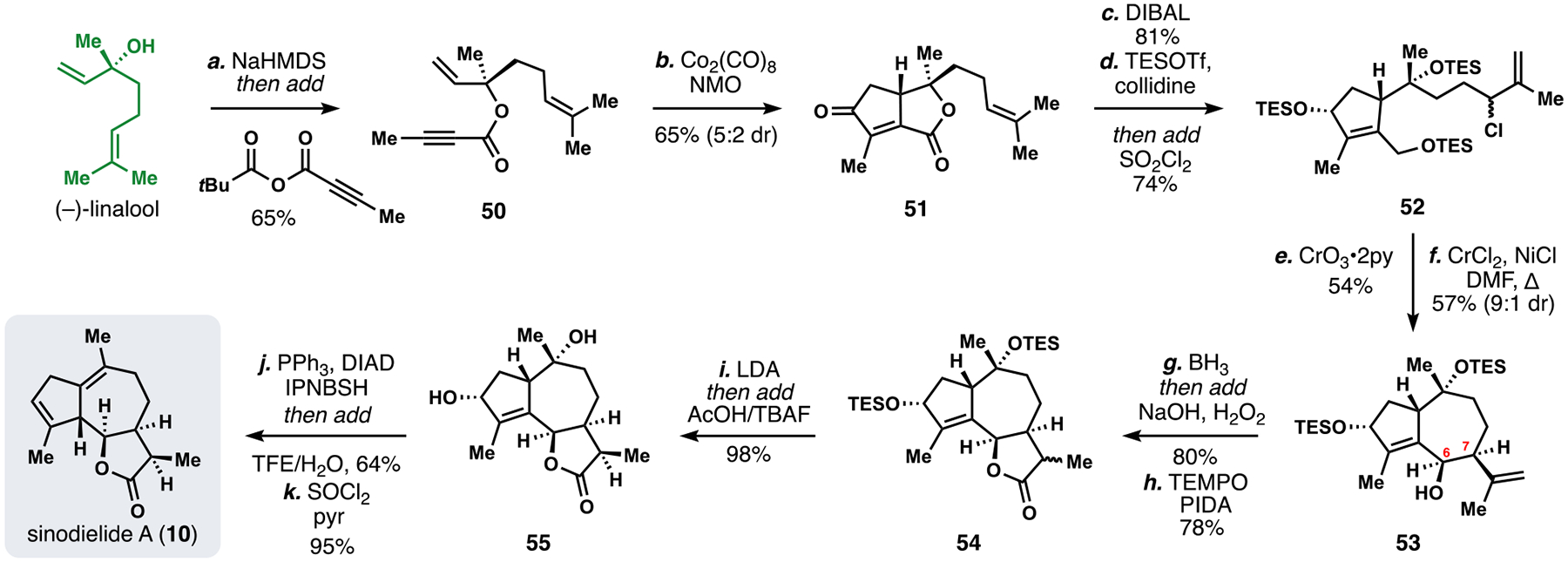
Total synthesis of sinodielide A (10) from (–)-linalool.
While this route was successful in generating a low oxidation state Apiaceae member, several hurdles remained to access more hydroxylated members (vide supra). Our investigations in this direction first lead us synthesize the stereochemically irregular Asteraceae family member mikanokryptin (Scheme 6).5e
Scheme 6.
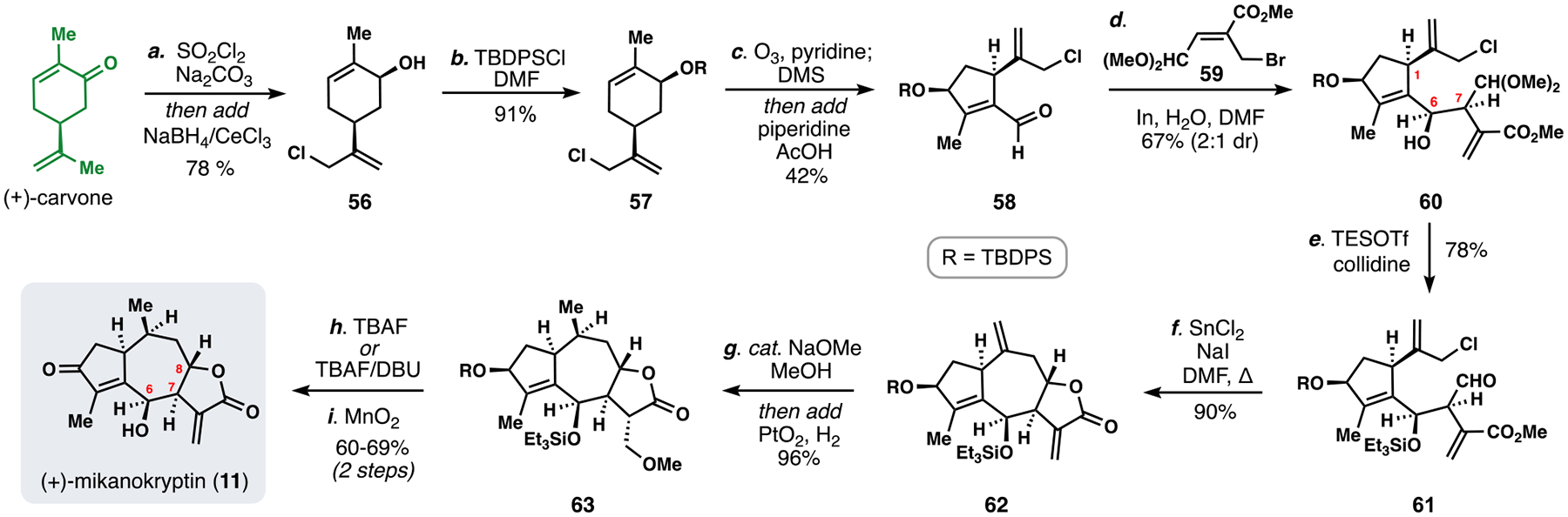
Total synthesis of mikanokryptin (11) from carvone.
Like 10, mikanokryptin (11) has a cis arrangement between substituents at C-6 and C-7, However the presence of C-8 oxygenation and a reactive exocyclic methylene lactone differentiates 11, and the latter functionality is presumably responsible for much of the reported biological activity of Asteraceae.35 We devised a double allylation strategy to accomplish our isoprene-based disconnection of guaianolides from a cyclic 10-carbon terpene and a 5-carbon piece (Figure 6B).
We developed a scalable, 3-step pathway to aldehyde 58, the requisite 10-carbon fragment, from (+)-carvone (Scheme 6).5e Allylic chlorination and Luche reduction gave 56, followed by protection with TBDPSCl, and a final ozonolysis/aldol sequence delivered 58. To achieve the first allylation, aldehyde 58 and allylic bromide 59 were coupled in the presence of indium to deliver 60 on a multigram scale.
Mild deacetalization with concomitant silylation generated 61 setting up the second key C–C bond-forming event – once more we turned to a Barbier-type coupling protocol. In-situ conversion of the allylic chloride to an allylic iodide allowed for a mild and high-yielding SnCl2-mediated cyclization, which generated lactone 62. A one-pot conjugate addition of methoxide served to protect the exo-methylene lactone prior to hydrogenation (PtO2, H2) of the other exocyclic alkene, ultimately delivering 63. Fluoride-induced deprotection with TBAF also elicited E1cB elimination of methanol restoring the reactive exocyclic methylene moiety. Finally, allylic oxidation with MnO2 generated 11 in only 9 steps overall. This pathway allowed for gram quantities of the target to be prepared and enabled further biological interrogation of the molecular targets of 11.37
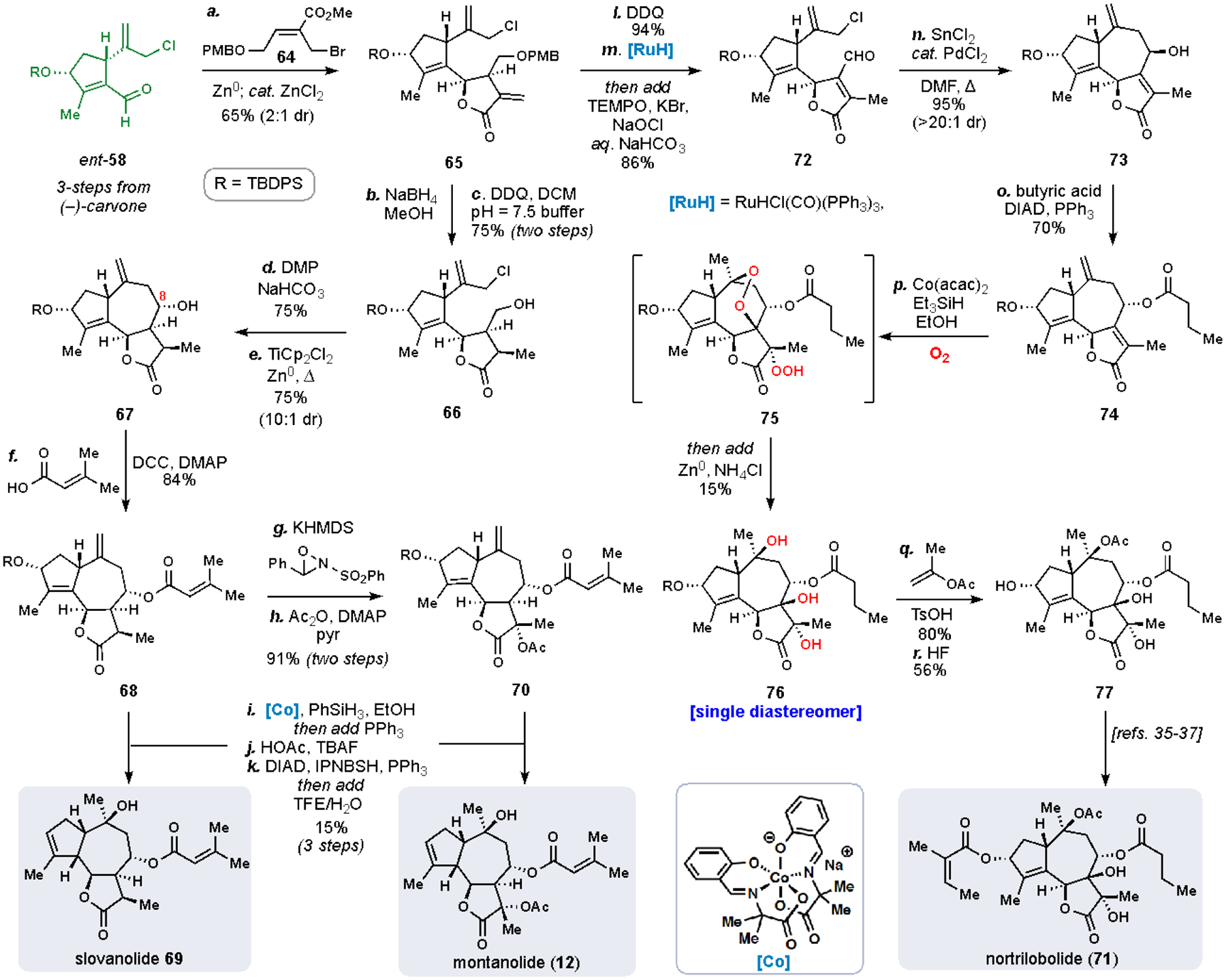
Finally, we sought to modify our second-generation route to access other Apiaceae family members with slovanolide-type stereochemical configurations such as 69, montanolide (12), and the heavily oxidized nortrilobolide (71) (Scheme 7).35b To achieve these goal, two modifications to our synthetic strategy were necessary. First, the 10-carbon aldehyde fragment was needed in the opposite enantiomeric series (see ent-58), and second, the initial allylation with the 5-carbon allylic bromide would need to proceed with alternative diastereoselectivity (Scheme 7). Whereas the In(III)-mediated allylation to yield 60 gave a configuration with all hydrogens being β-disposed, we found that slovanolide stereochemistry required Zn(0)-mediated allylation conditions to prepare 65 with the desired stereochemistry at C-6 from ent-58 and allylic bromide 64. Next, the exo-methylene lactone of 65 was reduced with NaBH4, and PMB deprotection (DDQ) revealed primary alcohol 66. After oxidation of 66 to the corresponding aldehyde, a reductive titanocene-mediated intramolecular allylation gave 67 with the correct C-8 configuration and once again, leveraged our previously adopted Barbier-type strategy. The C-8 alcohol was subsequently esterified with 3,3-dimethylacrylic acid to generate the requisite side chain found in 69 and 12. Cobalt-catalyzed Mukaiyama hydration of the exocyclic olefin, followed by deprotection, and finally reductive allylic transposition gave slovenolide 69. A modified sequence from common tricyclic intermediate 68 also provided the more highly oxidized montanolide (12). Oxidation of the lactone with Davis’ oxaziridine and subsequent acetylation yielded 70, which underwent a similar end-game procedure– hydration, deprotection, and allylic transposition– to provide 12.
With the realization of syntheses of mikanokryptin (11), slovanolide 69, and montanolide (12), we looked toward the most heavily oxidized Apiaceae subtypes, typified by nortribolide (71). In studying the relative configuration of the various hydroxyl stereocenters in 71, we recognized that a polyoxygenation cascade using molecular oxygen might be capable of constructing this motif, similarly to our work with the cardamom peroxide (Scheme 2). We began by adapting our prior synthesis to target butenolide 74, a key precursor to the proposed oxygenation cascade (Scheme 7). Starting from exo-methylene lactone 65, PMB removal was followed by a one-pot isomerization of the exocyclic olefin with catalytic quantities of RuHCl(CO)(PPh3) and oxidation of the primary alcohol to give aldehyde 72. A Pd-catalyzed, SnCl2-mediated allylation then closed the seven membered ring and generated tricycle 73 (d.r. 20:1). Inversion of the secondary alcohol was achieved under Mitsunobu conditions with butyric acid to give 74, which was poised to undergo the tandem hydroperoxidation cascade. After extensive investigation, we found that a Co(acac)2-based system provided desired product 76 as a single diastereomer after reductive workup with Zn(0) presumably via peroxide 75. Finally, acetylation and desilylation provided 77, an intermediate previously utilized to prepare 71,38–40 thus completing a 12-step formal synthesis to 71.
Our work on guaianolides highlights how multiple polyprenyl-based analyses can be leveraged in the development of synthetic solutions to a variety of related, but sterochemically distinct, terpene family members. Moreover, we note that even a single chiral pool building block in concert with a variety of parameter adjustments to a given reaction manifold (in this case allylation) can be used to generate an assortment of different stereochemical motifs as typified by syntheses of 11, 12, 69, 71 all from building block 58 or its enantiomer.
TOTAL SYNTHESIS OF ILLICIUM SESQUITERPENES
Phytochemicals from members of the Illlicum genus have centuries of documented use as both flavors and medicines.41 A wide range of highly oxygenated sesquiterpenes with a myriad of biological properties, ranging from small molecule neurotrophins to potent neurotoxins, are commonly found in these plants. An important mediator of many of these effects is the sesquiterpene lactone family (>50 members), which contains a 5,6-fused seco-prezizaane ring system (Figure 7). Such compounds have proven popular as synthetic targets over the past several decades.42 Substantial isolation efforts by Fukuyama has led to the biosynthetic hypothesis shown in Figure 7, wherein the cedrane carbocation (see 78), a well-documented biosynthetic intermediate, serves as a late-stage precursor to the seco-prezizaane scaffold via an alkyl shift and C–C bond scission process.41 This informed our choice to consider the [C5]3 building block (+)-cedrol as a starting material in devising a route to the illicium sesquiterpenes (Figure 7). Moreover, its low cost, high abundance, and correct absolute stereochemistry further add to the attractiveness of this route. While (+)-cedrol is potentially well suited for conversion to the seco-prezizaane ring system, the substantial number of C–H oxidations needed to convert this unfunctionalized feedstock chemical to pseudoanisatin (14) and majucin (15) posed formidable challenges.
Figure 7.
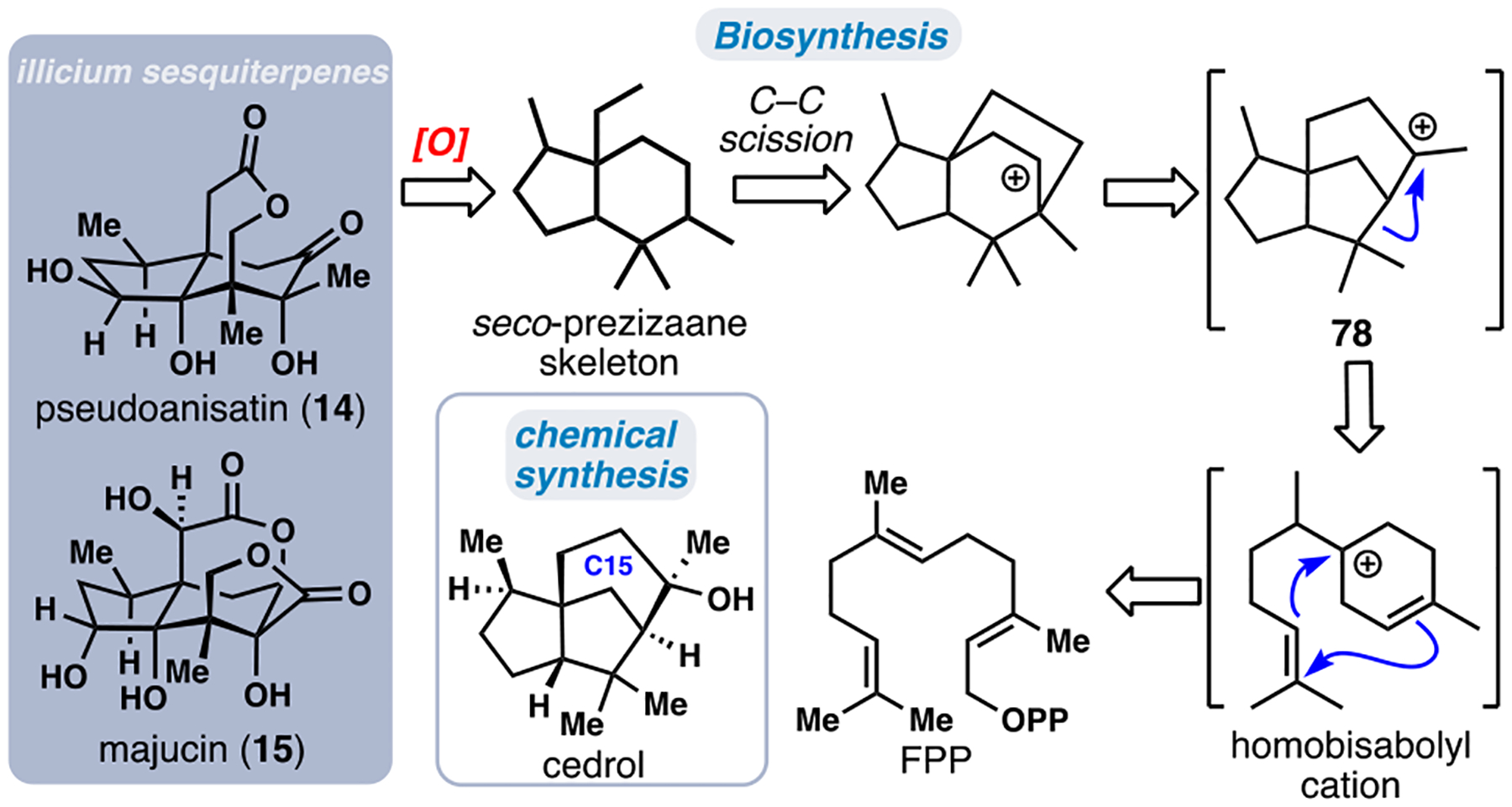
Representative illicium sesquiterpenes and Fukuyama’s proposed biosynthesis proceeding through the cedrane cation.
We initially targeted pseudoanisatin (14) owing to its moderately less-oxidized seco-prezizaane core (Scheme 8).5d Using Suárez’s protocol (I2/PhI(OAc)2, floodlamp),43 we oxidized an unactivated methyl group of cedrol. This was followed by opening of the strained tetrahydrofuran ring (79) to form 80 via methylation–elimination with Meerwein’s salt (Me3OBF4) and proton sponge. The alkene in methoxycedrene (80) was then oxidatively cleaved with in-situ generated RuO4 giving keto-acid 81, which underwent direct intramolecular acyloxylation when heated to 150 °C with CuBr2 and t-BuOH to give lactone 82. Notably, many known conditions for ketone α-oxidation failed to produce this compound.
Scheme 8.
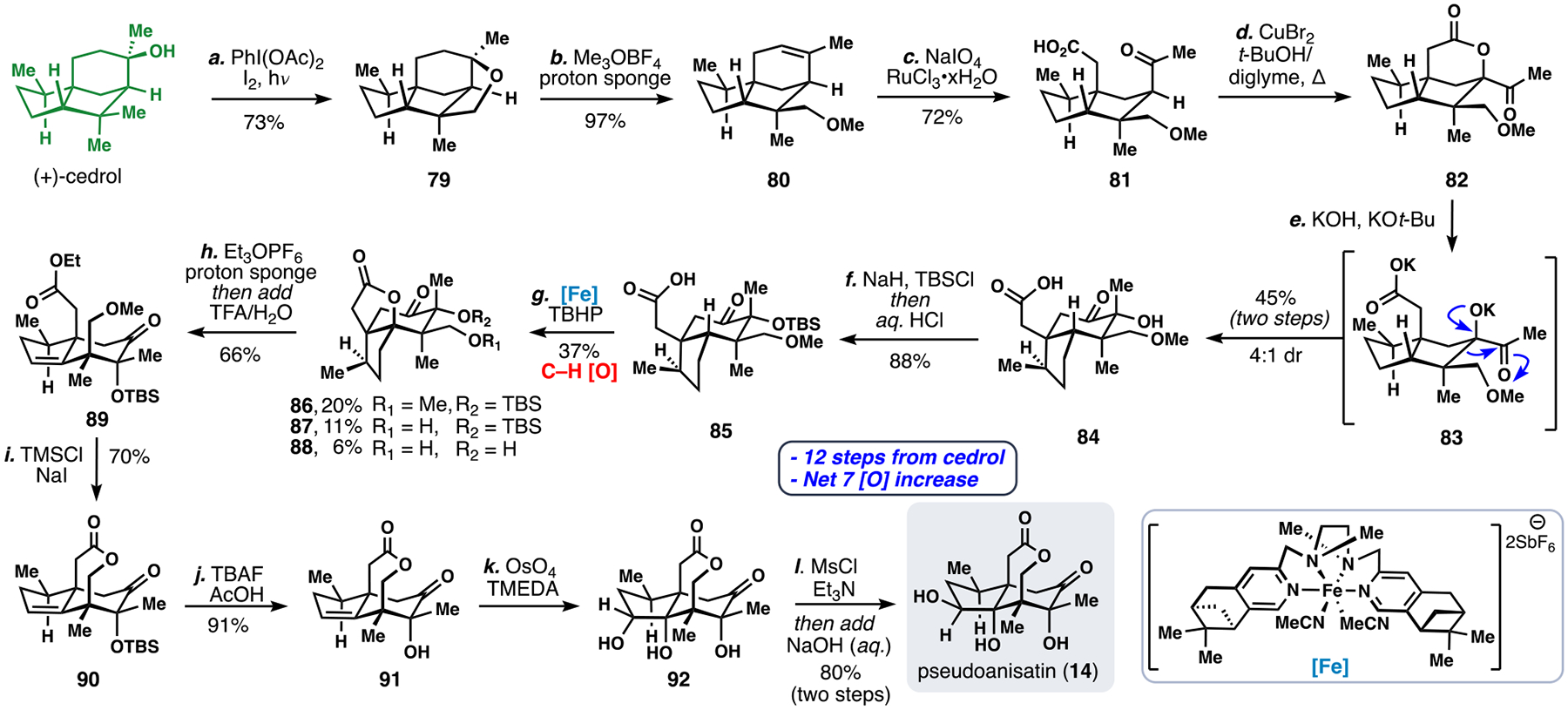
Semisynthesis of pseudoanisatin from (+)-cedrol using an oxidative approach.
With access to lactone 82, we investigated methods to convert the 5,5-fused cedrane ring system into the 5,6-fused seco-prezizaane skeleton, a key biomimetic disconnection. However, rather than adopt Nature’s biomimetic Wagner-Meerwein rearrangement, we turned toward an abiotic, anionic α-ketol rearrangement to accomplish a similar task.44 After extensive experimentation we discovered that treating 82 with t-BuOK/KOH in DMSO promoted both lactone hydrolysis (generating 83) and subsequent bond migration to give 84 (45%, 4:1 dr), thus forging the seco-prezizaane core in only 5 steps from cedrol. Next, the tertiary alcohol of 84 was protected (NaH, TBSCl) to give 85, which was suited to iron-catalyzed, acid-directed C-H oxidation of the unactivated tertiary methine center.45 Subjecting acid 85 to the iron complex shown and TBHP afforded products oxidized products 86-88 in 37% combined yield.46 Despite significant attempts to optimize the formation of 86, extensive oxidative degradation led to relatively low mass balances in this challenging transformation. Nevertheless, the major product (lactone 86) was carried on to di-ester 89, via lactone ethylation and elimination (Et3OPF6, proton sponge). Next, in-situ formed TMSI (TMSCl, NaI) cleanly dealkylated the methyl ether yielding compound 90 which possesses the hallmark ε-lactone of pseudoanisatin. Desilylation with TBAF then gave free alcohol 91, which was dihydroxylated according to Donahoe’s hydroxyl-directed protocol to generate triol 92.47 Finally, treatment with MsCl followed by NaOH gave pseudoanisatin (14). Overall, seven net C–H oxidations were accomplished in converting (+)-cedrol to 14.
In approaching the synthesis of more complex illiciums such as majucin (15), additional C–H activations are necessary as many illicium members contain an additional fused γ-lactone ring not present in 14 (Scheme 9). Moreover, the previous low-yielding, Fe-mediated C–H oxidation step prompted us to develop alternative solutions to the functionalization of inert C–H bonds. We again began our efforts by employing a Suárez-type oxidation of the unactivated methyl group; however, this time we elected to open the strained THF ring via acetylation to generate acetoxycedrene (93). Diverging from past work, 93 was converted to alcohol 94 via a hydroboration/oxidation and a subsequent diastereoselective reduction. Inspired by work from Waegell, we utilized this newly-formed hydroxyl group to effect Suárez oxidation of the neighboring methine C-H bond to construct ether 95.48 In contrast to the previous iron conditions, this C-H oxidation was exceptionally high yielding (93%). Treatment of 95 with in-situ generated RuO4 (RuCl3•xH2O, KBrO3) accomplished a remarkable triple oxidation reaction, cleaving a C–C bond and delivering 96.48 Next, we confronted the majucin-type γ-lactone (compare 14 to 15), which required exhaustive oxidation of a methyl group. Gratifyingly, this could be achieved under anhydrous Riley oxidation conditions (SeO2, 4Å MS, Δ) to elicit quadruple oxidation of all of the C–H bonds alpha to the ketone group in 96. The crude carboxylic acid intermediate was methylated (K2CO3, Me2SO4) to give ester 97. L-Selectride promoted ketone 1,2-reduction of 97 as a diastereomeric mixture of allylic alcohols, which when treated with basic methanol, converged to enol 98 via an ester hydrolysis/translactonization/alkene isomerization cascade process.
Scheme 9.
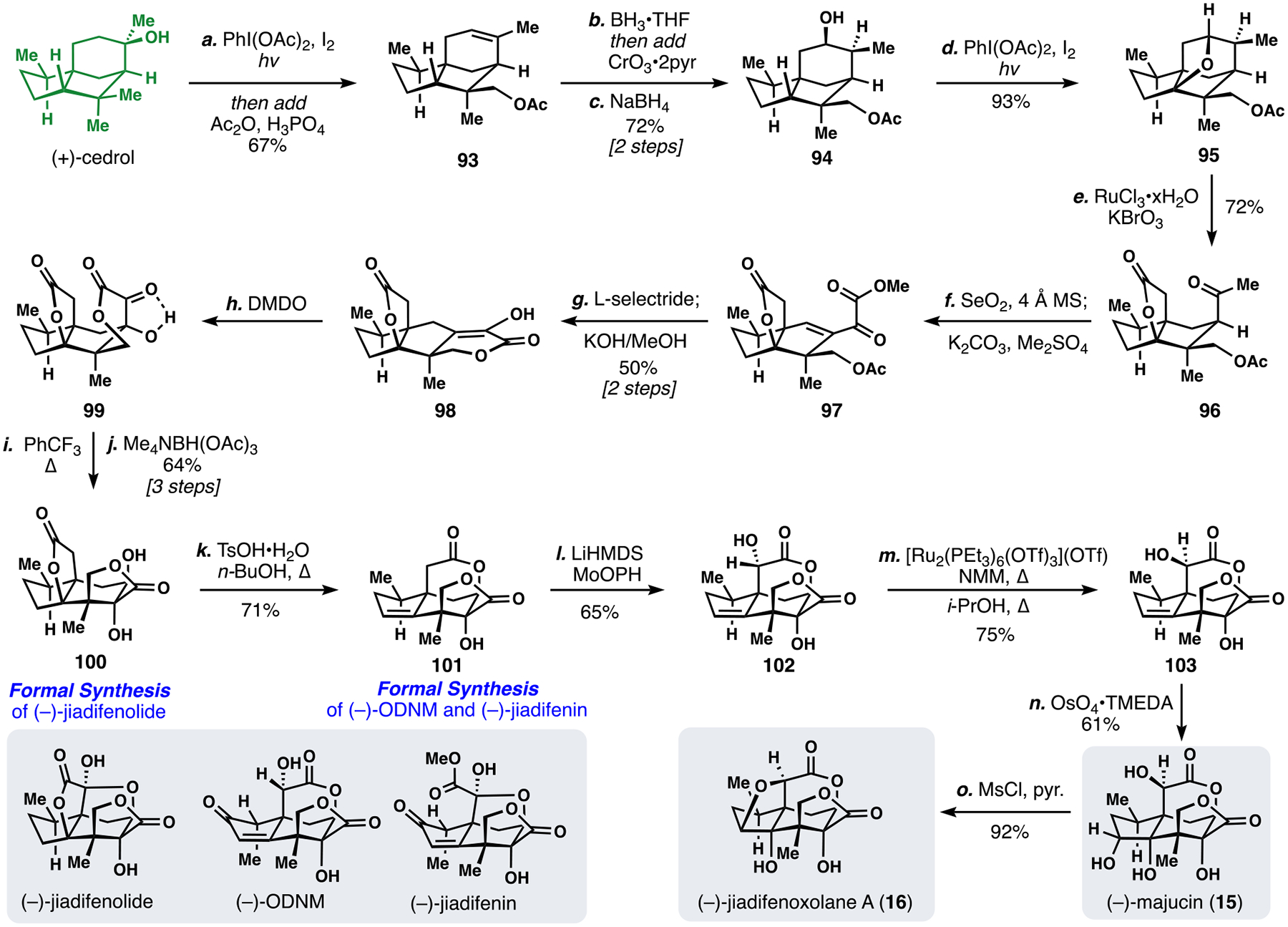
Second-generation synthesis of multiple illicium sesquiterpenes.
To rearrange the 5,5-fused ring system found in tetracycle 98 into the seco-prezizaane core we turned toward our previous α-ketol rearrangement strategy (see 83) with modifications to accommodate the additional ring. We first oxidized enol lactone 98 to a single, unstable, α-hydroxyketone diastereomer (99) using DMDO. While this sensitive intermediate was unstable to acidic and basic conditions, heating to 170 °C elicited bond reorganization to the majucin core. A directed reduction (Me4NBH(OAc)3) of the α-ketol group then furnished known tetracyclic diol 100. Notably the synthesis of 100 represents a formal synthesis to (–)-jiadifenolide.42 Next, we converted the γ-lactone ring into a δ-lactone system by treatment with acid (TsOH/n-BuOH, Δ), which formed trisubstituted alkene intermediate 101; notably this also achieved the formal syntheses of (–)-ODNM and (–)-jiadifenin.42 The enolate of 101 was oxidized with MoOPH to prepare hydroxy lactone 102, which could be formally epimerized under Hartwig’s conditions using the depicted ruthenium complex.49 Finally, 103 was converted into majucin (15) via directed dihydroxylation with OsO4•TMEDA. In addition, jiadifenoxolane A (16) could be generated from this material via an intramolecular displacement. Overall, our work on the illicium sesquiterpenes highlights both the power and challenges of strategies predicated on oxidizing C–H bonds and establishes (+)-cedrol as a versatile synthetic platform for sesquiterpene construction.
CONCLUSION
In this account we provide insight into our design and thought process toward planning the synthesis of a variety of disparate complex terpene structures. Most of these projects did not commence with a clear retrosynthetic outline or a desire to apply a certain reaction type or methodology. Rather they were initiated by pattern-based mapping of commercially available [C5]n building blocks in concert with plausible biosynthetic speculation.50,51 Nevertheless, the resulting syntheses proved concise and required extensive problem solving along the way. This allowed for the exploration of interesting chemical reactivity that would be awkward to study in more purely methodological settings. We believe these features capture the essence of total synthesis and why it remains an exciting and satisfying scientific endeavor.52,53
Acknowledgements
We are grateful to all Maimone laboratory members past and present for their contributions to these and other projects. We are appreciative to the NIH for funding our terpene synthetic program (R01GM116952). T. J. M. is an Arthur C. Cope Scholar and acknowledges unrestricted grant support from Bristol-Myers Squibb, Novartis, Amgen, and Eli Lilly. D. Q. T. and C. H. M. thank the NSF for providing graduate research fellowships (DGE-1106400).
Biographies
Tom Maimone received a B.S. degree in chemistry from UC-Berkeley in 2004 wherein he was introduced to chemical research in the laboratory of Prof. Dirk Trauner. From 2005–2009 he was a member of Prof. Phil Baran’s research group at Scripps, and from 2009–2012, an NIH postdoctoral fellow in Prof. Steve Buchwald’s lab at MIT. In 2012, he began his independent career at UC-Berkeley where he is currently an associate professor engaged in the study of natural products chemistry.
Claire S. Harmange Magnani received an A.B./A.M. degree in Chemistry and Physics at Harvard University in 2015 performing research under the supervision of Prof. Andrew G. Myers. She is a current graduate student in the Maimone group focusing on natural product synthesis.
Danny Thach received his B.S. in chemistry from The University of Texas at Austin in 2016, wherein he conducted research in the lab of Prof. Guangbin Dong. In 2016, he began his graduate studies in the Maimone lab working on the total synthesis of ophiobolin sesterterpenes.
Karl Haelsig received his B.S. degree in chemistry in 2014 from the University of Washington, conducting catalysis research under the guidance of Prof. Gojko Lalic. He is currently a graduate student in the Maimone lab, conducting research on the synthesis of pyrrole-containing alkaloids.
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1).(a) Zeng T; Liu Z; Liu H; He W; Tang X; Xie L; Wu R Exploring Chemical and Biological Space of Terpenoids. J. Chem. Inf. Model 2019, 59, 3667; [DOI] [PubMed] [Google Scholar]; (b) Breitmaier E Terpenes: Flavors, Fragrances, Pharmaca, Pheromones WILEY-VCH, Weinheim, 2006. [Google Scholar]
- 2).Christianson DW Structural Biology and Chemistry of the Terpenoid Cyclases. Chem. Rev 2006. 106, 3412. [DOI] [PubMed] [Google Scholar]
- 3).Maimone TJ; Baran PS Modern Synthetic Efforts Toward Biologically Active Terpenes. Nat. Chem. Bio 2007, 3, 396. [DOI] [PubMed] [Google Scholar]
- 4).Yoder RA; Johnston JN A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids. Chem. Rev 2005, 105, 4730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).(a) Hu X; Maimone TJ Four-step Synthesis of the Antimalarial Cardamom Peroxide via an Oxygen Stitching Strategy. J. Am. Chem. Soc 2014, 136, 5287; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhao Y-M; Maimone TJ Short, Enantioselective Total Synthesis of Chatancin. Angew. Chem. Int. Ed 2015, 54, 1223; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brill ZG; Grover H; Maimone TJ Enantioselective Synthesis of an Ophiobolin Sesterterpene via a Programmed Radical Cascade. Science, 2016, 352, 1078; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hung K; Condakes M; Morimoto T; Maimone TJ Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Syntheses of (+)-Pseudoanisatin. J. Am. Chem. Soc 2016, 138, 16616; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Hu X, Xu S, Maimone TJ A Double Allylation Strategy for Gram-Scale Guaianolide Production: Total Synthesis of (+)-Mikanokryptin. Angew. Chem. Int. Ed 2017, 56, 1624; [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Condakes M; Hung K; Harwood SL; Maimone TJ Total Syntheses of (–)-Majucin and (–)-Jiadifenoxolane A, Complex Majucin-type Illicium Sesquiterpenes. J. Am. Chem. Soc 2017, 139, 17783; [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Hung K; Condakes ML; Novaes LFT; Morikawa T; Harwood S Yang Z; Maimone TJ Development of a Terpene Feedstock-Based Oxidative Approach to the Illicium Sesquiterpenes. J. Am. Chem. Soc 2019, 141, 3083; [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Hu X; Musacchio A; Shen X; Tao Y; Maimone TJ Total Syntheses of Guaianolide Sesquiterpenes from Asteraceae and Apiacea. J. Am. Chem. Soc 2019, 141, 14904; [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Thach DQ; Brill ZG; Grover HK; Esguerra KV; Thompson JK; Maimone TJ Total Synthesis of (+)-6-epi-Ophiobolin A. Angew. Chem. Int. Ed 2020, 59, 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6).(a) Ting CP; Maimone TJ Total Synthesis of Hyperforin. J. Am. Chem. Soc 2015, 137, 10516; [DOI] [PubMed] [Google Scholar]; (b) Ting CP; Maimone TJ The Total Synthesis of Hyperforin. Synlett, 2016, 27, 1443; [DOI] [PubMed] [Google Scholar]; (c) Ting CP; Xu G; Zeng X; Maimone TJ Annulative Methods Enable a Total Synthesis of the Complex Meroterpene Berkeleyone A. J. Am. Chem. Soc 2016, 138, 14868; [DOI] [PubMed] [Google Scholar]; (d) Xu G; Elkin M; Tantillo DJ; Newhouse TR; Maimone TJ Traversing Biosynthetic Carbocation Landscapes in the Synthesis of Andrastin and Terretonin Meroterpenes. Angew. Chem. Int. Ed 2017, 56, 12498; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Shen X; Ting CP; Xu G; Maimone TJ Programmable Meroterpene Synthesis. Nat. Commun 2020, 11, 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).(a) Ruzicka L Isoprene rule and the biogenesis of terpenic compounds. Experientia 1953, 9, 357; [DOI] [PubMed] [Google Scholar]; (b) Eschenmoser A Leopold Ruzicka – From the Isoprene Rule to the Question of Life’s Origin. Chimia 1990, 44, 1–21; [Google Scholar]; (c) Hillier SG; Lathe R Terpenes, hormones and life: isoprene rule revisited. J. Endocrinol 2019, 242, R9–R22. [DOI] [PubMed] [Google Scholar]
- 8).Brill ZG; Condakes ML; Ting CP; Maimone TJ Navigating the Chiral Pool in the Total Synthesis of Complex Terpene Natural Products. Chem. Rev 2017, 117, 11753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9).Sugano M; Shindo T; Sato A; Iijima Y; Oshima T; Kuwano H; Hata T Chatancin, a PAF antagonist from a soft coral, Sarcophyton sp. J. Org. Chem 1990, 55, 5803. [Google Scholar]
- 10).Soucy P; L’Heureux A; Toro A; Deslongchamps P Pyranophane Transannular Diels–Alder Approach to (+)-Chatancin: A Biomimetic Asymmetric Total Synthesis. J. Org. Chem 2003, 68, 9983. [DOI] [PubMed] [Google Scholar]
- 11).Hare SR; Farnham JM; Tantillo DJ Putative biosynthetic cycloadditions en route to the diterpenoid (+)-chatancin. Tetrahedron 2017, 73, 4227. [Google Scholar]
- 12).Krenske EH; Perry EW; Jerome SV; Maimone TJ; Baran PS; Houk KN Why a Proximity-Induced Diels–Alder Reaction Is So Fast. Org. Lett 2012, 14, 3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13).Aigner J; Gössinger E; Kahlig H; Menz G; Pflugseder K Total Synthesis of Chatancin. Angew. Chem. Int. Ed 1998, 37, 2226. [DOI] [PubMed] [Google Scholar]
- 14).He C; Xuan J; Rao P; Xie P-P; Hong X; Lin X; Ding H Total Syntheses of (+)‐Sarcophytin, (+)‐Chatancin, (−)‐3‐Oxochatancin, and (−)‐Pavidolide B: A Divergent Approach. Angew. Chem. Int. Ed 2019, 58, 5100. [DOI] [PubMed] [Google Scholar]
- 15).Fettes A; Carreira EM Leucascandrolide A: Synthesis and Related Studies. J. Org. Chem 2003, 68, 9274. [DOI] [PubMed] [Google Scholar]
- 16).Sato M; Sakaki J-I; Sugita Y; Yasuda S; Sakoda H; Kaneko C Two Lactone Formation Reactions From 1,3-Dioxin-4-ones Having Hydroxyalkyl Group at the 6-Position: Difference in Ring Opening and Closure. Tetrahedron, 1991, 47, 5689. [Google Scholar]
- 17).Kamchonwongpaisan S; Nilanonta C; Tarnchompoo B; Thebtaranonth C; Thebtaranonth Y; Yuthavong Y; Kongsaeree P; Clardy J An Antimalarial Peroxide from Amomum krervanh Pierre. Tetrahedron Lett 1995, 36, 1821. [Google Scholar]
- 18).Czechowski T; Larson TR; Catania TM; Harvey D; Brown GD; Graham IA Artemisia annua mutant impaired in artemisinin synthesis demonstrates importance of nonenzymatic conversion in terpenoid metabolism. Proc. Natl. Acad. Sci 2016, 113, 15150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19).Korshin EE; Bachi MD Online. In: Synthesis of Cyclic Peroxides, Patai’s Chemistry of Functional Groups Wiley; 2009: 1e117. [Google Scholar]
- 20).Hu X; Lim P; Fairhurst R; Maimone TJ Synthesis and Study of the Antimalarial Cardamom Peroxide, Tetrahedron, 2018, 74, 3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21).(a) Isayama S; Mukaiyama T A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 1071; [Google Scholar]; (b) Mukaiyama T; Isayama S; Inoki S; Kato K; Yamada T; Takai T Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II). Chem. Lett 1989, 449; [Google Scholar]; (c) Inoki S; Kato K; Takai T; Isayama S; Yamada T Mukaiyama T Bis(trifluoroacetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins Selective Formation of Alcohols from Olefins. Chem Lett 1989, 515. [Google Scholar]
- 22).(a) Magnus P; Payne AH; Waring MJ; Scott DA; Lynch V Conversion of α,β-unsaturated ketones into α-hydroxy ketones using an Mn(III) catalyst, phenylsilane and dioxygen: Acceleration of conjugate hydride reduction by dioxygen. Tetrahedron Lett 2000, 41, 9725; [Google Scholar]; (b) Magnus P; Waring MJ; Scott DA Conjugate reduction of α,β-unsaturated ketones using an MnIII catalyst, phenylsilane and isopropyl alcohol. Tetrahedron Lett 2000, 41, 9731; [Google Scholar]; (c) Magnus P; Scott DA; Fielding MR Direct conversion of α,β-unsaturated nitriles into cyanohydrins using Mn(dpm)3 catalyst, dioxygen and phenylsilane. Tetrahedron Lett 2001, 42, 4127; [Google Scholar]; (d) Magnus P; Fielding MR Acceleration of the reduction of aldehydes and ketones using Mn(dpm)3 catalyst and phenylsilane in the presence of dioxygen. Tetrahedron Lett 2001, 42, 6633. [Google Scholar]
- 23).For reviews on ophiobolins, see:; (a) Au TK; Chick WSH; Leung PC The Biology of Ophiobolins. Life Sci 2000, 67, 733; [DOI] [PubMed] [Google Scholar]; (b) Tian W; Deng Z; Hong K The Biological Activities of Sesterterpenoid-Type Ophiobolins. Mar. Drugs 2017, 15, 229; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Masi M; Dasari R; Evidente A; Mathieu V; Kornienko A Chemistry and Biology of Ophiobolin A and its Congeners. Bioorg. Med. Chem. Lett 2019, 29, 859. [DOI] [PubMed] [Google Scholar]
- 24).Petasis NA; Patane MA The Synthesis of Carbocyclic Eight-Membered Rings. Tetrahedron 1992, 48, 5757. [Google Scholar]
- 25).Rowley M; Tsukamoto M; Kishi Y Total Synthesis of (+)-Ophiobolin C. J. Am. Chem. Soc 1989, 111, 2735. [Google Scholar]
- 26).(a) Tsuna K; Noguchi N; Nakada M Convergent Total Synthesis of (+)-Ophiobolin A. Angew. Chem. Int. Ed 2011, 50, 9452; [DOI] [PubMed] [Google Scholar]; (b) Tsuna K; Noguchi N; Nakada M Enantioselective Total Synthesis of (+)-Ophiobolin A. Chem. Eur. J 2013, 19, 5476. [DOI] [PubMed] [Google Scholar]
- 27).Chiba R; Minami A; Gomi K; Oikawa H Identification of Ophiobolin F Synthase by a Genome Mining Approach: A Sesterterpene Synthase from Aspergillus clavatus Org. Lett 2013, 15, 594. [DOI] [PubMed] [Google Scholar]
- 28).(a) Charette AB; Juteau H; Lebel H; Molinaro C Enantioselective Cyclopropanation of Allylic Alcohols with Dioxaborolane Ligands: Scope and Synthetic Applications. J. Am. Chem. Soc 1998, 120, 11943; [Google Scholar]; (b) Charette AB; Naud J Regioselective opening of substituted (cyclopropylmethyl)lithiums derived from cyclopropylmethyl iodides. Tetrahedron Lett 1998, 39, 7259. [Google Scholar]
- 29).Seebach D; Beck AK; Heckel A TADDOLs, Their Derivatives, and TADDOL Analogues: Versatile Chiral Auxiliaries. Angew. Chem. Int. Ed 2001, 40, 92. [PubMed] [Google Scholar]
- 30).Roberts BP Polarity-reversal catalysis of hydrogen-atom abstraction reactions: concepts and applications in organic chemistry. Chem. Soc. Rev 1999, 28, 25. [Google Scholar]
- 31).Mukaiyama T; Matsuo J-I; Kitagawa H New and One-Pot Synthesis of α,β-Unsaturated Ketones by Dehydrogenation of Various Ketones with N-tert-Butyl Phenylsulfinimidoyl Chloride. Chem. Lett 2000, 29, 1250. [Google Scholar]
- 32).Daniewski AR, Liu W, A Novel Silylcopper Catalyst for the Reductive Bromination of Hajos Dione. Improved Preparation of a CD Synthon for the Synthesis of Vitamin D. J. Org. Chem 2001, 66, 626. [DOI] [PubMed] [Google Scholar]
- 33).For related examples, see:; a) Eilbracht P et al. Tandem Reaction Sequences under Hydroformylation Conditions: New Synthetic Applications of Transition Metal Catalysis. Chem. Rev 1999, 99, 3329; [DOI] [PubMed] [Google Scholar]; b) Airiau E et al. Microwave-Assisted Domino Hydroformylation/Cyclization Reactions: Scope and Limitations. Synthesis 2010, 2901. [Google Scholar]
- 34).Hung K; Hu X; Maimone TJ Total Synthesis of Complex Terpenes Employing Radical Cascade Processes, Nat. Prod. Rep 2018, 35, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35).(a) Chadwick M; Trewin H; Gawthrop F; Wagstaff C Sesquiterpenoids Lactones: Benefits to Plants and People. Int. J. Mol. Sci 2013, 14, 12780; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Drew DP; Krichau N; Reichwald K; Simonsen HT Guaianolides in apiaceae: perspectives on pharmacology and biosynthesis. Phytochem. Rev 2009, 8, 581; [Google Scholar]; (c) Simonsen HT; Weitzel C; Christensen SB Guaianolide sesquiterpenoids: Pharmacology and biosynthesis In Natural Products; Ramawat KG, Merillon JM, Eds.; Springer-Verlag: Berlin, 2013; Vol. 5, p 3069; [Google Scholar]; (d) Santana A; Molinillo JMG; Macías FA Trends in the Synthesis and Functionalization of Guaianolides. Eur. J. Org. Chem, 2015, 2093–2110. [Google Scholar]
- 36).For recent biosynthetic investigations, see:; (a) Andersen TB; Martinez-Swatson KA; Rasmussen SA; Boughton BA; Jørgensen K; Andersen-Ranberg J; Nyberg N; Christensen SB; Simonsen HT Localization and in-Vivo Characterization of Thapsia garganica CYP76AE2 Indicates a Role in Thapsigargin Biosynthesis. Plant Physiol 2017, 174, 56; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ramirez AM; Saillard N; Yang T; Franssen MCR; Bouwmeester HJ; Jongsma MA Biosynthesis of Sesquiterpene Lactones in Pyrethrum (Tanacetum cinerariifolium). PLoS ONE, 2013, 8(5): e65030. doi: 10.1371/journal.pone.0065030. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Q; Kashkooli AB; Manzano D; Pateraki I; Richard L; Kolkman P; Lucas MF; Guallar V; de Vos RCH; Franssen MCR; van der Krol A; Bouwmeester H Kauniolide synthase is a P450 with unusual hydroxylation and cyclization-elimination activity. Nat. Commun 2018, 9, 4657; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) de Kraker J-W; Franssen MCR; Joerink M; de Groot A; Bouwmeester HJ Bio-synthesis of Costunolide, Dihydrocostunolide, and Leucodin. Demonstration of Cytochrome P450-Catalyzed Formation of the Lactone Ring Present in Sesquiterpene Lactones of Chicory. Plant Physiol 2002, 129, 257; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pickel B; Drew DP; Manczak T; Weitzel C; Simonsen HT; Ro DK Identifica-tion and characterization of a kunzeaol synthase from Thapsia garganica: implications for the biosynthesis of the pharmaceutical thapsigargin. Biochem. J 2012, 448, 261. [DOI] [PubMed] [Google Scholar]
- 37).Berdan CA; Ho R; Lehtola HS; To M; Hu X; Huffman TR; Petri Y; Altobelli CR; Demeulenaere SG; Olzmann JA; Maimone TJ; Nomura DK Parthenolide Covalently Targets and Inhibits Focal Adhesion Kinase in Breast Cancer Cells, Cell Chem. Biol 2019, 26, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Chu H; Smith JM; Felding J; Baran PS Scalable Synthesis of (–)-Thapsigargin. ACS. Cent. Sci 2017, 3, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39).Chen D; Evans PA A Concise, Efficient and Scalable Total Synthesis of Thapsigargin and Nortrilobolide from (R)-(–)-Carvone. J. Am. Chem. Soc 2017, 139, 6046. [DOI] [PubMed] [Google Scholar]
- 40).Doan NTQ; Crestey F; Olsen CE; Christensen SB Chemo- and Regioselective Functionalization of Nortrilobolide: Application for Semisynthesis of the Natural Product 2-Acetoxytrilobolide. J. Nat. Prod 2015, 78, 1406. [DOI] [PubMed] [Google Scholar]
- 41).For general reviews, see:; (a) Illicium, Pimpinella and Foeniculum; Jodral MM, Ed.; Medicinal and Aromatic Plants – Industrial Profiles; CRC Press: Boca Raton, FL, USA, 2004; Vol. 40; [Google Scholar]; (b) Fukuyama Y; Huang J-M Chemistry and neurotrophic activity of seco-prezizaane- and anislactone-type sesquiterpenes from Illicium species In Bioactive Natural Products (Part L); Studies in Natural Products Chemistry; Atta-ur-Rahman, Ed.; 2005, Vol. 32, p 395. [Google Scholar]
- 42).Condakes ML; Novaes LFT; Maimone TJ Contemporary Synthetic Strategies Toward Seco-Prezizaane Sesquiterpenes from Illicium Species, J. Org. Chem 2018, 83, 14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43).Concepción JI; Francisco CG; Hernández R; Salazar JA; Suárez E INTRAMOLECULAR HYDROGEN ABSTRACTION. IODOSOBENZENE DIACETATE, AN EFFICIENT AND CONVENIENT REAGENT FOR ALKOXY RADICAL GENERATION. Tetrahedron Lett 1984, 25, 1953. [Google Scholar]
- 44).Paquette LA; Hofferberth JE The α‐Hydroxy Ketone (α‐Ketol) and Related Rearrangements. Organic Reactions 2004, 62, 477. [Google Scholar]
- 45).(a) Chen M; White MC A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis. Science 2007, 318, 783. [DOI] [PubMed] [Google Scholar]; (b) Sun C-L; Li B-J; Shi Z-J Chem. Rev 2011, 111, 1293. [DOI] [PubMed] [Google Scholar]; (c) White MC; Zhao J Aliphatic C–H Oxidations for Late-Stage Functionalization J. Am. Chem. Soc 2018, 140, 13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46).Goméz L; Garcia-Bosch I; Company A; Benet-Buchholz J; Polo A; Sala X; Ribas X; Costas M Stereospecific C-H oxidation of H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem. Int. Ed 2009, 48, 5720. [DOI] [PubMed] [Google Scholar]
- 47).(a) Donohoe TJ; Moore PR; Waring MJ; Newcombe NJ Tetrahedron Lett 1997, 38, 5027. [Google Scholar]; (b) Donohoe TJ; Mitchell L; Waring MJ; Helliwell M; Bell A; Newcombe NJ Tetrahedron Lett 2001, 42, 8951. [Google Scholar]; (c) Donohoe TJ Synlett 2002, 8, 1223. [Google Scholar]
- 48).(a) Brun P; Waegell B Intramolecular heterocyclization of alcohols with a cedranic skeleton. Oxidation by lead tetraacetate and by mercury oxide and bromine Tetrahedron 1976, 32, 1137. [Google Scholar]; (b) Tenaglia A; Terranova E; Waegell B Ruthenium-catalyzed carbon-hydrogen bond activation. Oxyfunctionalization of nonactivated carbon-hydrogen bonds in the cedrane series with ruthenium tetraoxide generated in situ. J. Org. Chem 1992, 57, 5523. [Google Scholar]
- 49).Hill CK; Hartwig JF Site-selective oxidation, amination and epimerization reactions of complex polyols enabled by transfer hydrogenation. Nat. Chem 2017, 9, 1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50).Wilson RM; Danishefsky SJ Pattern Recognition in Retrosynthetic Analysis: Snapshots in Total Synthesis. J. Org. Chem 2007, 72, 4293. [DOI] [PubMed] [Google Scholar]
- 51).Corey EJ; Cheng X-M The Logic of Chemical Synthesis; Wiley: New York, 1995. [Google Scholar]
- 52).Nicolaou KC; Snyder SA The Essence of Total Synthesis. Proc. Natl. Acad. Sci 2004, 101, 11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53).For a recent discussion, see:; Baran PS Natural Product Total Synthesis: As Exciting as Ever and Here To Stay. J. Am. Chem. Soc 2018, 140, 4751. [DOI] [PubMed] [Google Scholar]