

SUMMARY
Adverse environmental conditions can affect rates of animal developmental progression and lead to temporary developmental quiescence (diapause), exemplified by the dauer larva stage of the nematode Caenorhabditis elegans. Remarkably, patterns of cell division and temporal cell fate progression in C. elegans larvae are not affected by changes in developmental trajectory. However, the underlying physiological and gene regulatory mechanisms that ensure robust developmental patterning despite substantial plasticity in developmental progression are largely unknown. Here, we report that diapause inducing pheromones correct heterochronic developmental cell lineage defects caused by insufficient expression of let-7 family microRNAs in C. elegans. Moreover, two conserved endocrine signaling pathways, DAF-7/TGF-β and DAF-2/Insulin, that confer on the larva diapause/non-diapause alternative developmental trajectories, interact with the nuclear hormone receptor, DAF-12, to initiate and regulate a rewiring of the genetic circuitry controlling temporal cell fates. This rewiring includes engagement of certain heterochronic genes, lin-46, lin-4 and nhl-2, that are previously associated with an altered genetic program in post-diapause animals, in combination with a novel ligand-independent DAF-12 activity, to downregulate the critical let-7 family target Hunchback-like-1 (HBL-1). Our results show how pheromone or endocrine signaling pathways can coordinately regulate both developmental progression and cell fate transitions in C. elegans larvae under stress, so that the developmental schedule of cell fates remains unaffected by changes in developmental trajectory.
Keywords: pheromones, ascarosides, endocrine signaling, dauer larva, heterochronic genes, microRNAs, let-7, developmental robustness
Graphical Abstract
eTOC Blurb
Ilbay and Ambros show that pheromones and nutritional signals coordinately regulate developmental progression and cell fate transitions during C. elegans development and this coordination involves activation of an alternative temporal downregulation program for a critical let-7 family target, HBL-1.
INTRODUCTION
Despite the vast complexity of animal development, developmental processes are remarkably robust in the face of environment and physiological stresses. Multicellular animals develop from a single cell through a temporal and spatial elaboration of events that include cell division, differentiation, migration, and apoptosis. Early developmental cell lineages rapidly diverge functionally and spatially, and continue to follow distinct paths towards building diverse parts of the animal body. Marvelously, the sequence and synchrony of these increasingly complex programs of cell fate progression are precisely coordinated, regardless of various environmental and physiological stresses that the animal may encounter in its natural environment.
The nematode C. elegans develops through four larval stages, each of which consists of an invariant set of characteristic developmental events [1]. During larval development, stem cells and blast cells divide and progressively produce progeny cells with defined stage-specific fates. The timing of cell fate transitions within individual postembryonic cell lineages is regulated by genes of the heterochronic pathway, whose products include cell fate determinant transcription factors, as well as microRNAs (miRNAs) and other regulators of these transcription factors [2,3]. In mutants defective in the activity of one or more heterochronic genes, the synchrony between cell fates and developmental stages is lost in certain cell lineages, which results in dissonance in the relative timing of developmental events across the animal and consequently morphological abnormalities.
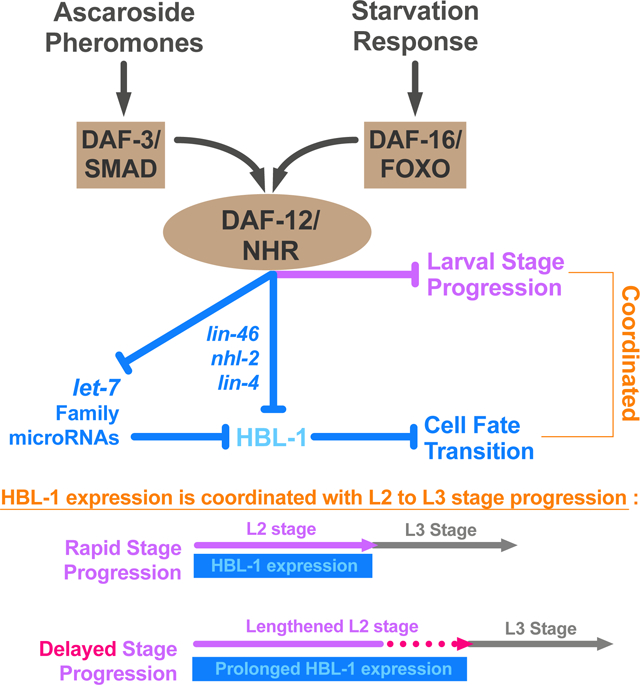
During C. elegans larval development, lateral hypodermal stem cells (‘seam cells’) express stage-specific proliferative or self-renewal behavior (Figure 1A). Particularly, whilst seam cells divide asymmetrically at each larval stage (L1–L4), giving rise to a new seam cell and a differentiating hypodermal (hyp7) cell, at the L2 stage, certain seam cells also undergo a single round of symmetric cell division, resulting in an increase in the number of seam cells on each side of the animal from ten to sixteen. This L2-specific proliferative cell fate is driven by a transcription factor, HBL-1, which specifies expression of the L2 cell fate, and also prevents the expression of the L3 cell fates [4,5]. Therefore, in order to allow progression to L3 cell fates, HBL-1 must be downregulated by the end of the L2 stage. If HBL-1 is not properly downregulated, for example in mutants defective in upstream regulatory genes, seam cells inappropriately execute L2 cell fates at later stages, resulting in an enlarged and developmentally retarded population of seam cells in adult worms. Three let-7 family miRNAs (mir-48/84/241) are redundantly required for proper temporal downregulation of HBL-1 [6]. Larvae lacking all three let-7 family miRNAs reiterate L2 cell fates in later stages of development. The degree of reiteration, hence the severity of the phenotype, varies depending on genetic and environmental factors [7], and can be quantified by counting the number of seam cells in young adult worms.
Figure 1. Temporal fates of hypodermal seam cells are robust against changes in developmental trajectory induced by crowding or starvation.
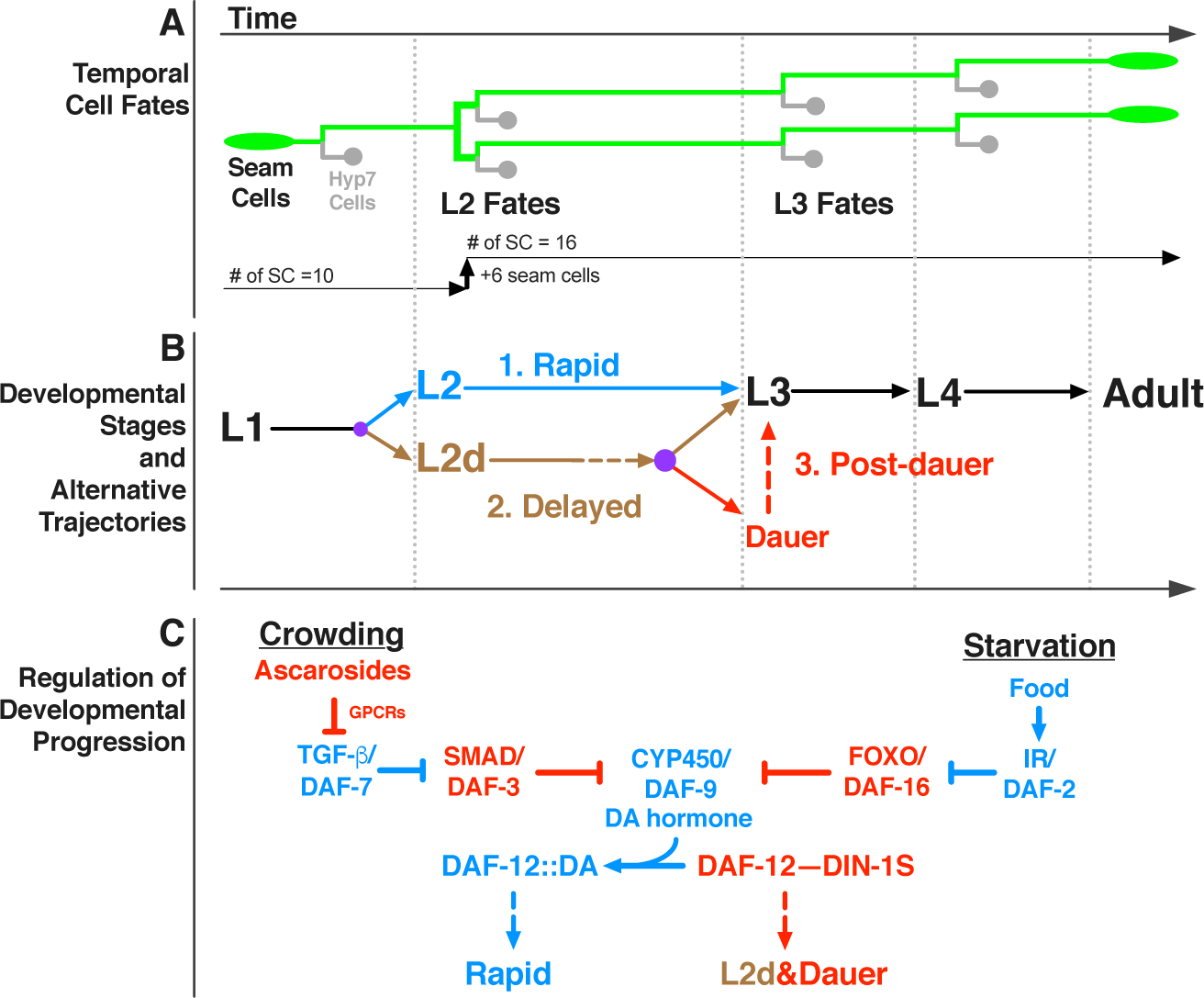
(A) Lineage diagram showing temporal (stage-specific) hypodermal seam cell fates. Seam cells (green) divide asymmetrically at each larval stage, renewing themselves while giving rise to hyp7 cells (gray). At the L2 stage, seam cells undergo a single round of symmetric cell division, resulting in an increase in their number. Note that only six out of ten seam cells undergo symmetric cell division which increases the total number of seam cells on each side of the worm from ten to sixteen. (B) Developmental stages and three distinct developmental trajectories: 1. Continuous, unipotent, and rapid progression define the L2 trajectory (blue); 2. Continuous but bipotent and delayed progression define the L2d trajectory (brown), 3. Developmental progression is interrupted by a diapause in the dauer-interrupted/post-dauer trajectory (brown followed by red). Time axis indicates the order of events in time (not proportional to absolute time). Lateral dotted lines indicate the molts between stages. Purple dots represent decision points between different trajectory options. (C) Regulation of developmental progression. Under favorable conditions dafachronic acid (DA) hormone is abundant and DA-bound DAF-12 promotes rapid development. Crowding or starvation induces L2d and dauer formation by repressing TGF-β/DAF-7 signals or insulin signaling (IR/DAF-2: insulin receptor), respectively. Activated effectors of these signaling pathways (DAF-3 or DAF-16) inhibit the biosynthesis of DA; and the unliganded DAF-12 interacts with DIN-1S, which together promote L2d and dauer formation.
C. elegans is a free-living nematode whose environment is prone to fluctuations between conditions that are favorable and unfavorable for completion of development [8]. Under favorable conditions (such as abundant food) C. elegans larvae develop rapidly and continuously progress through the four larval stages to the adult (Figure 1B, rapid). However, when the conditions are not favorable (for example, in the face of declining resources owing to high population density), the larva at the end of the L2 stage can elect to enter a developmentally arrested diapause, called the dauer larva, which is nonfeeding, stress-resistant, and long-lived [9]. When conditions improve, the dauer larva resumes development to the reproductive adult (Figure 1B, post-dauer). The DAF-7/TGF-β and DAF-2/insulin endocrine signaling pathways are the two major signaling pathways that regulate C. elegans dauer larva diapause. These two pathways act in parallel to integrate information about population density and nutritional status by co-modulating the biosynthesis of the dafachronic acid (DA) hormone. DA is the ligand of a nuclear hormone receptor, DAF-12, which opposes dauer formation when it is DA-bound and forms a repressor complex with DIN-1S and promotes dauer formation when it is unliganded (Figure 1C) [10].
The order and sequence of temporal cell fates in the various C. elegans larval cell lineages are robustly maintained regardless of developmental trajectory: for example, blast cells properly transition from L2 to L3 fates whether the larva develops rapidly and continuously, or instead traverses dauer diapause, which imposes a lengthy (even months-long) interruption of the L2 to L3 transition (Figure 1A&B). Interestingly, cell fate transition defects of many heterochronic mutants are modified (either suppressed or enhanced) when larval development is interrupted by dauer diapause, suggesting that the genetic regulatory pathways regulating temporal cell fate progression are modified depending on whether the animal develops continuously vs undergoes dauer-interrupted development [11–13]. The mechanisms by which temporal cell fate specification pathways are modified in association with the dauer larva trajectory are poorly understood, especially with regards to how modifications to the regulatory networks controlling temporal cell fate transitions may be coupled to particular steps in the specification and/or execution of the dauer larva diapause trajectory. Of particular interest is the question of whether and how the dauer-promoting signals that are monitored by L1 and L2d larvae might act prior to dauer commitment to directly modify gene regulatory mechanisms controlling temporal cell fate transitions.
To investigate the impact of dauer-inducing environmental and endocrine signals on the regulatory network controlling temporal cell fate transitions, we employed experimental conditions that induce the dauer formation program, but also efficiently prevent dauer commitment. We call these conditions “L2d-inducing” because the presence of both dauer-inducing and commitment-preventing conditions results in worm populations growing continuously (without dauer arrest) but where all animals traverse the lengthened bi-potential L2d stage (Figure 1B, L2d/delayed) [14,15].
We found that L2d-inducing pheromones suppress heterochronic defects caused by insufficient expression of let-7 family microRNAs, suggesting that these pheromones that enable the dauer life history option also activate a program alternative to let-7 family microRNAs in controlling stage-specific temporal cell fate progression. We found that the two major endocrine signaling pathways that regulate dauer formation in response to pheromones and food signals, the DAF-7/TGF-β and DAF-2/Insulin respectively, also mediate the effect of these same signals on temporal cell fates under L2d-inducing conditions. Moreover, we identified a previously undescribed ligand-independent activity of the nuclear hormone receptor DAF-12 that is responsible for activating the alternative program of cell fate specification in the L2d. This alternative program is responsible for correcting let-7 family insufficiency phenotypes and it requires the activities of certain heterochronic genes, lin-46, lin-4 and nhl-2, that are previously associated with an altered genetic program in post-diapause animals. This alternative program associated with L2d is coupled to a previously described reduction in the DAF-12-regulated expression of let-7 family microRNAs [16]. Hence, the overall L2d response is a “rewiring” program consisting two major operations: 1) repression of let-7 family microRNA expression, and 2) activation of an alternative program to downregulate the let-7 family target (Hunchback-like-1) HBL-1.
Our results show that environmental signals and downstream endocrine signaling pathways are capable of coordinately regulating developmental progression and cell fate transitions in C. elegans. We propose that this capability confers elasticity to C. elegans development, whereby the proper developmental schedule of cell fates remains unaffected by changes or uncertainties in developmental trajectory.
RESULTS
We developed three approaches to efficiently uncouple L2d from dauer commitment and thereby produce worm populations developing continuously through the bi-potential pre-dauer L2d phase directly to the L3, without dauer arrest. In the first approach, we employed the pheromone cocktail formula described by Butcher et al. [17], which contains three ascaroside molecules (ascr#2, ascr#3, and ascr#5) that synergistically induce L2d and dauer arrest [17] (Figure S1A). At sufficiently high doses, the ascaroside cocktail can induce 100% dauer formation (Figure S1C). Previous findings showed that the presence of food can antagonize pheromones and prevent dauer formation [14]. However, it was not clear if the food signals also prevented the L2d. We observed that the presence of live bacteria food, or the presence of dafachronic acid (DA) hormone, could efficiently prevent dauer formation while not preventing the L2d, evidenced by dramatically slowed second stage larval development. Therefore, to obtain animal populations traversing the L2d without committing to dauer arrest, we allowed larvae to develop in the presence of a combination of the ascaroside cocktail along with DA hormone [18] (Figure S1B and S1C). Our second approach also uses the ascaroside cocktail, but to eliminate the need for DA hormone, dauer-commitment defective daf-12(rh61) mutant worms are employed. In the third approach, to genetically induce L2d, we combined daf-12(rh61) with a temperature-sensitive daf-7 mutant that mimics the L2d-inducing pheromone conditions, or with a temperature-sensitive daf-2 mutant that mimics the L2d-inducing starvation conditions.
L2d-inducing ascarosides reduce the reliance on the let-7 family microRNAs for proper L2-to-L3 cell fate transition
Wild-type larvae robustly execute L2 stage cell fates and transition to L3 stage cell fates (thus # of seam cell=16 in young adult worms) regardless of developmental trajectory (Figure 2, rows 1–3). mir-48/84/241(0) mutant larvae reiterate L2 stage cell fates at later stages due to prolonged HBL-1 expression, resulting in extra (>16) seam cells in young adult animals (Figure 2, row 4). We found that when mir-48/84/241(0) mutant larvae developed through L2d -- induced by a combination of the ascaroside cocktail and the DA hormone -- the extra seam cell phenotype was substantially (albeit partially) suppressed (Figure 2, row 4 vs 6). To compare the strength of this L2d suppression with the previously described post-dauer suppression of the let-7 family phenotypes [13], we used the ascaroside cocktail but this time without the DA hormone. Under these conditions mir-48/84/241(0) larvae arrested as dauers; and as described previously [13] this resulted in complete suppression of the extra seam cell phenotype in post-dauer adults (Figure 2, row 4 vs 5). Therefore, the L2d suppression is weaker than the post-dauer suppression (Figure 2, row 6 vs 5), and unlike dauer arrest, L2d-inducing ascarosides do not completely eliminate the need for let-7 family microRNAs for proper L2-to-L3 cell fate transition. Nonetheless, the partial suppression of the extra seam cell phenotype of let-7 family microRNAs suggests that the L2d-inducing ascarosides rewire the genetic regulatory pathway controlling temporal cell fate progression in a way to reduce the reliance on the let-7 family microRNAs for proper L2-to-L3 cell fate transition.
Figure 2. Sensitized genetic backgrounds reveal that L2d-inducing environmental and endocrine signals impact the regulation of temporal cell fates.
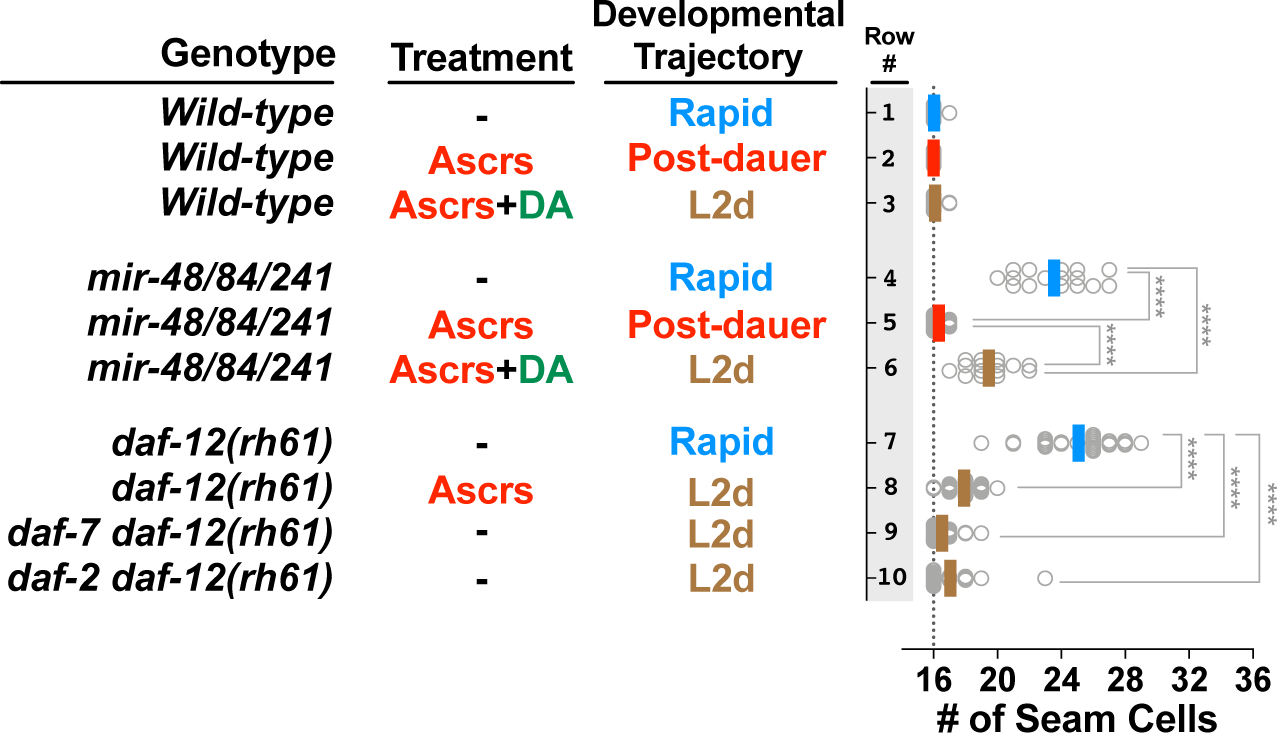
Genotypes are indicated in the first column; treatments and corresponding developmental trajectories are indicated in the second and third columns, respectively. Each dot in the plots to the right shows the number of seam cells on one side (left side or right side, observed interchangeably) of a single young adult animal, and solid lines (color code matching the developmental trajectory) indicate the average seam cell number of the animals scored for each condition. Wild-type animals have sixteen seam cells per side (vertical dotted line), regardless of developmental trajectory (lines 1–3). Experiments involving temperature sensitive alleles of daf-2 and daf-7 (lines 6 and 7) are performed at a permissive temperature (20°C) that allows continuous (L2d-to-L3 without dauer arrest) development. The student’s t-test is used to calculate statistical significance (p): n.s.(not significant) p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S1 and S2 and Table S1.
L2d-inducing ascarosides or L2d-inducing mutations of daf-7 and daf-2 suppress heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs in daf-12(rh61) mutants.
The daf-12(rh61) mutation combines three important properties which makes this mutation uniquely useful for studying the effects of L2d-inducing conditions on the regulation of temporal cell fates. These properties are: 1) daf-12(rh61) animals reiterate expression of L2 cell fates owing to reduced (insufficient) let-7 family levels, 2) daf-12(rh61) larvae are unable to execute dauer larvae commitment or arrest [19], enabling the use of dauer-promoting conditions to obtain populations of daf-12(rh61) animals undergoing an L2d-direct-to-L3 continuous development trajectory; 3) daf-12(rh61) animals are insensitive to the DA hormone (due to lack of the DAF-12 ligand binding domain) [18,19], and so the levels of let-7 family microRNAs are expected to be unresponsive to experimentally administered ascarosides, which are understood to regulate wild type DAF-12 activity by affecting the level of DA [10].
We observed that the presence of exogenous ascaroside cocktail during larval development almost completely suppressed the extra seam cell phenotype of daf-12(rh61) mutants (Figure 2, row 7 vs 8). To test the possibility that an unexpected elevation in the let-7 family levels could be responsible for the suppression of the heterochronic phenotypes of daf-12(rh61) animals in the presence of the ascaroside cocktail, we quantified the levels of let-7 family microRNAs in the absence and presence of the ascarosides (Figure S2). No elevation in the levels of these microRNAs in response to the ascaroside cocktail was evident (Figure S2). Therefore, the suppression of the heterochronic phenotypes of daf-12(rh61) mutants in the presence of the ascaroside cocktail is unlikely to result from restoration of normal levels of mir-48/84/241 or an elevation of the other members of the let-7 family microRNAs (Figure S2).
Similar to the ascaroside cocktail, conditional dauer-constitutive mutants of daf-7 (mimicking high ascarosides) or daf-2 (mimicking starvation) that allow continuous (L2d-to-L3 without dauer arrest) development at permissive temperatures [20], almost completely suppressed the extra seam cell phenotype of daf-12(rh61) mutants (Figure 2, row 7 vs 9 or 10). These results indicate that genetically induced L2d, whether by activation of the ascaroside response pathway (daf-7(lf)), or by activating the starvation response pathway (daf-2(lf)), results in an L2d-associated rewiring of the regulatory networks controlling temporal cell fate progression.
Ascarosides suppress the heterochronic phenotypes of daf-12(rh61) via srg-36/37-encoded GPCR signaling upstream of DAF-7/TGF-β-DAF-3 signaling.
Each of the individual ascarosides in the cocktail (Ascrs#2,3,5) has been shown previously to be alone sufficient to induce dauer formation, although with reduced potency compared to the combined cocktail [17,21]. Consistent with their individual capacities to induce L2d and dauer formation, we observed that each ascaroside ascr#2, ascr#3, and ascr#5 alone could suppress the extra seam cell phenotype of daf-12(rh61) mutants (Figure S3A). In the case of ascr#2 or ascr#3 alone, the suppression was partial, while for ascr#5 alone, the suppression was similar to the full cocktail (Figure S3A, rows 6 to 10). ascr#5 was the most potent of the three ascarosides in terms of both percent dauer formation of wild type larvae (Figure S3B) and suppression of the extra seam cell phenotype of daf-12(rh61) (Figure S3A, row 10 vs 8 and 9).
It has been shown previously that induction of dauer formation by ascarosides involves sensing of environmental ascaroside levels by specific G-protein coupled receptors (GPCRs) expressed in chemosensory neurons, wherein they repress DAF-7/TGF-β signals [22–25]. To test whether these GPCRs were also required for the suppression of the heterochronic phenotypes of daf-12(rh61) mutants, we employed mutations of srg-36 and srg-37, which encode GPCRs that are expressed in the ASI neurons and that are redundantly required for perceiving ascr#5 signal in the context of dauer induction [24]. We observed that for srg-36(0) srg-37(0); daf-12(rh61) compound mutants, ascaroside (in this case ascr#5) failed to suppress the extra seam cell phenotype daf-12(rh61) (Figure 3A, compare row 1 vs 2 with row 3 vs 4). Moreover, we found that the TGF-β signaling effector daf-3, which is thought to function downstream of SRG-36/37, is required for the suppression of daf-12(rh61) by asaroside (Figure 3B). These results indicate that the same GPCRs that mediate dauer formation in response to asaroside are also required for mediating the effects of asaroside on temporal cell fates, and supports a common pathway for suppression of daf-12(rh61) by ascaroside and dauer induction, involving activation of SRG-36/37 GPCRs and the potential downstream TGF-β effector DAF-3.
Figure 3. DAF-7/TGF-β and DAF-2/Insulin signaling pathways act in parallel to modulate a ligand-independent activity of DAF-12 that is responsible for correcting heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs.
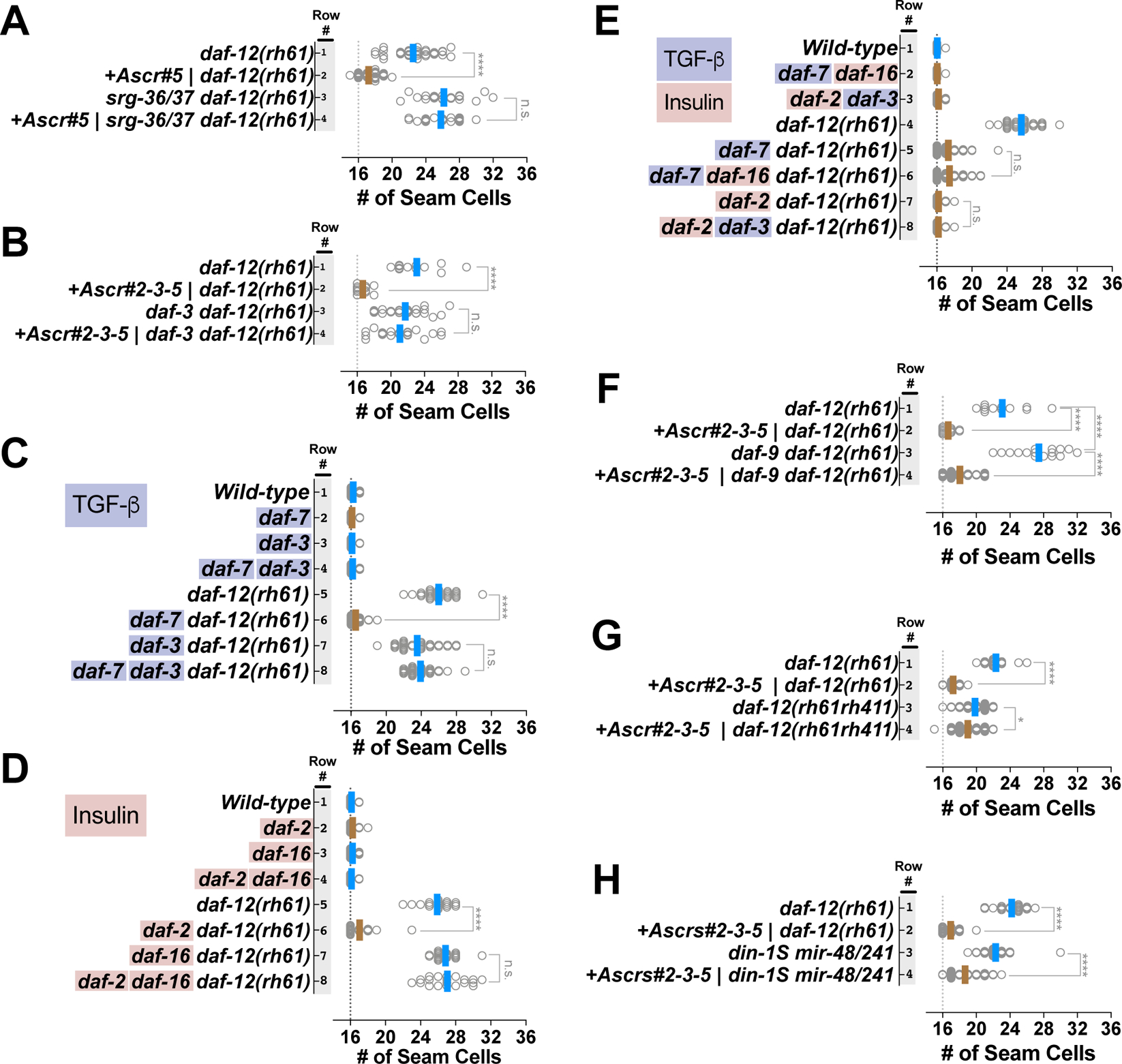
Number of seam cells in young adult animals of various mutants cultured on ascaroside or control plates (A, B and F. G), or on standard NGM plates (C-E). Each dot in the plots shows the number of seam cells of a single young adult animal, and solid lines indicate the average seam cell number of the animals scored for each condition (blue lines: rapid trajectory; brown lines: L2d trajectory). (A) Ascarosides suppress daf-12(rh61) via srg-36/37-encoded GPCR signaling upstream of DAF-7/TGF-β-DAF-3 signaling. (B) daf-3 activity is required for suppression of daf-12(rh61) by ascarosides. (C-E) DAF-7/TGF-β and DAF-2/Insulin signaling pathway act in parallel to mediate the suppression of daf-12(rh61). (F, G) Ligand-independent activity of daf-12 is required for the ascaroside-mediated L2d rewiring of the pathways regulating temporal cell fates. (H) The DAF-12 corepressor DIN-1S is not required for the ascaroside-mediated suppression of heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs. Suppression of extra seam cell phenotype of daf-12(rh61) is shown as a measure of the strength of the ascaroside conditions tested for din-1S(lf); mir-48/241(lf) animals. The student’s t-test is used to calculate statistical significance (p): n.s.(not significant) p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Figure S3 and Table S1.
DAF-7/TGF-β and DAF-2/Insulin signaling pathways act in parallel to mediate the suppression of the heterochronic phenotypes of daf-12(rh61).
As shown above, genetic activation of dauer-inductive signaling by daf-7(lf) or daf-2(lf) mutations is sufficient for suppression of daf-12(rh61) (Figure 2). In the context of dauer formation, DAF-7/TGF-β primarily mediates ascaroside signaling, and DAF-2/Insulin primarily mediates assessment of nutritional status [10,22,26]. To determine if the known downstream effectors of DAF-7/TGF-β and DAF-2/Insulin signaling that mediate dauer formation are also required for mediating the L2d rewiring caused by daf-7(lf) or daf-2(lf) [27,28], we generated compound mutants carrying daf-12(rh61) in combination with mutations that impair these effectors of the TGF-β or insulin signaling pathways, and determined the number of seam cells in young adults. We found that the downstream effector of the TGF-β signaling pathway, daf-3, and the downstream effector of the insulin signaling pathway, daf-16, were required for the suppression mediated by the daf-7(lf) mutation and the daf-2(lf) mutation, respectively (Figure 3C and 3D). These results are consistent with the finding that daf-3 was also required for the ascaroside-mediated suppression of daf-12(rh61) (Figure 3B).
To determine whether the TGF-β and insulin signaling pathways act in parallel to modulate temporal cell fates, we tested for crosstalk between these pathways in the context of suppression of daf-12(rh61) phenotypes. Specifically, we determined whether daf-16(lf) could alter the suppression of daf-12(rh61) phenotypes by daf-7(lf), and conversely, whether daf-3(lf) could alter the suppression of daf-12(rh61) phenotypes by daf-2(lf). We found that daf-16 was not required for daf-7-mediated suppression (Figure 3E, rows 5–6), and daf-3 was not required for daf-2-mediated suppression (Figure 3E, rows 7–8), indicating that, similar to their regulation of dauer diapause, the TGF-β and insulin signaling pathways act in parallel in the context of the L2d rewiring of the genetic regulatory pathways controlling larval cell fate progression.
Ligand-independent activity of daf-12 is required for the ascaroside-mediated L2d rewiring of the pathways regulating temporal cell fates.
Ascaroside (TGF-β) signaling and nutritional status (insulin) signaling converge to induce dauer larva arrest by down regulating DA production and hence reducing the levels of liganded DAF-12. Since we find that dauer-inducing conditions (ascarosides; loss of daf-7 or daf-2) can suppress the heterochronic phenotypes of the DA-insensitive daf-12(rh61) mutant, it would appear that the TGF-β and insulin signaling pathways may regulate cell fate transitions by repressing a hypothetical DAF-12-independent function of DA. If that were the case, inhibiting or preventing DA production would mimic the effect of ascarosides and suppress the extra seam cell phenotype of daf-12(rh61). To test this possibility, we employed genetic ablation of DA production. daf-9 encodes a CYP450 that is responsible for DA production [18]. Accordingly, daf-9(lf) mutants are dauer-constitutive due to lack of DA [18]. To test if ascarosides act by inhibiting DA production during L2d rewiring, we generated double mutants containing daf-9(lf) and daf-12(rh61). We observed that these double mutants lacking daf-9 in the daf-12(rh61) background had an even stronger extra seam cell fate phenotype than daf-12(rh61) mutants (Figure 3F, row 1 vs 3), and that this phenotype was suppressed in the presence of ascarosides (Figure 3F, row 3 vs 4). These results indicate that the ascaroside-induced L2d rewiring does not involve inhibition of DA biosynthesis, nor does rewiring require DA production or daf-9 activity. The enhancement of the extra seam cell phenotype of daf-12(rh61) phenotype in the daf-12(rh61); daf-9(lf) double mutant could reflect DAF-12-independent functions of DA or DA-independent functions of DAF-9.
The finding that ascaroside-mediated L2d rewiring did not involve the DA hormone raised the question as to whether the DA receptor, DAF-12 is required for the L2d rewiring. To determine whether daf-12 is required for ascaroside-induced suppression of retarded seam cell phenotypes, we tested whether ascarosides could suppress the phenotypes of daf-12(rh61rh411), a daf-12 null allele [19]. daf-12(rh61rh411) animals display a milder extra seam cell phenotype than daf-12(rh61) [19], presumably because of a milder reduction of let-7 family microRNAs compared to daf-12(rh61) [16]. We observed that the ascaroside conditions that resulted in a very potent suppression of the extra seam cell phenotype of daf-12(rh61) animals resulted in only a very modest (albeit statistically significant) suppression of the daf-12(rh61rh411) phenotype (Figure 3G, compare changes in the average number of seam cells in row 1 vs 2 with 3 vs 4). This result suggests that ascaroside-induced L2d rewiring of the pathways regulating temporal cell fates largely requires daf-12 function, and therefore represents a novel ligand-independent regulation of daf-12 by TGF-β and insulin signaling.
The DAF-12 corepressor DIN-1S is not required for the ascaroside-mediated suppression of heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs
When DA is absent, DAF-12 interact with DIN-1S, and together they form a repressive complex that is necessary for dauer formation [18,29]. din-1S(lf) suppresses heterochronic phenotypes of daf-12(rh61) [29], likely by relieving repression of let-7 family microRNA transcription. Therefore, it was possible that the ascaroside-mediated suppression of the heterochronic phenotypes of daf-12(rh61) could reflect ascaroside-induced down regulation of din-1S activity. To assess the potential involvement of din-1S in ascaroside-mediated suppression of the phenotypes caused by reduced let-7 family microRNAs, we tested for ascaroside suppression of a compound mutant lacking mir-48/241 and din-1S (Figure 3H). We observed that din-1(lf) did not prevent ascaroside suppression of the mir-48/241 extra seam cell phenotypes (Figure 3H, row 3 vs 4), indicating that DIN-1S is not required for ascaroside-mediated L2d rewiring.
Heterochronic genes previously associated with the altered HBL-1 down-regulation program in post-dauer animals are required for the L2d suppression of heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs
In animals that arrested as dauer larvae and then later resumed development through post dauer larval stages, the genetic programming of temporal cells fates differed substantially from animals that developed continuously [13]. In particular, proper cell fate progression through dauer larvae arrest and post-dauer development rests on an altered HBL-1 down-regulation program. These differences in HBL-1 down regulation include, 1) reallocation of roles for lin-4 microRNA and let-7 family microRNAs, and 2) alterations in the relative impacts of LIN-46 and the microRNA modulatory factor NHL-2 [13]. For example, animals deficient for lin-4 exhibited stronger retarded developmental timing phenotypes when traversing developmental arrest followed by post-dauer development compared to animals of the same genotype that developed rapidly and continuously. Similarly, animals carrying loss of function mutations of nhl-2 or lin-46 exhibited enhanced retarded phenotypes after post dauer development. nhl-2 encodes an RNA binding protein that functions as a microRNA positive modulator [30], and lin-46 encodes a protein that acts downstream of the LIN-28 RNA binding protein [31]. These results suggested that the rewiring of developmental cell fate progression in dauer-traversing larvae involves alterations in the post-transcriptional regulation of HBL-1 expression.
To confirm that L2d inducing conditions resulted in HBL-1 down-regulation, we tagged hbl-1 at its endogenous locus with mScarlet-I using CRISPR/Cas9 genome editing (See STAR Methods), and monitored the level of HBL-1::mScarlet-I expression in developing larvae. We compared HBL-1 expression in L2 and L3 stage daf-12(rh61) larvae to L2d and L3 stage larvae of the suppressed daf-7(lf); daf-12(rh61) double mutants (Figure 4A). At the L2/L2d stage, HBL1 expression in daf-12(rh61) and daf-7(lf); daf-12(rh61) were comparable (Figure 4A, L2/L2d). At the L3 stage, however, whereas HBL-1 was over expressed in both seam and hyp7 cells of daf-12(rh61) animals, HBL-1 was absent in the seam cells of daf-7(lf); daf-12(rh61) animals (Figure 4A, L3). This downregulation of HBL-1 expression in seam cells of daf-7(lf); daf-12(rh61) mutants is consistent with the suppression of extra seam cell phenotypes of daf-12(rh61) by daf-7(lf).
Figure 4. Heterochronic genes associated with the altered HBL-1 down-regulation program in post-dauer animals are required for the L2d suppression of heterochronic phenotypes caused by insufficient expression of let-7 family microRNAs.
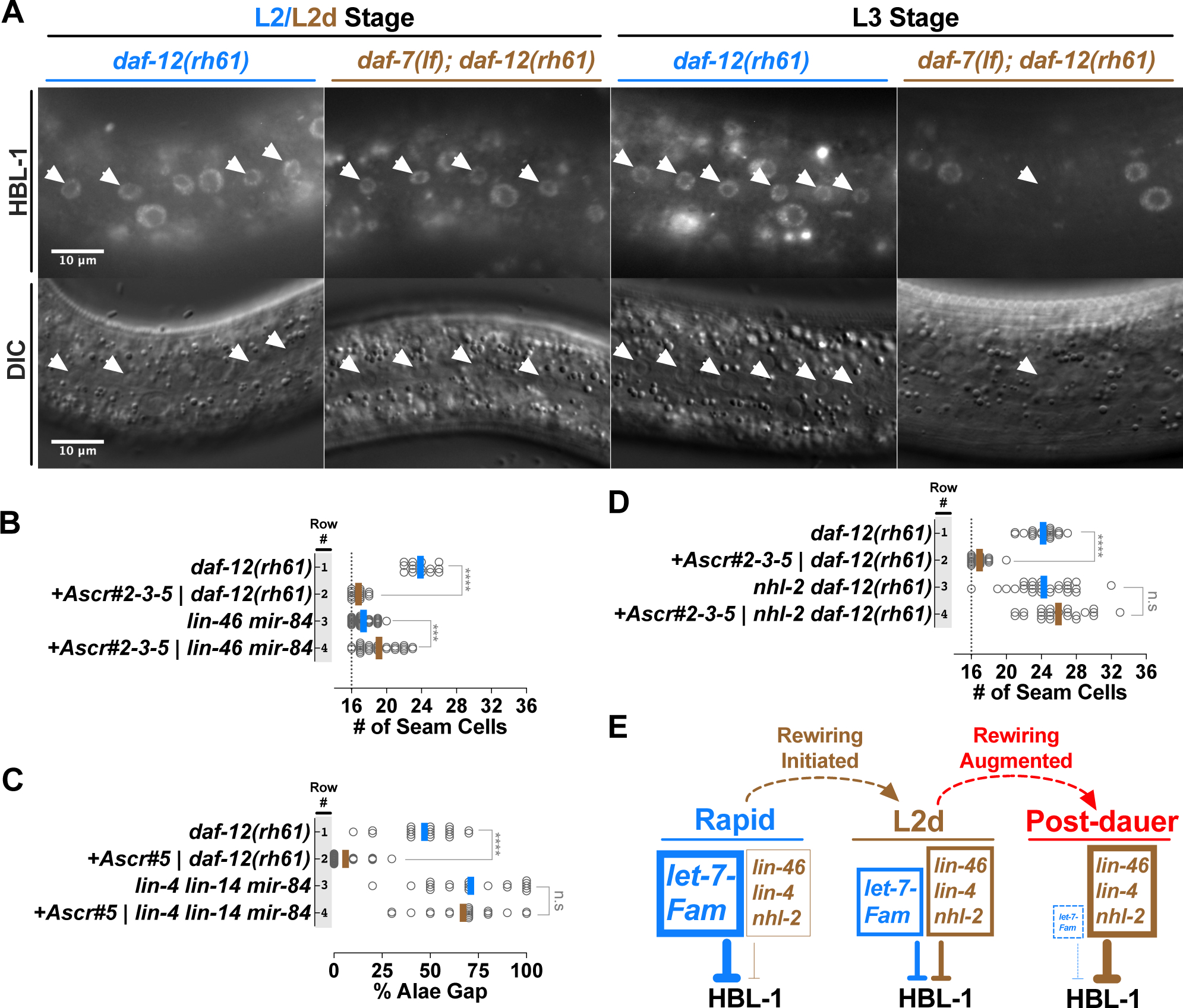
(A) Upper row: fluorescent images showing HBL-1 expression in L2/L2d and L3 stage hypodermal seam (white arrowheads) and hyp7 (all other) nuclei in daf-12(rh61) and daf-7(lf); daf-12(rh61) animals. Lower row: corresponding DIC images of the hypodermis. It should be noted that, consistent with the variability in the extra seam cell phenotype of daf-12(rh61) animals, HBL-1 expression at the L3 stage daf-12(rh61) animals displays variability across seam cells of individual worms. For example, HBL-1 expression may be present and absent in two neighboring seam cells, which presumably expresses L2 and L3 cell fates, respectively. (B) Ascaroside conditions that suppress the extra seam cell phenotype of daf-12(rh61) enhance the extra seam cell phenotype of larvae lacking lin-46 and mir-84. (C) Ascaroside conditions that suppress the gapped alae (a consequence of retarded seam cell development that is manifested in young adults) phenotype do not suppress the gapped alae phenotype of lin-4; lin-14; mir-84 animals. (D) nhl-2 activity is required for ascaroside-mediated suppression of daf-12(rh61) (E) A model for the L2d rewiring and its potential augmentation during dauer arrest. Under L2d-inducing conditions, let-7 family microRNAs are downregulated and also become less important. The reduction in the let-7 level and importance is coupled to enhanced roles for the heterochronic genes previously associated with the altered HBL-1 downregulation program in post-dauer animals, involving lin-46, lin-4, and nhl-2. This shift in the reliance on the let-7 family microRNAs to the reliance on the alternative program for proper HBL-1 down-regulation (hence for proper L2-to-L3 cell fate progression) constitutes the L2d rewiring. In post-dauer animals, consistent with an augmentation of the L2d rewiring program, the reliance on the altered HBL-1 down-regulation program further increases while the let-7 family microRNAs become dispensable for proper HBL-1 down-regulation. It should be noted that we do not know the mechanisms (e.g. elevated levels vs enhanced activities) of increased roles for lin-46, nhl-2, or lin-4 during L2d or post-dauer development. The student’s t-test is used to calculate statistical significance (p): n.s.(not significant) p>0.05, *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. See also Table S1.
To test whether the previously described genetic requirements for lin-46, lin-4, and nhl-2 to downregulate HBL-1 in the dauer/post-dauer context [13], also apply during L2d development, we examined the phenotypes of the relevant mutant strains during development through ascaroside-induced L2d, but in this case without dauer commitment or arrest. We observed that ascarosides failed to suppress the retarded phenotypes of animals that were lacking lin-46 or lin-4 in combination with mir-84(lf) (to provide a sensitized background, blunting the expression of let-7 family microRNAs), or that were lacking nhl-2 in the daf-12(rh61) background. (Figure 4B–D). Moreover, for doubly-mutant animals carrying both mir-84(lf) and lin-46(lf) mutations, ascaroside-induced L2d enhanced the retarded phenotypes (Figure 4B), similar to the enhancement reported for mir-84(lf); lin-46(lf) animals that developed through dauer arrest and post-dauer development [13]. Similarly, ascarosides failed to suppress the gapped alae phenotype of animals lacking lin-4 and mir-84 (Figure 4C), consistent with the previous finding that this phenotype of lin-4(lf); mir-84(lf) animals was enhanced for post dauer animals [13]. Lastly, ascarosides failed to suppress the extra seam cell phenotypes of nhl-2: daf-12(rh61) animals (Figure 4D), indicating that nhl-2 is required for ascaroside-mediated suppression of daf-12(rh61); analogous to its role in post-dauer enhancement of the retarded phenotypes of let-7 family microRNAs [13]. These results suggest that the L2d rewiring includes the activation of an alternative HBL-1 downregulation program, which involves lin-46, lin-4 and nhl-2, and that this alternative program accounts for the reduced reliance on let-7 family microRNAs for proper L2-to-L3 cell fate transition. These results also suggest that the genetic circuitry controlling cell fate progression via HBL-1 in larvae undergoing L2d development is similar to the circuitry associated with dauer larvae arrest, consistent with a rewiring mechanism that is initiated during L2d and augmented during dauer arrest (Figure 4E).
DISCUSSION
Environmental and physiological stress signals can challenge the progression of C. elegans larval development, causing the larva to choose one of three distinct alternative developmental trajectories: 1) a rapid and continuous trajectory without the option for dauer arrest, 2) continuous development through an extended, bipotent L2d trajectory, wherein the option for dauer arrest is enabled, but not necessarily selected, and 3) development through the L2d trajectory followed by dauer larva arrest (Figure 5A). Regardless of which trajectory is chosen by the larva, the same sequence of stage-specific cell fates is robustly expressed (Figure 1A and Figure 2 rows 1–3). The findings reported here illuminate how genetic regulatory networks that specify larval cell fate progression can accommodate these alternative life histories, and the physiological and environmental stresses that induce them.
Figure 5. Coordinate regulation of developmental progression and cell fate transitions in C. elegans larvae.
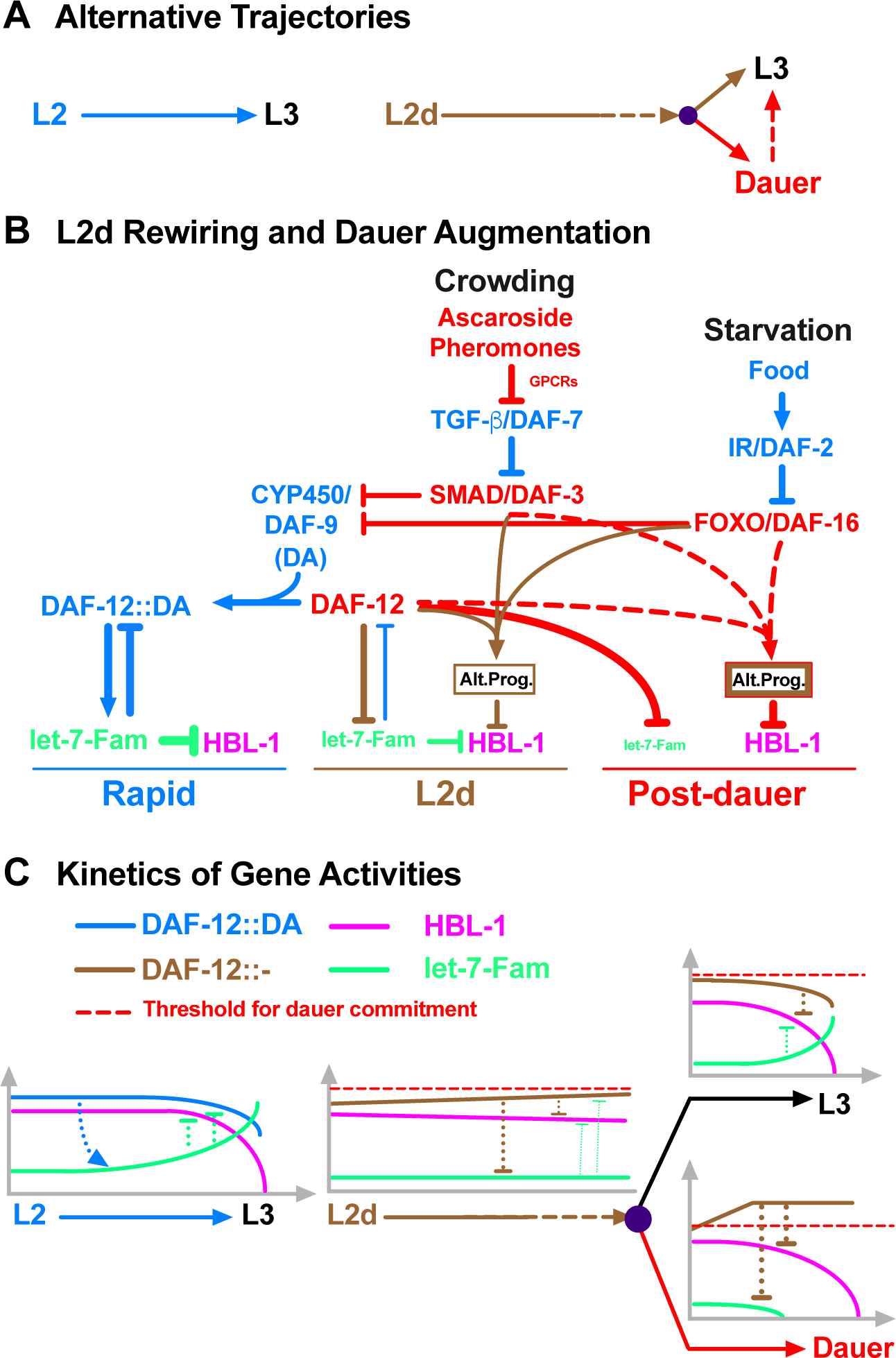
(A) Alternative trajectories. The L2-to-L3 transition is rapid and deterministic (once committed to the L2 stage, the larvae do not have the dauer option), whereas the L2d-to-L3 transition is slower and bipotential. In both cases, HBL-1 is present throughout the L2/L2d stage but it is downregulated by the beginning of the L3 stage (Figure S4). (B) Pheromone and endocrine signals engage DAF-12 to initiate and regulate the rewiring of the HBL-1 down-regulation. In response to crowding and starvation, TGF-β and insulin signaling pathways, respectively, modulate the ligand-dependent DAF-12 activity to repress the transcription of let-7 family microRNAs, and, at the same time, cooperate with DAF-12 in a ligand independent manner to activate the alternative HBL-1 downregulation program (Alt. Prog.). The alternative program of L2d and dauer-interrupted trajectories are similar, but the alternative program of dauer-interrupted trajectory is stronger either due to an enhancement of the alternative program of L2d (depicted as thicker brown border line) and/or due to employment of additional factors (depicted as red border line) after the L2d larvae commit to dauer formation. (C) DAF-12 ensures properly delayed but robust HBL-1 down-regulation during L2d-to-L3 transition by coordinating the repression of let-7 family microRNAs with the activation of the alternative HBL-1 downregulation program. During rapid, L2 development, DAF-12 activates the transcription of let-7 family microRNAs, which in turn, negatively regulate DAF-12, eliminating the dauer option. During slow, bipotential, L2d development, DAF-12 represses let-7 family microRNAs, which otherwise prevent the accumulation of DAF-12. If the unliganded DAF-12 reaches the threshold, larvae commit to dauer formation, if not, larvae commit to continuous development. While mediating this decision, which necessitates the repression of let-7 family microRNAs (for maintaining the dauer option) and delaying the downregulation of HBL-1 (for postponing L3 cell fates), DAF-12 cooperates with DAF-3 or DAF-16 to activate the alternative HBL-1 downregulation program (Alt. Prog) to ensure robust HBL-1 downregulation during the L2d-to-L3 transition. See also Figure S4.
Central to the coordination of temporal cell fates and life history choices in C. elegans is the nuclear hormone receptor transcription factor DAF-12 [19], and its ligand, dafachronic acid (DA) [18]. DAF-12 in the unliganded form is essential for the dauer larva trajectory, while ligand-bound DAF-12 inhibits the dauer larva program. At the same time, DAF-12 and DA control the L2-to-L3 cell fate transitions by regulating the expression of let-7 family microRNAs, which are required to downregulate HBL-1 and thereby specify the proper timing of expression of L3 cell fates [6,16,18,32].
The let-7 family microRNAs also regulate the abundance of DAF-12 via a feedback loop that has been proposed to help ensure robust coordination of cell fates with dauer arrest [16]. Under favorable conditions, when DA is abundant, DA-bound DAF-12 promotes continuous development, and also activates accumulation of let-7 family microRNAs during the L2 stage, which in turn attenuate the accumulation of DAF-12 (thereby eliminating the dauer option) and also down regulate HBL-1 to enable rapid progression from L2 to L3 cell fates. Conversely, under unfavorable conditions, DA production is low and unliganded DAF-12 promotes the L2d/dauer program and represses the expression of let-7 family microRNAs. In turn the low level of let-7 family microRNAs allows the accumulation of DAF-12 during L2d, maintaining the dauer option [16].
It was not previously clear how HBL-1 might be down regulated during L2d considering the repressed state of the let-7 family microRNAs. We propose that the L2d program instigated by unliganded DAF-12 includes, in addition to the repression of let-7 family microRNAs, also the activation of an alternative mechanism of HBL-1 downregulation (Figure 5B), which is responsible for the suppression of daf-12(rh61) by ascarosides (Figure 2, row 8), and by L2d-inducing mutants of daf-7 and daf-2 (Figure 2, rows 9 and 10). The L2d suppression of daf-12(rh61) phenotypes led us to propose that during wild type L2d, when DA is low, DAF-12 is unliganded and its activity is hence not unlike that of the ligand-binding defective mutant DAF-12(RH61). In this model, we propose that daf-12(rh61) animals constitutively run an “L2d-like program” for regulating temporal cell fates, which is discordant with rapid continuous development, but compatible with (and hence phenotypically suppressed by) the environmental and endocrine signals that induce the L2d trajectory.
Our conclusion that a DA-independent function of DAF-12 is required for the modulation of temporal cell fates during L2d is derived from our finding that the mild retarded phenotype of the daf-12 null allele, daf-12(rh61rh441), was not suppressed by ascarosides, in contrast to the stronger retarded phenotype of daf-12(rh61), which is efficiently suppressed (Figure 3G). Consistent with our results showing the requirement for daf-12 for L2d rewiring, a daf-2(lf) mutant was reported to not suppress (indeed, to enhance) the extra seam cell phenotypes of daf-12(lf) [33].
We find that the DA-independent function of DAF-12 that modulates temporal cell fates during the L2d can be activated by either of the two upstream transcription factors, DAF-3, which mediates responses to TGF-β signaling from ascaroside-sensing neurons, and DAF-16, which mediates DAF-2/IGF signaling. We hypothesize that DAF-12 cooperates with DAF-3 or DAF-16 in a DA-independent manner to activate and modulate the alternative HBL-1 downregulation program (Figure 5B). Although the immediate action of DAF-12, together with DAF-3 or DAF-16, would likely be transcriptional, ultimately the alternative pathway for HBL-1 down regulation appears to be post-transcriptional, as we found that suppression of daf-12(rh61) or mir-84(lf) by L2d involves contributions from lin-4 and let-7 family microRNAs, in addition to the miRISC cofactor NHL-2 [30] and also LIN-46, which is thought to function post transcriptionally [31]. Interestingly, the alternative HBL-1 downregulation program appears to be activated in seam cells but not in hyp7 cells (Figure 4A), which is distinctly different from the let-7 family regulation of HBL-1 occurring both in the seam and hyp7 cells.
Prior to this study, it was not clear whether the inhibition of dauer larva formation by exogenously-supplied DA reflects prevention of L2d in addition to a block of dauer-commitment. We observed that exogenous DA hormone at levels sufficient to prevent ascaroside-induced dauer formation does not prevent the extension of second larval stage development characteristic of L2d. Also, we observed that daf-12(rh61) animals, which are DA-insensitive and dauer-commitment defective nevertheless can readily undergo L2d. These observations suggest that L2d is initiated independently of DAF-12-DA activity, and are consistent with our model that a DA-independent function of DAF-12, together with activated DAF-3 or DAF-16, promote expression of the L2d program.
The studies described here involve animals that traverse the bi-potential L2d stage, but elect the option of resuming L3 development instead of dauer larva arrest. Previous studies showed that for animals that enter dauer diapause arrest and later are induced to emerge from dauer arrest and complete post-dauer L3 and L4 development, a similar change in the HBL-1 down-regulation program is evident [13]. Here we show that the genetic circuitry controlling cell fate progression via HBL-1 in larvae undergoing L2d development is similar to the circuitry previously identified for animals that arrest as dauer larvae [13]. In particular, we find that lin-4 and NHL-2 have similarly prominent roles in both the L2d-only and the post-dauer trajectories. Moreover, for both the L2d-to-L3 trajectory, and the L2d-to-dauer-to-postdauer trajectory, an enhanced role for lin-46 is evident, compared to the rapid continuous development trajectory. Our finding of an increased importance of LIN-46 during the L2d is consistent with a previous report that a daf-2(lf) mutation could enhance the retarded phenotype of sea-2(lf) [33], whose phenotype presumably reflects elevated lin-28 activity, and hence reduced lin-46 activity.
These results, that the rewiring of HBL-1 down regulation for the L2d-only trajectory is largely similar to that for dauer-postdauer trajectory, are consistent with the fact that dauer larvae arrest is always preceded by L2d. Therefore, the rewiring associated with traversing post-dauer likely is initiated during L2d. The observation that suppression of the retarded phenotypes of certain mutants is more potent for the L2d-dauer-postdauer trajectory compared to the L2d-only trajectory (Figure 2, row 5 vs 6), suggests that rewiring may be partially implemented during L2d, and more fully engaged in association with dauer commitment (Figure 5B).
Our observations of the kinetics of HBL-1 down regulation during the L2 and L2d stages (Figure S4) suggest that HBL-1 levels are maintained at relatively high levels for much of the L2 or L2d stage, and down-regulated near the end of the stage (Figure 5C and Figure S4). This suggests that down-regulation of HBL-1, and hence commitment to L3 cell fate specification, may be gated by some event(s) coupled to the completion of the L2 or L2d stage. This would be consistent with a model wherein most of the L2d is occupied with rewiring of pathways upstream of HBL-1, followed by implementation of HBL-1 down regulation at the end of the L2d, in association with commitment to the L3 cell fate.
In summary, we show that environmental and endocrine stress signals that regulate developmental progression and alternative developmental trajectories rewire the genetic regulatory network controlling temporal cell fate transitions during larval development of C. elegans. Our findings provide insight into how the regulation of temporal cell fate transitions can be adapted to accommodate stressful conditions that challenge developmental progression. Coordinate regulation of developmental progression and temporal cell fates seem to confer elasticity to C. elegans development, wherein the proper developmental schedule of cell fate transitions is unaffected by variations in developmental rate or developmental trajectory.
C. elegans larvae possess other life history options, in addition to the delayed developmental progression associated with the L2d-dauer trajectory. For example, appropriate pheromone signals and nutritional status can result in an acceleration of C. elegans development [34–36]. Moreover, larvae can temporarily suspend developmental progression at specific checkpoints in the late L2, L3, or L4 stages in response to acute starvation [37]. We hypothesize that these signals that either accelerate development or result in programmed developmental arrest would also be associated with appropriate rewirings of the genetic regulatory pathways regulating temporal cell fate progression, so that cell fate transitions are appropriately synchronized with the dynamics of larval stage progression. Lastly, rewiring mechanisms induced by environmental signals, such as that described here, might also be adapted for the evolution of morphological plasticity or polyphenism observed in certain nematodes [38,39] and other animals [40].
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents should be directed to and will be fulfilled by the Lead Contact, Victor Ambros, (victor.ambros@umassmed.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C. elegans culture conditions
C. elegans strains used in this study and corresponding figures in the paper are listed in Table S1. All C. elegans strains used in this study, which include new compound strains and new alleles generated in this paper, are also listed in the Key Resources Table. C. elegans strains were maintained at 20°C on nematode growth media (NGM) and fed with the E. coli HB101 strain (See Key Resources Table).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| E.Coli: OP50 | Caenorhabditis Genetics Center (CGC) | N/A |
| E.Coli: HB101 | Caenorhabditis Genetics Center (CGC) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ascr#2 (C6; daumone-2) | Schroeder Laboratory (Cornell) | CAS: 946524-24-9 |
| Ascr#3 (C9; daumone-3) | Schroeder Laboratory (Cornell) | CAS: 946524-26-1 |
| Ascr#5 (C3) | Schroeder Laboratory (Cornell) | CAS: 1086696-26-5 |
| Δ4-dafachronic acid (DA) | Cayman Chemical, item no. 14100 | CAS: 23017-97-2 |
| Critical Commercial Assays | ||
| Real-time-PCR-based TaqMan for C. elegans microRNAs | Applied Biosystems | N/A |
| Experimental Models: Organisms/Strains | ||
| C.elegans: Strain VT1367: maIs105[pCol-19::gfp] V | Ambros Laboratory | VT1367 |
| C.elegans: Strain VT1453: mir-48 mir-241(nDf51) maIs105 V; mir-84(n4037) X | Ambros Laboratory | VT1453 |
| C.elegans: Strain VT791: maIs105 V; daf-12(rh61) X | Ambros Laboratory | VT791 |
| C.elegans: Strain VT2962: daf-7(e1372) III; maIs105 V; daf-12(rh61) X | This paper | VT2962 |
| C.elegans: Strain VT3568: daf-2(e1370) III; maIs105 V; daf-12(rh61) X | This paper | VT3568 |
| C.elegans: Strain VT3135: maIs105 V; srg-36srg-37(kyIR95) daf-12(rh61) X | This paper | VT3135 |
| C.elegans: Strain VT2972: maIs105 V; daf-3(mgDf90) daf-12(rh61) X | This paper | VT2972 |
| C.elegans: Strain VT1308: daf-7(e1372) III; maIs105 V | Ambros Laboratory | VT1308 |
| C.elegans: Strain VT1755: maIs105 V; daf-3(mgDf90) X | Ambros Laboratory | VT1755 |
| C.elegans: Strain VT1747: daf-7(e1372) III; maIs105 V; daf-3(mgDf90) X | Ambros Laboratory | VT1747 |
| C.elegans: Strain VT2984: daf-7(e1372) III; maIs105 V; daf-3(mgDf90) daf-12(rh61) X | This paper | VT2984 |
| C.elegans: Strain VT1776: daf-2(e1370) III; maIs105 V | Ambros Laboratory | VT1776 |
| C.elegans: Strain VT3566: daf-16(mgDf50) I; maIs105 V | This paper | VT3566 |
| C.elegans: Strain VT3567: daf-16(mgDf50) I; daf-2(e1370) III; maIs105 V | This paper | VT3567 |
| C.elegans: Strain VT3569: daf-16(mgDf50) I; maIs105 V; daf-12(rh61) X | This paper | VT3569 |
| C.elegans: Strain VT3570: daf-16(mgDf50) I; daf-2(e1370) III; maIs105 V; daf-12(rh61) X | This paper | VT3570 |
| C.elegans: Strain VT3682: daf-16(mgDf90) I; daf-7(e1372) III; maIs105 V | This paper | VT3682 |
| C.elegans: Strain VT3681: daf-2(e1370) III; maIs105 V; daf-3(mgDf90) X | This paper | VT3681 |
| C.elegans: Strain VT3684: daf-16(mgDf90) I; daf-7(e1372) III; maIs105 V; daf-12(rh61) X | This paper | VT3684 |
| C.elegans: Strain VT3683: daf-2(e1370) III; maIs105 V; daf-3(mgDf90) daf-12(rh61) X | This paper | VT3683 |
| C.elegans: Strain VT2952: maIs105 V; daf-9(m540) daf-12(rh61) X | This paper | VT2952 |
| C.elegans: Strain VT3061: wIs51[pScm::gfp] V; daf-12(rh61rh411) X | This paper | VT3061 |
| C.elegans: Strain VT3057: din-1(dh127) II; mir-48 mir-241(nDf51) maIs105 V | This paper | VT3057 |
| C.elegans: daf-12(rh61) hbl-1(ma430[hbl-1::mScarlet-I) X | This paper | VT3894 |
| C.elegans: daf-7(e1372) III; daf-12(rh61) hbl-1(ma430[hbl-1::mScarlet-I) X | This paper | VT3895 |
| C.elegans: Strain VT2086: lin-46(ma164) maIs105 V; mir-84(n4037) X | Ambros Laboratory | VT2086 |
| C.elegans: Strain VT1065: lin-4(e912); lin-14(n179) mir-84(n4037) X | Ambros Laboratory | VT1065 |
| C.elegans: Strain VT3030: nhl-2(ok818) III; maIs105 V; daf-12(rh61) X | This paper | VT3030 |
| C.elegans: Strain VT3751: maIs105(pCol-19::gfp) V; hbl-1(ma430[hbl-1::mScarlet-I]) X | This paper | VT3571 |
| C.elegans: Strain VT3893: daf-7(e1372) III; hbl-1(ma430[hbl-1::mScarlet-I]) X | This paper | VT3893 |
| Recombinant DNA | ||
| Plasmid: pOI191. Worm mScarlet from pSEM91 is converted to mScarlet-I [42] by introducing the T74I (ACC to AtC) substitution (and subcloned to generate an HR template for tagging of hbl-1). | This paper | pOI191 |
| Plasmid: pOI83. pRB1017 [43] tracr sequence is modified to the more efficient (F+E) form | This paper | pOI83 |
| Plasmid: pOI89. hbl-1 sgRNA for C-terminal tagging of hbl-1 cloned into pOI83 | This paper | pOI89 |
| Plasmid: pOI91. unc-22 sgRNA [41] cloned into pOI83 | This paper | pOI91 |
| Software and Algorithms | ||
| GraphPad Prism 8 | GraphPad Software Inc. (https://www.graphpad.com/scientific-software/prism/) | RRID:SCR_002798 |
| ZEN (blue edition) | Carl Zeiss Microscopy. (https://www.zeiss.com/microscopy/us/products/microscope-software/zen.htm) |
RRID:SCR_013672 |
| ImageJ - Fiji | Open Source: (https://fiji.sc/) | RRID:SCR_002285 |
METHOD DETAILS
Dauer and L2d-inducing plates
For experiments involving the administration of ascarosides and/or DA, we adopted the protocol described by Butcher et. al. [17] with modifications. Namely, C. elegans was fed with the E. coli OP50 strain on plates containing 3 mL of 1% agarose (SeaKem® LE agarose, Cat#50004) with nematode growth media (NGM) without peptone. Synthetic ascarosides (kindly provided by the labs of Frank Schroeder and Jagan Srinivasan) were dissolved in ethanol (stock concentrations: ascr#2 5.69 mM, ascr#3 3.81 mM, and ascr#5 4.09 mM) and added to the melted agarose prior to plate-pouring to achieve the desired final concentration (3 μM, if not specified) (See Key Resources Table). Plates were seeded the next day with E. coli strain OP50 as follows: OP50 was grown in liquid Luria Broth (LB) media until the culture reached OD600=0.6–0.7. Then, the bacterial culture was pelleted by spinning at 3500 rpm for 10 minutes. The pellet was washed twice with a volume of sterile water equal to the LB culture volume. Finally, the pellet was resuspended in a volume of sterile water equal to one-fifth of the initial LB culture volume. 50 μLs of this washed and 5x concentrated OP50 culture were used to seed ascaroside plates. To prepare ascaroside plates also containing Δ4-dafachronic acid (DA; 1 mg of DA dissolved in ethanol, see Key Resources Table) 50 μL of water containing DA at specified concentrations was added onto the lawn of bacteria.
Analysis of extra seam cell phenotypes
Gravid adult animals were washed off from NGM plates and collected in 2 mL of water. 0.84 ml of freshly prepared 2:1 mixture of bleach (6% sodium hypochlorite, Fisher Chemical SS2901) and 5N sodium hydroxide (prepared by dissolving Fisher S318500 in distilled water) was added onto the worms in 2 ml of water. Worms were incubated in this solution containing bleach and sodium hydroxide for 3 minutes. To pellet the released eggs, 8 mLs of water was added, and the tube was spun for 20 seconds at 600 × g. The supernatant is carefully removed and the pelleted eggs were washed three times with 10 ml of sterile M9 buffer. Eggs in M9 buffer were pipetted on control or treatment plates and cultured at 20°C (unless otherwise specified) until they reached the adult stage. The worms were scored at the young adult stage for the number of seam cells using fluorescence microscopy with the help of the maIs105 [pCol-19::gfp] transgene that marks the lateral hypodermal cell nuclei or the wIs51[pScm::gfp] transgene that marks the seam cell nuclei.
TaqMan assays for microRNA quantification
Synchronized L1 larvae of daf-12(rh61) were raised on control or ascr#2-3-5 plates at 20°C. Larvae that reached the L2/L2d-to-L3 molt were identified and picked under a dissecting microscope and collected in M9 media within two hours. For each experimental condition, three biological samples were collected, containing approximately 200 larvae per sample. Collected worms were snap-frozen and kept at −80°C until RNA extraction. RNA was extracted using the Trizol reagent (Invitrogen).
Two microliters of 30 ng/L RNA samples were used for reverse transcription, and multiplex miRTaqman reactions were carried out according to the manufacturer’s instructions, and using an ABI 7900-HT Fast-Real Time PCR System (See Key Resources Table). MicroRNAs were assayed in three technical replicates for each biological sample. Five highly expressed microRNAs that have not been reported to be environmentally regulated (mir-1, lin-4, mir-52, mir-53, and mir-58), were used as a control microRNA set. The average CT of the control microRNA set was used to normalize the CTs obtained from let-7 family microRNAs.
Tagging of hbl-1 at its endogenous locus
A mixture of plasmids encoding SpCas9 (pOI90, 70 ng/μL), and single guide RNAs (sgRNAs) targeting the site of interest (pOI89, 20 ng/μL) and the unc-22 gene (pOI91, 10 ng/μL) as co-CRISPR marker [41], a donor plasmid (pOI191, 20 ng/μL) containing the mScarlet-I sequence [42] flanked by homology arms, and a rol-6(su1006) containing plasmid (pOI124, 50 ng/μL) as co-injection marker was injected into the germlines of ten young adult worms. F1 roller and/or twitcher animals (around 200 worms) were cloned and screened by PCR amplification (Primers 12&15; Table S2) for the presence of the expected homologous recombination (HR) product. F2 progeny of F1 clones positive for the HR-specific PCR amplification product were screened for homozygous HR edits by PCR amplification of the locus using primers that flanked the HR arms used in the donor plasmid (Primers 15&16; Table S2). Finally, the genomic locus spanning the HR arms and mScarlet-I DNA was sequenced using Sanger sequencing. A single worm with a precise HR edited locus was cloned and backcrossed twice before used in the experiments. This HR edited allele, which contains a linker and the mScarlet-I sequence integrated inframe with hbl-1 (See Methods S1), is named as ma430 [hbl-1::mScarlet-I].
Plasmid DNA purification and microinjection of worms
Plasmid DNA for tagging hbl-1 was purified using the ZR Plasmid Miniprep Kit (Zymo Research, Cat#: 11–308AC) with a modified protocol which includes a phenol:chloroform (Phenol:ChCl3::IAA, pH=7.9, Ambion, Cat#AM9730) extraction step before loading the samples onto the columns. First, 600 μLs of the bacterial supernatant (Step 5 of the kit’s protocol) was mixed with an equal volume of phenol: chloroform and vortexed for 10 seconds. Then, this mixture was centrifuged at 16.000xg for 5 minutes and 500 μLs of the aqueous (top, transparent) layer was transferred onto the columns. The plasmid DNA on the columns were washed with both endo-free wash and wash buffers as described in the kit’s protocol, and eluted with distilled water. Plasmid DNA was mixed at the final concentrations listed above for each plasmid in 20 μL of water. 3 μL of this injection mix was loaded into a glass injection needle using a microloader pipette tips (Eppendorf, 5242956003). Glass injection needles were pulled using a KOPF vertical pipette puller (model 720) and capillary glass tubes (FHC, Inc., borosil 30-30-0). Worms were immobilized on 2% agarose pads (by gently pressing the worms with a worm pick towards the agarose surface) covered with a drop of oil (Halocarbon, CAS#9002-83-9). Agarose pads were previously prepared by spreading a drop (~50–100 μLs) of 2% agarose (in water) between two cover slides (e.g. Fisherbrand 12–544E, 24X500-1.5). The immobilized worms were injected under a Zeiss Axiovert 35 inverted microscope equipped with a Leitz mechanical micromanipulator. Injected worms were washed off from agarose pads and transferred to NGM plates in M9 media using a P200 micropipette. Injected worms (P0) were cultured at 25°C in separate NGM plates for 2–3 days before F1 twitchers/rollers were cloned for culturing before genotyping.
Cloning of sgRNA plasmids
All plasmids in the injection mix had the same plasmid backbone which was derived from pRB1017 [43]. sgRNA encoding plasmids were derived from pRB1017 (first to generate pOI83) by modifying the tracr encoding sequence to (F+E) form of the tracr [44] (using the Q5 Site-Directed Mutagenesis kit, NEB Cat#E0554, and Primers 1&2; Table S2), which was reported to increase the CRISPR efficiency in C. elegans [45]. pOI89 and pOI91 sgRNA encoding plasmids were generated by cloning annealed primer pairs into the BsaI cloning sites of pOI83 (Primers 3–6; Table S2).
Cloning of pOI191 HR template plasmid
The Golden Gate Assembly Kit (NEB Cat#E1600) is used to fuse two PCR fragments: mScarlet (PCR amplified from pSEM91 [46] using primers 7&8 in Table S2) and a DNA fragment containing left and right HR arms fused to the pRB1017 plasmid backbone (PCR amplified from pOI115 using primers 9&10 in Table S2, which was previously generated by assembling four PCR fragments containing a left HR arm, a GFP, a right HR arm, and the backbone of pRB1017). Single colony purified plasmid DNA was used to identify colonies containing the precise assembly of the HR arms and mScarlet. pOI191 was derived from this mScarlet clone (pOI186) by mutating a single nucleotide to convert mScarlet to mScarlet-I using the Q5 Site-Directed Mutagenesis kit (NEB Cat#E0554 and Primers 11&12; Table S2).
Microscopy imaging of C. elegans larva
All DIC and fluorescent images are obtained using a ZEISS Imager Z1 equipped with ZEISS Axiocam 503 mono camera, and the ZEN Blue software (See Key Resources Table). Prior to imaging, worms were anesthetized with 0.2 mM levamisole in M9 buffer and mounted on 2% agarose pads. To be able to compare fluorescent signal intensities across different genetic backgrounds or larval stages, images of all larvae were taken using the same microscopy setting. These images were then stitched together (e.g. Figure 4A) using the ImageJ Fiji software and the brightness and contrast of the montaged images were adjusted to enhance the visualization of the fluorescent signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
Each circle on the genotype vs. number of seam cells plots shows the observed number of seam cells on one side of a single young adult worm. ≥20 worms for each genotype or condition are analyzed and the average number of seam cells are denoted by lateral bars in the genotype vs number of seam cell plots. The student’s t-test is used to calculate statistical significance when comparing different genotypes or conditions. The GraphPad Prism 8 software (See Key Resources Table) is used to plot the graphs and for statistical analysis.
Supplementary Material
Highlights.
Ascr#5 corrects heterochronic cell fate defects of daf-12(rh61) mutants
Ascarosides reduce the developmental reliance on let-7 family microRNAs
DAF-12 mediates an L2d rewiring of networks controlling temporal cell fates
Developmental progression and temporal cell fates are coordinately regulated
ACKNOWLEDGMENTS
We thank the Schroeder (Cornell) and Srinivasan (WPI) laboratories for providing ascarosides, the Bargmann (Rockefeller) laboratory for sharing the srg-36/37 mutant strains. We thank the members of the Mello (UMass Medical) and Ambros laboratories for helpful discussions and the members of the Ambros laboratory and Micah Belew for critical reading of the manuscript. This research was supported by funding from NIH grants R01GM088365 and R01GM034028 (V.A.) Some C. elegans strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Sulston JE, and Horvitz HR (1977). Post-embryonic cell lineages of the nematode, Caenorhabditis elegans. Dev. Biol 56, 110–156. [DOI] [PubMed] [Google Scholar]
- 2.Ambros V, and Horvitz HR (1984). Heterochronic mutants of the nematode Caenorhabditis elegans. Science 226, 409–16. [DOI] [PubMed] [Google Scholar]
- 3.Ambros V (2011). MicroRNAs and developmental timing. Curr. Opin. Genet. Dev 21, 511–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lin SY, Johnson SM, Abraham M, Vella MC, Pasquinelli A, Gamberi C, Gottlieb E, and Slack FJ (2003). The C. elegans hunchback homolog, hbl-1, controls temporal patterning and is a probable MicroRNA target. Dev. Cell 4, 639–650. [DOI] [PubMed] [Google Scholar]
- 5.Abrahante JE, Daul AL, Li M, Volk ML, Tennessen JM, Miller EA, and Rougvie AE (2003). The Caenorhabditis elegans hunchback-like gene lin-57/hbl-1 controls developmental time and is regulated by microRNAs. Dev. Cell 4, 625–37. [DOI] [PubMed] [Google Scholar]
- 6.Abbott AL, Alvarez-Saavedra E, Miska EA, Lau NC, Bartel DP, Horvitz HR, and Ambros V (2005). The let-7 MicroRNA family members mir-48, mir-84, and mir-241 function together to regulate developmental timing in Caenorhabditis elegans. Dev. Cell 9, 403–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ren Z, and Ambros VR (2015). Caenorhabditis elegans microRNAs of the let-7 family act in innate immune response circuits and confer robust developmental timing against pathogen stress. Proc. Natl. Acad. Sci 112, E2366–E2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Frézal L, and Félix M-A (2015). C. elegans outside the Petri dish. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu PJ (2007). Dauer. WormBook. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fielenbach N, and Antebi A (2008). C. elegans dauer formation and the molecular basis of plasticity. Genes Dev. 22, 2149–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu Z, and Ambros V (1991). Alternative temporal control systems for hypodermal cell differentiation in Caenorhabditis elegans. Nature 350, 162–165. [DOI] [PubMed] [Google Scholar]
- 12.Euling S, and Ambros V (1996). Reversal of cell fate determination in Caenorhabditis elegans vulval development. Development 122, 2507–2515. [DOI] [PubMed] [Google Scholar]
- 13.Karp X, and Ambros V (2012). Dauer larva quiescence alters the circuitry of microRNA pathways regulating cell fate progression in C. elegans. Development 139, 2177–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Golden JW, and Riddle DL (1984). The Caenorhabditis elegans dauer larva: Developmental effects of pheromone, food, and temperature. Dev. Biol 102, 368–378. [DOI] [PubMed] [Google Scholar]
- 15.Avery L (2014). A Model of the Effect of Uncertainty on the C elegans L2/L2d Decision. PLoS One 9, e100580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hammell CM, Karp X, and Ambros V (2009). A feedback circuit involving let-7-family miRNAs and DAF-12 integrates environmental signals and developmental timing in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A 106, 18668–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Butcher RA, Ragains JR, Kim E, and Clardy J (2008). A potent dauer pheromone component in Caenorhabditis elegans that acts synergistically with other components. [DOI] [PMC free article] [PubMed]
- 18.Motola DL, Cummins CL, Rottiers V, Sharma KK, Li T, Li Y, Suino-Powell K, Xu HE, Auchus RJ, Antebi A, et al. (2006). Identification of Ligands for DAF-12 that Govern Dauer Formation and Reproduction in C. elegans. Cell 124, 1209–1223. [DOI] [PubMed] [Google Scholar]
- 19.Antebi A, Yeh WH, Tait D, Hedgecock EM, and Riddle DL (2000). daf-12 encodes a nuclear receptor that regulates the dauer diapause and developmental age in C. elegans. Genes Dev. 14, 1512–27. [PMC free article] [PubMed] [Google Scholar]
- 20.Swanson MM, and Riddle DL (1981). Critical Periods in the Development of the Caenorhabditis elegans Dauer Larva. [DOI] [PubMed]
- 21.Butcher R.a, Fujita M, Schroeder FC, and Clardy J (2007). Small-molecule pheromones that control dauer development in Caenorhabditis elegans. Nat. Chem. Biol 3, 420–422. [DOI] [PubMed] [Google Scholar]
- 22.Ren P, Lim CS, Johnsen R, Albert PS, Pilgrim D, and Riddle DL (1996). Control of C. elegans larval development by neuronal expression of a TGF-beta homolog. Science 274, 1389–1391. [DOI] [PubMed] [Google Scholar]
- 23.Kim K, Sato K, Shibuya M, Zeiger DM, Butcher R.a, Ragains JR, Clardy J, Touhara K, and Sengupta P (2009). Two chemoreceptors mediate developmental effects of dauer pheromone in C. elegans. Science 326, 994–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGrath PT, Xu Y, Ailion M, Garrison JL, Butcher R.a, and Bargmann CI (2011). Parallel evolution of domesticated Caenorhabditis species targets pheromone receptor genes. Nature 477, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Park D, O’Doherty I, Somvanshi RK, Bethke A, Schroeder FC, Kumar U, and Riddle DL (2012). Interaction of structure-specific and promiscuous G-protein-coupled receptors mediates small-molecule signaling in Caenorhabditis elegans. Proc. Natl. Acad. Sci. U. S. A 109, 9917–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kimura KD, Tissenbaum HA, Liu Y, and Ruvkun G (1997). daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science 277, 942–6. [DOI] [PubMed] [Google Scholar]
- 27.Patterson GI, Koweek A, Wong A, Liu Y, and Ruvkun G (1997). The DAF-3 Smad protein antagonizes TGF-beta-related receptor signaling in the Caenorhabditis elegans dauer pathway. Genes Dev. 11, 2679–2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ogg S, Paradis S, Gottlieb S, Patterson GI, Lee L, Tissenbaum HA, and Ruvkun G (1997). The Fork head transcription factor DAF-16 transduces insulin-like metabolic and longevity signals in C. elegans. Nature 389, 994–999. [DOI] [PubMed] [Google Scholar]
- 29.Ludewig AH, Kober-Eisermann C, Weitzel C, Bethke A, Neubert K, Gerisch B, Hutter H, and Antebi A (2004). A novel nuclear receptor/coregulator complex controls C. elegans lipid metabolism, larval development, and aging. Genes Dev. 18, 2120–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammell CM, Lubin I, Boag PR, Blackwell TK, and Ambros V (2009). nhl-2 Modulates microRNA activity in Caenorhabditis elegans. Cell 136, 926–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pepper AS-R, McCane JE, Kemper K, Yeung DA, Lee RC, Ambros V, and Moss EG (2004). The C. elegans heterochronic gene lin-46 affects developmental timing at two larval stages and encodes a relative of the scaffolding protein gephyrin. Development 131, 2049–2059. [DOI] [PubMed] [Google Scholar]
- 32.Bethke A, Fielenbach N, Wang Z, Mangelsdorf DJ, and Antebi A (2009). Nuclear hormone receptor regulation of microRNAs controls developmental progression. Science 324, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang X, Zhang H, and Zhang H (2011). The zinc-finger protein SEA-2 regulates larval developmental timing and adult lifespan in C. elegans. Development 138, 2059 LP–2068. [DOI] [PubMed] [Google Scholar]
- 34.Aprison EZ, and Ruvinsky I (2016). Sexually Antagonistic Male Signals Manipulate Germline and Soma of C. elegans Hermaphrodites. Curr. Biol 26, 2827–2833. [DOI] [PubMed] [Google Scholar]
- 35.Ludewig AH, Gimond C, Judkins JC, Thornton S, Pulido DC, Micikas RJ, Döring F, Antebi A, Braendle C, and Schroeder FC (2017). Larval crowding accelerates C. elegans development and reduces lifespan. PLOS Genet. 13, e1006717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacNeil LT, Watson E, Arda HE, Zhu LJ, and Walhout AJM (2013). Diet-Induced Developmental Acceleration Independent of TOR and Insulin in C. elegans. Cell 153, 240–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schindler AJ, Baugh LR, and Sherwood DR (2014). Identification of Late Larval Stage Developmental Checkpoints in Caenorhabditis elegans Regulated by Insulin/IGF and Steroid Hormone Signaling Pathways. PLoS Genet. 10, e1004426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiontke K, and Fitch DHA (2010). Phenotypic Plasticity: Different Teeth for Different Feasts. Curr. Biol 20, R710–R712. [DOI] [PubMed] [Google Scholar]
- 39.Susoy V, Herrmann M, Kanzaki N, Kruger M, Nguyen CN, Rödelsperger C, Röseler W, Weiler C, Giblin-Davis RM, Ragsdale EJ, et al. (2016). Large-scale diversification without genetic isolation in nematode symbionts of figs. Sci. Adv 2, e1501031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simpson SJ, Sword GA, and Lo N (2011). Polyphenism in Insects. Curr. Biol 21, R738–R749. [DOI] [PubMed] [Google Scholar]
- 41.Kim H, Ishidate T, Ghanta KS, Seth M, Conte DJ, Shirayama M, and Mello CC (2014). A co-CRISPR strategy for efficient genome editing in Caenorhabditis elegans. Genetics 197, 1069–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bindels DS, Haarbosch L, van Weeren L, Postma M, Wiese KE, Mastop M, Aumonier S, Gotthard G, Royant A, Hink MA, et al. (2016). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53. [DOI] [PubMed] [Google Scholar]
- 43.Arribere JA, Bell RT, Fu BXH, Artiles KL, Hartman PS, and Fire AZ (2014). Efficient marker-free recovery of custom genetic modifications with CRISPR/Cas9 in Caenorhabditis elegans. Genetics 198, 837–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS, et al. (2013). Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ward JD (2015). Rapid and precise engineering of the Caenorhabditis elegans genome with lethal mutation co-conversion and inactivation of NHEJ repair. Genetics 199, 363–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.El Mouridi S, Lecroisey C, Tardy P, Mercier M, Leclercq-Blondel A, Zariohi N, and Boulin T (2017). Reliable CRISPR/Cas9 Genome Engineering in Caenorhabditis elegans Using a Single Efficient sgRNA and an Easily Recognizable Phenotype. G3 (Bethesda). 7, 1429–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.