

CONSPECTUS:
Electrochemistry has been used as a tool to drive chemical reactions for over two centuries. With the help of an electrode and a power source, chemists are bestowed with an imaginary reagent whose potential can be precisely dialed in. The theoretically infinite redox range renders electrochemistry capable of oxidizing or reducing some of the most tenacious compounds (e.g., F− to F2 and Li+ to Li0). Meanwhile, a granular level of control over the electrode potential allows for the chemoselective differentiation of functional groups with minute differences in potential. These features make electrochemistry an attractive technique for the discovery of new modes of reactivity and transformations that are not readily accessible with chemical reagents alone. Furthermore, the use of an electrical current in place of chemical redox agents improves the cost-efficiency of chemical processes and reduces byproduct generation. Therefore, electrochemistry represents an attractive approach to meet the prevailing trends in organic synthesis and has seen increasingly broad use in the synthetic community over the past several years.
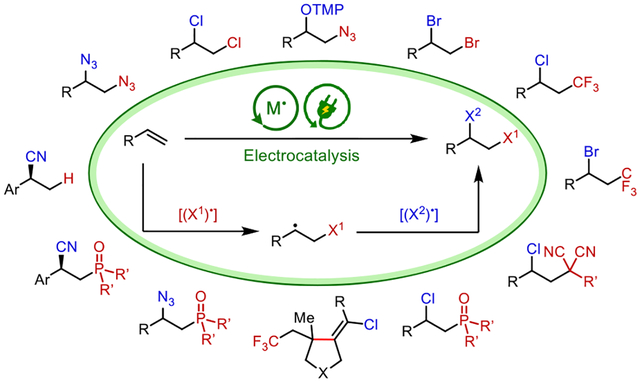
While electrochemical oxidation or reduction can provide access to reactive intermediates, redox-active molecular catalysts (i.e., electrocatalysts) can also enable the generation of these intermediates at reduced potentials with improved chemoselectivity. Moreover, electrocatalysts can impart control over the chemo-, regio-, and stereoselectivities of the chemical processes that take place after electron transfer at electrode surfaces. Thus, electrocatalysis has the potential to significantly broaden the scope of organic electrochemistry and enable a wide range of new transformations. Our initial foray into electrocatalytic synthesis led to the development of two generations of alkene diazidation reactions, using transition-metal and organic catalysis, respectively. In these reactions, the electrocatalysts play two critical roles; they promote the single-electron oxidation of N3− at a reduced potential and complex with the resultant transient N3• to form persistent reactive intermediates. The catalysts facilitate the sequential addition of 2 equiv of azide across the alkene substrates, leading to a diverse array of synthetically useful vicinally diaminated products.
We further applied this electrocatalytic radical mechanism to the heterodifunctionalization of alkenes. Anodically coupled electrolysis enables the simultaneous anodic generation of two distinct radical intermediates, and the appropriate choice of catalyst allowed the subsequent alkene addition to occur in a chemo- and regioselective fashion. Using this strategy, a variety of difunctionalization reactions, including halotrifluoromethylation, haloalkylation, and azidophosphinoylation, were successfully developed. Importantly, we also demonstrated enantioselective electrocatalysis in the context of Cu-promoted cyanofunctionalization reactions by employing a chiral bisoxazoline ligand. Finally, by introducing a second electrocatalyst that mediates oxidatively induced hydrogen atom transfer, we expanded scope of electrocatalysis to hydrofunctionalization reactions, achieving hydrocyanation of conjugated alkenes in high enantioselectivity. These developments showcase the generality of our electrocatalytic strategy in the context of alkene functionalization reactions. We anticipate that electrocatalysis will play an increasingly important role in the ongoing renaissance of synthetic organic electrochemistry.
Graphical Abstract
1. INTRODUCTION
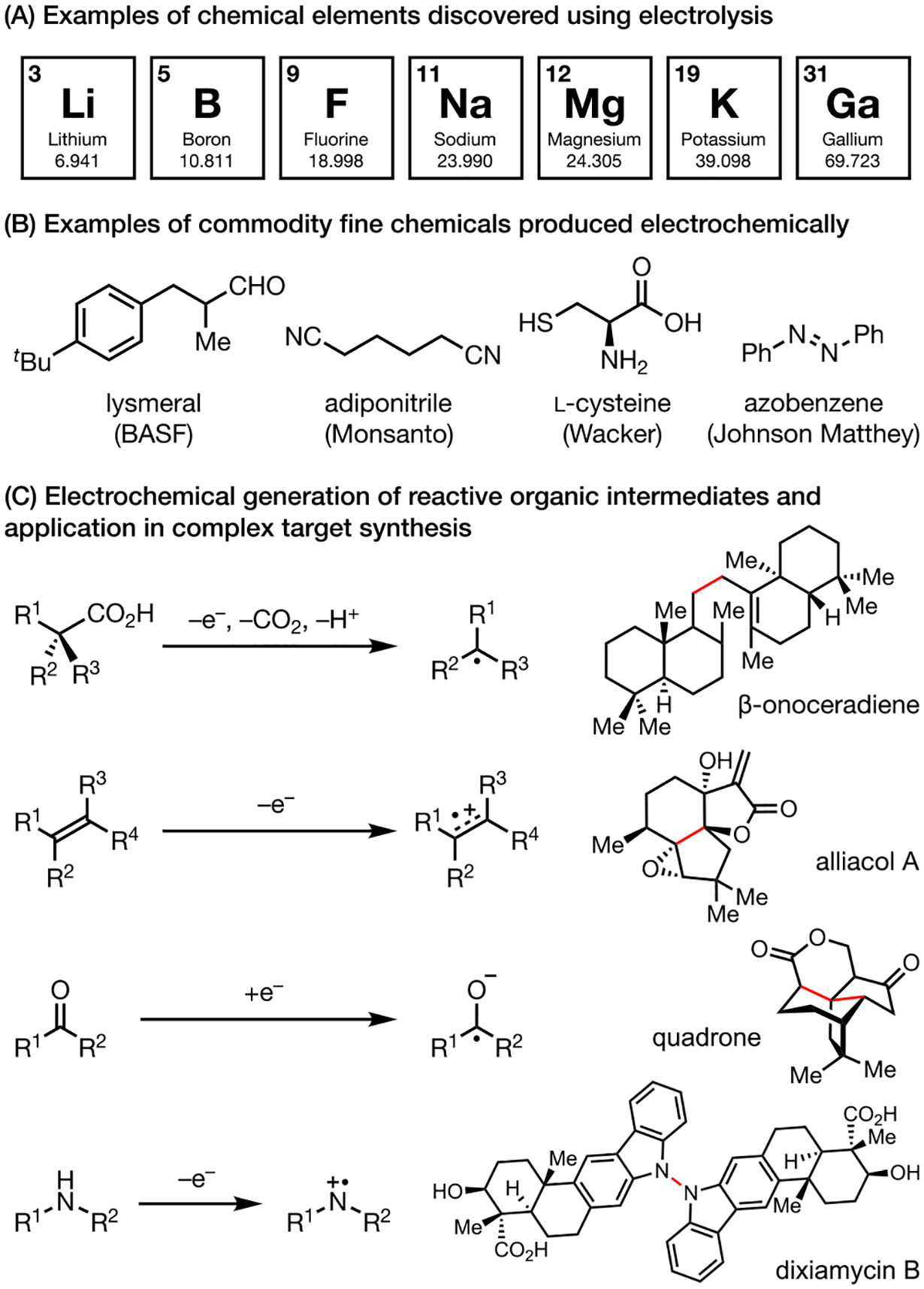
Electrochemistry, though historically perceived as a niche field, has played key roles in driving major scientific discoveries since the 1700s. For example, electrochemical techniques allowed scientists like Henry Moissan1 and Sir Humphry Davy2 to discover new elements, such as Li, Na, and F, that would have otherwise been very difficult, if not impossible, to access using chemical reagents at the time (Scheme 1A). Since Michael Faraday’s discovery of the laws of electrolysis in 18333 and Hermann Kolbe’s demonstration of electrochemical oxidation of valeric acid in 1848,4 great advances have been made in employing electrical current to drive chemical reactions (Scheme 1B).5 In essence, all chemical reactions are movements of electrons from high potential to low potential, from a filled molecular orbital to an empty molecular orbital. Analogously, an electrical current is also a flow of electrons from a high potential to a low potential. Therefore, electrochemistry is one of the most intimate ways for chemists to interact with a microscopic chemical reaction at the flip of a switch, literally.
Scheme 1.
Electrochemically Driven Reactions: From Element Discovery to Complex Target Synthesis
Throughout the years, electrochemistry has expanded from its roots as a tool for element discovery to the synthesis of complex molecules and beyond.6 With the help of a power source and an electrode, chemists are bestowed with an imaginary reagent whose potential can be dialed in at will. As such, electrochemistry in theory offers an infinite redox range and can drive the most difficult of electron movements like the oxidation of fluoride to fluorine. Additionally, the magnitude and duration of the applied potential can be precisely regulated to control both the identity and flux of intermediates formed on the electrodes, something that cannot easily be done with chemical reagents. Indeed, organic chemists have exploited these properties to generate highly reactive intermediates in a controlled fashion for strategic bond formation (Scheme 1C).7
While electrochemistry enables the activation of some of the most tenacious compounds, it relies predominantly on the applied potential and electrode kinetics to control electron transfer events. The lack of tunable molecular recognition elements between the electrode and target reactants makes it challenging to regulate the chemoselectivity of electrochemical reactions in complex systems. Furthermore, the electrode cannot exert control over the selectivity of chemical steps that follow electron transfer.8 Thus, direct electrolysis methods frequently rely on substrate engineering to regulate reactivity and selectivity.9 This is particularly problematic in complex organic molecules where control over chemo- and stereoselectivity is desired. However, such a limitation can be addressed by the introduction of redox-active molecular catalysts (i.e., an electrocatalyst).10
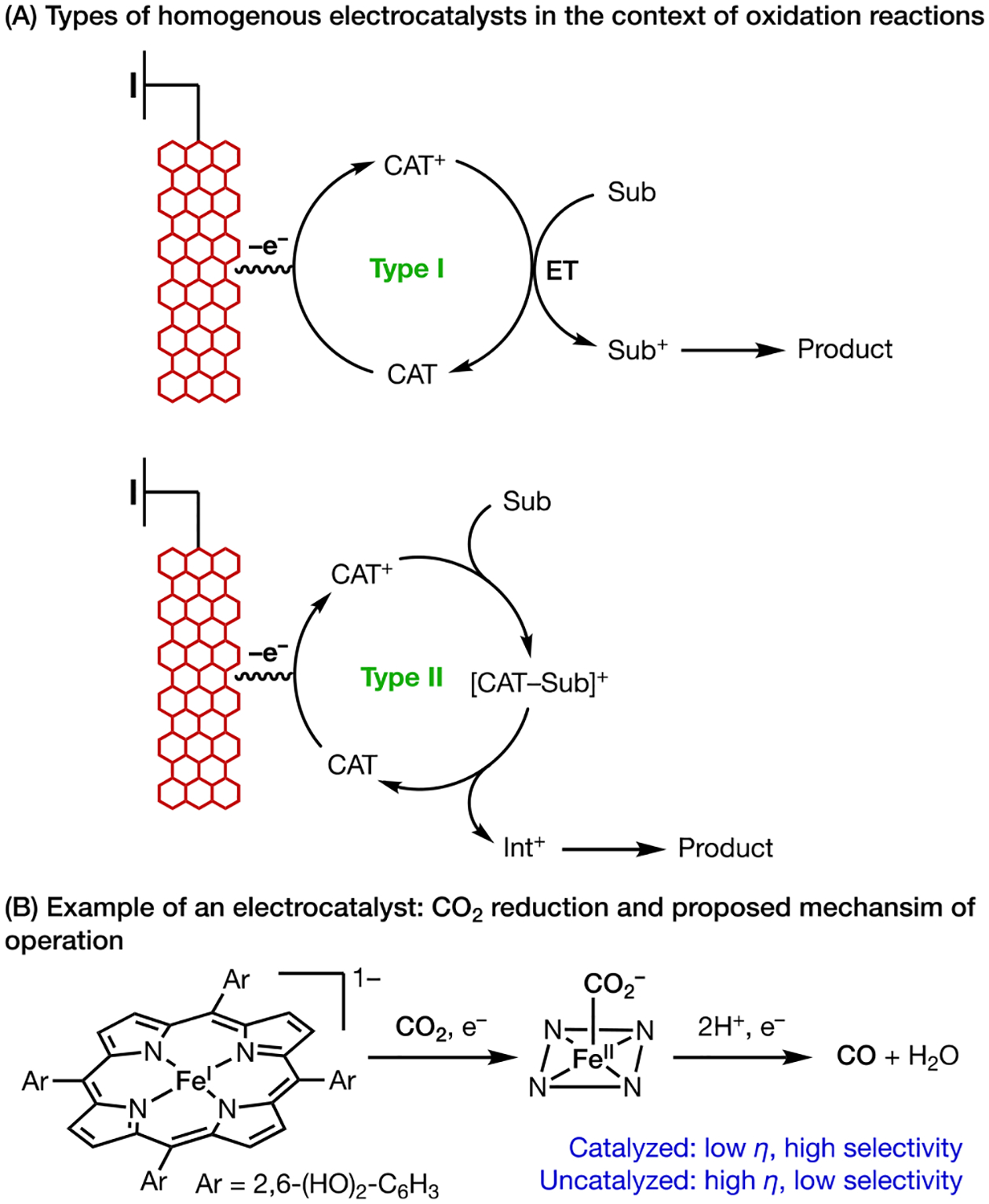
Electrocatalysts can be categorized into two general types based on their role in the chemical steps following electron transfer (Scheme 2A). Type I includes redox mediators that serve the sole purpose of shuttling electrons between the electrode surface and substrate. This type of electrocatalyst accelerates redox reactions that would otherwise be hindered by slow electron-transfer kinetics with electrode surfaces. Type I electrocatalysts have been extensively discussed in recent reviews10a and will not be a focus of this Account. Type II electrocatalysts serve to shuttle both electrons and chemical information to the organic substrates, thus enhancing both reaction rates and chemical selectivity.11 Type II electrocatalysts have been developed for applications in the energy sector, namely for catalytic water splitting12 and CO2 reduction.13 The power of electrocatalysis is exquisitely demonstrated in iron-porphyrin-catalyzed CO2 reduction (Scheme 2B), in which the explicit molecular recognition of CO2 with a metal-carboxylate complex decreases the overpotential for activating an otherwise inert molecule and improves product selectivity.14 In contrast to the extensive studies of electrocatalysis in energy conversion transformations, the development of electrocatalysts specifically for organic transformations still remains largely underexplored.
Scheme 2.
Types of Electrocatalysts and Their Application in Energy Conversion Transformations
The desire to explore novel modes of reactivity and improve the sustainability of synthetic organic chemistry has spawned renewed interest in electrochemically driven reactions within the organic chemistry community. The development of electrocatalytic transformations lies at the center of the Renaissance of synthetic electrochemistry. In this Account, we discuss our laboratory’s recent forays into electrocatalysis for new reaction discovery, with emphases on the mechanistic hypotheses underpinning reaction development as well as the use of mechanistic information and special characteristics of electrochemistry to explore new chemical spaces.
2. THE TRILOGY OF ELECTROCHEMICAL ALKENE AZIDATION
Owing to the importance of nitrogen-containing molecules in many areas of chemistry, new methods that construct C–N bonds in an efficient and selective fashion are highly sought after.15 In this context, azide (N3−) is an attractive agent for the formation of C–N bonds and has widely been used in nucleophilic substitution reactions. The resultant organic azides (R–N3) are valuable synthetic intermediates and can be further transformed to various functional groups including amines, amides, and N-heterocycles.16 A lesser known but important characteristic of N3− is its redox behavior: N3− can readily undergo single-electron oxidation to form azidyl (N3•),17 which is an electrophilic radical that can engage in various C–N bond forming reaction.18 By dialing in a suitable potential, electrochemistry provides direct access to N3• under mild conditions without the use of a chemical oxidant. Therefore, we sought to employ electrocatalysis to discover new azidation reactions and expand the scope of C–N bond formation.
2.1. Mn-Catalyzed Diazidation
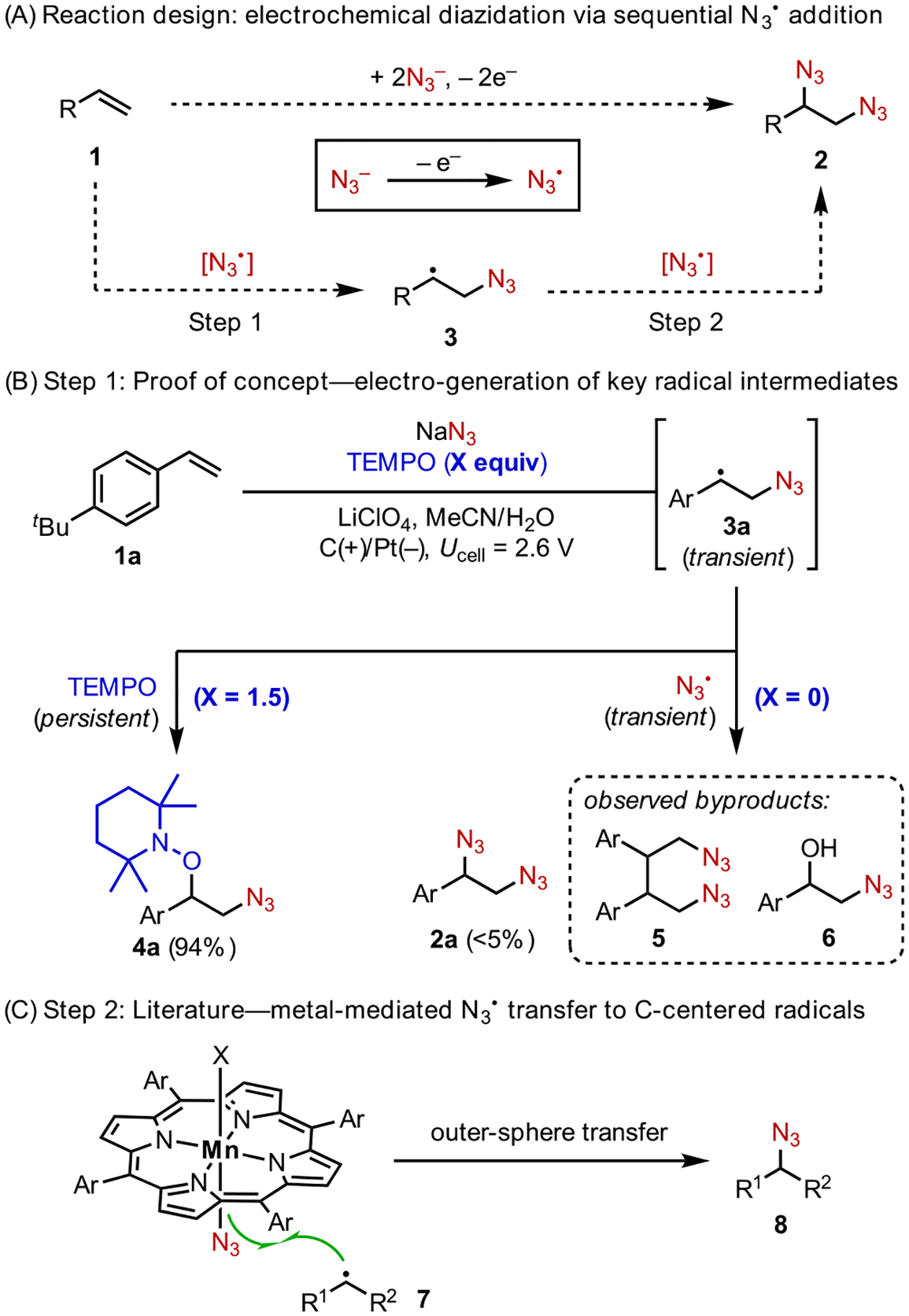
Our early explorations focused on the development of alkene diazidation reactions.19 This transformation would grant access to vicinally diaminated products, which are prevalent moieties in bioactive natural products, pharmaceuticals, and molecular catalysts.20 To achieve alkene diazidation using electrochemistry, we envisioned the following mechanistic design principle (Scheme 3A). The anodic oxidation of N3− (either directly on the electrode or mediated by an electrocatalyst) furnishes N3•,21 which then adds across an alkene 1 in an anti-Markovnikov fashion, producing a C-centered radical 3 (step1). Finally, capture of 3 with another equivalent of N3• completes the desired diazidation (step 2).
Scheme 3.
Electrocatalytic Diazidation: Mechanistic Design Principle
Control experiments utilizing a radical trap lent support to the feasibility of step 1. Electrolysis of a mixture of alkenes 1a, NaN3, and TEMPO led to the formation of azidooxygenated products 4a (Scheme 3B). However, attempts to achieve diazidation in the absence of TEMPO failed to generate an appreciable amount of 1,2-diazides 2a upon full consumption of 1a. This result is expected given that both radicals involved in step 2, 3a and N3•, are transient species; their cross-coupling is statistically unlikely.
The successful coupling of 3 and TEMPO arises from the persistent character of TEMPO (cf. the persistent radical effect22). We hypothesized that the identification of a suitable catalyst that could bind and stabilize N3• while maintaining its radical character would provide a solution to the elusive second C–N bond formation step. Indeed, recent literature shows that redox-active transition metals, such as Mn23 or Cu,24 could form azide complexes to facilitate N3• transfer to C-centered radicals, forging C–N bonds (Scheme 3C).
We surveyed a collection of transition metal complexes in the electrochemical alkene diazidation and discovered that MnBr2 catalyzed the desired reaction in high efficiency (Scheme 4A).25 The application of a moderate cell voltage (Ucell = 2.3 V, anodic potential Eanode ~ 0.5 V vs Fc+/0) gave rise to diazides 2 in high yield. The generality of our reaction was established across a broad range of alkenes with diverse substitution patterns and electronic properties. This expansive substrate generality arises from the radical nature of the reaction mechanism, which is generally less sensitive to the steric and electronic properties of the alkene than mechanisms that involve cationic or anionic intermediates. Another attractive feature of this reaction is its exceptional compatibility with functional groups that are prone to oxidation, such as aldehydes, amines, and sulfides. This feature stems from the mild reaction conditions granted by electrocatalysis, which require a minimal anodic potential to turn over the catalyst.
Scheme 4.
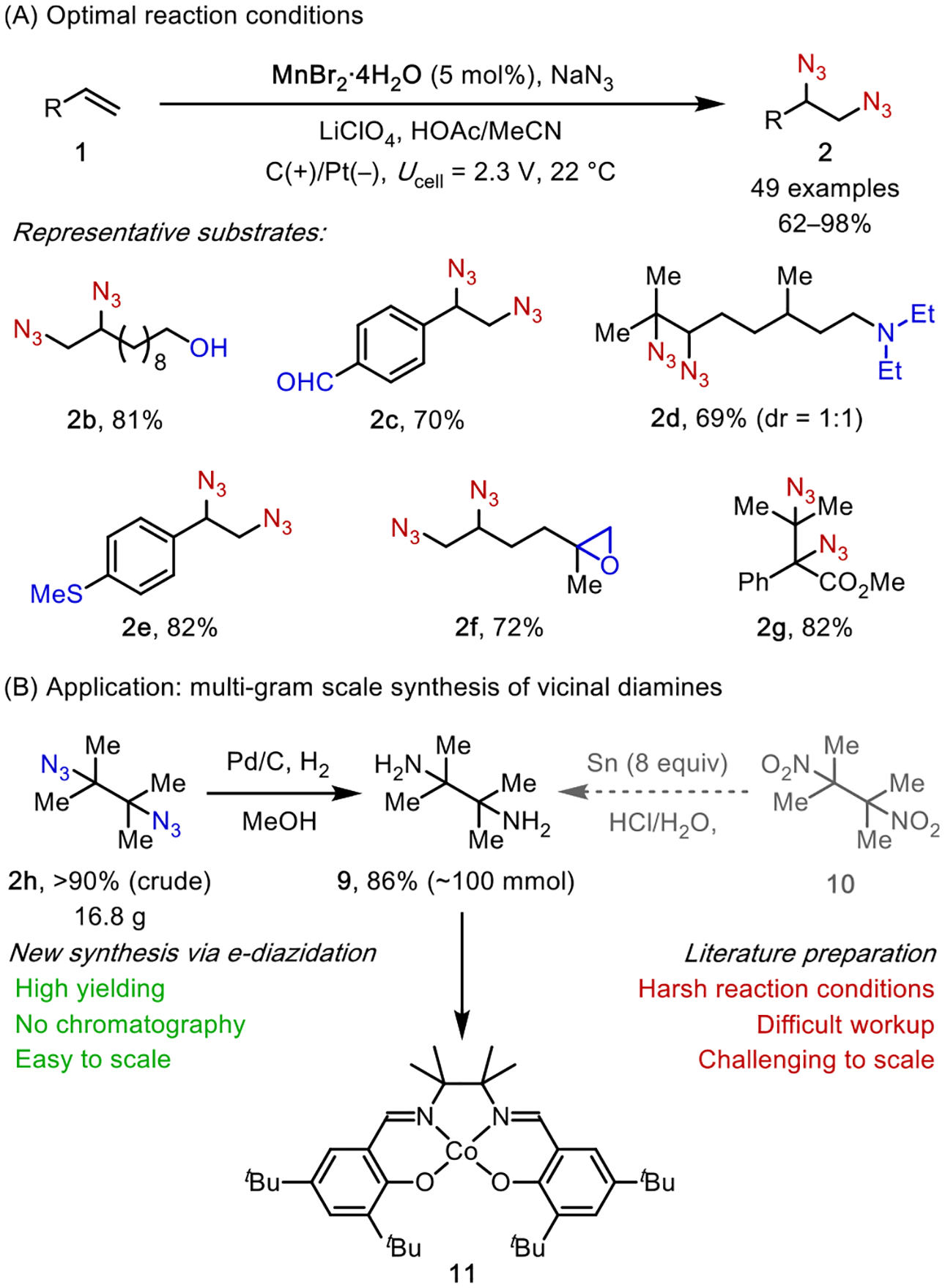
Electrocatalytic Diazidation: Reaction Development
Cyclic voltammetry (CV) analysis (Scheme 5A) of the reaction system revealed that Mn catalyzes the oxidation of N3− to N3•, which induces a cathodic shift of the oxidation wave by ca. 150 mV. Controlled potential electrolysis at 0.5 V gave the desired diazide in excellent yield, showing that the direct anodic oxidation of N3− (Ep/2 = 0.7 V) contributes minimally to the observed reactivity. The CV data along with knowledge26 of the radical reactivity of MnIII led us to propose an electrocatalytic mechanism mediated by the MnIII/II redox couple (Scheme 5B). Specifically, the catalyst is activated on the anode to generate [MnIII]-N3 as the latent N3• source, and this catalytic intermediate delivers the N3 group, first to the alkene and then to the resultant C-centered radical 3, to complete diazidation.
Scheme 5.
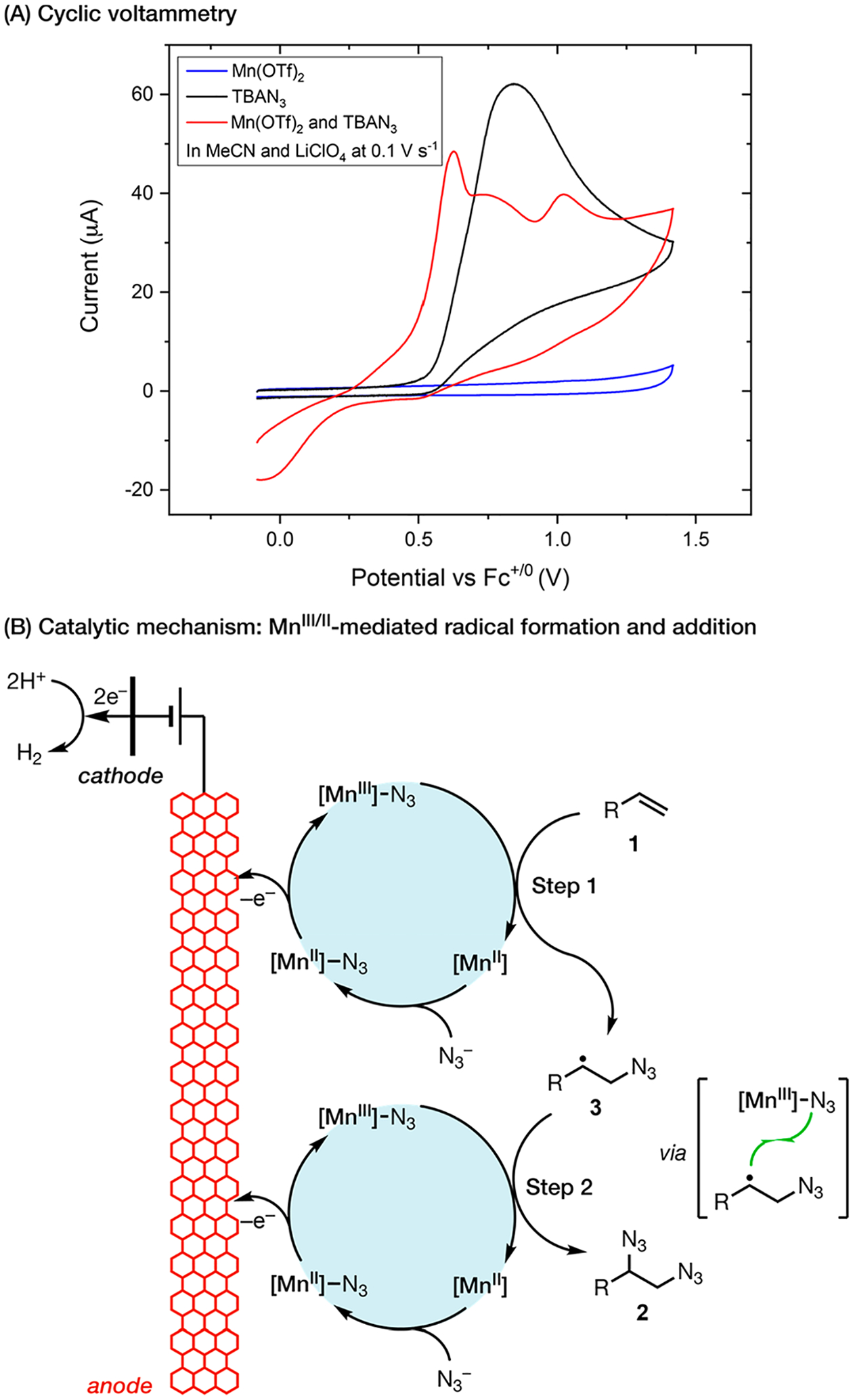
Electrocatalytic Diazidation: Reaction Mechanism
The synthetic utility of this reaction was demonstrated in a 100 mmol scale synthesis of 2,3-dimethylbutane-2,3-diazide (2h), which afforded ~17 g of the product in high yield (Scheme 4B). The crude product can be directly subjected to reduction, furnishing 2,3-dimethylbutane-2,3-diamine (9) in high purity. In fact, our laboratory has routinely employed this two-step protocol to synthesize diamine-derived catalysts (e.g., 11), replacing the standard literature preparation that relies on laborious reduction of 2,3-dimethyl-2,3-dinitrobutane (10) by Sn.27
2.2. TEMPO-Mediated Azidooxygenation
While studying alkene diazidation, we discovered that TEMPO can intercept C-centered radicals 3 resulting from N3• addition to the alkene (Scheme 6A).25a This reaction provides vicinally azidooxygenated products, which contain two versatile functional handles that can be furnished into useful products including vicinal aminoalcohols and α-azidoketones.28 The initial mechanistic hypothesis entailed the anodic oxidation of N3− to N3• followed by sequential addition of N3• and TEMPO to the alkene (Scheme 6B, pathway 1). However, this intuition-based mechanism was not fully consistent with experimental data. CV data (Figure 1A) showed that the redox potential of N3•/− (Ep/2 = 0.45 V) is higher than that of TEMPO+/• (E1/2 = 0.25 V). Controlled potential electrolysis at0.35 V promoted azidooxygenation in comparable efficiency. Therefore, the direct anodic generation of N3• is unlikely to contribute to the observed azidooxygenation.
Scheme 6.
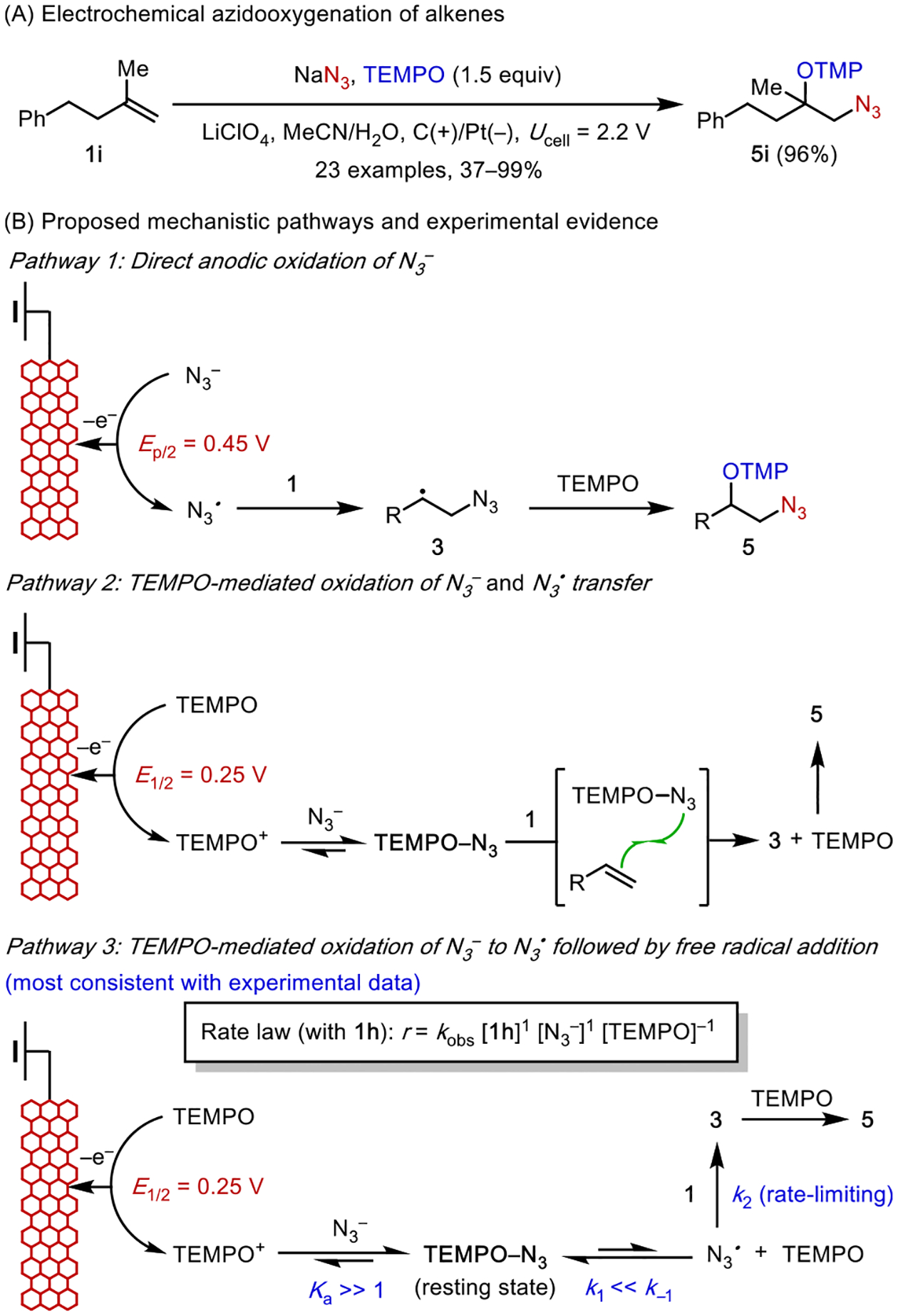
Electrocatalytic Azidooxygenation: Mechanistic Study
Figure 1.
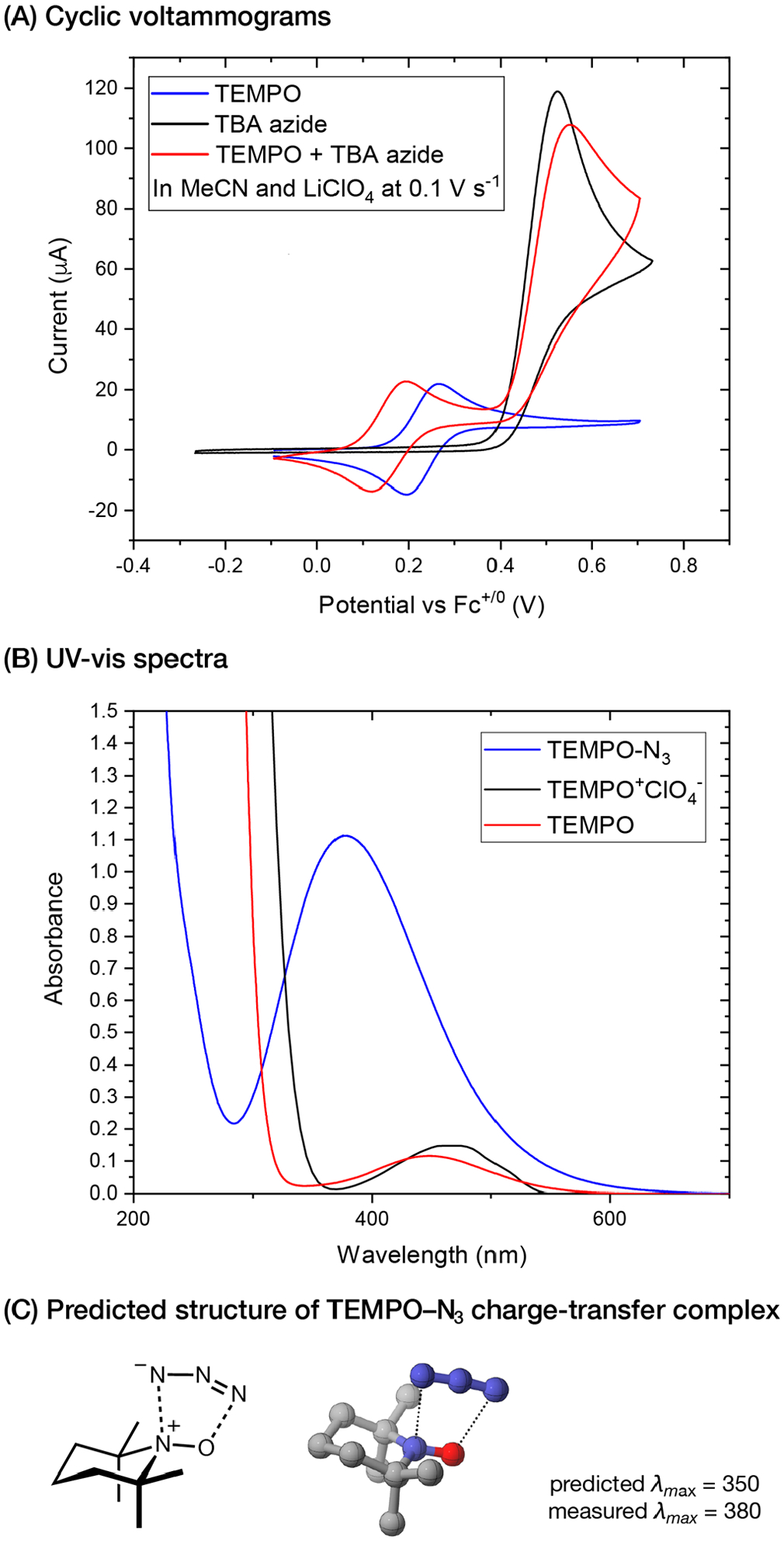
Electrocatalytic azidooxygenation: discovery of a charge-transfer complex.
This analysis led us to propose a new mechanism, in which the oxidation of N3− to N3• is mediated by anodically generated TEMPO+ (Scheme 6B, pathways 2–3). This oxidation step, however, is thermodynamically unfavorable by about 200 mV (~4.6 kcal/mol). Although an outer-sphere electron transfer pathway is energetically feasible, experimental evidence is more consistent with an inner-sphere pathway in which TEMPO+ and N3− react to form a charge-transfer complex (CTC). Electrolysis of the azidooxygenation reaction produces a dark red color, which is also observed when mixing independently prepared TEMPO+ClO4− solution with NaN3. UV-vis study of this mixture (Figure 1B) confirmed the formation of a new species with an absorbance profile substantially different from that of TEMPO and TEMPO+.29 In addition, CV data showed that the peak potential of the TEMPO+/• redox wave displayed a cathodic shift upon addition of NaN3 (Figure 1A). The magnitude of this shift follows a Nernstian dependence, which indicates the reversible formation of a 1:1 complex.30 In the absence of an alkene, this TEMPO-N3 CTC undergoes decomposition, converting to TEMPO and N2. Due to the instability of TEMPO-N3, we have yet to unequivocally elucidate its structure. However, DFT computation in combination with spectroscopic measurements provided insight into this structural uncertainty (Figure 1C). The predicted structure displays an unusual two-site binding interaction between the N3 group and the aminoxyl motif of TEMPO with a slightly bent N3 geometry (N–N–N angle = 173°).
In the presence of an alkene (e.g., 1i), the TEMPO-N3 complex reacts rapidly to yield the azidooxygenated product (5i). Two potential mechanistic pathways could account for the reaction between TEMPO-N3 and 3 (Scheme 6B). In the first mechanism, TEMPO-N3 directly transfers N3 to the alkene in the form of a radical, generating a C-centered radical 3 and TEMPO before radical termination to form 5i (pathway2). Alternatively, the CTC dissociates into TEMPO and N3• prior to their additions to the alkene (pathway 3). Kinetic studies together with computational data ruled out pathway 2 and supported pathway 3, indicating a reversible homolytic fragmentation of TEMPO-N3 preceding the rate-limiting N3• addition to 3. Thus, the formation of the CTC lowers the barrier to electron transfer and promotes the oxidation of N3− by TEMPO+. The function of TEMPO-N3 resembles that of [MnIII]-N3 in Mn-catalyzed diazidation (vide supra); by reversibly capturing and releasing azide, TEMPO-N3 behaves as a stabilized persistent N3• source.
2.3. Aminoxyl Radical-Catalyzed Diazidation
During the substrate scope study of the azidooxygenation reaction,28 we discovered that some sterically hindered alkenes gave rise to a small quantity of 1,2-diazide side products (e.g., 2i, Scheme 7A). Control experiments showed that TEMPO is necessary for the formation of the diazides. We envisioned that the competing azidooxygenation and diazidation share the same pathway for the formation of C-centered radicals 3 but bifurcate thereafter (Scheme 7B). Specifically, the radical combination of 3 and TEMPO, two sterically encumbered species, is slow. Thus, 3 reacts with TEMPO-N3 in a competing pathway in a manner akin to the reaction between 3 and [MnIII]-N3, which delivers 1,2-diazide 2i and liberates TEMPO. In principle, TEMPO could be reoxidized on the anode to render the reaction catalytic. This proposed mode of action by TEMPO is unprecedented31 but mechanistically conceivable.
Scheme 7.
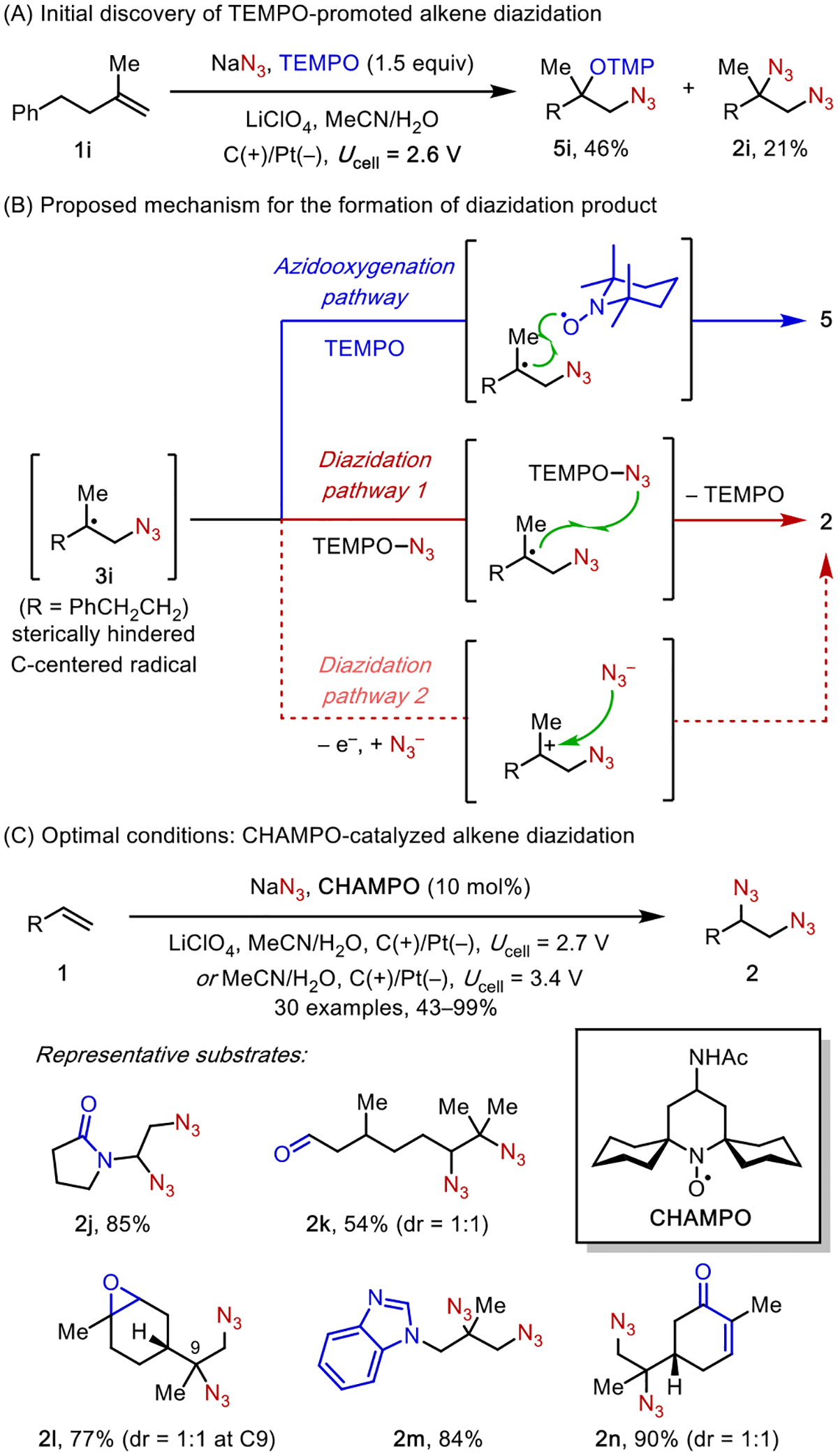
Aminoxyl-Catalyzed Electrochemical Diazidation
These unexpected results led us to hypothesize that, with the right reaction conditions, the TEMPO-consuming azidooxygenation pathway could be suppressed in favor of TEMPO-catalyzed diazidation. Consequently, we discovered that using a novel designer catalyst CHAMPO, a more sterically hindered aminoxyl radical, under low temperatures, the azidooxygenation reaction was largely inhibited, allowing diazidation to predominate. This reaction provided a broad scope of structurally diverse 1,2-diazides in good yields (Scheme 7C).32
Experiments using various cationic (1p) and radical probe (1o) substrates (Scheme 8A) lent strong support to the proposed mechanism (Scheme 8B) in which the second azide addition to the C-centered radical 3 proceeds via radical transfer promoted by the CHAMPO-N3 CTC. An alternative mechanism in which 3i was oxidized to the corresponding carbocation followed by nucleophilic trapping with N3− is unlikely to occur (Scheme 7B, diazidation pathway 2).
Scheme 8.
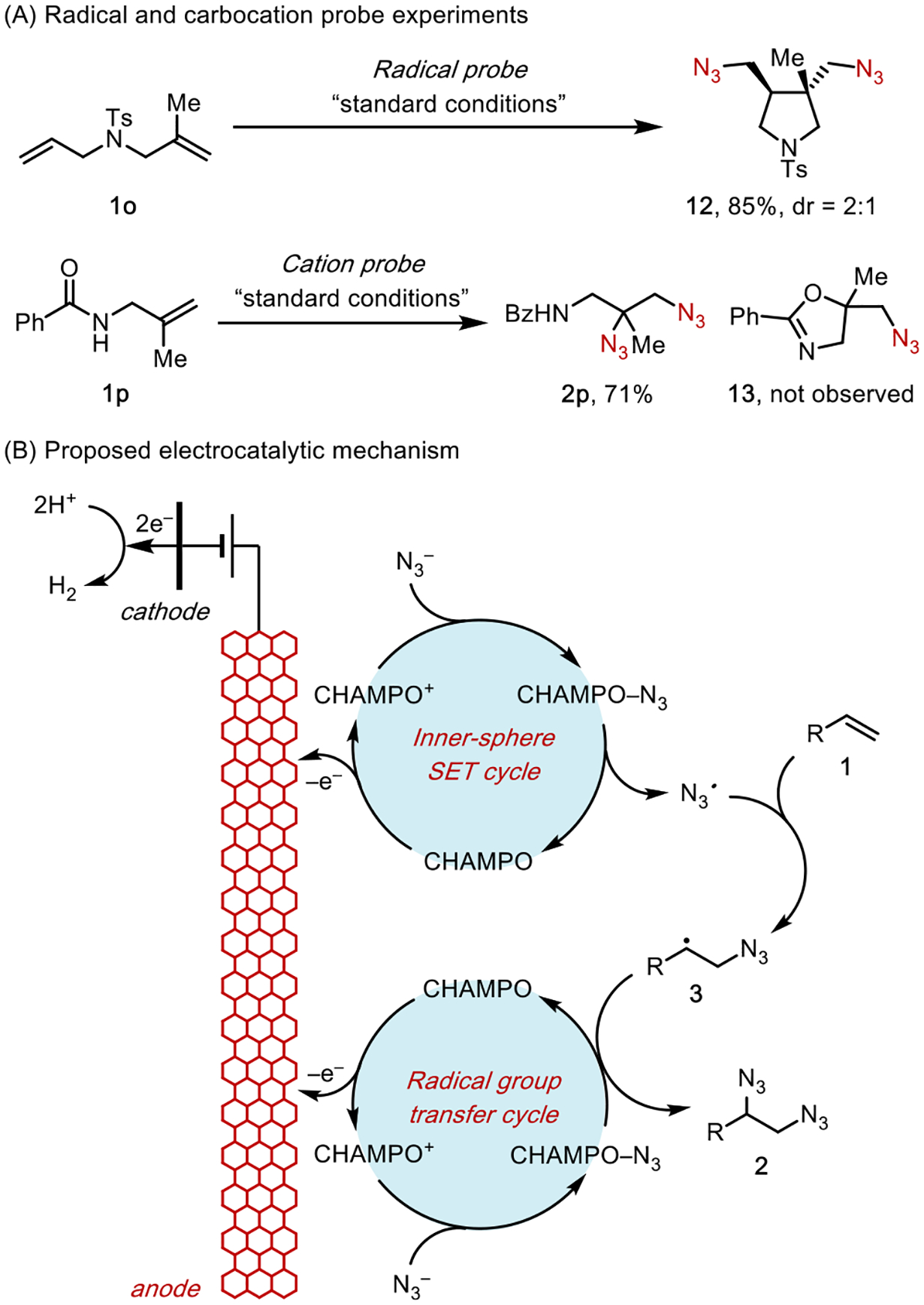
Mechanism of Aminoxyl-Catalyzed Diazidation
Aside from being a conceptual advance in aminoxyl radical catalysis, this second-generation diazidation method also presents several attractive features from a practical perspective. For example, the reaction can be carried out under transitionmetal-free and pH-neutral conditions, eliminating the formation of potentially hazardous metal azides and hydrazoic acid. In addition, an exogenous supporting electrolyte is unnecessary, as NaN3 is soluble in the reaction medium of MeCN and H2O, making the solution conductive.
2.4. Extension to Dihalogenation
The proposed mechanism for Mn-catalyzed alkene diazidation led us to hypothesize that this mode of activity could be generalized to discover other alkene difunctionalization reactions. Specifically, with an appropriate anion X−, the [MnIII]-X complex could behave as a latent X• equivalent and promote the radical addition of X groups across an alkene substrate. To this end, we developed the electrocatalytic dichlorination of alkenes using MgCl2 as the chlorine source (Scheme 9A).33 Under conditions similar to the diazidation, a wide range of alkenes were transformed to the 1,2- dichloroalkanes in high efficiency. Control experiments showed that excluding Mn led to full consumption of the alkene with little conversion to the desired product. In this case, highly reactive Cl• is likely generated directly on the anode, leading to uncontrolled side reactions of the alkene. Indeed, CV data revealed that the addition of MnII lowers the potential needed to oxidize Cl−. Thus, this electrocatalyst not only lowers the energy barrier to the formation of the Cl• equivalent species, but it also moderates its reactivity toward alkene addition in a selective fashion.
Scheme 9.
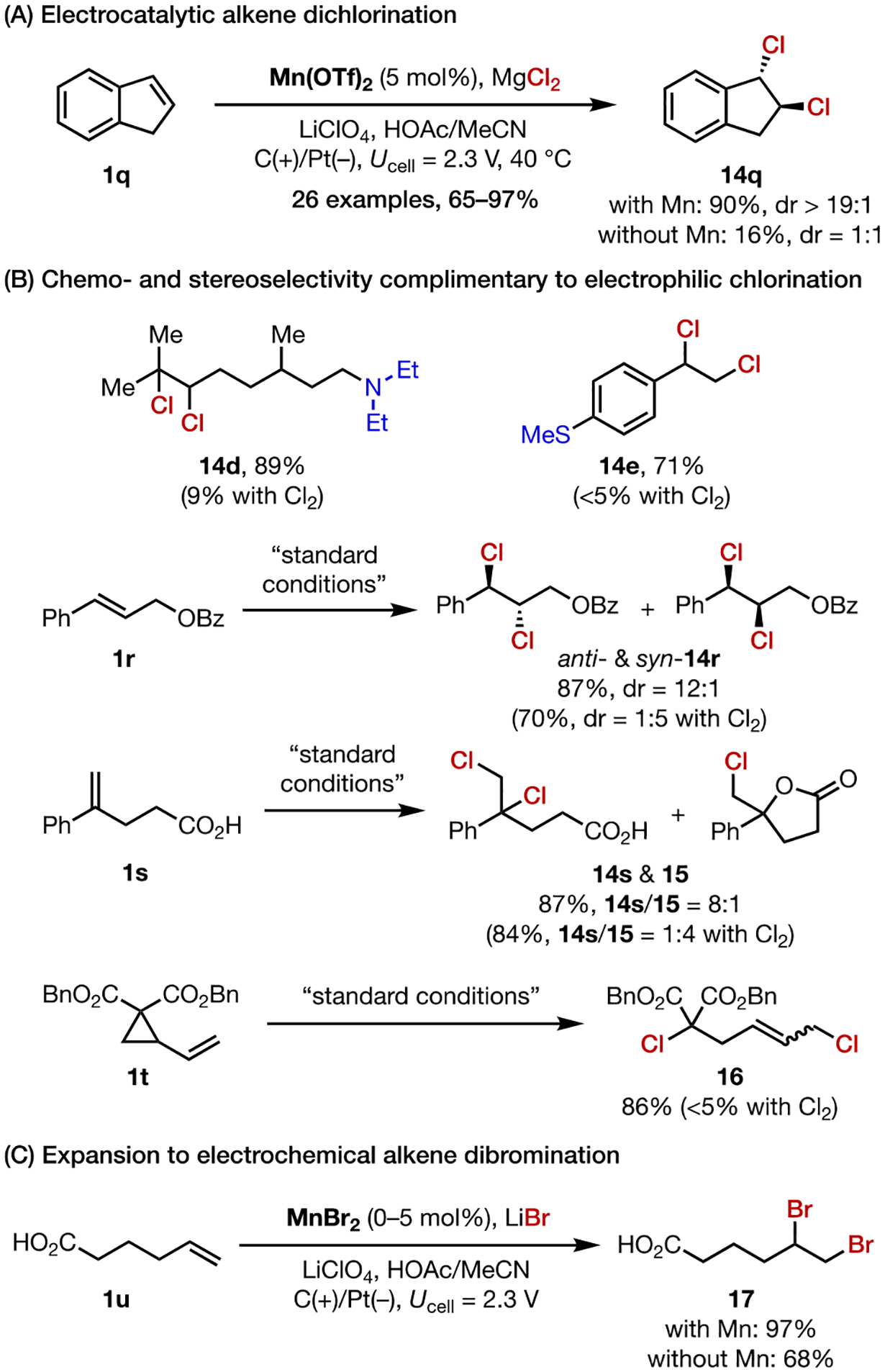
Electrocatalytic Dihalogenation
Alkene dichlorination reactions are traditionally carried out using electrophilic Cl sources such Cl2. The use of MgCl2 in combination with an electrical current presents a more chemoselective (Scheme 9B) and practical method for making the same products. In addition, this electrochemical dichlorination proceeds via a radical mechanism and does not rely on the chloronium-mediated pathway thought to be operative in electrophilic chlorination reactions. Therefore, we employed this methodology to achieve chemo- and stereoselectivity patterns that were previously challenging (Scheme 9B).
We further extended the scope of this general strategy to the dibromination of alkenes using LiBr (Scheme 9C).34 However, because Br• is both easier to generate (EBr•/Br− = ~0.7 V) and more persistent than Cl•,35 typical dibromination reactions do not require Mn as the electrocatalyst.
3. ELECTROCATALYTIC ALKENE HETERODIFUNCTIONALIZATION AND ENANTIOSELECTIVE ELECTROCATALYSIS
The heterodifunctionalization of alkenes, in which two distinct functional groups are added to a C=C bond in a single synthetic operation, is an efficient transformation that rapidly increases the structural and functional complexity of unsaturated molecules. Our early successes in electrochemical diazidation and dihalogenation reactions provided us with a platform to further expand the scope of electrocatalysis to heterodifunctionalization. Achieving this objective required the simultaneous generation of two open-shell intermediates and control over their chemo- and regioselective additions to the alkene (Scheme 10A).
Scheme 10.
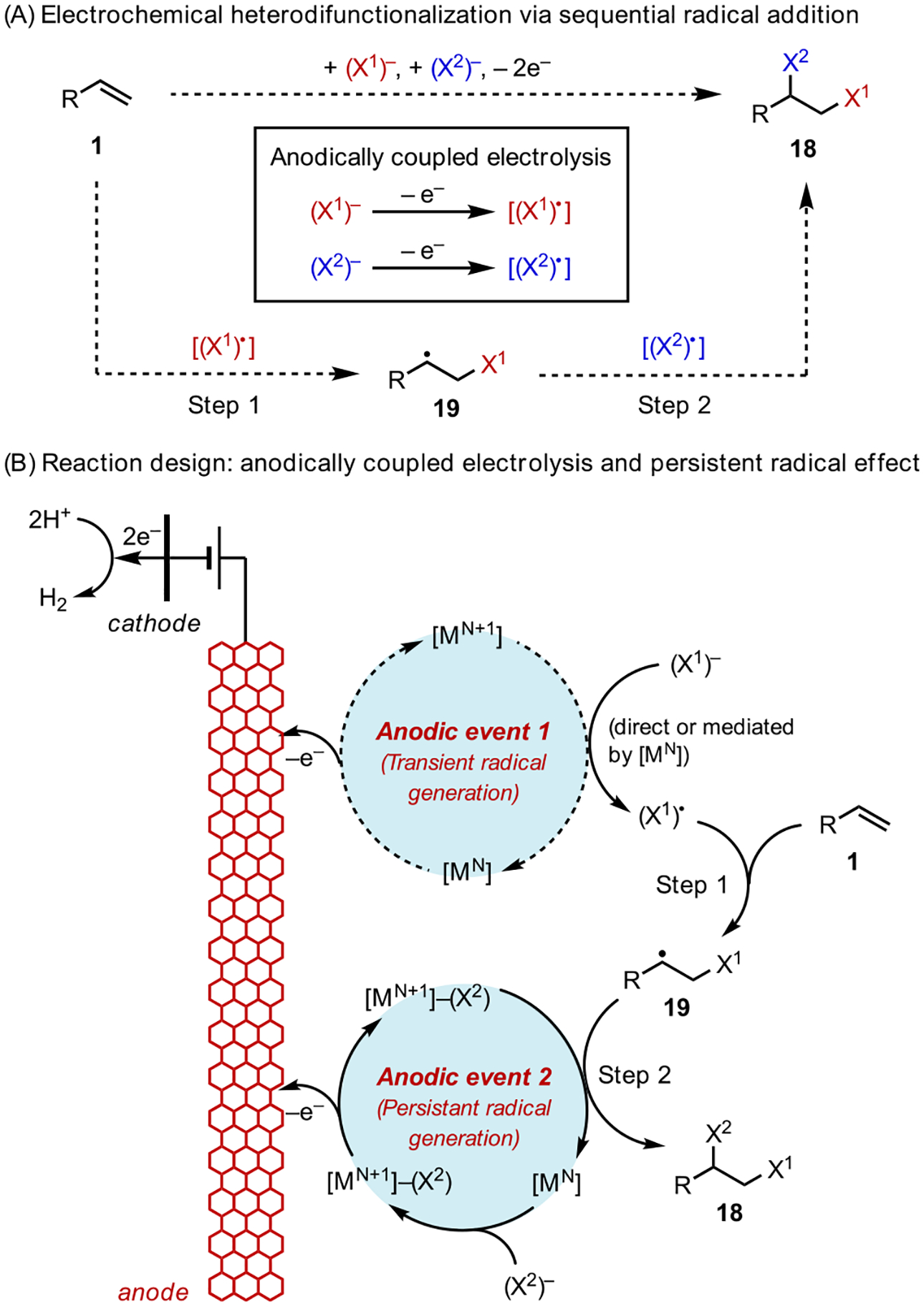
Electrocatalytic Heterodifunctionalization: Anodically Coupled Electrolysis and the Persistent Radical Effect
To this end, we advanced a new strategy, anodically coupled electrolysis (ACE; Scheme 10B), for the heterodifunctionalization of alkenes.36 In this strategy, the electrode surface accommodates multiple concurrent redox reactions upon the application of an appropriate potential, and the resultant reactive intermediates react in a convergent manner to yield the desired product. This design principle is reminiscent of paired electrolysis,37 in which a combination of anodic oxidation and cathodic reduction reactions are coupled in an electrolytic cell to produce two reactive intermediates that converge into the same reaction system.
The ACE strategy along with knowledge of the persistent radical effect22 guided our development of the heterodifunctionalization reaction. We envisioned that, with an appropriate pair of reagents, (X1)− and (X2)− in combination with a metal catalyst, [MN], two parallel anodic reactions will take place. (X1)− will undergo single-electron oxidation, either directly on the anode or mediated by the catalyst, to form a transient free radical (X1)•. Meanwhile, (X2)− will undergo catalyst-assisted anodic oxidation to form an (X2)• equivalent in the form of a persistent metal complex, [MN+1]-X2. Upon generation of this pair of radical intermediates, the selectivity of the alkene addition process is dictated by the persistent radical effect. Specifically, transient X1• is more reactive than persistent [MN+1]-X2 but has an exceedingly low concentration. As such, alkene 1 will preferentially react with X1• to produce a C-centered radical 19, which is also a transient species. Because the reaction between two transient radicals is statistically unlikely, 19 will preferentially react with [MN+1]-X2, thus giving rise to the heterodifunctionalized product 18 in a chemo- and regioselective fashion.
3.1. Chlorotrifluoromethylation
We first implemented the strategy of ACE in the chlorotrifluoromethylation of alkenes (Scheme 11).36 This transformation is synthetically attractive, as the resulting CF3 and Cl functional groups are useful in both medicinal and materials chemistry, and the alkyl chloride could further engage in C–C and C–N bond forming reactions. We employed readily available reagents including CF3SO2Na and MgCl2 as the sources for CF3• and Cl•, respectively, and Mn(OAc)2 as the electrocatalyst. CV data indicated that the generation of transient CF3• and persistent [MnIII]-Cl occurs readily on a carbon anode at similar oxidation potentials (Figure 2), supporting the feasibility of ACE toward desired heterodifunctionalization.
Scheme 11.
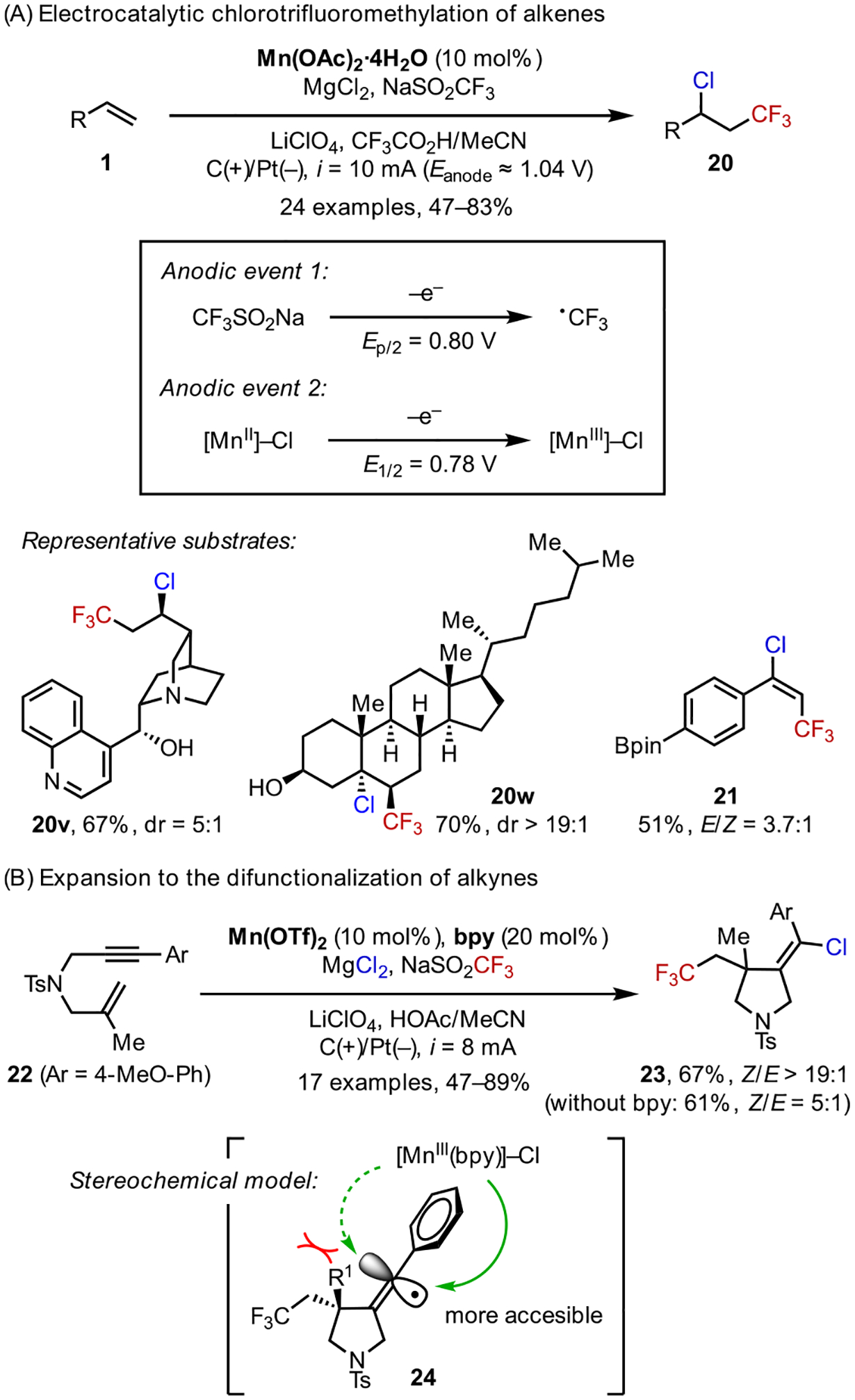
Electrocatalytic Chlorotrifluoromethylation
Figure 2.
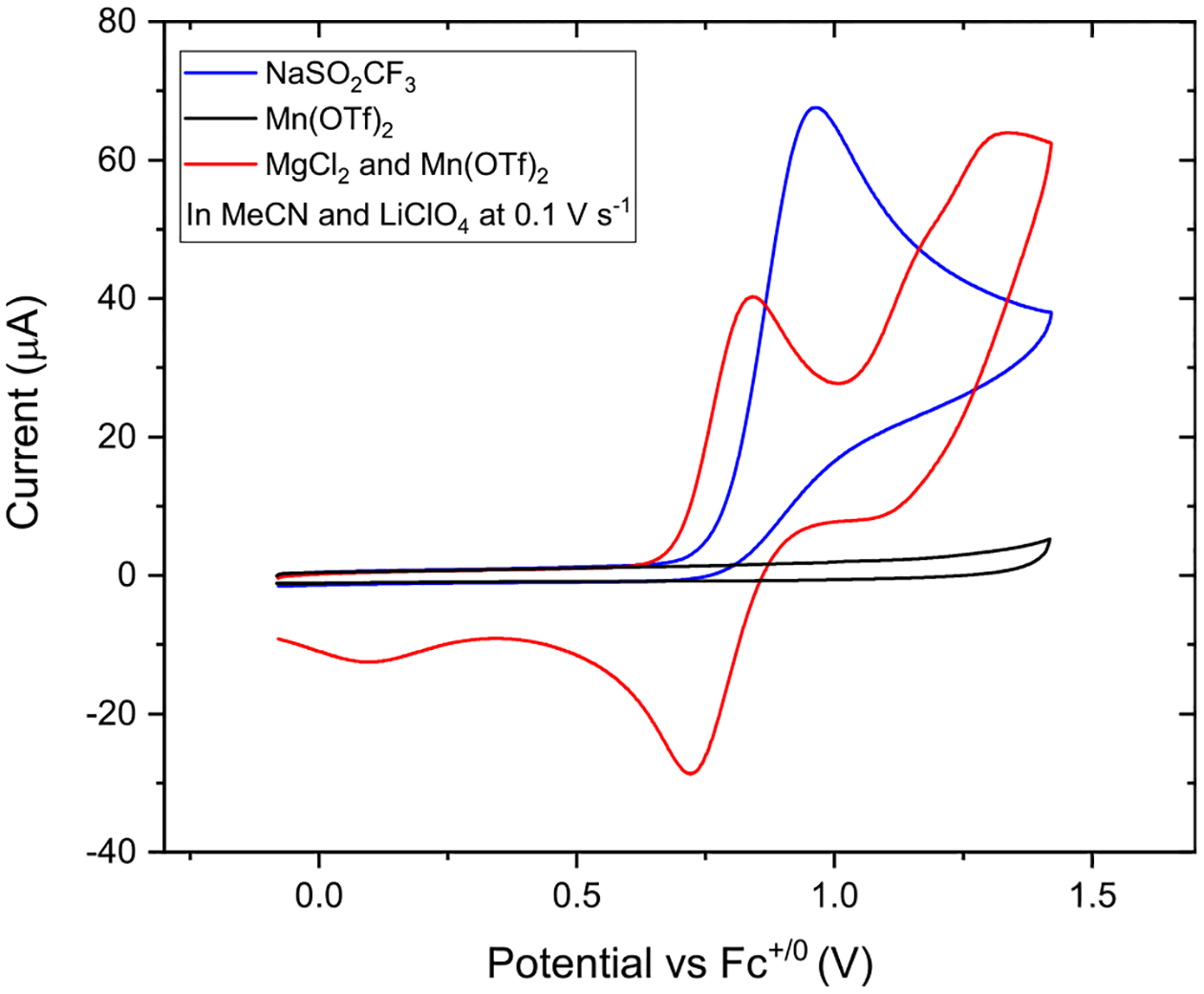
Cyclic voltammetry supporting anodically coupled electrolysis
Like our previous reaction systems, the optimized reaction conditions are broadly applicable to the functionalization of a diverse suite of alkenes. In particular, complex substrates bearing oxidatively labile functional groups (20v, 20w) can be chemoselectively chlorotrifluoromethylated, showing that this reaction could be applied to the late-stage modification of complex molecules. We further extended this reaction method to the difunctionalization of alkynes (21). In particular, electrocatalytic ene-yne cyclization was achieved for the synthesis of chlorotrifluoromethylated pyrrolidines (Scheme 11B).38 Notably, the introduction of bipyridine as a ligand for Mn has a marked influence on the stereochemistry of the Cl• transfer step from [MnIII]-Cl to alkenyl radical 24, improving the diastereoselectivity from 5:1 to >19:1 favoring the (E)- alkene product.
3.2. Other Heterodifunctionalizations
The ACE strategy was shown to be quite modular and applicable to a broad range of alkene difunctionalization reactions with appropriate reagents and catalysts. For example, using activated carbonyl compounds (e.g., malononitriles and α-cyanoacetates), Mn-catalyzed chloro- and bromoalkylation of alkenes could be achieved, forging vicinal C–C and C-halogen bonds in a single synthetic manipulation (Scheme 12A).39 Mechanistic data are consistent with a dual electrocatalytic mechanism, in which the oxidation of both reagents, malononitriles (25) and Cl−, are mediated by MnIII/II redox cycling. The resultant pair of radical intermediates, transient 27 and persistent [MnIII]-Cl, undergo subsequent additions to alkenes 1 in a chemo- and regioselective manner.
Scheme 12.
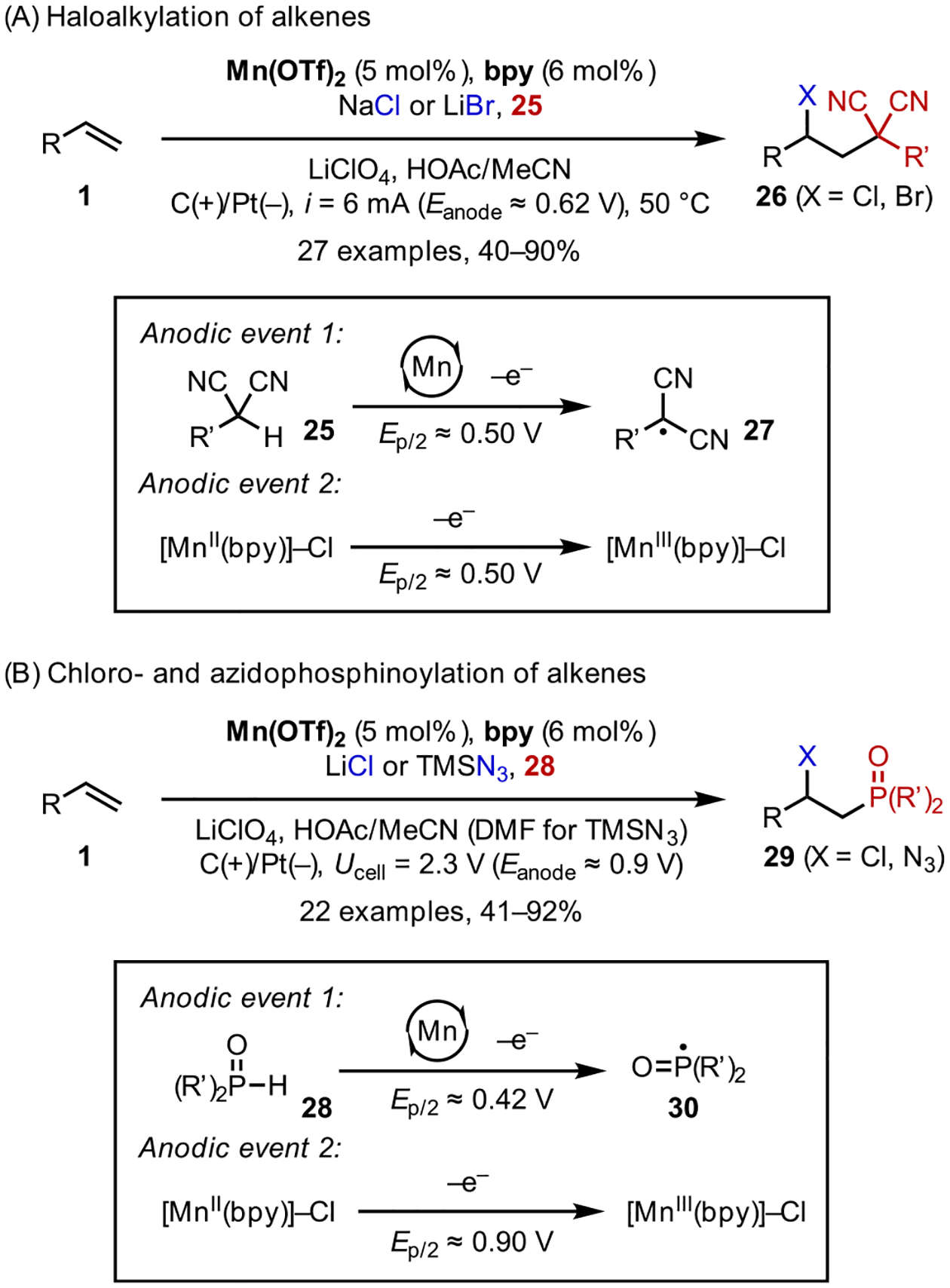
Modular Electrocatalytic Heterodifunctionalizations
CV studies suggested that MnIII is also capable of oxidizing secondary phosphine oxides (28) to the corresponding electrophilic P-centered radical (30). We harnessed this reactivity in the development of chloro- and azidophosphi-noylation reactions using the same electrocatalytic design principle (Scheme 12B).40
3.3. Enantioselective Cyanofunctionalization
Having established the feasibility of electrocatalysis for the heterodifunctionalization of alkenes, we aimed to develop enantioselective variants of these transformations. In spite of the rapid growth of synthetic electrochemistry in recent years, enantioselective electrocatalytic reactions remain rare.41 In the realm of alkene functionalization, early reports by Torii et al. and Moeller et al. showed that canonical Sharpless dihydroxylation and Jacobsen epoxidation reactions could be made electrocatalytic by using an anode to replace the terminal oxidants.42 Our proposed mechanism for the heterodifunctionalization suggests that the use of a chiral ligand could control the stereochemistry of the catalyst-mediated radical combination step (i.e., the formation of C–X2 bond in Scheme 10), thus rendering the overall transformation enantioselective. Indeed, strong ligand effects on the reaction diastereoselectivity have been demonstrated in our electrochemical ene-yne cyclization that proceeds via a similar mechanism (Scheme 11B).
To this end, we developed highly enantioselective cyanofunctionalization of vinylarenes using Cu electrocatalysis (Scheme 13).43 Using cyanophosphinoylation reactions as an example, the envisioned mechanism entailed the parallel electrochemical generation of transient phosphinoyl radical (30) and persistent [CuII]-CN complex as the CN• equivalent.44 The subsequent reaction of these open-shell intermediates with alkene 1 completes the difunctionalization. In this mechanism, the Cu-mediated CN transfer to C-centered radicals 31 likely occurs in an inner-sphere fashion, which is different from the outer-sphere mechanism that is commonly proposed for similar group transfer processes mediated by [MnIII]-X (X = N3, Cl).45 Specifically, [CuII]-CN undergoes single-electron oxidative addition to 31 to form a formally CuIII intermediate 33, which then undergoes reductive elimination to construct the C-CN bond. Computational data in the literature46 suggested that the oxidative addition is reversible and that the reductive elimination is rateand enantio-determining. This inner-sphere mechanism enforces a strong stereochemical communication between the catalyst and the substrate assembly in the transition state and make this reaction suitable for enantioselective catalysis.
Scheme 13.
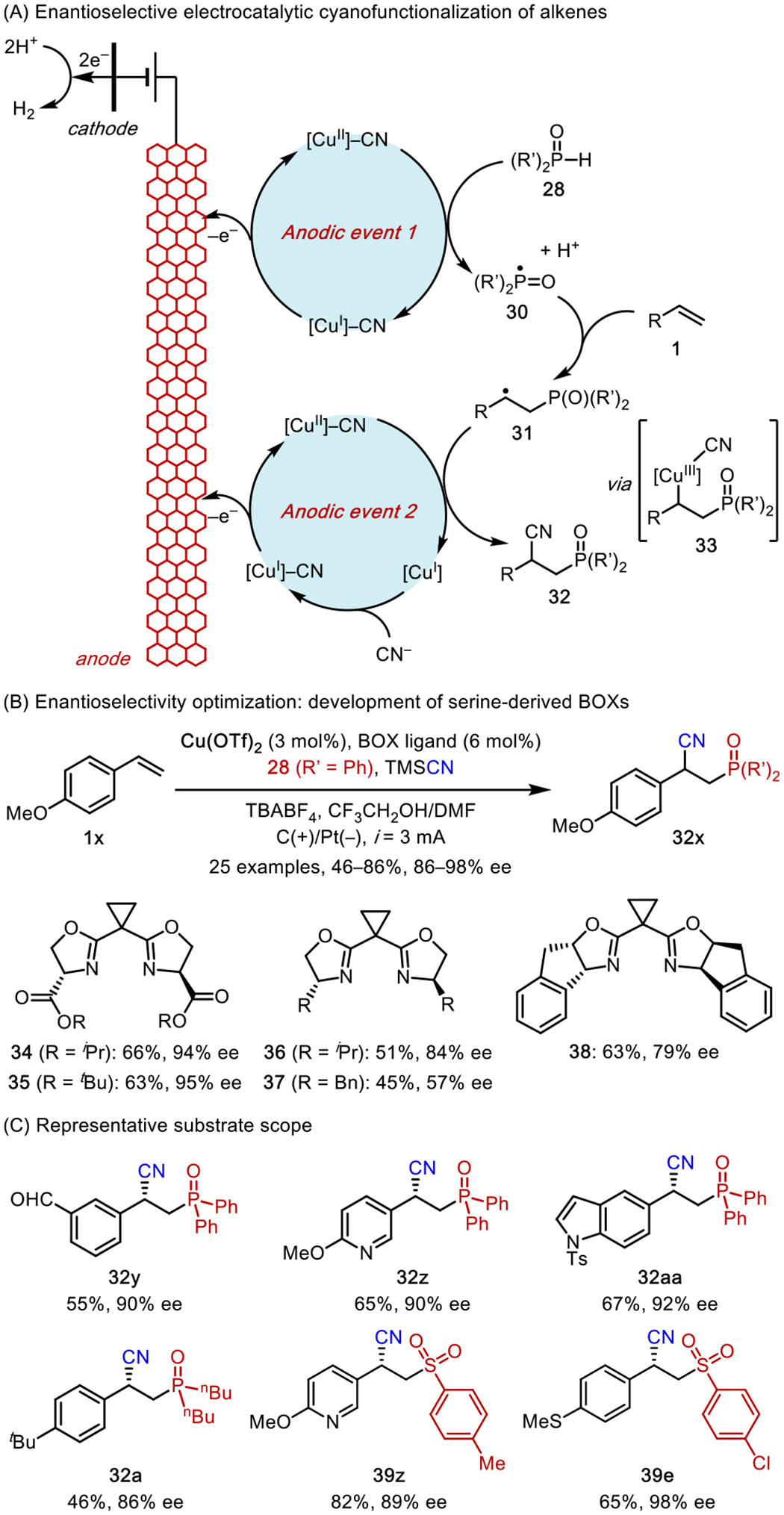
Enantioselective Electrocatalysis: Cyanofunctionalization of Vinylarenes
A major challenge in Cu electrocatalysis lies in the susceptibility of Cu ions to undergo electroreduction on the cathode, resulting in catalyst deactivation. Thus, electrochemical reactions mediated by Cu are often carried out in a divided cell.47 Our optimization led to an efficient cyanophosphinoylation reaction that can be conducted in a more practical undivided cell, in which the Cu catalyst remains in solution during the course of electrolysis. To further render this reaction enantioselective, we surveyed a variety of chiral ligands that are commonly employed in Cu asymmetric catalysis. However, for reasons not yet fully understood, the highest enantioselectivity for the formation of product 32x was 84% ee (12:1 er) using bisoxazoline (BOX) ligand 36. In further optimizations, we synthesized serine-derived BOX ligands (sBOXs) with pendant ester substituents and found that these new ligands provided major improvements, with 35 promoting the reaction in 95% ee (39:1 er). DFT computation revealed that the second-sphere ester groups play critical roles in inducing high enantioselectivity (vide infra).
Finally, we expanded the reaction scope to the cyanosulfinylation of vinylarenes using sulfinic acids (39e, 39z). The successful development of enantioselective electrocatalysis further demonstrated the robustness of the electrocatalytic strategy as a potentially general mechanistic platform for the vicinal difunctionalization of simple alkenes.
4. ENANTIOSELECTIVE HYDROCYANATION VIA DUAL ELECTROCATALYSIS
Hydrofunctionalization reactions offer a complementary technique to difunctionalization in adding complexity to feedstock chemicals and complex bioactive molecules containing alkenes. Therefore, we became interested in expanding our electrocatalytic strategy to the realm of hydrofunctionalization, with the primary objective of enabling new reactivity using the unique features of electrochemistry. We employed electrochemical hydrofunctionalization to the enantioselective hydrocyanation of alkenes, a long-standing synthetic problem that has not been addressed with known chemistries. Despite seminal contributions in the past few decades,48 including elegant developments in Ni-H chemistry, a broadly applicable and highly enantioselective strategy for the direct hydrocyanation of alkenes remains elusive.
We recently disclosed a dual electrocatalytic approach for the asymmetric hydrocyanation of conjugated alkenes (Scheme 14).49 Our mechanistic design principle entails two distinct catalytic cycles operating simultaneously and synergistically on the anode, providing H• and CN• equivalents that are subsequently added to the alkene under catalyst control. This reaction design was inspired by recent literature on metal-hydride hydrogen-atom transfer catalysis, in which a catalytically generated metal-hydride species could enable HAT to an alkene to initiate diverse hydrofunctionalization reactions.50 We sought to couple this reactivity with Cu-mediated radical cyanation that we employed in cyanofunctionalization reactions (section 3.3). A critical challenge in this reaction design was to find an oxidant equivalent to turn over both catalysts yet remain compatible with both catalytic systems. We reasoned that electrochemistry could provide a means to power the desired hydrofunctionalization reaction (Scheme 14B).
Scheme 14.
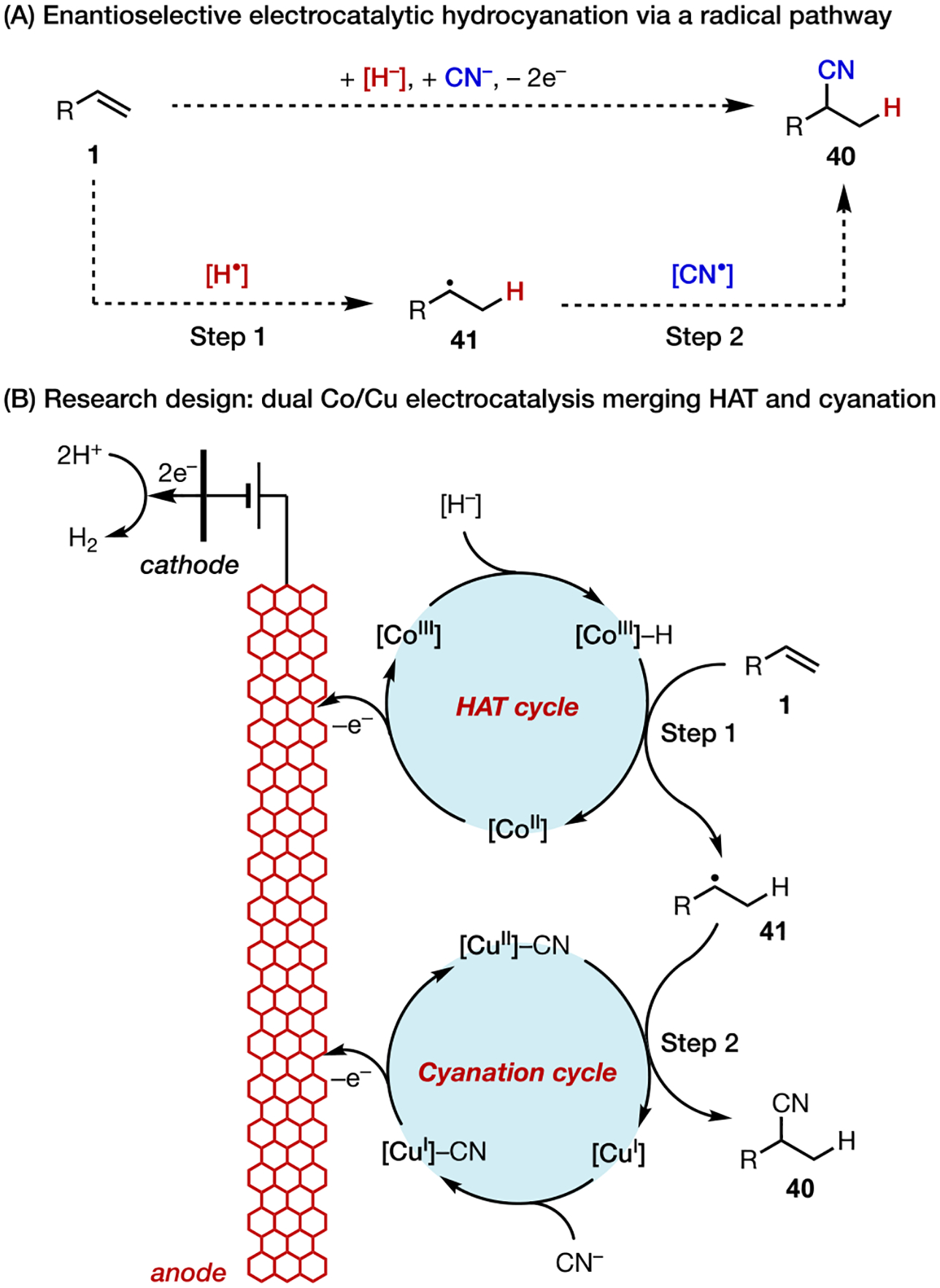
Enantioselective Hydrocyanation via Dual Electrocatalysis
Using Co(salen) (11) and Cu(sBOX) as the catalysts, the optimal reaction operates under very mild conditions (Eanode ~0.2 V) and is broadly applicable to the functionalization of conjugated alkenes, including alkenylarenes, dienes, enynes, and allenes (Scheme 15A). We also demonstrated the use of potential control in the optimization of challenging substrates that are prone to overoxidation (e.g., 40ac). In contrast, this hydrocyanation cannot be promoted at the same level of efficiency, chemoselectivity, or enantioselectivity using a chemical oxidant under otherwise identical conditions (Scheme 15B). Therefore, electrochemistry, which uses electrons as traceless redox equivalents at a precisely controlled potential, allowed us to explore new chemical space and provide solutions to pertinent synthetic challenges.
Scheme 15.
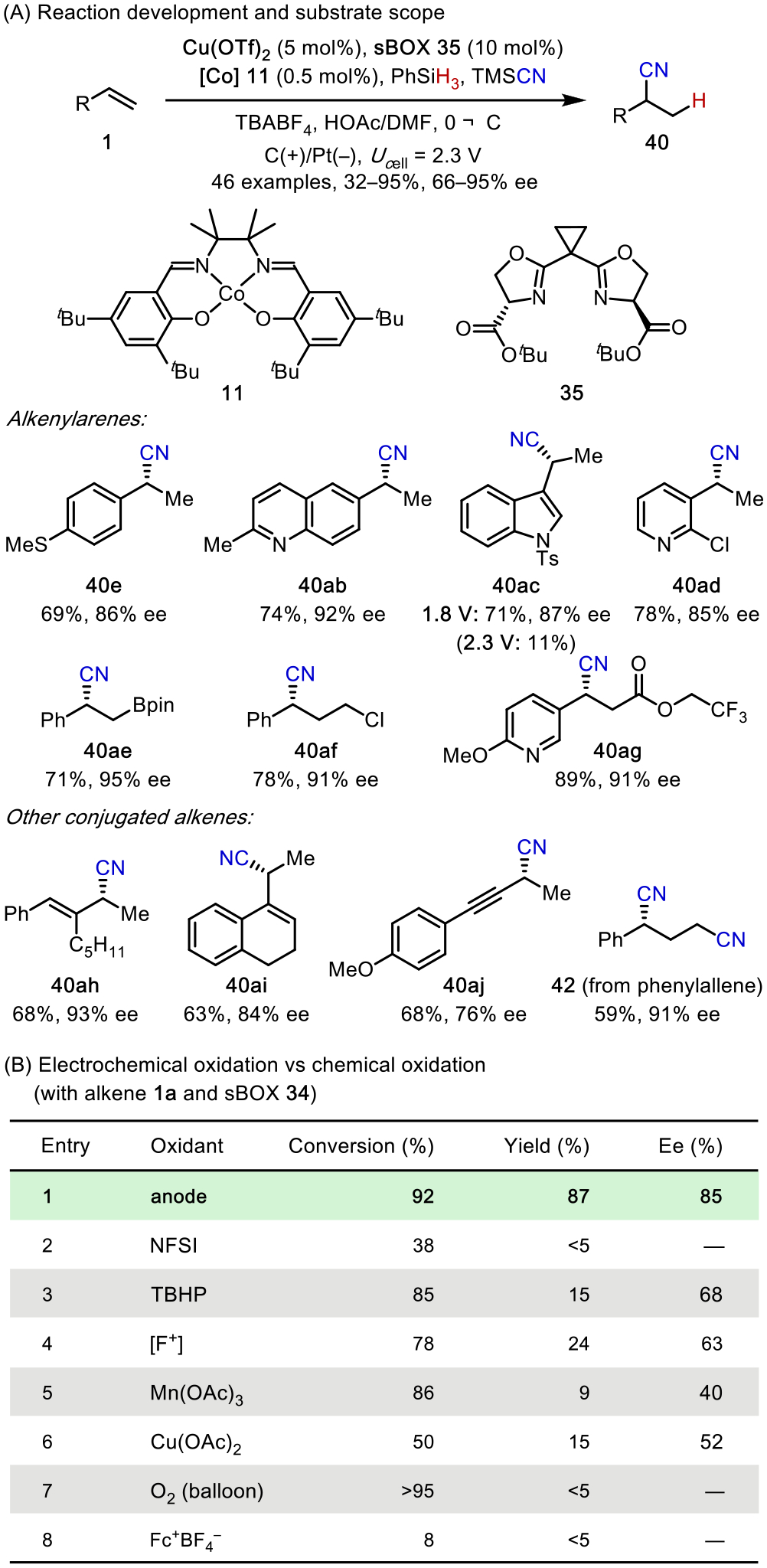
Enantioselective Hydrocyanation: Reaction Development and Comparison with Chemical Oxidation
Finally, we employed DFT computation to understand the mechanism of enantio-induction by Cu catalysts with sBOX ligands (Figure 3). In contrary to canonical BOX ligands that rely predominantly on steric interactions to impart stereocontrol,51 sBOX ligands with their second-sphere ester groups employ a combination of repulsive and attractive noncovalent interactions to achieve high enantioselectivity. During the enantio-determining C-CN formation, the acidic α-H of an ester group on the ligand engages in a C–H···π interaction with the aryl group of the alkene substrate. This interaction dictates the geometry of both major and minor transition states (TSR and TSS, respectively) by positioning the alkyl substituent on the substrate toward the ester group on the opposite half of the catalyst, which leads to more severe steric interactions in TSS than TSR We hypothesize that this combination of attractive and repulsive interactions allows sBOX ligands to achieve higher levels of enantio-induction than the more common BOX ligands.
Figure 3.
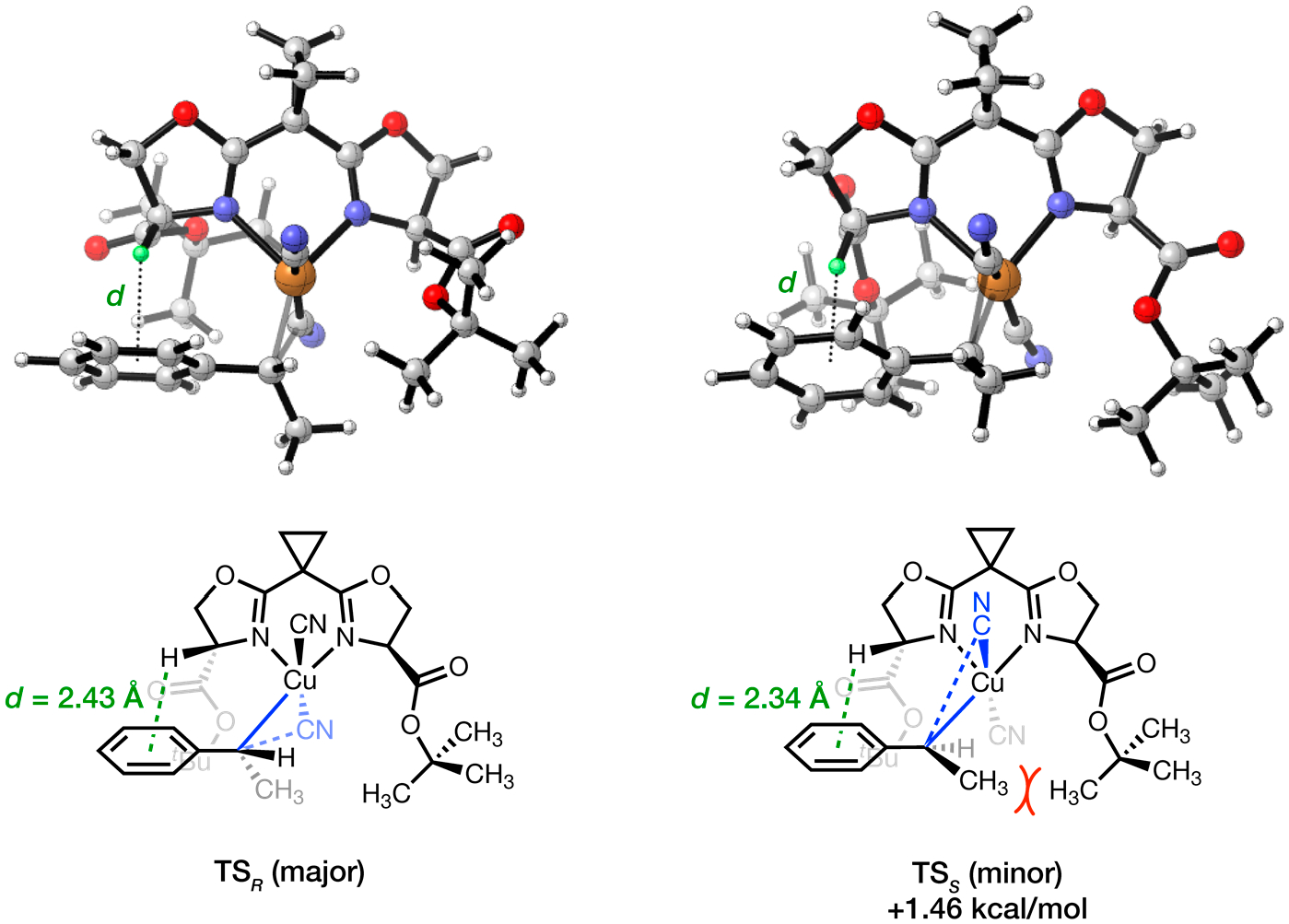
Enantioselective hydrocyanation: stereochemical model using sBOX ligands.
5. OUTLOOK
Recent developments in our lab in the area of alkene difunctionalization have demonstrated the power of Type II electrocatalysts to generate diverse radical intermediates under mild conditions. These electrocatalytic reactions often exhibit exceptionally broad substrate scope and avoid the use of stoichiometric oxidants. Exploring mechanistic design principles such as anodically coupled electrolysis allowed us to deconvolute complex reactions into modular components that can be easily modified to achieve desired transformations. Utilizing the persistent radical effect, we have gained access to a variety of vicinally difunctionalized and hydrofunctionalized products from simple alkenes by employing appropriate electrocatalysts and reagents. We also achieved highly enantioselective electrocatalysis with the development of new chiral ligands. We anticipate that future development of new electrocatalysts and reaction strategies will continue to power the discovery of new transformations and solve a multitude of synthetic problems that are challenging to existing redox strategies.
ACKNOWLEDGMENTS
Financial support is provided by Cornell University and NIGMS (R01GM130928). We are especially grateful for our colleagues and collaborators who made much of the work discussed in this article possible. S.L. acknowledges the Sloan Foundation for a Sloan Research Fellowship. We dedicate this article to Prof. Eric Jacobsen on the occasion of his 60th birthday.
Biographies
Juno C. Siu was born in the outskirts of Hong Kong where he developed his passion for green chemistry. He received his M.Sci. in Chemistry from University College London in 2016 where he worked with Prof. Darren Caruana on the development of organic redox flow batteries. He then moved to Cornell University to join Prof. Song Lin’s group as one of the group’s founding batch of graduate students. His current research interests revolve around electrochemically driven radical reactions.
Niankai Fu obtained his B.S. at Hubei University in 2009 and Ph.D. in organic chemistry under the guidance of Prof. Sanzhong Luo at the Institute of Chemistry, Chinese Academy of Sciences in 2014. In July 2016, he began his postdoctoral appointment with Prof. Song Lin at Cornell University where his research is focused on synthetic electrocatalysis.
Song Lin is an Assistant Professor of Chemistry and Howard Milstein Faculty Fellow at Cornell University. He obtained B.S. at Peking University in 2008 and Ph.D. at Harvard University, where he carried out graduate research with Prof. Eric Jacobsen. After postdoctoral training with Prof. Christopher Chang at UC Berkeley, he joined Cornell University as a faculty member in 2016. The Lin Laboratory is interested in developing new catalytic strategies for organic reaction discovery with a particular emphasis on electrochemistry and radical catalysis.
Footnotes
The authors declare no competing financial interest.
DEDICATION
Dedicated to the memory of Prof. Jun-ichi Yoshida.
REFERENCES
- (1).Moissan H Action d’un courant électrique sur l’acidefluorhydriqueanhydre. Comptesrendushebdomadaires des séances de l’Académie des sciences 1886, 102, 1543–1544. [Google Scholar]
- (2).Davy HI The Bakerian Lecture, on Some New Phenomena of Chemical Changes Produced by Electricity, Particularly the Decomposition of the Fixed Alkalies, and the Exhibition of the New Substances Which Constitute Their Bases; and on the General Nature of Alkaline Bodies. Phil. Trans. R. Soc. London 1808, 98, 1–44. [Google Scholar]
- (3).Ehl RG; Ihde AJ Faraday’s electrochemical laws and the determination of equivalent weights. J. Chem. Educ 1954, 31 (5), 226–232. [Google Scholar]
- (4).Kolbe H Zersetzung der Valeriansäuredurch den elektrischen Strom. Annalen der Chemie und Pharmacie. 1848, 64, 339–341. [Google Scholar]
- (5).(a) Moeller KD Synthetic Applications of Anodic Electrochemistry. Tetrahedron 2000, 56, 9527–9554. [Google Scholar]; (b) Yoshida J; Kataoka K; Horcajada R; Nagaki A Modern Strategies in Electroorganic Synthesis. Chem. Rev 2008, 108, 2265–2299. [DOI] [PubMed] [Google Scholar]
- (6).(a) Yan M; Kawamata Y; Baran PS Synthetic Organic Electrochemical Methods Since 2000: On the Verge of a Renaissance. Chem. Rev 2017, 117, 13230–13319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Botte GG Electrochemical Manufacturing in the Chemical Industry. Electrochem. Soc. Interface 2014, 49–55. [Google Scholar]; (c) Waldvogel SR; Janza B Renaissance of Electrosynthetic Methods for the Construction of Complex Molecules. Angew. Chem., Int. Ed 2014, 53, 7122–7123. [DOI] [PubMed] [Google Scholar]; (d) Sperry JB; Wright DL The Application of Cathodic Reductions and Anodic Oxidations in the Synthesis of Complex Molecules. Chem. Soc. Rev 2006, 35, 605–621. [DOI] [PubMed] [Google Scholar]; (e) Kärkäs MK Electrochemical strategies for C-H functionalization and C-N bond formation. Chem. Soc. Rev 2018, 47, 5786–5865. [DOI] [PubMed] [Google Scholar]
- (7).(a) Sowell CG; Wolin RL; Little RD Electroreductive Cyclization Reactions. Stereoselection, Creation of Quaternary Centers in Bicyclic Frameworks, and a Formal Total Synthesis of Quadrone. Tetrahedron Lett. 1990, 31, 485–488. [Google Scholar]; (b) Corey EJ; Sauers RR The Synthesis of Pentacyclosqualene (8,8′-Cycloönocerene) and the α- and β-Onoceradienes. J. Am. Chem. Soc 1959, 81, 1739–1743. [Google Scholar]; (c) Mihelcic J; Moeller KD Anodic Cyclization Reactions: the Total Synthesis of Alliacol A. J. Am. Chem. Soc 2003, 125, 36–37. [DOI] [PubMed] [Google Scholar]; (d) Rosen BR; Werner EW; O’Brien AG; Baran PS Total Synthesis of Dixiamycin B by Electrochemical Oxidation. J. Am. Chem. Soc 2014, 136, 5571–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Watkins BF; Behling JR; Kariv E; Miller LL A chiral electrode. J. Am. Chem. Soc 1975, 97, 3549–3550. [Google Scholar]; (b) Attard GA Electrochemical Studies of Enantioselectivity at Chiral Metal Surfaces. J. Phys. Chem. B 2001, 105, 3158–3167. [Google Scholar]; (c) Abe S; Nonaka T; Fuchigami T Electroorganic Reactions on Organic Electrodes.1. Asymmetric Reduction of Prochiral Activated Olefins on a Poly-Lvaline-Coated Graphite. J. Am. Chem. Soc 1983, 105, 3630–3632. [Google Scholar]; (d) Yutthalekha T; Wattanakit C; Lapeyre V; Nokbin S; Warakulwit C; Limtrakul J; Kuhn A Asymmetric Synthesis Using Chiral-Encoded Metal. Nat. Commun 2016, 7, 12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Moeller KD Using Physical Organic Chemistry to Shape the Course of Electrochemical Reactions. Chem. Rev 2018, 118, 4817–4833. [DOI] [PubMed] [Google Scholar]
- (10).(a) Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]; (b) Jutand A Contribution of Electrochemistry to Organometallic Catalysis. Chem. Rev 2008, 108, 2300–2347. [DOI] [PubMed] [Google Scholar]
- (11).(a) Sauer GS; Lin S An Electrocatalytic Approach to the Radical Difunctionalization of Alkenes. ACS Catal. 2018, 8, 5175–5187. [Google Scholar]; (b) Sauermann N; Meyer TH; Qiu Y; Ackermann L Electrocatalytic C-H Activation. ACS Catal. 2018, 8, 7086–7103. [Google Scholar]
- (12).(a) Kärkäs MD; Verho O; Johnston EV; Åkermark B Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev 2014, 114, 11863–12001. [DOI] [PubMed] [Google Scholar]; (b) Thoi VS; Sun Y; Long JR; Chang CJ Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev 2013, 42, 2388–2400. [DOI] [PubMed] [Google Scholar]
- (13).Appel AM; Bercaw JE; Bocarsly AB; Dobbek H; Dubois DL; Dupuis M; Ferry JG; Fujita E; Hille R; Kenis PJA; Kerfeld CA; Morris RH; Peden CHF; Portis AR; Ragsdale SW; Rauchfuss TB; Reek JNH; Seefeldt LC; Thauer RK; Waldrop GL Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev 2013, 113, 6621–6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For an example, see:; Costentin C; Drouet S; Robert M; Saveant J-M A Local Proton Source Enhances CO2 Electroreduction to CO by a Molecular Fe Catalyst. Science 2012, 338, 90–94. [DOI] [PubMed] [Google Scholar]
- (15).Hartwig JF Evolution of a fourth generation catalyst for the amination and thioetherification of aryl halides. Acc. Chem. Res 2008, 41, 1534–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ruiz-Castillo P; Buchwald SL Applications of palladium-catalyzed C-N cross-coupling reactions. Chem. Rev 2016, 116, 12564–12649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bräse S; Banert K Organic Azides: Syntheses and Applications; Wiley: Chichester, U.K., 2009. [Google Scholar]
- (17).(a) Ward GA; Wright CMJ Investigation of the anodic oxidation of azide ion on platinum electrodes. J. Electroanal. Chem 1964, 8, 302–309. [Google Scholar]; (b) Bryant JI; Kemp MD Simultaneous Polarographic Determination of Lead and Azide Ions of Lead Azides in Aqueous Media. Anal. Chem 1960, 32, 758–760. [Google Scholar]; (c) Alfassi ZB; Harriman A; Huie RE; Mosseri S; Neta P The redox potential of the azide/azidyl couple. J. Phys. Chem 1987, 91, 2120–2122. [Google Scholar]
- (18).Workentin M; Wagner B; Lusztyk J; Wayner D Azidyl Radical Reactivity. N6.bul.- as a Kinetic Probe for the Addition Reactions of Azidyl Radicals with Olefins. J. Am. Chem. Soc 1995, 117, 119–126. [Google Scholar]
- (19).For representative previous examples on alkene diazidation using chemical oxidants, see:; (a) Yuan YA; Lu DF; Chen YR; Xu H Iron-Catalyzed Direct Diazidation for a Broad Range of Olefins. Angew. Chem., Int. Ed 2016, 55, 534–538. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Snider BB; Lin H An Improved Procedure for the Conversion of Alkenes and Glycals to 1,2-Diazides UsingMn(OAc)3·2H2O in Acetonitrile Containing Trifluoroacetic Acid. Synth. Commun 1998, 28, 1913–1922. [Google Scholar]; (c) Magnus P; Roe MB; Hulme C New trialkylsilyl enol ether chemistry: direct 1,2-bis-azidonation of triisopropylsilyl enol ethers: an azido-radical addition process promoted by TEMPO. J. Chem. Soc., Chem. Commun 1995, 263–265. [Google Scholar]; (d) Fristad WE; Brandvold TA; Peterson JR; Thompson SR Conversion of alkenes to 1,2-diazides and 1,2-diamines. J. Org. Chem 1985, 50, 3647–3649. [Google Scholar]
- (20).(a) Lucet D; Le Gall TL; Mioskowski C The Chemistry of Vicinal Diamines. Angew. Chem., Int. Ed 1998, 37, 2580–2627. [DOI] [PubMed] [Google Scholar]; (b) Cardona F; Goti A Metal-catalysed 1,2-diamination reactions. Nat. Chem 2009, 1, 269–275. [DOI] [PubMed] [Google Scholar]
- (21).Schafer H Oxidative Addition of the Azide Ion to Olefins. A Simple Route to Diamines. Angew. Chem., Int. Ed. Engl 1970, 9, 158–159. [Google Scholar]
- (22).(a) Fischer H The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev 2001, 101, 3581–3610. [DOI] [PubMed] [Google Scholar]; (b) Studer A The Persistent Radical Effect in Organic Synthesis. Chem. - Eur. J 2001, 7, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (23).Huang X; Bergsten TM; Groves JT Manganese-Catalyzed Late-Stage Aliphatic C-H Azidation. J. Am. Chem. Soc 2015, 137, 5300–5303. [DOI] [PubMed] [Google Scholar]
- (24).Bunescu A; Ha TM; Wang Q; Zhu J Copper-Catalyzed Three-Component Carboazidation of Alkenes with Acetonitrile and Sodium Azide. Angew. Chem., Int. Ed 2017, 56, 10555–10558. [DOI] [PubMed] [Google Scholar]
- (25).(a) Fu N; Sauer GS; Saha A; Loo A; Lin S Metalcatalyzed electrochemical diazidation of alkenes. Science 2017, 357, 575–579. [DOI] [PubMed] [Google Scholar]; (b) Fu N; Sauer GS; Lin S A general, electrocatalytic approach to the synthesis of vicinal diamines. Nat. Protoc 2018, 13, 1725–1743. [DOI] [PubMed] [Google Scholar]; (c) Parry JB; Fu N; Lin S Electrocatalytic Difunctionalization of Olefins as a General Approach to the Synthesis of Vicinal Diamines. Synlett 2018, 29, 257–265. [Google Scholar]
- (26).(a) Snider BB Manganese(III)-Based Oxidative Free-Radical Cyclizations. Chem. Rev 1996, 96, 339–364. [DOI] [PubMed] [Google Scholar]; (b) Fristad WE; Brandvold TA; Peterson JR; Thompson SR Conversion of alkenes to 1,2-diazides and 1,2-diamines. J. Org. Chem 1985, 50, 3647–3649. [Google Scholar]; (c) Snider BB; Duvall JR Termination of Mn(III)- Based Oxidative Cyclizations by Trapping with Azide. Org. Lett 2004, 6, 1265–1268. [DOI] [PubMed] [Google Scholar]
- (27).Hirel C; Vostrikova KE; Pecaut J; Ovcharenko VI; Rey P Nitronyl and Imino Nitroxides: Improvement of Ullman’s Procedure and Report on a New Efficient Synthetic Route. Chem. - Eur. J 2001, 7, 2007–2014. [DOI] [PubMed] [Google Scholar]
- (28).Siu JC; Sauer GS; Saha A; Macey RL; Fu N; Chauvire T; Lancaster KL; Lin S Electrochemical Azidooxygenation of Alkenes Mediated by a TEMPO-N3Charge-Transfer Complex. J. Am.Chem. Soc 2018, 140, 12511–12520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Samuni A; Goldstein S; Russo A; Mitchell J; Krishna M; Neta P Kinetics and Mechanism of Hydroxyl Radical and OH-Adduct Radical Reactions with Nitroxides and with Their Hydroxylamines. J. Am. Chem. Soc 2002, 124, 8719–8724. [DOI] [PubMed] [Google Scholar]
- (30).Sandford C; Edwards MA; Klunder KJ; Hickey DP; Li M; Barman K; Sigman MS; White HS; Minteer SD A synthetic chemist’s guide to electroanalytical tools for studying reaction mechanisms. Chem. Sci 2019, 10, 6404–6422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Siu JC; Parry JB; Lin S Aminoxyl-Catalyzed Electrochemical Diazidation of Alkenes Mediated by a Metastable Charge-Transfer Complex. J. Am. Chem. Soc 2019, 141, 2825–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fu N; Sauer GS; Lin S Electrocatalytic Radical Dichlorination of Alkenes with Nucleophilic Chlorine Sources. J. Am. Chem. Soc 2017, 139, 15548–15553. [DOI] [PubMed] [Google Scholar]
- (34).Siu JC; Lin S Unpublished results.
- (35).Beitz T; Bechmann W; Mitzner R Investigations of reactions of selected azaarenes with radicals in water. 2. Chlorine and bromine radicals. J. Phys. Chem. A 1998, 102, 6766–6771. [Google Scholar]
- (36).Ye K; Pombar G; Fu N; Sauer GS; Keresztes I; Lin S Anodically Coupled Electrolysis for the Heterodifunctionalization of Alkenes. J. Am. Chem. Soc 2018, 140, 2438–2441. [DOI] [PubMed] [Google Scholar]
- (37).(a) Frontana-Uribe BA; Little R; Ibanez JG; Palma A; Vasquez-Medrano RC Organic Electrosynthesis: A Promising Green Methodology in Organic Chemistry. Green Chem. 2010, 12, 2099–2119. [Google Scholar]; (b) Llorente MJ; Nguyen BH; Kubiak CP; Moeller KD Paired Electrolysis in the Simultaneous Production of Synthetic Intermediates and Substrates. J. Am. Chem. Soc 2016, 138, 15110–15113. [DOI] [PubMed] [Google Scholar]; (c) Zhao H-B; Xu P; Song J; Xu H-C Cathode Material Determines Product Selectivity for Electrochemical C-H Functionalization of Biaryl Ketoximes. Angew. Chem., Int. Ed 2018, 57, 15153–15156. [DOI] [PubMed] [Google Scholar]
- (38).Ye K-Y; Song Z; Sauer GS; Harenberg JH; Fu N; Lin S Synthesis of Chlorotrifluoromethylated Pyrrolidines by Electrocatalytic Radical Ene-Yne Cyclization. Chem. - Eur. J 2018, 24, 12274–12279. [DOI] [PubMed] [Google Scholar]
- (39).Fu N; Shen Y; Allen AR; Song L; Ozaki A; Lin S Mn-catalyzed Electrochemical Chloroalkylation of Alkenes. ACS Catal. 2019, 9, 746–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lu L; Fu N; Lin S Three-Component Chlorophosphinoylation of Alkenes via Anodically Coupled Electrolysis. Synlett 2019, 30, 1199–1203. [Google Scholar]
- (41).Lin Q; Li L; Luo S Asymmetric Electrochemical Catalysis. Chem. - Eur. J 2019, 25, 10033. [DOI] [PubMed] [Google Scholar]
- (42).(a) Nguyen BH; Redden A; Moeller KD Sunlight, electrochemistry, and sustainable oxidation reaction. Green Chem. 2014, 16, 69–72. [Google Scholar]; (b) Torii S; Liu P; Tanaka H Electrochemical Os-Catalyzed Asymmetric Dihydroxylation of Olefins with Sharpless’ Ligand. Chem. Lett 1995, 24, 319–320. [Google Scholar]; (c) Tanaka H; Kuroboshi M; Takeda H; Kanda H; Torii S Electrochemical asymmetric epoxidation of olefins by using an optically active Mn-salen complex. J. Electroanal. Chem 2001, 507, 75–81. [Google Scholar]
- (43).Fu N; Song L; Liu J; Shen Y; Siu JC; Lin S New Bisoxazoline Ligands Enable Enantioselective Electrocatalytic Cyanofunctionalization of Vinylarenes. J. Am. Chem. Soc 2019, 141, 14480–14485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Wang F; Chen P; Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 2018, 51, 2036–2046. [DOI] [PubMed] [Google Scholar]
- (45).Liu W; Groves JT Manganese-catalyzed C-H Halogenation. Acc. Chem. Res 2015, 48, 1727–1735. [DOI] [PubMed] [Google Scholar]
- (46).Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).(a) Merchant RR; Oberg KM; Lin Y; Novak AJE; Felding J; Baran PS Divergent Synthesis of Pyrone Diterpenes via Radical Cross Coupling. J. Am. Chem. Soc 2018, 140, 7462–7465. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Seavill P; Holt KB; Wilden JD Electrochemical preparation and applications of copper(i)acetylides: a demonstration of how electrochemistry can be used to facilitate sustainability in homogeneous catalysis. Green Chem. 2018, 20, 5474–5478. [Google Scholar]; (c) Tsuchida K; Kochi T; Kakiuchi F Copper-Catalyzed Electrochemical Chlorination of1,3-Dicarbonyl Compounds Using Hydrochloric Acid. Asian J. Org. Chem 2013, 2, 935. [Google Scholar]
- (48).(a) Saha B; RajanBabu TV Nickel(0)-catalyzed asymmetric hydrocyanation of 1,3-dienes. Org. Lett 2006, 8, 4657–4659. [DOI] [PubMed] [Google Scholar]; (b) RajanBabu TV; Casalnuovo AL Tailored ligands for asymmetric catalysis. J. Am. Chem. Soc 1992, 114, 6265–6266. [Google Scholar]; (c) Wilting J; Janssen M; Müller C; Vogt D The enantioselective step in the Nickelcatalyzed hydrocyanation of 1,3-cyclohexadiene. J. Am. Chem. Soc 2006, 128, 11374–11375. [DOI] [PubMed] [Google Scholar]; (d) Falk A; Göderz A-L; Schmalz H-G Enantioselective nickel-catalyzed hydrocyanation of vinylarenes using chiral phosphine-phosphite ligands and TMS-CN as a source of HCN. Angew. Chem., Int. Ed 2013, 52, 1576–1580. [DOI] [PubMed] [Google Scholar]; During the preparation of our manuscript (ref 49), a clever strategy entailing tandem Rh-catalyzed hydroformylation and functional group transformation was reported:; (e) Li X; You C; Yang J; Li S;Zhang D; Lv H; Zhang X Asymmetric Hydrocyanation of Alkenes without HCN. Angew. Chem., Int. Ed 2019, 58, 10928–10931. [DOI] [PubMed] [Google Scholar]
- (49).Song L; Fu N; Ernst BG; Lee WH; Frederick MO; DiStasio RA; Lin S Dual Electrocatalysis Enables Enantioselective Hydrocyanation of Conjugated Alkenes. ChemRxiv 2019, DOI: 10.26434/chemrxiv.9784625.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).(a) Crossley SWM; Martinez RM; Obradors C; Shenvi RA Mn, Fe, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chem. Rev 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green SA; Crossley SWM; Matos JLM; Vasquez-Cespedes S; Shevick SL; Shenvi RA The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res 2018, 51, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).(a) Cornejo A; Fraile JM; García JI; Gil MJ; Martínez-Merino V; Mayoral JA; Salvatella L Asymmetric versus C2-Symmetric Ligands: Origin of the Enantioselectivity in RutheniumPybox-Catalyzed Cyclopropanation Reactions. Angew. Chem., Int. Ed 2005, 44, 458–461. [DOI] [PubMed] [Google Scholar]; (b) García JI; Jimenéz-Osés G; Martínez-Merino V; Mayoral JA; Pires E; Villalba I QM/MM Modeling of Enantioselective Pybox-Ruthenium- and Box-Copper-Catalyzed Cyclopropanation Reactions: Scope, Performance, and Applications to Ligand Design. Chem. - Eur. J 2007, 13, 4064–4073. [DOI] [PubMed] [Google Scholar]