

Abstract
Promiscuous and allosteric drug interactions with cytochrome P450 3A4 (CYP3A4) are ubiquitous but incompletely understood at the molecular level. A classic allosteric CYP3A4 drug interaction includes the benzodiazepine midazolam (MDZ). MDZ exhibits homotropic and heterotropic allostery when metabolized to 1′-hydroxy and 4-hydroxy metabolites in varying ratios. The combination of hydrogen−deuterium exchange mass spectrometry (HDX-MS) and Gaussian accelerated molecular dynamics (GaMD) simulations of CYP3A4 in lipid nanodiscs and in a lipid bilayer, respectively, reveals MDZ-dependent changes in dynamics in a membrane environment. The F-, G-, and intervening helices, as well as the loop preceding the β1-sheets, display the largest observed changes in HDX. The GaMD suggests a potential allosteric binding site for MDZ in the F′- and G′-regions, which undergo significant increases in HDX at near-saturating MDZ concentrations. The HDX-MS and GaMD results confirm that changes in dynamics are most significant near the developing consensus allosteric site, and these changes are distinct from those observed previously with the nonallosteric inhibitor ketoconazole. The results suggest that the allosteric MDZ remains mobile in its binding site at the Phe-cluster. The results further suggest that this binding site remains dynamic or changes the depth of insertion in the membrane.
Graphical Abstract
Membrane-bound cytochrome P450 3A4 (CYP3A4) dominates drug metabolism in humans and displays remarkable substrate promiscuity, which is presumed to be a result of conformational plasticity. The plasticity of CYP3A4 has been documented via numerous crystal structures that reveal significant changes in active site structure and rearrangement of structural elements remote from the active site.1,2 The ligand-dependent induced fit3 is clearly operative with CYP3A4, wherein different protein conformations distinct from ligand-free CYP3A4 are observed when structurally distinct ligands bind. Despite the wealth of structural information, our understanding of CYP3A4-ligand interactions remains incomplete because the static models are based on the crystalline state in the absence of a membrane, which is likely to modulate the ligand-dependent conformational ensembles.4 Furthermore, although elegant advances have been made in computational methods applied to CYPs,5,6 very few experimental approaches report directly on the dynamics of CYP3A4 in a membrane environment. As a result, our understanding of the detailed mechanisms by which drugs enter or leave the active site is limited, and the time scales of protein dynamics or solvent reorientation in relevant access channels that link the membrane or bulk solvent to the active site are not established. By analogy with the structural plasticity indirectly suggested by CYP3A4 crystal structures, ligand-dependent protein and solvent dynamics are expected. It is possible, even likely, that different ligands bind and dissociate from the active site via different routes, but there are insufficient data to address this possibility.
Among the uncertainties regarding ligand-dependent CYP dynamics is the effect of allosteric ligands on the dynamics of CYP3A4. The allosteric behavior of CYPs, in general, is enigmatic because it parallels the substrate and catalytic promiscuity. In this regard and others, CYP3A4 allostery is distinct from traditional allostery wherein specific allosteric ligands are exploited to regulate the function of an active site. With CYP3A4, many ligands have substrate-dependent allosteric effects. The nature of such a promiscuous and substrate-dependent allosteric site is unknown. Several historical and recent studies suggest a set of overlapping allosteric sites near the Phe-cluster of CYP3A4 that provide for promiscuous allostery.7–10 Hypothetically, the structural or dynamic properties of this allosteric site would be different from a ligand-specific allosteric site that regulates the function of substrate-specific enzymes.
Hydrogen–deuterium exchange mass spectrometry (HDX-MS) provides high-resolution information about the time-dependent exposure of peptide backbone amides of the folded protein state to solvent.11,12 Structured regions involved in hydrogen bonding networks (helices or sheets) and regions buried in the protein exchange more slowly than unstructured regions (loops) and solvent-exposed areas. Although HDX-MS is straightforward for small cytosolic proteins, it remains challenging and less commonly used for membrane proteins,13,14 and there are very few examples of H/DX with membrane proteins in a membrane bilayer.15 We previously have initiated HDX-MS with CYP3A4 in nanodiscs and demonstrated that the lipid bilayer composition and structure could affect CYP3A4 dynamics.15 Here we extend the previous work and monitor the HDX of CYP3A4 POPC nanodiscs in the presence of the classic CYP3A4 substrate midazolam (MDZ).
MDZ is metabolized to 1′-hydroxy MDZ (1′-OH MDZ) and 4-hydroxy MDZ (4-OH MDZ) by CYP3A4 (Figure 1A), and the product ratio exhibits homotropic and heterotropic allosteric effects in vitro16 and in vivo,17 indicative of multiple ligand binding. The 1′-OH/4-OH MDZ ratio decreases with increasing MDZ concentrations. Mutagenesis work by Khan and co-workers supports the hypothesis that a discrete secondary binding site exists proximal to the catalytic binding site near the Phe-cluster.7 Additionally, NMR work done by Roberts and co-workers indicates two MDZ molecules in or near the active site.16 Furthermore, the equilibrium binding experiments described here indicate cooperative isotherms, consistent with multiple MDZ binding. As a result of the long-appreciated behavior of MDZ with CYP3A4, the current dogma is that CYP3A4 binds multiple MDZ molecules, as with other ligands, and here we aim to interrogate the structural dynamics of CYP3A4 when bound to MDZ in order to better characterize the allosteric sites.
Figure 1.
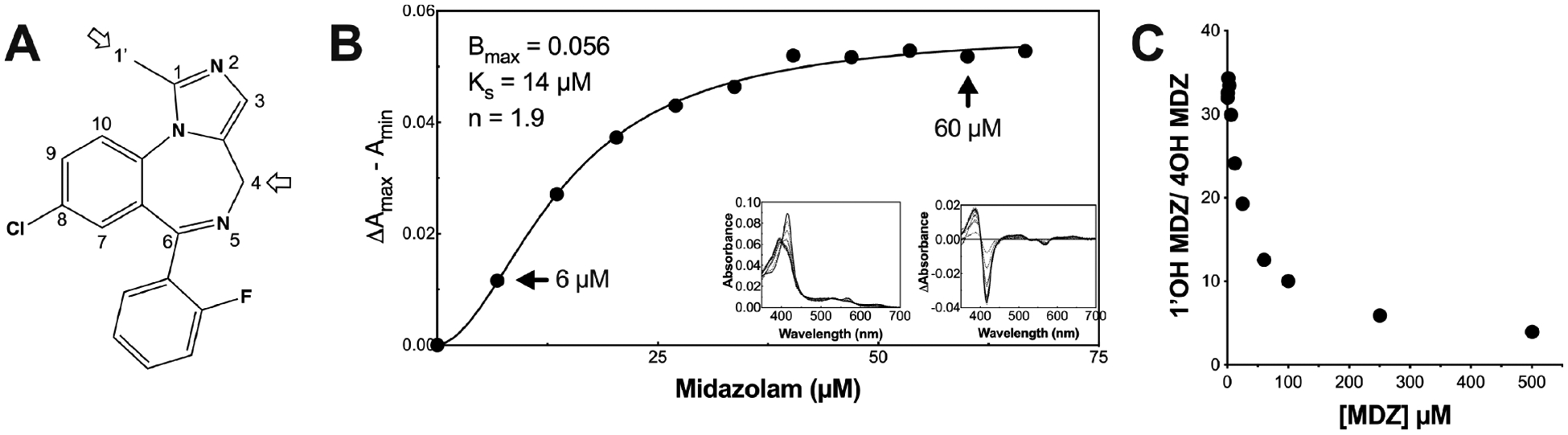
MDZ structure, equilibrium titration into CYP3A4 nanodiscs, and product ratio vs MDZ concentration. (A) MDZ structure with the indicated primary (C1′) and secondary (C4) hydroxylation sites. (B) Binding isotherm for MDZ with CYP3A4 nanodiscs, based on optical difference spectra as described in the methods section. The black line is the fit to the Hill equation, and black points are the optical titration data. The left inset and right inset are the absolute and difference spectra at varying concentrations of MDZ, respectively. The arrows indicate the two concentrations used in the HDX experiments, 6 or 60 μM MDZ. (C) 1′-OH MDZ/4OH MDZ product ratio vs MDZ concentration (μM).
Toward this end, we performed Gaussian accelerated molecular dynamics simulations (GaMD) for CYP3A4 in a POPC lipid bilayer under different ligation states to aid in our interpretation of the MDZ-induced changes in HDX. The HDX-MS and GaMD are reasonably cross-correlated, and they identify regions of CYP3A4 that have altered dynamics upon MDZ binding. Furthermore, the results suggest important differences between the structure of CYP3A4 in the crystalline state vs in a membrane, when MDZ is bound. The combination of several structural and dynamic techniques appears necessary for a complete understanding of CYP3A4 interactions with ligands.
MATERIALS AND METHODS
Protein Expression and Purification (CYP3A4, MSP1D1, and rCPR).
Recombinant CYP3A4 was expressed in Escherichia coli C41(DE3) cells transformed with a pCWori + vector encoding genes for ampicillin resistance and the Nf14 construct of CYP3A4,18,19 preceding a C-terminal histidine tag, as previously described.20 A single isolated colony was used to inoculate an overnight culture of Luria–Bertani (LB) broth containing 100 μg/mL ampicillin. The overnight culture was used to inoculate Terrific broth media containing 100 μg/mL ampicillin, 1 mM thiamine, and trace elements in a ratio of 1:100. After 4 h of growth at 37 °C, or at an optical density at 660 nm of ~0.4, with shaking at 220 rpm, 0.5 mL of 1 M δ-aminolevulinic acid (δ-ALA) and 0.5 mL of 1 M isopropyl β-D-thiogalactoside (IPTG) were added to each flask to induce CYP3A4 expression after both temperature and speed of shaking were reduced to 27 °C and 125 rpm, respectively. After 40 additional hours of growth, cells were harvested via centrifugation of each culture for 30 min at 4 °C at 4800 rpm. The resulting pellets were frozen at −80 °C until purification.
CYP3A4 growth cell pellets were thawed and gently resuspended in ice-cold CYP3A4 resuspension buffer (100 mM KPi, pH 7.4, 20% glycerol, 10 mM β-mercaptoethanol (BME), 3% Emulgen 911, 50 μM testosterone (TST), 20 mM imidazole (IMZ), supplemented with protease inhibitor cocktail for His-tagged proteins (EDTA-free), benzonase, and 2 mg/mL lysozyme) then stirred at 4 °C for 1 h before homogenizing several times through a hand-held Potter-Elvehjem homogenizer with a PTFE pestle attached.
A second round of stirring at 4 °C for 4 h, followed by several more passes with the hand-held homogenizer, greatly improved the yield. The lysate was centrifuged for 1 h at 4 °C at 37 000 rpm with a Beckman Ti45 rotor. The resulting pellet was discarded, and the supernatant was loaded onto a 30 mL Ni-NTA affinity column (Qiagen) equilibrated with wash buffer 1 (50 mM KPi, pH 7.4, 20% glycerol, 2 mM BME, 50 μM TST, 0.2% Anapoe C12E10, 300 mM KCl, 20 mM IMZ). The column was then washed with 250 mL of wash buffer 1, then 500 mL of wash buffer 2 (50 mM KPi, pH 7.4, 20% glycerol, 2 mM BME, 50 μM TST, 0.1% cholate, 300 mM KCl, 100 mM glycine, 20 mM IMZ), followed by a final 500 mL of wash with wash buffer 3 (50 mM KPi, pH 7.4, 20% glycerol, 2 mM BME, 50 μM TST, 0.1% cholate, 40 mM IMZ). TST is included in the early steps of purification because it stabilizes CYP3A4 but can be removed subsequently. Protein was eluted with elution buffer (50 mM KPi, pH 7.4, 20% glycerol, 2 mM BME, 0.1% cholate, and an IMZ gradient from 50−500 mM). P450 containing fractions were diluted 1:1 with an ice-cold solution of 20% glycerol and 0.1% cholate to reduce the KPi concentration before loading to a 20 mL hydroxyapatite (HA) column equilibrated with 10 mM KPi, pH 7.4, 20% glycerol, 2 mM BME, and 0.1% cholate. The column was then washed with 1000 mL of HA wash buffer (25 mM KPi, 20% glycerol, 2 mM BME). Protein was eluted with 400 mM KPi, pH 7.4, 20% glycerol, and 2 mM BME. P450 containing fractions were dialyzed with a 10 kDa MWCO SnakeSkin dialysis tubing (Thermo) into 100 mM KPi, pH 7.4, 20% glycerol, 1 mM EDTA, and 0.5 mM TCEP. Protein was stored at −80 °C until further use. Purity was >95% as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). CYP3A4 concentration was determined by “dithionite difference spectra,” which is the difference between Fe3+-H2O vs Fe2+-CO, using an extinction coefficient difference of 99 mM−1 cm−1,21,22 for the difference between 490–450 nm. The method is similar to those published by Omura and Sato (Fe2+ vs Fe2+-CO) without the anaerobic conditions.23 No P420 was detected from the difference absorbance spectrum.
The membrane scaffold protein, MSP1D1, was expressed and purified as previously described.15,24 MSP1D1 was heterologously expressed in E. coli BL21-Gold (DE3) cells transformed with a pET expression vector encoding genes for kanamycin resistance and MSP1D1 with an N-terminal histidine tag. An overnight culture was grown in LB media containing 50 μg/mL kanamycin. The overnight culture was used to inoculate Terrific broth media containing 100 μg/mL ampicillin and 1 mM thiamine in a ratio of 1:100. After 4 h of growth at 37 °C with shaking at 220 rpm, 0.5 mL of 1 M IPTG was added to each flask to induce MSP1D1 expression. After 3 h of growth, cells were harvested via centrifugation of each culture for 30 min at 4 °C at 4800 rpm. The resulting pellets were frozen at −80 °C until purification.
MSP1D1 growth cell pellets were thawed and gently resuspended in ice-cold MSP1D1 resuspension buffer (20 mM KPi, pH 7.4, 1% Triton X-100, supplemented with protease inhibitor cocktail for His-tagged proteins (EDTA-free), benzonase, and 2 mg/mL lysozyme) and stirred at 4 °C for 1 h. Cells were lysed by a French Press, and the lysate was centrifuged for 45 min at 4 °C at 22 000 rpm with a Beckman Ti42 rotor. The resulting pellet was discarded, and the supernatant was loaded to a 20 mL Ni-NTA affinity column (Qiagen) equilibrated with wash buffer 1 (40 mM Tris, pH 8.0, 300 mM NaCl, 1% Triton X-100). The column was then washed with 250 mL of wash buffer 1, then 100 mL of wash buffer 2 (40 mM Tris, pH 8.0, 300 mM NaCl, 50 mM cholate, 20 mM IMZ), and finally with 100 mL of wash buffer 3 (40 mM Tris, pH 8.0, 300 mM NaCl, 50 mM IMZ). Protein was eluted with 40 mM Tris, pH 7.4, 300 mM NaCl, and 400 mM IMZ. MSP1D1-containing fractions were dialyzed into 20 mM Tris, pH 7.4, 100 mM NaCl, and 0.5 mM EDTA and then filtered with a 0.2 μm filter. The protein concentration was spectrally determined using the calculated extinction coefficient for MSP1D1 absorbance at 280 nm (ε280 = 21 mM−1 cm−1).
To aid in 3A4 nanodisc purification, the N-terminal histidine tag of MSP1D1 was cleaved by Pro-TEV Plus Protease (Promega Corporation) and further purified with a Ni-NTA column, hereafter referred to as MSP1D1(−). The protein concentration was spectrally determined using the calculated extinction coefficient for MSP1D1(−) absorbance at 280 nm (ε280nm = 18.2 mM−1 cm−1).
Recombinant rat NADPH-cytochrome P450 (oxido)-reductase (rCPR) was expressed as previously described.20,25 Briefly, cultures were grown as described for CYP3A4, except that LB media supplemented with 1 μg/mL riboflavin, 1 mM thiamine, and 100 μg/mL ampicillin were used. After 4 h of growth at 37 °C with shaking at 220 rpm, the expression was induced by the addition of 1 mM IPTG, and cultures were further grown. The temperature and speed of shaking were reduced to 27 °C and 125 rpm. After 16 additional hours of growth, cells were harvested via centrifugation of each culture for 30 min at 4 °C at 4800 rpm. The resulting pellets were frozen at −80 °C until purification.
rCPR growth cell pellets were thawed and gently resuspended in ice-cold CPR resuspension buffer (75 mM Tris, pH 8.0, 0.25 M sucrose, 0.5 mM EDTA, 200 μM FMN, 0.02 mg/mL lysozyme, 0.5 μg/mL DNaseI, and 50 μM dithiothreitol [DTT], supplemented with protease inhibitor cocktail for His-tagged proteins [EDTA-free]), and the mixture then stirred at 4 °C for 1 h before passage through a French Press. The lysate was centrifuged for 1 h at 4 °C at 37 000 rpm with a Beckman Ti45 rotor. The resulting supernatant was discarded and the pellet resuspended with homogenization/wash buffer (50 mM Tris, pH 7.7, 0.1 mM EDTA, 50 μM DTT, 10% glycerol, and 0.15% Triton X-100) and then passed through a hand-held homogenizer, followed by centrifugation at 4 °C at 37 000 rpm with a Beckman Ti45 rotor for 30 min. The resulting yellow/green supernatant was loaded to a 2′,5′-ADP Sepharose column equilibrated with wash buffer. The column was washed with 20 column volumes of wash buffer. Protein was eluted with elution buffer (50 mM Tris, pH 7.7, 10% glycerol, 0.1 mM EDTA, 0.05 mM DTT, and an NADP+ gradient from 0−6 mM). rCPR-containing fractions were dialyzed with a 10 kDa MWCO SnakeSkin dialysis tubing (Thermo) into 100 mM KPi, pH 7.5, 20% glycerol, and 0.1 mM EDTA. Protein was stored at −80 °C until further use. Purity was >95% as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). The rCPR concentration was determined spectrally, by measuring the difference in absorbance between 456 and 550 nm of the purified protein in a solution of 0.01 mM K3Fe(CN)6 in 100 mM KPi using an extinction coefficient of 21.1 mM−1 cm−1.26
Self-Assembly of Nanodiscs.
CYP3A4 nanodiscs were prepared as previously described.15,27 A chloroform POPC stock solution was dried under a gentle stream of nitrogen, and the remaining solvent was removed by a vacuum desiccator overnight. Here, 0.1% Emulgen 911 was used to solubilize CYP3A4, MSP1D1(−), and POPC, which were mixed in a 0.1:1:65 molar ratio in a disc-forming buffer (DFB, 100 mM KPi, pH 7.4, 50 mM NaCl) with 20 mM sodium cholate in a bottle and placed on a Thermo Scientific Digital Bottle Roller for 1 h at 4 °C. Amberlite XAD-2 resin beads were added to the mixture at a final concentration 1 g/mL to remove the detergent and thus initiate nanodisc self-assembly to proceed on the bottle roller overnight at 4 °C. The resulting solution was removed, and beads were washed with 2 volumes of DFB and beads removed from the resulting solution. CYP3A4 nanodiscs were purified after Ni-NTA chromatography, followed by size exclusion chromatography on a Superdex 200 10/300 GL column (GE Healthcare). Purified CYP3A4 nanodiscs were stored at −80 °C until HDX experiments.
Equilibrium Binding Titrations.
Steady-state titrations were performed using a dual-beam OLIS/Aminco DW2a spectrophotometer (OLIS, Bogart, GA) maintained at 25 °C. The sample cuvette contained CYP3A4 nanodiscs at 0.73 μM in 50 mM HEPES, pH 7.4, and 50 mM NaCl. The reference cuvette contained exchange buffer. MDZ was added to both sample and reference cuvettes in 1 μL increments. Spectral titrations were done in triplicate, and spectra were analyzed as the difference relative to ligand-free spectra. The differences in absorbance between λmax and λmin were plotted as a function of ligand concentration and fit to the Hill equation using IGOR pro 6.35A (Wavemetrics, Lake Oswego, OR):
| (1) |
where Amax is the maximum absorbance, n is the Hill coefficient, and Ks is the spectral binding affinity. Analysis of intermediate states of occupancy of CYP3A4 binding sites via sequential binding models was not possible in the absence of knowledge of spectral amplitudes for the separate binding events.28
Reconstitution System and Kinetic Assays.
Human CYP3A4 and rat CPR were reconstituted in a 1:1 ratio as described by Shaw et al.,29 except a 100× lipid stock containing 2 mg/mL POPC lipid was used in place of the lipid mixture. The final concentrations of the enzyme in each 100 μL incubation were 100 nM CYP3A4 and 100 nM rCPR. For each incubation, 80 μL of the lipid/protein reconstitution mixture was added to a 200 μL 96-well thin-walled PCR plate, followed by the addition of 10 μL of MDZ stock. All incubations contained 2% methanol. The PCR plate was placed in a hot water bath set to 37 °C and the temperature allowed to equilibrate for 5 min. The reaction was initiated by the addition of 10 μL of 5 mg/mL NADPH. After 5 min, the reaction was quenched by the addition of an equal volume (100 μL) quench solution (1:1 mixture of ACN/MeOH containing 1′-OH MDZ-D4 as an internal standard). The plate was sealed and centrifuged for 10 min at 4 °C at 1200 rpm using a Sorvall Legend XTR tabletop centrifuge. The supernatant was transferred to a separate 96-well plate for LC-MS/MS analysis. A standard curve was created by adding known amounts of MDZ to identical incubation conditions without CYP3A4, rCPR, and NADPH.
LC-MS/MS Method for MDZ Metabolism.
Analyses of 1′-OH MDZ and 4-OH MDZ activity were performed using an API 4000 QTrap (AB Sciex LLC, Framingham, MA) coupled with a Waters H Class UPLC System (Milford, MA). The metabolites were separated using a Waters Sunfire C18 column (50 × 2.1 mm, 5 μm particle size) with 0.1% formic acid in water and 0.1% formic acid in acetonitrile for mobile phases at 0.75 mL/min. The gradient used for the separation is the following: 0.0–0.8 min 100% A, 1.5 min 80% A, 3 min 60% A, 3.5 min 100% B, 3.6–6 min 100% A. The MRM transitions for this assay were 342 → 203 for 1′-OH MDZ, 342 → 297 for 4-OH MDZ, and 346 → 203 for 1′-OH MDZ-D4. 1′-OH MDZ-D4 was used as an internal standard for both 1′-OH MDZ and 4-OH MDZ. Integration and quantification were performed with AB Sciex Analyst 1.6 software using standard curves that ranged from 1 to 1000 nM for both metabolites.
Hydrogen–Deuterium Exchange.
Deuterium exchange experiments were performed in duplicate with CYP3A4 nanodiscs, as described previously.15,30 A 15 μL aliquot (per incubation) of ligand-bound or ligand-free CYP3A4 nanodiscs was incubated at 25 °C for 5 min prior to dilution into 105 μL of ligand-containing or ligand-free D2O buffer at 25 °C for 10 s, 30 s, 1.5 s, 4.5 s, and 13.5 min. The final concentration during each exchange was 0.92 μM CYP3A4 nanodisc, 50 mM HEPES pD 7.4, 50 mM NaCl, 83% D2O, 2% methanol, and MDZ at 0 μM, 6 μM, or 60 μM. The exchange was quenched by equal volume (120 μL) ice-cold 0.8% formic acid for a final pH of 2.5, followed by nanodisc disassembly initiated by the addition of a 4 mM ice-cold sodium cholate to nanodisc concentration. The sample was then rapidly transferred to a tube containing 200 μL of ice-cold immobilized pepsin beads (Pierce) and vortexed frequently for a total of 5 min. In the last minute of digest, 10 μL of 300 mg/mL ZrO2 HybridSPE Phospholipid beads (Supelco) in 0.8% formic acid was added to the digest to facilitate lipid removal.31 After the digest was complete, the sample was filtered through a chilled 0.45 μm Spin-X microcentrifuge tube filter (Costar) to remove pepsin beads, precipitated sodium cholate, and ZrO2 beads. The filtered sample was aliquoted and immediately flash-frozen in liquid nitrogen and stored at −80 °C until analysis. Undeuterated samples were prepared as described above without D2O.
Mass Spectrometry and Data Analysis of Hydrogen–Deuterium Exchange Samples.
Samples were thawed on ice over 5 min then injected with an ice-cold Hamilton syringe onto a Waters HDX Manager coupled to a Synapt G2-Si QTOF mass spectrometer, operating in positive ion mode with ion mobility enabled as described previously.15,32 Peptides were trapped on a Vanguard BEH Shield RP18 1.7 μm trap column (2.1 × 5 mm; Waters), flowing 0.1% formic acid (FA) with 0.025% trifluoroacetic acid (TFA) at 200 μL/min for 3 min, after which the sample was resolved over a Hypersil 1.9 μm C18 column (1 × 50 mm; Thermo Scientific) using an acetonitrile (ACN) gradient of 8−50% solvent B at 40 μL/min for 8.5 min (solvent A 0.1% FA, 0.025% TFA, and 2% ACN in water; solvent B, 0.1% FA in ACN). Toward the end of the gradient, the sample flow was diverted to waste to prevent the eluting cholate from entering the MS. Two rapid gradient steps (5–95% solvent B for 30 s) at the end of each sample run were added to minimize carryover between injections. The syringe, loop, and trap column were washed between injections with five subsequent injections of 10% FA, 30% trifluoroethanol, 80% methanol, 2:1 isopropyl alcohol/ACN, and 80% ACN.33 Peptide identification was performed using PLGS Version 3 (Waters) by analyzing ion-mobility MSE data against a library containing sequences of CYP3A4, MSP1D1, and porcine pepsin using previously published criteria.15 The relative deuterium uptake processed by DynamX Version 3 (Waters) and HX-Express v234 is reported and not corrected for back-exchange.
System Setup and Conventional Molecular Dynamics.
The same N-terminally modified version of the 2.05 Å resolution CYP3A4 structure (PDB ID: 1TQN) previously described6 with the same sequence as that used in the HDX experiments was used as the starting point for all simulations. Protonation states of amino acids were assigned using Poisson–Boltzmann-based PropKa35–38 calculations at pH 7.4. Curated CYP3A4 was then inserted into a 100 Å × 100 Å POPC membrane constructed with the Membrane Builder plugin of VMD.39 POPC and water molecules overlapping and within 0.8 Å of the protein were removed. The system was solvated with a 10 Å layer of TIP3P water in the ± z directions. Potassium and chloride ions were added to an ionic strength of 0.15 M, thereby ensuring electrical neutrality. The final models included 253 POPC, 69 chloride ions, 56 potassium ions, and 19686 TIP3P water molecules.
All molecular dynamics procedures were performed with the NAMD 2.12 code40 using the CHARM3639 force field for POPC, CHARMM2741 for the protein and heme, and parameters for MDZ were from CGenFF.42 The ligand-free CYP3A4 system was minimized for 10 000 steps, followed by equilibration of the lipids for 1 ns by freezing the positions of the lipid phosphate atoms as well the water, ions, and protein atoms. Melting of the lipid tails was followed by a 2 ns constant temperature and pressure (NPT) simulation with a harmonic restraint (5 kcal mol−1 Å−2) applied to the protein Cα atoms. Bonds to hydrogen atoms were restrained with the SHAKE algorithm, and a 2 fs time step was used. The Langevin piston thermostat with a 0.5 ps−1 damping constant was used to maintain a constant temperature. The Nosé–Hoover Langevin piston method43 was used with a target pressure of 1.01325 bar, oscillation period of 50 fs, decay period of 25 fs, and piston target temperature of 300 K. After completion of the preparative simulations, the complete CYP3A4 system was equilibrated in the NPT ensemble for an additional 100 ns. The end point of this simulation was used to generate both MDZ-containing systems. For the single-MDZ-containing system, MDZ was introduced only in the active site. In the two-MDZ system, the ligands were introduced into the active site as well as to a water-filled cavity bound by the β–β2 loop and the F′ helix (Figure S1). Both systems were minimized for 10 000 steps and subject to a two ns NPT simulation, wherein the S119 OγH-MDZ N2 (2.6 Å) and Fe-MDZ-C1 (4.9 Å) distances of the active site MDZ were restrained with a force constant of 5 kcal mol−1 Å−2. These restraints successfully reproduced the binding orientation assumed by MDZ in the crystal structure,2 and both served as starting points for an additional 100 ns unrestrained NPT equilibration.
Gaussian Accelerated Molecular Dynamics.
The three 100 ns unrestrained cMD simulations also served to collect the potential statistics (Vmax, Vmin, Vavg, and σV) necessary to calculate the GaMD boost potential.44,45 The end point of this simulation was used to spawn four 0.5 μs GaMD simulations with independently randomized velocities at 300 K. GaMD simulations were performed in “dual-boost” mode, meaning that boost potentials were added to both the dihedral and total system potential energies by setting the reference energy to the lower bound, E = Vmax. The average and standard deviation of the potential energy was calculated every 200 ps. The upper limit for the dihedral and total boost potential energies was set to 6 kcal mol−1 (~10kBT). Trajectory frames were saved every 200 ps for further analysis. Only the latter 0.4 μs of each trajectory was subject to subsequent analyses.
RESULTS AND DISCUSSION
MDZ Displays Complex Equilibrium Binding Behavior to CYP3A4 Nanodiscs.
In order to assess the binding affinity of MDZ to CYP3A4 in nanodiscs, the CYP3A4 heme spin state was monitored at varying MDZ concentrations via UV/vis spectroscopy (Figure 1). The spectral titrations report on the ligation state of the ferric heme iron at the active site. In the resting state, the heme iron of CYP3A4 is predominantly in a hexacoordinate low-spin state with water in the sixth axial position. Displacement of the water upon binding of MDZ results in a transition to a pentacoordinate high-spin state, termed a type I spectral change. Figure 1B shows the characteristic type I difference spectra induced by MDZ and the resultant binding isotherm, consistent with previous reports. A fit to the Hill eq (eq 1, methods) resulted in a Ks value of 14 ± 1.1 μM and a Hill coefficient of 1.9. For similar titrations in phosphate buffer and in the absence of a membrane, the data were fit to hyperbolic binding isotherms, and the recovered KD varied with the phosphate concentration.2,46
For CYP3A4 in nanodiscs, our titration data do not fit well to hyperbolic binding, and significant cooperativity is apparent. A comparison of the data fit to a hyperbolic binding isotherm or Eadie–Hofstee plots is shown in Figure S2. Because the spectral titrations report only on the displacement of the water at the heme iron, they are insufficient to determine the binding pose of MDZ and the stoichiometry. Importantly, unlike sigmoidal velocity vs [substrate] plots from catalytic turnover, sigmoidal binding isotherms at equilibrium demand multiple ligand binding.47 Thus, in accordance with the existing dogma, we consider two MDZ binding sites for CYP3A4 in lipid nanodiscs. The arrows in Figure 1B indicate the concentrations of MDZ used in the H/DX experiments below. The turnover experiments are intended only to demonstrate the functional consequences of changing ratios of [CYP * MDZ] vs [CYP * MDZ * MDZ]. Obviously, the data indicate that the concentration dependence of the heme spin state in lipid nanodiscs is different from the concentration dependence of the product ratio in the turnover conditions, which include CPR and lack the membrane. It is important to note that the conformational status of CYP3A4 varies in nanodiscs vs detergent or lipid solution. In some solutions, CYP3A4 forms oligomers with functional and conformational differences compared to monomers, including differences in allosteric properties.48,49 Thus, the quantitative and qualitative differences between the binding isotherms we observe in nanodiscs vs the isotherms observed by others in the absence of lipid bilayers are expected, and this precludes comparison of the catalytic data with the spectral binding data in terms of conformational changes that dictate regioselectivity.
MDZ Metabolism by CYP3A4.
In order to provide context for the multiple MDZ binding and to relate these studies to the previously appreciated behavior of CYP3A4, we confirmed the concentration-dependent switch in regioselectivity of MDZ hydroxylation. Determination of kinetic constants, KM and Vmax, using CYP3A4 and CPR followed the procedure of Shaw et al.29 with the exception that only POPC lipid was used in the reconstitution system to match better the HDX-MS experiments where POPC nanodiscs were used. Data for the rate of product formation fit well to the Michaelis–Menten equation (Figure S1C), and the change in product ratio vs concentration is shown in Figure 1C. The results are consistent with, but modestly different from, other reports with similar reconstitution systems containing recombinant enzymes7,46 without the inhibition seen in human liver microsomes and supersomes.16,50,51 The KM values for 1′-OH MDZ and 4-OH MDZ formation were 17 and 389 μM, respectively. The Vmax values for 1′-OH MDZ and 4-OH MDZ formation were 3.5/min and 1.5/min, respectively. The different environments, with or without the redox partner or membrane, could contribute to differences in KM values and Ks based on spectral titration. The choice of MDZ concentrations for the H/DX MS studies below was based on the spectral titrations in nanodiscs.
HDX-MS Profile of CYP3A4 Nanodiscs.
CYP3A4 nanodiscs in different ligand-bound states (±MDZ) were analyzed by HDX-MS. After optimization of incubation and quench conditions, a sequence coverage of 88% with 70 unique peptides for CYP3A4 was achieved. Deuterium uptake profiles for the ligand-free state are consistent with previously published results.13 More detailed analyses and comparison with the MDZ-bound states are provided below. Individual deuterium uptake plots for specific peptides are shown in Figure S3.
sMDZ Binding Induces Changes in HDX-MS.
HDX was performed at two concentrations of MDZ, 6 μM or 60 μM. Although the HDX does not reveal changes due specifically to the first vs the second ligand, the binding data do suggest that, at 60 μM MDZ, the overwhelming majority of CYP3A4 has two MDZ molecules bound. A general observation based on the results is that, for the majority of peptides, there is no effect of 6 μM MDZ, suggesting that neither the first or second MDZ binding event causes dramatic changes in HDX for any peptides. At 60 μM MDZ, the MDZ-dependent changes in CYP3A4 dynamics are not limited to the active site but expand to distal regions of the enzyme.
Deuterium uptake plots for peptides with significant changes in HDX are shown in Figure 2.
Figure 2.
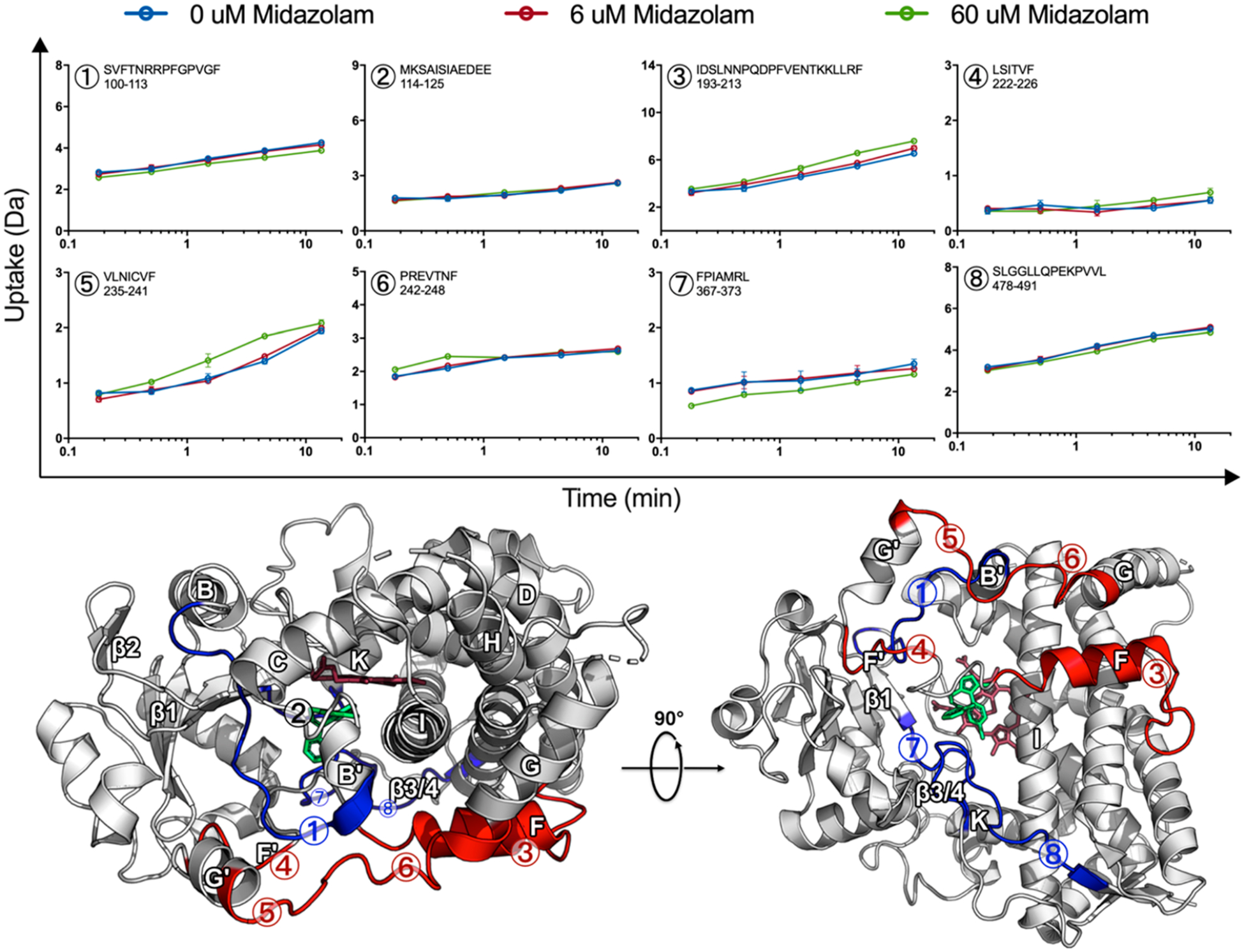
MDZ-induced changes are globally distributed. Uptake plots for selected peptides, 0 μM MDZ (blue), 6 μM MDZ (red), and 60 μM MDZ (green) with error bars representing the standard deviation for duplicate measurements. Differences are mapped onto the MDZ-bound CYP3A4 crystal structure (PDB ID: 5TE8), where the MDZ-induced increase and decrease in HDX for the 60 μM MDZ state are highlighted red and blue, respectively. Regions where no change was observed in HDX are highlighted in tan or white, while regions with no data are highlighted in black. The heme and MDZ are shown by stick representation in dark red and green, respectively.
The differences are mapped onto the MDZ-bound CYP3A4 crystal structure (PDB ID: 5TE8),2 where an increase or decrease in HDX relative to the ligand-free state is highlighted in red or blue, respectively. It should be emphasized that these changes are modest but reproducible across multiple CYP3A4 nanodisc preparations and separate experiments performed over the course of several years. The data shown are from same-day duplicate incubations that were injected onto the MS using optimized LC/MS methods such that all peptides were recovered from a single injection to allow for quantitative comparisons. The error for each time point is shown in the deuterium uptake plots and is usually smaller than the data points. Statistical significance was established by paired t tests for each time point across the different ligation states. Data for peptides 1, 3, and 5–8 yielded p < 0.05 for the comparison of 60 μM MDZ to 0 μM MDZ data. Data for peptide 4 approached p = 0.07 for the last two time points. There were no significant differences in the time points in peptide 2. Importantly, data for overlapping peptides, when available, displayed the same trends in differences, thus supporting the significance of the observed differences.
Regions affected by MDZ binding are globally distributed rather than confined to the active site. In our work, HDX indicates that the largest effects on dynamics upon multiple MDZ binding occur at the F- and G-helices and the intervening loops (Figure 2, peptides 3–6), as well as the loop connecting the K-helix and β1-sheet (peptide 7). There are modest effects at the BC loop as well as on the backside of the protein at the β3/4-sheets (peptides 1 and 8, respectively), which become more protected. Although no difference in HDX was observed at the C-terminus of the B–C loop (peptide 2), we include the uptake for this peptide in Figure 2 because it contains Ser-119, which forms a hydrogen bond with MDZ in the crystal structure.2
At the active site, peptides with residues in contact with the single MDZ in the crystal structure display changes in our HDX experiments at 60 μM MDZ. Noteworthy contacts in the crystal structure are present in peptides 2, 7, and 8 at the following residues: (A) Ser-119 from peptide 2 forms a hydrogen bond with N2 of MDZ, (B) Ile-369 and Ala-370 in peptide 7 contact the chlorophenyl ring of MDZ, and (C) Leu-482 in peptide 8 contacts the fluorophenyl group of MDZ. A decrease in HDX was observed for peptides 7 and 8, while no difference was observed for peptide 2. However, it must be emphasized that exchange at amino acid side chains in proteins is not quenched at a low pH, as an exchange at the backbone amide hydrogen bonds is. As a result, the exchange of deuterons at amino acid side chains is invisible to HDX, and only peptide amide hydrogen bonds are probed. Additionally, peptide 2 is a highly protected region in the ligand-free state, and any potential protective effect from MDZ might be masked in the time scale of the experiment. Notably, the region is also influenced by KTZ binding, which will be discussed in greater detail below.
At the membrane interface, the addition of 60 μM MDZ caused an increase in HDX at the F-, G-, and intervening helices (peptides 3−6), while a modest protective effect was observed at the opposite side of the membrane at the β3/4-sheets (peptide 8). Conformational changes at the membrane interface observed in the GaMD simulations will be discussed in greater detail below.
GaMD Simulations with MDZ in the Catalytic and Allosteric Sites.
The CYP3A4-POPC membrane system used for simulations with MDZ was similar to the system described previously to simulate TST binding to CYP3A4.6 While available, features of the structure of the CYP3A4-MDZ crystal structure (PDB ID: 5TE8) are not consistent with the ability of CYP3A4 to bind multiple ligands. In the structure, the F–F′ loop collapses into the active site and packs against MDZ, precluding binding of a second ligand. This distinct conformational feature was attributed to the MDZ-induced fit; however, the observed collapse of a hydrophobic region into the active site is also likely due in part to the high ionic strength required to obtain the crystal. Binding studies at similarly high ionic strengths, supporting hydrophobic collapse, also occur in solution, yielding hyperbolic binding curves consistent with a single MDZ binding site. Kd values of other ligands also shift to lower values with increasing ionic strength, demonstrating that the phenomenon is not unique to MDZ. We attribute the distinct conformational features of the 5TE8 structure, in part, to ionic strength-induced hydrophobic collapse rather than the ligand-induced fit. This proposal is further supported by the inability to crystallize the complex at a lower ionic strength due to MDZ’s diffusion from the crystal, as noted by the authors. Had these structures been reported, we speculate that they would have revealed a more open active site. Therefore, we used the ligand-free 1TQN structure of CYP3A4 as it was obtained at an ionic strength comparable to the solution studies described herein and had a more open active site consistent with multiple ligand binding. The steric clash of the F–F′ loop and the allosteric MDZ is shown in Figure S5, where the local structure of the 5TE8 structure is overlaid with 3 snapshots from MD simulations with 2 MDZ starting from the 1TQN crystal structure.
For the MD studies here, 100 ns of conventional MD (cMD) simulations were performed without a ligand, with one MDZ oriented in the active site, and with MDZ oriented in the active site, as well as in a probable entrance channel (Figure S1). Prior to the cMD simulations, the MDZ occupying the catalytic site was initially subject to a short simulation, where it was restrained to reproduce the orientation of MDZ observed in the crystal structure.2 The end points of the MDZ-free simulation as well those with one and two MDZ served as the initial configurations for each independent Gaussian accelerated molecular dynamics (GaMD) simulations. GaMD affords sampling of rare events such as infrequent conformational changes or ligand binding events that occur on longer time scales not accessible by cMD. GaMD achieves more extensive conformational sampling by lowering the energetic barriers and thereby increasing the likelihood of escape from energetic basins.52
Comparison of the backbone amide nitrogen root-mean-square deviations calculated for each group of GaMD trajectories reveals only a modest correlation between the MDZ-induced changes in RMSD (Figure 3) and the regions that exhibit a change in HDX in the mass spectrometry experiments (Figure 2). Apparently, the second MDZ affects backbone dynamics in several spatially distributed peptides.
Figure 3.
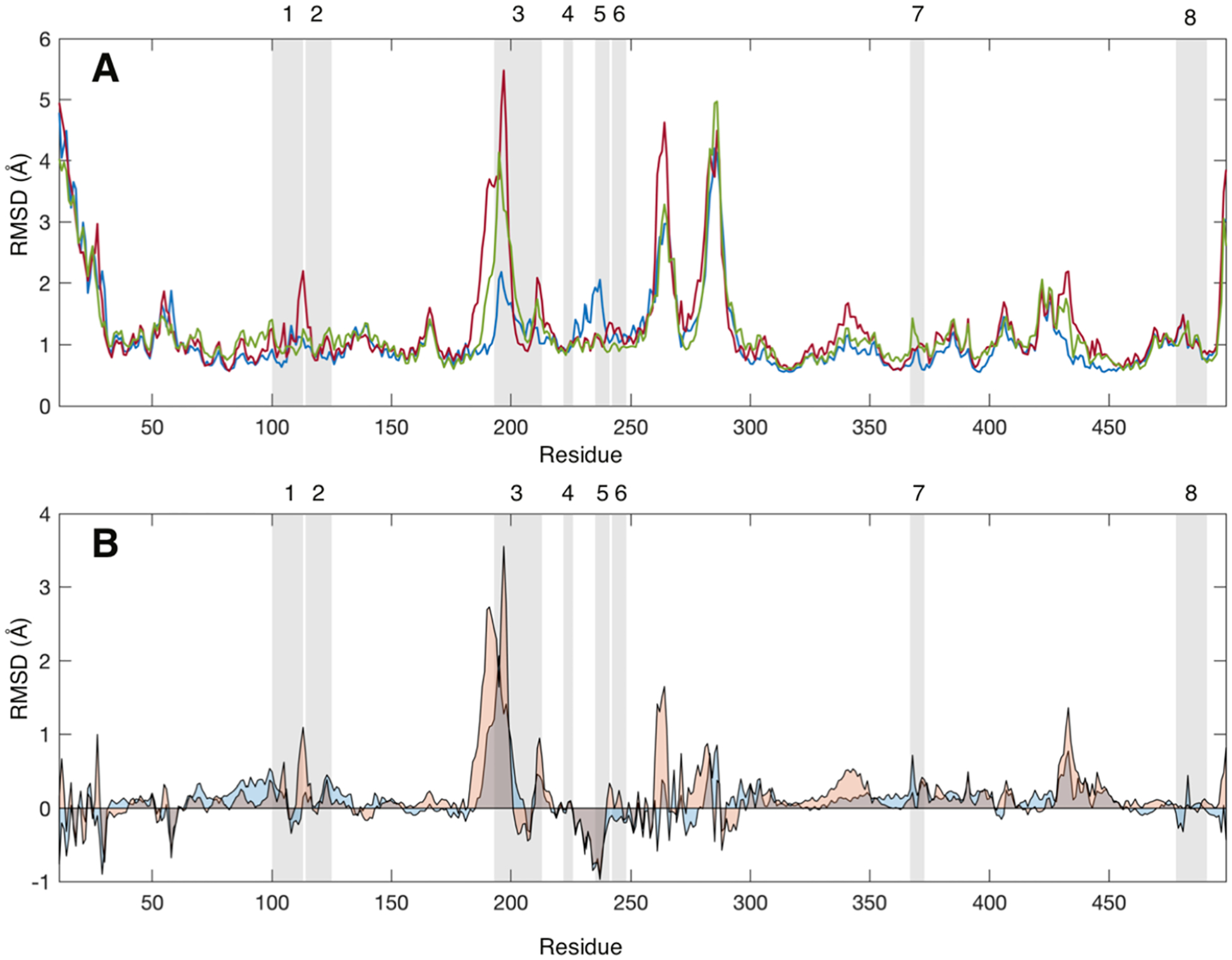
(A) Average root-mean-square deviation (RMSD, Å) values for each CYP3A4 residue without a ligand (blue), with one MDZ (red), and with two MDZ (green) bound in the active site. (B) Average RMSD values for each CYP3A4 with one MDZ (blue) or two MDZ (orange) less the average values for the ligand-free enzyme. RMSD values were calculated relative to the average structure over the entire trajectory. Gray-shaded regions highlight the peptides identified in the mass spectrometry experiments. RMSD values were calculated using the CA, CB C, O, N, HA, HA1, HA2, and HN atoms for all frames in the latter 0.4 μs of each simulation.
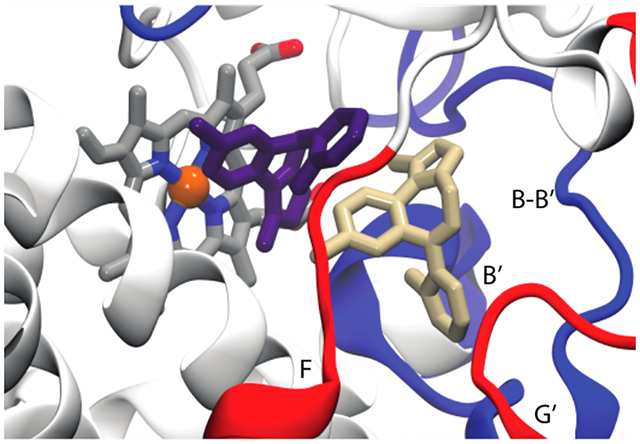
In contrast, there is a correlation between the peptides affected in H/DX experiments and those contributing to the allosteric MDZ site in Figure 4. Of the very few peptides that show a change in H/DX (peptides 1, 3, 4, 5, 6, and 7), essentially all are contained within or directly adjacent to the binding site for the second MDZ. However, there are additional peaks or troughs in Figure 2B that are not in the binding site, some of which correspond to peptides for which we do not have coverage in the H/DX experiments. The effects of MDZ on the RMSD of these peptides are interesting inasmuch as they are varied with respect to the direction of change. For example, peptide 3 exhibits a large increase in RMSD, with either one or two MDZs present, and an increase in HDX at later times with two MDZs. In contrast, peptide 7 and the edge of peptide 1 exhibit an MDZ-dependent increase in RMSD, but they exhibit a decrease in HDX. Peptide 5 yields a decrease in RMSD but a pronounced increase in HDX. It is interesting that there are more “positive differences” in Figure 2B for the doubly bound state than negative differences, suggesting the enzyme is more dynamic in these peptides when two MDZ’s are bound.
Figure 4.
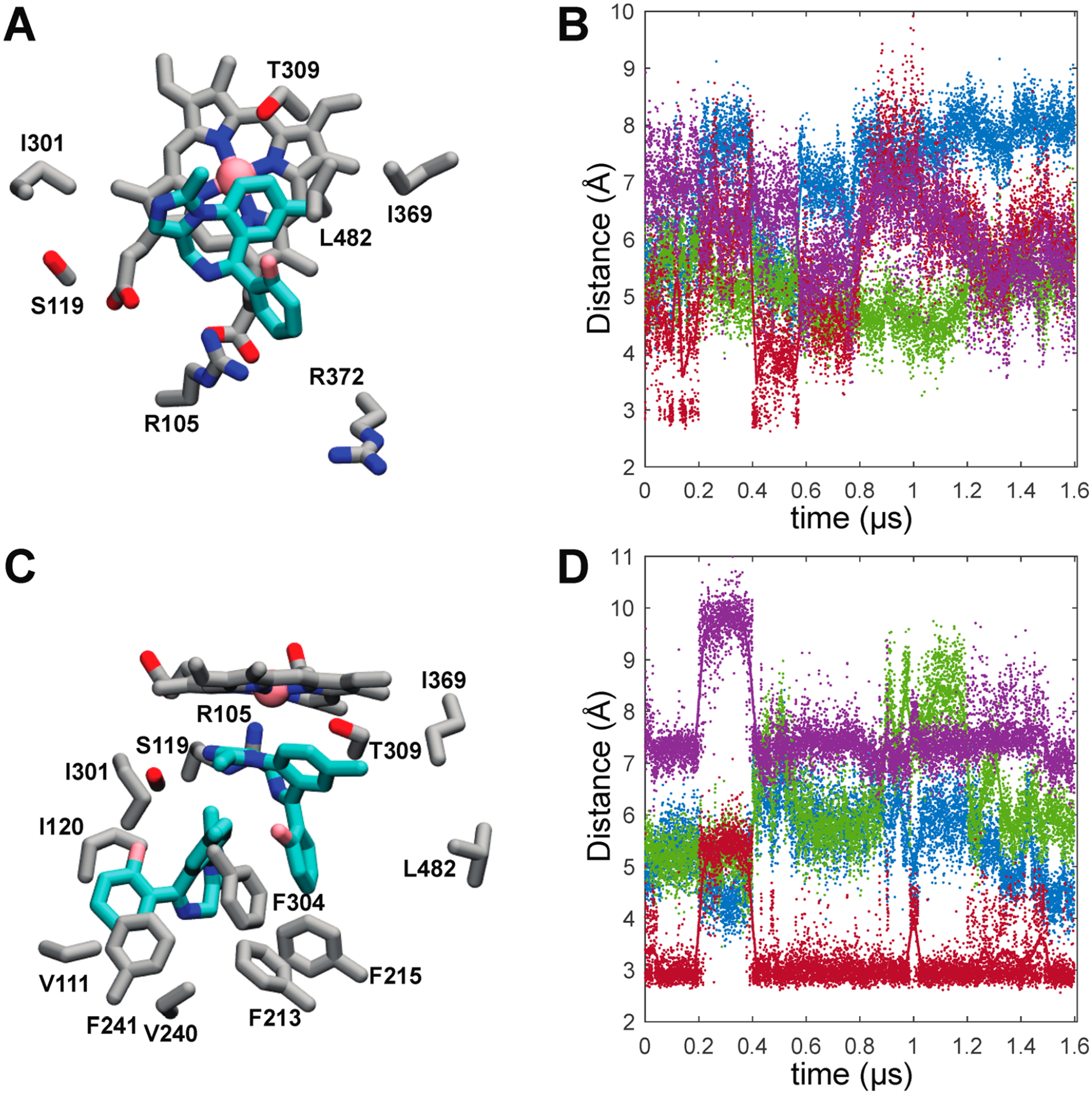
Representative MDZ binding modes and time dependences of active site MDZ-CYP3A4 interactions. (A) Representative MDZ binding mode from the single-MDZ system with side chains of residues that contact MDZ in 90% or more trajectory frames. (B) Time dependence of active site MDZ-CYP3A4 distances in the single-MDZ simulations. (C) Representative MDZ binding modes from the double-MDZ simulation with side chains of residues that contact each MDZ in 90% or more trajectory frames. (D) Time dependence of the active site MDZ-CYP3A4 distances with two MDZ bound. In plots B and D, MDZ N2-Ser119 Oγ (red), MDZ N5-Ser119 Oγ (purple), MDZ C4-Heme Fe (green), and the MDZ C1-Heme Fe (blue) distances are illustrated with solid points.
The differences in the direction of the effects of MDZ on HDX vs RMSD in some cases amplify the uncertainty about the factors that control H/D exchange rates in a membrane environment where a local charge may be a critical determinant and could oppositely affect backbone dynamics and H/D exchange.53 An additional source of the distinction between the approaches is that the changes reflected in the MD simulation are sampling time regimes that are outside the scope of the HDX experiments54 and vice versa. As shown by others,54,55 it is possible, even expected, that MD and HDX could exhibit apparently opposite effects of ligands because they sample such different time scales. For example, ligands could damp motion on fast time scales for some peptides but increase slow domain motions that increase HDX in the same peptides on longer time scales. Therefore, for the sake of locating the allosteric MDZ site, the most relevant comparison from MD and HDX is which peptides are affected, rather than the direction of the change. The overlap of peptides that are affected in the HDX at 60 μM MDZ and those affected in the GaMD upon the addition of the second MDZ suggests that the observed changes in HDX at 60 μM MDZ are due primarily to occupancy of the second, allosteric, site.
An essential point is that all of the CYP3A4 residues that make close contact with MDZ in the crystal structure also do so with overwhelming frequency in the simulations despite starting the simulations from a different structure (Figure 4A). However, when one MDZ is bound, it is highly mobile and rarely assumes the same binding mode that was observed in the crystal. This mobility is evident in Figure 4B that illustrates the distances between the MDZ nitrogens and Ser-119 as well as the observed sites of oxidation (1′ and 4) and the heme iron. The hallmark of the CYP3A4-MDZ crystal structure is an apparent hydrogen bond between the N2 of MDZ and the Ser-119 OγH. Despite enforcing this hydrogen bond at the outset of the simulations to establish the crystallographic binding mode, this hydrogen bond is only transiently maintained, appearing in less than ~25% of the combined trajectories with an average distance of 5.6 ± 1.2 Å. The MDZ N5 is not oriented for hydrogen bonding to Ser-119 either, though it occasionally hydrogen bonds with the guanidinium side chain of Arg-105 (Figure S5). As evidenced by the sparsity of the S119 hydrogen bond, the absence of the collapsed F–F′ loop and the more open active site provides an expanded volume for the single MDZ to explore.
When a second MDZ is introduced into a possible entrance channel bound by the β1−β2 loop and F′ helix, it rapidly moves into a binding site adjacent to the first MDZ surrounded by residues of the B–C loop and the Phe-cluster (Figure 4C). Many of the residues in contact with the second MDZ such as Ile-120, Ile-301, Phe-241, and Phe-304 were also predicted to partially define an intermediate state on TST’s binding path.6 It was proposed that the intermediate site is also an allosteric site that could be occupied at high TST concentrations. The introduction of a second MDZ into the active site substantially attenuates the mobility of the first. However, more remarkable is the recovery of the MDZ N2-Ser119 OγH hydrogen bond observed in the crystal, evidenced by an average distance between the relevant oxygen and nitrogen atoms of 3.4 ± 0.8 Å (Figure 4D). Recovery of the S119 hydrogen bond observed in the crystal structure is readily explained by comparing the 5TEQ structure with snapshots from the MD simulations (Figure S4). The hydrophobic collapse of the F–F′ loop in the 5TEQ structure orients several residues, most notably Leu-216, into the same volume occupied by the allosteric MDZ. Despite their structural dissimilarity, the addition of a second MDZ enforces the same binding mode and protein interactions and similarly limits the orientational heterogeneity of the first MDZ as filling this space with residues of the F–F′ loop.
The simulations reveal MDZ-dependent, correlated changes in interactions between several CYP3A4 regions, namely, regions including peptides 1, 4, and 5. The B–B′ loop, including peptide 1, undergoes distinct conformational changes in the presence of the second MDZ. This is at least partially attributable to direct interactions with the MDZ occupying the allosteric site where it primarily packs against the Val-111 side chain (Figure 4C) in more than 90% of the trajectory and transiently accepts an apparent hydrogen bond at the N5 position from the Gly-109 backbone amide.
What is more striking is that the MDZ occupation of the allosteric site stabilizes a bidentate hydrogen bond between Glu-234 on the G′-helix and the backbone amides of Val-111 and Gly-112. (Figure 5A,B) This interaction impacts the local fold of the B−B′ loop (Figure 5C–F) and induces changes in dynamics that propagate to peptides 4 and 5 to impact the interactions of these regions with the membrane. Figure 5G,H illustrates the cumulative probabilities of finding the mass-weighted centers of peptides 4 and 5 in the bilayer. For reference, these are superimposed on the average positions of the POPC membrane acylglycerols and phosphates. Remarkably, while the occupation of the catalytic site alone results in a small disengagement of peptide 4 from the membrane and small changes in the position of peptide 5, the occupation of the allosteric site by a second MDZ shifts both of these peptides deeper into the membrane, but they do not become immobilized.
Figure 5.
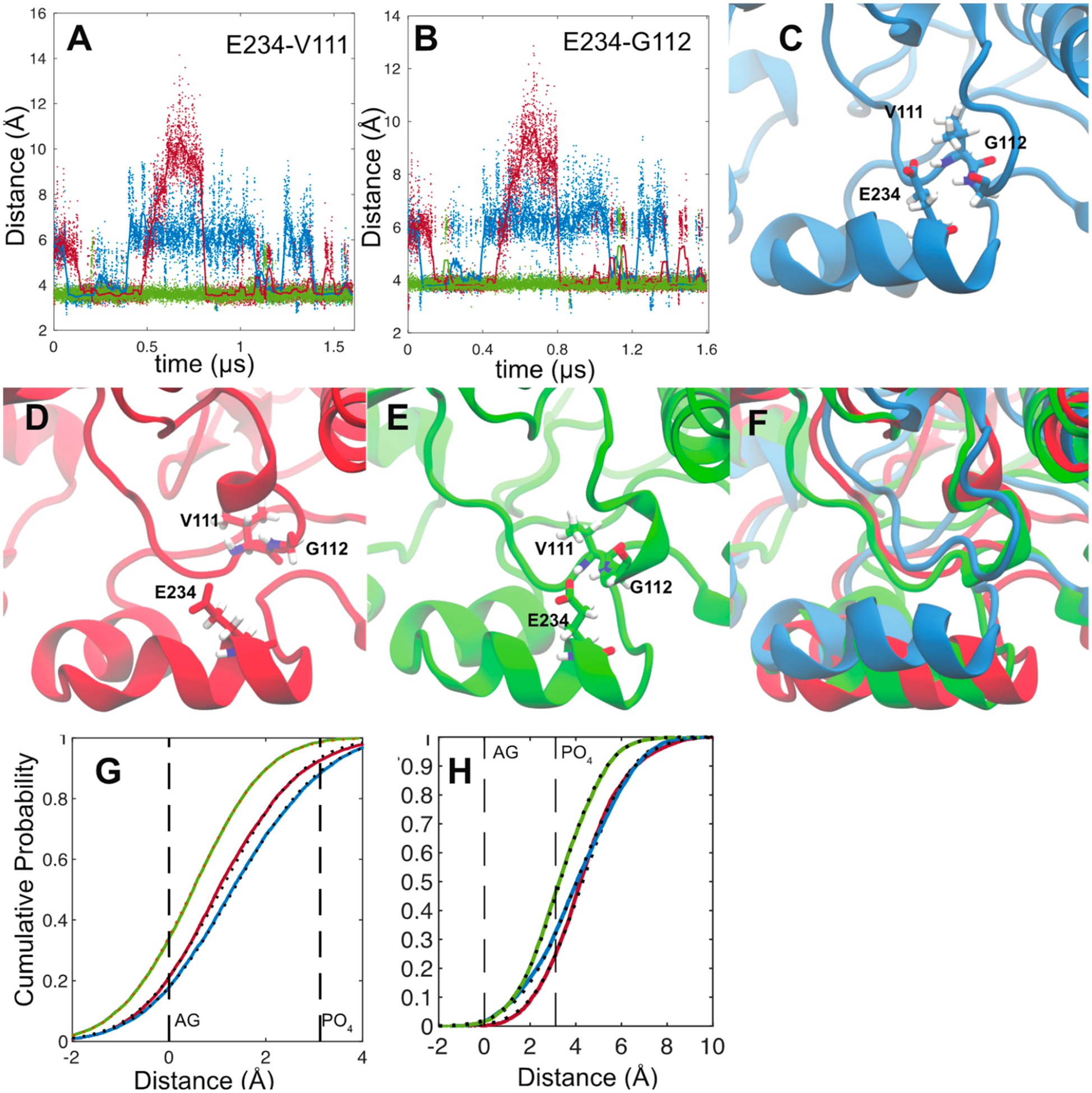
Distances between the Glu-234 carboxylate center of mass (calculated using Cδ and both O atoms) and the backbone amide nitrogen of Val-111 (A) and Gly-112 (B). Distances from the ligand-free (blue), single-MDZ (red), and double-MDZ (green) simulations are illustrated as solid points. Solid lines represent a 30 ns moving average of the distances. Representative snapshots from ligand-free (C), single-MDZ (D), and double-MDZ (E) simulations. The three snapshots are superimposed in panel F. Cumulative probability plots of the distance of peptide 4 (222–226) (G) and peptide 5 (235–241) (H) centers of mass from the average position of the POPC acylglycerols (AGs) and phosphates (PO4). Distance data for from the ligand-free (blue), single-MDZ (red), and double-MDZ (green) simulations are illustrated with solid points. Fits to a normal distribution are illustrated with black-hashed lines. Peptide centers of mass were calculated using the weighted masses of the peptide backbone C, O, N, H, Cα, and Cβ atoms. AG centers of mass were calculated using the C1, C2, C3, O11, O21, and O31 atoms. Phosphate centers of mass were calculated using the P, O11, O12, O13, and O14 atoms. AG and phosphate atom numbers are from the CHARMM 36 lipid topology.
The striking result that emerges from the GaMD is that the first MDZ bound at the active site is highly mobile and makes transient interactions with several active site residues. Upon addition of the second MDZ that binds in the allosteric site, the active site MDZ becomes significantly less mobile. In contrast, the allosteric MDZ remains dynamic. The RMSD for the distance between the center of mass of each MDZ is shown in Figure 6. The allosteric MDZ is highly mobile, and its movement toward and away from the active is coupled to rearrangements in the Phe-cluster, whereas the active site MDZ remains relatively static. Thus, the GaMD results suggest that the ligand in allosteric sites remains dynamic.
Figure 6.
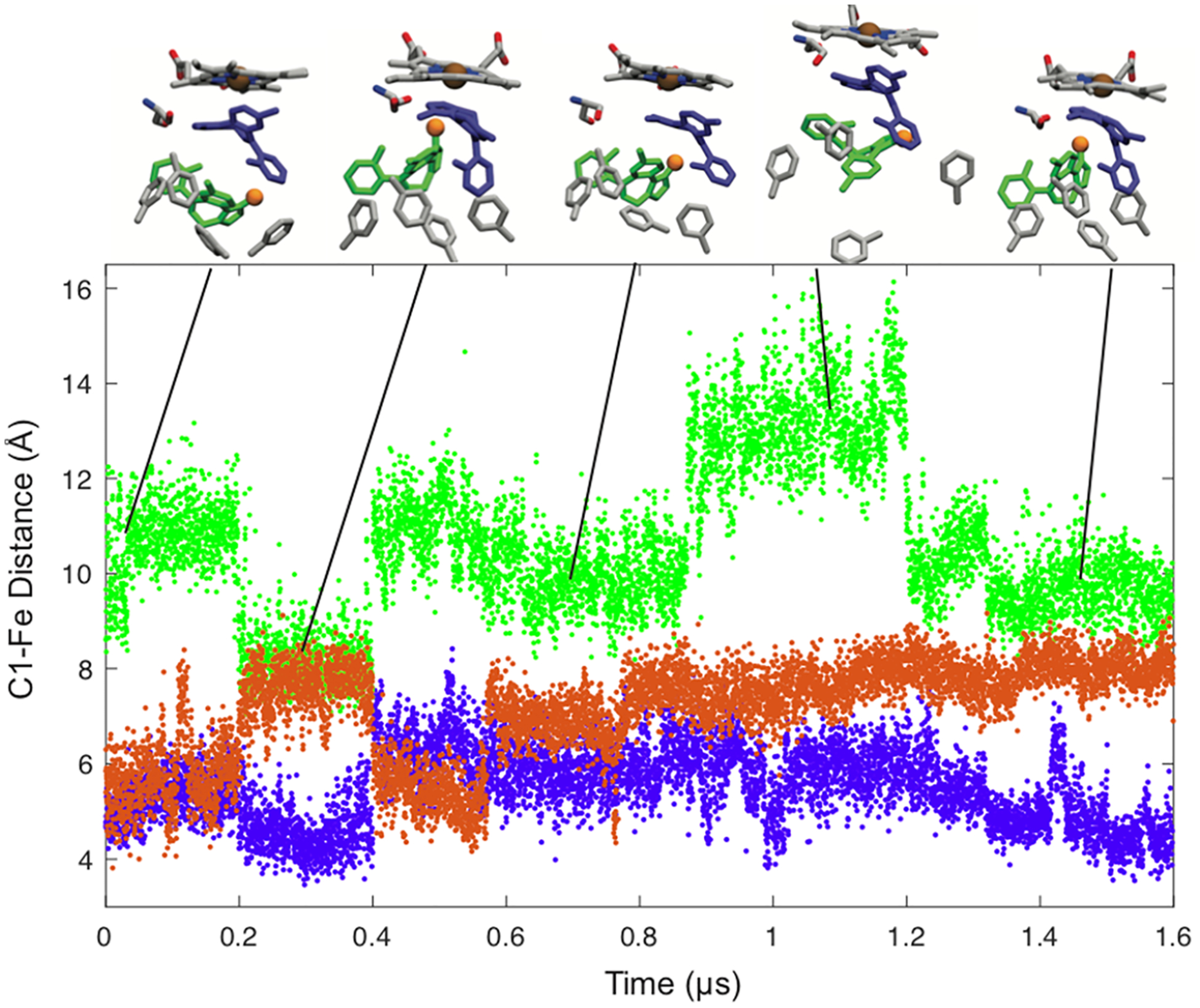
Distances between MDZ C1′ and the heme iron atom in the simulation with one MDZ (orange) and the simulation with an MDZ near the heme (purple) and in the allosteric site (green). Snapshots of the two-MDZ simulations at 0.08, 0.30, 0.66, 1.1, and 1.5 us are the inset. The positions of Ser119 and residues of the Phe-cluster (213, 215, 241, 304) are shown with the position of the C1 in the allosteric site MDZ highlighted with an orange sphere.
Comparison of MDZ and Ketoconazole effects on HDX.
A comparison between the MDZ and previously documented KTZ-induced changes in HDX reveal that these ligands differently affect the dynamics of CYP3A4. The regions with the largest changes in HDX are mapped onto the ligand-free CYP3A4 crystal structure in Figure 7 (PDB ID: 1TQN). Peptides at the F- and G- helices that are exclusively altered by MDZ (increase in HDX) or KTZ (decrease in HDX) are highlighted in red or blue, respectively. Both ligands cause a decrease in uptake at the loop preceding the β1-sheets for both ligands, which is highlighted in purple. At the F′-helix, MDZ binding results in an increase in HDX, while the opposite effect is observed when KTZ is bound, highlighted in green. It is interesting that HDX is dramatically different at the F- and G- helices, given their potential role in ligand entry/exit channels.
Figure 7.
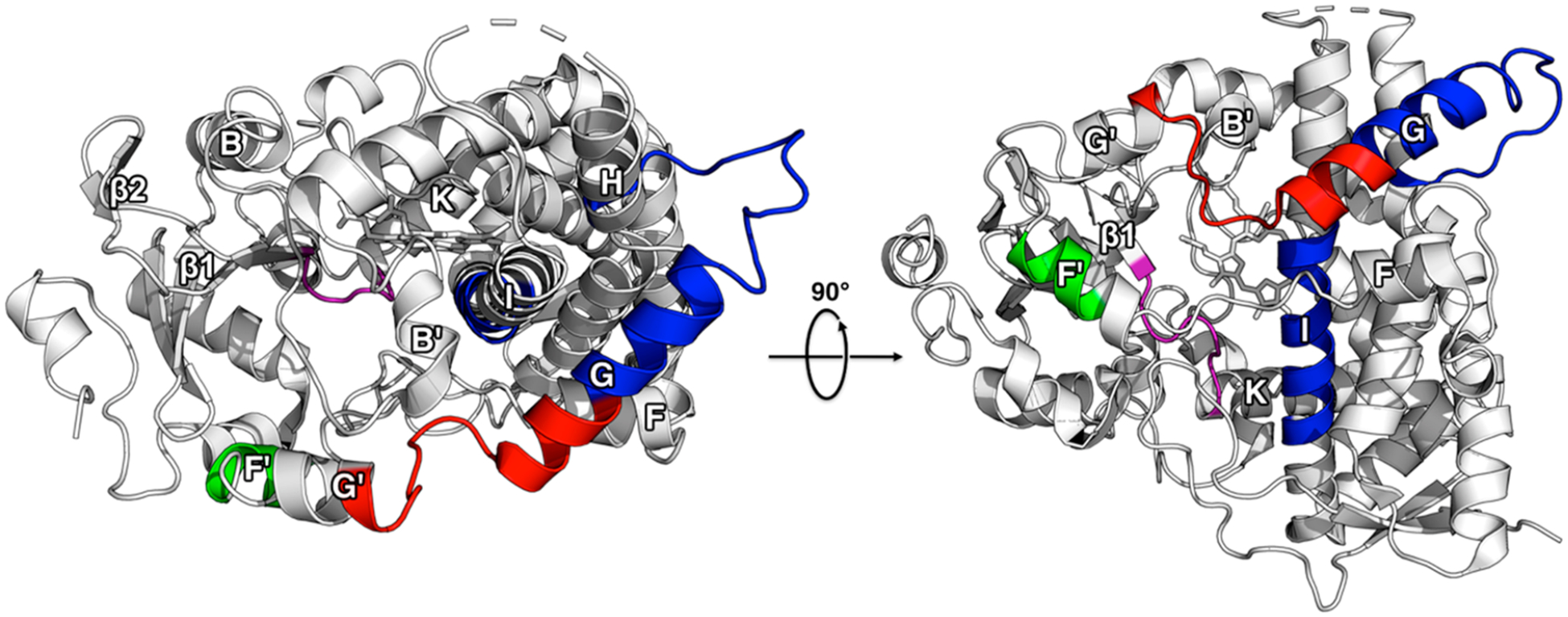
Comparison of HDX changes from MDZ and KTZ binding. The changes are mapped onto the ligand-free crystal structure of CYP3A4 (PDB ID: 1TQN). Regions where KTZ binding induces a decrease in HDX relative to ligand-free CYP3A4 are highlighted in blue, green, and purple. Regions where MDZ binding induces an increase in HDX relative to ligand-free CYP3A4 are highlighted in red and green, while regions where a decrease in HDX occurs are highlighted in purple.
CONCLUSIONS
The location and dynamic nature of the allosteric MDZ site are noteworthy. The GaMD and the HDX suggest that the Phe-cluster and its local environment comprise a dynamic site between the lipid bilayer and the active site. This site has been suggested to accommodate several allosteric ligands and to mediate allosteric effects.7–9 The effects of the allosteric MDZ on the Phe-cluster are essentially opposite to the effects caused by KTZ in this region, based on H/DX. Apparently, the promiscuity that is well appreciated for the active site of CYP3A4 is paralleled by the promiscuity of the allosteric site. Interestingly, allosteric sites used for regulation of enzymes in well-defined metabolic pathways are typically highly specific. The current results suggest a possible mechanistic basis for a promiscuous allosteric site, wherein ligand binding, either at the active site or the allosteric site, increases the local dynamics of the latter and generates a highly fluid binding site directly adjacent to the active site. The fact that an allosteric site can remain dynamic or fluid even when occupied by a ligand, as suggested by the HDX and GaMD, is consistent with a highly promiscuous allostery, which seems to be the case with CYP3A4. Alternatively, the changes in H/DX for peptides comprising the allosteric site could be due to their movement deeper into the membrane, which changes the local electro-static environment and affects the H/DX, or a combination of both electrostatics and dynamics.
Supplementary Material
ACKNOWLEDGMENTS
We would like to thank Miklos Guttman and Amanda Clouser for thoughtful discussions.
Funding
This work was supported by grants from the National Institutes of Health awarded to W.M.A. (R01GM130810) and J.C.H. (R01GM114168) as well as a generous allocation of computational resources from the Ohio Supercomputer Center.
ABBREVIATIONS
- δ-ALA
delta-aminolevulinic acid
- ACN
acetonitrile
- AG
acylglycerols
- BME
β-mecaptoethanol
- CHAPS
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- cMD
conventional molecular dynamics
- CYP3A4
cytochrome p450 3A4
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- FA
formic acid
- GaMD
Gaussian accelerated molecular dynamics
- HA
hydroxyapatite (type I)
- HDX-MS
hydrogen–deuterium exchange mass spectrometry
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- IMZ
imidazole
- IPTG
isopropyl β-D-1-thiogalactopyranoside
- KPi
potassium phosphate
- KTZ
ketoconazole
- LB
Luria–Bertani
- MDZ
midazolam
- MDZ
midazolam
- MSP1D1
membrane scaffold protein 1D1
- PO4
phosphates
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- rCPR
rat NADPH-cytochrome P450 oxido-reductase
- TCEP
tris(2-carboxyethyl)-phosphine
- TFA
trifluoroacetic acid
- TST
testosterone
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.9b01001.
Initial position of the second MDZ for two-MDZ simulations; binding data fit to the Michaelis–Menten equation and Eadie–Hofstee plot; Michaelis–Menten plots for 1′-OH and 4-OH MDZ formation; deuterium uptake profiles for all peptides; overlay of CYP3A4 5TE8 stricture with one MDZ bound and snapshots from MD simulations showing steric clash in the 5TE8 structure and allosteric MDZ in the MD; distances between the Arg-105 Cζ and the MDZ1 N5 and center of the MDZ1 fluorophenyl ring (PDF)
Accession Codes
Cytochrome P450 3A4 (CYP3A4): P08684.
The authors declare no competing financial interest.
Contributor Information
Michelle Redhair, Department of Medicinal Chemistry, University of Washington, Seattle, Washington 98195-7610, United States.
John C. Hackett, Department of Physiology and Biophysics and the Massey Cancer Center, School of Medicine, Virginia Commonwealth University, Richmond, Virginia 23298-0035, United States
Robert D. Pelletier, Department of Medicinal Chemistry, University of Washington, Seattle, Washington 98195-7610, United States
William M. Atkins, Department of Medicinal Chemistry, University of Washington, Seattle, Washington 98195-7610, United States.
REFERENCES
- (1).Ekroos M, and Sjögren T (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A 103, 13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Sevrioukova IF, and Poulos TL (2017) Structural basis for regiospecific midazolam oxidation by human cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A 114, 486–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Vogt AD, and Di Cera E (2012) Conformational selection or induced fit? A critical appraisal of the kinetic mechanism. Biochemistry 51, 5894–5902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).McClary WD, Sumida JP, Scian M, Paco L, and Atkins WM (2016) Membrane Fluidity Modulates Thermal Stability and Ligand Binding of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 55, 6258–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Baylon JL, Lenov IL, Sligar SG, and Tajkhorshid E (2013) Characterizing the membrane-bound state of cytochrome P450 3A4: structure, depth of insertion, and orientation. J. Am. Chem. Soc 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hackett JC (2018) Membrane-embedded substrate recognition by cytochrome P450 3A4. J. Biol. Chem 293, 4037–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Khan KK, He YQ, Domanski TL, and Halpert JR (2002) Midazolam oxidation by cytochrome P450 3A4 and active-site mutants: an evaluation of multiple binding sites and of the metabolic pathway that leads to enzyme inactivation. Mol. Pharmacol 61, 495–506. [DOI] [PubMed] [Google Scholar]
- (8).Davydov DR, Rumfeldt JA, Sineva EV, Fernando H, Davydova NY, and Halpert JR (2012) Peripheral ligand-binding site in cytochrome P450 3A4 located with fluorescence resonance energy transfer (FRET). J. Biol. Chem 287, 6797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, and Sligar SG (2015) Mechanism of Drug-Drug Interactions Mediated by Human Cytochrome P450 CYP3A4Monomer. Biochemistry 54, 2227–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Denisov IG, Baylon JL, Grinkova YV, Tajkhorshid E, and Sligar SG (2018) Drug-Drug Interactions between Atorvastatin and Dronedarone Mediated by Monomeric CYP3A4. Biochemistry 57, 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Englander SW (2006) Hydrogen exchange and mass spectrometry: A historical perspective. J. Am. Soc. Mass Spectrom 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Marcsisin SR, and Engen JR (2010) Hydrogen exchange mass spectrometry: what is it and what can it tell us? Anal. Bioanal. Chem 397, 967–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Redhair M, Clouser AF, and Atkins WM (2019) Hydrogen-deuterium exchange mass spectrometry of membrane proteins in lipid nanodiscs. Chem. Phys. Lipids 220, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Wilderman PR, Shah MB, Liu T, Li S, Hsu S, Roberts AG, Goodlett DR, Zhang Q, Woods VL Jr., Stout CD, and Halpert JR (2010) Plasticity of cytochrome P450 2B4 as investigated by hydrogen-deuterium exchange mass spectrometry and X-ray crystallography. J. Biol. Chem 285, 38602–38611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Treuheit NA, Redhair M, Kwon H, McClary WD, Guttman M, Sumida JP, and Atkins WM (2016) Membrane Interactions, Ligand-dependent Dynamics, and Stability of Cytochrome P4503A4 in Lipid Nanodiscs. Biochemistry 55, 1058–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Roberts AG, Yang J, Halpert JR, Nelson SD, Thummel KT, and Atkins WM (2011) The Structural Basis for Homotropic and Heterotropic Cooperativity of Midazolam Metabolism by Human Cytochrome P450 3A4. Biochemistry 50, 10804–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yang J, Atkins WM, Isoherranen N, Paine MF, and Thummel KE (2012) Evidence of CYP3A Allosterism In Vivo: Analysis of Interaction Between Fluconazole and Midazolam. Clin. Pharmacol. Ther 91, 442–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Gillam E, Guo ZY, and Guengerich FP (1994) Expression of modified human cytochrome P450 2E1 in Escherichia coli purification, and spectral and catalytic properties. Arch. Biochem. Biophys 312, 59–66. [DOI] [PubMed] [Google Scholar]
- (19).Domanski TL, Liu J, Harlow GR, and Halpert JR (1998) Analysis of four residues within substrate recognition site 4 of human cytochrome P450 3A4: role in steroid hydroxylase activity and alpha-naphthoflavone stimulation. Arch. Biochem. Biophys 350, 223–232. [DOI] [PubMed] [Google Scholar]
- (20).Woods CM, Fernandez C, Kunze KL, and Atkins WM (2011) Allosteric activation of cytochrome P450 3A4 by α-naphthoflavone: branch point regulation revealed by isotope dilution analysis. Biochemistry 50, 10041–10051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Matsubara T, Koike M, Touchi A, Tochino Y, and Sugeno K (1976) Quantitative determination of cytochrome P-450 in rat liver homogenate. Anal. Biochem 75, 596–603. [DOI] [PubMed] [Google Scholar]
- (22).Rutten AA, Falke HE, Catsburg JF, Topp R, Blaauboer BJ, van Holsteijn I, Doorn L, and van Leeuwen FX (1987) Interlaboratory comparison of total cytochrome P-450 and protein determinations in rat liver microsomes. Reinvestigation of assay conditions. Arch. Toxicol 61, 27–33. [DOI] [PubMed] [Google Scholar]
- (23).Omura T, and Sato R (1964) The carbon monoxide-binding pigment of liver microsomes. I. Evidence for its hemoprotein nature. J. Biol. Chem 239, 2370–2378. [PubMed] [Google Scholar]
- (24).Baas BJ, Denisov IG, and Sligar SG (2004) Homotropic cooperativity of monomeric cytochrome P450 3A4 in a nanoscale native bilayer environment. Arch. Biochem. Biophys 430, 218–228. [DOI] [PubMed] [Google Scholar]
- (25).Shen AL, Porter TD, Wilson TE, and Kasper CB (1989) Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J. Biol. Chem 264, 7584–7589. [PubMed] [Google Scholar]
- (26).French JS, and Coon MJ (1979) Properties of NADPH-cytochrome P-450 reductase purified from rabbit liver microsomes. Arch. Biochem. Biophys 195, 565–577. [DOI] [PubMed] [Google Scholar]
- (27).Nath A, Grinkova YV, Sligar SG, and Atkins WM (2007) Ligand binding to cytochrome P450 3A4 in phospholipid bilayer nanodiscs: the effect of model membranes. J. Biol. Chem 282, 28309–28320. [DOI] [PubMed] [Google Scholar]
- (28).Lefurgy ST, and Leyh TS (2012) Analytical expressions for the homotropic binding of ligand to protein dimers and trimers. Anal. Biochem 421, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Shaw PM, Hosea NA, Thompson DV, Lenius JM, and Guengerich FP (1997) Reconstitution premixes for assays using purified recombinant human cytochrome P450, NADPH-cytochrome P450 reductase, and cytochrome b5. Arch. Biochem. Biophys 348, 107–115. [DOI] [PubMed] [Google Scholar]
- (30).Hebling CM, Morgan CR, Stafford DW, Jorgenson JW, Rand KD, and Engen JR (2010) Conformational Analysis of Membrane Proteins in Phospholipid Bilayer Nanodiscs by Hydrogen Exchange Mass Spectrometry. Anal. Chem 82, 5415–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Adhikary S, Deredge DJ, Nagarajan A, Forrest LR, Wintrode PL, and Singh SK (2017) Conformational dynamics of a neurotransmitter:sodium symporter in a lipid bilayer. Proc. Natl. Acad. Sci. U. S. A 114, E1786–E1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Li MJ, Guttman M, and Atkins WM (2018) Conformational dynamics of P-glycoprotein in lipid nanodiscs and detergent micelles reveal complex motions on a wide time scale. J. Biol. Chem 293, 6297–6307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fang J, Rand KD, Beuning PJ, and Engen JR (2011) False EX1 signatures caused by sample carryover during HX MS analyses. Int. J. Mass Spectrom 302, 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Weis DD, Engen JR, and Kass IJ (2006) Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom 17, 1700–1703. [DOI] [PubMed] [Google Scholar]
- (35).Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, and Baker NA (2007) PDB2PQR: expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 35, W522–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Bas DC, Rogers DM, and Jensen JH (2008) Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins: Struct., Funct., Genet 73, 765–783. [DOI] [PubMed] [Google Scholar]
- (37).Kieseritzky G, and Knapp EW (2008) Improved pK(a) prediction: combining empirical and semimicroscopic methods. J. Comput. Chem 29, 2575–2581. [DOI] [PubMed] [Google Scholar]
- (38).Olsson MHM, Søndergaard CR, Rostkowski M, and Jensen JH (2011) PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput 7, 525–537. [DOI] [PubMed] [Google Scholar]
- (39).Klauda JB, Venable RM, Freites JA, O’Connor JW, Tobias DJ, Mondragon-Ramirez C, Vorobyov I, MacKerell AD, and Pastor RW (2010) Update of the CHARMM all-atom additive force field for lipids: validation on six lipid types. J. Phys. Chem. B 114, 7830–7843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, and Schulten K (2005) Scalable molecular dynamics with NAMD. J. Comput. Chem 26, 1781–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).MacKerell AD, Feig M, and Brooks CL (2004) Extending the treatment of backbone energetics in protein force fields: limitations of gas-phase quantum mechanics in reproducing protein conformational distributions in molecular dynamics simulations. J. Comput. Chem 25, 1400–1415. [DOI] [PubMed] [Google Scholar]
- (42).Vanommeslaeghe K, and MacKerell AD (2012) Automation of the CHARMM General Force Field (CGenFF) I: bond perception and atom typing. J. Chem. Inf. Model 52, 3144–3154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Feller SE, Zhang Y, Pastor RW, and Brooks BR (1995) Constant pressure molecular dynamics simulation: The Langevin piston method. J. Chem. Phys 103, 4613–4621. [Google Scholar]
- (44).Wang Y, Harrison CB, Schulten K, and McCammon JA (2011) Implementation of Accelerated Molecular Dynamics in NAMD. Comput. Sci. Discovery 4, 015002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Pang YT, Miao Y, Wang Y, and McCammon JA (2017) Gaussian Accelerated Molecular Dynamics in NAMD. J. Chem. Theory Comput 13, 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Hosea NA, Miller GP, and Guengerich FP (2000) Elucidation of distinct ligand binding sites for cytochrome P450 3A4. Biochemistry 39, 5929–5939. [DOI] [PubMed] [Google Scholar]
- (47).Qian H (2008) Cooperativity and specificity in enzyme kinetics: a single-molecule time-based perspective. Biophys. J 95, 10–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Davydov DR, Fernando H, Baas BJ, Sligar SG, and Halpert JR (2005) Kinetics of dithionite-dependent reduction of cytochrome P450 3A4: heterogeneity of the enzyme caused by its oligomerization. Biochemistry 44, 13902–13913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Davydov DR, Baas BJ, Sligar SG, and Halpert JR (2007) Allosteric mechanisms in cytochrome P450 3A4 studied by high-pressure spectroscopy: pivotal role of substrate-induced changes in the accessibility and degree of hydration of the heme pocket. Biochemistry 46, 7852–7864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Kronbach T, Mathys D, Umeno M, Gonzalez FJ, and Meyer UA (1989) Oxidation of midazolam and triazolam by human liver cytochrome P450IIIA4. Mol. Pharmacol 36, 89–96. [PubMed] [Google Scholar]
- (51).Williams JA, Ring BJ, Cantrell VE, Jones DR, Eckstein J, Ruterbories K, Hamman MA, Hall SD, and Wrighton SA (2002) Comparative metabolic capabilities of CYP3A4, CYP3A5, and CYP3A7. Drug Metab. Dispos 30, 883–891. [DOI] [PubMed] [Google Scholar]
- (52).Miao Y, Feher VA, and McCammon JA (2015) Gaussian Accelerated Molecular Dynamics: Unconstrained Enhanced Sampling and Free Energy Calculation. J. Chem. Theory Comput 11, 3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Shaw BF, Arthanari H, Narovlyansky M, Durazo A, Frueh DP, Pollastri MP, Lee A, Bilgicer B, Gygi SP, Wagner G, and Whitesides GM (2010) Neutralizing positive charges at the surface of a protein lowers its rate of amide hydrogen exchange without altering its structure or increasing its thermostability. J. Am. Chem. Soc 132, 17411–17425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Hamuro Y (2017) Determination of Equine Cytochrome c Backbone Amide Hydrogen/Deuterium Exchange Rates by Mass Spectrometry Using a Wider Time Window and Isotope Envelope. J. Am. Soc. Mass Spectrom 28, 486–497. [DOI] [PubMed] [Google Scholar]
- (55).Goswami D, Devarakonda S, Chalmers MJ, Pascal BD, Spiegelman BM, and Griffin PR (2013) Time window expansion for HDX analysis of an intrinsically disordered protein. J. Am. Soc. Mass Spectrom 24, 1584–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.