

Abstract
The GTPase OPA1 and the AAA-protease OMA1 serve well-established roles in mitochondrial stress responses and mitochondria-initiated cell death. In addition to its role in mitochondrial membrane fusion, cristae structure, and bioenergetic function, OPA1 controls apoptosis by sequestering cytochrome c (cyt c) in mitochondrial cristae. Cleavage of functional long OPA1 (L-OPA1) isoforms by OMA1 inactivates mitochondrial fusion and primes apoptosis. OPA1 cleavage is regulated by the prohibitin (PHB) complex, a heteromeric, ring-shaped mitochondrial inner membrane scaffolding complex composed of PHB1 and PHB2. In neurons, PHB plays a protective role against various stresses, and PHB deletion destabilizes OPA1 causing neurodegeneration. While deletion of OMA1 prevents OPA1 destabilization and attenuates neurodegeneration in PHB2 KO mice, how PHB levels regulate OMA1 is still unknown. Here, we investigate the effects of modulating neuronal PHB levels on OMA1 stability and OPA1 cleavage. We demonstrate that PHB promotes OMA1 turnover, effectively decreasing the pool of OMA1. Further, we show that OMA1 binds to cardiolipin (CL), a major mitochondrial phospholipid. CL binding promotes OMA1 turnover, as we show that deleting the CL-binding domain of OMA1 decreases its turnover rate. Since PHB is known to stabilize CL, these data suggest that PHB modulates OMA1 through CL. Furthermore, we show that PHB decreases cyt c release induced by tBID and attenuates caspase 9 activation in response to hypoxic stress in neurons. Taken together, our results suggest that PHB-mediated CL stabilization regulates stress responses and cell death through OMA1 turnover and cyt c release.
Subject terms: Neuroscience, Neurological disorders
Introduction
The prohibitin (PHB) complex is an inner membrane megaDalton-sized scaffold composed of the highly conserved proteins PHB1 and PHB2. Several functions have been proposed for PHB, including scaffolding mitochondrial proteins, regulating mitochondrial morphology and cristae architecture, signaling for mitophagy, and stabilizing mitochondrial lipids [1, 2]. Specifically, studies have demonstrated that PHB is involved in mitochondrial lipid maturation and binds the phospholipid cardiolipin (CL) [3, 4], a major component of the mitochondrial inner membrane (IMM). PHB was also shown to stabilize the respiratory chain supercomplexes and promote respiratory functions in neural cells [3]. Another function of PHB is thought to be the stabilization of the GTPase optic atrophy 1 (OPA1), which plays a fundamental role in regulating inner membrane fusion and cristae structure [5–9]. Proteolytic cleavage of long OPA1 (L-OPA1) isoforms into short OPA1 (S-OPA1) isoforms regulates OPA1 function. Ablation of PHB2 destabilizes L-OPA1, disrupting cristae structure and resulting in neurodegeneration [10]. On the other hand, increasing PHB is beneficial, as potent neuroprotection is conferred by PHB overexpression in neurons subjected to various stressors, in vitro and in vivo [3, 11–13]. The ATP-independent mitochondrial protease OMA1 cleaves OPA1 in response to stress [14–17]. However, whether PHB regulates OMA1 activity and L-OPA1 cleavage under stress is unknown. Here, to address the role of PHB in regulating OMA1 activity and OPA1 cleavage in basal conditions and under mitochondrial stress, we modulate the expression of PHB in neurons and assess the effects on OPA1 and OMA1. Surprisingly, we find that PHB overexpression increases basal L-OPA1 processing and does not preserve L-OPA1 from stress-induced cleavage. However, we show that PHB regulates OMA1 turnover, revealing a novel role of PHB in mitochondrial proteostasis. We also show that OMA1 binds to CL and that CL binding promotes OMA1 turnover, suggesting that PHB regulation of OMA1 could occur through its CL-stabilizing function.
Results
PHB does not affect depolarization-induced OPA1 cleavage in neurons, despite regulating OMA1 levels
Ablation of the PHB complex by PHB2 knockout was shown to destabilize L-OPA1 [10]. However, whether increasing PHB affects OPA1 processing is unknown. Surprisingly, we found that transient overexpression of both subunits of the PHB complex, PHB1 and PHB2, in mouse neuroblastoma Neuro-2a (N2a) cells results in a decrease in L-OPA1 content relative to vector control cells at baseline (Fig. 1a, b). Moreover, upon treatment with the membrane potential (ΔΨm) dissipating protonophore carbonyl cyanide m-chlorophenyl hydrazone (CCCP), processing of L-OPA1 was similar in PHB1/2 overexpressing cells and vector controls. This result was confirmed in another neuron-like cell line (PC12 cells) stably overexpressing PHB1 [3] exposed to oxidative stress. PHB1-expressing PC12 cells showed decreased L-OPA1 at baseline and after treatment with H2O2 relative to control cells (Supplementary Fig. 1A, B). In addition, we found that the differentiation of PHB1-expressing PC12 cells induced by nerve growth factor (NGF) was increased relative to vector controls, with no change in neurite length, indicating that a slight decrease in L-OPA1 is not detrimental to neurons (Supplementary Fig. 1C–E).
Fig. 1. PHB modulates OPA1 and OMA1 levels and attenuates cytochrome c release, but does not prevent CCCP-induced OPA1 processing.

a Representative western blot of OPA1 long (L-OPA1) and short (S-OPA1) isoforms in N2a cells transiently overexpressing PHB1/2 (PHB) or empty vector. Cells were treated with 10 µM CCCP or vehicle (DMSO). β-actin was used as a loading control. b The ratio of L-OPA1 to S-OPA1 was quantified from n = 4 experiments. *p < 0.05, Student’s t test. c Cytochrome c content in mitochondrial pellets (P) or released into supernatant (S) measured by western blots of mitochondria isolated from N2a cells overexpressing PHB or vector control incubated for 30 min with 100 nM tBID at 30 °C. TIM23 served as a mitochondrial protein loading control. d Quantification of cytochrome c localization in tBID treated mitochondria relative to total cytochrome c detected, n = 5 experiments. *p < 0.05 S relative to P fraction, Student’s t test. e Schematic representation of C-terminal OMA1-Flag constructed and proposed activating cleavage site (dashed line) and resulting OMA1 isoforms. f OMA1 activation visualized by western blot of lysates prepared from N2a cells transiently expressing a C-terminal OMA1-Flag and treated with 10 µM CCCP or vehicle (DMSO). Blots were probed with antibodies to OPA1, Flag, and β-actin as a loading control. g Western blot of whole cell lysates from N2a cells overexpressing a C-terminal OMA1-Flag construct plus PHB1/2, siRNA targeting PHB1, or scrambled (Scr) control siRNA. Blots were probed with specific antibodies to Flag, PHB1, PHB2, and β-actin as a loading control. h Quantification of relative OMA1-Flag band intensity normalized by β-actin from n = 4 experiments. *p < 0.05, **p < 0.01 relative to Scr controls, Student’s t test.
Cleavage of L-OPA1 and disruption of OPA1 oligomers have been shown to cause release of proapoptotic cytochrome c from IMM cristae [7, 9, 18–20]. Given the effect we observed of PHB on L-OPA1 stability, we investigated if PHB also regulates cytochrome c release. Isolated mitochondria from N2a cells overexpressing PHB1/2 and vector controls were treated with tBID to induce cytochrome c release [7, 21]. PHB1/2 overexpression attenuated tBID-induced cytochrome c release from mitochondria (Fig. 1c, d). These data suggest that PHB overexpression prevents the induction of apoptosis, despite increasing OPA1 proteolytic processing, dissociating the antiapoptotic effect of PHB from its effect on OPA1.
The protease that cleaves OPA1 is OMA1, which is activated by proteolytic cleavage under stress conditions [14–17]. Many proteases are produced as zymogens, inactive proenzymes, and their activity is regulated by continuous turnover until a stimulus removes the inhibitory prodomain to activate the enzyme [22]. OMA1 appears to be regulated in this manner, as it is generated as a proenzyme and becomes active when stress stimulates its autocatalytic cleavage to remove the C-terminal prodomain, generating a very short-lived active enzyme [14, 17], which is difficult to detect (Fig. 2a). To assess the effects of modulating PHB levels on the processing and turnover of OMA1 at baseline and under mitochondrial depolarization, we utilized a C-terminally Flag-tagged OMA1 construct (Fig. 1e) due to the unavailability of reliable antibodies targeting the inactive protein (Supplementary Fig. 2A, B). In vehicle treated cells, this construct expressed a band migrating at ~43 kDa, which has been proposed to be the inactive pro-OMA1 [14, 17] (Fig. 1f). Upon CCCP treatment, this OMA1 band disappeared, while L-OPA1 was cleaved (Fig. 1f), indicating that OMA1 was activated and demonstrating that OMA1-Flag performed as expected. Using an established OMA1 KO mouse embryonic fibroblast line [23] and N2a cells silenced for OMA1, we confirmed that OMA1 is the protease responsible for cleaving OPA1 in response to CCCP (Supplementary Fig. 2A, C), as previously described [14–17].
Fig. 2. OMA1 turnover is regulated by PHB.

a Western blot of lysates from OMA1 KO MEFs transiently transfected with an internally Flag-tagged OMA1 construct and PHB1/2 or vector control probed with specific antibodies to Flag, PHB1, and β-actin as a loading control. b OMA1-Flag bands were quantified relative to β-actin from n = 5 experiments. *p < 0.05, **p < 0.01, Student’s t test. c Western blot of N2a cells transfected with internal Flag-OMA1 treated with 10 µM CCCP for 30 min or vehicle (DMSO). d OMA1-Flag bands were quantified relative to β-actin from n = 5 experiments. ***p < 0.001, Student’s t test. e Western blot of N2a cells transfected with empty vector or full length (FL) or deletion (Δ245–305) internal OMA1-Flag constructs. Hyphen indicates an empty lane to control for background signal. Blots were probed with specific antibodies to Flag and β-actin as a loading control. f Schematic representation of internally Flag-tagged OMA1 constructs with cleavages sites for the MTS and inhibitory C-terminus (activating).
To ascertain the effect of PHB expression on OMA1, we transiently transfected OMA1-Flag together with PHB1/2 or siRNA targeting PHB1, in N2a cells (Fig. 1g). PHB1/2 overexpression reduced the content of OMA1 by more than 50%, while PHB1 silencing, which as expected also decreases PHB2 [24], resulted in an accumulation of pro-OMA1 (Fig. 1g, h). These data demonstrate that PHB regulates the levels of OMA1.
PHB regulates OMA1 processing and degradation
OMA1 is cleaved at its N-terminus to remove the mitochondrial-targeting sequence (MTS) and, upon mitochondrial depolarization, at its C-terminus to activate the proteolytic function [14, 17]. Therefore, to obtain a comprehensive view of OMA1 cleavage, which is not feasible with only the C-terminal Flag construct, we generated a second OMA1 construct with an internal Flag tag after amino acid 212, immediately following the transmembrane domain of the protein (Fig. 2f). In OMA1 KO MEFs, this construct produced the expected bands for full-length OMA1, containing the MTS (~60 kDa), and pro-OMA1 (~43 kDa), but not the band corresponding to the proposed active S-OMA1 (~37 kDa) [17] (Fig. 2a). Instead, we detected Flag-immunoreactive bands between 15 and 20 kDa (Fig. 2a), suggestive of degradation products (deg-OMA1). With this OMA1 construct, we showed that PHB1/2 overexpression decreases the levels of both pro-OMA1 and deg-OMA1 (Fig. 2a, b). Treatment of N2a cells expressing this OMA1 construct with CCCP resulted in the loss of pro-OMA1, indicating OMA1 processing into its active form, and confirming that inserting an internal Flag tag did not impair sensitivity of the protein to depolarization (Fig. 2c, d). CCCP also increased the proportion of deg-OMA1 to total OMA1, while the total amount of OMA1 was unchanged by CCCP treatment. To further investigate if deg-OMA1 is the result of a discrete cleavage event of pro-OMA1 we generated a third internal Flag-OMA1 construct, in which we deleted the region containing a presumptive cleavage site, based on the size of the cleavage products of the other constructs (deletion of amino acids 245–305, Fig. 2f). Deleting this internal site of OMA1 did not prevent the generation of deg-OMA1 fragments in N2A cells (Fig. 2e), suggesting that deg-OMA1 is not the result of a discrete cleavage event, but more likely results from C-terminal degradation of OMA1, which produces multiple Flag-immunoreactive bands. These data indicate that processing of OMA1 generates previously unreported deg-OMA1 fragments, and demonstrate that PHB regulates the total content of OMA1.
PHB regulates OPA1 and OMA1 processing in primary cortical neurons
Maintenance of L-OPA1 is essential for neuronal viability, and loss of OPA1 results in neurodegeneration [25, 26]. Since we find that PHB regulates OPA1 and OMA1 in neuronal cell lines, we sought to determine if PHB also governs the processing of these proteins in primary neurons. First, we observed a decrease in L-OPA1 in primary neurons expressing PHB1/2 by lentiviral transduction (Fig. 3a, b). Second, we found that overexpression of PHB1/2 did not prevent L-OPA1 processing in neurons treated with CCCP (Fig. 3a, b). Third, by transduction with lentivirus expressing internal OMA1-Flag (Fig. 2f) we found that inactive pro-OMA1 levels were decreased in primary neurons overexpressing PHB1/2 (Fig. 3c, d). At baseline, deg-OMA1 was detected in neurons, demonstrating that these fragments of OMA1 are consistently observed across different cell models. When neurons were treated with CCCP, the processing of pro-OMA1 to deg-OMA1 in PHB1/2 overexpressing neurons was significantly less than in vector transduced cells (Fig. 3c, e). Taken together these results validate the findings obtained in cell lines, showing that PHB regulates the processing and turnover of OMA1 and OPA1 in primary neurons.
Fig. 3. PHB regulates OMA1 cleavage in neurons.

a Western blot of primary cortical neurons transduced with lentivirus to overexpress PHB1/2 or vector control treated with 10 µM CCCP. Blots were probed with specific antibodies to PHB1, PHB2, OPA1, and β-actin as a loading control. b The ratio of L-OPA1 to S-OPA1 was quantified from n = 5 experiments. **p < 0.01, Student’s t test. c Western blot of primary cortical neurons transduced with lentivirus containing internally Flag-tagged OMA1 and either PHB1/2 or vector control treated with 10 µM CCCP. Blots were probed with specific antibodies to Flag and β-actin as a loading control. d Quantification of pro-OMA1-Flag band (~43 kDa) relative to untreated vector control. n = 3 experiments. *p < 0.05, Student’s t test. e Quantification of deg-OMA1-Flag band (~18 kDa) relative to untreated vector control. n = 3 experiments. *p < 0.05, Student’s t test. f Quantification of neuronal viability for each condition after 4 h OGD plus 24 h reoxygenation. n = 5 experiments. The fraction of alive, calcein positive cells is expressed relative to sham-treated controls. **p < 0.01 relative to vector. g Caspase 9 activity was assayed in neuronal lysate after 4 h OGD plus reoxygenation for the indicated time and expressed as a percentage of vector sham-treated condition. n = 3. *p < 0.05. h Mitochondrial length in µm measured in neurons expressing mito-DsRed2 plus PHB1/2 or vector during OGD and after reperfusion quantified using MetaMorph. n = 4 experiments.
Proteolytic processing of L-OPA1 has been observed in models of neuronal hypoxia [27–30]. To recapitulate pathophysiological features of ischemia-reperfusion injury, we subjected neurons to oxygen and glucose deprivation (OGD), a more disease relevant model system than mitochondrial stress induced by CCCP. Primary neurons transduced with PHB1/2 were protected from OGD-induced cell death assessed 24 h after reoxygenation (Fig. 3f). Decreased cell death corresponded to decreased activation of apoptosis measured by caspase 9 activity 8 h after reoxygenation (Fig. 3g). Furthermore, as anticipated, neurons exposed to OGD stress developed decreased average mitochondrial length, but surprisingly PHB overexpression did not prevent mitochondrial fragmentation (Fig. 3h). Together, these data suggest that PHB protects neurons from hypoxia-induced cell death despite increased OPA1 cleavage at baseline (Fig. 3a, b) and indicate that PHB neuroprotection and mitochondrial morphological changes are dissociated events in the context of ischemia-reperfusion injury.
PHB regulation of OMA1 processing is not dependent on ΔΨm or Yme1L
We show here that PHB regulates OMA1 processing from pro-OMA1 to deg-OMA1, and it is also known that OMA1 activity can be regulated by ΔΨm [14, 15, 17]. Therefore, to determine if PHB levels regulate OMA1 by altering ΔΨm we modulated its expression in N2a cells and assessed ΔΨm by TMRM fluorescence. Neither PHB1/2 overexpression nor PHB1 downregulation by siRNA significantly altered ΔΨm (Fig. 4a, b). Furthermore, since the mitochondrial protease Yme1L has been shown to cleave OMA1 under certain stress conditions [31], we assessed the effects of modulating PHB on the levels of Yme1L. We show that PHB1/2 overexpression and PHB1 silencing do not alter Yme1L levels in N2A cells (Fig. 4c, d). Moreover, to further demonstrate that Yme1L does not mediate the effects of PHB on OMA1, we silenced Yme1L in N2a cells and assessed the effects on PHB1/2 regulation of OMA1 processing from pro-OMA1 to deg-OMA1. Yme1L downregulation did not affect PHB1/2 overexpression effects on pro-OMA1, but it increased the levels of deg-OMA1, suggesting that Yme1L is involved in the elimination of deg-OMA1. Overall, these results indicate that PHB regulation of OMA1 processing is not dependent on ΔΨm or Yme1L, and suggest that autocatalytic turnover of OMA1 can be activated in a PHB1/2-dependent and ΔΨm-independent manner.
Fig. 4. PHB does not alter mitochondrial membrane potential or Yme1L levels.

a Membrane potential was measured in N2a cells transiently expressing empty vector, PHB1/2, scrambled siRNA control, or siRNA targeting PHB1 using 50 nM TMRM fluorescence (left panels). Background fluorescence was measured after treatment with 10 µM CCCP for 30 min (right panels). Scale bar = 25 µm. b Quantification of average TMRM fluorescence minus background fluorescence after CCCP. n = 5 vect, n = 12 PHB, n = 5 Scr, and n = 12 siPHB wells. c Western blots of whole cell lysates from N2a cells expressing empty vector, PHB1/2, scrambled siRNA control, or siRNA targeting PHB1. Blots were probed with specific antibodies to Yme1L and β-actin as a loading control. d Quantification of relative Yme1L band intensity normalized by β-actin from n = 4 independent experiments. e Western blots of whole cell lysates from N2a cells transfected with internally Flag-tagged OMA1 plus siRNA targeting Yme1L or scrambled siRNA control. Blots were probed with specific antibodies to Flag, Yme1L, PHB1, and β-actin as a loading control.
OMA1 binds CL and is part of high-molecular-weight complexes in mitochondria
PHB binds CL and regulates its levels in mitochondria [3, 4]. PHB is also essential to stabilize large molecular weight protein complexes in the IMM [3]. Furthermore, OMA1 contains an N-terminal hydrophobic region and positively charged residues that are required for depolarization-induced activation [14], which could act as a sensor via ionic interactions with lipid membranes. We and others have demonstrated that PHB regulates CL content in mitochondria. Thus, to test if OMA1 binds CL, we obtained native protein lysates from N2a mitochondria expressing the internal OMA1-Flag construct. We showed that OMA1 was pulled down by CL-coated beads, but not by control beads without CL (Fig. 5b). Moreover, a OMA1 deletion mutant devoid of the putative CL-binding domain [14] (Δ144–163, Fig. 5a), failed to bind CL-coated beads (Fig. 5b).
Fig. 5. OMA1 binds CL and forms HMW complexes that co-migrate with PHB.
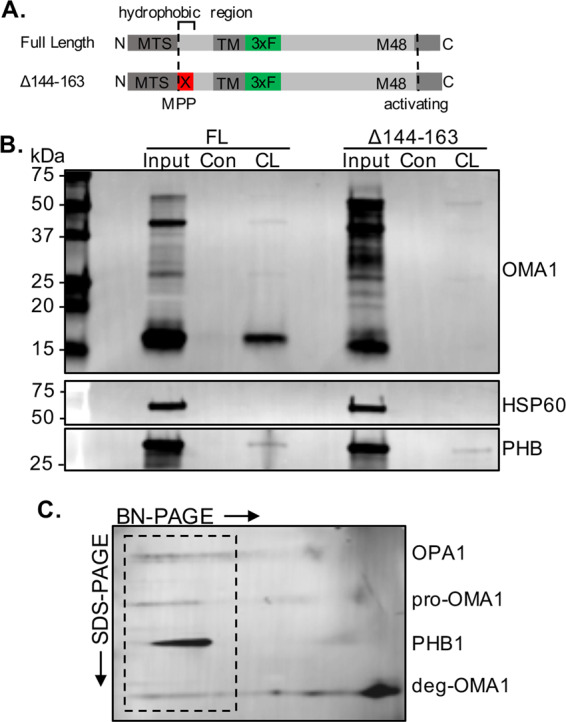
a Schematic representation of full length (FL) and Δ144–163 deletion mutant internally Flag-tagged OMA1 constructs. b Western blot of CL-binding proteins pulled down using CL-conjugated beads from crude mitochondria isolated from N2a cells expressing either full length (FL) or CL-binding domain deletion (Δ144–163) internally Flag-tagged OMA1 constructs. Blots were probed with specific antibodies to Flag, PHB1 as a positive CL-binding control, and HSP60 as a non-CL-binding control. c 2D BN-PAGE of digitonin-treated mitochondria from N2a cells expressing internally Flag-tagged OMA1. Blot probed with specific antibodies to OPA1, Flag, and PHB1. OPA1, Flag, and PHB immunoreactivity align in high-molecular-weight complexes (dashed box).
PHB1/2 form large molecular weight complexes in mitochondria and previous studies have reported that OMA1 is a part of large protein complexes in yeast and mammalian cells [14, 32]. Given the functional interaction between PHB and OMA1, we investigated the participation of OMA1 and PHB in high-molecular-weight complexes with OPA1. We separated native proteins from N2a cells expressing Flag-tagged OMA1 by 2D blue native polyacrylamide gel electrophoresis (BN-PAGE). We detected pro-OMA1 (~43 kDa) in high-molecular-weight complexes whose migration overlapped with both PHB and OPA1 (Fig. 5c, outlined region). Interestingly, a fraction of deg-OMA1 (~15–20 kDa) co-migrated with PHB, OPA1, and pro-OMA1 in the high-molecular-weight complexes. Deg-OMA1 was also detected in smaller complexes corresponding to the size of previously reported structures [14, 32], in addition to a large portion of monomeric form. Taken together, these results confirm that OMA1 binds CL and suggest that it is part of a large molecular complex, which includes PHB1/2 and OPA1.
Discussion
Our results identify a role for the PHB complex in regulating OMA1 turnover in cell lines and neurons. While the precise molecular mechanisms of this regulation remain to be fully elucidated, we hypothesize that PHB mediates OMA1 processing from pro-OMA1 to deg-OMA1 through its CL-binding and stabilizing properties [3, 4], as we report the novel finding that OMA1 binds CL and participates in high-molecular-weight complexes, which may also contain PHB1/2 and OPA1. OMA1 activation occurs in depolarized mitochondria by an autocatalytic cleavage [14], but OMA1 can also be cleaved by the protease Yme1L in certain stress conditions [31]. However, since Yme1L levels were unchanged by altering PHB expression and silencing of Yme1L did not alter the effects of PHB on pro-OMA1, Yme1L is unlikely to mediate the effect of PHB on OMA1.
Using an internally Flag-tagged OMA1 construct, we observed OMA1 cleavage products defined here as deg-OMA1, which accumulate in response to mitochondrial depolarization. Deletion mutant studies suggested that deg-OMA1 likely derives from C-terminal degradation of the protein. Interestingly, data suggest that Yme1L is involved in the turnover of deg-OMA1, as deg-OMA1 accumulates in Yme1L silenced cells.
The prodomain of OMA1 is required for its activation in response to ΔѰm loss, suggesting a functional role beyond inhibiting peptidase activity [14]. Zymogen prodomains can also regulate the localization and compartmentalization of enzymes [22]. Here, we showed that OMA1 binds CL, raising the possibility that the N-terminal prodomain sequesters OMA1 in CL microdomains in the IMM. A functional relationship between CL and OMA1 is supported by data demonstrating that OMA1 is activated by loss of DNAJC19, a CL structuring protein related to the PHB complex [4]. Interestingly, depletion of DNAJC19 does not change ΔѰm [4], similar to PHB depletion, suggesting that OMA1 stability is affected by CL-binding IMM proteins, and can be regulated independent of ΔѰm. Further, OMA1 contains a domain that mediates its stability and function [14], which we show to be critical for CL binding. Both pro-OMA1 and deg-OMA1 are decreased by PHB overexpression. Thus, we could speculate that PHB regulates OMA1 through CL interactions, by promoting turnover and decreasing the pool of pro-OMA1 available to be activated under mitochondrial stress. In this way, PHB could fine-tune OMA1 levels as a stress-responsive rheostat.
Despite leading to increased L-OPA1 processing, PHB overexpression decreases cytochrome c release induced by tBID from isolated mitochondria and attenuates apoptosis in neurons exposed to OGD. These results suggest that PHB mediates a protective effect independently of preventing OPA1 cleavage. The mechanisms by which PHB prevents apoptosis remain to be fully elucidated, but some hypotheses can be generated from our results. It is well-established that CL binds cytochrome c and is required for its cristae localization [33, 34]. Oxidation and loss of CL, resulting in cytochrome c release, are among the first steps in the induction of apoptosis [35, 36]. Because PHB binds to and stabilizes CL in mitochondria [3, 4] and reduces reactive oxygen species production [13], the role of PHB in regulating pro-OMA1, cytochrome c release, and cell death may be mediated by a common mechanism involving CL stabilization.
Interestingly, decreasing pro-OMA1 did not prevent OPA1 cleavage induced by CCCP, demonstrating that OMA1 is maintained at levels that exceed the amount required to maximally cleave OPA1 upon mitochondrial depolarization. This excess pro-OMA1 points to functions other than OPA1 processing, but very few have been identified thus far. In addition to itself and OPA1, OMA1 has been reported to cleave the protein UQCC3 upon mitochondrial depolarization [37]. UQCC3 binds CL and is involved in the stability of respiratory chain supercomplexes, a function we and others have reported for PHB [3, 24]. Further, OMA1 is required for the stability of supercomplexes and maximal respiratory output from mitochondria [32]. Taken together, these observations implicate OMA1 as a critical regulator of mitochondrial functions beyond OPA1 cleavage.
In summary, we uncover a previously unappreciated role of PHB in the regulation of the mitochondrial stress-responsive protease OMA1 in neurons. Using a model of neuronal hypoxia, we also demonstrate that modulation of PHB expression regulates apoptosis, independently of OPA1 and mitochondrial fission. These data expand our molecular understanding of the role of PHB in mitochondrial function and provide novel insights into the regulation of mitochondrial cell death pathways.
Materials and methods
Cell culture and primary cortical neuron cultures
The N2a mouse neuroblastoma cell line (ATCC® CCL-131™) was cultured in DMEM with 10% fetal bovine serum at 37 °C in a 5% CO2 incubator. PC12 cells stably overexpressing PHB1 were previously described [3]. The generation of OMA1 knockout mouse embryonic fibroblasts was previously described [23]. Cells were tested periodically for mycoplasma contamination by PCR. Cells were passaged regularly and plated for individual experiments by dissociating them with 0.25% Trypsin-EDTA up to 20 total passages. Where indicated, cells were transfected with Lipofectamine 3000 (Thermo Fisher) according to manufacturer’s protocol and assayed at 48 h post transfection. Cells were treated with 10 µM carbonyl cyanide m-chlorophenyl hydrazine (CCCP) or DMSO (vehicle control) in serum-free media where indicated.
Experimental protocols using timed pregnancies were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medicine. Pregnant C57BL/6 mice at embryonic day 16.5 (E16.5) were euthanized under deep anesthesia. Primary cortical neurons were isolated and dissociated as described previously [13]. Briefly, cortices from embryos were removed and washed in ice-cold HBSS before incubation with 0.25% trypsin and 0.15 µg/mL DNase. Cortical tissue was triturated with 30 passages through a P1000 tip to dissociate cells. Cells were spun down and resuspended in Neurobasal Media plus B27 supplement (Thermo Fisher). Cells were then plated on poly-L-ornithine coated dishes and kept at 37 °C in a 5% CO2 incubator. Media was changed completely at 1 day in vitro (DIV) and half media was replaced every 2 days thereafter. To transduce primary neurons, lentiviral particles were added to neurons at a multiplicity of infection (MOI) of 3 on 5 DIV. Cells were incubated with lentivirus overnight before half media was exchanged. Neurons were used at 12–14 DIV. For transient overexpression or knock down experiments N2a cells were transfected with Lipofectamine 3000 (Thermo Fisher) according to manufacturer’s protocols. Oligonucleotides used for Yme1L and PHB1 silencing and negative control were from Thermo Fisher Silencer Select library.
Cloning and lentivirus production
Plasmids in the pcDNA3.1 backbone were used for transient transfection of cell lines and in pLenti lentivector under the control of a CMV promoter for lentiviral production. cDNA clones of mouse PHB1, PHB2, and flag-tagged OMA1 sequences were obtained from OriGene. Restriction sites were added to inserts by overlap extension PCR before ligation into digested plasmid DNA. Internally Flagged OMA1 constructs were manufactured by Genscript and ligated into either the pLenti backbone or pcDNA3.1. Constructs were verified by sequencing.
Lentiviral particles were produced in HEK293T cells using a second-generation system following a standard protocol [38]. Briefly, HEK293T cells were transfected with the pLenti vector containing cDNA inserts, packaging vector pCMV-Δ8.91, and VSVG envelope vector pMD2.G using Lipofectamine 3000. After 6 h, media was changed to B27 supplemented Neurobasal (Thermo Fisher). Culture media containing lentivirus was collected after 24 h and replaced with fresh media for another 24 h before a second collection. The resulting lentivirus was pooled, spun at 1000 × g to remove debris, filtered through a 0.45 µm polyvinylidene difluoride (PVDF) filter, and frozen in aliquots at −80 °C until use.
Oxygen-glucose deprivation (OGD) model
To model ischemia-like conditions, primary neurons were exposed to glucose-free buffered saline solution (BSS; 138 mM NaCl, 5 mM KCl, 1.2 mM NaH2PO4, 20 mM NaHCO3, 1.8 mM CaCl2, 0.9 mM MgCl2, 15 mM HEPES) bubbled with a 95% nitrogen, 5% CO2 gas mixture for 5 min to remove oxygen. Cells were placed in a Billups-Rothenberg modular incubation chamber flushed with the same gas mixture for 6 min. Sham controls received the same number of media changes but were incubated with BSS containing 10 mM glucose in a normoxic incubator. Reoxygenation was accomplished by replacing BSS or BSS with glucose with regular glucose-containing culture media and returning cells to a normoxic incubator.
Neuronal viability and mitochondrial morphology imaging
To assess neuronal viability, cells were stained with 1 µM calcein red-orange (Thermo Fisher) and 1 µg/mL Hoescht (Thermo Fisher) for 30 min and washed twice with phosphate buffered saline (PBS). Neuronal viability was assessed using a Nikon TE2000 inverted fluorescence microscope. Neurons were identified from other cell types by their polar morphology and viable cells counted based on the presence of calcein fluorescence and intact, uniform nuclei stained by Hoescht. Nonviable neurons were apparent in bright field images but were not stained or weakly stained by calcein and contained fragmented, pyknotic nuclei. The fraction of viable cells to total cells was expressed relative to untreated or sham treated, parallel control cultures transduced with the same lentivectors.
For mitochondrial length measurements, primary neurons transduced with a dsRed2 fluorescent protein targeted to mitochondria (mito-dsRed2) at 5 DIV were imaged at 12 DIV with or without OGD/reoxygenation. Neurons plated on poly-L-ornithine coated No. 1.0 coverglass were fixed in 4% paraformaldehyde/0.1 M phosphate buffer for 15 min and mounted on slides using Fluoromount G (Electron Microscopy Sciences). Mitochondria were visualized using a Leica TCS SP5 confocal laser-scanning microscope (Mannheim, Germany). Images were captured with a 63× oil-immersion objective with a 4× optical zoom. Z-stack images (400 Hz, 1024 × 1024) at 0.1 µM intervals were captured throughout the thickness of a cell. Mitochondrial length was measured using MetaMorph software (Molecular Devices).
Crude mitochondrial preparation
N2a cells were washed once with ice-cold PBS and scraped from plates in ice-cold mannitol-sucrose buffer (MS-EGTA; 225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 1 mM EGTA, 0.4 mg/mL fatty-acid free BSA, pH 7.4). Cells were pelleted at 800 × g for 5 min and homogenized in a glass/Teflon homogenizer in MS-EGTA buffer. Remaining intact cells were pelleted with another spin at 800 × g and supernatant containing mitochondria was collected and spun at 10,000 × g for 15 min at 4 °C. The resulting pellet was washed once in MS-EGTA and spun again at 10,000 × g for 15 min. Mitochondrial fractions were resuspended in a buffer containing 225 mM mannitol, 75 mM sucrose, 5 mM HEPES, 0.1 mM EGTA, and 0.1 mg/mL fatty-acid free BSA. Protein content was measured using the BCA Protein Assay (Pierce).
Western bot analyses
Whole cell lysates were prepared by lysing cells in RIPA buffer (50 mM Tris HCl pH 7.4, 150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate, 1.0 mM EDTA, 0.1% SDS) plus 1× protease inhibitors (Complete Mini; Roche). Protein content in samples was assessed using a BCA Protein Assay Kit (Pierce) and equal amounts of proteins were loaded on Tris-Glycine gels and separated by SDS-PAGE. Proteins were transferred to PVDF membranes and blocked with 5% BSA followed by immunoblotting with indicated antibodies overnight at 4 °C. Antibodies used were anti-OPA1 (1:1000, BD Biosciences 612606), anti-β-actin (1:3000, Cell Signaling Technology, 4970), anti-Flag-M2 (1:1000, Sigma, F1804), anti-PHB1 (1:1000, clone # ll-14–10, Thermo Fisher, MA5-12858), anti-PHB2 (1:1000, Cell Signaling Technology, 14085), anti-cytochrome c (1:500, A-8, Santa Cruz Biotechnology, sc-13156), anti-Yme1L (1:1000, ProteinTech, 11510-1-AP). To detect antibody binding, blots were probed with anti-mouse (1:10,000, LI-COR Biosciences, 926-68072) or anti-rabbit IgG secondary antibodies (1:10,000, LI-COR Biosciences, 926–32213) for 1 h at room temperature then imaged using the LI-COR CLx imaging system. Band intensities were quantified using Image Studio software (vs3.1, LI-COR Biosciences). In the Yme1L silencing experiments, the protein was detected with enhanced chemiluminescence in a Chemidoc (BioRad), using SuperSignal West Femto substrate (Thermo Fisher, 34095).
Blue native polyacrylamide gel electrophoresis
Crude mitochondria preparations from N2a cells were used for 2D blue native polyacrylamide gel electrophoresis (BN-PAGE) performed as described previously [39]. Briefly, mitochondria were lysed in digitonin (8 mg/ml) for 20 min followed by a clearing spin at 20,000 × g. The resulting supernatant was collected and 10× sample buffer (750 mM aminocaproic acid, 50 mM Bis-Tris, 0.5 mM EDTA, 5% Coomassie Brilliant Blue G) was added. Solubilized native proteins were separated in the first dimension on 4–12% NativePAGE gradient gels (Thermo Fisher). Individual sample lanes from these gels were removed and incubated in 1× Laemmli sample buffer containing β-mercaptoethanol. The resulting gel strips containing denatured proteins were loaded onto 12% Tris-Glycine gels and separated by conventional SDS-PAGE. Separated proteins were transferred to PVDF membranes for immunoblotting.
Caspase 9 activity assay
Primary neurons were washed once in ice-cold PBS and homogenized in lysis buffer (5 mM MgCl2, 1.5 mM EDTA, 1 mM EGTA, 25 mM HEPES pH 7.4, 2 mM DTT, 0.1% Triton-X, 1× protease inhibitor cocktail), incubated on ice for 10 min, and centrifuged at 13,000 × g for 5 min. The supernatant was collected and mixed with an equal volume of 2× assay buffer (20 mM HEPES pH 7.4, 100 mM NaCl, 1 mM EDTA, 0.1% CHAPS, 10% sucrose, 10 mM DTT) containing 5 µM of the caspase 9 substrate AC-LEHD-AFC. Samples were incubated for 1 h at 37 °C before fluorescence was read by plate reader at 405 nm excitation and 505 emission.
Cytochrome c release assay
Crude mitochondria (50 µg) were suspended in isolation buffer containing 1× protease inhibitor and 10 µM Boc-D-FMK. Mitochondria were treated with a final concentration of 100 nM caspase 8-cleaved BID (tBID) or tBID dilution buffer (25 mM HEPES-KOH pH 7.4, 100 mM KCl, 0.1 mg/mL fatty-acid free BSA) as a control for 30 min at 30 °C. After treatment, mitochondrial fractions were separated by spinning at 10,000 × g for 15 min and resuspended in 1× SDS sample buffer. 4× SDS sample buffer was added to a final 1× concentration in the supernatant collected after mitochondria were spun down. Samples were resolved by SDS-PAGE and analyzed by western blotting as described above.
CL-binding assay
CL-binding proteins were pulled down with CL-coated beads (Echelon Biosciences) from 100 µg of crude mitochondria isolated from N2a cells. Mitochondria were lysed in 1.0% NP-40, 150 mM NaCl, 10 mM HEPES, pH 7.4 on ice for 30 min, and the resulting lysate was incubated overnight with CL-coated or uncoated control beads. Beads were washed five times with 0.5% NP-40, 150 mM NaCl, 10 mM HEPES, pH 7.4, and bound proteins were eluted in 2× SDS sample buffer. Eluted samples and 25 µg of unbound mitochondrial input lysate were resolved by SDS-PAGE and immunoblotted as indicated.
Statistics
All experiments were repeated at least three times (exact n is provided in the figure legends) and the statistical significance was evaluated. Data in bar graphs are presented as mean ± standard error of the mean (SEM). Statistical differences between experimental and control groups were evaluated by unpaired two-tailed Student’s t test. The threshold for significance was set at p < 0.05.
Supplementary information
Acknowledgements
We are grateful to Drs Carlos Lopez-Otin and Pedro M. Quiros (University of Oviedo) for kindly sharing with us the OMA1+/+ and OMA1−/− MEFs. This work was supported by grants 1R01NS095692 (to GM and CI) and 1R01NS067078 (to PZ).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Footnotes
Edited by N. Chandel
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Giovanni Manfredi, Phone: +646-962-8172, Email: gim2004@med.cornell.edu.
Costantino Iadecola, Phone: 646-962-8279, Email: coi2001@med.cornell.edu.
Supplementary information
The online version of this article (10.1038/s41418-019-0469-4) contains supplementary material, which is available to authorized users.
References
- 1.Merkwirth C, Langer T. Prohibitin function within mitochondria: essential roles for cell proliferation and cristae morphogenesis. Biochim Biophys Acta. 2009;1793:27–32. doi: 10.1016/j.bbamcr.2008.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Osman C, Merkwirth C, Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J Cell Sci. 2009;122:3823–30. doi: 10.1242/jcs.037655. [DOI] [PubMed] [Google Scholar]
- 3.Anderson CJ, Kahl A, Qian L, Stepanova A, Starkov A, Manfredi G, et al. Prohibitin is a positive modulator of mitochondrial function in PC12 cells under oxidative stress. J Neurochem. 2018;146:235–50. doi: 10.1111/jnc.14472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richter-Dennerlein R, Korwitz A, Haag M, Tatsuta T, Dargazanli S, Baker M, et al. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014;20:158–71. doi: 10.1016/j.cmet.2014.04.016. [DOI] [PubMed] [Google Scholar]
- 5.Cogliati S, Frezza C, Soriano ME, Varanita T, Quintana-Cabrera R, Corrado M, et al. Mitochondrial cristae shape determines respiratory chain supercomplexes assembly and respiratory efficiency. Cell. 2013;155:160–71. doi: 10.1016/j.cell.2013.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Del Dotto V, Mishra P, Vidoni S, Fogazza M, Maresca A, Caporali L, et al. OPA1 isoforms in the hierarchical organization of mitochondrial functions. Cell Rep. 2017;19:2557–71. doi: 10.1016/j.celrep.2017.05.073. [DOI] [PubMed] [Google Scholar]
- 7.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, et al. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–89. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 8.Olichon A, ElAchouri G, Baricault L, Delettre C, Belenguer P, Lenaers G. OPA1 alternate splicing uncouples an evolutionary conserved function in mitochondrial fusion from a vertebrate restricted function in apoptosis. Cell Death Differ. 2007;14:682–92. doi: 10.1038/sj.cdd.4402048. [DOI] [PubMed] [Google Scholar]
- 9.Varanita T, Soriano ME, Romanello V, Zaglia T, Quintana-Cabrera R, Semenzato M, et al. The Opa1-dependent mitochondrial cristae remodeling pathway controls atrophic, apoptotic, and ischemic tissue damage. Cell Metab. 2015;21:834–44. doi: 10.1016/j.cmet.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Merkwirth Carsten, Martinelli Paola, Korwitz Anne, Morbin Michela, Brönneke Hella S., Jordan Sabine D., Rugarli Elena I., Langer Thomas. Loss of Prohibitin Membrane Scaffolds Impairs Mitochondrial Architecture and Leads to Tau Hyperphosphorylation and Neurodegeneration. PLoS Genetics. 2012;8(11):e1003021. doi: 10.1371/journal.pgen.1003021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kahl Anja, Anderson Corey J, Qian Liping, Voss Henning, Manfredi Giovanni, Iadecola Costantino, Zhou Ping. Neuronal expression of the mitochondrial protein prohibitin confers profound neuroprotection in a mouse model of focal cerebral ischemia. Journal of Cerebral Blood Flow & Metabolism. 2017;38(6):1010–1020. doi: 10.1177/0271678X17720371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurinami H, Shimamura M, Ma T, Qian L, Koizumi K, Park L, et al. Prohibitin viral gene transfer protects hippocampal CA1 neurons from ischemia and ameliorates postischemic hippocampal dysfunction. Stroke. 2014;45:1131–8. doi: 10.1161/STROKEAHA.113.003577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou P, Qian L, D’Aurelio M, Cho S, Wang G, Manfredi G, et al. Prohibitin reduces mitochondrial free radical production and protects brain cells from different injury modalities. J Neurosci. 2012;32:583–92. doi: 10.1523/JNEUROSCI.2849-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baker MJ, Lampe PA, Stojanovski D, Korwitz A, Anand R, Tatsuta T, et al. Stress-induced OMA1 activation and autocatalytic turnover regulate OPA1-dependent mitochondrial dynamics. EMBO J. 2014;33:578–93. doi: 10.1002/embj.201386474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Head B, Griparic L, Amiri M, Gandre-Babbe S, Van Der Bliek AM. Inducible proteolytic inactivation of OPA1 mediated by the OMA1 protease in mammalian cells. J Cell Biol. 2009;187:959–66. doi: 10.1083/jcb.200906083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korwitz A, Merkwirth C, Richter-Dennerlein R, Tröder SE, Sprenger HG, Quirós PM, et al. Loss of OMA1 delays neurodegeneration by preventing stress-induced OPA1 processing in mitochondria. J Cell Biol. 2016;212:157–66. doi: 10.1083/jcb.201507022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang K, Li H, Song Z. Membrane depolarization activates the mitochondrial protease OMA1 by stimulating self-cleavage. EMBO Rep. 2014;15:576–85. doi: 10.1002/embr.201338240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Arnoult D, Grodet A, Lee YJ, Estaquier J, Blackstone C. Release of OPA1 during apoptosis participates in the rapid and complete release of cytochrome c and subsequent mitochondrial fragmentation. J Biol Chem. 2005;280:35742–50. doi: 10.1074/jbc.M505970200. [DOI] [PubMed] [Google Scholar]
- 19.Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, et al. Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem. 2003;278:7743–6. doi: 10.1074/jbc.C200677200. [DOI] [PubMed] [Google Scholar]
- 20.Yamaguchi R, Perkins G. Dynamics of mitochondrial structure during apoptosis and the enigma of Opa1. Biochim Biophys Acta. 2009;1787:963–72. doi: 10.1016/j.bbabio.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei MC, Lindsten T, Mootha VK, Weiler S, Gross A, Ashiya M, et al. tBID, a membrane-targeted death ligand, oligomerizes BAK to release cytochrome c. Genes Dev. 2000;14:2060–71. [PMC free article] [PubMed] [Google Scholar]
- 22.Demidyuk IV, Shubin AV, Gasanov EV, Kostrov SV. Propeptides as modulators of functional activity of proteases. Biomol Concepts. 2010;1:305–22. doi: 10.1515/bmc.2010.025. [DOI] [PubMed] [Google Scholar]
- 23.Quirós PM, Ramsay AJ, Sala D, Fernández-Vizarra E, Rodríguez F, Peinado JR, et al. Loss of mitochondrial protease OMA1 alters processing of the GTPase OPA1 and causes obesity and defective thermogenesis in mice. EMBO J. 2012;31:2117–33. doi: 10.1038/emboj.2012.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jian C, Xu F, Hou T, Sun T, Li J, Cheng H, et al. Deficiency of PHB complex impairs respiratory supercomplex formation and activates mitochondrial flashes. J Cell Sci. 2017;130:2620–30. doi: 10.1242/jcs.198523. [DOI] [PubMed] [Google Scholar]
- 25.Burté F, Carelli V, Chinnery PF, Yu-Wai-Man P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11:11–24. doi: 10.1038/nrneurol.2014.228. [DOI] [PubMed] [Google Scholar]
- 26.Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet. 2012;46:265–87. doi: 10.1146/annurev-genet-110410-132529. [DOI] [PubMed] [Google Scholar]
- 27.Baburamani AA, Hurling C, Stolp H, Sobotka K, Gressens P, Hagberg H, et al. Mitochondrial Optic Atrophy (OPA) 1 processing is altered in response to neonatal hypoxic-ischemic brain injury. Int J Mol Sci. 2015;16:22509–26. doi: 10.3390/ijms160922509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanderson TH, Raghunayakula S, Kumar R. Neuronal hypoxia disrupts mitochondrial fusion. Neuroscience. 2015;301:71–78. doi: 10.1016/j.neuroscience.2015.05.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wappler Edina A., Institoris Adam, Dutta Somhrita, Katakam Prasad V. G., Busija David W. Mitochondrial Dynamics Associated with Oxygen-Glucose Deprivation in Rat Primary Neuronal Cultures. PLoS ONE. 2013;8(5):e63206. doi: 10.1371/journal.pone.0063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens K, Park JH, Gourley S, Jones H, Kristian T. Mitochondrial dynamics: cell-type and hippocampal region specific changes following global cerebral ischemia. J Bioenerg Biomembr. 2014;47:13–31. doi: 10.1007/s10863-014-9575-7. [DOI] [PubMed] [Google Scholar]
- 31.Kelly Rainbolt T, Lebeau J, Puchades C, Luke Wiseman R. Reciprocal degradation of YME1L and OMA1 adapts mitochondrial proteolytic activity during stress. Cell Rep. 2016;14:2041–9. doi: 10.1016/j.celrep.2016.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohovych I, Fernandez MR, Rahn JJ, Stackley KD, Bestman JE, Anandhan A, et al. Metalloprotease OMA1 fine-tunes mitochondrial bioenergetic function and respiratory supercomplex stability. Sci Rep. 2015;5:1–14. doi: 10.1038/srep13989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Iverson SL, Enoksson M, Gogvadze V, Ott M, Orrenius S. Cardiolipin is not required for bax-mediated cytochrome c release from yeast mitochondria. J Biol Chem. 2004;279:1100–7. doi: 10.1074/jbc.M305020200. [DOI] [PubMed] [Google Scholar]
- 34.Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S. Cytochrome c release from mitochondria proceeds by a two-step process. Proc Natl Acad Sci. 2002;99:1259–63. doi: 10.1073/pnas.241655498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, et al. Cytochrome C acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat Chem Biol. 2005;1:223–32. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 36.Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587–602. doi: 10.1016/S0306-4522(02)00512-2. [DOI] [PubMed] [Google Scholar]
- 37.Desmurs M, Foti M, Raemy E, Vaz FM, Martinou J-C, Bairoch A, et al. C11orf83, a mitochondrial cardiolipin-binding protein involved in bc 1 complex assembly and supercomplex stabilization. Mol Cell Biol. 2015;35:1139–56. doi: 10.1128/MCB.01047-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ding B. Kilpatrick DL. Chapter 12 Lentiviral vector production, titration, and transduction of primary neurons. Methods Mol Bio. 2013;1018:119–31. [DOI] [PubMed]
- 39.Wittig I, Braun HP, Schägger H. Blue native PAGE. Nat Protoc. 2006;1:418–28. doi: 10.1038/nprot.2006.62. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.