

SUMMARY
Postnatal brain circuit assembly is driven by temporally regulated intrinsic and cell-extrinsic cues that organize neurogenesis, migration, and axo-dendritic specification in post-mitotic neurons. While cell polarity is an intrinsic organizer of morphogenic events, environmental cues in the germinal zone (GZ) instructing neuron polarization and their coupling during postnatal development are unclear. We report that oxygen tension, which rises at birth, and the von Hippel–Lindau (VHL)–hypoxia-inducible factor 1α (Hif1α) pathway, regulate polarization and maturation of post-mitotic cerebellar granule neurons (CGNs). At early postnatal stages with low GZ vascularization, Hif1α restrains CGN-progenitor cell-cycle exit. Unexpectedly, cell-intrinsic VHL-Hif1α pathway activation also delays the timing of CGN differentiation, germinal zone exit, and migration initiation through transcriptional repression of the partitioning-defective (Pard) complex. As vascularization proceeds, these inhibitory mechanisms are downregulated, implicating increasing oxygen tension as critical switch for neuronal polarization and cerebellar GZ exit.
Graphical Abstract
eToc Blurb
While many signaling pathways modulate neuronal differentiation and migration, Kullmann et al. show immature regions of brain are hypoxic and neuronal maturation proceed only when Hif1 inhibition of neuronal polarity and germinal zone exit recedes. Hif1 antagonism of neuronal polarity controls migration initiation by altering adhesion to germinal niche extracellular matrix.
INTRODUCTION
Mammalian brain lamination depends on the ability of neurons to exit the cell cycle, migrate, and coalesce into the layers that are neuronal circuitry’s fundamental building blocks (Ayala et al., 2007;Cooper, 2013;Hatten, 2002). Many pediatric neurologic disorders and brain cancers involve neuronal migration dysregulation or the timing of progenitor germinal zone (GZ) exit, that directly impact lamina formation (Hatten and Roussel, 2011;Metin et al., 2008). Interestingly, a picture is emerging that cell polarity signaling pathways are intimately involved in these processes by directing neurogenesis (Falk et al., 2017;Taverna et al., 2014), axon-dendrite specification (Barnes and Polleux, 2009;Lewis et al., 2013), radial migration (Cooper, 2013;Singh and Solecki, 2015), or cellular recognition via adhesion (Rasin et al., 2007;Solecki, 2012). However, mechanisms upstream of polarity pathways coordinating neural development and impacted in disease are poorly understood.
The cerebellum is a suitable system to investigate this issue. Studies by our laboratory in the mouse cerebellar granule neuron (CGN) model system show that polarity regulation extends beyond canonical cell signaling (McCaffrey and Macara, 2009;Namba et al., 2015)). For example, parallel transcriptional (e.g., Zeb1 (Singh et al., 2016)) and ubiquitination (e.g., Siah2 (Famulski et al., 2010)) pathways act in a switch-like manner linking polarity to neuronal differentiation and migration initiation by controlling the onset of partitioning-defective (Pard)-complex genes or proteins expression. Despite advances in our knowledge of these cell-intrinsic mechanisms endowing differentiating neurons with new polarity-dependent cell biological activities (Singh and Solecki, 2015;Laumonnerie and Solecki, 2018;Uzquiano et al., 2018), key challenges remain to our understanding of how polarity signaling is affected by dynamic cell-extrinsic environmental conditions.
We investigated the role of oxygen (O2) homeostasis in CGN development given putative links between polarity regulators to O2 levels in other cell systems (Bui et al., 2009;Krishnamachary et al., 2006;Nakayama et al., 2004;Nakayama and Ronai, 2004). In many brain regions, O2 levels affect neural stem cells (Lange et al., 2016;Mazumdar et al., 2010;Morrison et al., 2000;Studer et al., 2000) (reviewed by (Mohyeldin et al., 2010;Panchision, 2009;Simon and Keith, 2008)), and specific vascular relations with germinal niches in vivo were described for some lineages (Javaherian and Kriegstein, 2009;Lange et al., 2016;Palmer et al., 2000;Shen et al., 2004;Shen et al., 2008;Tavazoie et al., 2008). Also, O2 tension is critical to healthy brain development; both elevated and reduced O2 is linked to altered neurodevelopment and cognitive impairment (Salmaso et al., 2014). The hypoxia-inducible factor 1α (Hif1α) pathway is the primary adaptive mechanism used by cells to respond to O2 (Kaelin, 2008;Kaelin and Ratcliffe, 2008). In normoxia, the prolyl hydroxylase (PHD) enzymes Egl nine homologs 1–3 (EGLN1–3, Ivan and Kaelin, 2017) inhibit the Hif1α transcription factor by creating a proline-based binding site on Hif1α for the von Hippel–Lindau (VHL) E3 ubiquitin ligase (Gossage et al., 2015) targeting Hif1α for degradation. In hypoxia, this repression is relieved and Hif1α activates target-gene expression (Semenza et al., 2006). While the Hif1α pathway is well characterized molecularly, a key challenge in basic neuroscience is to identify the pathways that are affected by O2 homeostasis during brain maturation, such as those restraining the onset of neuronal differentiation or morphogenesis.
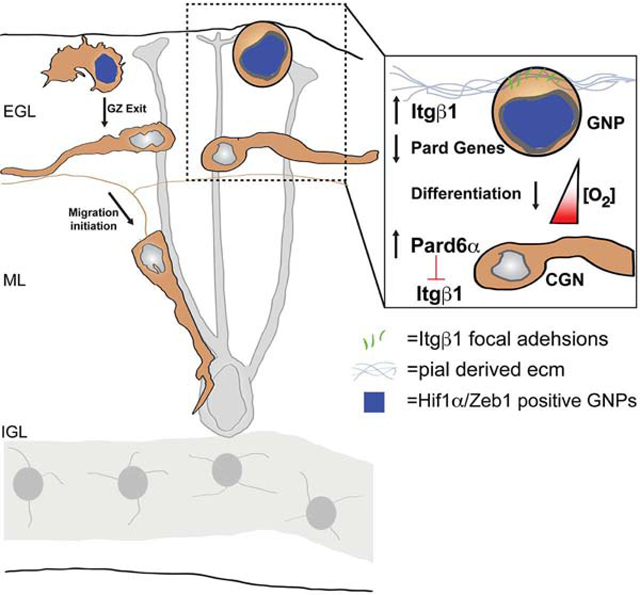
We hypothesized that O2 controlled the onset of CGN differentiation and GZ exit in a manner involving the regulation of cell polarity. We found that the postnatal mouse cerebellum is poorly vascularized and that granule neuron precursors (GNPs) in the external germinal layer (EGL) express Hif1α. In vivo Hif1α pathway manipulation modulates GNP proliferation and events occurring concomitantly with neuronal differentiation such as GZ exit and migration initiation. Epistasis analysis revealed that Hif1α controls the onset of neuronal polarization and GZ exit via the Zeb1 transcriptional repressor that suppresses the expression of the polarity genes Pard3 and Pard6α (Singh et al., 2016) to ultimately modulate levels of GNP integrin β−1 (Itgβ1) adhesion to extracellular matrix (ECM) that is enriched in their GZ niche. Thus, we provide an integrated model of how O2 controls polarity-dependent cell interactions with niche substrates, enhancing our understanding of normal brain development and the cell biological mechanisms that go awry when O2 homeostasis is perturbed.
RESULTS
Oxygen Tension and Hif1α Pathway Activation Change Dynamically in the EGL
To test the role of O2 homeostasis in GNP and CGN development, we examined features of the niche where these cells develop, like the relative O2 levels, vascularization timing and Hif1α expression. Organs and cells lower than 2% O2 can be identified by intraperitoneal pimonidazole injections. While pimonidazole adducts were detectable in the cerebellar cortex at postnatal day 5 (P5) and P9, pimonidazole binding was reduced at P15 in vivo (Figure 1A), suggesting that hypoxia is relieved later in development. Indeed, vascularization is low at P5 and P9 and there is a marked increase in the vasculature at P15 as demonstrated by immunostaining of the endothelial glucose transporter 1 (GluT1) in CLARITY cleared tissue (Figure 1B, GluT1 levels doubled in all layers from P5 to P15). We were curious if the relative vascular disparity in the EGL and molecular layer (ML) to the internal granule layer (IGL) was a local difference or if it extended across brain regions and iDisco+ cleared the GluT1 in postnatal hindbrain, midbrain, and cerebellum. We computationally analyzed the entire 3D volume of these regions and used the 100-μm region in the vicinity of an Atoh1EGFP/EGFP staining (Rose et al., 2009) to mark the location of immature CGNs in the EGL and ML. The 3D vascular volumes measured via Ilastik machine learning (Kreshuk et al., 2011) showed that the EGL and ML have half the vascular volume of not only the IGL but also the hindbrain and midbrain (Figure 1C, Supplemental Movie 1, [t-test vascular volume fraction EGL+ML vs. IGL P=0.0004, vs. Hindbrain=0.00004, vs. Midbrain=0.019]), indicating that brain regions near the cerebellum are well vascularized. Finally, we surveyed Hif1α expression: At P7, Hif1α staining is coincident with the proliferation marker Ki67 in the external granule layer (EGL) but is reduced in p27Kip1-positive postmitotic CGNs (Figure 1D). At P15, Hif1α is diminished in the cerebellar cortex. Peak Hif1α expression during GNP neurogenesis led us to examine if this period correlated to a hypoxic environment. Western blots of lysates from different developmental time-points with Hif1α and pimonidazole antibodies showed both hypoxia markers were abundant between P2 and P9 but abated after P9 (see Figure 1E for quantitation, 60±6.8% reduction in Hif1α expression compared to P7 levels, [t-test, P<0.0001], 59.7±12.5% reduction in pimonidazole values normalized to actin levels when compared to P7 values (t-test, P<0.05). Taken together, these results show proliferating GNPs expressing Hif1α reside in hypoxic regions and a vascularization burst occurs at P9, a time when CGN differentiation begins.
Figure 1. Cerebellar Hypoxia Accompanies GNP Proliferation.
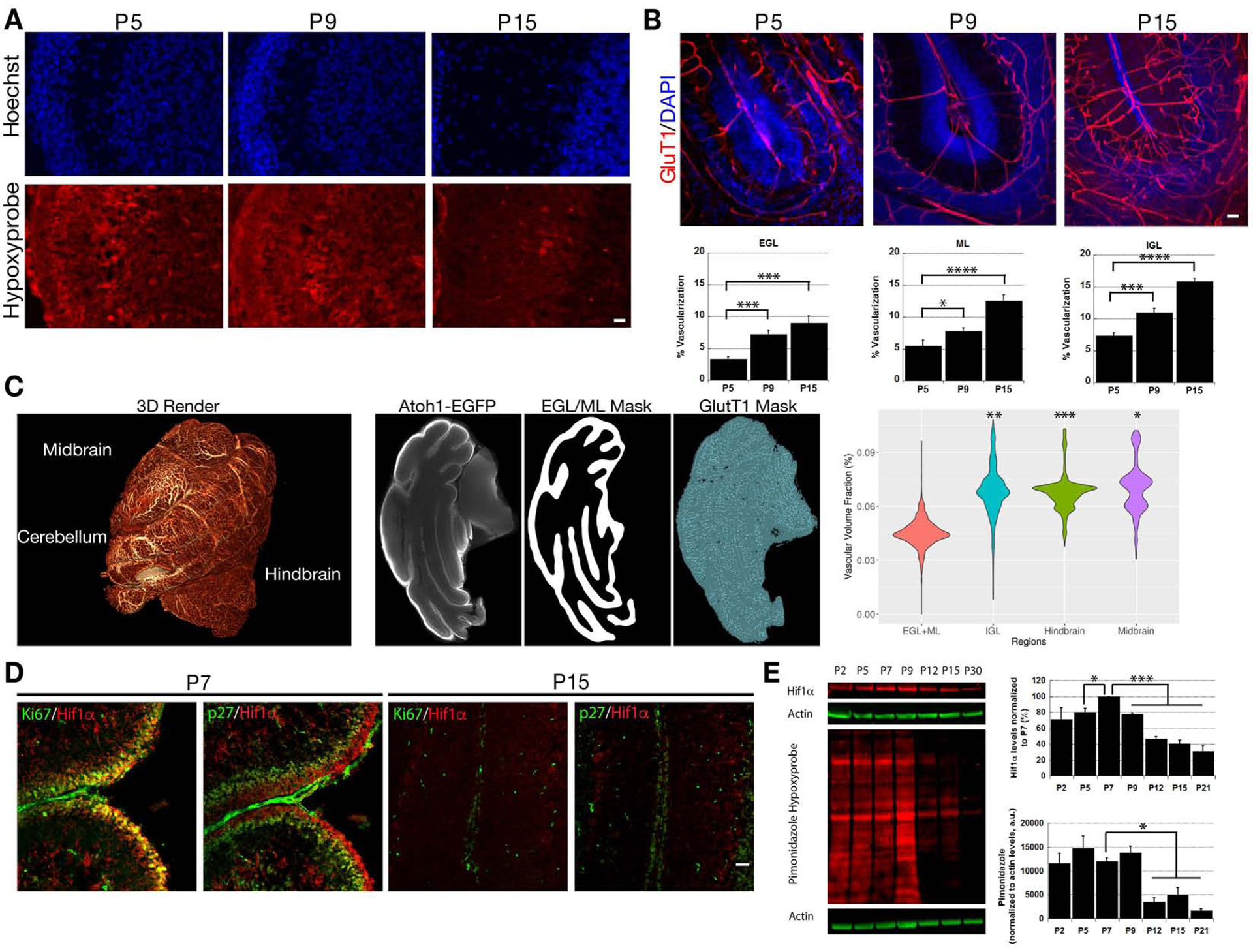
(A) Immunostaining of P5 and P9 cerebella reveals pimonidazole (red) binding in all layers that abates by P15. (B) GluT1 staining (red) is sparse at P5 and P9 but covers wider areas at P15. Bar charts show % vascularization of the EGL, ML and IGL layers that increased after P5 (t-test, P<0.05 vs. P5 levels). (C) Vascularization analysis in iDisco-cleared P7 cerebellum, hindbrain, and midbrain (see 3D rendering, left panel). The middle panels are representative examples of the Atoh1-EGFP signal used to segment the EGL/ML, an EGL/ML segmentation mask, and segmented vasculature. A violin plot compares the EGL/ML, hindbrain, and midbrain vascular volumes (n=3). (D) Immunostaining of P7 cerebella shows Hif1α (red) expression coincides with Ki67 expression (green) but is complementary to p27Kip1 expression (green). The Hif1α signal abates by P15. Scale bar=25 μm. (E) Western blots and bar diagrams at lower right reveal high Hif1α and pimonidazole adduct levels from P2 and P9 that abate after P9 (Hif1α t-test, P<0.001, Pimonidazole t-test, P<0.05). Actin (green) was used for normalization.
The Hif1α Pathway Modulates GNP Neurogenesis in Vivo
As Hif1α is expressed in Ki67-positive GNPs (Figure 1), we assayed if altering Hif1α pathway activity influenced their proliferation. Thus, we generated Atoh1-Cre:Hif1αflx/flx and Atoh1-Cre:VHLflx/flx mice that lacked Hif1α or VHL, respectively, in the CGN lineage. To test for the successful Hif1α deletion, we purified P7 CGNs and incubated them for 24h in 20% or 2% O2 . Hif1αflx/flx CGNs cultured in 2% O2 expressed Hif1α, but we detected a 75.6±5.8% reduction in levels of Hif1α in Atoh1-Cre:Hif1αflx/flx CGNs by Western blot (Figure 2A). We next found a small but consistent decrease in the cerebellar size of Atoh1-Cre:Hif1αflx/flx mice at P7, P10, and P15 when compared to control mice (Figure 2B, 15%, 11% and 9% volume change respectively for each timepoint; t-test P<0.01, 0.02 and 0.07 respectively). We analyzed if reduced cerebellar size was due to diminished GNP proliferation by 5-ethynyl-2’-deoxyuridine (EdU) incorporation and phospho- histone H3 (pH3) staining. While the number of EdU+ GNPs was slightly lower in Hif1α-deficient CGNs (Hif1αflx/flx: 268±17 cells/mm;Atoh1-Cre:Hif1αflx/flx 246±11 cells/mm; P > 0.05); the number of pH3+ cells was reduced by 23±6% (Figure 2C), suggesting that GNP proliferation was impacted. To assess actively cycling, proliferating GNPs, we calculated the ratio of cells that had been recently born but still capable for proliferation (EdU+/Ki67+ cells) and those that had already exited the cell cycle (EdU+/Ki67− cells). We found a 53±15% increase in EdU+/Ki67− cells in Atoh1-Cre:Hif1αflx/flx mice, indicating a larger fraction of Hif1α-deleted GNPs had exited cell-cycle and begun differentiation (t-test, P<0.05; Figure 2D,E). Hif1α pathway activation in Atoh1-Cre:VHLflx/flx mice led to increased EdU incorporation into GNPs (VHLflx/flx: 218±1 cells/μm; Atoh1-Cre:VHLflx/flx: 274±3 cells/μm; P < 0.01) and in the number of pH3+ cells in the EGL (39±5% increase), as compared to that of VHLflx/flx mice (t-test, P<0.02; Figure 2C and 2D). There were fewer cell-cycle exit events in the EGL of Atoh1-Cre:VHLflx/flx mice because the percentage of EdU+/Ki67− cells was reduced by 30±5% (t-test, p<0.02; Figure 2E). The increase in Hif1α-pathway activity in Atoh1-Cre:VHLflx/flx mice is accompanied by a slight alteration in apoptosis in the P7 cerebellum, mainly in the IGL (Supplemental Figure 1A). These results show that not only does the cerebellum have a changing O2 environment in development but the Hif1α pathway functions to adjust the levels of actively cycling progenitor-cells within the EGL GZ niche.
Figure 2. Altering the Hif1α Pathway Tunes GNP Proliferation.
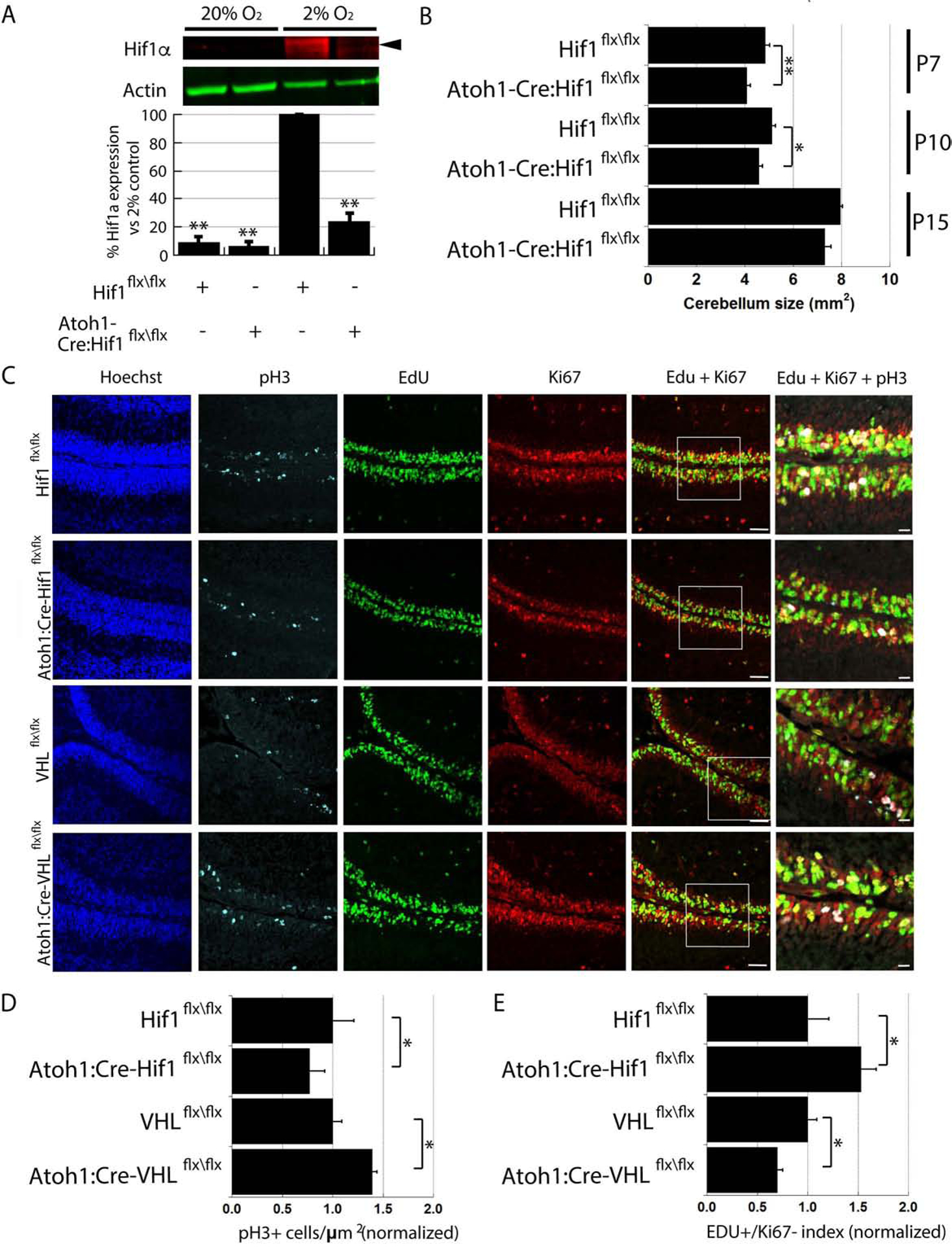
(A) Western blots of Atoh1-Cre:Hif1αflx/flx CGNs grown in culture in 2% O2 show 75.6±5.8% (mean±sem) reduced Hif1α (red [t-test, P<0.01]) compared to Hif1αflx/flx CGNs. Actin (green) is loading control. (B) Graphs show Atoh1-Cre:Hif1αflx/flx cerebella are smaller than controls (P7 Hif1αflx/flx: 4.8±0.2 mm2, Atoh1-Cre:Hif1αflx/flx 4.1±0.2 mm2, t-test, P < 0.01; P10 Hif1αflx/flx: 5.1±0.1 mm2, Atoh1-Cre:Hif1αflx/flx : 4.6±0.2 mm2, t-test, P < 0.05; P15 Hif1αflx/flx: 8.0±0.1 mm2, Atoh1-Cre:Hif1αflx/flx : 7.3±0.3 mm2, t-test, P < 0.07). (C) pH3 (cyan), EdU (green), and Ki67 (red) immunostaining in P7 cerebella from control, Atoh1-Cre:Hif1flx/flx and Atoh1-Cre:VHLflx/flx mice. (D) pH3-positive (pH3+) GNPs decrease 23±6% in Atoh1-Cre:Hif1flx/flx mice and increase 39±13% in Atoh1-Cre:VHLflx/flx mice compared controls (P < 0.05). Ratio of EdU+/Ki67− cells increased 53±15% in Atoh1-Cre:Hif1αflx/flx mice (P < 0.02) and decreased 30±5% in Atoh1-Cre:VHLflx/flx mice (P < 0.05).
The Hypoxia and Hif1α Pathway Regulate the Timing of CGN GZ Exit and Migration
Having demonstrated that the Hif1α pathway influences the proportions of proliferating GNPs and differentiating CGNs residing in the EGL, we next sought to investigate if alterations in O2 level or Hif1α also regulated events that occur after differentiating CGNs become postmitotic, such as GZ exit or the initiation of radial migration. For these experiments, where migration is linked with CGN maturation, we used ex vivo organotypic cerebellar slices that were electroporated with an H2B-mCherry–labeled expression construct and cultured under a range of O2 levels including: standard culture condition of 20% O2, hypoxia (2%, 5%, 10% or 15% O2), or hyperoxia (40% O2). Previous work established that GNPs in ex vivo cerebellar slices exhibit differentiation and migration kinetics indistinguishable to those seen in vivo (Famulski et al., 2010;Ruiz de Almodovar et al., 2010). After incubating slices ex vivo for 24 or 48h, we analyzed the distribution of CGNs through the GZ and other cerebellar layers by measuring the distance of H2B-mCherry–positive CGNs from the pial surface of the slices (Figure 3A). Hypoxia increased the numbers of cells remaining in their GZ depending on the relative O2 level (e.g. a 2–40% gradient), while higher O2 levels supported maximal displacement toward the IGL. At 24h, cells in slices incubated at 20% O2 showed an average distance from the pial surface of 61.5±2.1 μm , While cells in slices incubated at 2% O2 displayed an average distance of 39.6±4.1 μm (t-test, P<0.002; Figure 3A). At 48h there was a significant difference between average distances of cells in slices incubated at 2% O2 (66.1±7.0 μm) and 20% O2 (93.0±5.3 μm, t-test, P<0.01). Slices incubated in 40% O2 trended to more IGL-directed migration (100.7±0.4 μm), but replicate variability diminished statistical significance (t-test, P<0.15, vs 20% O2). To examine CGN migration kinetics in hypoxia, we used long-term time-lapse microscopy to longitudinally image CGN motility in ex vivo slices, starting 18h after the slices were placed in culture (Figure 3B). Time-lapse imaging revealed that higher amounts of GZ resident cells in slices cultured in 2% O2 than in those cultured in 20% O2 (see Supplemental Movies 2 & 3). Dynamic layer-occupancy calculations showed that while the population of cells in the outer EGL (oEGL) of slices cultured in 20% O2 decreased over the observation period, the rate of oEGL exit was diminished in 2% O2. Similarly, the increases in layer occupancy in the ML and IGL were diminished in 2% O2, and the efficiency of motility (measured in terms of how many neurons persisted along migration directions) was most disrupted in these layers (Figure 3B).
Figure 3. Oxygen Tension Tunes CGN GZ Exit and Migration.
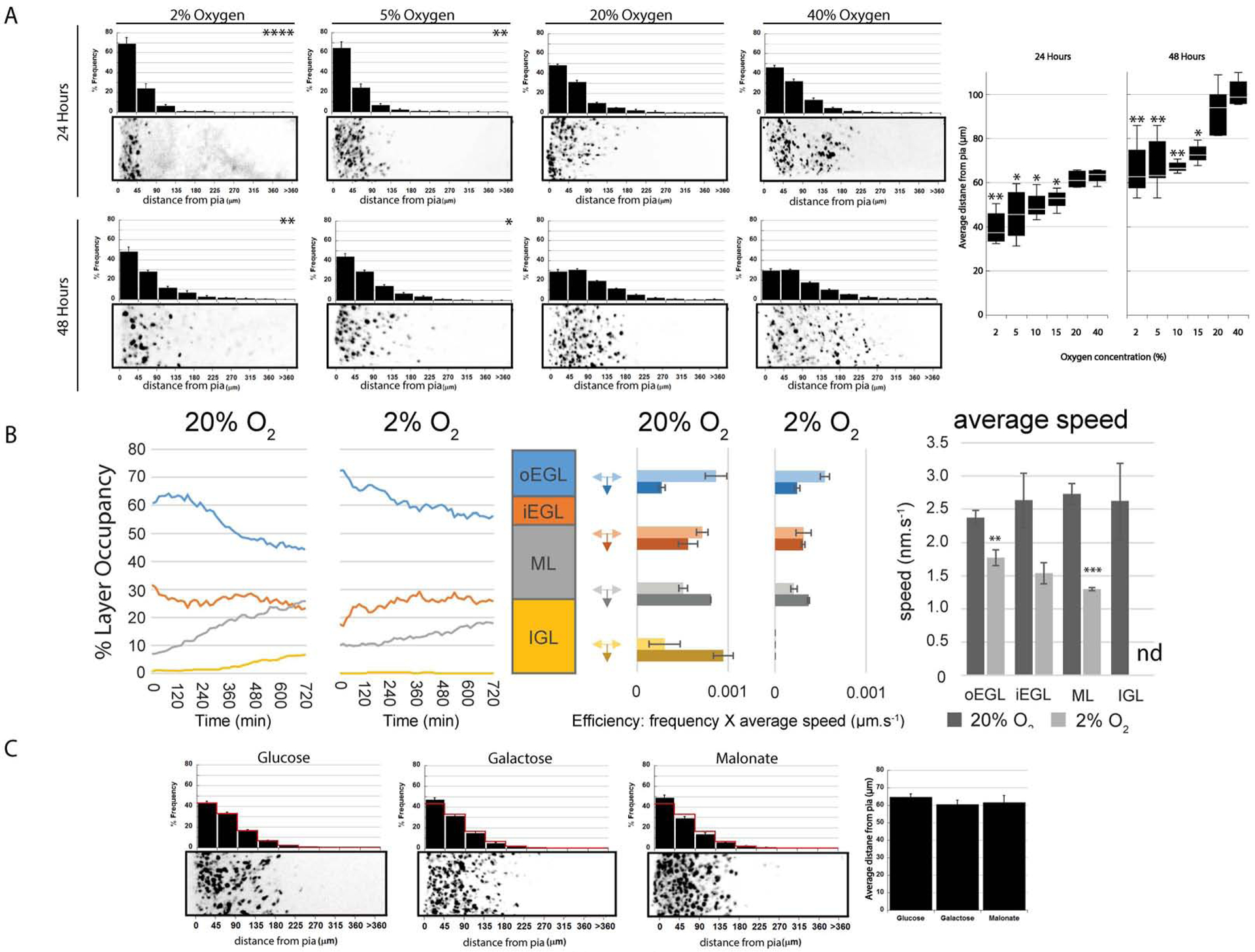
(A) Ex vivo cerebellar slices were incubated 24 or 48h at listed O2 levels and the distance of labeled CGNs from the pial surface (left in each image) analyzed. The histograms above each image show a binned distance distribution (n > 1,300 [24h] or >2,000 [48h] cells analyzed); χ2 test showed that 2% or 5% O2 decreased migration (P < 0.04 vs. 20% O2). Box plot (far right) shows over a broader range of O2 levels below 20% migration was decreased (P < 0.03 for all). (B) Analysis of ex vivo slices live-cell imaging at 20% or 2% O2. The left graph shows % layer occupancy over time in outer EGL (blue), inner EGL (orange), ML (grey), and IGL (yellow). The middle plots are migration efficiency plots of migration speed frequency for inward or tangential migration for each layer. (C) Inhibiting glycolysis or the TCA cycle had no effect on CGN migration (; , P > 0.05; , P > 0.05).
Hypoxic insults reduce the ability of cells to produce energy equivalents through oxidative phosphorylation and, therefore, they increase the abundance of glycolytic enzymes (Knobloch and Jessberger, 2017). Because less efficient energy production through glycolysis predominates in hypoxia, we wondered if reduced energy production limited GZ exit in hypoxia. Accordingly, we incubated ex vivo slices in medium supplemented with glucose , as the control condition; in medium supplemented with galactose to prevent energy production through glycolysis ; and in the presence of 1 mM malonate (; Figure 3C), a succinate dehydrogenase TCA electron transport chain inhibitor. Inhibiting energy production through glycolysis or oxidative phosphorylation alone had no influence on migration.
Given that hypoxia regulated either the timing or kinetics of GZ exit and subsequent migration to the IGL, we next assessed the role of the Hif1α pathway. We performed Hif1α gain-of-function studies by ectopically expressing a hydroxylation-deficient, and thereby stabilized, form of Hif1α (Hif1αHD) (Kaelin, 2008;Kaelin and Ratcliffe, 2008) in GNPs in ex vivo cerebellar slices for 48h. Expression of Hif1αHD in GNPs led to a dose-dependent increase in GZ occupancy at 48h (Figure 4A upper-left panels, ; 1 μg/μ L Hif1αHD , P < 0.05; 4 μg/μL Hif1αHD , P < 0.001 χ2 test and P,0.02 t-test). To characterize Hif1 function we introduced inactive or active Cre recombinase into the Hif1αflx/flx GNPs. Not only did electroporation of Cre recombinase into ex vivo slices from Hif1αflx/flx mice lead to more migration to the IGL of CGNs in slices cultured under standard O2 conditions (Figure 4A, lower-left panels), Hif1α deletion also reversed enhanced GZ occupancy seen in hypoxia (Figure 4A, lower-right panels, at 2% O2 , , χ2 and t-test both P < 0.0002). Finally, we tested if enhanced migration of Hif1α-deficient CGNs represented an alteration in the timing of migration or enhanced general motility. Longitudinal time-lapse imaging starting 18h after the slices were placed in ex vivo culture revealed that most Hif1α-deficient CGNs had already undergone GZ exit by the start of imaging (Figure 4B, dynamic layer-occupancy graph in left panel, see Supplemental Movies 4 & 5) and that these cells had no changes in average migration speed (Figure 4B, migration speeds in right panel). Thus, Hif1α deletion not only reduced the proportion of GNPs in the EGL in vivo but also accelerated the onset of GZ exit and migration initiation in post-mitotic CGNs.
Figure 4. Hif1α Deletion Regulates GZ Exit.
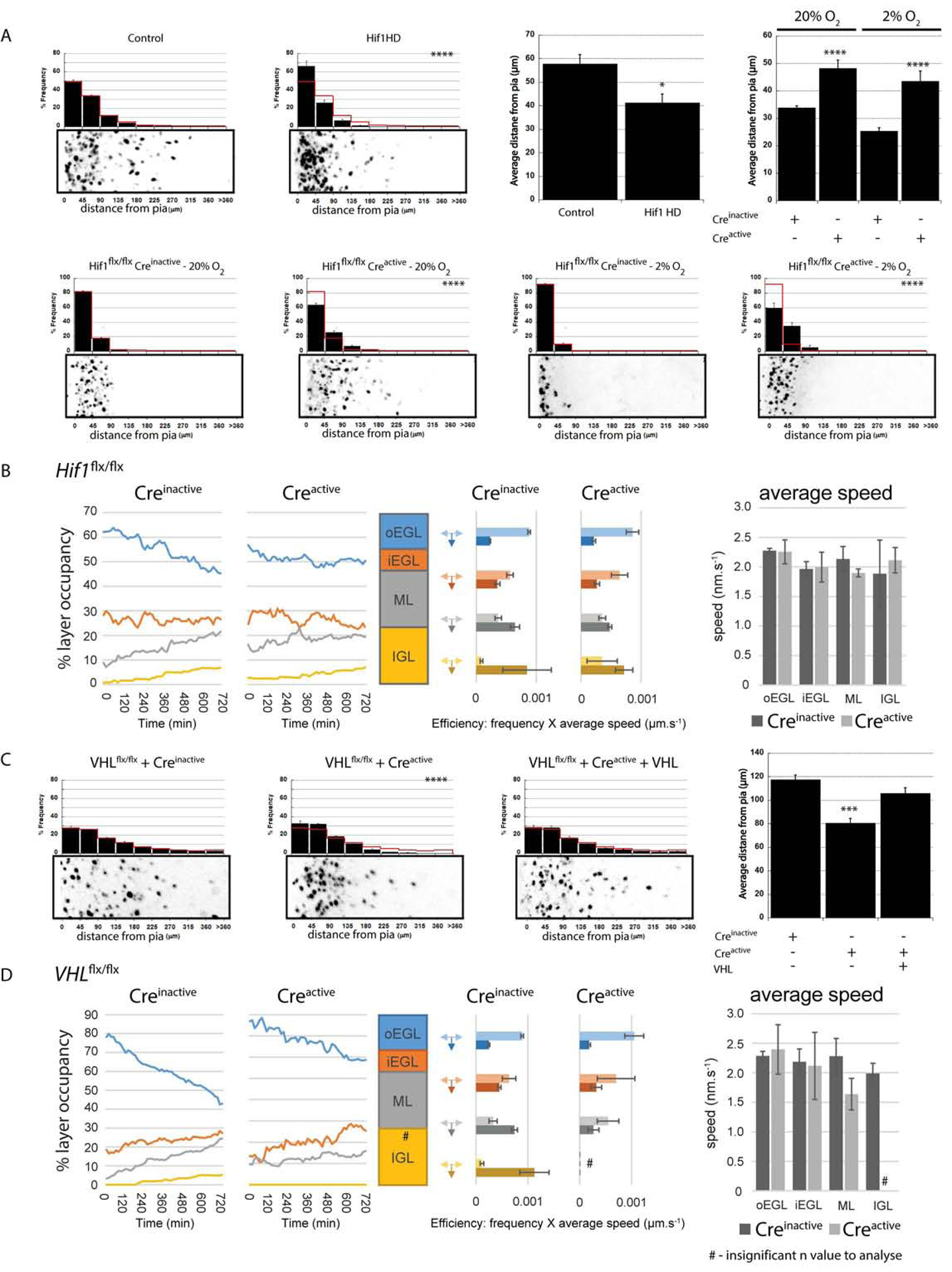
(A) Hydroxylation-deficient Hif1α (Hif1 HD) expression impairs ex vivo CGN migration (41.7±3.6 μm vs. 58.2±4.0 μm in a LacZ controls [χ2 test, P<0.0001, 48h t-test P<0.02]). Expressing Cre recombinase in Hif1αflx/flx cerebella led to precocious migration in 20% O2 (, , P < 0.001) and rescued the 2% O2 migration deficit (, , χ2 and t-test P < 0.0002). (B) Longitudinal time-lapse analysis of ex vivo migration kinetics in Hif1αflx/flx CGNs transfected with Creinactive or Creactive (see Fig. 3B for plot labels). Layer occupancy plots for Hif1αflx/flx CGNs and Hif1α−/− CGNs shows Hif1α−/− CGNs leave the GZ early, but Hif1αflx/flx CGNs quickly catch up (C) Expressing Cre recombinase in VHLflx/flx cerebella reduced ex vivo CGN migration (, , P < 0.0001 [χ2 test] or P < 0.001 [t-test]), but was restored by VHL cDNA (; P < 0.0001 [χ2 test] or P < 0.004 [t-test] vs Creactive). (D) Longitudinal time-lapse analysis of ex vivo migration kinetics in VHLflx/flx CGNs transfected with Creinactive or Creactive (see Fig. 3B for plot labels). Percentage layer occupancy plots show VHL−/− CGNs have delayed GZ exit compared to VHLflx/flx CGNs.
Under normoxic conditions, Hif1α is degraded through the action of the VHL ubiquitin ligase and PHD enzymes, which are mediators of the O2 homeostasis machinery (Kaelin, 2008;Kaelin and Ratcliffe, 2008). Thus, we assayed if deleting VHL or PHDs regulated GZ exit or migration initiation. We electroporated P7 EGLs of cerebella harvested from VHLflx/flx or EGN1flx/flx mice with inactive or active Cre recombinase and examined the distribution of labeled CGNs. As with the ectopic expression of Hif1α or 2% O2 culture, GZ occupancy was increased when we deleted VHL or EGLN1 (; ; P < 0.001 [χ2 test] or P < 0.001 [t-test]) (Figure 4C and Supplemental Figure 2 for EGLN1 deletion or gain of function). In these cases, GZ exit and subsequent CGN displacement towards the IGL was restored by introducing VHL or EGLN1 cDNA, respectively (; P < 0.001 [χ2 test] and P < 0.004 [t-test] vs Creactive [Figure 4C]). Deletion of Hif1α in VHL-deficient CGNs restored displacement to the IGL to control levels, confirming the Hif1α dependence of the VHL phenotype (Supplemental Figure 3A). Longitudinal time-lapse imaging of VHL-deficient cells, starting 18 h after they were placed in ex vivo culture, revealed persistent GZ occupancy and delayed transition to the ML and IGL (Figure 4D; see dynamic layer-occupancy graph in left panel, see Supplemental Movies 6 & 7) with no changes in overall cell motility, showing that VHL loss of function delays the onset of GZ exit and the initiation of radial migration. Taken together, these results show that the low O2 levels in the postnatal cerebellar environment and, more specifically, the Hif1α pathway modulate progenitor-cell neurogenesis in vivo but also control the timing of the exit of GNPs from the germinal niche to initiate radial migration.
Given the apparent dual role of the Hif1α pathway in modulating GNP neurogenesis and the ensuing timing of GZ exit and migration initiation, we also investigated crosstalk with the Sonic hedgehog (Shh) mitogen signaling cascade, a primary regulator of GNP neurogenesis. To delete VHL alone or in combination with the Patched (Ptch) Shh receptor, we electroporated P7 EGLs of cerebella harvested from VHLflx/flx or VHLflx/flx;Ptchflx/flx mice with inactive or active Cre recombinase. Analysis of labeled CGN distribution in ex vivo slices revealed that Ptch deletion does not enhance VHL loss of function phenotypes (Supplemental Figure 3B). VHL and Ptch deletion activates the Hif1α or Shh signaling respectively as both are negative regulators of their respective pathways, therefore no additional or synergistic effects in this epistasis experiment place Hif1α- and Shh-signaling in a common pathway modulating GNP neurogenesis. To address the Shh-Hif1α relationship in the context of hypoxia, we performed an additional pharmacological epistasis experiment using a small molecule inhibitor of GLI1 and GLI2 transcription factors (e.g. GANT68) that are the transcriptional effectors of the canonical, transcriptional arm of the Shh signaling cascade. Inhibition of GLI transcription factors does not rescue the 2% O2 GZ occupancy phenotype (Supplemental Figure 3C), showing that while Shh and Hif1α are part of a similar genetic pathway controlling GNP proliferation, hypoxia can regulate the timing of CGN migration independently of Shh signaling.
The Hifα Pathway Controls CGN Differentiation, Migration timing, and Polarity via Zeb1
Having learned that the Hif1α pathway supports GNP proliferation while restraining GZ exit and CGN migration initiation, we sought to identify molecular targets in order to determine how hypoxia or the Hif1α pathway mechanistically controls this balance. Our first clue regarding the mechanism came from comparing the transcriptomes of purified CGNs from P0 and P15 to CGNs cultured under hypoxia (Supplemental Figure 4). These analyses showed that P0 CGNs and purified CGNs cultured in hypoxia exhibited similar degrees of immature gene expression, as compared to mature CGNs purified at P15, suggesting that low O2 levels maintained CGNs in an immature, undifferentiated state. We gained insight into how O2 levels and the Hif1α pathway control CGN differentiation by examining their relation to the Zeb1 transcription factor, an inhibitor of CGN differentiation (Singh et al., 2016). Interestingly, Zeb1 expression is not only activated by ischemic insults in the adult mouse brain (Bui et al., 2009) but is also activated Hif1α in renal carcinoma (Krishnamachary et al., 2006). We reasoned that Zeb1 might be involved in the O2-homeostasis regulation of GNP differentiation or GZ exit. We first examined Zeb1 expression via immunostaining in ex vivo cerebellar slices where O2 levels were manipulated. Slices incubated in 2% O2 for 48h showed an abundance of Zeb1-positive cells in the EGL GZ niche, when compared to slices incubated in 20% O2 (Figure 5A). Next, we prepared RNA from P7 GNPs nucleofected with LacZ (control), Hif1α, or Zeb1 expression vectors and qRT-PCR analysis of CGNs overexpressing Hif1α not only confirmed the expected increase in known Hif1α targets such as Bnip3, Hk2, VEGFa, PHD2, and PHD3, but also showed a marked increase in Zeb1 transcript expression, implicating Zeb1 as a mediator of Hif1α regulation of GNP maturation (Figure 5B). We also observed reduced NeuN expression and enhanced Zeb1 expression in CGNs from P7 Atoh1-Cre:VHLflx/flx mice, where the Hif1α pathway is activated by the loss of VHL (Supplemental Figure 1B). Indeed, Zeb1 expression continues in the IGL of VHL-deficient CGNs at P15, a time-point at which its expression is extinguished in control animals (Supplemental Figure 1C).
Figure 5. Zeb1 is Activated by Hif1α.
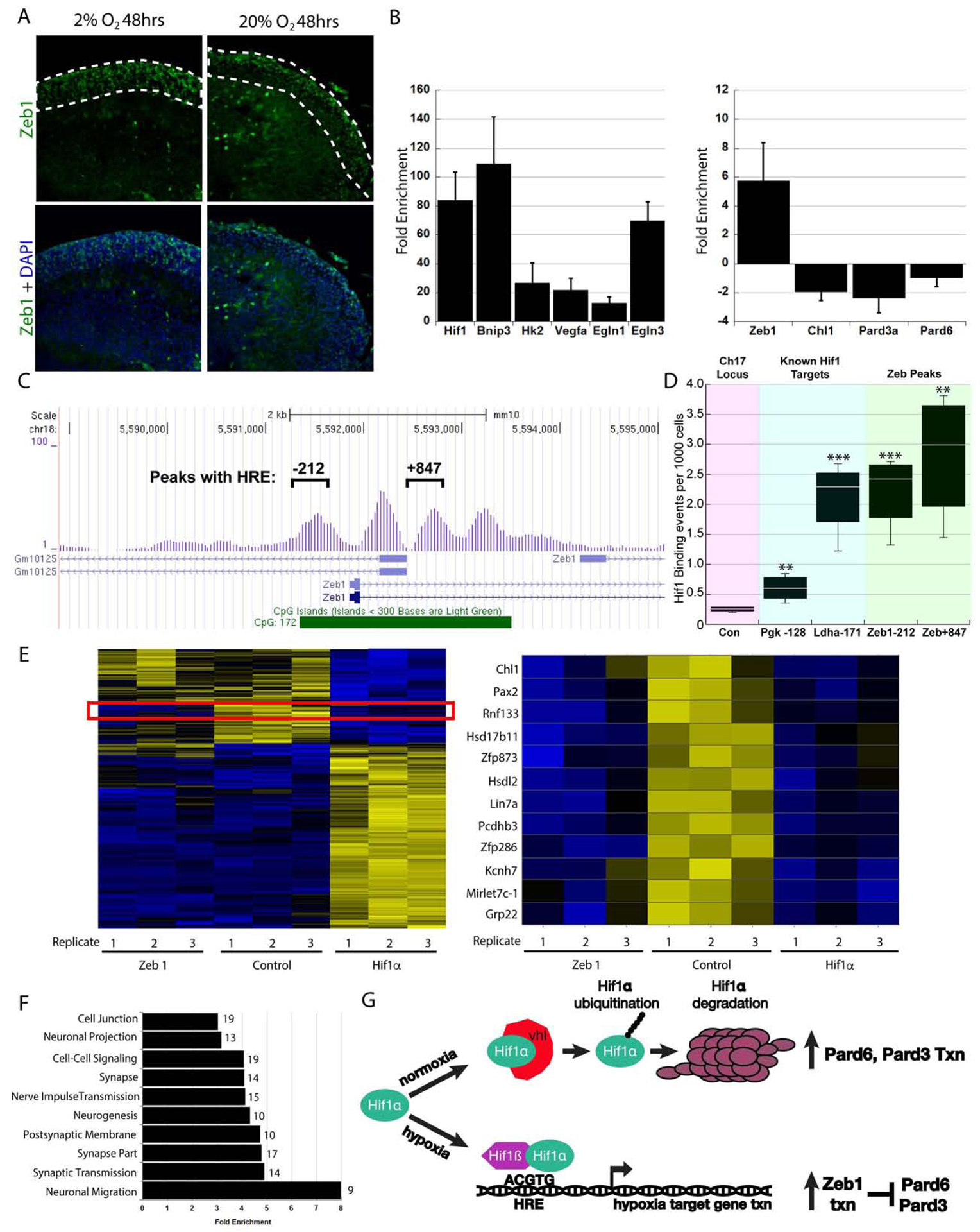
(A) Immunostaining of ex vivo cerebellar slices cultured in 2% O2 for 48h shows increased Zeb1 staining (green) in the EGL. (B) qRT-PCR of granule cells overexpressing Hif1αHD shows upregulation of Hif1α targets and Zeb1 while Pard3, Pard6α and Chl1 were repressed. (C) Visual representation of Hif1α ChIP-seq enrichment in the vicinity of the Zeb1 locus. Exons are blue rectangles and line with hatched arrows are introns. Hif1α peaks at −212 and +847 relative to the Zeb1 TSS harbor conserved HREs. (D) Hif1α ChIP PCR validation of −212 and +847 HREs. Relative binding graph (events per 1000 cells) to control locus, Pgk and Ldha genes, and Zeb1 −212 and +847. (E) Microarray Heat maps of CGNs electroporated with LacZ (control), Zeb1, or Hif1α. A red box marks the transcript subset repressed by Zeb1 and Hif1α (magnified at right) (F) Cluster analysis using gene ontology terms and DAVID algorithms for gene classes regulated by Zeb1 and Hif1α. (G) Model for the genetic interactions between Hif1α, Zeb1 and the Pard proteins in hypoxia or normoxia
We assessed if Hif1α regulation of Zeb1 was direct by determining if Hif1α binds the Zeb1 locus. Hif1α CHiP-seq performed using chromatin harvested from FACS isolated Atoh1-EGFP positive GNPs revealed four peaks of Hif1α enriched reads denoting direct binding to a ~2.0 kb region surrounding the transcriptional start site of the Zeb1 gene (Figure 5C). Two of these peaks harbor evolutionarily conserved Hif1α response elements (e.g., mouse vs. human) and were subjected to Hif1α CHiP-PCR validation. Importantly, the peaks centered at −212 and +847 had equal or better binding than known/validated Hif1α binding site in genes like Vegf, Ldha and Pgk1 that also harbored peaks in the Hif1α CHiP-Seq dataset (Figure 5D, control genomic region vs. −212 and +847 TTest P value < 0.01). Taken together these results suggest that hypoxia regulates Zeb1 expression by direct binding of Hif1α to the Zeb1 promoter.
To examine how the Hif1α pathway controlled CGN differentiation, we performed a comparative analysis of the transcriptomes of cells overexpressing Hif1α or Zeb1 via Affymetrix DNA arrays to gain insight into common mechanisms of GNP differentiation regulation. Our global analysis of transcripts revealed distinct patterns of gene expression, as would be expected for cells overexpressing the transcriptional repressor Zeb1 or the transcriptional activator Hif1α. For example, the expression of a large group of glucose metabolism genes that are activated by Hif1α was unchanged in both the LacZ-expressing control and Zeb1-overexpressing cells (Figure 5E, left panel). Nevertheless, a group of transcripts were repressed by both Zeb1- and Hif1α-expressing CGNs. Interestingly, two of the top 5 hits included two previously characterized Zeb1 targets, Chl1 and Lin7a, that we showed can promote CGN differentiation (Singh et al., 2016) (Figure 5E). By clustering a large group of transcripts that are downregulated by both Zeb1 and Hif1α to biological processes, using gene ontology terms, we discovered that many of the genes repressed in both sets of transcriptomes were related to classical hallmarks of neuronal differentiation, such as neurogenesis, neuronal projection, and synaptic maturation, with the highest fold enrichment occurring for genes already known to be necessary and sufficient for neuronal migration (DCC, CTTNBP2, NDE1, ATOH1, NAV1, NEUROD4, SYNE1, NEUROG2, and CHL1 [Figure 5F]). qRT-PCR validation also showed that Zeb1 target genes such as Chl1, Pard3, and Pard6α were downregulated in the Hif1α gain-of-function study (Figure 5B). Indeed, reduced Chl1, Pard3, and Pard6α expression was also observed in CGNs from P7 Atoh1-Cre:VHLflx/flx mice (Supplemental Figure 1B). We recently showed these genes to be the most critical Zeb1-repressed targets that control GNP polarization, differentiation, and GZ exit (Singh et al., 2016). Taken together, these results show that Hif1α negatively regulates a group of genes implicated in neuronal maturation, and they suggest a working model that Hif1α mechanistically mediates the timing of GZ exit via Zeb1 inhibition of polarity or migration genes (Figure 5G).
Since Zeb1 is necessary and sufficient to maintain the GNP state and GZ occupancy in a manner requiring Pard-complex transcriptional repression (Singh et al., 2016), we sought to assess if Hif1α controlled CGN migration initiation through Zeb1 or its target genes. To do this, we first tested if Zeb1 loss of function could rescue the observed 2% O2 GZ exit delay. Electroporation of a validated, published mir30 based Zeb1 shRNA into the EGL of ex vivo cerebellar slices restored CGN displacement to the IGL in 2% O2, when Hif1αHD was overexpressed or when VHL was deleted via Cre-mediated excision, suggesting that Zeb1 acts downstream of VHL and Hif1α and mediates the 2% O2 migration delay (Figure 6A). Finally, we co-electroporated a Zeb1 cDNA and a Cre-encoding expression vector into cerebellar slices prepared from Hif1αflx/flx cerebella. Zeb1 overexpression prevented precocious GZ exit and migration elicited by Hif1α deletion, showing that elevated Zeb1 expression is sufficient to restore GZ occupancy to Hif1α-deficient cells (Figure 6A). Not only do these results show that Zeb1 expression is activated by both hypoxia and elevated Hif1α activity, but epistasis analysis reveals that Zeb1 is necessary for Hif1α to promote GNP GZ occupancy.
Figure 6. Zeb1 Loss of Function or Pard Complex Gain of Function Rescues Hypoxia and Hif1α-Pathway Inhibition of GZ Exit.
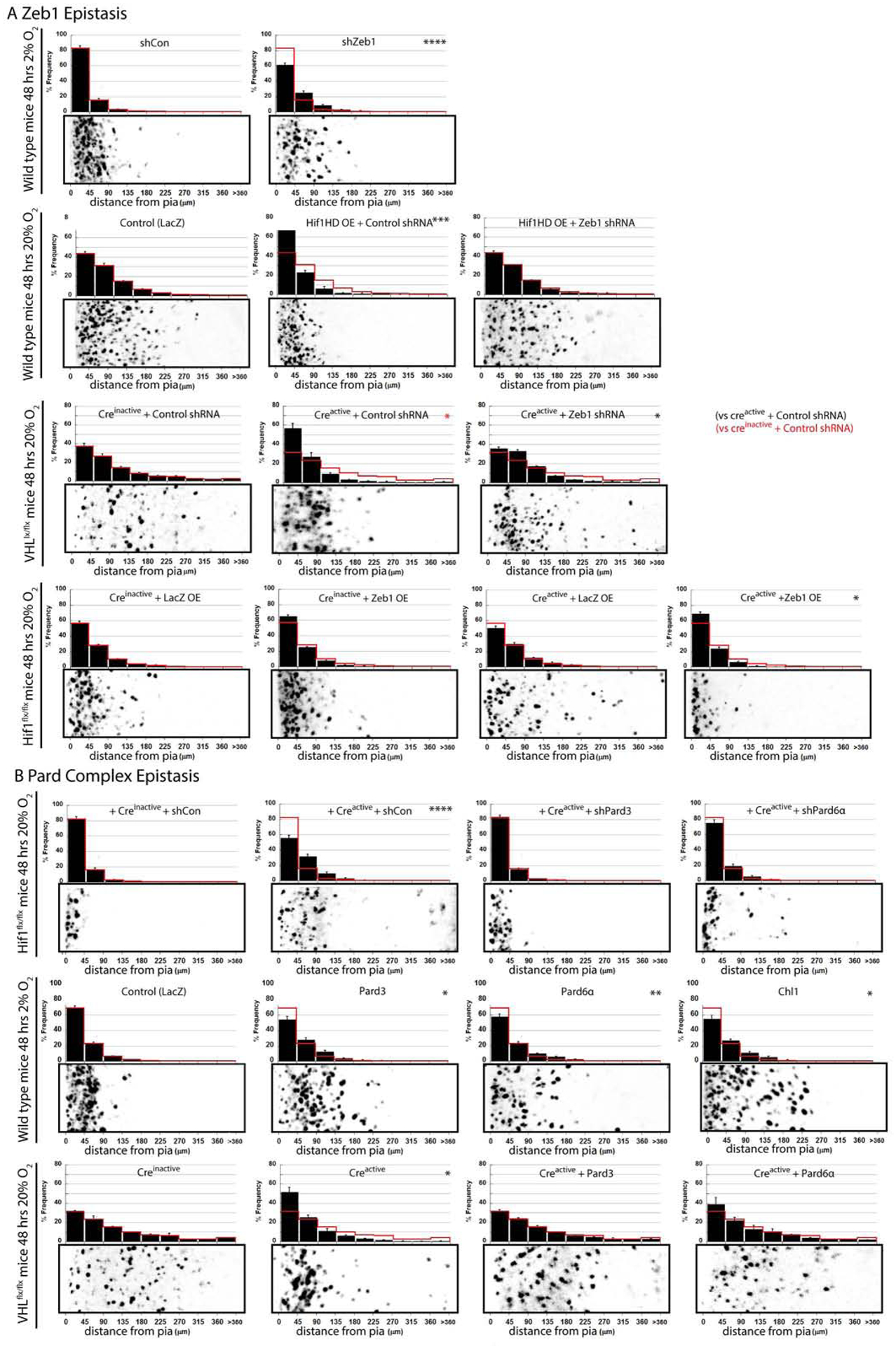
(A) Zeb1 silencing rescues O2 migration deficit (shControl at 20% : shControl at 2% ; shZeb1 at 2% ; t-test and χ2 test: P < 0.001), Hif1αHD over-expression (; Hif1αHD ; Hif1αHD ; t-test and χ2 test: P < 0.01) or VHL deletion (; ; ; χ2 test: P<0.02). Zeb1 expression inhibited Hif1α deletion precocious migration (; (t-test; P<0.03); ; ; t-test: P < 0.001, χ2 test; P<0.03). (B) Silencing Pard3 or Pard6α in 2% O2 abolished the Hif1α deletion rescue of migration in hypoxia (; ; , t-test and χ2 test: P < 0.001; , t- test and χ2 test: P < 0.01). Pard3, Pard6α, or Chl1 expression rescued the 2% O2 migration deficit (; , t-test and χ2 test P < 0.05; , t-test and χ2 test P < 0.01; , t-test and χ2 test P < 0.05) Pard6α and Pard3 expression rescued the VHL- deletion GZ exit phenotype (; ; t-test and χ2 test: P<0.01; ; t-test and χ2 test: P>0.3; ; t-test and χ2 test: P>0.9)
Given that Hif1α pathway activation activated Zeb1 expression and suppressed Zeb1 target-gene expression (Figure 5B,G), we assayed if Zeb1 downstream targets (Pard6α, Pard3, and Chl1, Singh et al., 2016) were involved in Hif1α pathway modulation of GZ occupancy or the 2% O2 delay of displacement to the IGL. Silencing the Pard-complex proteins Pard6α or Pard3 in Hif1α-deficient CGNs inhibited the displacement to the IGL seen in ex vivo slices incubated in 2% O2 where Hif1α had been genetically deleted (Figure 6B), showing that cell polarity gene function is required for GZ exit to proceed when CGNs are relieved of Hif1α pathway–activity in hypoxia. We next tested if Pard6α, Pard3, or Chl1 gain of function altered displacement to the IGL in cerebellar slices where the Hif1α pathway was activated by hypoxia, Hif1α gain of function, or VHL deletion (Figure 6B). While LacZ-expressing CGNs remained in the EGL GZ under all three experimental conditions, Pard6α, Pard3, and Chl1 rescued displacement to the IGL in all three modes of Hif1α pathway activation (Figure 6B; see Supplemental Figure 5 for Hif1αHD overexpression experiment plus compiled analysis of epistasis experiments and Supplemental Figure 2 for similar experiments in EGLN1-deficient CGNs). Finally, we performed a structure function analysis on the Par6α protein to determine if the interaction with classical binding partners is required for enhanced polarity signaling to rescue a hypoxia phenotype. Singly introducing a panel Pard6α cDNA mutants into ex vivo slices cultured at 2% O2 revealed that the Pard6α PDZ domain, the aPKC- and Pard3-binding sites were essential for Pard6α to rescue delayed GZ exit, suggesting Pard6α acts in a concert with aPKC, Pard3 and ligands for its PDZ domain to support the initiation of CGN migration (Supplemental Figure 6). Surprisingly, a nuclear localization signal was also essential for Pard6α to rescue delayed CGN migration, however the CRIB Cdc42 binding site was dispensable. The results of these epistasis and structure-function studies show that Hif1α and hypoxia restraint of GNP differentiation involves functional antagonism of neuronal polarization through a subset of classical Pard complex components. Enhanced polarity protein expression, mimicking the natural rise in polarity gene expression accompanying CGN differentiation, restores GZ exit even in the presence of high Hif1α pathway activity. Therefore, cerebellar hypoxia activates the Hif1α pathway, which in turn stimulates Zeb1-mediated Pard-complex repression, thus gating the exit of GNPs from their GZ niche.
Antagonism Between Hif1α-Zeb1 and the Pard Complex Controls GZ Occupancy via Integrin Extracellular Matrix Adhesion
Having found that Hif1α and Zeb1 cooperate to inhibit Pard complex–dependent GZ exit, we sought the downstream mechanisms by which O2 homeostasis controlled GZ occupancy. Zeb1 is a key regulator of epithelial mesenchymal transition (EMT) in epithelia and developing GNPs. In many EMT paradigms, extracellular matrix (ECM) contacts driven by Itgβ1 receptors are favored over cell-cell adhesions such as tight or adherens junctions that control morphogenic movements of these cells (Baum et al., 2008;Shook and Keller, 2003). Spurred by classical electron microscopy studies that found that GNPs maintain extensive contacts with the pial-derived ECM until they become postmitotic (Hausmann and Sievers, 1985) and control GNP mitogen responses (Blaess et al., 2004), we investigated if integrin receptors were involved in GNP GZ exit. While Zeb1 functions as a transcriptional repressor, we noted a 5.5 fold increase in Itgβ1 mRNA expression in Zeb1-overexpressing CGNs (Figure 7A). We also surveyed the expression of Itgβ1 in the cerebellum: at P7, Itgβ1 expression was high in oEGL GNPs that co-expressed Zeb1 as well as focal adhesion kinase (FAK) phosphorylated on residue tyrosine 397 (phospho–FAK Y397), a marker reporting integrin activation and high mechanical tension when cells interact with ECM (Paszek et al., 2005;Figure 7B). Given that Hif1α activates Zeb1 expression, we tested if hypoxia or Hif1α pathway activation affected integrin activity in GNPs or CGNs. Compared to control cells incubated under standard culture conditions (117.1±21.1 cells/mm2 cell density), purified CGNs incubated in 2% O2 bound more to a pure laminin substrate (179.0 cells/mm2 cell density, t-test, P< 0.0002), an Itgβ1 ECM ligand found at a high level in the basal lamina produced by pial fibroblasts (Figure 7C). Moreover, phospho–FAK Y397 was more abundant in vivo in the EGLs of mice where VHL had been conditionally deleted via Atoh1-Cre than in the EGLs of control mice, suggesting that integrin signaling is elevated when the Hif1α pathway is activated (Figure 7D). The potential correlation between hypoxia or high Hif1α pathway activity with elevated integrin adhesion led us to test if Itgβ1 was involved in GZ exit. Cre-mediated deletion of floxed Itgβ1 alleles rescued GNP GZ exit defects in ex vivo slices cultured in 2% O2 (Figure 7E, , , t-test, P<0.004). Moreover, Itgβ1 gain of function via overexpression of a constitutively active V737N receptor–clustering mutant (Paszek et al., 2005) in ex vivo cerebellar slices was sufficient to restrain GNP GZ exit at 20% O2 (Figure 7F, Control , While Itgβ1 V737N OE ). Thus, not only does Itgβ1 gain of function phenocopy Hif1α gain of function or hypoxia phenotypes, but Itgβ1 loss is sufficient to restore GZ exit in hypoxia.
Figure 7. The Antagonistic Relationship Between Hif1α-Zeb1 and Pard6α Regulates Itgβ1 adhesion (.
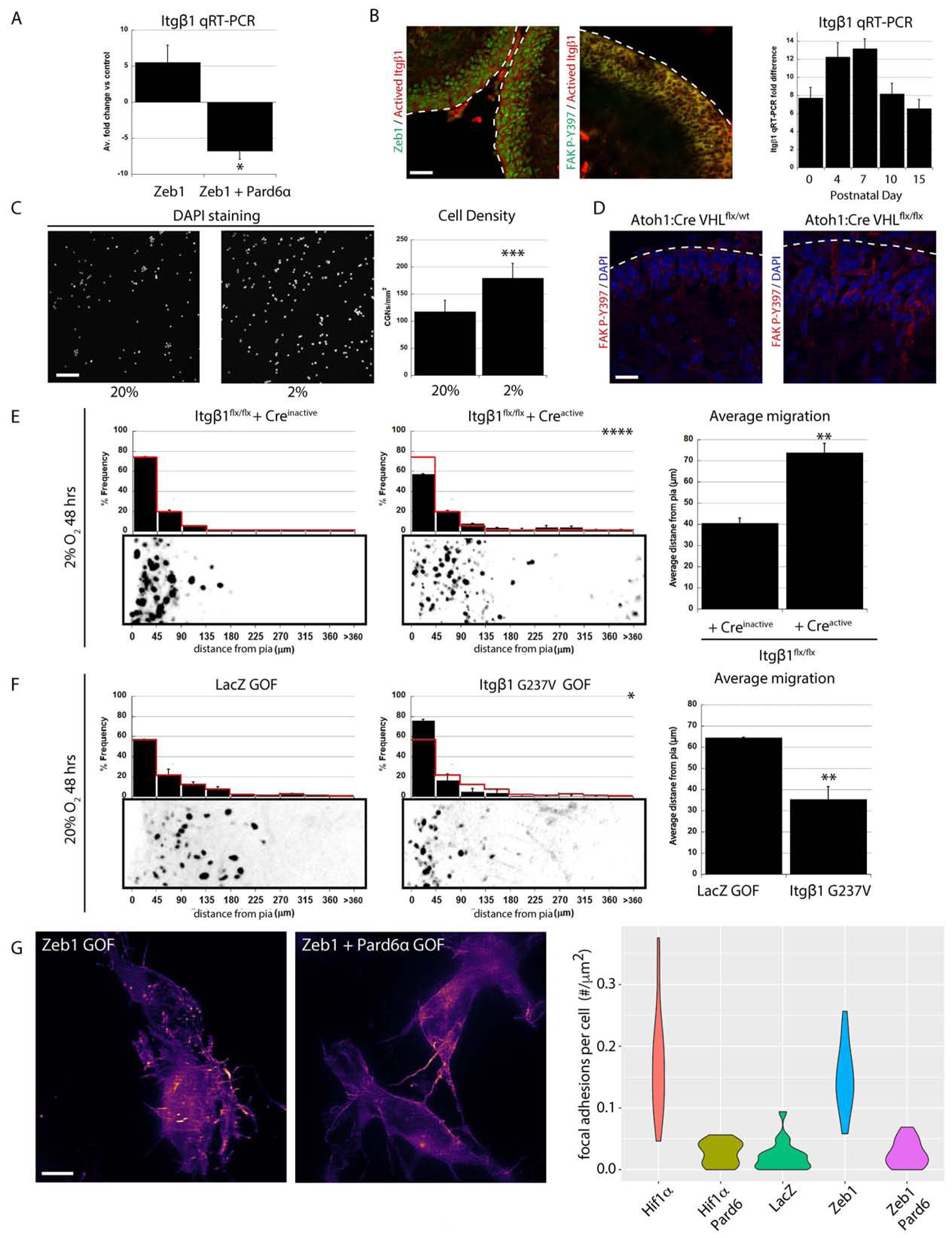
A) qRT-PCR of FACS sorted CGNs show that Zeb1 expression induces Itgβ1 mRNA expression (5.5±2.4 fold increase vs controls ) and Pard6α expression reduces Itgβ1 expression 6.8±1.1 fold (P<0.02). (B) Immunostaining shows activated Itgβ1 expression (red) in P7 GNPs is coincident with Zeb1 (left) and phospho-FAK Y397 (middle). qRT-PCR on GNPs shows that Itgβ1 mRNA expression peaks at P7. (C) Low O2 tension increases CGN adhesion to laminin (CGNs visualized by Dapi, n = 3, 117.1±21.1 cells/mm2 at 20% O2 and 179.0±27.2 cells/mm2 at 2% O2, t-test, P< 0.0005) (D) FAK-P-Y397 immunostaining (red) is enhanced in GNPs from Atoh1-Cre:VHLflx/flx cerebella. (E) Itgβ1 deletion rescues 2% O2 migration deficit (, , P < 0.005 and 0.0001 [by t-test and χ2 test respectively]). (F) Itgβ1 G737V over-expression impairs GZ exit ex vivo (, , P < 0.01 [by t-test and χ2 test]). (G) LLS-SIM reveals that Hif1α and Zeb1 stimulate CGN focal adhesion formation on laminin (detected by LASP1-Emerald puncta), a phenotype rescued by Pard6α expression (see violin plot for quantitation).
We gained insights into Itgβ1 regulation during GZ exit through our analysis of the changes in gene expression that accompany Pard6α rescue of Zeb1-overexpression phenotypes. Our previous work showed that Pard6α rescues GNP differentiation and GZ exit defects linked to elevated Zeb1 expression but does not restore the expression of genes repressed by Zeb1 (Singh et al. 2016). Surprisingly, qRT-PCR showed that restored Pard6α expression in the context of Zeb1 gain of function caused Itgβ1 mRNA expression to decline to levels below those in unmanipulated CGNs (Figure 7A). To confirm the protein expression in these mRNA analyses in GNPs, we FACS isolated pure progenitors from Atoh1-EGFP knock-in animals and incubated them at 2% O2 in the presence or absence of echinomycin, a small molecule Hif1α antagonist (Supplemental Figure 7A, B). Hif1α inhibition at 2% O2 decreased the number of Atoh1-EGFP positive GNPs (Supplemental Figure 7C, D). Western blotting revealed that Hif1α inhbition decreased Zeb1 and BNIP3 expression, increased α-internexin (a CGN differentiation marker) and Pard6α expression and not only diminished Itgα6 laminin receptor expression but also levels of the phospho–FAK Y397 marker for integrin engagement (Supplemental Figure 7E, F), supporting the Hif1α pathway modulation of polarity and integrin expression or engagement model.
Pard6α inhibition of Zeb1 induced Itgβ1 expression suggested that Pard6α functions to inhibit Itgβ1-mediated adhesion. To assess this possibility directly, we developed an assay to image GNP integrin-laminin focal contacts, using a LIM and SH3 protein 1 fusion protein (LASP1-Emerald). LASP1 is one of the few integrin focal adhesion adaptor proteins expressed in GNPs (according to the RIKEN brain transcriptome database (Sato et al., 2008)) that also serves as a reporter for Itgβ1 focal adhesion to ECM substrates in non-neuronal cells (Lin et al., 2004). The resolution and signal-to-noise ratio of spinning-disk or confocal microscopy was insufficient to capture GNP focal adhesions or their dynamics (data not shown). Therefore, to perform our LASP1-Emerald phenotypic screen, we implemented the Structured Illumination (SIM) mode of Lattice Light-Sheet (LLS) microscopy, which uses ultrathin light sheets derived from optical lattices to rapidly section cells grown in culture with minimal phototoxicity and provide near-isotropic imaging in the x, y, and z dimensions. Combinations of LASP1-Emerald, Hif1α, Zeb1, and Pard6α expression vectors were nucleofected into purified CGNs, cultured on laminin-coated coverslips, and LLS-SIM time-lapse microscopy was used to assess the focal adhesion levels. After applying a Weka machine-learning approach (Arganda-Carreras et al., 2017) to quantitate the GNP focal adhesions, we found that both Hif1α and Zeb1 gain of function alone led to a five- to six-fold increase in LASP1 focal adhesion density on a laminin substrate, when compared to control cells (Figure 7G). Interestingly, Pard6α expression in the context of Hif1α or Zeb1 gain of function reversed LASP1 focal adhesion density to control levels. Finally, we assessed if enhanced neuronal polarity modulated Itgβ1 mediated delay in GZ exit. Expression of Pard6α rescued GZ exit in GNPs over-expressing Itgβ1 V737N receptor–clustering mutant (Paszek et al., 2005) in ex vivo cerebellar slices (Supplemental Figure 6C). Taken together, these results show not only does oxygen tension regulation of Hif1α and Zeb1 promote Itgβ1-based contacts that by themselves are sufficient to promote GNP GZ occupancy but also that Pard6α facilitates GZ exit in part by inhibiting ECM adhesion.
DISCUSSION
Neuronal differentiation involves a complex interplay of extrinsic cues with intrinsic signaling pathways that support morphologic specialization, migration, and subsequent synaptic integration of post-mitotic neurons. Despite the intense focus on the function of genetically encoded morphogens, neurotrophic factors, or cell-surface cues on neuronal polarization and differentiation, the impact of environmental cues, such as O2 tension, on these processes has been relatively unexplored at the cellular or molecular levels. Earlier studies uncovered varied relationships of developing neural cells to the vasculature. In many cases, the vasculature has been thought to provide positive trophic support for neural stem cells (Javaherian and Kriegstein;2009;Palmer et al., 2000;Shen et al., 2004;Shen et al., 2008;Tavazoie et al., 2008), but a lack of vasculature was proposed to be critical to generating appropriate levels of cerebral cortical neural stem cell proliferation via the Hif1α pathway controlling the metabolic status of the stem cell population (Lange et al., 2016). Moreover, the Hif1α pathway controls oligodendrocyte maturation in a manner that couples angiogenesis with optimal vascularization (Yuen et al., 2014). The profile of Hif1α expression, pimonidazole labeling, and vascularization in the developing cerebellum indicates a switch in Hif1α pathway activity, whereby an early epoch in GNP proliferation occurs in a hypoxic environment, after which the period of CGN differentiation arises with a period of vascularization. The EGL is unique in that this secondary germinal matrix, which is fated to vanish during development, is never fully vascularized by comparison with regions where mature neurons reside, such as the IGL or the mid- and hindbrains. This surprising finding introduces an innovative concept regarding the relation of developing neural cells to the vasculature: not only is poorly developed vasculature in specific lamina an arbiter of the overall maturation of neurons residing in that layer but, given that hypoxia inhibits GZ exit and migration, environmental conditions within the layer itself are a barrier to subsequent lamination that must be overcome. More studies are required to understand how cell types in the developing cerebellum communicate to regulate the onset of vascularization. While oligodendrocyte precursors are the source of signals that promote white-matter angiogenesis (Yuen et al., 2014), it is unknown what cell types contribute towards ML angiogenesis.
A number of studies implicated hypoxia as a regulator of adult or embryonic neural stem cell self-renewal and quiescence (Lange et al., 2016;Mazumdar et al., 2010;Mohyeldin et al., 2010;Morrison et al., 2000;Panchision, 2009;Simon and Keith, 2008;Studer et al., 2000). To our knowledge, the role of the Hif1α pathway or hypoxia in regulating neuronal differentiation and, especially, polarization is unexplored. We now provide multiple levels of evidence based on Hif1α pathway gain or loss of function to support the hypothesis of Hif1α not only regulates neuronal differentiation but also key milestones of the post-mitotic state: First, the Hif1α pathway is necessary and sufficient to maintain peak levels of actively cycling GNPs in vivo. Indeed, the loss of Hif1α increased the fraction of GNPs exiting the cell cycle even when mitogens such as sonic hedgehog were present. Second, the Hif1α pathway controls the timing of GZ exit and radial migration initiation, which occurs when CGNs enter the postmitotic phase of their differentiation. Third, hypoxia and Hif1α elevate Zeb1 expression while repressing Pard6α, Pard3, and other genes required for neuronal maturation. Fourth, Hif1α pathway activation maintains the affinity of CGNs for Itgβ1 substrates, which is a hallmark of GZ-resident CGNs. While earlier studies implicated Hif1α in indirect effects on non-neuronal cell differentiation through paracrine or autocrine modulation of BMP or Wnt signaling (Mazumdar et al., 2010), our findings identified a well-defined cluster of Hif1α-regulated genes that represent a cell-autonomous pathway controlling neuronal cell differentiation.
Our work highlights a significant conceptual shift in the understanding of cell polarity regulation. The field had focused on post-translational rheostat-like polarity signaling mechanisms based on polarity protein phosphorylation and asymmetric subcellular partitioning of signaling complexes (McCaffrey and Macara, 2009;Namba et al., 2015). Recent vertebrates studies revealed diverse polarity regulatory mechanisms, including global switch-like mechanisms whereby polarity competency is modulated through expression levels of polarity genes, as in polarity inhibition by microRNAs, ubiquitin ligases (Cheng et al., 2011;Famulski et al., 2010;Laumonnerie and Solecki, 2018), and now Hif1α-Zeb1–mediated transcriptional repression. Unexpectedly, Hif1α regulation of Pard-complex function via Zeb1 is the first known example of cellular polarity modulation by O2 tension during development, in the nervous system or elsewhere. Our data suggest a model where low O2 tension in the cerebellum before P10 leads to elevated Hif1α and Zeb1 activity that antagonizes Pard-complex gene expression in immature cells of the CGN lineage (see Graphical Abstract). Subsequent elevation of O2 tension, presumably through postnatal vascularization, leads to diminished repression of polarity genes by Hif1α-Zeb1, leading to GZ exit and migration initiation. Mechanistically, our results also show that the Pard complex allows GNPs to release Itgβ1 focal adhesive contacts with pial-derived ECM substrates, a function that complements its role in promoting Jam-C–dependent CGN-CGN or CGN–Bergmann glial adhesion, which allows these cells to exit their germinal niche (Famulski et al., 2010;Singh et al., 2016). Thus, the Pard complex not only regulates adhesive affinities to postmitotic neurons or glial during GZ exit and migration, but also promotes an O2 tension-dependent loosening of GNP adhesion to GZ niche ECM promoting GZ occupancy. Interestingly, Hif1α and Itgβ1 loss of function both reduce GNP sensitivity to mitogen signaling (Blaess et al., 2004;Nguyen et al., 2018), suggesting that investigation of the O2 tension–cell polarity–ECM interactions will yield insights into not only how these interactions maintain appropriate levels of neuronal differentiation but also provide more insights into how O2 shapes neuronal circuit formation.
The finding that O2 tension regulates the cell polarization status of developing neurons has implications beyond GZ occupancy and radial migration initiation; it suggests that crucial polarity-controlled events in neurons, such as spindle orientation during neural stem-cell division, axon-dendrite specification, or adhesive events that promote synaptogenesis, may also be regulated by O2 tension (Singh and Solecki, 2015;Uzquiano et al., 2018). This paradigm is relevant to neurodevelopmental abnormalities associated with pre-term birth, where perinatal hypoxia leads to underdevelopment and damage to sensitive populations of immature neurons and glial cells (Salmaso et al., 2014). It will be interesting if a review of clinical neuroimaging studies notes neuronal heterotopias in the brains of children with vascular defects. It is worth noting the classical function of polarity signaling cascades in non-neural tissues is to maintain epithelial or vascular endothelial tight and adherens junctions; thus, hypoxic inhibition of cell polarity may also be related to the vascular leakage and hemorrhage associated with hypoxic brain insults. Hypoxic inhibition of polarity signaling cascades not only provides a molecular entry point to mechanistically dissect the pathogenesis of perinatal hypoxic insults but also suggests cell polarity promoting treatments may be useful to support healthy brain development in preterm infants.
STAR METHODS
Lead Contact and Materials Availability
Further information and requests for reagents may be directed and will be fulfilled by the lead contact David J. Solecki (david.solecki@stjude.org). All unique/stable reagents generated in this study are available from the corresponding author.
Experimental Models and Subject Details
Mice
All mouse lines were maintained in standard conditions (e.g. pathogen free and continuous access to food/water) in accordance with guidelines established and approved by the Institutional Animal Care and Use Committee at St. Jude Children’s Research Hospital (Protocol no. 483). Atoh1-cre/Esr1*14Fsh/J, Egln1tm1Kael/J, B6.129-Hif1atm3Rsjo/J, C;129S-Vhltm1Jae/J and B6.129S-Atoh1tm4.1Hzo/J mouse strains were obtained from The Jackson Laboratory. Animals were collected at postnatal days five through twenty-one for studies detailed in the methods section below or as denoted in experimental details in the figures. Male and female animals were used for described experiments as thus far no effect of sex on the timing of GZ exit or migration initiation has been observed.
Methods Details
Blinding was not used in data collection.
Plasmid vectors
All cDNAs encoding proteins of interest were commercially synthesized and subcloned into pCIG2 by GenScript (Piscataway, NJ). Expression plasmids for Pard3, Pard6α, and fluorescent fusion proteins such as pCIG2 H2B-mCherry and pCIG2 GPI-pHluorin were subcloned as previously described in Singh et al., 2016. See Key Resource Table for full recombinant DNA list.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Pard6a (clone C-3) (1:100) | Santa Cruz Biotechnology | Cat# sc-365323, RRID:AB_10846183 |
| Rabbit monoclonal anti-Integrin alpha 6 (clone EPR18124) (1:200) | Abcam | Cat# ab181551 |
| Rabbit polyclonal anti-Fak (1:200) | Abcam | Cat# ab131435, RRID:AB_11154758 |
| Rabbit monoclonal anti-Phospho-FAK (Tyr397) (clone 31H5L17) (1:200) | Thermo Fisher Scientific | Cat# 700255, RRID:AB_2532307 |
| Rabbit polyclonal anti-Zeb1 (1:1000) | Sigma-Aldrich | Cat# HPA027524, RRID:AB_1844977 |
| Rabbit monoclonal anti-Bnip3/Nix (clone D4R4B) (1:500) | Cell Signaling Technology | Cat# 12396, RRID:AB_2688036 |
| Rabbit polyclonal anti-alpha internexin (1:1000) | Abcam | Cat# ab7259, RRID:AB_305807 |
| Goat polyclonal anti-GFP (1:500) | Abcam | Cat# ab6673, RRID:AB_305643 |
| Mouse monoclonal anti-activated beta1 integrin HUTS4 | Millipore | Catalog #: MAB2079Z |
| Rabbit polyclonal anti-Hif1 alpha (1:100) | ThermoFisher | Cat# PA1–16601 RRID:AB 2117128 |
| Mouse monoclonal anti-Human Ki-67 (1:50) | BD Pharmingen | Cat# 556003 RRID:AB_396287 |
| Rabbit polyclonal anti-Glut1 (1:500) | Millipore | Cat# 07–1401 RRID:AB_1587074 |
| Mouse monoclonal anti-p27 Kip1 (1:100) | Cell Signaling | Cat# 3698 RRID:AB_2077832 |
| Rabbit polyclonal anti-phospho Histone H3 (1:500) | Millipore | Cat# 06–570 RRID:AB_310177 |
| Rabbit polyclonal anti-Caspase 3 (1:100) | Cell Signaling | Cat# 9664 RRID:AB_2070042 |
| Mouse polyclonal anti-CHL-1 (1:100) | R&D Systems | Cat# AF2147 RRID:AB_2079332 |
| Mouse monoclonal anti-NeuN (1:100) | Millipore | Cat# MAB377 RRID:AB_177621 |
| Rabbit polyclonal anti-Partitioning-defective 3 (1:100) | Millipore | Cat# 07–330 RRID:AB_11213581 |
| Chicken polyclonal anti-GFP (1:100) | Aves Labs, Inc | Cat# GFP-1020 RRID:AB_10000240 |
| Hoeschst 33342 Staining Dye/Dapi (1:500) | Abcam | Cat# ab228551 |
| Donkey anti-chicken 649 (1:500) | Jackson Immuno Labs | Cat# 703-606-155 RRID:AB 2340380 |
| Donkey anti-rabbit 555 (1:500) | Invitrogen | Cat# A31572 RRID:AB_162543 |
| Donkey anti-mouse 488 (1:750) | Invitrogen | Cat# A32766 RRID:AB_2762823 |
| Donkey anti-rabbit 488 (1:750) | Invitrogen | Cat# A32790 RRID:AB_2762833 |
| Donkey anti rabbit 649 (1:750) | Invitrogen | Cat# A32795 RRID:AB_2762835 |
| Donkey anti-mouse 555 (1:750) | Invitrogen | Cat# A32773 RRID:AB_2762848 |
| Click-IT Edu AlexaFluor 488 Imaging Kit | Invitrogen | Cat# C10337 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Echinomycin | Tocris | Cat# 5520 |
| Mouse Laminin | EMD Millipore, Chemicon | Cat# CC095 |
| Gant 61 | Tocris | Cat# 3191 |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Microarray Data: ArrayExpress E-MTAB-8761 | E-MTAB-8761 | |
| Experimental Models: Cell Lines | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: Atoh1-eGFP: B6.129S-Atoh1tm4.1Hzo/J | The Jackson Laboratory | JAX: 013593 |
| Mouse: Ptch1: B6N.129-Ptch1tm1Hahn/J | The Jackson Laboratory | JAX: 012457 |
| Mouse: Hif1a: B6.129-Hif1atm3Rsjo/J | The Jackson Laboratory | JAX: 007561 |
| Mouse: Vhl: B6.129S4(C)-Vhltm1Jae/J | The Jackson Laboratory | JAX: 012933 |
| Mouse: Egln1: Egln1tm1Kael/J | The Jackson Laboratory | JAX: 009672 |
| Mouse: Itgb1: B6;129-Itgb1tm1Efu/J | The Jackson Laboratory | JAX: 004605 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| pCIG2 Histone2B-mCherry | Laboratory Stocks | |
| pCIG2 Inactive Cre T2A Histone 2B mCherry | “ | |
| pCIG2 Active Cre T2A Histone 2B mCherry | “ | |
| pCIG2 HifaHD cDNA | “ | |
| pCIG2 Zeb1 cDNA | “ | |
| pCIG2 Pard6α cDNA and point mutants | ||
| pCIG2 Pard3A cDNA | “ | |
| pCIG2 Chl1 cDNA | “ | |
| pCIG2 mir30 Zeb1 shRNA | “ | |
| pCIG2 mir30 Pard3A shRNA | “ | |
| pCIG2 mir30 Pard6 α shRNA | “ | |
| Software and Algorithm Section | ||
| Image Studio 5.2 | Biotechnology |
https://www.licor.com/bio/image-studio/, RRID:SCR_015795 |
| Amira | ThermoFisher | https://www.thermofisher.com/us/en/home/industrial/electron-microscopy/electron-microscopy-instruments-workflow-solutions/3d-visualization-analysis-software/amira-life-sciences-biomedical.html |
| Slidebook | Intelligent Imaging Innovations | https://www.intelligent-imaging.com/slidebook |
Preparation and nucleofection of CGNs
Briefly, cerebella were dissected from the brains of P7 mice and the pial layer removed, then the tissue was treated with a Neural Tissue Dissociation Kit (Miltenyi Biotec) and triturated into a single-cell suspension by using fine-bore Pasteur pipettes. The suspension was layered onto a discontinuous Percoll gradient and separated by centrifugation. The small-cell fraction was then isolated. The resulting cultures routinely contained 95% CGNs and 5% glia. Expression vectors encoding fluorescently labeled nuclear proteins and pCIG2 expressing proteins of interest were introduced into granule neurons via Amaxa nucleofection, using an Amaxa Mouse Neuron Nucleofector Kit in accordance with the manufacturer’s instructions and program A005. The concentration of pCIG2 expression vectors used was determined such that there would be at least a two-fold increase in protein expression. After cells had been allowed to recover from the nucleofection for 10 min, they were plated in 6-cm dishes coated with low concentrations of poly-L-ornithine to facilitate the attachment of neurons to glial processes.
Ex vivo cerebellar electroporation, organotypic slice culture, and imaging
P7 cerebella were dissected, soaked in endotoxin-free plasmid DNA suspended in Hank’s balanced salt solution (1–3 μg/μL of each DNA was generally used, with pCIG2-mCherryH2B being electroporated as a nuclear marker for migrating CGNs), transferred to a CUY520-P5 platinum-block petri-dish electrode (Protech International), and electroporated with a CUY21EDIT square-wave electroporator (90 V, 5 pulses, 50-ms pulse, 500-ms interval) (Protech International). Electroporated cerebella were embedded in 4% low-melting-point agarose and 300-μm sagittal cerebellar slices were prepared using a VT1200 Vibratome (Leica Microsystems). Slices were transferred to Millicell tissue culture inserts (Millipore) and incubated in basal Eagle medium supplemented with 2 mM L-glutamine, 0.5% glucose, 50 U/mL penicillin-streptomycin, and 1× B27 and 1× N2 supplements (Invitrogen) at the air-medium interface for the times indicated in the figures.
To measure the migration distance of CGNs, cerebellar slices were fixed with 4% paraformaldehyde and mounted on slides by using ProLong Gold (Invitrogen). The migration distance was measured as the distance between the cerebellar pial surface and the center of individual cell nuclei marked by mCherry-H2B. Measurements were made using a self-written program in the Amira software. Statistical analysis used Microsoft Excel, and graphs were prepared with KaleidaGraph v4.03 (Synergy Software). For live-imaging analysis of the migration of H2B-mCherry–labeled CGNs, slice cultures were transferred at 18 h to the humidified chamber of the spinning-disk confocal microscope described above. Z-stacks (60–80 μm in width, approximately 20 sections per stack) were collected at multiple x, y stage positions every 15 min for 24–48 h.
Slice movie analysis
The nuclei of neurons were tracked manually over the course of the movie by using Slidebook 6 (Intelligent Imaging Innovations) and a Marianas Spinning Disk confocal microscope. The distance from the edge (DFE) of the slice was computed using Amira 6.2 (FEI, Thermo Fisher Scientific) and added as an intensity channel to the respective movie. Only movies in which the overall XY drift was limited to 5 μm were considered in the analysis. Nuclear coordinates (X, Y, DFE) tracked at each time-point were exported to Excel (Microsoft) in order to plot the distance distribution for each time-point and calculate the instant speed average and average distance variation. Angles were calculated from the distance variation and instant speed vectors at each time-point. Data were divided into layers based on extrapolated measurements from histologic sections at P7, i.e., oEGL: 0–30 μm; iEGL: 30–50 μm; ML: 50–100 μm; and IGL: >100 μm, based on their DFE at a given time-point.
Lattice light-sheet structured illumination microscopy (LLS-SIM).
Images were acquired on a lattice light-sheet microscope in the Developmental Neurobiology Department’s Neuroimaging Laboratory. The specialized optics of this instrument have been described by Chen and coworkers (Chen et al., 2014). The optics used for the experiments described herein included the following: 560-and 488-nm laser lines, with maximum power of 500 mW and 300 mW, respectively, and a quad-band emission filter to resolve spectrally the imaged channels.
The LLS was aligned each morning, allowing at least 4 h for thermal equilibration after the heating apparatus was activated. All the cell samples were imaged in Fluorobrite low-fluorescence medium supplemented with 10% heat-inactivated horse serum, to which the spherical aberration correction parameter of the objective was adjusted. Cells were seeded on 5-mm coverslips and mounted in custom-fabricated sample holders for imaging. Images were acquired with the Slidebook software package, using a custom-designed 15 μm–square light-sheet pattern in the 5-phase structured illumination z-galvo and objective scan mode, with both colors being captured at each z position. The acquired images were background subtracted, then high-resolution SIM images were generated using an open-source, GPU-accelerated SIM reconstruction software with freshly acquired optical transfer functions (OTFs) generated on each day of imaging (Chen et al., 2014;Rego and Shao, 2015). Weka-mediated machine learning in Fiji was then used to analyze the numbers of LASP1-Emerald focal adhesions in maximum-intensity projections of LLS-SIM 3D volumes to determine the effects of Zeb1 or Hif1α overexpression on integrin-mediated adhesion.
Gene expression analyses: Flow cytometry, RNA isolation, RT-PCR, and Affymetrix arrays
To obtain a pure population of GNPs expressing the protein of interest, GNPs isolated from cerebella of postnatal day 7 (P7) mice were nucleofected with pCIG2 H2B-mCherry (to label cells red [control]) or mCherry with Hif1α or Zeb1, maintained in culture for 24h, triturated into a single-cell suspension, and stained with DAPI. Sorting of viable mCherry-positive cells was carried out in the Shared Resource flow cytometry facility at St. Jude, using a BD Aria III SORP sorter. A bandpass 610/20 filter was used to detect mCherry signals during excitation at 561 nm. The cells were directly collected in the lysis buffer, and RNA was extracted with an Ambion RNAqueous Kit (Austin, TX). In accordance with the manufacturer’s instructions, each sample was isolated in 40 μL of elution buffer and treated with DNase (Ambion) to eliminate any genomic contamination. The quantity and quality of the isolated RNA was checked using an Agilent 2100 Bioanalyzer with RNA 6000 Nano Chips (Agilent Technologies, Santa Clara, CA). Primer sets for each gene were designed with Primer Express Software (Applied Biosystems, Foster City, CA) and synthesized (IDT, Coralville, IA). RNA amplification and reverse transcription were performed with the WT-Ovation RNA Amplification System (NuGEN) in accordance with the manufacturer’s instructions. Two-step real-time RT-PCR was performed with an ABI PRISM 7900 Sequence Detection System, using the SYBR Green PCR Master Mix (Applied Biosystems). Data were normalized to the 18S ribosomal RNA and actin expression levels in each sample.
RNA quality and microarray analyses were performed by the microarray core facility at St Jude. RNA quality was analyzed with an Agilent 2100 Bioanalyzer, and all samples had an RIN of less than 8. A 100-ng sample of total RNA was processed using an Affymetrix 3′ IVT Express Kit. Biotin-labeled cRNAs were hybridized to the Affymetrix GeneChip HT MG-430 PM array, then washed, stained, and scanned on a GeneTitan instrument (Affymetrix). Data were summarized using Affymetrix Expression Console software (v1.1) to apply the robust multi-array average (RMA) algorithm (ArrayExpress accession no. E- MTAB-3557). The arrays were RMA normalized and batch corrected with R/ComBat. Unsupervised hierarchical clustering analysis and principal component analysis was performed with Spotfire and GeneMaths. Differentially expressed genes were analyzed using a linear models algorithm (R/Limma). Differentially expressed genes in Hif1α- and Zeb1-overexpressing cells were selected by using an FDR-corrected P-value (q-value) of 0.05 and a fold change of 1.5 as the cutoffs.
GO analysis was performed with the DAVID Bioinformatics Resource for the common upregulated genes in Hif1α- and Zeb1-overexpressing cells.
Cerebellar immunohistochemistry
Postnatal brains were fixed by immersion in 4% paraformaldehyde at 4°C overnight, followed by cryoprotection in PBS containing 30% sucrose. Histologic sagittal sections were cut on a cryostat and pre-blocked for 1 h in PBS with 0.1% Triton X-100 and 10% normal donkey serum. Sections were incubated overnight at 4°C with the primary antibodies. This was follow ed by incubation for 1 h with the appropriate Alexa Fluor–labeled secondary antibody (Invitrogen; diluted 1:1000) before mounting. For cell proliferation assays, mice were injected intraperitoneally with 50 mg/kg EdU, and EdU incorporation was assayed with the Click-iT assay (Invitrogen) in accordance with the manufacturer’s instructions. To detect organs and cells low in O2, mice were injected intraperitoneally with 60 mg/kg pimonidazole hydrochloride 2 h before they were euthanized, and binding of the compound was visualized with the Hypoxyprobe-1 Omni kit. GluT1 staining required tissue to be cleared with the CLARITY method and was essentially performed as described at http://wiki.claritytechniques.org.
iDisco clearing
EGFP-Atoh1–expressing p7 brains were stained to visualize their vasculature by using the iDisco protocol (https://idisco.info/idisco-protocol/). Briefly, the P7 brains were dissected and trimmed down to leave the hindbrain, cerebellum, and midbrain intact. The brains were fixed in PFA and then underwent pretreatment and bleaching with methanol/dichloromethane (DCM) and H2O2 in accordance with the protocol. Permeabilization and blocking steps were carried out for 3 days each at 37°C. Samples were then immunolabeled with chick anti-GFP antibody (EMD Millipore, diluted 1:1000) (to label EGFP-Atoh1) and rabbit anti-GLUT-1 (Aves, diluted 1:500) (to label blood vessels). Both primary and appropriate secondary antibodies (Alexa donkey anti-rabbit 555 [LifeTechnologies, diluted 1:500] and donkey anti-chick 647 [Jackson Immunoresearch, diluted 1:500]) were incubated with the brains for 6 days at 37°C. Samples were then cleared using the methanol/DCM/dibenzyl ether (DBE) treatment outlined in the protocol.
Imaging of staining was carried out on a LaVision Ultramicroscope II, using 3-μm step sizes at 1.2× magnification. Images were processed and rendered in Arivis 4D (Arivis AG).
Atoh1-eGFP sorted cells analysis of polarity protein expression.
Pups homozygous for Atoh1-eGFP (B6.129S-Atoh1tm4.1Hzo/J, The Jackson Laboratory, JAX: 013593) were collected at P7, and cerebellar granule neurons were isolated following the dissociation methods described above. Progenitors were sorted based on their high GFP intensity with a cell sorter (FACSAria Fusion, BD Biosciences, 85μm nozzle 35PSI). Sample for CHiP PCR or CHiP-Seq were snap frozen. GNPs used for validation of Zeb1, Pard protein and integrin related protein expression profiles were cultured for 24h on Laminin (2μg.μm−2, EMD Millipore, Chemicon, Cat# CC095) at 2% O2 in presence of DMSO or Echinomycin (Fresh batch for each replicates, Tocris, Cat# 5520). Proteins were isolated from cell lysate and reduced in Laemmlli buffer (Sigma-Aldrich, Cat# S3401) prepared to then loaded on gel for Western Blot analysis (antibody and concentration provided in the key resource table) using the quantitative Li-COR Biotechnology system. Band intensity was measured and analyzed with Image Studio (Li-COR Biotechnology) and each signal was normalized using total protein stain (Revert™ 700 Total Protein Stain Kits, Li-COR Biotechnology, Cat# P/N 926–11016).
Expression analysis for Hif1α protein or Hypoxyprobe levels
Nuclear and cytoplasmic protein fractions were extracted with NE-PER Nuclear and Cytoplasmic Extraction Reagent (Thermo Fisher Scientific). Protein lysates were denatured for 5 min with NuPAGE sample-reducing agent (LifeTech) and loading buffer heated to 75°C. Samples were subject ed to electrophoresis on SDS-PAGE 4–12% Bis-Tris gels (LifeTech). Proteins were electroblotted onto polyvinylidene fluoride membranes by using a Gel Transfer Device (Invitrogen). Membranes were dried, activated with methanol, blocked for 1 h at room temperature with Odyssey Blocking buffer diluted 1:2 in TBS, and incubated with the indicated antibodies at 4°C overnight. Odyssey secondary IR antibodies (diluted 1:10000) were used for detecting proteins with the Odyssey Infrared Imaging System.
Chromatin Immunoprecipitation
Atoh1-EGFP positive GNPs were fixed with 1% formaldehyde for 15 min and quenched with 0.125 M glycine. Chromatin was isolated by the addition of lysis buffer, followed by disruption with a Dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp. Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by ethanol precipitation. An aliquot of chromatin (10 ug) was precleared with protein A agarose beads (Invitrogen). Genomic DNA regions of interest were isolated using 5 μg of antibody against HIF1a (Abcam, ab2185, Lot GR3239692–1). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation. Quantitative PCR (QPCR) reactions were carried out in triplicate on specific genomic regions using SYBR Green Supermix (Bio-Rad). The resulting signals were normalized for primer efficiency by carrying out QPCR for each primer pair using Input DNA.
Illumina ChIP Sequencing
Illumina sequencing libraries were prepared from the ChIP and Input DNAs by the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. Steps were performed on an automated system (Apollo 342, Wafergen Biosystems/Takara). After a final PCR amplification step, the resulting DNA libraries were quantified and sequenced on Illumina’s NextSeq 500 (75 nt reads, single end). Reads were aligned to the mouse genome (mm10) using the BWA algorithm (default settings). Duplicate reads were removed and only uniquely mapped reads (mapping quality >= 25) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library, and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in bigWig files. Peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of p-value= 1e-7. Peaks that were on the ENCODE blacklist of known false ChIP-Seq peaks were removed. Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations.
Quantitation and Statistical analysis
Standard image processing and analysis were carried out either with Amira (Thermo Fisher) or Slidebook (Intelligent Imaging Innovations) software. Analyzed metrics of quantitated data are expressed as the mean ± SEM. Student’s t-test was used for comparing two groups, and the one-way analysis of variance and Holm-Sidak post-hoc test was used for multiple comparisons, with the level of statistical significance being set at P < 0.01 unless otherwise specified. In migration rescue assays, if the rescuing conditions resulted in a χ2-test P-value greater than 0.8 when compared to controls and a t-test P-value of less than 0.01 when compared to Zeb1 overexpression or hypoxia mimicking condition alone, they were considered to have achieved rescue. See main text and all figure legends for statistical details of each experiment. Common statistical assumptions were used as a guideline for statistical tests. For example, t-tests was used for normally distributed metrics like average migration distance, while χ2 tests were used to compared skewed population migration distributions.
Data and Code Availability
All array data was submitted to ArrayExpress and has the E-MTAB-8761 accession number. Original imaging data available upon request.
Additional Resources
None to report.
Supplementary Material
Supplemental Movie 1, Related to Figure 1 3D render and animated tour of an iDisco cleared P7 cerebellum from an Atoh1EGFP/EGFP knockin animal stained with antibodies against EGFP (magenta) and Glut1 (cyan). Note: cyan label micro-blood vessels are generally located in progenitor free zones of the developing cerebellum. Rendering and animation were generated as a volume view in arivisAG Vision4D software.
Supplemental Movie 2, Related to Figure 3 Representative time-lapse sequence of CGNs labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 3, Related to Figure 3 Representative time-lapse sequence of CGNs labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 2% O2. Low O2 tension blocks the transition of CGNs to the ML and IGL. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 4, Related to Figure 4 Representative time-lapse sequence of Hif1αflx/flx CGNs expressing Creinactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 5, Related to Figure 4 Representative time-lapse sequence of Hif1αflx/flx CGNs expressing Creactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Hif1α deletion leads to early population of the ML and IGL of the cerebellar slice compared to Creinactive expressing cells. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 6, Related to Figure 4 Representative time-lapse sequence of VHLflx/flx CGNs expressing Creinactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 7, Related to Figure 4 Representative time-lapse sequence of VHLflx/flx CGNs expressing Creactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. VHL deletion leads to retention of GNP or CGNs in the outer layer of the cerebellar slice compared to Creinactive expressing cells. Time lapse encompasses roughly twelve hours of imaging time.
Highlights.
An extended postnatal phase of cerebellar hypoxia aids GNP proliferation via Hif1α
Hif1α controls CGN GZ exit and Pard complex function by activating Zeb1 expression
Hif1α–Zeb1 antagonism of Pard6α modulates Itgβ1 adhesion that promotes GZ occupancy.
Hif1α control of polarity is a basis to dissect the pathology of hypoxic insults.
ACKNOWLEDGEMENTS:
We thank David Finkelstein for bioinformatics support. Keith A. Laycock, PhD, ELS edited the manuscript. JK was supported by Deutsche Forschungsgemeinschaft fellowship KU3253/1-1. Solecki laboratory is funded by the American Lebanese Syrian Associated Charities (ALSAC) and by grants 1R01NS066936 and R01NS104029 from the National Institute of Neurological Disorders (NINDS). The Rowitch laboratory is funded by NINDS P01NS08351.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS: None
REFERENCES
- Arganda-Carreras I, Kaynig V, Rueden C, Eliceiri KW, Schindelin J, Cardona A, and Sebastian Seung H (2017). Trainable Weka Segmentation: a machine learning tool for microscopy pixel classification. Bioinformatics 33, 2424–2426. [DOI] [PubMed] [Google Scholar]
- Ayala R, Shu T, and Tsai LH (2007). Trekking across the brain: the journey of neuronal migration. Cell 128, 29–43. [DOI] [PubMed] [Google Scholar]
- Barnes AP, and Polleux F (2009). Establishment of axon-dendrite polarity in developing neurons. Annual review of neuroscience 32, 347–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum B, Settleman J, and Quinlan MP (2008). Transitions between epithelial and mesenchymal states in development and disease. Seminars in cell & developmental biology 19, 294–308. [DOI] [PubMed] [Google Scholar]
- Blaess S, Graus-Porta D, Belvindrah R, Radakovits R, Pons S, Littlewood-Evans A, Senften M, Guo H, Li Y, Miner JH, et al. (2004). Beta1-integrins are critical for cerebellar granule cell precursor proliferation. J Neurosci 24, 3402–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui T, Sequeira J, Wen TC, Sola A, Higashi Y, Kondoh H, and Genetta T (2009). ZEB1 links p63 and p73 in a novel neuronal survival pathway rapidly induced in response to cortical ischemia. PloS one 4, e4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA 3rd, Liu Z, et al. (2014). Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science 346, 1257998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng PL, Lu H, Shelly M, Gao H, and Poo MM (2011). Phosphorylation of E3 ligase Smurf1 switches its substrate preference in support of axon development. Neuron 69, 231–243. [DOI] [PubMed] [Google Scholar]
- Cooper JA (2013). Cell biology in neuroscience: mechanisms of cell migration in the nervous system. The Journal of cell biology 202, 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk S, Bugeon S, Ninkovic J, Pilz GA, Postiglione MP, Cremer H, Knoblich JA, and Gotz M (2017). Time-Specific Effects of Spindle Positioning on Embryonic Progenitor Pool Composition and Adult Neural Stem Cell Seeding. Neuron 93, 777–791 e773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Famulski JK, Trivedi N, Howell D, Yang Y, Tong Y, Gilbertson R, and Solecki DJ (2010). Siah regulation of Pard3A controls neuronal cell adhesion during germinal zone exit. Science 330, 1834–1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossage L, Eisen T, and Maher ER (2015). VHL, the story of a tumour suppressor gene. Nature reviews Cancer 15, 55–64. [DOI] [PubMed] [Google Scholar]
- Hatten ME (2002). New directions in neuronal migration. Science 297, 1660–1663. [DOI] [PubMed] [Google Scholar]
- Hatten ME, and Roussel MF (2011). Development and cancer of the cerebellum. Trends Neurosci 34, 134–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausmann B, and Sievers J (1985). Cerebellar external granule cells are attached to the basal lamina from the onset of migration up to the end of their proliferative activity. J Comp Neurol 241, 50–62. [DOI] [PubMed] [Google Scholar]
- Ivan M, and Kaelin WG Jr. (2017). The EGLN-HIF O2-Sensing System: Multiple Inputs and Feedbacks. Molecular cell 66, 772–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaherian A, and Kriegstein A (2009). A stem cell niche for intermediate progenitor cells of the embryonic cortex. Cereb Cortex 19 Suppl 1, i70–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG Jr. (2008). The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nature reviews Cancer 8, 865–873. [DOI] [PubMed] [Google Scholar]
- Kaelin WG Jr., and Ratcliffe PJ (2008). Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Molecular cell 30, 393–402. [DOI] [PubMed] [Google Scholar]
- Knobloch M, and Jessberger S (2017). Metabolism and neurogenesis. Current opinion in neurobiology. [DOI] [PubMed] [Google Scholar]
- Kreshuk A, Straehle CN, Sommer C, Koethe U, Cantoni M, Knott G, and Hamprecht FA (2011). Automated detection and segmentation of synaptic contacts in nearly isotropic serial electron microscopy images. PloS one 6, e24899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamachary B, Zagzag D, Nagasawa H, Rainey K, Okuyama H, Baek JH, and Semenza GL (2006). Hypoxia-inducible factor-1-dependent repression of E-cadherin in von Hippel-Lindau tumor suppressor-null renal cell carcinoma mediated by TCF3, ZFHX1A, and ZFHX1B. Cancer Res 66, 2725–2731. [DOI] [PubMed] [Google Scholar]
- Lange C, Turrero Garcia M, Decimo I, Bifari F, Eelen G, Quaegebeur A, Boon R, Zhao H, Boeckx B, Chang J, et al. (2016). Relief of hypoxia by angiogenesis promotes neural stem cell differentiation by targeting glycolysis. The EMBO journal 35, 924–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnerie C, and Solecki DJ (2018). Regulation of Polarity Protein Levels in the Developing Central Nervous System. J Mol Biol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL Jr., Courchet J, and Polleux F (2013). Cell biology in neuroscience: Cellular and molecular mechanisms underlying axon formation, growth, and branching. The Journal of cell biology 202, 837–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin YH, Park ZY, Lin D, Brahmbhatt AA, Rio MC, Yates JR 3rd, and Klemke RL (2004). Regulation of cell migration and survival by focal adhesion targeting of Lasp-1. The Journal of cell biology 165, 421–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazumdar J, Mazumdar J, O'Brien WT, O'Brien WT, Johnson RS, Johnson RS, LaManna JC, LaManna JC, Chavez JC, Chavez JC, et al. (2010). O2 regulates stem cells through Wnt/β-catenin signalling. Nature cell biology 12, 1007–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCaffrey LM, and Macara IG (2009). Widely conserved signaling pathways in the establishment of cell polarity. Cold Spring Harb Perspect Biol 1, a001370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metin C, Vallee RB, Rakic P, and Bhide PG (2008). Modes and mishaps of neuronal migration in the mammalian brain. The Journal of neuroscience : the official journal of the Society for Neuroscience 28, 11746–11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohyeldin A, Garzón-Muvdi T, and Quiñones-Hinojosa A (2010). Oxygen in Stem Cell Biology: A Critical Component of the Stem Cell Niche. Cell Stem Cell 7, 150–161. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Csete M, Groves AK, Melega W, Wold B, and Anderson DJ (2000). Culture in Reduced Levels of Oxygen Promotes Clonogenic Sympathoadrenal Differentiation by Isolated Neural Crest Stem Cells. Journal of Neuroscience 20, 7370–7376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, et al. (2004). Siah2 regulates stability of prolyl-hydroxylases, controls HIF1alpha abundance, and modulates physiological responses to hypoxia. Cell 117, 941–952. [DOI] [PubMed] [Google Scholar]
- Nakayama K, and Ronai Z (2004). Siah: new players in the cellular response to hypoxia. Cell Cycle 3, 1345–1347. [DOI] [PubMed] [Google Scholar]
- Namba T, Funahashi Y, Nakamuta S, Xu C, Takano T, and Kaibuchi K (2015). Extracellular and Intracellular Signaling for Neuronal Polarity. Physiol Rev 95, 995–1024. [DOI] [PubMed] [Google Scholar]
- Nguyen V, Sabeur K, Maltepe E, Ameri K, Bayraktar O, and Rowitch DH (2018). Sonic Hedgehog Agonist Protects Against Complex Neonatal Cerebellar Injury. Cerebellum 17, 213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer TD, Willhoite AR, and Gage FH (2000). Vascular niche for adult hippocampal neurogenesis. The Journal of comparative neurology 425, 479–494. [DOI] [PubMed] [Google Scholar]
- Panchision DM (2009). The role of oxygen in regulating neural stem cells in development and disease. Journal of cellular physiology 220, 562–568. [DOI] [PubMed] [Google Scholar]
- Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, Reinhart-King CA, Margulies SS, Dembo M, Boettiger D, et al. (2005). Tensional homeostasis and the malignant phenotype. Cancer cell 8, 241–254. [DOI] [PubMed] [Google Scholar]
- Rasin MR, Gazula VR, Breunig JJ, Kwan KY, Johnson MB, Liu-Chen S, Li HS, Jan LY, Jan YN, Rakic P, et al. (2007). Numb and Numbl are required for maintenance of cadherin-based adhesion and polarity of neural progenitors. Nature neuroscience 10, 819–827. [DOI] [PubMed] [Google Scholar]
- Rego EH, and Shao L (2015). Practical structured illumination microscopy. Methods Mol Biol 1251, 175–192. [DOI] [PubMed] [Google Scholar]
- Rose MF, Ren J, Ahmad KA, Chao HT, Klisch TJ, Flora A, Greer JJ, and Zoghbi HY (2009). Math1 is essential for the development of hindbrain neurons critical for perinatal breathing. Neuron 64, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Almodovar C, Coulon C, Salin PA, Knevels E, Chounlamountri N, Poesen K, Hermans K, Lambrechts D, Van Geyte K, Dhondt J, et al. (2010). Matrix-binding vascular endothelial growth factor (VEGF) isoforms guide granule cell migration in the cerebellum via VEGF receptor Flk1. J Neurosci 30, 15052–15066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salmaso N, Jablonska B, Scafidi J, Vaccarino FM, and Gallo V (2014). Neurobiology of premature brain injury. Nature neuroscience 17, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Sekine Y, Saruta C, Nishibe H, Morita N, Sato Y, Sadakata T, Shinoda Y, Kojima T, and Furuichi T (2008). Cerebellar development transcriptome database (CDT-DB): profiling of spatio-temporal gene expression during the postnatal development of mouse cerebellum. Neural Netw 21, 1056–1069. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Shimoda LA, and Prabhakar NR (2006). Regulation of gene expression by HIF-1. Novartis Found Symp 272, 2–8; discussion 8–14, 33–16. [PubMed] [Google Scholar]
- Shen Q, Goderie SK, Jin L, Karanth N, Sun Y, Abramova N, Vincent P, Pumiglia K, and Temple S (2004). Endothelial cells stimulate self-renewal and expand neurogenesis of neural stem cells. Science 304, 1338–1340. [DOI] [PubMed] [Google Scholar]
- Shen Q, Wang Y, Kokovay E, Lin G, Chuang S-M, Goderie SK, Roysam B, and Temple S (2008). Adult SVZ stem cells lie in a vascular niche: a quantitative analysis of niche cell-cell interactions. Cell Stem Cell 3, 289–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shook D, and Keller R (2003). Mechanisms, mechanics and function of epithelial-mesenchymal transitions in early development. Mechanisms of development 120, 1351–1383. [DOI] [PubMed] [Google Scholar]
- Simon MC, and Keith B (2008). The role of oxygen availability in embryonic development and stem cell function. Nature reviews Molecular cell biology 9, 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, Howell D, Trivedi N, Kessler K, Ong T, Rosmaninho P, Raposo AA, Robinson G, Roussel MF, Castro DS, et al. (2016). Zeb1 controls neuron differentiation and germinal zone exit by a mesenchymal-epithelial-like transition. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S, and Solecki DJ (2015). Polarity transitions during neurogenesis and germinal zone exit in the developing central nervous system. Front Cell Neurosci 9, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solecki DJ (2012). Sticky situations: recent advances in control of cell adhesion during neuronal migration. Curr Opin Neurobiol 22, 791–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer L, Csete M, Lee S-H, Kabbani N, Walikonis J, Wold B, and McKay R (2000). Enhanced Proliferation, Survival, and Dopaminergic Differentiation of CNS Precursors in Lowered Oxygen. Journal of Neuroscience 20, 7377–7383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavazoie M, Van der Veken L, Silva-Vargas V, Louissaint M, Colonna L, Zaidi B, Garcia-Verdugo JM, and Doetsch F (2008). A specialized vascular niche for adult neural stem cells. Cell Stem Cell 3, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna E, Gotz M, and Huttner WB (2014). The cell biology of neurogenesis: toward an understanding of the development and evolution of the neocortex. Annu Rev Cell Dev Biol 30, 465–502. [DOI] [PubMed] [Google Scholar]
- Uzquiano A, Gladwyn-Ng I, Nguyen L, Reiner O, Gotz M, Matsuzaki F, and Francis F (2018). Cortical progenitor biology: key features mediating proliferation versus differentiation. J Neurochem. [DOI] [PubMed] [Google Scholar]
- Yuen TJ, Silbereis JC, Silbereis JC, Griveau A, Griveau A, Chang SM, Chang SM, Daneman R, Daneman R, et al. (2014). Oligodendrocyte-Encoded HIF Function Couples Postnatal Myelination and White Matter Angiogenesis. Cell 158, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Movie 1, Related to Figure 1 3D render and animated tour of an iDisco cleared P7 cerebellum from an Atoh1EGFP/EGFP knockin animal stained with antibodies against EGFP (magenta) and Glut1 (cyan). Note: cyan label micro-blood vessels are generally located in progenitor free zones of the developing cerebellum. Rendering and animation were generated as a volume view in arivisAG Vision4D software.
Supplemental Movie 2, Related to Figure 3 Representative time-lapse sequence of CGNs labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 3, Related to Figure 3 Representative time-lapse sequence of CGNs labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 2% O2. Low O2 tension blocks the transition of CGNs to the ML and IGL. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 4, Related to Figure 4 Representative time-lapse sequence of Hif1αflx/flx CGNs expressing Creinactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 5, Related to Figure 4 Representative time-lapse sequence of Hif1αflx/flx CGNs expressing Creactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Hif1α deletion leads to early population of the ML and IGL of the cerebellar slice compared to Creinactive expressing cells. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 6, Related to Figure 4 Representative time-lapse sequence of VHLflx/flx CGNs expressing Creinactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. Time lapse encompasses roughly twelve hours of imaging time.
Supplemental Movie 7, Related to Figure 4 Representative time-lapse sequence of VHLflx/flx CGNs expressing Creactive labeled with H2B-mCherry (nucleus, red) and GPI-pHluorin (external face of plasma membrane, green) migrating in an ex vivo cerebellar slice preparation incubated at 20% O2. VHL deletion leads to retention of GNP or CGNs in the outer layer of the cerebellar slice compared to Creinactive expressing cells. Time lapse encompasses roughly twelve hours of imaging time.
Data Availability Statement
All array data was submitted to ArrayExpress and has the E-MTAB-8761 accession number. Original imaging data available upon request.