

Abstract
Introduction
Insulin regulates renal glucose production and utilization; both these fluxes are increased in type 2 diabetes (T2D). Whether insulin also controls urinary glucose excretion is not known.
Methods
We applied the pancreatic clamp technique in 12 healthy subjects and 13 T2D subjects. Each participant received a somatostatin infusion and a variable glucose infusion to achieve (within 1 hour) and maintain glycemia at 22 mmol/L for 3 hours; next, a constant insulin infusion (240 pmol/min/kg) was added for another 3 hours. Urine was collected separately in each period for glucose and creatinine determination.
Results
During saline, glucose excretion was lower in T2D than controls in absolute terms (0.49 (0.32) vs 0.69 (0.18) mmol/min, median (IQR), p=0.01) and as a fraction of filtered glucose (16.2 (6.4) vs 19.9 (7.5)%, p<0.001). With insulin, whole-body glucose disposal rose more in controls than T2D (183 (48) vs 101 (48) µmol/kgFFM/min, p<0.0003). Insulin stimulated absolute and fractional glucose excretion in controls (p<0.01) but not in T2D. Sodium excretion paralleled glucose excretion. In the pooled data, fractional glucose excretion was directly related to whole-body glucose disposal and to fractional sodium excretion (r=0.52 and 0.54, both p<0.01). In another group of healthy controls, empagliflozin was administered before starting the pancreatic clamp to block sodium-glucose cotransporter 2 (SGLT2). Under these conditions, insulin still enhanced both glucose and sodium excretion.
Conclusions
Acute exogenous insulin infusion jointly stimulates renal glucose and sodium excretion, indicating that the effect may be mediated by SGLTs. This action is resistant in patients with diabetes, accounting for their increased retention of glucose and sodium, and is not abolished by partial SGLT2 inhibition by empagliflozin.
Keywords: insulin, kidney, glucose transport, sodium glucose cotransporter
Significance of this study.
What is already known about this subject?
Insulin regulates renal glucose production and utilization; this action is resistant in type 2 diabetes. Whether insulin also controls urinary glucose excretion is not known.
What are the new findings?
Exogenous insulin infusion under clamped hyperglycemic conditions enhances glycosuria; this effect is lost in patients with diabetes.
As insulin-induced glycosuria is paralleled by enhanced sodium excretion, inhibition of sodium-glucose cotransporters (SGLT) is a potential mechanism.
Resistance to this renal tubular action of insulin might contribute to the hyperglycemia and sodium retention of type 2 diabetes.
How might these results change the focus of research or clinical practice?
Results demonstrated an insulin effect on urinary glucose excretion and point to a possible interference with the renal glucose transport through the SGLT system. Thus, new studies must investigate the molecular basis of this insulin action and the possible therapeutic interactions between insulin and SGLT2 inhibitors.
Introduction
The kidneys partake of glucose metabolism both by releasing glucose into the circulation (via gluconeogenesis, principally in the cortex) and by taking up glucose for energy production and glycogen storage (predominantly in the medulla).1–3 Together, the kidneys account for ~15% of whole-body glucose disposal (WBGD); both de novo production and utilization of glucose are regulated by insulin.4 5 The bulk of renal energy expenditure takes place in the proximal tubules, which are charged with the reabsorption of, among other solutes, ~200 g of glucose and over 600 g of sodium (Na+) daily delivered through glomerular filtration. Active reabsorptive processes are energized by the Na+-K+-ATPase (Na-K pump) located in the basolateral membrane, the foldings of which are closely surrounded by mitochondria.6 Sodium-glucose cotransporters (SGLT) on the luminal surface of tubular cells—SGLT2 in the S1 and S2 segments, SGLT1 in the S3 segment—execute reabsorption along the sodium gradient created by the Na-K pump. Normally, this process takes place mostly through SGLT2 (with a 1:1 sodium:glucose stoichiometry).7 For sodium, proximal tubular reabsorption is only 4.4% of the filtered load, and its energy cost is ~0.4 mol/day of adenosine triphosphate (ATP) (200 g/day, or 12% of whole kidney ATP consumption).8 Several other sodium transporters, located along the entire nephron, as well as paracellular ion diffusion ensure that, under normal circumstances, urinary sodium excretion eventually balances out sodium intake.9 In contrast, as there are no other major glucose transporters downstream to the proximal tubule, virtually all filtered glucose is reabsorbed proximally until the glucose transport maximum is exceeded. Coupling of sodium and glucose transport therefore is incomplete and topographically confined.
Whereas the actions of insulin on renal glucose metabolism have been rather extensively investigated in humans,1–5 little is known about the effects of insulin on renal glucose handling.10 An early study in humans suggested an inhibitory influence of insulin on glucose reabsorption.11 In that study, glycemia was raised—by primed intravenous glucose infusions— to 500–800 mg/dL (28–44 mmol/L) for three consecutive 20 min periods for the measurement of glucose transport maximum (TmG), after which insulin was infused at 2 U/min (~7 nmol/min/m2 of body surface area, presumably yielding plasma insulin concentrations >20 000 pmol/L12) for another 20 min. Under these conditions, a 10% reduction in TmG was recorded in 12 patients with insulin-treated diabetes. This observation, however, has not been followed up on either in animal models or in humans. The present study was therefore designed to determine the physiological effect of insulin on renal glucose reabsorption in normal subjects and in patients with type 2 diabetes (T2D) under steady-state conditions of plasma glucose and insulin concentrations (with the use of the pancreatic clamp technique). The concomitant effects on sodium and potassium excretion were also measured. Since SGLT2 inhibition effectively reduces glucose reabsorption, in a separate series of experiments in healthy subjects we tested whether any effect of insulin on renal glucose handling was mediated through SGLT; this was done by applying physiological hyperinsulinemia on the background of acute, partial SGLT2 blockade (using the highly selective SGLT2 inhibitor, empagliflozin13).
Methods
Subjects
The study was carried out at two centers, University of Pisa, Italy, and University of Campinas, Brazil. Thirteen patients with T2D under lifestyle treatment or stable doses of oral hypoglycemic drugs were recruited from the outpatient clinics; inclusion criteria were HbA1c 6.5%–10.0%, and not on treatment with insulin, thiazolidinediones or SGLT2 inhibitors. All glucose-lowering drugs were stopped 48 hours before the study. Twenty healthy subjects volunteered to serve as a control group. Inclusion criteria for all participants were: ages 18–60 years, body mass index <40 kg/m2 stable during 3 months before screening, estimated glomerular filtration rate (eGFR) ≥60 mL/min/1.73 m2, absence of clinical evidence of cancer, chronic inflammatory diseases, major organ (liver, kidney, heart, pulmonary) failure or previous bariatric surgery. Excluding diabetes, all subjects were in good health as assessed by screening laboratory measurements, ECG, medical history, and physical examination. All participants were on a free diet, and none was taking drugs that would alter renal Na handling. Anthropometric and metabolic characteristics of the study participants are described in online supplementary table 1.
bmjdrc-2020-001178supp004.pdf (98.5KB, pdf)
Study design
All experiments were carried out in the metabolic unit with the use of the pancreatic clamp technique. In the main protocol, after an overnight (10–12 hours) fast an indwelling catheter was placed in an antecubital vein for the infusion of test substances; another catheter was placed retrograde at the hand dorsum for blood drawing while the hand was heated at 55°C to allow arterialization of venous blood. From time −60 min until the end (+360 min), somatostatin was infused at a rate of 400 µg/hour to paralyze endogenous insulin secretion. Simultaneously, a glucose infusion was initiated at a rate calculated to raise plasma glucose concentrations to ~22 mmol/L in ~60 min (equilibration period); thereafter, the glucose infusion was frequently adjusted to clamp glycemia at this plateau for the following 6 hours using an ad hoc developed algorithm.14 After a control period of 3 hours (saline period, 0–180 min) an infusion of regular insulin was started with a priming dose of 960 pmol/min/m2 for 6 min followed by a continuous infusion rate of 240 pmol/min/m2 (insulin period, 180–360 min). This protocol was completed by all patients with T2D and 12 controls (online supplementary table 1). In another healthy volunteer, the main protocol was applied by inverting the order of saline and insulin infusion.
Empagliflozin protocol (Empa)
In seven healthy volunteers (with anthropometric and biochemical characteristics similar to those of the main control group, data not shown), the pancreatic clamp was modified as follows: (A) empagliflozin (25 mg orally) was administered at time −120 min, thereby allowing for a baseline empagliflozin period between −120 and −60 min, and (B) plasma glucose concentrations were clamped at 10 mmol/L instead of 22 mmol/L. The saline and insulin periods were the same as in the main protocol.
Blood and urine sampling
Plasma glucose was measured at 10 min intervals; samples for insulin, sodium and potassium measurements were collected every hour throughout the study period. Accurate urine collection was performed separately during the Empa (−120 to −60 min), equilibration (−60 to 0 min), saline (0–180 min), and insulin (180–360 min) periods by spontaneous micturition. Overall fluid balance ([saline+glucose solutions]−[urine output+blood draw]) averaged 23±603 mL in the control and 24±916 mL in the T2D subjects of the main protocol.
Measurements
Fat-free mass (FFM) was evaluated using a Body Composition Analyzer Model TB-300 (Tanita, Japan). Bedside plasma glucose concentrations were measured by the glucose oxidase technique (on a YSI 2300 Stat Plus Glucose Analyzer, OH, or Beckman Glucose Analyzers, Fullerton, CA). Urine glucose concentrations were determined (using Beckman Glucose Analyzers), plasma insulin was measured by an electrochemiluminescence assay (COBAS e411, Roche, Indianapolis, USA), plasma and urine creatinine were measured by colorimetry by the modified Jaffe method (Beckman Glucose Analyzers). Plasma and urinary sodium and potassium were assayed by conductivity using an ion-selective electrode method (Beckman Glucose Analyzers).
Calculations
Areas under time-concentration curves (AUC) were calculated by the trapezium rule over the saline (60–180 min) and insulin (240–360) periods; mean concentrations were then calculated by dividing AUCs by the corresponding time intervals. During the clamp, the exogenous glucose infusion rate (GIR) was corrected for the plasma glucose changes by assuming a glucose distribution space of 200 mL/kg body weight. Given the very high-target glucose concentrations, endogenous glucose production was assumed to be completely suppressed throughout the clamp.15 Whole-body tissue glucose disposal was obtained as the difference between GIR and urinary glucose excretion over corresponding time intervals. Urine flow rate was obtained as the ratio of urine volume to time interval of collection. Urinary solute excretion rate was obtained as the product of urine flow rate and urine solute concentration. Solute filtration rate was obtained as the product of creatinine clearance (in mL/min) and plasma solute concentration. Solute reabsorption was calculated as the difference between filtered load and urinary excretion; fractional solute excretion was calculated as the ratio of urinary excretion to filtered load.
Fluid balance was calculated as follows: ([intravenous glucose+oral water+intravenous saline]−[diuresis+insensible water loss]), separately for the initial hour, the 3-hour saline infusion and the 3-hour insulin infusion period. Insensible water loss was estimated as 0.5 mL/min/kg.16
Statistics
Data are represented as mean±SD or median (IQR) for normally or non-normally distributed variables, respectively. Group differences between controls and T2D subjects were tested by the unpaired t-test or the Mann-Whitney test. Within each group, comparison of saline and insulin periods was performed using Wilcoxon signed-rank test. Paired data were also analyzed across group and period using two-way analysis of variance for repeated measures with rank transformation. Simple and multiple regressions were carried out by standard methods. Statistical analyses were performed using Statistical Analysis System for Windows V.9.4 (SAS Institute). A p≤0.05 was considered statistically significant.
Results
Main protocol
Plasma glucose concentrations were raised to ~22 mmol/L within 1 hour and clamped at that level for the following 6 hours, similarly during saline and insulin infusion and in T2D and controls. Plasma insulin concentrations were maintained at basal levels during saline infusion, and were raised ~12-fold during insulin infusion, with no differences between the two groups (online supplementary figure 1). During insulin infusion, WBGD rose from 36 (16) to 183 (48) in controls and from 27 (11) to 101 (48) µmol/min/kgFFM (p=0.0003 for the interaction of group and insulin) in T2D subjects (online supplementary figure 2), thereby quantifying the insulin resistance of the latter group (online supplementary table 2).
bmjdrc-2020-001178supp001.pdf (99.2KB, pdf)
bmjdrc-2020-001178supp002.pdf (75.7KB, pdf)
During hyperglycemia and saline infusion, urinary glucose excretion was lower in T2D than control subjects, both in absolute terms and as a fraction of the filtered glucose load (p<0.01 and p=0.006, respectively). During matched hyperglycemia and insulin infusion, urine output and creatinine clearance increased in both groups, whereas urinary glucose excretion increased in controls but not in T2D subjects, both in absolute (table 1) and fractional terms (figure 1).
Table 1.
Effect of insulin on the renal handling of glucose and Na+ in controls and patients with type 2 diabetes (T2D)*
| Controls | T2D | ANOVA | |||||
| Saline | Insulin | Saline | Insulin | P value† | P value‡ | P value§ | |
| Urine flow (mL/min) | 6.5 (3.6) | 7.1 (3.0) | 6.1 (2.8) | 8.1 (4.8)** | ns | 0.0012 | ns |
| Creatinine clearance (mL/min) | 163 (62) | 165 (61) | 148 (28) | 168 (32)¶ | ns | 0.0014 | ns |
| Glucose filtration rate (mmol/min) | 3.68 (1.51) | 3.55 (1.26) | 3.20 (0.64) | 3.61 (0.65)¶ | ns | – | 0.0352 |
| Glucose reabsorption (mmol/min) | 2.84 (1.25) | 2.64 (0.90) | 2.63 (0.68) | 2.94 (0.60) | ns | ns | 0.002 |
| Glucose excretion rate (mmol/min) | 0.69 (0.18) | 0.83 (0.41)¶ | 0.49 (0.32) | 0.53 (0.30) | 0.0071 | – | 0.0128 |
| Fractional glucose excretion (%) | 19.9 (7.5) | 25.4 (6.7)** | 16.2 (6.4) | 16.1 (6.4) | 0.0003 | – | 0.0064 |
| Na+ filtration rate (mEq/min) | 22.4 (11.5) | 22.6 (9.9) | 19.4 (4.7) | 22.0 (4.0)¶ | ns | – | 0.03 |
| Na+ excretion rate (mEq/min) | 0.26 (0.14) | 0.36 (0.26) | 0.17 (0.12) | 0.18 (0.07) | 0.0082 | – | (0.058) |
| Fractional Na+ excretion (%) | 1.12 (0.70) | 1.66 (1.31) | 0.88 (0.66) | 0.85 (0.24) | 0.0091 | ns | ns |
| K+ filtration rate (mEq/min) | 0.81 (0.35) | 0.71 (0.28)†† | 0.72 (0.15) | 0.74 (0.15) | ns | 0.01 | ns |
| K+ excretion rate (mEq/min) | 0.08 (0.06) | 0.04 (0.03)** | 0.06 (0.04) | 0.05 (0.01)†† | ns | 0.006 | ns |
| Fractional K+ excretion (%) | 8.4 (6.7) | 5.2 (2.0)†† | 9.1 (7.4) | 6.6 (1.5)†† | ns | 0.005 | ns |
*Entries are median (IQR).
†Group effect;
‡Period effect;
§Interaction (group×period) by repeated measures ANOVA.
¶P<0.001;
**P<0.01;
††P<0.05 for the paired comparison between saline and insulin by Wilcoxon signed-rank test in each group.
ANOVA, analysis of variance; ns, not significant.
Figure 1.
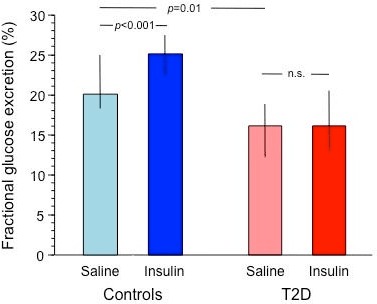
Fractional glucose excretion during saline (0–180 min) and insulin periods (180–360 min) in control subjects (blue bars) and patients with type 2 diabetes (red bars) receiving the main protocol. Plots represent median and IQR. T2D, type 2 diabetes.
In combined data analysis across group and saline/insulin infusion, glucose excretion was significantly lower in T2D than control subjects, and in T2D subjects it failed to increase with insulin. In the pooled data from both groups, insulin-induced fractional glucose excretion and WBGD were directly related to one another (figure 2A).
Figure 2.
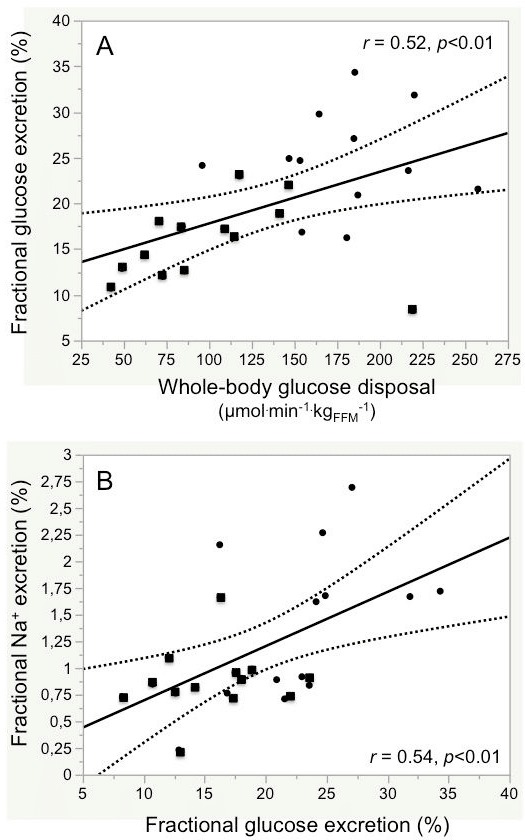
Association between fractional glucose excretion and whole-body glucose uptake (A) and fractional sodium excretion (B) in control subjects (filled circles) and patients with type 2 diabetes (filled squares) combined.
Plasma Na+ concentrations were stable in both study groups throughout the protocol table 1 (online supplementary table 2). During hyperglycemia and saline infusion, urinary Na+ excretion was lower in T2D than control subjects in absolute terms (p<0.05), and trended to be lower also as a fraction of the filtered Na+ load (p=0.07). During matched hyperglycemia and insulin infusion, Na+ excretion tended to rise in controls but not in the T2D group; across group and infusion, Na+ excretion was consistently higher in controls than T2D subjects (table 1). In the pooled data from both groups, Na+ excretion was directly associated with glucose excretion (figure 2B). Furthermore, the insulin-induced changes in Na+ excretion and fractional Na+ excretion were well correlated with the corresponding insulin-induced changes in glucose excretion (online supplementary figure 3). With high insulin, plasma K+ concentrations decreased significantly in both groups (online supplementary table 2) as did urinary K+ excretion, in absolute terms as well as a fraction of the filtered load (table 1).
bmjdrc-2020-001178supp003.pdf (902.3KB, pdf)
Empagliflozin protocol
In the seven healthy participants who received 25 mg of empagliflozin at time −120 min, plasma glucose at time 0 was unchanged but plasma insulin had dropped significantly. During the subsequent 6 hours of pancreatic clamp (plasma glucose=9.9±0.5 mmol/L), plasma insulin averaged 28 (17) pmol/L with saline infusion and 649 (318) pmol/L with exogenous insulin infusion (similar to the main protocol). WBGD rose from 13.8 (1.6) to 68.0 (20.0) µmol/min/kgFFM in the transition from saline to insulin infusion (p=0.02) (online supplementary table 3). Under fasting conditions, empagliflozin raised both glucose and Na+ excretion, in absolute as well as fractional terms. During hyperglycemia and insulin infusion, both glucose and Na+ excretion were higher than at matched hyperglycemia and saline infusion, and K+ excretion was markedly reduced (table 2).
Table 2.
Effect of insulin on the renal handling of glucose, Na+ and K+ in healthy subjects during SGLT2 inhibition*
| Baseline | Empa | P value† | Saline | Insulin | P value‡ | |
| Glucose filtration rate (mmol/min) | 0.69 (0.10) | 0.99 (0.33) | ns | 1.55 (0.45) | 1.54 (0.63) | ns |
| Glucose excretion rate (µmol/min) | 0.2 (0.3) | 146 (89) | 0.04 | 581 (212) | 588 (294) | 0.03 |
| Fractional glucose excretion (%) | 0.03 (0.04) | 20.2 (7.0) | 0.04 | 35.8 (6.7) | 37.3 (7.2) | 0.05 |
| Na+ filtration rate (mEq/min) | 24.6 (4.6) | 27.0 (9.6) | ns | 22.5 (5.8) | 23.1 (5.8) | 0.04 |
| Na+ excretion rate (mEq/min) | 0.06 (0.01) | 0.21 (0.07) | 0.04 | 0.19 (0.12) | 0.22 (0.18) | 0.05 |
| Fractional Na+ excretion (%) | 0.27 (0.05) | 0.80 (0.20) | 0.04 | 0.90 (0.42) | 0.90 (0.70) | ns |
| K+ filtration rate (mEq/min) | 0.83 (0.08) | 1.08 (0.56) | ns | 0.77 (0.30) | 0.55 (0.19) | 0.02 |
| K+ excretion rate (mEq/min) | 0.09 (0.06) | 0.11 (0.03) | ns | 0.07 (0.02) | 0.03 (0.01) | 0.03 |
| Fractional K+ excretion (%) | 10.8 (6.28) | 8.5 (5.28) | ns | 9.5 (5.1) | 4.0 (2.1) | 0.03 |
*Entries are median (IQR).
†For empa period versus baseline.
‡For the paired comparison between saline and insulin (Wilcoxon signed-rank test).
ns, not significant; SGLT2, sodium-glucose cotransporter 2.
In a multivariate regression model including plasma glucose and insulin concentrations (as continuous variables) as well as SGLT2 inhibition (as a nominal variable) from all 19 healthy participants, both absolute glucose excretion and fractional glucose excretion were independently related to insulin levels (partial r’s of 0.33 and 0.42, p=0.05 and p<0.02, respectively). The model predicts an increase in glucose excretion of 125 µmol/min—and an increase in fractional excretion from 20% to 25%—as plasma insulin is raised from 20 to 400 pmol/L.
As a time control, in one of the healthy subjects participating in the main protocol the study was repeated by omitting the exogenous insulin infusion during the 180–360 min time period. Despite a slight increase in endogenous plasma insulin concentrations during the latter time period, urinary glucose excretion fell (from 0.94 to 0.84 mmol/min) as did fractional glucose excretion (from 23.9% to 19.7%); in the standard protocol in this subject, glucose excretion had risen from 0.72 to 0.99 mmol/min and fractional glucose excretion from 17.3% to 23.8%.
In another healthy volunteer, the main protocol was applied by inverting the order of saline and insulin infusions. As plasma insulin dropped from 471 to 267 pmol/L during the second half of the clamp (180–360 min), glucose excretion decreased from 0.96 to 0.74 mmol/min while fractional glucose excretion remained constant (from 24.0% to 24.2%).
Overall fluid balance during the 8 hours of the experiment was positive in both control and T2D participants in the main protocol (+807 (981) and 1104 (738) mL, respectively, p=ns), whereas it was neutral in the empagliflozin experiment (−24 (721) mL). The change in fluid balance between the insulin and saline periods was larger in the healthy controls (+1194 [1,294] mL) than in the patients with T2D (+367 [740] mL, p=0.003), and was positive in the empagliflozin experiment (+547 (655) mL) (online supplementary table 4).
Discussion
The main finding of the present study is that under hyperglycemic conditions physiological hyperinsulinemia stimulates urinary glucose excretion in proportion to insulin sensitivity. Because the insulin-induced increment in glycosuria was paralleled by an increase in sodium excretion, the SGLT cotransporters are one potential target of this action of insulin. Because inhibition of SGLT2 with empagliflozin did not fully prevent the glycosuric effect of insulin, it is possible that insulin may inhibit SGLT1. However, since inhibition of SGLTs by empagliflozin is incomplete (averaging 30%–40% in humans13), insulin may only partially inhibit SGLT2 (or both SGLT2 and SGLT1). In either case, this cotransport system is insulin resistant in T2D. These conclusions require qualification.
First, we tested the effect of insulin under hyperglycemic conditions to ensure complete urine collection and sufficient glycosuria to allow accurate measurement of effects in vivo. In anesthetized, instrumented animals, single-nephron micropuncture can detect much smaller effects.17 Second, our study design, by allowing enough time for stabilization of glucose and insulin concentrations, created steady-state conditions of substrate and stimulus; hence, the results may quantitatively differ from those pertaining to time-varying combinations of glycemia and insulinemia, that is, during feeding. Third, chronic rather than acute hyperinsulinemia—as prevails in insulin-resistant states and obese T2D—may alter the size of the observed effect. Finally, it is possible that supraphysiological insulin levels—as used in the early study11—may overcome the insulin resistance of sodium-glucose cotransport of patients with T2D.
Several lines of established evidence cohere with the physiological mechanism here revealed. With regard to the population sample we included in this study, the current results confirm that glucose reabsorption is inherently increased in T2D, as originally reported by Farber and colleagues in 1951,11 and later documented by DeFronzo et al,18 with the use of ascending glucose infusions. Thus, in our group of relatively well-controlled patients with T2D—comparable to those in the above-mentioned studies—fractional glucose excretion was ~20% lower than in healthy controls. Not previously reported is the concomitant reduction in sodium excretion (figure 2B), which likely subtends the characteristic volume sensitivity of the blood pressure of patients with diabetes.19 Several explanations for the increased glucose reabsorption in T2D have been invoked. In human embryonic kidney (HEK) cells, chronic incubation with high glucose concentrations increased plasma membrane expression of SGLT2 in a protein kinase A-dependent way.20 In human-cultured proximal tubular cells, pharmacological insulin concentrations (1250 µUI/mL), but not glucose, increased SGLT2 expression and protein.21 In HEK cells, insulin enhanced SGLT2, but not SGLT1, glucose transport, possibly by stimulating the translocation of SGLT2 from an intracellular pool to the S1 and S2 brush border membrane.22 Renal tubular expression of SGLT2 has been reported to be increased in diabetic Akita mice,23 in diabetic rats,24 25 in human exfoliated proximal tubular epithelial cells isolated from fresh urine in four patients with diabetes,26 and in formalin-fixed paraffin-embedded (FFPE) tissue specimens from patients with diabetic nephropathy (but not from db/db mice).27 In contrast, using FFPE biopsy specimens from patients with T2D and preserved renal function, Norton et al28 and Solini et al29 did not find any increase in SGLT2 expression. On the other hand, the expression of the insulin receptor and the levels of tyrosine phosphorylated receptor are reduced in insulin-resistant rat models30 and in renal biopsy specimens from patients with diabetes.31 Notably, in the latter study downregulation of insulin receptor levels was prominent in proximal tubules. Moreover, a target of renal insulin signaling, namely the gluconeogenic enzyme phosphoenolpyruvate carboxykinase, which is localized to compartments near the apical membrane of proximal tubules, showed increased expression and activity. Thus, it is still possible that in T2D modulation of renal glucose-sodium reabsorption involves changes in the expression of the cotransporters (perhaps as a function of the clinical phenotype, ie, age, duration of disease, glycemic control, and so on). However, the present in vivo findings and the bulk of experimental evidence converge on the conclusion that the constitutively higher glucose reabsorption of patients with T2D is a functional manifestation of renal insulin resistance.
With regard to the effect of insulin, strong support is provided by the early studies of DeFronzo et al32 using single-nephron micropuncture in the dog, which showed that physiological hyperinsulinemia inhibits proximal tubular sodium reabsorption. In those experiments, the overall antinatriuretic effect of insulin was therefore ascribed to segments distal to the proximal tubule. Subsequent studies applying the euglycemic insulin clamp in healthy subjects33 34 confirmed these findings. Likewise, O’Hare et al35 found a reduction in sodium excretion during high-dose insulin clamp in healthy subjects but not in obese non-diabetic individuals.35
It must be emphasized that, despite efforts to maintain fluid balance the pancreatic clamp protocol was associated with an expansion of the extracellular fluid volume—as documented by a positive fluid balance (online supplementary table 4)—a drop in plasma Na+ from its normal levels (138–140 mEq/L), and an increase in urine output and eGFR (table 2). These hemodynamic changes can by themselves lead to an increased loss of both Na+ and glucose through the urine,36 and mask insulin-induced antinatriuresis. Furthermore, we estimated that the change in fluid balance between the insulin and saline periods was more positive in the controls than in the patients with T2D, mostly because of the higher exogenous GIR in the former than the latter. In consequence, the higher insulin-induced urinary excretion of solutes in controls than patients would not involve a hormonal effect on proximal tubule cotransporters. While this purely hemodynamic mechanism is apparently confirmed by our data (showing higher glucose/Na losses with insulin in controls but not T2D), it does not necessarily follow that the absolute and the fractional excretion of glucose or sodium should be selectively increased in insulin-sensitive subjects (table 1). At any rate, we cannot exclude that this mechanism may be responsible, in all or in part, for the observed effect of insulin under the experimental conditions of our study.
Also important is to consider that sodium-glucose cotransport on the luminal side of the proximal renal tubule is driven by the Na-K pump at the basolateral membrane.37 As insulin generally stimulates Na-K pump activity, sodium-glucose cotransport should be enhanced rather than reduced under hyperinsulinemic conditions. The situation, however, is more complex as the Na-K pump powers the activity of other sodium transporters (eg, sodium-hydrogen exchangers (NHE3), sodium-phosphate cotransporters, and so on9), which are also subject to multiple influences (eg, fluid drag38). Moreover, in diabetic rodents raised,39 40 decreased,41–43 or unchanged44 levels of Na-K pump activity have been described. Most of these studies were carried out in streptozotocin models of diabetes, but, to our knowledge, only one study used insulin-resistant rats.39 Insulin receptors are richly expressed in renal tubules, both on the basolateral and brush border membrane.30 In the work of Mikaelian et al,42 the reduction in Na-K pump activity was associated with a reduction of insulin binding to its receptor. A study using cultured human tubular cells showed a glucose effect to reduce Na-K pump activity and membrane protein.45 In cultured proximal tubular cells of rats a short exposure to insulin raised46 47 and a longer (24 hours) exposure reduced Na-K pump activity.48 Another study also demonstrated a dual time-dependent insulin action on Na-K pump activity, rising in the first 30 min, returning to preinsulin levels within 2 hours and decreasing after 48 hours of exposure.49 More in general, the activity of the Na-K pump in the proximal tubule is modulated by a number of stimulatory (low angiotensin II, glucocorticoids, α1 and ß-adrenergic agonists) and inhibitory factors (dopamine, parathyroid hormone and high angiotensin II), which could not be controlled in our experimental set-up. Therefore, from our results we cannot infer a reduced ability of insulin to act on the Na-K pump with any degree of certainty.
Acute SGLT2 inhibition with empagliflozin caused the expected increase in glucose and Na excretion, which was amplified by the subsequent induction of hyperglycemia. Under these conditions, superimposing hyperinsulinemia still led to a small increase in glucose and sodium excretion (table 2). This result is compatible with an insulin action on SGLT1 or a partial inhibitory effect on SGLT2 (or a combination of the two). However, the small number of subjects in this protocol coupled with the intrinsic intersubject variability of in vivo excretion rates (~30% in our hands) does not allow a more precise identification of the target of insulin action in the proximal tubule. On the other hand, in both protocols the changes in plasma K+ concentrations and urinary excretion rates were fully as expected given that insulin is a potent stimulus for K+ uptake into extrarenal (liver and muscle) tissues.50
In summary (figure 3), the novel finding is that exogenous insulin infusion jointly reduces the reabsorption of glucose and sodium. This action is defective in insulin-resistant patients with T2D, and functionally accounts for the increased glucose reabsorption that is typical of T2D. While this insulin effect may result from a purely hemodynamic mechanism, the present data and the bulk of previous experimental evidence support the interpretation that inhibition of SGLTs in the proximal tubule is an additional mechanism. Further studies are required to characterize the molecular basis of this insulin action.
Figure 3.
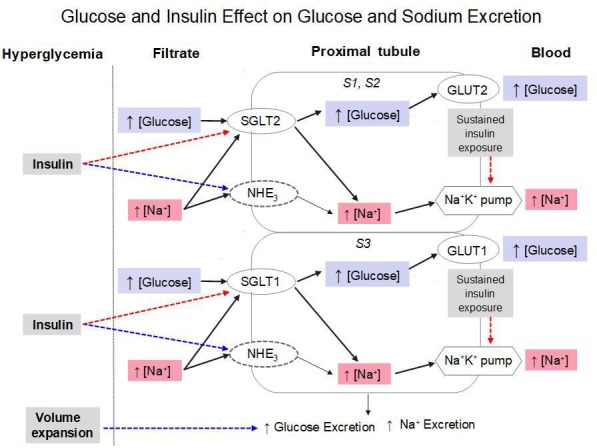
Scheme putting the study findings into context. In the presence of acute hyperglycemia, volume expansion per se can increase glucose and sodium excretion (blue dotted line). The effect of added hyperinsulinemia is to inhibit SGLT-mediated glucose and sodium reabsorption (red dotted line). Also indicated are the stimulatory action of insulin on the sodium-hydrogen cotransporter (NHE3) and a possible inhibitory action of prolonged insulin exposure on the Na+K+ pump. See text for further explanation. GLUT, glucose transporter; SGLT, sodium-glucose cotransporters.
Footnotes
EF and RP-M contributed equally.
Contributors: EF designed the experiments. EF, R-PM and EM analyzed the data and wrote the paper. R-PM, MS, ER, ALS, VBC, CA and EM conducted the experiments. All authors read and approved the final version of the manuscript. EF is the guarantor of this manuscript.
Funding: This work was supported in part through an investigator-initiated grant by Boehringer-Ingelheim Italia (IIS 1245.115). R-PM received financial support from the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brasil (CAPES) Finance Code 001.
Competing interests: EF has participated in scientific advisory boards for Boehringer Ingelheim, Eli Lilly, and Sanofi; has been involved in ad hoc consulting for Janssen, AstraZeneca, and Mitsubishi Tanabe; has participated in occasional speaking engagements for AstraZeneca, Novo Nordisk, Sanofi, Mitsubishi Tanabe, Eli Lilly, Boehringer Ingelheim, and Merck Sharp & Dohme; and has received research grant support from Boehringer Ingelheim and AstraZeneca.
Patient consent for publication: Not required.
Ethics approval: Studies were conducted according to Declaration of Helsinki principles; participants gave their written informed consent, and the study was approved by both ethics committees (EudraCT 2015-005727-80, CAAE 44961615.6.0000.540).
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request, the reasonable request being an additional analysis plan to test a new hypothesis formulated by a named investigator.
References
- 1.Alsahli M, Gerich JE. Renal glucose metabolism in normal physiological conditions and in diabetes. Diabetes Res Clin Pract 2017;133:1–9. 10.1016/j.diabres.2017.07.033 [DOI] [PubMed] [Google Scholar]
- 2.Gerich JE, Meyer C, Woerle HJ, et al. . Renal gluconeogenesis: its importance in human glucose homeostasis. Diabetes Care 2001;24:382–91. 10.2337/diacare.24.2.382 [DOI] [PubMed] [Google Scholar]
- 3.Mather A, Pollock C. Glucose handling by the kidney. Kidney Int 2011;79:S1–6. 10.1038/ki.2010.509 [DOI] [PubMed] [Google Scholar]
- 4.Cersosimo E, Garlick P, Ferretti J. Insulin regulation of renal glucose metabolism in humans. Am J Physiol 1999;276:E78–84. 10.1152/ajpendo.1999.276.1.E78 [DOI] [PubMed] [Google Scholar]
- 5.Cersosimo E, Garlick P, Ferretti J. Regulation of splanchnic and renal substrate supply by insulin in humans. Metabolism 2000;49:676–83. 10.1016/S0026-0495(00)80048-7 [DOI] [PubMed] [Google Scholar]
- 6.Féraille E, Doucet A. Sodium-potassium-adenosinetriphosphatase-dependent sodium transport in the kidney: hormonal control. Physiol Rev 2001;81:345–418. 10.1152/physrev.2001.81.1.345 [DOI] [PubMed] [Google Scholar]
- 7.Wright EM, Loo DDF, Hirayama BA. Biology of human sodium glucose transporters. Physiol Rev 2011;91:733–94. 10.1152/physrev.00055.2009 [DOI] [PubMed] [Google Scholar]
- 8.Ferrannini E. Sodium-Glucose co-transporters and their inhibition: clinical physiology. Cell Metab 2017;26:27–38. 10.1016/j.cmet.2017.04.011 [DOI] [PubMed] [Google Scholar]
- 9.Palmer LG, Schnermann J. Integrated control of Na transport along the nephron. Clin J Am Soc Nephrol 2015;10:676–87. 10.2215/CJN.12391213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pereira-Moreira R, Muscelli E. Effect of insulin on proximal tubules handling of glucose: a systematic review. J Diabetes Res 2020:8492467 10.1155/2020/8492467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Farber SJ, Berger EY, Earle DP. Effect of diabetes and insulin of the maximum capacity of the renal tubules to reabsorb glucose. J Clin Invest 1951;30:125–9. 10.1172/JCI102424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrannini E, Natali A, Bell P, et al. . Insulin resistance and hypersecretion in obesity. European group for the study of insulin resistance (EGIR). J Clin Invest 1997;100:1166–73. 10.1172/JCI119628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferrannini E, Muscelli E, Frascerra S, et al. . Metabolic response to sodium-glucose cotransporter 2 inhibition in type 2 diabetic patients. J Clin Invest 2014;124:499–508. 10.1172/JCI72227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muscelli E, Mari A, Natali A, et al. . Impact of incretin hormones on beta-cell function in subjects with normal or impaired glucose tolerance. Am J Physiol Endocrinol Metab 2006;291:E1144–50. 10.1152/ajpendo.00571.2005 [DOI] [PubMed] [Google Scholar]
- 15.DeFronzo RA, Ferrannini E. Regulation of hepatic glucose metabolism in humans. Diabetes Metab Rev 1987;3:415–59. 10.1002/dmr.5610030204 [DOI] [PubMed] [Google Scholar]
- 16.Guyton AC, Hall JE. The body fluids compartments: extracellular and intracellular fluids; interstitial fluids and edema. textbook of medical physiology. Philadelphia: W.B.Saunders Company, 2000. [Google Scholar]
- 17.Agus ZS, Puschett JB, Senesky D, et al. . Mode of action of parathyroid hormone and cyclic adenosine 3',5'-monophosphate on renal tubular phosphate reabsorption in the dog. J Clin Invest 1971;50:617–26. 10.1172/JCI106532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeFronzo RA, Hompesch M, Kasichayanula S, et al. . Characterization of renal glucose reabsorption in response to dapagliflozin in healthy subjects and subjects with type 2 diabetes. Diabetes Care 2013;36:3169–76. 10.2337/dc13-0387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ferrannini E, Cushman WC. Diabetes and hypertension: the bad companions. Lancet 2012;380:601–10. 10.1016/S0140-6736(12)60987-8 [DOI] [PubMed] [Google Scholar]
- 20.Beloto-Silva O, Machado UF, Oliveira-Souza M. Glucose-Induced regulation of NHEs activity and SGLTs expression involves the pKa signaling pathway. J Membr Biol 2011;239:157–65. [DOI] [PubMed] [Google Scholar]
- 21.Nakamura N, Matsui T, Ishibashi Y, et al. . Insulin stimulates SGLT2-mediated tubular glucose absorption via oxidative stress generation. Diabetol Metab Syndr 2015;7:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghezzi C, Wright EM. Regulation of the human Na+-dependent glucose cotransporter hSGLT2. Am J Physiol Cell Physiol 2012;303:C348–54. 10.1152/ajpcell.00115.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vallon V, Gerasimova M, Rose MA, et al. . Sglt2 inhibitor empagliflozin reduces renal growth and albuminuria in proportion to hyperglycemia and prevents glomerular hyperfiltration in diabetic Akita mice. Am J Physiol Renal Physiol 2014;306:F194–204. 10.1152/ajprenal.00520.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tojo A, Hatakeyama S, Kinugasa S, et al. . Angiotensin receptor blocker telmisartan suppresses renal gluconeogenesis during starvation. Diabetes Metab Syndr Obes 2015;8:103–13. 10.2147/DMSO.S78771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Osorio H, Coronel I, Arellano A, et al. . Ursodeoxycholic acid decreases sodium-glucose cotransporter (SGLT2) expression and oxidative stress in the kidney of diabetic rats. Diabetes Res Clin Pract 2012;97:276–82. 10.1016/j.diabres.2012.02.022 [DOI] [PubMed] [Google Scholar]
- 26.Rahmoune H, Thompson PW, Ward JM, et al. . Glucose transporters in human renal proximal tubular cells isolated from the urine of patients with non-insulin-dependent diabetes. Diabetes 2005;54:3427–34. 10.2337/diabetes.54.12.3427 [DOI] [PubMed] [Google Scholar]
- 27.Wang XX, Levi J, Luo Y, et al. . Sglt2 protein expression is increased in human diabetic nephropathy: SGLT2 protein inhibition decreases renal lipid accumulation, inflammation, and the development of nephropathy in diabetic mice. J Biol Chem 2017;292:5335–48. 10.1074/jbc.M117.779520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Norton L, Shannon CE, Fourcaudot M, et al. . Sodium-Glucose co-transporter (SGLT) and glucose transporter (GLUT) expression in the kidney of type 2 diabetic subjects. Diabetes Obes Metab 2017;19:1322–6. 10.1111/dom.13003 [DOI] [PubMed] [Google Scholar]
- 29.Solini A, Rossi C, Mazzanti CM, et al. . SGLT)2 and SGLT1 renal expression in patients with type 2 diabetes. Diabetes Obes Metab 2017;19:1289–94. [DOI] [PubMed] [Google Scholar]
- 30.Tiwari S, Halagappa VKM, Riazi S, et al. . Reduced expression of insulin receptors in the kidneys of insulin-resistant rats. J Am Soc Nephrol 2007;18:2661–71. 10.1681/ASN.2006121410 [DOI] [PubMed] [Google Scholar]
- 31.Gatica R, Bertinat R, Silva P, et al. . Altered expression and localization of insulin receptor in proximal tubule cells from human and rat diabetic kidney. J Cell Biochem 2013;114:639–49. 10.1002/jcb.24406 [DOI] [PubMed] [Google Scholar]
- 32.DeFronzo RA, Goldberg M, Agus ZS. The effects of glucose and insulin on renal electrolyte transport. J Clin Invest 1976;58:83–90. 10.1172/JCI108463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Skøtt P, Hother-Nielsen O, Bruun NE, et al. . Effects of insulin on kidney function and sodium excretion in healthy subjects. Diabetologia 1989;32:694–9. 10.1007/BF00274259 [DOI] [PubMed] [Google Scholar]
- 34.Stenvinkel P, Bolinder J, Alvestrand A. Effects of insulin on renal haemodynamics and the proximal and distal tubular sodium handling in healthy subjects. Diabetologia 1992;35:1042–8. 10.1007/BF02221679 [DOI] [PubMed] [Google Scholar]
- 35.O'Hare JA, Minaker KL, Meneilly GS, et al. . Effect of insulin on plasma norepinephrine and 3,4-dihydroxyphenylalanine in obese men. Metabolism 1989;38:322–9. 36. 10.1016/0026-0495(89)90118-2 [DOI] [PubMed] [Google Scholar]
- 36.Verbalis JG. Whole-Body volume regulation and escape from antidiuresis. Am J Med 2006;119:S21–9. 10.1016/j.amjmed.2006.05.004 [DOI] [PubMed] [Google Scholar]
- 37.Hediger MA, Rhoads DB. Molecular physiology of sodium-glucose cotransporters. Physiol Rev 1994;74:993–1026. 10.1152/physrev.1994.74.4.993 [DOI] [PubMed] [Google Scholar]
- 38.Wang T, Weinbaum S, Weinstein AM. Regulation of glomerulotubular balance: flow-activated proximal tubule function. Pflugers Arch 2017;469:643–54. 10.1007/s00424-017-1960-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fakhruddin S, Alanazi WA, Alhamami HN, et al. . Hyperglycaemia induced by chronic i.p. and oral glucose loading leads to hypertension through increased Na+ retention in proximal tubule. Exp Physiol 2018;103:236–49. 10.1113/EP086604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moreira-Rodrigues M, Quelhas-Santos J, Serrão P, et al. . Glycaemic control with insulin prevents the reduced renal dopamine D1 receptor expression and function in streptozotocin-induced diabetes. Nephrol Dial Transplant 2010;25:2945–53. 10.1093/ndt/gfq150 [DOI] [PubMed] [Google Scholar]
- 41.Al-Numair KS, Veeramani C, Alsaif MA, et al. . Influence of kaempferol, a flavonoid compound, on membrane-bound ATPases in streptozotocin-induced diabetic rats. Pharm Biol 2015;53:1372–8. 10.3109/13880209.2014.982301 [DOI] [PubMed] [Google Scholar]
- 42.Mikaelian NP, Gurina AE, Terent'ev AA. Dysfunction of membrane-receptor system of blood cells and kidney tissue in experimental diabetes mellitus. Bull Exp Biol Med 2013;154:610–3. 10.1007/s10517-013-2011-x [DOI] [PubMed] [Google Scholar]
- 43.Shi Q, Zeng J, Dong Y, et al. . Concurrent impairment of (Na++K+)-ATPase activity in multi-organ of type-1 diabetic NOD mice. J Diabetes Complications 2013;27:29–33. 10.1016/j.jdiacomp.2012.07.004 [DOI] [PubMed] [Google Scholar]
- 44.Javorková V, Mézešová L, Vlkovičová J, et al. . Influence of sub-chronic diabetes mellitus on functional properties of renal Na(+), K(+)-ATPase in both genders of rats. Gen Physiol Biophys 2010;29:266–74. 10.4149/gpb_2010_03_266 [DOI] [PubMed] [Google Scholar]
- 45.Galuska D, Pirkmajer S, Barrès R, et al. . C-peptide increases Na, K-ATPase expression via PKC- and MAP kinase-dependent activation of transcription factor ZEB in human renal tubular cells. PLoS One 2011;6:e28294. 10.1371/journal.pone.0028294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y, Chen C, Fu C, et al. . Angiotensin II type 2 receptor inhibits expression and function of insulin receptor in rat renal proximal tubule cells. J Am Soc Hypertens 2018;12:135–45. 10.1016/j.jash.2017.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang Y, Ren H, Lu X, et al. . Inhibition of D4 dopamine receptors on insulin receptor expression and effect in renal proximal tubule cells. J Am Heart Assoc 2016;5:pii: e002448. 10.1161/JAHA.115.002448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang J, Cui Z, He D, et al. . Insulin increases D5 dopamine receptor expression and function in renal proximal tubule cells from Wistar-Kyoto rats. Am J Hypertens 2009;22:770–6. 10.1038/ajh.2009.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banday AA. Chronic insulin treatment phosphorylates the renal Na-K-ATPase α1-subunit at serine 16/23 and reduces its activity involving PI3-kinase-dependent PKC activation. Am J Physiol Renal Physiol 2016;311:F958–66. 10.1152/ajprenal.00355.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bia MJ, DeFronzo RA. Extrarenal potassium homeostasis. Am J Physiol 1981;240:F257–68. 10.1152/ajprenal.1981.240.4.F257 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
bmjdrc-2020-001178supp004.pdf (98.5KB, pdf)
bmjdrc-2020-001178supp001.pdf (99.2KB, pdf)
bmjdrc-2020-001178supp002.pdf (75.7KB, pdf)
bmjdrc-2020-001178supp003.pdf (902.3KB, pdf)