

Abstract
Hemostasis is the normal process that produces a blood clot at a site of vascular injury. Mice are widely used to study hemostasis and abnormalities of blood coagulation because their hemostatic system is similar in most respects to that of humans, and their genomes can be easily manipulated to create models of inherited human coagulation disorders. Two of the most widely used techniques for assessing hemostasis in mice are the tail bleeding time (TBT) and saphenous vein bleeding (SVB) models. Here we discuss the use of these methods in the evaluation of hemostasis, and the advantages and limits of using mice as surrogates for studying hemostasis in humans.
Introduction
The term hemostasis is commonly used to indicate the cessation of bleeding at a site of injury by formation of a sealing blood clot [1–3]. More broadly, hemostasis implies that the coagulation response is limited to the site of the injury, allowing blood flow to continue normally in the intact circulation. The hemostatic response is one of several homeostatic mechanisms that prevent acute changes in blood volume and pressure, allowing an organism to maintain normal tissue perfusion.
Formation of a blood clot involves a tightly orchestrated set of interactions between cellular and plasma constituents of the blood, and cellular and extracellular components of the blood vessel (1–3). While in vitro and ex vivo techniques to study protein-protein, cell-cell, and cell-protein interactions have been instrumental to our understanding of the biochemistry of blood coagulation, the complexity of the system requires animal models to put these findings into proper context.
Here, we discuss the use of mice as surrogates for humans in studying hemostasis, with an emphasis on models that measure duration and extent of bleeding. We focus on the use of these assays to assess inherited or acquired defects in plasma coagulation, but the techniques are applicable to studying platelet contributions as well.
Advantages of using mice to study hemostasis
Inbred mouse strains have been used for decades to study the physiology and pathology of blood coagulation [4–8]. Mice offer several advantageous features that, combined with their relatively low-cost of upkeep, have established them as important model organisms in hemostasis and thrombosis research. The blood plasmas of mice and humans contain similar complements of coagulation factors and regulatory proteins [5–7]. In general, human plasma coagulation proteins function adequately when infused into mice. With some notable exceptions, hemostatic reactions involving murine blood vessels and platelets are similar to their human counterparts.
Our ability to manipulate the mouse genome to create total or conditional knockouts of a gene of interest has been key to identifying and studying the contributions of various proteins to hemostasis and to a variety of pathologic processes. Even when gene deletion causes embryonic death, strategies to restrict the deficiency state to the adult organism or transgenic approaches to express low levels of protein sufficient to prevent embryonic lethality are available [3,5,9,10]. Knock-in and over-expression strategies facilitate in vivo structure-function analyses, disease modeling, and creation of “humanized” mice to study drugs that specifically target human proteins.
Limitations of mice for studying human blood coagulation
There are important physiologic, anatomic, and biochemical differences between mice and humans that must be considered when employing mice as surrogates for humans in hemostasis studies. The three orders of magnitude size difference results in large differences in mechanical forces exerted on mouse and human tissues. The greater propensity for humans with hemophilia (deficiency of factor VIII [fVIII] or factor IX [fIX]) to bleed into joints (hemarthrosis) than do fVIII or fIX deficient mice is at least partly explained by the different magnitudes of force exerted on the tissues of 70 kilogram bipeds and 0.025 kilogram quadrupeds [11–14]. Along similar lines, it is our experience that simple skin incisions are usually well tolerated in hemophiliac mice in the absence of factor replacement, as long as care is taken not to nick underlying structures. However, larger skin injuries, such as punch biopsy wounds are prone to severe delayed bleeding unless small amounts of the missing factor are provided [15,16].
There are notable differences in the coagulation mechanisms of mice and humans that complicate certain analyses. One of the best characterized involves the protease-activated receptors (PARs) expressed on platelet surfaces [17–20]. Human platelets express PAR-1 and PAR-4, and cleavage of either by thrombin promotes platelet activation. Murine platelets express PAR-4 and also PAR-3, which functions as a cofactor for PAR-4, but do not express PAR-1. This situation has created significant difficulties in pre-clinical evaluations of anti-platelet agents that target PAR-1.
While mice and humans have similar complements of plasma coagulation factors, some proteins may serve different functions in the two species. For example, in humans factor XI (fXI) deficiency causes a variable tendency to bleed excessively with trauma or surgery [21,22]. A similar phenotype has been noted in fXI deficient cattle, dogs and cats [23–25]. FXI deficient mice, on the other hand, have not demonstrated a consistent propensity to bleed abnormally with any hemostatic challenge [26–28, and unpublished observations]. In humans, most fXI circulates in plasma in complex with high molecular weight kininogen (HK) [29]. In mice, most fXI is bound to glycosaminoglycans on blood vessels [30]. These findings raise the possibility that the primary function of fXI in mice is not hemostasis, and brings into question the suitability of the mouse as a model for human fXI deficiency.
Mouse Models of Hemostasis
Several approaches have been used to study hemostasis in mice, but most are not well standardized. Here we concentrate on two of the most widely used and best characterized models, the tail bleeding time (TBT) and saphenous vein bleeding (SVB) models. We will briefly discuss models based on injury to the liver or cuticle. Differences in mouse strain and methods of anesthesia may influence bleeding in the TBT and SVB models [31–33]. When we describe our experiences with a model, they are based on work with C57Bl/6 mice anesthetized with pentobarbital or ketamine.
Tail Bleeding Time (TBT) Model
TBT assays are the most widely used approach for assessing hemostasis in mice [8,34–41]. The assays are relatively simple to perform, with a small impact on the health of the animal. In the basic model, a razor or scalpel is used to transect the tail at a predetermined distance from the tip (1 – 5 mm), or at a point with a specific circumference [38–40]. The bleeding tail stump is usually immersed in normal saline warmed to 37 °C, and time to cessation of bleeding is determined. Blood loss is assessed by measuring hemoglobin in the saline. Alternatively, the transected tail may be left exposed to air, with frequent wicking of blood using filter paper. The hemoglobin on the filter paper can then be quantified.
In our hands, wild type (WT) mice usually bleed for 150 to 300 seconds after tail transection (Figure 1A) [27]. However, there can be significant variability, and regular users of the TBT are familiar with the occasional control animal with prolonged bleeding. Tail transection causes injury to multiple types of tissues (skin, bone, connective tissue) including three large blood vessels (a central artery and two lateral veins) each of which may respond differently to injury. Furthermore, the anatomy of the tail varies substantially between individual animals. These factors likely contribute to variation in injury severity and subsequent bleeding that is not amenable to technique standardization.
Figure 1. Effects of factor IX or factor XI deficiency on the tail bleeding time and saphenous vein bleeding models.
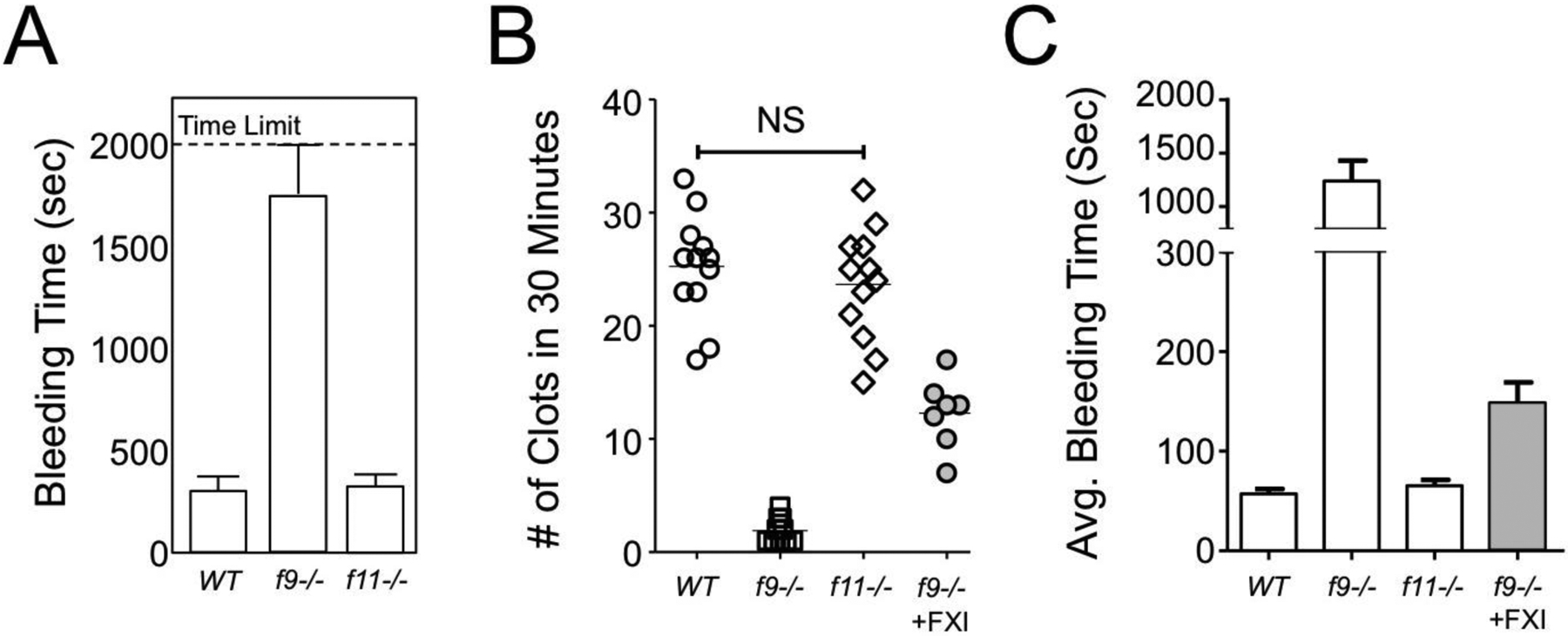
(A) Groups of ten C57Bl/6 wild type (WT), fIX deficient (f9−/−) or fXI deficient (f11−/−) mice were tested with a standard tail bleeding time assay. Mice were not allowed to bleed for more than 2000 seconds. Data are shown as means of times to final termination of bleeding +/− 1 SD. (B and C) Groups of twelve WT (○), fIX−/− (◻) and f11−/− (◇) mice were tested in the saphenous vein bleeding model. Data for number of clots in 30 minutes is shown in panel B, and the average duration of each bleeding episode (+/− 1 SD) is shown in panel C. Note that there is no significant difference between WT and fXI−/− mice. The gray circles in panel B and gray bar in panel C show the effect of an infusion of human fXI into f9−/− mice calculated to double the plasma fXI concentration.
We found that transecting the tail at a location where it is 2 mm wide (as determined by a template) is associated with less variability than transecting at a specific distance from the tail tip. While a recent study found that the type of anesthetic used (pentobarbital, ketamine or isoflurane) did not affect the TBT in wild type mice [new 42], we and others have noted that injury-induced thrombosis varies considerably depending on anesthetic type [27, new 42]. This may reflect differences in drug effects on vascular tone, blood velocity through certain vascular beds, platelet reactivity, and perhaps plasma enzymatic reactions. Furthermore, while bleeding in wild type mice may not be affected appreciably by anesthetic types, this may not hold for mice with hemostatic defects. For this reason, we use a single anesthetic (pentobarbital) for our work with the TBT.
Bleeding in the TBT assay is not continuous. That is, it consists of bleeding punctuated by periods where hemostasis appears to have been achieved. Broze and colleagues proposed that this is due to cycles of vasoconstriction and vasodilatation [31]. When evaluating bleeding after tail transection, then, one should record both the time to initial cessation of bleeding and the time at which bleeding stops without rebleeding. A modification of the basic TBT model that replaces tail transection with an incision along one of the lateral tail veins has been adopted by several groups [31,43]. This technique causes less severe bleeding that is more reproducible than with standard tail transection.
The TBT is sensitive to deficiencies of plasma coagulation factors associated with severe bleeding phenotypes in humans. Mice lacking fVIII or fIX (models of human hemophilia A and hemophilia B, respectively) usually exsanguinate after tail transection unless the tail tip is cauterized (Figure 1A) [14,27,44,45]. Bleeding usually stops shortly after tail transection in hemophiliac animals in a manner that appears similar to WT mice. However, subsequent rebleeding in the hemophiliac mice is severe and usually fatal without intervention. We observed a similar bleed-stop-rebleed pattern in fIX deficient mice after puncturing the carotid artery with a 25-gauge needle (unpublished observation). In the TBT, bleeding in fVIII [31,42–44,46–48] and fIX deficient [31,49,50] mice responds to factor replacement and other therapeutic interventions used to treat humans with hemophilia.
While fXI deficient humans may experience excessive trauma-induced bleeding, fXI deficient mice are indistinguishable from WT mice in the TBT model (Figure 1A) [27,31,51,52]. The plasmas of mice lacking individual components of the plasma contact activation system (factor XII [fXII], prekallikrein and HK) display significant defects in surface initiated clotting assays in vitro; however, their TBTs are comparable to those of WT mice [53–56]. This is consistent with the absence of a bleeding diathesis in humans lacking fXII, prekallikrein or HK.
Prolonged TBTs have been observed in WT mice treated with anticoagulants including heparin [27,31], warfarin [41], and small molecule inhibitors of thrombin [57–59] and factor Xa [60]; the anti-platelet agents aspirin [27,31] and clopidogrel [39]; and with over-expression of the coagulation regulator protein C [61].
Mice with platelet abnormalities equivalent to the human conditions Glanzmann thrombasthenia (caused by mutations affecting either subunit of the GP IIb/IIIa complex) and Bernard-Soulier syndrome (caused by mutations of one of the three subunits that make up the GP Ib-IX-V complex) demonstrate severe hemostatic defects in the TBT [31,62–64]. The effects of disrupting other platelets receptors/proteins affect the TBT to varying degrees and have been reviewed elsewhere [65].
Saphenous Vein Bleeding (SVB) Model
The SVB model was developed by Whinna with the goal of establishing an assay to study hemostasis in hemophiliac mice that has better sensitivity and lower variability than the TBT [66–68]. Bleeding in the SVB model can be adjusted to show a graded response to plasma factor concentrations between 1 and 20% of normal by changing the degree of injury. This allows the assay to more closely mirror the varying degrees of bleeding observed in patients with severe (<1%), moderate (1 to 5%) or mild (>5%) hemophilia. Monroe adapted the model for the study of anticoagulation therapy [69].
Mice under general anesthesia are placed in a supine position with the hind limbs gently restrained to a procedure mat warmed to 37 °C. The ventral surface of the hind limb is shaved, and a longitudinal skin incision is made along the length of the limb exposing the saphenous vein. The vein is punctured once with a 23-gauge needle to create an entry hole. After initial bleed stops (1–2 minutes), a blade of a Student Vannas spring scissor is inserted through the entry hole into the distal part of the vessel and a longitudinal incision is made. We make an ~1 mm incision, but the length can be varied to change the bleeding severity. With an ~1 mm incision, bleeding in fIX deficient mice correlates with plasma fIX levels between 1% and 20% of normal. Blood is wicked away from the injury site using gauze until bleeding stops. The clot is then gently disrupted to restart bleeding. The goal is to remove the clot with minimal manipulation of the blood vessel. We disrupt clots by gently running gauze along the clot surface in the direction of blood flow. Others use the tip of a 30-gauge needle to dislodge the clot. Each time bleeding stops, the clot is disrupted to restart blood flow. The vessel is observed for 30 minutes, and normal saline is applied periodically to prevent the surgical field from drying. The number of clots formed over the course of 30 minutes (Figure 1B) and the duration of each bleed (Figure 1C) are recorded. The total amount of blood lost may also be determined.
While we find the SVB model is subject to less intra-operator variability than the TBT model, it is more technically demanding. We observed considerable inter-operator variability, likely reflecting subtle differences in technique. Differences in the size of the vessel opening alters the sensitivity of the assay to plasma coagulation factor levels, and is likely one source of variability. The manner in which clots are disrupted also alters the hemostatic response [28,70].
The SVB model was designed to be sensitive to fVIII or fIX deficiencies and bleeding is reduced by factor replacement and other treatment modalities used to treat hemophiliacs [67,71–74]. Untreated fIX-deficient mice have relatively few clotting events (Figure 1B, 1 to 3 over 30 minutes, compared to 25 to 30 for WT mice), and each bleeding event is substantially longer than in WT mice (Figure 1C). While the SVB model, like the TBT, does not detect a difference between fXI deficient and WT mice in our hands [28], administering a large amount of human fXI to mice lacking fIX (hemophilia B) increases the number of clots and reduces the duration of bleeding (Figure 1B and 1C, data highlighted in gray) [28]. This is likely due to fXIa activation of factors V and X that partially bypass the need for fIX. [75]. This supports the impression that the SVB model is useful for studying novel therapeutic strategies in hemophilia other than factor replacement. The SVB model has also been quite useful for assessing the effects of anticoagulation therapy on hemostasis [69,76,77].
Recently, a variation on the SVB model has been described that incorporates intravital fluorescence microscopy to study clot formation in real time [78]. Labeled antibodies to fibrin and platelets are infused before saphenous vein injury is inflicted with a laser. The laser punches a hole in the vessel wall without transecting it. There is no need for a longitudinal incision in the vessel, as the purpose of the incision in the surgical model is to prevent vasoconstriction, which is not an issue with the laser. The effects of repeated injury to the same site may be assessed. Alternatively, separate injuries can be created along the length of the vessel. In addition to the standard parameters, platelet fibrin formation and adhesion/aggregation are observed in real time. The surgical SVB model is sensitive to changes in both coagulation factors and platelet adhesion/aggregation [79]. In contrast, the laser-based model detects subtle differences in platelet function and von Willebrand factor activity, but is relatively insensitive to plasma factor deficiencies but does [78,80]. The reasons for the differences in the two versions of the SVB model have not been established. In some respects, the laser-based model behaves like the template bleeding times used for many years in clinical practice, with sensitivity to platelet defects and relative (but not absolute) insensitivity to defects in thrombin generation. Perhaps surgical injury causes a greater degree of tissue factor exposure to blood than does laser injury.
Liver Laceration Model
This model has been used in a variety of species, most often to assess the anti-hemostatic effects of novel therapeutic agents, and to test strategies for anticoagulant reversal [81]. We used it to investigate hemostasis in fXI-deficient mice and in WT mice treated with antibodies to fXI or fXII (unpublished data). In our version of the model, mice are placed on a warming pad in a supine position. The abdomen is shaved, and the liver is exposed via an anterior right subcostal incision. The left lobe of the liver is externalized by applying gentle pressure posteriorly, and a 5-mm transverse incision is made with a #10 scalpel blade starting one cm from the lobe’s inferior edge, and parallel to the inferior edge. Blood is collected into a small tray and the total blood loss determined [81–83]. Heparin and other anticoagulants increase bleeding in this model. Standardizing the degree of injury (particularly the depth of the wound) is a challenge, as is collecting all blood issuing from the wound.
Cuticle Bleeding Model
The nails of many mammals contain a vascular central region called the quick. Cutting into the quick produces bleeding that can be measured. The original model was developed to study therapeutic agents in hemophiliac dogs, and showed a correlation between intensity of therapy and reduced bleeding [84,85]. It has been adapted for other species, most notably rabbits and, less frequently, mice. In mice, the entire cuticle of the middle digit is removed with sharp scissors, and blood is collected with filter paper [86,87]. It is difficult to determine if a consistent injury is being inflicted in mice because of the small size of the digits. There is probably a component of crush injury in addition to the laceration. Furthermore, it is not clear that this model is more reproducible than other models, and some of the groups that reported its initial use have transitioned to other models for subsequent studies.
Conclusions
Mice are invaluable tools for assessing the contributions of plasma, platelet, and blood vessel constituents to normal and pathologic coagulation. Murine bleeding models are commonly used to assess prospective anticoagulants, and to determine the ability of pro-hemostatic therapies to correct bleeding defects. Because of its relative ease of use, the TBT is widely used to evaluate hemostasis in mice. We feel the SVB model is an improvement on the TBT that is particularly useful for studying hemophilia and anticoagulation therapy (and its reversal). While these assays are used routinely in pre-clinical studies, they have limitations as models of human bleeding disorders.
First, the requirement for certain hemostatic factors varies depending on the types of tissue injured, as clearly demonstrated by the propensity of hemophiliacs to bleed preferentially into certain areas of the body. When considering the utility of the TBT and SVB models then, the suitability of mouse tail and saphenous vein tissue as models for bleeding in other areas must be considered. Taking an example we are particularly familiar with, humans lacking fXI tend to bleed most severely after trauma to the nasopharynx, mouth and genitourinary tract [21,22]. These tissues have high intrinsic levels of fibrinolytic activity that prematurely degrade clots, and it is hypothesized that fXI-supported thrombin generation stabilizes clots in this setting [22]. Such conditions may not be reproduced with the TBT or SVB models, perhaps partly explaining why fXI and WT mice respond similarly in these assays.
Second, some proteins identified as coagulation factors in humans may serve a different role in mice. The core of the vertebrate thrombin generation mechanism is comprised of a group of vitamin K-dependent proteases and their cofactors [1–3]. It appears that all vertebrates require these proteins for normal hemostasis [88]. However, the roles of ancillary proteins may differ. Again, taking the example of fXI, we identified important roles for this protein in occlusive thrombus formation [27,89] and inflammation [90] in mice, but have not identified a condition under which it is required for hemostasis [27,28]. The fact that most mouse fXI is bound to blood vessels and is not in plasma suggests adaptation for novel functions not relevant to human physiology [30]. Such issues will become more important as antithrombotic strategies targeting fXI move into clinical trials [89,91]. The take home lesson is that absence of bleeding in a laboratory animal model may not accurately reflect the responses of human patients to various drugs and conditions.
Acknowledgement
The authors wish to acknowledge support from award HL140025 from the National Heart, Lung and Blood Institute, a Bridge Grant from the American Society of Hematology, and post-doctoral award 8POST34030076 from the American Heart Association. D. Gailani receives consultant’s fees from several pharmaceutical companies.
Bibliography
- 1.Fredenburgh JC, Weitz JI, Overview of hemostasis and thrombosis, in Hoffman R, Benz EJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, Salama ME, Abutalib SY. Hematology: basic principles and practice (7th edition). 2018. Elsevier, Philadelphia, PA, pgs1831–1842. [Google Scholar]
- 2.Negrier C, Shima M, Hoffman M. The central role of thrombin in bleeding disorders. Blood Rev. (in press). [DOI] [PubMed] [Google Scholar]
- 3.Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger for thrombosis. Arterioscler. Thromb. Vasc. Biol 2018;38:709–725. [DOI] [PubMed] [Google Scholar]
- 4.Hogan KA, Weiler H, Lord ST. Mouse models in coagulation. Thromb. Haemost 2002;87:563–574. [PubMed] [Google Scholar]
- 5.Mackman N Mouse models in haemostasis and thrombosis. Thromb. Haemost 2004;92:440–443 [DOI] [PubMed] [Google Scholar]
- 6.Emeis JJ, Jirouskova M, Muchitsch EM, Shet AS, Smyth SS, Johnson GJ. A guide to murine coagulation factor structure, function, assays, and genetic alterations. J Thromb Haemost. 2007. April;5(4):670–9. [DOI] [PubMed] [Google Scholar]
- 7.McManus MP, Gailani D: Mouse models of coagulation factor deficiencies, in Animal Models of Diseases: Translational Medicine Perspective for Drug Discovery and Development. Bentham Scientific Publishers, pp 67–121, 2012. [Google Scholar]
- 8.Brake MA, Ivanciu L, Maroney SA, Martinez ND, Mast AE, Westrick RJ. Assessing Blood Clotting and Coagulation Factors in Mice. Curr Protoc Mouse Biol. 2019. June;9(2):e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tai SJ, Herzog RW, Margaritis P, Arruda VR, Chu K, Golden JA, Labosky PA, High KA. A viable mouse model of factor X deficiency provides evidence for maternal transfer of factor X. J Thromb Haemost. 2008. February;6(2):339–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun WY, Coleman MJ, Witte DP, Degen SJ. Rescue of prothrombin-deficiency by transgene expression in mice. Thromb Haemost. 2002. December;88(6):984–91. [PubMed] [Google Scholar]
- 11.Carcao M, Moorehead P, Lilllicrap D. Hemophilia A and B, in Hoffman R, Benz EJ, Silberstein LE, Heslop HE, Weitz JI, Anastasi J, Salama ME, Abutalib SY. Hematology: basic principles and practice (7th edition). 2018. Elsevier, Philadelphia, PA, pgs 2023–2033. [Google Scholar]
- 12.Yen CT, Fan MN, Yang YL, Chou SC, Yu IS, Lin SW. Current animal models of hemophilia: the state of the art. Thromb J. 2016. October 4;14(Suppl 1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lozier JN, Nichols TC. Animal models of hemophilia and related bleeding disorders. Semin Hematol. 2013. April;50(2):175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sabatino DE, Nichols TC, Merricks E, Bellinger DA, Herzog RW, Monahan PE. Animal models of hemophilia Prog Mol Biol Transl Sci. 2012;105:151–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoffman M, Harger A, Lenkowski A, Hedner U, Roberts HR, Monroe DM. Cutaneous wound healing is impaired in hemophilia B. Blood. 2006. November 1;108(9):3053–60. [DOI] [PubMed] [Google Scholar]
- 16.Gao G, Mashausi DS, Negi H, Li D, Li D. A new mouse model for wound healing in hemophilia A. Int. J. Clin. Exp. Patjol 2015;8:3015–21. [PMC free article] [PubMed] [Google Scholar]
- 17.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost. 2005. August;3(8):1800–14. [DOI] [PubMed] [Google Scholar]
- 18.De Candia E Mechanisms of platelet activation by thrombin: a short story. Thromb. Res 2012;129:250–6. [DOI] [PubMed] [Google Scholar]
- 19.Nieman MT. Protease-activated receptors in hemostasis. Blood 2016;128:169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Posma JJ, Grover SP, Hisada Y, Owens AP 3rd, Antoniak S, Spronk HM, Mackman N. Roles of Coagulation Proteases and PARs (Protease-Activated Receptors) in Mouse Models of Inflammatory Diseases. Arterioscler Thromb Vasc Biol. 2019. January;39(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.James P, Salomon O, Mikovic D, et al. Rare bleeding disorders - bleeding assessment tools, laboratory aspects and phenotype and therapy of FXI deficiency. Haemophilia. 2014;20(Suppl 4):71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Exp. Rev. Hematol 2016;9:629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gentry PA: The relationship between factor XI coagulant and factor XI antigen in cattle. Can J Comp Med 48:58, 1984 [PMC free article] [PubMed] [Google Scholar]
- 24.Knowles C, Giger U, Dodds WJ, Brooks M: Factor XI deficiency in Kerry Blue Terriers. J Am Vet Med Assoc 205:1157, 1994 [PubMed] [Google Scholar]
- 25.Troxel MT, Brooks MB, Esterline ML. Congenital factor XI deficiency in a domestic shorthair cat. J. Am. Anim. Hosp. Assoc 2002;38:549–53. [DOI] [PubMed] [Google Scholar]
- 26.Gailani D, Lasky NM, Broze GJ Jr. A murine model of factor XI deficiency. Blood Coagul Fibrinolysis. 1997. March;8(2):134–44. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Cheng, Xu L, Feuerstein GZ, Hsu MY, Smith PL, Seiffert DA, Schumacher WA, Ogletree ML, Gailani D. Effects of factor IX or factor XI deficiency on ferric chloride-induced carotid artery occlusion in mice. J Thromb Haemost. 2005. April;3(4):695–702. [DOI] [PubMed] [Google Scholar]
- 28.Mohammed BM, Cheng Q, Matafonov A, Monroe DM, Meijers JCM, Gailani D. Factor XI promotes hemostasis in factor IX-deficient mice. J Thromb Haemost. 2018. October;16(10):2044–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thompson RE, Mandle R Jr, Kaplan AP. Association of factor XI and high molecular weight kininogen in human plasma. J Clin Invest. 1977. December;60(6):1376–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mohammed BM, Cheng Q, Matafonov A, Verhamme IM, Emsley J, McCrae KR, McCarty OJT, Gruber A, Gailani D. A non-circulating pool of factor XI associated with glycosaminoglycans in mice. J Thromb Haemost. 2019. September;17(9):1449–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Broze GJ, Yin ZF, Lasky N. A tail vein bleeding time model and delayed bleeding in hemophiliac mice. Thromb Haemost. 2001. April;85(4):747–8. [PubMed] [Google Scholar]
- 32.Joover-Plow J, Shchurin A, Hart E, Sha J, Hill AE, Singer JB, Nadeau JH. Genetic background determines response to hemostasis and thrombosis. BMC Blood Disord. 2006;6:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene TK, Schiviz A, Hoellriegl W, Poncz M, Muchitsch EM; Animal Models Subcommittee of the Scientific And Standardization Committee Of The ISTH. J Thromb Haemost. 2010. December;8(12):2820–2. Towards a standardization of the murine tail bleeding model. [DOI] [PubMed] [Google Scholar]
- 34.Dejana E, Callioni A, Quintana A, de Gaetano G. Bleeding time in laboratory animals. II - A comparison of different assay conditions in rats. Thromb Res. 1979;15:191–7. [DOI] [PubMed] [Google Scholar]
- 35.Dejana E, Villa S, de Gaetano G. Bleeding time in rats: a comparison of different experimental conditions. Thromb Haemost. 1982. August 24;48(1):108–11. [PubMed] [Google Scholar]
- 36.Wang JP, Hsu MF, Hsu TP, Teng CM. Antihemostatic and antithrombotic effects of capsaicin in comparison with aspirin and indomethacin. Thromb Res. 1985. March 15;37(6):669–79. [DOI] [PubMed] [Google Scholar]
- 37.Kaiser B, Markwardt F. Antithrombotic and haemorrhagic effects of the naturally occurring thrombin inhibitor hirudin. Folia Haematol Int Mag Klin Morphol Blutforsch. 1988;115(1–2):41–6. [PubMed] [Google Scholar]
- 38.Greene TK, Schiviz A, Hoellriegl W, Poncz M, Muchitsch EM; Animal Models Subcommittee of the Scientific And Standardization Committee Of The ISTH. Towards a standardization of the murine tail bleeding model. J Thromb Haemost. 2010. December;8(12):2820–2. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Jennings NL, Dart AM, Du XJ. Standardizing a simpler, more sensitive and accurate tail bleeding assay in mice. World J. Exp. Med 2012;2:30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molina ES, Fujita A, Sogayar MC, Demasi MA. A quantitative and humane tail bleeding assay for efficacy evaluation of antihaemophilic factors in haemophilia A mice. Haemophilia. 2014;20(6):e392–8. [DOI] [PubMed] [Google Scholar]
- 41.Saito MS, Lourenço AL, Kang HC, Rodrigues CR, Cabral LM, Castro HC, Satlher PC. New approaches in tail‐bleeding assay in mice: improving an important method for designing new anti‐thrombotic agents. Int. J. Exp. Pathol 2016;97:285–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sashindranath M, Sturgeon SA, French S, Craenmehr DDD, Selan C, Freddi S, Johnson C, Cody SH, Nesbitt WS, Hamilton JR, Nandurkar HH. The mode of anesthesia influences outcome in mouse models of arterial thrombosis. Res Pract Thromb Haemost. 2019. February 15;3(2):197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansen PB, Tranholm M, Haaning J, Knudsen T. Development of a tail vein transection bleeding model in fully anaesthetized haemophilia A mice: characterization of two novel FVIII molecules. Haemophilia. 2016;22(4):625–31. [DOI] [PubMed] [Google Scholar]
- 44.Bi L, Lawler AM, Antonarakis SE, High KA, Gearhart JD, Kazazian HH Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nature Genetics 1995;10:119–121. [DOI] [PubMed] [Google Scholar]
- 45.Wang L, Zoppè M, Hackeng TM, Griffin JH, Lee KF, Verma IM. A factor IX-deficient mouse model for hemophilia B gene therapy. Proc Natl Acad Sci (USA) 1997. October 14; 94(21): 11563–11566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elm T, Karpf DM, Øvlisen K, Pelzer H, Ezban M, Kjalke M, Tranholm M. Pharmacokinetics and pharmacodynamics of a new recombinant FVIII (N8) in haemophilia A mice. Haemophilia. 2012. January;18(1):139–45. [DOI] [PubMed] [Google Scholar]
- 47.Gu R, Liu L, Xie L, Gai W, Cao S, Meng Z, Gan H, Wu Z, Li J, Zheng Y, Zhu X, Dou G. Pharmacokinetics and pharmacodynamics of SCT800, a new recombinant FVIII, in hemophilia A mice. Acta Pharmacol Sin. 2016. March;37(3):408–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karpf DM, Sørensen BB, Hermit MB, Holmberg HL, Tranholm M, Bysted BV, Groth AV, Bjørn SE, Stennicke HR. Prolonged half-life of glycoPEGylated rFVIIa variants compared to native rFVIIa. Thromb Res. 2011. August;128(2):191–5. [DOI] [PubMed] [Google Scholar]
- 49.Kundu RK, Sangiorgi F, Wu LY, Kurachi K, Anderson WF, Maxson R, Gordon EM. Targeted inactivation of the coagulation factor IX gene causes hemophilia B in mice. Blood. 1998. July 1;92(1):168–74. [PubMed] [Google Scholar]
- 50.Lin HF, Maeda N, Smithies O, Straight DL, Stafford DW. A coagulation factor IX-deficient mouse model for human hemophilia B. Blood. 1997. November 15;90(10):3962–6. [PubMed] [Google Scholar]
- 51.Tucker EI, Verbout NG, Leung PY, Hurst S, McCarty OJT, Gailani D, Gruber A. Inhibition of factor XI activation attenuates inflammation and coagulopathy while improving the survival of mouse polymicrobial sepsis. Blood. 2012. May 17;119(20):4762–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.van Montfoort ML, Knaup VL, Marquart JA, Bakhtiari K, Castellino FJ, Hack CE, Meijers JCM. Two novel inhibitory anti-human factor XI antibodies prevent cessation of blood flow in a murine venous thrombosis model. Thromb Haemost. 2013. November;110(5):1065–73. [DOI] [PubMed] [Google Scholar]
- 53.Renné T, Pozgajová M, Grüner S, Schuh K, Pauer HU, Burfeind P, Gailani D, Nieswandt B. Defective thrombus formation in mice lacking coagulation factor XII. J Exp Med. 2005. July 18;202(2):271–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Merkulov S, Zhang WM, Komar AA, Schmaier AH, Barnes E, Zhou Y, Lu X, Iwaki T, Castellino FJ, Luo G, McCrae KR. Deletion of murine kininogen gene 1 (mKng1) causes loss of plasma kininogen and delays thrombosis. Blood. 2008. February 1;111(3):1274–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Revenko AS, Gao D, Crosby JR, Bhattacharjee G, Zhao C, May C, Gailani D, Monia BP, MacLeod AR. Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood. 2011. November 10;118(19):5302–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bird JE, Smith PL, Wang X, Schumacher WA, Barbera F, Revelli J-P, Seiffert D. Effects of plasma kallikrein deficiency on haemostasis and thrombosis in mice: murine ortholog of the Fletcher trait. Thromb Haemost. 2012. June;107(6):1141–50. [DOI] [PubMed] [Google Scholar]
- 57.Berny-Lang MA, Hurst S, Tucker EI, Pelc LA, Wang RK, Hurn PD, Di Cera E, McCarty OJT, Gruber A. Thrombin mutant W215A/E217A treatment improves neurological outcome and reduces cerebral infarct size in a mouse model of ischemic stroke. Stroke. 2011. June;42(6):1736–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shaw MA, Kombrinck KW, McElhinney KE, Sweet DR, Flick MJ, Palumbo JS, Cheng M, Esmon NL, Esmon CT, Brill A, Wagner DD, Degen JL, Mullins ES. Limiting prothrombin activation to meizothrombin is compatible with survival but significantly alters hemostasis in mice. Blood. 2016. April;128(5):721–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li Y-Z, Gong G-Q, Yang W-H, Wang X-H, Jiang M-L, Zhou Y, Yang X-Z, Xu Y-G, He G-W. Antithrombotic activity of HY023016, a novel Dabigatran prodrug evaluated in animal thrombosis models. Thromb Res. 2013. May;131(5):425–35. [DOI] [PubMed] [Google Scholar]
- 60.Wagner N-M, Dressel T, Schäfer K, Konstantinides S. Effect of the factor Xa inhibitor rivaroxaban on arterial thrombosis in wild-type and apolipoprotein E-deficient mice. Thromb Res. 2012. November;130(5):793–8. [DOI] [PubMed] [Google Scholar]
- 61.Lee KFE, Lu B, Roussel JC, Murray-Segal LJ, Salvaris EJ, Hodgkinson SJ, Hall BM, d’Apice AJF, Cowan PJ, Gock H. Protective effects of transgenic human endothelial protein C receptor expression in murine models of transplantation. Am J Transplant. 2012. September;12(9):2363–72. [DOI] [PubMed] [Google Scholar]
- 62.Hynes RO, Hodivala-Dilke KM. Insights and questions arising from studies of a mouse model of Glanzmann thrombasthenia. Thromb Haemost. 1999. August;82(2):481–5. [PubMed] [Google Scholar]
- 63.Kato K, Martinez C, Russell S, Nurden P, Nurden A, Fiering S, Ware J. Genetic deletion of mouse platelet glycoprotein Ibβ produces a Bernard-Soulier phenotype with increased α-granule size. Blood. 2004. October 15;104(8):2339–44. [DOI] [PubMed] [Google Scholar]
- 64.Kanaji S, Kuether EL, Fahs SA, Schroeder JA, Ware J, Montgomery RR, Shi Q. Correction of murine Bernard-Soulier syndrome by lentivirus-mediated gene therapy. Mol Ther. 2012. March;20(3):625–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jirouskova M, Shet AS, Johnson GJ. A guide to murine platelet structure, function, assays, and genetic alterations. J Thromb Haemost. 2007. April;5(4):661–9. [DOI] [PubMed] [Google Scholar]
- 66.Buyue Y, Whinna HC, Sheehan JP. The heparin-binding exosite of factor IXa is a critical regulator of plasma thrombin generation and venous thrombosis. Blood. 2008. October 15;112(8):3234–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pastoft AE, Lykkesfeldt J, Ezban M, Tranholm M, Whinna HC, Lauritzen B. A sensitive venous bleeding model in haemophilia A mice: effects of two recombinant FVIII products (N8 and Advate(®)). Haemophilia. 2012. September;18(5):782–8. [DOI] [PubMed] [Google Scholar]
- 68.Feng D, Whinna, Monroe D, Stafford DW. FVIIa as used pharmacologically is not TF dependent in hemophilia B mice. Blood. 2014. March 13;123(11):1764–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Monroe DM, Hoffman M. A mouse bleeding model to study oral anticoagulants. Thromb Res. 2014. May;133 Suppl 1:S6–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ay C, Hisada Y, Cooley BC, Mackman N. Factor XI-deficient mice exhibit increased bleeding after injury to the saphenous vein. J Thromb Haemost. 2017;15(9):1829–33. [DOI] [PubMed] [Google Scholar]
- 71.Cooley B, Funkhouser W, Monroe D, Ezzell A, Mann DM, Lin F-C, Monahan PE, Stafford DW. Prophylactic efficacy of BeneFIX vs Alprolix in hemophilia B mice. Blood. 2016. 14;128(2):286–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Keshava S, Sundaram J, Rajulapati A, Pendurthi UR, Rao LVM. Pharmacological concentrations of recombinant factor VIIa restore hemostasis independent of tissue factor in antibody-induced hemophilia mice. J Thromb Haemost. 2016. March;14(3):546–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Feng D, Whinna H, Monroe D, Stafford DW. FVIIa as used pharmacologically is not TF dependent in hemophilia B mice. Blood. 2014. March 13;123(11):1764–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pastoft AE, Ezban M, Tranholm M, Lykkesfeldt J, Lauritzen B. Prolonged effect of a new O-glycoPEGylated FVIII (N8-GP) in a murine saphenous vein bleeding model. Haemophilia. 2013. November;19(6):913–9. [DOI] [PubMed] [Google Scholar]
- 75.Matafonov A, Cheng Q, Geng Y, Verhamme IM, Umunakwe O, Tucker EI, Sun MF, Serebrov V, Gruber A, Gailani D. Evidence for factor IX-independent roles for factor XIa in blood coagulation. J Thromb Haemost. 2013. December;11(12):2118–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hoffman M, Volovyk Z, Monroe DM. Reversal of dabigatran effects in models of thrombin generation and hemostasis by factor VIIa and prothrombin complex concentrate. Anesthesiology 2015; 122: 353–62. [DOI] [PubMed] [Google Scholar]
- 77.Tatsumi K, Antoniak S, Subramaniam S, Gondouin B, Neidich SD, Beck MA, Mickelson J, Monroe DM, Bastarache JA, Mackman N. Anticoagulation increases alveolar hemorrhage in mice infected with influenza A. Physiol Rep 2016; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Getz TM, Piatt R, Petrich BGg, Monroe D, Mackman N, Bergmeier W. Novel mouse hemostasis model for real-time determination of bleeding time and hemostatic plug composition. J Thromb Haemost. 2015. March;13(3):417–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nimjee SM, Dornbos D, Pitoc GA, Wheeler DG, Layzer JM, Venetos N, Huttinger A, Talentino SE, Musgrave NJ, Moody H, Rempel RE, Jones C, Carlisle K, Wilson J, Bratton C, Joseph ME, Khan S, Hoffman MR, Sommerville L, Becker RC, et al. Preclinical Development of a vWF Aptamer to Limit Thrombosis and Engender Arterial Recanalization of Occluded Vessels. Mol Ther 2019; 27: 1228–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Akbar H, Duan X, Piatt R, Saleem S, Davis AK, Tandon NN, Bergmeier W, Zheng Y. Small molecule targeting the Rac1-NOX2 interaction prevents collagen-related peptide and thrombin-induced reactive oxygen species generation and platelet activation. J Thromb Haemost 2018; 16: 2083–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bajaj MS, Ogueli GI, Kumar Y, Vadivel K, Lawson G, Shanker S, Schmidt AE, Bajaj SP. Engineering Kunitz Domain 1 (KD1) of Human Tissue Factor Pathway Inhibitor-2 to Selectively Inhibit Fibrinolysis PROPERTIES OF KD1-L17R VARIANT. J Biol Chem. 2011. February 11;286(6):4329–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sheffield WP, Eltringham-Smith LJ, Gataiance S, Bhakta V. A plasmin-activatable thrombin inhibitor reduces experimental thrombosis and assists experimental thrombolysis in murine models. J Thromb Thrombolysis. 2015. May 1;39(4):443–51. [DOI] [PubMed] [Google Scholar]
- 83.Sheffield WP, Eltringham-Smith LJ, Bhakta V. A factor XIa-activatable hirudin-albumin fusion protein reduces thrombosis in mice without promoting blood loss. BMC Biotechnology. 2018. April 5;18(1):21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Giles AR, Tinlin S, Greenwood R. A canine model of hemophilic (factor VIII:C deficiency) bleeding. Blood 1982; 60: 727–30. [PubMed] [Google Scholar]
- 85.Brinkhous KM, Sandberg H, Garris JB, Mattsson C, Palm M, Griggs T, Read MS. Purified human factor VIII procoagulant protein: comparative hemostatic response after infusions into hemophilic and von Willebrand disease dogs. Proc Natl Acad Sci USA 1985; 82: 8752–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yarovoi HV, Kufrin D, Eslin DE, Thornton MA, Haberichter SL, Shi Q, Zhu H, Camire R, Fakharzadeh SS, Kowalska MA, Wilcox DA, Sachais BS, Montgomery RR, Poncz M. Factor VIII ectopically expressed in platelets: efficacy in hemophilia A treatment. Blood. 2003. December 1;102(12):4006–13.12881300 [Google Scholar]
- 87.Greene TK, Wang C, Hirsch JD, Zhai L, Gewirtz J, Thornton MA, Miao HZ, Pipe SW, Kaufman RJ, Camire RM, Arruda VR, Kowalska MA, Poncz M. In vivo efficacy of platelet-delivered, high specific activity factor VIII variants. Blood. 2010. December 23;116(26):6114–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Doolittle RF. Step-by-step evolution of vertebrate blood coagulation. Cold Spring Harb Symp Quant Biol. 2009;74:35–40. [DOI] [PubMed] [Google Scholar]
- 89.Gailani D, Gruber A. Factor XI as a Therapeutic Target. Arterioscler Thromb Vasc Biol. 2016. July;36(7):1316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bane CE Jr, Ivanov I, Matafonov A, Boyd KL, Cheng Q, Sherwood ER, Tucker EI, Smiley ST, McCarty OJ, Gruber A, Gailani D. Factor XI Deficiency Alters the Cytokine Response and Activation of Contact Proteases during Polymicrobial Sepsis in Mice. PLoS One. 2016. April 5;11(4):e0152968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tillman BF, Gruber A, McCarty OJT, Gailani D. Plasma contact factors as therapeutic targets. Blood Rev. 2018. November;32(6):433–448. [DOI] [PMC free article] [PubMed] [Google Scholar]