

Abstract
Understanding the processes that shape neutral and adaptive genomic variation is a fundamental step to determine the demographic and evolutionary dynamics of pest species. Here, we use genomic data obtained via restriction site‐associated DNA sequencing to investigate the genetic structure of Moroccan locust (Dociostaurus maroccanus) populations from the westernmost portion of the species distribution (Iberian Peninsula and Canary Islands), infer demographic trends, and determine the role of neutral versus selective processes in shaping spatial patterns of genomic variation in this pest species of great economic importance. Our analyses showed that Iberian populations are characterized by high gene flow, whereas the highly isolated Canarian populations have experienced strong genetic drift and loss of genetic diversity. Historical demographic reconstructions revealed that all populations have passed through a substantial genetic bottleneck around the last glacial maximum (~21 ka BP) followed by a sharp demographic expansion at the onset of the Holocene, indicating increased effective population sizes during warm periods as expected from the thermophilic nature of the species. Genome scans and environmental association analyses identified several loci putatively under selection, suggesting that local adaptation processes in certain populations might not be impeded by widespread gene flow. Finally, all analyses showed few differences between outbreak and nonoutbreak populations. Integrated pest management practices should consider high population connectivity and the potential importance of local adaptation processes on population persistence.
Keywords: ddRadSeq, demographic inference, environmental association analyses, genetic structure, local adaptation, pest species
In this study, we use genomic data (ddRADseq) to infer historical demographic trends and determine the relative role of neutral versus selective processes on shaping spatial patterns of genomic variation in the Moroccan locust, a pest species of great economic importance. Our study revealed low levels of genetic differentiation at regional scales, signatures of divergent selection in several loci, and a demographic history characterized by peaks of effective population sizes matching with warm periods (interglacials) during the Pleistocene. All our analyses also showed very little differences between traditionally outbreak and nonoutbreak populations in patterns of genomic diversity and structure, demographic profiles, or signatures of selection. Overall, our study suggests that integrated management practices aimed at reducing the negative impacts of this locust species on agriculture should consider population connectivity at contrasting spatial scales, the potential role of local adaptation processes on population persistence and likely future range expansions, and increased outbreak frequency in response to ongoing climate warming.

1. INTRODUCTION
The genetic makeup of a species is influenced by complex interactions between neutral and selective forces, life‐history characteristics, and contemporary and past environmental conditions that collectively shape the evolutionary and demographic fate of its populations (Bernatchez et al., 2018). From a neutral perspective, recently developed analytical methods allow inferring complex demographic processes from genomic data and obtaining robust estimates of population size changes and gene flow at different time scales (Liu & Fu, 2015; Miles et al., 2017; Sherpa et al., 2018). Historical demographic reconstructions, linking population size changes with past environmental fluctuations, can help to predict future demographic trends of species under hypothetical climate change scenarios (Brown et al., 2016; Espindola et al., 2012; Fordham, Brook, Moritz, & Nogués‐Bravo, 2014), whereas contemporary estimates of population genetic connectivity and spatially explicit landscape genetic analyses are useful to obtain baseline information on dispersal rates (Crossley, Rondon, & Schoville, 2019a) and identify corridors for gene flow (Crossley, Rondon, & Schoville, 2019b; Karsten, Addison, Jansen van Vuuren, & Terblanche, 2016; Qin et al., 2018; Venkatesan & Rasgon, 2010; Zepeda‐Paulo et al., 2010). From an adaptive perspective, new molecular tools bring the opportunity to infer selection at specific loci displaying patterns of variation contrasting to those characterizing genomic regions only affected by neutral processes such as mutation, genetic drift, migration, and demographic changes (Berdan, Mazzoni, Waurick, Roehr, & Mayer, 2015; Luikart, England, Tallmon, Jordan, & Taberlet, 2003). Identifying loci that exhibit a significant reduction of within‐population genetic variability and higher divergence across populations consistent with disruptive selection (Vasemägi & Primmer, 2005), coupled with environment association analyses (EAA) linking variation in allele frequencies with ecological gradients (Rellstab, Gugerli, Eckert, Hancock, & Holderegger, 2015), can help to determine local adaptation processes in response to specific selective forces such as those imposed by climate (Crossley, Chen, Groves, & Schoville, 2017; Dowle et al., 2017; Dudaniec, Yong, Lancaster, Svensson, & Hansson, 2018; Guo, Li, & Merilä, 2016), pesticide application (Crossley et al., 2017; Gassmann, Onstad, & Pittendrigh, 2009; Leftwich, Bolton, & Chapman, 2016), or host plant use (Gassmann et al., 2009; Simon et al., 2015; Soria‐Carrasco et al., 2014). Gene flow is generally accepted to constrain local adaptation whereas strong divergent selection is expected to prevent interpopulation realized gene flow (Lenormand, 2002). Thus, joined inference of both neutral and selective phenomena can provide a more comprehensive understanding on the relative role of dispersal and local adaptation processes on structuring genetic variation (Dudaniec et al., 2018; Guo et al., 2016).
Pest species, either invasive or native, are responsible of considerable economic losses worldwide and, as a result, public, private, and nonprofit organizations annually invest vast amounts of resources to prevent or mitigate their negative impacts (Enserink, 2004; Skaf, Popov, Roffey, Scorer, & Hewitt, 1990). In turn, such management practices often have undesirable side effects on wildlife, natural ecosystems, and human health (e.g., Baker & Wilkinson, 1990; Carson, 2002). For these reasons, understanding the population dynamics, dispersal routes, demographic history, and idiosyncratic evolutionary processes of pest species is a fundamental step to predict their future impacts and develop informed, less pernicious, and more targeted management practices (Abrol, 2014; Lankau, Jørgensen, Harris, & Sih, 2011). Population genetic approaches have proven useful to address several of the abovementioned aspects, and their potential has exponentially grown in the last years by the generalization of high‐throughput sequencing techniques and the possibility of inferring both neutral and adaptive evolutionary processes at an unprecedented resolution (Crossley et al., 2017; Wang et al., 2014). The application of genomic tools is particularly important if we consider that most pest species often show large effective population sizes, high dispersal potential, shallow genetic differentiation, and fluctuating and complex demographic dynamics that are difficult to study using traditional capture–mark–recapture approaches or standard genetic methods (Bekkevold, Gross, Arula, Helyar, & Ojaveer, 2016; Chapuis et al., 2008, 2011; Ibrahim, Sourrouille, & Hewitt, 2000; Kirk, Dorn, & Mazzi, 2013).
Locusts are a paradigmatic case of pest species with cyclical outbreaks that cause considerable agricultural losses and remission periods during which local populations either disappear or persist at very low densities (Chapuis et al., 2009; Enserink, 2004; Latchininsky, 1998; Skaf et al., 1990). The prevalence of outbreak and solitary phases varies geographically, with populations from some areas recurrently becoming agricultural pests while those from nonoutbreaking regions often occur at low numbers forming harmless populations (Chapuis et al., 2009; Latchininsky, 1998). An intriguing example of this demographic stochasticity is the case of the mysterious Rocky Mountain locust Melanoplus spretus, a devastating pest species endemic from North American prairies during the 19th century that went inexplicably extinct within a 30‐year interval (Chapco & Litzenberger, 2004; Lockwood, 2004). Extreme demographic oscillations charactering locust populations are expected to have a considerable impact on genetic variation and the potential of species to respond to selection and evolve local adaptations. On the one hand, population crashes in the transition phase from gregarious to solitary forms are likely to leave genetic signatures of demographic bottlenecks that are probably ephemeral and blurred by genetic admixture after population expansions during outbreak periods (Chapuis et al., 2008; Ibrahim, 2001; Ibrahim et al., 2000). On the other hand, local adaptation processes in response to spatially varying selective pressures could be impeded by high gene flow (Babin, Gagnaire, Pavey, & Bernatchez, 2017; Lenormand, 2002; Pujolar et al., 2014) or restricted to isolated populations that are not swamped by gene flow from outbreaking populations (Chapuis et al., 2014). Previous single‐locus and microsatellite‐based studies on different locust species have found very shallow patterns of genetic structure at local/regional scales (e.g., Chapuis et al., 2011), no genetic differentiation between gregarious and solitary phase populations (Chapuis et al., 2008), higher levels of gene flow among outbreaking populations than among recession or nonoutbreak populations (Chapuis et al., 2009; Ibrahim, 2001; Ibrahim et al., 2000), and a little impact of recession periods on genetic diversity (Chapco & Litzenberger, 2004; Chapuis et al., 2014; Ibrahim et al., 2000). However, with the exception of the recent sequencing and annotation of the locust Locusta migratoria genome (Wang et al., 2014) and the identification of some genes linked to physiology, phase change, and dispersal capacity (Bakkali & Martín‐Blázquez, 2018; Ernst et al., 2015; Martín‐Blázquez, Chen, Kang, & Bakkali, 2017; Wang et al., 2014), high‐resolution genomic data have not been yet employed to determine fine‐spatial scale patterns of genetic structure, perform robust demographic inferences in outbreak and nonoutbreak populations, and assess the potential role of selective processes on shaping spatial patterns of genetic variation in these organisms of great economic importance (e.g., Crossley et al., 2017).
The Moroccan locust, Dociostaurus maroccanus (Thunberg, 1815; Figure 1), is a xerophilous species distributed in most of the Western Palearctic, from the Canary Islands to south Kazakhstan (Cigliano, Braun, Eades, & Otte, 2019; Latchininsky, 1998). The species is characterized by its broad polyphagia, extreme voracity, enormous fecundity, extraordinarily fluctuating populating sizes, and high capability to migrate (del Cañizo & Moreno, 1949; el Ghadraoui, Petit, Picaud, & Yamani, 2002; Latchininsky, 1998; Uvarov, 1977). Its distribution is discontinuous and consists of fragmented nonoutbreaking populations in some areas and permanent foci of outbreak populations that cyclically become devastating agricultural pests (del Cañizo & Moreno, 1949; Latchininsky, 1998). It has been reported that Moroccan locusts can move distances of 70–100 km during their entire lifetime (rarely up to 200 km; Latchininsky, 1998), making possible the exchange of individuals between distant populations during swarming phases (Latchininsky, 1998). The species is considered a major agricultural pest of high economic importance, damaging pastures, and a wide variety of crops during outbreaks, which requires extensive control operations and chemical interventions with a tremendous cost year after year in affected countries (e.g., Arias‐Giralda, Jiménez‐Viñuelas, & Pérez‐Romero, 1997; Guerrero et al., 2019; Latchininsky, 2013).
Figure 1.

Adult male (top) and nymph (bottom) of Moroccan locust (Dociostaurus maroccanus) from Valle de Alcudia (Ciudad Real, Spain). Photograph by Pedro J. Cordero
Here, we focus on populations of the Moroccan locust from the Iberian Peninsula and the Canary Islands, which represent the west margin of the species’ distribution (Latchininsky, 2013). In the Iberian Peninsula, there are three main vast regions that are foci of population outbreaks and have suffered considerable damages to pastures and crops for centuries: Monegros (Aragón), La Serena (Extremadura) and Valle de Alcudia (Castilla‐La Mancha) (Alberola‐Roma, 2012; Arias‐Giralda, Morales‐Agacino, Cobos‐Suárez, Sopeña‐Mañas, & Martín‐Bernal, 1993). These regions are characterized by considerable cattle overgrazing, a factor that has been related to irruptive population growth in the Moroccan locust (Latchininsky, 1998; Louveaux, Mouhim, Roux, Gillon, & Barral, 1996). Besides, there are other regions from the Iberian Peninsula that represent historically outbreak areas that nowadays only sustain small populations or where the species has traditionally occurred at very low densities in isolated pockets of suitable habitat (Aragón, Coca‐Abia, Llorente, & Lobo, 2013; Latchininsky, 1998). The small size of some formerly outbreak populations has been hypothesized to be related with the expansion of agriculture and certain plowing techniques, the destruction and fragmentation of suitable breeding areas linked to land use changes, and the massive application of pesticides to control locusts’ populations (Latchininsky, 1998). On this respect, several Moroccan locust populations from outbreak areas have been considerably reduced by human intervention to the point that many of them have almost disappeared in the last decades (Aragón et al., 2013; Latchininsky, 1998). However, no study has been performed so far to understand the degree of genetic connectivity among populations of the Moroccan locust at regional scales, infer its past demographic history, or determine the potential role of local adaptation processes, information that might help to shed light on key aspects of the ecology, distribution, and evolutionary dynamics of this economically important species.
In this study, we use genomic data obtained via restriction site‐associated DNA sequencing (ddRADseq) to investigate the relative role of neutral versus selective processes on shaping genetic variation in the Moroccan locust, determine spatial patterns of genetic diversity and structure in solitary and outbreaking populations from the westernmost portion of the distribution of the species, and, ultimately, infer its past demographic history. First, we performed genome scans and environmental association analyses to identify putative loci under selection and evaluate the potential importance of local adaptation processes on shaping genetic differentiation at non‐neutral genomic regions. Second, we calculated different estimates of genetic diversity and performed a comprehensive suite of analyses of genetic structure to test the hypothesis of lower levels of genetic variation and increased genetic differentiation in solitary than outbreak populations (Chapuis et al., 2014). Finally, we used genomic data to infer the past demography of the studied populations. Specifically, we predict genomic signatures of recent demographic declines in solitary populations and formerly outbreaking populations that have undergone remarkable retreats during the last decades (Chapuis et al., 2014; Ibrahim et al., 2000) and, given the thermophilous character of the Moroccan locust, we also expect that the species has experienced historical bottlenecks during Pleistocene glacial periods and population expansions in interglacials (Meco et al., 2011).
2. MATERIALS AND METHODS
2.1. Population sampling
Between May and July 2011–2016, we prospected adequate habitats for the Moroccan locust (Dociostaurus maroccanus) (i.e., grazed grasslands, natural sparse vegetation, arid or semidesert steppes, and abandoned agricultural fields; Latchininsky, 1998; Latchininsky, 2013) in the Iberian Peninsula and the Canary Islands. We sampled a total of 21 localities representative of both outbreak and nonoutbreak populations (Figure 2; Table 1), a status defined according with our own field observations during sampling (i.e., densities of more than ~20 individuals/m2 were considered as outbreak populations) and corroborated with information provided by regional government authorities implementing pest management programs. Information about the outbreak or nonoutbreak status of the different sampling populations is presented in Table 1. Adult individuals were collected via sweep netting in an area not higher than 500 m2 around each sampling locality. Fresh whole adult specimens were placed in vials with 2–5 ml ethanol 96% and stored at −20°C until needed for genomic analyses. For this study, we analyzed a total of 5–8 adult individuals per locality (Table 1).
Figure 2.
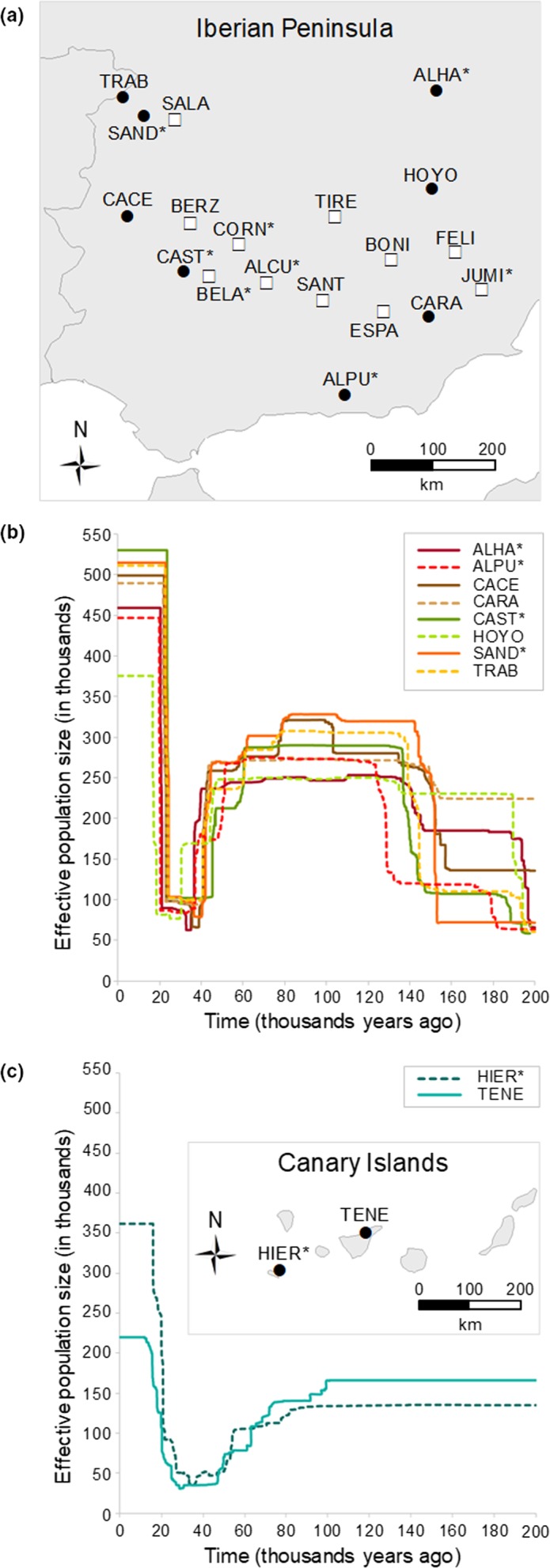
Geographic location of the studied populations of Moroccan locust in (a) the Iberian Peninsula and (c) the Canary Islands. Black dots indicate those populations analyzed with stairway plot (n = 8 individuals) and white squares the rest of the populations (n < 8 individuals). Panels (b) and (c) present the inferred demographic profiles for Iberian and Canarian populations, respectively. Lines show the median estimate of effective population size (N e) over time, assuming a mutation rate of 2.8 × 10–9 and 1‐year generation time. Populations with an asterisk indicate pest outbreaks during the sampling year. Population codes are described in Table 1.
Table 1.
Geographical location of the studied populations of Moroccan locust, population codes, sampling year, number of individuals per population (n), and scores for the three environmental principal components used in LFMM analyses (PC1, PC2, and PC3). Populations with an asterisk indicate pest outbreaks during the sampling year. Average values of genetic statistics across neutral loci are presented for major allele frequency (P), observed (H O) and expected (H E) heterozygosity, nucleotide diversity (π), and the Wright's inbreeding coefficient (F IS) for all positions (polymorphic and nonpolymorphic)
| Locality (Province) | Code | Year | n | Latitude | Longitude | PC1 | PC2 | PC3 | P | H O | H E | π | F IS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Trabanca (Salamanca) | TRAB | 2015 | 8 | 41.24275 | −6.40222 | −0.447 | −0.283 | 1.282 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Sando (Salamanca)* | SAND | 2015 | 8 | 40.96961 | −6.10954 | −0.573 | −0.279 | 0.630 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Salamanca (Salamanca) | SALA | 2015 | 5 | 40.93764 | −5.66806 | −0.608 | −0.335 | −0.726 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0006 |
| Alhama de Aragón (Zaragoza)* | ALHA | 2015 | 8 | 41.34571 | −1.92277 | −1.613 | −0.820 | −0.939 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Los Llanos de Cáceres (Cáceres) | CACE | 2015 | 8 | 39.53310 | −6.32455 | 1.072 | 0.887 | 0.094 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0008 |
| Puerto de la Berzocana (Cáceres) | BERZ | 2012 | 5 | 39.44998 | −5.42609 | −0.349 | 0.401 | 1.006 | 0.9994 | 0.0007 | 0.0008 | 0.0009 | 0.0005 |
| Castuera (Cáceres)* | CAST | 2015 | 8 | 38.74827 | −5.54450 | 1.036 | 1.186 | 0.466 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Cañada del Hoyo (Cuenca) | HOYO | 2015 | 8 | 39.93709 | −1.99317 | −1.787 | −0.467 | 0.010 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Laguna de Tirez (Toledo) | TIRE | 2015 | 7 | 39.54259 | −3.35907 | 0.144 | 0.660 | −1.063 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Raña Cornicabra (Ciudad Real)* | CORN | 2011 | 5 | 39.13134 | −4.73814 | 0.550 | 0.924 | −0.471 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0005 |
| Valle de Alcudia (Ciudad Real)* | ALCU | 2015 | 7 | 38.58958 | −4.35354 | 0.560 | 1.059 | 0.502 | 0.9994 | 0.0008 | 0.0009 | 0.0010 | 0.0006 |
| Belalcázar (Córdoba)* | BELA | 2015 | 5 | 38.67942 | −5.16172 | 1.125 | 1.311 | 0.342 | 0.9994 | 0.0008 | 0.0009 | 0.0010 | 0.0005 |
| El Bonillo (Albacete) | BONI | 2015 | 7 | 38.93758 | −2.56544 | −0.528 | 0.209 | −0.507 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0008 |
| La Felipa (Albacete) | FELI | 2015 | 5 | 39.03552 | −1.64832 | −0.299 | −0.185 | −1.642 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0005 |
| Santa Elena (Jaén) | SANT | 2015 | 5 | 38.33753 | −3.51923 | 0.594 | 0.935 | 0.098 | 0.9994 | 0.0008 | 0.0009 | 0.0010 | 0.0005 |
| Santiago de la Espada (Jaén) | ESPA | 2011 | 5 | 38.18128 | −2.66677 | −1.238 | −0.058 | 0.920 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0005 |
| Jumilla (Murcia)* | JUMI | 2015 | 5 | 38.49016 | −1.24497 | 0.228 | −0.237 | −1.815 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0005 |
| Caravaca de la Cruz (Murcia)* | CARA | 2015 | 8 | 38.10283 | −2.01999 | −0.208 | −0.053 | −0.895 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Alpujarra (Granada)* | ALPU | 2015 | 8 | 36.96846 | −3.21425 | −0.986 | −0.101 | 1.811 | 0.9994 | 0.0007 | 0.0009 | 0.0010 | 0.0007 |
| Tenerife (Santa Cruz de Tenerife) | TENE | 2016 | 8 | 28.41569 | −16.40991 | 1.343 | −2.257 | 1.449 | 0.9995 | 0.0006 | 0.0007 | 0.0008 | 0.0004 |
| El Hierro (Santa Cruz de Tenerife)* | HIER | 2016 | 8 | 27.77303 | −17.95796 | 1.984 | −2.497 | −0.555 | 0.9995 | 0.0006 | 0.0007 | 0.0008 | 0.0004 |
2.2. Genomic library preparation
We used NucleoSpin Tissue kits (Macherey‐Nagel, Durën, Germany) to extract and purify genomic DNA from the hind femur of each individual. Genomic DNA from 141 individuals of the 21 sampling localities (Table 1) was processed into three genomic libraries (47 individuals per library) using the double‐digestion restriction site‐associated DNA sequencing procedure (ddRADseq) described in Peterson, Weber, Kay, Fisher, and Hoekstra (2012). In brief, DNA was doubly digested with the restriction enzymes MseI and EcoR1 (New England Biolabs, Ipswich, MA, USA) and Illumina adaptors including unique 7‐bp barcodes were ligated to the digested fragments. Ligation products from individuals assigned to each library were pooled, size‐selected between 475 and 580 bp with a Pippin Prep (Sage Science, Beverly, MA, USA) machine, and amplified by PCR with 12 cycles using the iProofTM High‐Fidelity DNA Polymerase (BIO‐RAD, Hercules, CA, USA). Each library was sequenced in single‐read 150‐bp lane on an Illumina HiSeq2500 platform at The Centre for Applied Genomics (SickKids, Toronto, ON, Canada).
2.3. Genomic data analyses
We used the different programs distributed as part of the stacks v. 1.35 pipeline (process_radtags, ustacks, cstacks, sstacks, and populations) to assemble our sequences into de novo loci and call genotypes (Catchen, Amores, Hohenlohe, Cresko, & Postlethwait, 2011; Catchen, Hohenlohe, Bassham, Amores, & Cresko, 2013a; Hohenlohe et al., 2010). For the different filtering and assembling steps with stacks, we used the default parameters recommended by the authors (Catchen et al., 2011; Catchen, Hohenlohe, et al., 2013; Hohenlohe et al., 2010). Reads were demultiplexed and filtered for overall quality using the program process_radtags, retaining reads with a Phred score > 10 (using a sliding window of 15%), no adaptor contamination, and that had an unambiguous barcode and restriction cut site. Raw reads were screened for quality with fastqc v. 0.11.5 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc), and all sequences were trimmed to 129‐bp using seqtk (https://github.com/lh3/seqtk) in order to remove low‐quality reads near the 3´ ends. Filtered reads of each individual were assembled de novo into putative loci with the ustacks program. The minimum stack depth (m) was set to three, and we allowed a maximum distance of two nucleotide mismatches (M) to group reads into a “stack.” We used the “removal” (r) and “deleveraging” (d) algorithms to eliminate highly repetitive stacks and resolve over‐merged loci, respectively. Single nucleotide polymorphisms (SNPs) were identified at each locus, and genotypes were called using a multinomial‐based likelihood model that accounts for sequencing errors, with the upper bound of the error rate (ε) set to 0.2 (Catchen et al., 2011; Catchen, Hohenlohe, et al., 2013; Hohenlohe et al., 2010). A catalogue of loci was built using the cstacks program, with loci recognized as homologous across individuals if the number of nucleotide mismatches between consensus sequences (n) was ≤2. Each individual data were matched against this catalogue using sstacks program, and output files were exported in different formats for subsequent analyses using the program populations. For all downstream analyses, we exported only the first SNP per RAD locus (option write_single_snp) and retained loci with a minimum stack depth ≥ 5 (m = 5), that were sequenced in at least 50% of the individuals of each population (parameter r = 0.5), represented in ~66% of populations, and with a minimum minor allele frequency (MAF) ≥ 0.01 to reduce the number of false polymorphic loci due to sequencing errors. As demonstrated in previous studies, the choice of different filtering thresholds affecting the proportion of missing data (p and r in program populations) had little impact on the obtained inferences (e.g., González‐Serna, Cordero, & Ortego, 2018; González‐Serna, Cordero, & Ortego, 2019; Ortego, Gugger, & Sork, 2018). The resulting files were used for subsequent analyses or converted into other formats using the program pgdspider v.2.1.0.3 (Lischer & Excoffier, 2012).
2.4. Outlier loci detection and environmental association analyses
In a first step, we screened for loci not conforming to neutral expectations using two outlier detection approaches: the coalescent‐based FDIST method from arlequin (Excoffier & Lischer, 2010) and the Bayesian approach implemented in bayescan v.2.1 (Foll & Gaggiotti, 2008). The FDIST method in arlequin was run in two different ways, considering both the nonhierarchical (Beaumont & Nichols, 1996) and hierarchical (Excoffier, Hofer, & Foll, 2009) island models. The nonhierarchical island model was run using 200,000 simulations, 100 demes, and expected heterozygosity ranging from 0 to 1. The hierarchical island model was run grouping populations according to their geographical origin and the results obtained from genetic structure analyses (i.e., Iberian versus Canarian populations; see Results section), using the same settings as the nonhierarchical model, and considering three simulated groups (i.e., the number of defined population groups plus one, as recommended in Excoffier and Lischer (2010). P‐values were corrected for multiple testing using the p.adjust function in r (R Core Team, 2018) and loci significantly outside the neutral distribution at a false discovery rate (FDR) of 5% (i.e., q < 0.05) were considered as outliers. bayescan analyses were run under default settings (thinning interval size of 10; 20 pilot runs of 5,000 iterations; burn‐in length of 50,000 iterations), except for an increase of outputted iterations to 10,000. We used 10 (default) prior odds and adopted the same FDR to identify candidate loci putatively under selection as in arlequin analyses (FDR of 5%, q < 0.05). bayescan and arlequin analyses were run independently for two different genomic datasets, one considering all populations and another one only considering populations from the Iberian Peninsula (e.g., Guo et al., 2016).
In a second step, we performed environmental association analyses (EAA) using Latent Factor Mixed Models (LFMM) implemented in the r v.3.3.3 (R Core Team, 2018) package lea (Frichot & François, 2015). This approach uses a stochastic Monte Carlo Markov Chain algorithm to test for associations between environmental/ecological variables and allele frequencies while simultaneously controlling for background levels of population structure (Frichot, Schoville, Bouchard, & François, 2013). As environmental information, we used the 19 bioclimatic variables from the worldclim dataset interpolated to 30‐arcsec resolution (~1 km2 cell size) (Hijmans, Cameron, Parra, Jones, & Jarvis, 2005). These variables summarize information about temperature and precipitation and have been commonly used in exploratory analyses aimed to test the broad hypothesis of environment‐driven selection (e.g., François, Martins, Caye, & Schoville, 2016; Frichot & François, 2015; de Kort et al., 2014; Rellstab et al., 2015). We extracted the values for these variables from all adjacent cells around each sampling location (i.e., ~9 km2) using bilinear interpolations as implemented in arcgis 10.3 (ESRI, Redlands, CA, USA). To summarize and reduce strong redundancy among the 19 bioclimatic variables, we ran a principal component analysis (PCA) on all of them (e.g., François et al., 2016; Frichot & François, 2015; de Kort et al., 2014). The first three principal components (PCs) cumulatively accounted for 94.82% of the variance (PC1: 38.58%; PC2: 33.77%; PC3: 22.47%) and were retained for LFMM analyses (Table A1). The contribution of bioclimatic variables to each axis (i.e., the factor loadings for each PC) is presented in Table 1. The MCMC algorithm was implemented for each of the three PCs (i.e., PC1, PC2, and PC3), using 10,000 iterations, 5,000 as burning period, and 5 independent replicates of the analysis. As indicated above for bayescan and nonhierarchical arlequin analyses, LFMM analyses were run independently for all populations and only considering Iberian populations. The number of latent factors included in the model as a covariate to control for demographic history were defined on the basis of structure analyses (Pritchard, Stephens, & Donnelly, 2000; see Results section) and sparse non‐negative matrix factorization (snmf) analyses implemented in the r package lea (Frichot, Mathieu, Trouillon, Bouchard, & Francois, 2014). We set the number of latent factors (K) at K = 2 for analyses including all populations and K = 1 for analyses focused on Iberian populations. The z‐scores over the five replicates were combined and recalibrated using the genomic inflation factor (λ) (Frichot & François, 2015). Finally, we performed a FDR adjustment to control for multiple tests (FDR of 5%, q < 0.05) and identify putative loci under environmental selection for each of the three PCs summarizing bioclimatic variation.
2.5. Genetic structure
We employed three complementary approaches to exhaustively explore spatial patterns of genetic structure in our study system, including (a) principal component analyses (PCA) (Jombart, 2008); (b) classic structure analyses considering and not considering prior population information (Hubisz, Falush, Stephens, & Pritchard, 2009; Pritchard et al., 2000); and (c) the recently developed spatial method implemented in the r program construct (Bradburd, Coop, & Ralph, 2018). In all cases, genetic structure was analyzed only considering putatively neutral loci. To this end, outlier loci detected by arlequin and bayescan and SNPs identified by LFMM analyses as being putatively under environment‐driven selection (a total of 9,346 loci; see Section 3) were conservatively excluded to create datasets only containing neutral loci (e.g., Brauer, Hammer, & Beheregaray, 2016; Ortego et al., 2018). This yielded neutral datasets of 40,179 SNPs for all populations, 42,114 SNPs for Iberian populations, and 33,998 SNPs for Canarian populations.
2.5.1. Principal component analyses (PCA)
In order to visualize the major axes of population genetic differentiation, we performed individual‐based principal component analyses (PCA) using the r package adegenet (Jombart, 2008). Before running the PCA, we scaled and centered allele frequencies and replaced missing data with mean allele frequencies using the scalegen function as recommended by Jombart (2008). PCAs were run using all neutral SNPs for the two main datasets (all populations and Iberian Peninsula).
2.5.2. structure analyses
We inferred genetic structure at neutral loci using the Bayesian Markov chain Monte Carlo clustering method implemented in the program structure v.2.3.3 (Falush, Stephens, & Pritchard, 2003; Hubisz et al., 2009; Pritchard et al., 2000). We conducted structure analyses hierarchically, initially analyzing data from all populations jointly and, subsequently, running independent analyses for subsets of populations assigned to the same genetic cluster in the previous hierarchical level analysis (e.g., Coulon et al., 2008; González‐Serna et al., 2019). To make analyses computationally tractable, we ran structure using a single random subset of 10,000 unlinked neutral SNPs. According to previous studies, this number of loci is an order of magnitude higher than that necessary to obtain robust and reproducible results in structure (e.g., Catchen, Bassham, et al., 2013:1,000 loci; González‐Serna et al., 2019:1,250 loci). We ran structure using 200,000 MCMC iterations after a burn‐in step of 100,000 iterations, assuming correlated allele frequencies and admixture, and both considering and not considering prior population information (Hubisz et al., 2009). We performed 15 independent runs for each value of K. In order to ensure analysis convergence, we only retained the ten runs having the highest likelihood for each value of K (e.g., Yannic et al., 2018) and checked that all retained replicates reached a similar solution in terms of individual's probabilities of assignment to each genetic cluster (q‐values; Gilbert et al., 2012). As recommended, we used two statistics to identify the most likely number of genetic clusters (K) (Gilbert et al., 2012; Janes et al., 2017): log probabilities of Pr(X|K) (Pritchard et al., 2000) and the ΔK method (Evanno, Regnaut, & Goudet, 2005). These statistics were calculated as implemented in structure harvester (Earl & vonHoldt, 2012). Finally, we used clumpp v. 1.1.2 and the Greedy algorithm to align multiple runs of structure for the same K value (Jakobsson & Rosenberg, 2007) and distruct v. 1.1 (Rosenberg, 2004) to visualize as bar plots the individual's probabilities of membership to each inferred genetic cluster.
2.5.3. construct analyses
We used the spatial model implemented in r package construct v. 1.0.3 to infer patterns of genetic structure across the Iberian populations and determine whether genetic differentiation is a consequence of continuous (i.e., isolation‐by‐distance) or discrete (e.g., separation by geographic barriers, etc.) processes (Bradburd et al., 2018). Given that this approach is highly sensitive to missing data, we analyzed a database with no missing data (2,904 unlinked SNPs) as recommended by the authors (Bradburd et al., 2018). We ran construct analyses with 5,000 iterations and visually checked for convergence using trace plots (Bradburd et al., 2018). We used a fivefold cross‐validation approach to examine predictive accuracy across the range of tested K values and determine the best‐fit number of genetic clusters (Bradburd et al., 2018). As done for structure, we plotted individual coancestry coefficients for the different K values using distruct.
2.6. Geographical and environmental drivers of genetic differentiation
We tested for the presence of isolation‐by‐distance (IBD) and/or isolation‐by‐environment (IBE) patterns of genetic structure by analyzing the association between genetic differentiation (F ST) and geographic and environmental distances among Iberian populations (Sexton, Hangartner, & Hoffmann, 2014; Shafer & Wolf, 2013; Wang, 2013). Genetic differentiation (F ST) between all pairs of populations was calculated for the subset of neutral loci (see previous section) using the program populations from stacks (Table A2). Geographic distance between each pair of Iberian populations was calculated using geographic distance matrix generator v.1.2.3 (Ersts, 2018). Environmental distances were calculated for each PC (PC1, PC2, and PC3) obtained from a PCA on the 19 bioclimatic variables (see LFMM analyses above for details on PCA) using the “dist” function in r 3.3.3 (R Core Team, 2018). Genetic differentiation [F ST/(1 − F ST)] was tested against matrices of geographical (log10 transformed) and environmental distances using multiple matrix regressions with randomization (MMRR) as implemented in the “mmrr” function (Wang, 2013) in r 3.3.3 (R Core Team, 2018). Given that geographical/environmental distances are only expected to have positive effect on the degree of genetic differentiation between populations, we used one‐tailed hypothesis tests for making statistical decisions regarding the null hypothesis of no effect of independent variables on genetic differentiation (Ruxton & Neuhäuser, 2010; e.g., Berkman, Nielsen, Roy, & Heist, 2013; Yannic et al., 2018). We selected final models following a backward procedure, initially fitting all explanatory terms and progressively removing nonsignificant variables until all retained variables were significant. The significance of the variables excluded from the model was tested again until no additional term reached significance (e.g., Ortego, Aguirre, Noguerales, & Cordero, 2015a).
2.7. Genetic diversity and past demographic history
We only employed neutral loci for calculating genetic diversity statistics and performing demographic inference analyses (Luikart et al., 2003). We used the program populations from stacks to calculate some genetic statistics, including nucleotide diversity (π), observed (H O) and expected (H E) heterozygosity, major allele frequency (P), and the Wright's inbreeding coefficient (F IS) (Catchen, Hohenlohe, et al., 2013). Standardized multilocus heterozygosity (sMLH) was calculated for each individual using the r package inbreedr (Stoffel et al., 2016). sMLH is an individual‐based metric defined as the total number of heterozygous loci in an individual divided by the sum of average observed heterozygosities in the population, over the subset of loci successfully typed in the focal individual (Coltman, Pilkington, Smith, & Pemberton, 1999).
We used stairway plot (Liu & Fu, 2015) to reconstruct the demographic history of the studied populations, a novel model‐flexible method based on the site frequency spectrum (SFS) that does not require whole‐genome sequence data or reference genome information to infer changes in effective population size (N e) over time. These analyses were restricted to populations with eight genotyped individuals (see Figure 2 and Table 1), as the calculation of the SFS requires a downsampling procedure to remove missing data. These populations are representative of the distribution of the species across the study area (see Figure 2a). To compute the SFS for each population, we ran the program populations from stacks (Catchen, Hohenlohe, et al., 2013) in order to export the first SNP per RAD locus and retain loci with a minimum stack depth ≥ 5 (m = 5) and that were represented in at least 50% of the individuals of the focal population (r = 0.5). To remove all missing data for the calculation of the SFS and minimize errors with allele frequency estimates, each population was down‐sampled to 6 individuals using a custom Python script written by Qixin He and available on Dryad (Papadopoulou & Knowles, 2015). We ran stairway plot for each population fitting a flexible multi‐epoch demographic model, assuming the mutation rate per site per generation of 2.8 × 10–9 estimated for Drosophila melanogaster (Keightley, Ness, Halligan, & Haddrill, 2014), a one‐year generation time (Latchininsky, 1998), four different number of random breakpoints [(nseq‐2)/4, (nseq‐2)/2, (nseq‐2)*3/4, and nseq‐2], and 200 bootstrap replicates to estimate 95% confidence intervals.
3. RESULTS
3.1. Genomic data analyses
Illumina sequencing of ddRAD libraries generated > 358 millions of reads in total after first quality filtering using the program process_radtags. The number of reads per individual before and after different quality filtering steps is shown in Figure A1. Only one sample from population BONI was excluded for subsequent analyses due to low number of reads (Figure A1). The dataset obtained with stacks for all populations contained a total of 49,373 unlinked SNPs.
3.2. Outlier loci detection and environmental association analyses
For the dataset including all populations, we identified 318 (0.64%) outlier loci using arlequin (FDIST method) and 503 (1.02%) using bayescan (Figures 3a,b). When the analyses were restricted to Iberian populations, we identified 93 (0.18%) outlier loci using arlequin and 270 (0.52%) using Bayescan (Figure 3a,b). Several SNPs were commonly identified as outliers by both arlequin and bayescan analyses (all populations: 74 SNPs, 0.15%, Figure 3a; Iberian populations: 35 SNPs, 0.07%, Figure 3b). Most outlier loci were identified as being putatively under divergent selection in analyses based on both the datasets including all populations (arlequin: n = 275, 86.47%; bayescan: n = 467, 92.84%) and the one restricted to Iberian populations (arlequin: n = 76, 81.72%; bayescan: n = 269, 99.63%). Environmental association analyses in LFMM showed that a high number of loci were significantly associated with environmental variation (Figure 3c,d). For the dataset including all populations, LFMM detected 7,710 unique loci (15.62%) associated with at least one PC of environmental variation (PC1: 2,932 loci; PC2: 4,051 loci; PC3: 2,797 loci; Figure A2) and 196 of them (0.40%) showed significant associations with the three PCs (Figure 3c). Similarly, when the analyses were restricted to Iberian populations, LFMM detected 6,104 unique loci (11.87%) associated with at least one PC of environmental variation (PC1: 2,713 loci; PC2: 3,081 loci; PC3: 2,982 loci; Figure A2) of which 363 (0.71%) were shared across all PCs (Figure 3d). Only 42 loci (0.08%) for analyses based on all populations (Figure 3a) and 23 loci (0.04%) for analyses focused on Iberian populations (Figure 3b) showed associations with environmental variation in LFMM and were also identified as F ST outliers by arlequin and bayescan analyses.
Figure 3.
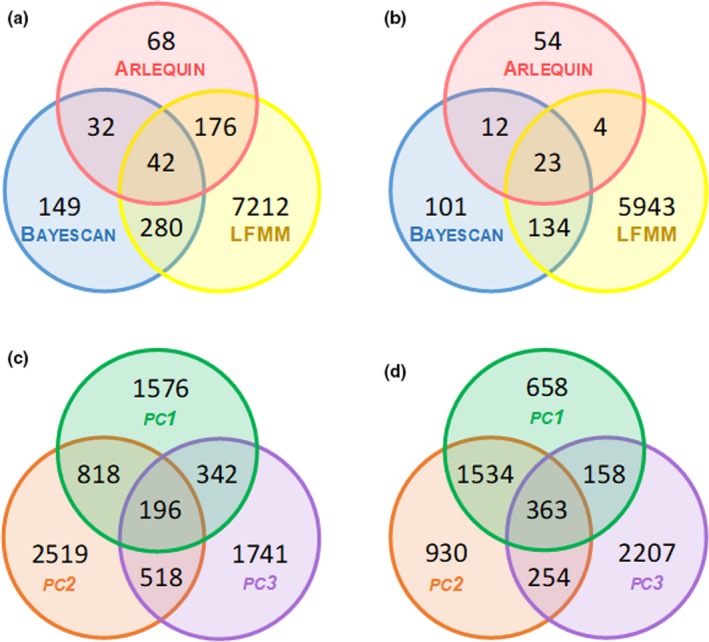
Venn diagrams showing the overlap of number of loci identified as being putatively under selection by arlequin (FDIST method) (red circles) and bayescan (blue circles) and presenting environmental associations according to LFMM (yellow circles) for analyses based on (a) all populations and (b) restricted to populations from the Iberian Peninsula. Panels (c–d) show the number of loci presenting associations with environmental PC1 (green circles), PC2 (orange circles), and PC3 (purple circles) according to LFMM analyses based on (c) all populations and (d) restricted to populations from the Iberian Peninsula.
3.3. Genetic structure
The PCA based on all populations showed a clear separation of Iberian and Canarian populations (Figure 4a). The PCA for the subset of Iberian populations showed that HOYO was the most differentiated population and some individuals from two other populations (ALHA and ALPU) also tended to stand out from the rest (Figure 4b). These results are in good agreement with those obtained from Bayesian clustering analyses. structure analyses for all populations and not considering prior population information identified K = 2 as the most likely clustering solution according to both the ΔK criterion and log probabilities of the data [LnPr (X|K)] (Figure A3a). These two clusters showed a very low degree of genetic admixture and split Canarian and Iberian populations (Figure 5c). Remarkably, genetic drift after divergence (parameter F in structure; Pritchard et al., 2000) for the cluster corresponding to the Canary Islands (F‐value = 0.419) was more than threefold the estimated for Iberian populations (F‐value = 0.121). structure analyses for K = 3 revealed further genetic structure and showed that one population from the Iberian Peninsula (BERZ) split from the rest of the mainland populations, albeit with a high degree of genetic admixture with the other Iberian populations (~10%–20%; Figure 5c). structure analyses restricted to Iberian populations identified that the most likely number of clusters was K = 2 according to the ΔK criterion, but LnPr (X|K) steadily increased up to K = 4 (Figure A3c). For K = 2, all the Iberian populations and individuals presented the same proportion of ancestry to the two inferred clusters (~20/80), indicating that they represent ghost clusters with no biological significance (see Chen, Durand, Forbes, & François, 2007; Guillot, Estoup, Mortier, & Cosson, 2005; Tonzo, Papadopoulou, & Ortego, 2019). However, structure analyses for K = 3 and K = 4 showed that BERZ and HOYO were assigned to two different genetic groups albeit with some degree of genetic admixture with the rest of Iberian populations (Figure 5c). The genetic clusters corresponding to these two populations had much higher estimates of genetic drift after divergence (BERZ: F‐value = 0.263; HOYO: F‐value = 0.084) than the one representing the rest of Iberian populations (F‐value = 0.021). Finally, structure analyses restricted to populations from the Canary Islands showed K = 2 as the most likely clustering solution according to both the ΔK criterion and LnPr (X|K) (Figure A3e). These two clusters separated HIER and TENE populations, which showed a very low degree of genetic admixture (Figure 5c). structure analyses ran considering prior population information (Hubisz et al., 2009) yielded qualitatively similar results, albeit in this case BERZ and HOYO presented a very small probability of assignment to their specific clusters in analyses focused on Iberian populations for K = 3 and K = 4 (see Figures A3 and A4). Finally, spatial analyses in construct focused on Iberian populations also showed strong admixture and no clear pattern of genetic structure. The predictive accuracy of construct analyses sharply increased from K = 1 to K = 2 (Figure A5a). However, layers (i.e., genetic clusters) beyond K = 1 contributed relatively little to total covariance (Figure A5b). This was particularly remarkable for K = 2, in which the second layer contributed less than 3% to total covariance (Figure A5b). Accordingly, the inferred genetic clusters for K > 1 presented high genetic admixture with little geographical congruence and only BERZ for K = 2–3 and ALHA for K = 3 tended to be assigned to different genetic clusters (Figure A6).
Figure 4.
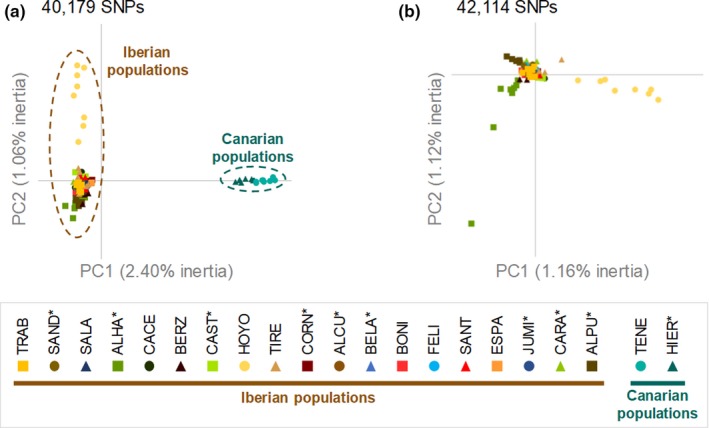
Principal component analyses (PCA) of genetic variation for populations of Moroccan locust for (a) all populations and (b) only considering populations from the Iberian Peninsula. Dashed brownish and greenish ellipses encircle Iberian and Canarian populations, respectively. The number of loci used in the different analyses is indicated in each panel. Populations with an asterisk indicate pest outbreaks during the sampling year. Population codes are described in Table 1
Figure 5.
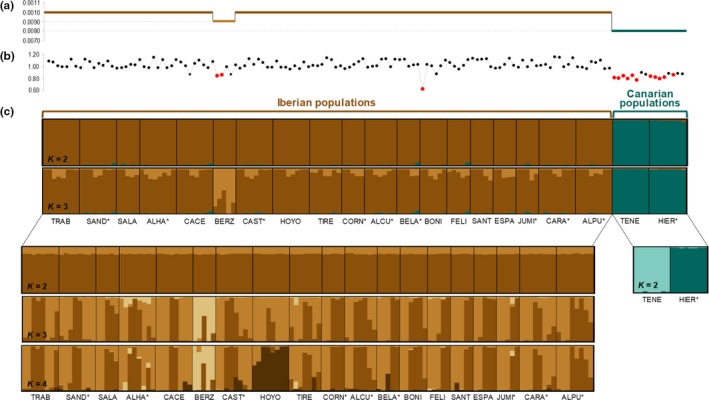
(a–b) Genetic diversity and (c) results of structure analyses not considering prior population information for the studied populations of Moroccan locust. Panel (a) shows nucleotide diversity (π) for each population calculated in stacks for all positions (polymorphic and nonpolymorphic). Panel (b) shows standardized multilocus heterozygosity (sMLH) of each individual, with values below the lowest 10th percentile shown in red. (c) Bar plots of structure analyses show the individual's probabilities of membership to each inferred genetic cluster for different K values. Each individual is represented by a vertical bar, which is partitioned into k colored segments showing the individual's probability of belonging to the cluster with that color. Thin vertical black lines separate individuals from different populations. structure analyses were performed at different hierarchical levels, analyzing all populations together and Iberian (brownish) and Canarian (greenish) populations separately. Populations with an asterisk indicate pest outbreaks during the sampling year. Population codes are described in Table 1.
3.4. Geographical and environmental drivers of genetic differentiation
Multiple matrix regression with randomization (MMRR) analyses showed that genetic differentiation [F ST/(1 − F ST)] among Iberian populations was not significantly associated with geographical or environmental (PC1, PC2, and PC3) distances (Table 2).
Table 2.
Multiple matrix regression with randomization (MMRR) for genetic differentiation at neutral loci [F ST/(1 − F ST)] in relation with geographical (log10 transformed) and environmental distances between each pair of Iberian populations. Environmental distances (PC1, PC2, and PC3) were estimated from a principal component analysis (PCA) on the 19 bioclimatic variables from the worldclim dataset. β: regression coefficient; t: t‐statistic; r 2: coefficient of determination; p: one‐tailed significance level
| β | t | r 2 | p | |
|---|---|---|---|---|
| Geographic distance (log10) | −.666 | −2.948 | .049 | .980 |
| Environmental PC1 | −.103 | −2.081 | .025 | .853 |
| Environmental PC2 | −.080 | −1.619 | .015 | .863 |
| Environmental PC3 | .025 | 0.492 | .001 | .397 |
3.5. Genetic diversity and past demographic history
Population genetic statistics (P, H O, H E, π, F IS) calculated in stacks for all positions (polymorphic and nonpolymorphic) are presented in Table 1. Population genetic diversity (π) and standardized multilocus heterozygosity (sMLH) for each individual are shown in Figure 5a. All estimates of population genetic diversity were significantly intercorrelated (H O – H E: r = .681, p < .001; H O – π: r = .681, p < .001; H E – π: r = .99, p < .001; Table 1). Estimates of genetic diversity (H O: r = .99, p < .001; H E: r = .99, p < .001; π: r = .95, p < .001) and differentiation (F ST: r = .99, p < .001; see previous section) obtained from a subset of five individuals randomly sampled from populations with eight genotyped individuals were highly correlated with those estimated based on the full datasets. This indicates that a relatively small number of genotyped individuals per population (i.e., as few as five individuals, our minimum sample size; Table 1) provides reliable estimates of genetic diversity and differentiation (see also Ortego, Gugger, & Sork, 2015b). All estimates of population genetic diversity were significantly lower in Canarian than in Iberian populations (one‐way anovas; all Ps < 0.001; Table 1) but did not significantly differ between outbreak and nonoutbreak populations (one‐way ANOVAs; all Ps > 0.2; Table 1). stairway plot analyses showed no clear demographic differences between outbreak and nonoutbreak populations, so we grouped the demographic profiles inferred for the different populations according to the two main geographical regions: Iberian Peninsula and Canary Islands (Figure 2). All Iberian populations showed an ancestral population increase around 150 ka BP, a severe demographic bottleneck around the last glacial maximum (LGM; ~21 ka BP) with a N e reduction by ~80% (from ~250,000 to ~50,000 diploid individuals), and an abrupt expansion at the onset of the Holocene to reach contemporary population sizes of around ~450,000 individuals (Figure 2b; Figure A7). The two Canarian populations presented a similar demographic profile than the Iberian ones although with a relatively less pronounced decline in N e during the last glacial period (Figure 2c). Canarian populations showed a long period of stability in the past (200–100 ka BP) followed by a demographic bottleneck during the last glacial period (100–21 ka BP) and a subsequent expansion at the onset of the Holocene to reach contemporary effective population sizes of 220,000–350,000 diploid individuals (Figure 2c; Figure A7).
4. DISCUSSION
We employed a large single‐nucleotide polymorphism dataset (~50,000 loci) to infer historical demographic trends and understand the neutral and selective processes shaping spatial patterns of genomic variation in the Moroccan locust, a grasshopper that has traditionally emerged as an devastating pest in overgrazed areas and caused extensive agricultural damage in Spain (e.g., Buj‐Buj, 1992) and many other areas across its ample distribution range (Latchininsky, 1998). Our analyses, focused on the Iberian Peninsula and remote populations from the Canary Islands, indicated that the species is characterized by widespread gene flow and low levels of genetic differentiation at regional scales. They also showed little differences in past demography and levels of genetic diversity and differentiation between outbreak and nonoutbreak populations. Finally, genome scans revealed that a small fraction (~0.2%–1.0%) of the sequenced loci are putatively under divergent selection and might be involved in local adaptation processes in response to the different ecological and environmental gradients experienced by the species.
4.1. Population genetic structure
Principal component analyses and Bayesian clustering analyses showed the presence of two well‐defined genetic groups corresponding to Iberian and Canarian populations. The large geographical distance between these two regions (1,700 km), their separation by oceanic water barriers to dispersal, and genetic drift in the small and highly isolated populations from the Canary Islands are probably the main drivers of observed patterns of genetic structure (e.g., Chapuis et al., 2009). According to this last point, Canarian populations presented lower levels of genetic diversity than most Iberian populations (Figure 5a; Table 1) and structure analyses showed that genetic drift after divergence for the cluster corresponding to the Canary Islands was much higher than that estimated for Iberian populations (Pritchard et al., 2000). PCA and hierarchical structure analyses for the two main genetic clusters showed that Canarian populations split into two very well‐defined genetic clusters corresponding to Tenerife and El Hierro Islands, whereas continental populations from Iberian Peninsula presented a much shallower genetic structure and considerable admixture across all analyses. structure analyses for Iberian populations showed the presence of two genetic clusters mostly corresponding to the nonoutbreaking and relatively isolated populations of BERZ and HOYO (Figure 5c). The genetic clusters of these two populations had much higher estimates of genetic drift after divergence than the one representing the rest of Iberian populations, which suggests that their relatively small sizes and/or lower connectivity with other populations are probably the main causes underlying their stronger genetic differentiation. Accordingly, one of these populations (BERZ) had the lowest levels of genetic diversity among all Iberian populations (Table 1). Apart from these two exceptions, we did not find clear differences in spatial patterns of genetic diversity and structure between outbreak and nonoutbreak populations. Levels of genetic differentiation among Iberian populations of D. maroccanus (mean F ST = 0.067; range = 0.051–0.102) were much lower than those reported in previous SNP‐based studies on nonpest cross‐backed grasshoppers (genus Dociostaurus) with narrower distributions such as D. crassiusculus (mean F ST = 0.129; range = 0.033–0.237; González‐Serna et al., 2018) and D. hispanicus (mean F ST = 0.189; range = 0.082–0.307; González‐Serna et al., 2019). As expected, levels of genetic differentiation among populations of the Moroccan locust were similar, albeit in some cases sensibly higher, than microsatellite‐based estimates obtained for other locusts and pest grasshoppers sampled at a similar spatial scale (Table A3). However, it should be noted that the different kinds of markers employed (i.e., SNPs versus microsatellites) make difficult the direct comparison of our estimates of genetic differentiation with those obtained in previous studies and, thus, small differences in absolute values must be interpreted with extreme caution (DeFaveri, Viitaniemi, Leder, & Merila, 2013; Ryynanen, Tonteri, Vasemagi, & Primmer, 2007). Overall, our results are in agreement with previous studies on other locust (e.g., Chapuis et al., 2008; Chapuis et al., 2011) and pelagic marine species (e.g., Als et al., 2011; Hoarau, Rijnsdorp, Veer, Stam, & Olsen, 2002) finding very low levels of genetic differentiation and indicate occasional genetic drift and differentiation in nonoutbreaking, solitarious or phase transition populations persisting at low densities in isolated pockets of suitable habitats (Chapuis et al., 2014; Ibrahim et al., 2000).
4.2. Past demographic history
Demographic reconstructions for the Moroccan locust using stairway plot revealed the presence of a remarkable genetic bottleneck during the last glacial maximum (~21 ka BP) followed by an abrupt expansion at the onset of the Holocene (Figure 2). Changes in effective population size (N e) through time did not qualitatively differ among populations, indicating that all of them have experienced parallel demographic histories. Demographic reconstructions for the recent past did not reveal more pronounced expansions in outbreak populations or population bottlenecks in solitarious or phase transition populations. It is also noteworthy that Canarian and Iberian population presented similar demographic profiles, with two main differences: Canarian populations consistently sustained lower effective population sizes over time and experienced less marked demographic declines during the last glacial period than Iberian populations. These results are congruent with the comparatively lower levels of genetic diversity of Canarian populations (Table 1) and compatible with the less severe impact of Pleistocene climatic oscillations at lower latitudes (Fernández‐Palacios et al., 2016; Hewitt, 1999; Snyder, 2016). The inferred demographic trends are also in agreement with fossil records of egg pods of D. maroccanus in sediments from the Canary Islands (Meco et al., 2011; Meco, Petit‐Maire, Ballester, Betancort, & Ramos, 2010). Paleontological evidence indicates that the abundance of the species has dramatically oscillated since the end of the Pliocene (3 Ma), with population peaks matching with warm interglacial periods of the Middle and Late Pleistocene (Meco et al., 2010, 2011; Snyder, 2016). The overall good correspondence between peaks of population size and warm periods inferred from both fossil records (Meco et al., 2010, 2011) and genomic data (present study) can be explained by the thermophilic nature of the species and the fact that its development and distribution are limited by low ambient temperatures (Aragón et al., 2013; Aragón & Lobo, 2012; Arias‐Giralda et al., 1997; Benlloch, 1947; Quesada‐Moraga & Santiago‐Álvarez, 2000).
4.3. Putative loci under selection and potential for local adaptation
Genome scans based on F ST outlier tests revealed that a small portion of the sampled genome (between 0.2% and 1.0%, depending on the dataset and method) is putatively under selection, whereas environmental association analyses (EAA) identified a much larger proportion of SNPs (12%–15%) with allele frequencies correlated with one or more environmental gradients. Beyond these differences in numbers, the specific loci identified by the two methods showed little overlap (Figure 3), which is in agreement with the results obtained by previous RADseq‐based studies (e.g., Dudaniec et al., 2018; Guo et al., 2016). Such differences have been interpreted to be a consequence of the contrasting sensitivities of each approach to deal with the effects of genetic drift and structure and by the better performance of EAA methods to detect weak or polygenic signatures of selection (De Mita et al., 2013; Frichot & François, 2015; de Villemereuil, Frichot, Bazin, François, & Gaggiotti, 2014). A BLAST search in an attempt to identify candidate genes associated with SNPs identified as outliers by the three methods yielded no significant alignment with available sequences at NCBI database (see also Jeffery et al., 2018). This might be explained by the very scarce genomic resources available for grasshoppers, with only one draft genome sequenced so far for a species (Locusta migratoria) (Wang et al., 2014) belonging to a different subfamily than the Moroccan locust (Cigliano et al., 2019). Also, the putative signals of selection in certain loci could be a consequence of genetic hitchhiking resulted from linkage disequilibrium between the identified outlier SNPs and nearby nonsequenced genes actually subjected to diversifying selection (Smith & Haigh, 1974). This is particularly relevant considering the extraordinarily large size charactering the genome of grasshoppers, which might have resulted in we have only sampled a relatively small representation of it (Camacho et al., 2015; Wang et al., 2014).
4.4. Conclusions and prospects
Overall, our study shows for the first time that populations of the economically important Moroccan locust present a shallow genetic differentiation and little differences in past demography, putative signatures of selection, and contemporary levels of genetic diversity and structure between outbreak and nonoutbreak populations. Outbreaks of Moroccan locust are usually linked to considerable cattle densities and overgrazing (Latchininsky, 1998; Louveaux et al., 1996), and it has been suggested that their frequency might increase in the future favored by global warming (Aragón et al., 2013). Although genetically nondifferentiated populations have often been interpreted as a single panmictic unit (e.g., Hoarau et al., 2002; Schrey, Schrey, Heist, & Reeve, 2008), the homogenizing effects of gene flow do not necessarily indicate demographic dependence or synchrony among populations (Chapuis et al., 2011) and evidence of selective processes inferred in this study suggests that some populations might experience idiosyncratic evolutionary dynamics (e.g., Guo et al., 2016; Pujolar et al., 2014). Future longitudinal and functional genomic studies could help to identify the proximate factors and specific genes underlying the observed putative signatures of selection (e.g., Bakkali & Martín‐Blázquez, 2018; Wang et al., 2014) and determine whether these are temporally stable or if, on the contrary, they are ephemeral and restricted to one or a few generations due to the homogenizing effects of gene flow (Babin et al., 2017; Laporte et al., 2016; Pujolar et al., 2014). The low levels of genetic differentiation among Iberian populations of the Moroccan locust revealed by our analyses indicate that even the application of thousands of SNP markers will not be able to identify the source of nascent outbreak populations at regional scales (see also Chapuis et al., 2009). However, these markers would be useful to determine the origin of eventual waves of long‐distance migrants from remote populations with genotypic differences. The high connectivity among populations of the Moroccan locust within the Iberian Peninsula also points out that the demographic dynamics of the species largely exceed regional and national boundaries. For this reason, and in order to avoid the implementation of local control actions with limited success and high environmental and economic costs, the development of future monitoring and sustainable management programs will require the coordination across the different administration levels and government authorities involved. Overall, the results of this study provide baseline information about some unknown aspects of the biology of the Moroccan locust with implications to develop integrated management practices aimed at reducing the negative impacts of this species that is expected to experience in the near future range expansions and increased outbreak intensity in response to ongoing climate warming and land use alterations (Abrol, 2014; Aragón et al., 2013; Benfekih, Chara, & Doumandji‐Mitche, 2002; Lankau et al., 2011).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
AUTHORS' CONTRIBUTIONS
MGS, PJC, and JO conceived the study and collected the samples. MGS and JO designed the study and analyses. MGS performed the laboratory work and analyzed the data guided by JO. MGS and JO led manuscript preparation with input from PJC.
Supporting information
Supplementary Material
ACKNOWLEDGEMENTS
We thank the unconditional support of Mercedes París and Vicenta Llorente during our visits to the entomological collections from the MNCN, José Miguel Aparicio for his help during sampling in the Canary Islands, María del Milagro Coca‐Abia for providing us samples from Zaragoza populations, Anna Papadopoulou for her great support with genomic data analyses, and three anonymous referees for helpful and constructive comments on an earlier version of this article. We also wish to thank to Carlos Muñoz‐Alcón and Antonio González, Benito Ortiz, and Manolo Pérez for taking us to some sampling locations from Salamanca and Canary Islands, respectively. Centro de Supercomputación de Galicia (CESGA) and Doñana's Singular Scientific‐Technical Infrastructure (ICTS‐RBD) provided access to computing resources. The respective administrative authorities from each study area (Andalucía, Aragón, Castilla‐La Mancha, Castilla y León, Cataluña, El Hierro, Extremadura, La Rioja, Madrid, Melilla, Murcia, Navarra, and Tenerife) provided us the corresponding permits for sampling. MGS was supported by a predoctoral scholarship from Junta de Comunidades de Castilla‐La Mancha and European Social Fund. This work received financial support from research grants CGL2011‐25053, CGL2014‐54671‐P, CGL2016‐80742‐R, and CGL2017‐83433‐P (cofunded by the Dirección General de Investigación y Gestión del Plan Nacional I+D+i and European Social Fund); PEII‐2014‐023‐P (cofunded by Junta de Comunidades de Castilla‐La Mancha and European Social Fund).
González‐Serna MJ, Cordero PJ, Ortego J. Insights into the neutral and adaptive processes shaping the spatial distribution of genomic variation in the economically important Moroccan locust (Dociostaurus maroccanus). Ecol Evol. 2020;10:3991–4008. 10.1002/ece3.6165
DATA AVAILABILITY STATEMENT
SNP datasets, input files for structure, PCA, construct, and stairway plot analyses are available for download from the Dryad Digital Repository (https://doi.org/10.5061/dryad.hx3ffbgb2).
REFERENCES
- Abrol D. P. (Ed.) (2014). Integrated pest management: Current concepts and ecological perspective (p. 576). San Diego, CA: Academic Press. [Google Scholar]
- Alberola‐Roma, A. (2012). Plagas de langosta y clima en la España del siglo XVIII. Relaciones, 129, 21–50. [Google Scholar]
- Als, T. D. , Hansen, M. M. , Maes, G. E. , Castonguay, M. , Riemann, L. , Aarestrup, K. , … Bernatchez, L. (2011). All roads lead to home: Panmixia of European eel in the Sargasso Sea. Molecular Ecology, 20(7), 1333–1346. 10.1111/j.1365-294X.2011.05011.x [DOI] [PubMed] [Google Scholar]
- Aragón, P. , Coca‐Abia, M. M. , Llorente, V. , & Lobo, J. M. (2013). Estimation of climatic favourable areas for locust outbreaks in Spain: Integrating species' presence records and spatial information on outbreaks. Journal of Applied Entomology, 137(8), 610–623. 10.1111/jen.12022 [DOI] [Google Scholar]
- Aragón, P. , & Lobo, J. M. (2012). Predicted effect of climate change on the invasibility and distribution of the western corn root‐worm. Agricultural and Forest Entomology, 14(1), 13–18. 10.1111/j.1461-9563.2011.00532.x [DOI] [Google Scholar]
- Arias‐Giralda, A. , Jiménez‐Viñuelas, J. , & Pérez‐Romero, A. (1997). Observaciones sobre el desarrollo embrionario y el avivamiento de Dociostaurus maroccanus (Thunb) en una finca de "La Serena" (Extremadura). Boletín De Sanidad Vegetal. Plagas, 23, 113–132. [Google Scholar]
- Arias‐Giralda, A. , Morales‐Agacino, E. , Cobos‐Suárez, J. M. , Sopeña‐Mañas, J. M. , & Martín‐Bernal, E. (1993). La langosta mediterránea Dociostaurus maroccanus (Thunberg). Boletín De Sanidad Vegetal. Plagas, 19(3), 1001–1011. [Google Scholar]
- Babin, C. , Gagnaire, P. A. , Pavey, S. A. , & Bernatchez, L. (2017). RAD‐seq reveals patterns of additive polygenic variation caused by spatially‐varying selection in the American eel (Anguilla rostrata). Genome Biology and Evolution, 9(11), 2974–2986. 10.1093/gbe/evx226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker S. R., & Wilkinson C. F. (Eds.) (1990). The effect of pesticides on human health. Princeton, NJ: Princeton Scientific Publishing. [Google Scholar]
- Bakkali, M. , & Martín‐Blázquez, R. (2018). RNA‐seq reveals large quantitative differences between the transcriptomes of outbreak and non‐outbreak locusts. Scientific Reports, 8(1), 9207 10.1038/s41598-018-27565-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaumont, M. A. , & Nichols, R. A. (1996). Evaluating loci for use in the genetic analysis of population structure. Proceedings of the Royal Society of London. Series B: Biological Sciences, 263(1377), 1619–1626. 10.1098/rspb.1996.0237 [DOI] [Google Scholar]
- Bekkevold, D. , Gross, R. , Arula, T. , Helyar, S. J. , & Ojaveer, H. (2016). Outlier loci detect intraspecific biodiversity amongst spring and autumn spawning herring across local scales. PLoS ONE, 11(4), e0148499 10.1371/journal.pone.0148499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfekih, L. , Chara, B. , & Doumandji‐Mitche, B. (2002). Influence of anthropogenic impact on the habitats and swarming risks of Dociostaurus maroccanus and Locusta migratoria (Orthoptera, Acrididae) in the Algerian Sahara and the semiarid zone. Journal of Orthoptera Research, 11(2), 243–250. [Google Scholar]
- Benlloch, M. (1947). Influencia de la humedad y la temperatura sobre la vitalidad y desarrollo de los huevos de langosta. Boletín De Patología Vegetal Y Entomología Agrícola, 15, 271–274. [Google Scholar]
- Berdan, E. L. , Mazzoni, C. J. , Waurick, I. , Roehr, J. T. , & Mayer, F. (2015). A population genomic scan in Chorthippus grasshoppers unveils previously unknown phenotypic divergence. Molecular Ecology, 24(15), 3918–3930. 10.1111/mec.13276 [DOI] [PubMed] [Google Scholar]
- Berkman, L. K. , Nielsen, C. K. , Roy, C. L. , & Heist, E. J. (2013). Resistance is futile: Effects of landscape features on gene flow of the northern bobwhite. Conservation Genetics, 14(2), 323–332. 10.1007/s10592-013-0471-1 [DOI] [Google Scholar]
- Bernatchez, S. , Xuereb, A. , Laporte, M. , Benestan, L. , Steeves, R. , Laflamme, M. , … Mallet, M. A. (2018). Seascape genomics of eastern oyster (Crassostrea virginica) along the Atlantic coast of Canada. Evolutionary Applications, 12(3), 587–609. 10.1111/eva.12741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradburd, G. S. , Coop, G. M. , & Ralph, P. L. (2018). Inferring continuous and discrete population genetic structure across space. Genetics, 210(1), 33–52. 10.1534/genetics.118.301333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brauer, C. J. , Hammer, M. P. , & Beheregaray, L. B. (2016). Riverscape genomics of a threatened fish across a hydroclimatically heterogeneous river basin. Molecular Ecology, 25(20), 5093–5113. 10.1111/mec.13830 [DOI] [PubMed] [Google Scholar]
- Brown, J. L. , Weber, J. J. , Alvarado‐Serrano, D. F. , Hickerson, M. J. , Franks, S. J. , & Carnaval, A. C. (2016). Predicting the genetic consequences of future climate change: The power of coupling spatial demography, the coalescent, and historical landscape changes. American Journal of Botany, 103(1), 153–163. 10.3732/ajb.1500117 [DOI] [PubMed] [Google Scholar]
- Buj‐Buj, A. (1992). Control de las plagas de langosta y modernización agrícola en la España de la segunda mitad del siglo XIX. Cuadernos Críticos De Geografía Humana. Universidad De Barcelona, 95(17), 1–67. [Google Scholar]
- Camacho, J. P. M. , Ruiz‐Ruano, F. J. , Martín‐Blázquez, R. , López‐León, M. D. , Cabrero, J. , Lorite, P. , … Bakkali, M. (2015). A step to the gigantic genome of the desert locust: Chromosome sizes and repeated DNAs. Chromosoma, 124(2), 263–275. 10.1007/s00412-014-0499-0 [DOI] [PubMed] [Google Scholar]
- Carson, R. (2002). Silent spring (40th anniversary ed.). Boston, MA: Houghton Mifflin. [Google Scholar]
- Catchen, J. M. , Amores, A. , Hohenlohe, P. A. , Cresko, W. , & Postlethwait, J. H. (2011). stacks: Building and genotyping loci de novo from short‐read sequences. G3: Genes|genomes|genetics, 1(3), 171–182. 10.1534/g3.111.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. , Bassham, S. , Wilson, T. , Currey, M. , O'Brien, C. , Yeates, Q. , & Cresko, W. A. (2013b). The population structure and recent colonization history of Oregon threespine stickleback determined using restriction‐site associated DNA‐sequencing. Molecular Ecology, 22(11), 2864–2883. 10.1111/mec.12330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catchen, J. M. , Hohenlohe, P. A. , Bassham, S. , Amores, A. , & Cresko, W. A. (2013a). stacks: An analysis tool set for population genomics. Molecular Ecology, 22(11), 3124–3140. 10.1111/mec.12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapco, W. , & Litzenberger, G. (2004). A DNA investigation into the mysterious disappearance of the rocky mountain grasshopper, mega‐pest of the 1800s. Molecular Phylogenetics and Evolution, 30(3), 810–814. 10.1016/s1055-7903(03)00209-4 [DOI] [PubMed] [Google Scholar]
- Chapuis, M. P. , Lecoq, M. , Michalakis, Y. , Loiseau, A. , Sword, G. A. , Piry, S. , & Estoup, A. (2008). Do outbreaks affect genetic population structure? A worldwide survey in Locusta migratoria, a pest plagued by microsatellite null alleles. Molecular Ecology, 17(16), 3640–3653. 10.1111/j.1365-294X.2008.03869.x [DOI] [PubMed] [Google Scholar]
- Chapuis, M.‐P. , Loiseau, A. , Michalakis, Y. , Lecoq, M. , Franc, A. , & Estoup, A. (2009). Outbreaks, gene flow and effective population size in the migratory locust, Locusta migratoria: A regional‐scale comparative survey. Molecular Ecology, 18(5), 792–800. 10.1111/j.1365-294X.2008.04072.x [DOI] [PubMed] [Google Scholar]
- Chapuis, M.‐P. , Plantamp, C. , Blondin, L. , Pagès, C. , Vassal, J.‐M. , & Lecoq, M. (2014). Demographic processes shaping genetic variation of the solitarious phase of the desert locust. Molecular Ecology, 23(7), 1749–1763. 10.1111/mec.12687 [DOI] [PubMed] [Google Scholar]
- Chapuis, M.‐P. , Popple, J.‐A. , Berthier, K. , Simpson, S. J. , Deveson, E. , Spurgin, P. , … Sword, G. A. (2011). Challenges to assessing connectivity between massive populations of the Australian plague locust. Proceedings of the Royal Society of London. Series B: Biological Sciences, 278(1721), 3152–3160. 10.1098/rspb.2010.2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, C. , Durand, E. , Forbes, F. , & François, O. (2007). Bayesian clustering algorithms ascertaining spatial population structure: A new computer program and a comparison study. Molecular Ecology Notes, 7(5), 747–756. 10.1111/j.1471-8286.2007.01769.x [DOI] [Google Scholar]
- Cigliano, M. M. , Braun, H. , Eades, D. C. , & Otte, D. (2019). Orthoptera species file. Version, 5.0/5.0. Retrieved from http://orthoptera.speciesfile.org/ [Google Scholar]
- Coltman, D. W. , Pilkington, J. G. , Smith, J. A. , & Pemberton, J. M. (1999). Parasite‐mediated selection against inbred soay sheep in a free‐living, island population. Evolution, 53(4), 1259–1267. 10.2307/2640828 [DOI] [PubMed] [Google Scholar]
- Coulon, A. , Fitzpatrick, J. W. , Bowman, R. , Stith, B. M. , Makarewich, C. A. , Stenzler, L. M. , & Lovette, I. J. (2008). Congruent population structure inferred from dispersal behaviour and intensive genetic surveys of the threatened Florida scrub‐jay (Aphelocoma coerulescens). Molecular Ecology, 17(7), 1685–1701. 10.1111/j.1365-294X.2008.03705.x [DOI] [PubMed] [Google Scholar]
- Crossley, M. S. , Chen, Y. H. , Groves, R. L. , & Schoville, S. D. (2017). Landscape genomics of Colorado potato beetle provides evidence of polygenic adaptation to insecticides. Molecular Ecology, 26(22), 6284–6300. 10.1111/mec.14339 [DOI] [PubMed] [Google Scholar]
- Crossley, M. S. , Rondon, S. I. , & Schoville, S. D. (2019a). Patterns of genetic differentiation in Colorado potato beetle correlate with contemporary, not historic, potato land cover. Evolutionary Applications, 12(4), 804–814. 10.1111/eva.12757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley, M. S. , Rondon, S. I. , & Schoville, S. D. (2019b). Effects of contemporary agricultural land cover on Colorado potato beetle genetic differentiation in the Columbia Basin and Central Sands. Ecology & Evolution, 9(16), 9385–9394. 10.1002/ece3.5489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kort, H. , Vandepitte, K. , Bruun, H. H. , Closset‐Kopp, D. , Honnay, O. , & Mergeay, J. (2014). Landscape genomics and a common garden trial reveal adaptive differentiation to temperature across Europe in the tree species Alnus glutinosa . Molecular Ecology, 23(19), 4709–4721. 10.1111/mec.12813 [DOI] [PubMed] [Google Scholar]
- De Mita, S. , Thuillet, A. C. , Gay, L. , Ahmadi, N. , Manel, S. , Ronfort, J. , & Vigouroux, Y. (2013). Detecting selection along environmental gradients: Analysis of eight methods and their effectiveness for outbreeding and selfing populations. Molecular Ecology, 22(5), 1383–1399. 10.1111/mec.12182 [DOI] [PubMed] [Google Scholar]
- de Villemereuil, P. , Frichot, É. , Bazin, É. , François, O. , & Gaggiotti, O. E. (2014). Genome scan methods against more complex models: When and how much should we trust them? Molecular Ecology, 23(8), 2006–2019. 10.1111/mec.12705 [DOI] [PubMed] [Google Scholar]
- DeFaveri, J. , Viitaniemi, H. , Leder, E. , & Merila, J. (2013). Characterizing genic and nongenic molecular markers: Comparison of microsatellites and SNPs. Molecular Ecology Resources, 13(3), 377–392. 10.1111/1755-0998.12071 [DOI] [PubMed] [Google Scholar]
- del Cañizo, J. , & Moreno, V. (1949). Biología y ecología de la langosta mediterránea o marroquí (Dociostaurus maroccanus Thunb.). Boletín De Patología Vegetal Y Entomología Agrícola, 17, 209–242. [Google Scholar]
- Dowle, E. J. , Bracewell, R. R. , Pfrender, M. E. , Mock, K. E. , Bentz, B. J. , & Ragland, G. J. (2017). Reproductive isolation and environmental adaptation shape the phylogeography of mountain pine beetle (Dendroctonus ponderosae). Molecular Ecology, 26, 6071–6084. 10.1111/mec.14342 [DOI] [PubMed] [Google Scholar]
- Dudaniec, R. Y. , Yong, C. J. , Lancaster, L. T. , Svensson, E. I. , & Hansson, B. (2018). Signatures of local adaptation along environmental gradients in a range‐expanding damselfly (Ischnura elegans). Molecular Ecology, 27(11), 2576–2593. 10.1111/mec.14709 [DOI] [PubMed] [Google Scholar]
- Earl, D. A. , & vonHoldt, B. M. (2012). structure harvester: A website and program for visualizing structure output and implementing the Evanno method. Conservation Genetics Resources, 4(2), 359–361. 10.1007/s12686-011-9548-7 [DOI] [Google Scholar]
- el Ghadraoui, L. , Petit, D. , Picaud, F. , & el Yamani, J. (2002). Relationship between labrum sensilla number in the Moroccan locust Dociostaurus maroccanus and the nature of its diet. Journal of Orthoptera Research, 11(1), 11–18. [Google Scholar]
- Enserink, M. (2004). Can the war on locusts be won? Science, 306(5703), 1880–1882. 10.1126/science.306.5703.1880 [DOI] [PubMed] [Google Scholar]
- Ernst, U. R. , van Hiel, M. B. , Depuydt, G. , Boerjan, B. , de Loof, A. , & Schoofs, L. (2015). Epigenetics and locust life phase transitions. The Journal of Experimental Biology, 218, 88–99. 10.1242/jeb.107078 [DOI] [PubMed] [Google Scholar]
- Ersts, P. J. (2018). Geographic distance matrix generator vol 1.2.3. American Museum of Natural History, Center for Biodiversity and Conservation. Retrieved from http://biodiversityinformatics.amnh.org
- Espindola, A. , Pellissier, L. , Maiorano, L. , Hordijk, W. , Guisan, A. , & Alvarez, N. (2012). Predicting present and future intra‐specific genetic structure through niche hindcasting across 24 millennia. Ecology Letters, 15(7), 649–657. 10.1111/j.1461-0248.2012.01779.x [DOI] [PubMed] [Google Scholar]
- Evanno, G. , Regnaut, S. , & Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: A simulation study. Molecular Ecology, 14(8), 2611–2620. 10.1111/j.1365-294X.2005.02553.x [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , Hofer, T. , & Foll, M. (2009). Detecting loci under selection in a hierarchically structured population. Heredity, 103(4), 285–298. 10.1038/hdy.2009.74 [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. (2010). arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Falush, D. , Stephens, M. , & Pritchard, J. K. (2003). Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics, 164(4), 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández‐Palacios, J. M. , Rijsdijk, K. F. , Norder, S. J. , Otto, R. , Nascimento, L. , Fernández‐Lugo, S. , … Whittaker, R. J. (2016). Towards a glacial‐sensitive model of island biogeography. Global Ecology and Biogeography, 25, 817–830. 10.1111/geb.12320 [DOI] [Google Scholar]
- Foll, M. , & Gaggiotti, O. (2008). A genome‐scan method to identify selected loci appropriate for both dominant and codominant markers: A Bayesian perspective. Genetics, 180(2), 977–993. 10.1534/genetics.108.092221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fordham, D. A. , Brook, B. W. , Moritz, C. , & Nogués‐Bravo, D. (2014). Better forecasts of range dynamics using genetic data. Trends in Ecology & Evolution, 29(8), 436–443. 10.1016/j.tree.2014.05.007 [DOI] [PubMed] [Google Scholar]
- François, O. , Martins, H. , Caye, K. , & Schoville, S. D. (2016). Controlling false discoveries in genome scans for selection. Molecular Ecology, 25(2), 454–469. 10.1111/mec.13513 [DOI] [PubMed] [Google Scholar]
- Frichot, E. , & François, O. (2015). LEA: An R package for landscape and ecological association studies. Methods in Ecology and Evolution, 6(8), 925–929. 10.1111/2041-210X.12382 [DOI] [Google Scholar]
- Frichot, E. , Mathieu, F. , Trouillon, T. , Bouchard, G. , & Francois, O. (2014). Fast and efficient estimation of individual ancestry coefficients. Genetics, 196(4), 973–983. 10.1534/genetics.113.160572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frichot, E. , Schoville, S. D. , Bouchard, G. , & François, O. (2013). Testing for associations between loci and environmental gradients using Latent Factor Mixed Models. Molecular Biology and Evolution, 30(7), 1687–1699. 10.1093/molbev/mst063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gassmann, A. J. , Onstad, D. W. , & Pittendrigh, B. R. (2009). Evolutionary analysis of herbivorous insects in natural and agricultural environments. Pest Management Science, 65, 1174–1181. 10.1002/ps.1844 [DOI] [PubMed] [Google Scholar]
- Gilbert, K. J. , Andrew, R. L. , Bock, D. G. , Franklin, M. T. , Kane, N. C. , Moore, J. S. , … Vines, T. H. (2012). Recommendations for utilizing and reporting population genetic analyses: The reproducibility of genetic clustering using the program structure . Molecular Ecology, 21(20), 4925–4930. 10.1111/j.1365-294X.2012.05754.x [DOI] [PubMed] [Google Scholar]
- González‐Serna, M. J. , Cordero, P. J. , & Ortego, J. (2018). Using high‐throughput sequencing to investigate the factors structuring genomic variation of a Mediterranean grasshopper of great conservation concern. Scientific Reports, 8(1), 13436 10.1038/s41598-018-31775-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Serna, M. J. , Cordero, P. J. , & Ortego, J. (2019). Spatiotemporally explicit demographic modelling supports a joint effect of historical barriers to dispersal and contemporary landscape composition on structuring genomic variation in a red‐listed grasshopper. Molecular Ecology, 28(9), 2155–2172. 10.1111/mec.15086 [DOI] [PubMed] [Google Scholar]
- Guerrero, A. , Ramos, V. E. , López, S. , Álvarez, J. M. , Domínguez, A. , Coca‐Abia, M. M. , … Quero, C. (2019). Enantioselective synthesis and activity of all diastereoisomers of (e)‐phytal, a pheromone component of the Moroccan locust, Dociostaurus maroccanus . Journal of Agricultural and Food Chemistry, 67(1), 72–80. 10.1021/acs.jafc.8b06346 [DOI] [PubMed] [Google Scholar]
- Guillot, G. , Estoup, A. , Mortier, F. , & Cosson, J. F. (2005). A spatial statistical model for landscape genetics. Genetics, 170(3), 1261–1280. 10.1534/genetics.104.033803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, B. , Li, Z. , & Merilä, J. (2016). Population genomic evidence for adaptive differentiation in the Baltic sea herring. Molecular Ecology, 25(12), 2833–2852. 10.1111/mec.13657 [DOI] [PubMed] [Google Scholar]
- Hewitt, G. M. (1999). Post‐glacial re‐colonization of European biota. Biological Journal of the Linnean Society, 68(1–2), 87–112. 10.1111/j.1095-8312.1999.tb01160.x [DOI] [Google Scholar]
- Hijmans, R. J. , Cameron, S. E. , Parra, J. L. , Jones, P. G. , & Jarvis, A. (2005). Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology, 25(15), 1965–1978. 10.1002/joc.1276 [DOI] [Google Scholar]
- Hoarau, G. , Rijnsdorp, A. D. , Van Der Veer, H. W. , Stam, W. T. , & Olsen, J. L. (2002). Population structure of plaice (Pleuronectes platessa L.) in northern Europe: Microsatellites revealed large‐scale spatial and temporal homogeneity. Molecular Ecology, 11(7), 1165–1176. 10.1046/j.1365-294X.2002.01515.x [DOI] [PubMed] [Google Scholar]
- Hohenlohe, P. A. , Bassham, S. , Etter, P. D. , Stiffler, N. , Johnson, E. A. , & Cresko, W. A. (2010). Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS Genetics, 6(2), e1000862 10.1371/journal.pgen.1000862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubisz, M. J. , Falush, D. , Stephens, M. , & Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Molecular Ecology Resources, 9(5), 1322–1332. 10.1111/j.1755-0998.2009.02591.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim, K. M. (2001). Plague dynamics and population genetics of the desert locust: Can turnover during recession maintain population genetic structure? Molecular Ecology, 10(3), 581–591. 10.1046/j.1365-294x.2001.01212.x [DOI] [PubMed] [Google Scholar]
- Ibrahim, K. M. , Sourrouille, P. , & Hewitt, G. M. (2000). Are recession populations of the desert locust (Schistocerca gregaria) remnants of past swarms? Molecular Ecology, 9(6), 783–791. 10.1046/j.1365-294x.2000.00932.x [DOI] [PubMed] [Google Scholar]
- Jakobsson, M. , & Rosenberg, N. A. (2007). clumpp: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics, 23(14), 1801–1806. 10.1093/bioinformatics/btm233 [DOI] [PubMed] [Google Scholar]
- Janes, J. K. , Miller, J. M. , Dupuis, J. R. , Malenfant, R. M. , Gorrell, J. C. , Cullingham, C. I. , & Andrew, R. L. (2017). The K=2 conundrum. Molecular Ecology, 26(14), 3594–3602. 10.1111/mec.14187 [DOI] [PubMed] [Google Scholar]
- Jeffery, N. W. , Bradbury, I. R. , Stanley, R. R. E. , Wringe, B. F. , Van Wyngaarden, M. , Lowen, J. B. , … DiBacco, C. (2018). Genomewide evidence of environmentally mediated secondary contact of European green crab (Carcinus maenas) lineages in eastern North America. Evolutionary Applications, 11(6), 869–882. 10.1111/eva.12601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jombart, T. (2008). adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics, 24(11), 1403–1405. 10.1093/bioinformatics/btn129 [DOI] [PubMed] [Google Scholar]
- Karsten, M. , Addison, P. , Jansen van Vuuren, B. , & Terblanche, J. S. (2016). Investigating population differentiation in a major African agricultural pest: Evidence from geometric morphometrics and connectivity suggests high invasion potential. Molecular Ecology, 25(13), 3019–3032. 10.1111/mec.13646 [DOI] [PubMed] [Google Scholar]
- Keightley, P. D. , Ness, R. W. , Halligan, D. L. , & Haddrill, P. R. (2014). Estimation of the spontaneous mutation rate per nucleotide site in a Drosophila melanogaster full‐sib family. Genetics, 196(1), 313–320. 10.1534/genetics.113.158758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, H. , Dorn, S. , & Mazzi, D. (2013). Molecular genetics and genomics generate new insights into invertebrate pest invasions. Evolutionary Applications, 6(5), 842–856. 10.1111/eva.12071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lankau, R. , Jørgensen, P. S. , Harris, D. J. , & Sih, A. (2011). Incorporating evolutionary principles into environmental management and policy. Evolutionary Applications, 4(2), 315–325. 10.1111/j.1752-4571.2010.00171.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laporte, M. , Pavey, S. A. , Rougeux, C. , Pierron, F. , Lauzent, M. , Budzinski, H. , … Bernatchez, L. (2016). RAD sequencing reveals within‐generation polygenic selection in response to anthropogenic organic and metal contamination in north Atlantic eels. Molecular Ecology, 25(1), 219–237. 10.1111/mec.13466 [DOI] [PubMed] [Google Scholar]
- Latchininsky, A. V. (1998). Moroccan locust Dociostaurus maroccanus (Thunberg, 1815): A faunistic rarity or an important economic pest? Journal of Insect Conservation, 2(3), 167–178. 10.1023/a:1009639628627 [DOI] [Google Scholar]
- Latchininsky, A. V. (2013). Locusts and remote sensing: A review. Journal of Applied Remote Sensing, 7(1), 1–19. 10.1117/1.JRS.7.075099 [DOI] [Google Scholar]
- Leftwich, P. T. , Bolton, M. , & Chapman, T. (2016). Evolutionary biology and genetic techniques for insect control. Evolutionary Applications, 9(1), 212–230. 10.1111/eva.12280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenormand, T. (2002). Gene flow and the limits to natural selection. Trends in Ecology & Evolution, 17(4), 183–189. 10.1016/S0169-5347(02)02497-7 [DOI] [Google Scholar]
- Lischer, H. E. , & Excoffier, L. (2012). pgdspider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics, 28(2), 298–299. 10.1093/bioinformatics/btr642 [DOI] [PubMed] [Google Scholar]
- Liu, X. , & Fu, Y.‐X. (2015). Exploring population size changes using SNP frequency spectra. Nature Genetics, 47(5), 555–559. 10.1038/ng.3254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockwood, J. A. (2004). Locust: The devastating rise and mysterious disappearance of the insect that shaped the American frontier (p. 320). New York, NY: Basic Books. [Google Scholar]
- Louveaux, A. , Mouhim, A. , Roux, G. , Gillon, Y. , & Barral, H. (1996). Effect of pastoral activities upon locust populations in the Siroua Massif (Morocco). Revue D'écologie: La Terre Et La Vie, 51(2), 139–151. [Google Scholar]
- Luikart, G. , England, P. R. , Tallmon, D. , Jordan, S. , & Taberlet, P. (2003). The power and promise of population genomics: From genotyping to genome typing. Nature Reviews Genetics, 4, 981 10.1038/nrg1226 [DOI] [PubMed] [Google Scholar]
- Martín‐Blázquez, R. , Chen, B. , Kang, L. , & Bakkali, M. (2017). Evolution, expression and association of the chemosensory protein genes with the outbreak phase of the two main pest locusts. Scientific Reports, 7(1), 6653 10.1038/s41598-017-07068-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J. , & Haigh, J. (2007). The hitch‐hiking effect of a favourable gene. Genetics Research, 89(5‐6), 391–403. 10.1017/S0016672308009579 [DOI] [PubMed] [Google Scholar]
- Meco, J. , Muhs, D. R. , Fontugne, M. , Ramos, A. J. , Lomoschitz, A. , & Patterson, D. (2011). Late Pliocene and quaternary Eurasian locust infestations in the Canary archipelago. Lethaia, 44(4), 440–454. 10.1111/j.1502-3931.2010.00255.x [DOI] [Google Scholar]
- Meco, J. , Petit‐Maire, N. , Ballester, J. , Betancort, J. F. , & Ramos, A. J. (2010). The Acridian plagues, a new Holocene and Pleistocene palaeoclimatic indicator. Global and Planetary Change, 72(4), 318–320. 10.1016/j.gloplacha.2010.01.007 [DOI] [Google Scholar]
- Miles, A. , Harding, N. J. , Bottà, G. , Clarkson, C. S. , Antão, T. , Kozak, K. , … Kwiatkowski, D. P. (2017). Genetic diversity of the African malaria vector Anopheles gambiae . Nature, 552, 96 10.1038/nature24995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortego, J. , Aguirre, M. P. , Noguerales, V. , & Cordero, P. J. (2015a). Consequences of extensive habitat fragmentation in landscape‐level patterns of genetic diversity and structure in the Mediterranean esparto grasshopper. Evolutionary Applications, 8(6), 621–632. 10.1111/eva.12273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortego, J. , Gugger, P. F. , & Sork, V. L. (2015b). Climatically stable landscapes predict patterns of genetic structure and admixture in the Californian canyon live oak. Journal of Biogeography, 42(2), 328–338. 10.1111/jbi.12419 [DOI] [Google Scholar]
- Ortego, J. , Gugger, P. F. , & Sork, V. L. (2018). Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). New Phytologist, 218(2), 804–818. 10.1111/nph.14951 [DOI] [PubMed] [Google Scholar]
- Papadopoulou, A. , & Knowles, L. L. (2015). Species‐specific responses to island connectivity cycles: Refined models for testing phylogeographic concordance across a Mediterranean Pleistocene aggregate island complex. Molecular Ecology, 24(16), 4252–4268. 10.1111/mec.13305 [DOI] [PubMed] [Google Scholar]
- Peterson, B. K. , Weber, J. N. , Kay, E. H. , Fisher, H. S. , & Hoekstra, H. E. (2012). Double digest RADseq: An inexpensive method for de novo SNP discovery and genotyping in model and non‐model species. PLoS ONE, 7(5), e37135 10.1371/journal.pone.0037135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K. , Stephens, M. , & Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics, 155(2), 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pujolar, J. M. , Jacobsen, M. W. , Als, T. D. , Frydenberg, J. , Munch, K. , Jónsson, B. , … Hansen, M. M. (2014). Genome‐wide single‐generation signatures of local selection in the panmictic European eel. Molecular Ecology, 23(10), 2514–2528. 10.1111/mec.12753 [DOI] [PubMed] [Google Scholar]
- Qin, Y.‐J. , Krosch, M. N. , Schutze, M. K. , Zhang, Y. , Wang, X.‐X. , Prabhakar, C. S. , … Li, Z.‐H. (2018). Population structure of a global agricultural invasive pest, Bactrocera dorsalis (Diptera: Tephritidae). Evolutionary Applications, 11(10), 1990–2003. 10.1111/eva.12701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada‐Moraga, E. , & Santiago‐Álvarez, C. (2000). Temperature related effects on embryonic development of the Mediterranean locust, Dociostaurus Maroccanus . Physiological Entomology, 25(2), 191–195. 10.1046/j.1365-3032.2000.00185.x [DOI] [Google Scholar]
- R Core Team . (2018). R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; Retrieved from https://www.R-project.org/. [Google Scholar]
- Rellstab, C. , Gugerli, F. , Eckert, A. J. , Hancock, A. M. , & Holderegger, R. (2015). A practical guide to environmental association analysis in landscape genomics. Molecular Ecology, 24(17), 4348–4370. 10.1111/mec.13322 [DOI] [PubMed] [Google Scholar]
- Rosenberg, N. A. (2004). distruct: A program for the graphical display of population structure. Molecular Ecology Notes, 4(1), 137–138. 10.1046/j.1471-8286.2003.00566.x [DOI] [Google Scholar]
- Ruxton, G. D. , & Neuhauser, M. (2010). When should we use one‐tailed hypothesis testing? Methods in Ecology and Evolution, 1(2), 114–117. 10.1111/j.2041-210X.2010.00014.x [DOI] [Google Scholar]
- Ryynanen, H. J. , Tonteri, A. , Vasemagi, A. , & Primmer, C. R. (2007). A comparison of biallelic markers and microsatellites for the estimation of population and conservation genetic parameters in Atlantic salmon (Salmo salar). Journal of Heredity, 98(7), 692–704. 10.1093/jhered/esm093 [DOI] [PubMed] [Google Scholar]
- Schrey, N. M. , Schrey, A. W. , Heist, E. J. , & Reeve, J. D. (2008). Fine‐scale genetic population structure of southern pine beetle (Coleoptera: Curculionidae) in Mississippi forests. Environmental Entomology, 37(1), 271–276. 10.1093/ee/37.1.271 [DOI] [PubMed] [Google Scholar]
- Sexton, J. P. , Hangartner, S. B. , & Hoffmann, A. A. (2014). Genetic isolation by environment or distance: Which pattern of gene flow is most common? Evolution, 68(1), 1–15. 10.1111/evo.12258 [DOI] [PubMed] [Google Scholar]
- Shafer, A. B. , & Wolf, J. B. (2013). Widespread evidence for incipient ecological speciation: A meta‐analysis of isolation‐by‐ecology. Ecology Letters, 16(7), 940–950. 10.1111/ele.12120 [DOI] [PubMed] [Google Scholar]
- Sherpa, S. , Rioux, D. , Goindin, D. , Fouque, F. , Francois, O. , & Despres, L. (2018). At the origin of a worldwide invasion: Unraveling the genetic makeup of the Caribbean bridgehead populations of the dengue vector Aedes aegypti . Genome Biology and Evolution, 10(1), 56–71. 10.1093/gbe/evx267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, J.‐C. , d'Alencon, E. , Guy, E. , Jacquin‐Joly, E. , Jaquiery, J. , Nouhaud, P. , … Streiff, R. (2015). Genomics of adaptation to host‐plants in herbivorous insects. Briefings in Functional Genomics, 14(6), 413–423. 10.1093/bfgp/elv015 [DOI] [PubMed] [Google Scholar]
- Skaf, R. , Popov, G. B. , Roffey, J. , Scorer, R. S. , & Hewitt, J. (1990). The desert locust: An international challenge [and discussion]. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 328(1251), 525–538. [Google Scholar]
- Snyder, C. W. (2016). Evolution of global temperature over the past two million years. Nature, 538, 226–228. 10.1038/nature19798 [DOI] [PubMed] [Google Scholar]
- Soria‐Carrasco, V. , Gompert, Z. , Comeault, A. A. , Farkas, T. E. , Parchman, T. L. , Johnston, J. S. , … Nosil, P. (2014). Stick insect genomes reveal natural selection's role in parallel speciation. Science, 344(6185), 738–742. 10.1126/science.1252136 [DOI] [PubMed] [Google Scholar]
- Stoffel, M. A. , Esser, M. , Kardos, M. , Humble, E. , Nichols, H. , David, P. , & Hoffman, J. I. (2016). inbreedr: An R package for the analysis of inbreeding based on genetic markers. Methods in Ecology and Evolution, 7(11), 1331–1339. 10.1111/2041-210X.12588 [DOI] [Google Scholar]
- Tonzo, V. , Papadopoulou, A. , & Ortego, J. (2019). Genomic data reveal deep genetic structure but no support for current taxonomic designation in a grasshopper species complex. Molecular Ecology, 28(17), 3869–3886. 10.1111/mec.15189 [DOI] [PubMed] [Google Scholar]
- Uvarov, B. (1977). Grasshoppers and locusts: A handbook of general Acridology (vol. 2): Behaviour, ecology, biogeography, population dynamics (Vol. 2, p. 613). London, UK: Centre for Overseas Pest Research. [Google Scholar]
- Vasemägi, A. , & Primmer, C. R. (2005). Challenges for identifying functionally important genetic variation: The promise of combining complementary research strategies. Molecular Ecology, 14(12), 3623–3642. 10.1111/j.1365-294X.2005.02690.x [DOI] [PubMed] [Google Scholar]
- Venkatesan, M. , & Rasgon, J. L. (2010). Population genetic data suggest a role for mosquito‐mediated dispersal of west Nile virus across the western United States. Molecular Ecology, 19(8), 1573–1584. 10.1111/j.1365-294X.2010.04577.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, I. J. (2013). Examining the full effects of landscape heterogeneity on spatial genetic variation: A multiple matrix regression approach for quantifying geographic and ecological isolation. Evolution, 67(12), 3403–3411. 10.1111/evo.12134 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Fang, X. , Yang, P. , Jiang, X. , Jiang, F. , Zhao, D. , … Kang, L. E. (2014). The locust genome provides insight into swarm formation and long‐distance flight. Nature Communications, 5, 2957 10.1038/ncomms3957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yannic, G. , Ortego, J. , Pellissier, L. , Lecomte, N. , Bernatchez, L. , & Cote, S. D. (2018). Linking genetic and ecological differentiation in an ungulate with a circumpolar distribution. Ecography, 41(6), 922–937. 10.1111/ecog.02995 [DOI] [Google Scholar]
- Zepeda‐paulo, F. A. , Simon, J.‐C. , Ramírez, C. C. , Fuentes‐contreras, E. , Margaritopoulos, J. T. , Wilson, A. C. C. , … Figueroa, C. C. (2010). The invasion route for an insect pest species: The tobacco aphid in the new world. Molecular Ecology, 19(21), 4738–4752. 10.1111/j.1365-294X.2010.04857.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
SNP datasets, input files for structure, PCA, construct, and stairway plot analyses are available for download from the Dryad Digital Repository (https://doi.org/10.5061/dryad.hx3ffbgb2).