

Summary
The trinuclear ruthenium amine Ruthenium Red (RuR) inhibits diverse ion channels including K2P potassium channels, TRPs, the calcium uniporter, CALHMs, ryanodine receptors, and Piezos. Despite this extraordinary array, there is limited information for how RuR engages targets. Here, using X-ray crystallographic and electrophysiological studies of an RuR-sensitive K2P, K2P2.1 (TREK-1) I110D, we show that RuR acts by binding an acidic residue pair comprising the ‘Keystone inhibitor site’ under the K2P CAP domain archway above the channel pore. We further establish that Ru360, a dinuclear ruthenium amine not known to affect K2Ps, inhibits RuR-sensitive K2Ps using the same mechanism. Structural knowledge enabled a generalizable design strategy for creating K2P RuR ‘super-responders’ having nanomolar sensitivity. Together, the data define a ‘finger in the dam’ inhibition mechanism acting at a novel K2P inhibitor binding site. These findings highlight the polysite nature of K2P pharmacology and provide a new framework for K2P inhibitor development.
Graphical Abstract
TOC blurb
Polynuclear ruthenium amines inhibit diverse ion channel classes. Pope et al. describe the structural basis of K2P potassium channel inhibition by Ruthenium Red(RuR) and Ru360, defining a ‘finger in the dam’ mechanism highlighting K2P polysite pharmacology and providing a framework for understanding polynuclear amine recognition.
Introduction
Ruthenium red (RuR) (Fletcher et al., 1961) (Figure 1A) is a trinuclear oxo-bridged ruthenium amine polycation with many biological applications (Clarke, 2002), including a ~50 year legacy of use as an inhibitor of diverse ion channels, such as select members of the K2P (KCNK) family (Braun et al., 2015; Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006), numerous TRP channels (Arif Pavel et al., 2016; Caterina et al., 1999; Caterina et al., 1997; Guler et al., 2002; Story et al., 2003; Strotmann et al., 2000; Voets et al., 2004; Voets et al., 2002), the mitochondrial calcium uniporter (MCU) (Chaudhuri et al., 2013; Kirichok et al., 2004; Moore, 1971; Rahamimoff and Alnaes, 1973), CALHM calcium channels (Choi et al., 2019; Dreses-Werringloer et al., 2013; Ma et al., 2012), ryanodine receptors (Ma, 1993; Smith et al., 1988), and Piezo channels (Coste et al., 2012; Zhao et al., 2016). Despite this remarkably wide range of ion channel targets and the recent boom in ion channel structural biology, structural understanding of how RuR acts on any ion channel is limited to a recent cryo-EM study of the CALHM2 channel that provides few molecular details regarding the coordination chemistry that underlies RuR binding (Choi et al., 2019). In the case of K2Ps, functional studies have established that a negatively charged residue at the base of the K2P extracellular dimerization domain that forms an archway over the channel pore, the CAP domain, comprises a key RuR sensitivity determinant in the natively RuR sensitive channels K2P9.1 (TASK-3) (Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006) and K2P10.1 (TREK-2) (Braun et al., 2015). Further, installation of a negatively charged amino acid at the equivalent CAP domain site in a non-RuR sensitive channel is sufficient to confer RuR sensitivity (Braun et al., 2015). The archway above the selectivity filter extracellular mouth made by the K2P CAP domain creates a pair of water-filled portals, the extracellular ion pathway (EIP), through which ions exit the channel under physiological conditions (Brohawn et al., 2012; Dong et al., 2015; Lolicato et al., 2017; Miller and Long, 2012). Although the EIP has been proposed as the site of RuR action (Braun et al., 2015; Gonzalez et al., 2013), the mechanism by which RuR inhibits K2Ps remains unresolved and to date there is no direct structural evidence indicating that the EIP can be targeted by RuR or any other class of small molecule or protein-based inhibitors.
Figure 1. Functional and structural analysis of the K2P2.1 I110D:RuR complex.
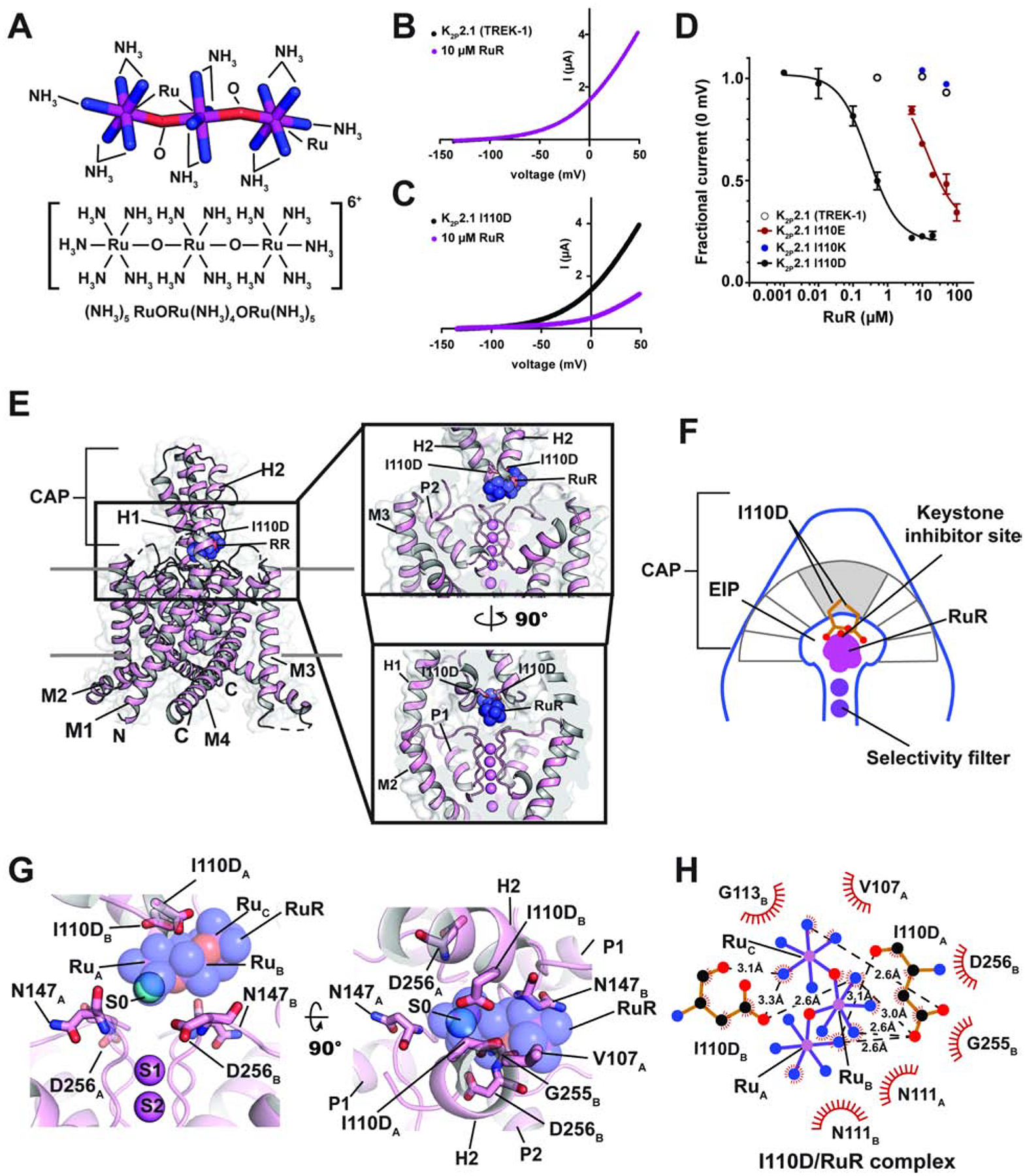
(A) Ruthenium Red (RuR) structure. (B) and (C) Exemplar TEVC recordings of (B) K2P2.1 (TREK-1) and (C) K2P2.1 I110D responses to 10 μM RuR (magenta). (D) Dose-response curves for K2P2.1 (TREK-1) (open white circles), K2P2.1 I110D (black), K2P2.1 I110E (red), and K2P2.1 I110K to RuR. (E) Structure of the K2P2.1 I110D:RuR complex. Inset shows the location of the RuR binding site. I110D is shown as sticks. (F) Cartoon depicting the Keystone inhibitor site. View is from the lower panel of ‘E’. The archway formed by the CAP is diagramed in grey. The keystone position is shaded. (G) Close up view of K2P2.1 I110D:RuR interactions. S0 ion (cyan) from the K2P2.1 I110D structure is indicated. Subscripts indicate Chain A and Chain B residues. RuR is shown in space filling in panels (E) and (G). (H) LigPLOT (Wallace et al., 1995) diagram of K2P2.1 I110D:RuR interactions showing ionic interactions (dashed lines) and van der Waals contacts (red) ≤5Å. See also Figures S1 and S2 and Table S1 and S2.
K2Ps produce an outward ‘leak’ potassium current that plays a critical role in stabilizing the resting membrane potential of diverse cell types in the nervous, cardiovascular, and immune systems (Enyedi and Czirjak, 2010; Feliciangeli et al., 2014; Renigunta et al., 2015). There are fifteen K2P subtypes comprising six subfamilies in which the channel monomers assemble into dimers wherein each subunit contributes two conserved pore forming domains to make the channel pore (Brohawn et al., 2012; Dong et al., 2015; Feliciangeli et al., 2014; Lolicato et al., 2017; Miller and Long, 2012; Rödström et al., 2019). A range of physical and chemical signals control K2P function (Enyedi and Czirjak, 2010; Feliciangeli et al., 2014; Renigunta et al., 2015) and various K2P subtypes have emerging roles in a multitude of physiological responses and pathological conditions such as action potential propagation in myelinated axons (Brohawn et al., 2019; Kanda et al., 2019), anesthetic responses (Heurteaux et al., 2004; Lazarenko et al., 2010), microglial surveillance (Madry et al., 2018), sleep duration (Yoshida et al., 2018), pain (Alloui et al., 2006; Devilliers et al., 2013; Vivier et al., 2017), arrythmia (Decher et al., 2017), ischemia (Heurteaux et al., 2004; Laigle et al., 2012; Wu et al., 2013), cardiac fibrosis (Abraham et al., 2018), depression (Heurteaux et al., 2006), migraine (Royal et al., 2019), intraocular pressure regulation (Yarishkin et al., 2018), and pulmonary hypertension (Lambert et al., 2018). Although there have been recent advances in identifying new K2P modulators (Bagriantsev et al., 2013; Lolicato et al., 2017; Pope et al., 2018; Su et al., 2016; Tian et al., 2019; Vivier et al., 2017; Wright et al., 2019) and in defining key structural aspects of K2P channel pharmacology (Dong et al., 2015; Lolicato et al., 2017; Schewe et al., 2019), as is the case with many ion channel classes, pharmacological agents targeting K2Ps remain poorly developed and limit the ability to probe K2P mechanism and biological functions (Sterbuleac, 2019).
Here, we present X-ray crystal structures of a K2P2.1 (TREK-1) mutant bearing a single change at the site that controls K2P channel RuR sensitivity, K2P2.1 I110D, alone and complexed with two different polynuclear ruthenium amines, RuR and the dinuclear ruthenium amine, Ru360 (Ying et al., 1991), an inhibitor of the mitochondrial calcium uniporter (Baughman et al., 2011; Kirichok et al., 2004; Oxenoid et al., 2016) not previously known to affect potassium channels. The structures show that the negatively charged residues at position 110 comprise ‘the Keystone inhibitor site’ on the ceiling of the CAP domain archway to which positively charged RuR and Ru360 bind through ionic interactions. This interaction holds the polybasic compounds directly over the mouth of the channel pore, blocks one EIP arm, and prevents channel function. Functional studies corroborated by a crystal structure of K2P2.1 I110D bound simultaneously to RuR and a small molecule activator of the channel selectivity filter ‘C-type gate’, ML335 (Lolicato et al., 2017), establish that polynuclear ruthenium amine inhibition of K2Ps is unaffected by C-type gate activation. Using molecular recognition principles derived from the structures of the K2P:RuR and K2P:Ru360 complexes, we demonstrate a general design strategy for endowing any K2P channel with nanomolar RuR sensitivity. Our work establishes that polynuclear ruthenium compounds act through a ‘finger in the dam’ mechanism to inhibit K2P function by binding under the CAP domain archway at the Keystone inhibitor site and blocking the selectivity filter pore. The structural definition of this new modulatory site demonstrates the importance of electronegativity and specific sidechain geometry for polynuclear amine molecular recognition, defines a new small molecule binding site that augments the rich, polysite pharmacology of K2P modulation, and opens a path for targeting the Keystone inhibitor site and EIP for the development of new K2P modulators.
Results
A single site in the K2P CAP domain confers RuR sensitivity
K2P2.1 (TREK-1) is the founding member of the thermo- and mechanosensitive subgroup of K2Ps (Douguet and Honore, 2019; Feliciangeli et al., 2014). Although this channel is resistant to RuR inhibition (Figure 1B and D, Table 1) (Braun et al., 2015), two electrode voltage clamp (TEVC) recordings in Xenopus oocytes of outward current inhibition by RuR under physiological ionic conditions showed that installation of a point mutation I110D at the base of the K2P2.1 (TREK-1) CAP domain conferred sub-micromolar RuR sensitivity to K2P2.1 (TREK-1) (IC50 = 0.287 ± 0.054 μM)(Figures 1C–D, Table 1). This inhibition followed a 1:1 RuR:channel stoichiometry, in agreement with K2P studies using other recording protocols (Braun et al., 2015; Czirjak and Enyedi, 2003), and validates previous studies showing that this point mutant renders K2P2.1 (TREK-1) sensitive to RuR (Braun et al., 2015). Importantly, the response of K2P2.1 I110D to RuR matched that of the closely-related, natively-RuR sensitive K2P10.1 (TREK-2) (Braun et al., 2015) in which there are native aspartate residues at the K2P2.1 Ile110 analogous site (Figures 1D and S1A, Table 1) (IC50 = 0.287 ± 0.054 and 0.23 ± 0.06 μM for K2P2.1 I110D and K2P10.1 (TREK-2) (Braun et al., 2015), respectively), suggesting that the I110D change to K2P2.1 (TREK-1) captures the essence of the requirements for RuR inhibition.
Table 1.
IC50 values for RuR and Ru360
| Inhibitor | Construct | EC50 (μM) | n | |
|---|---|---|---|---|
| RuR | K2P2.1 (TREK-1) | K2p2.1 (TREK-1) | >100 | 3 |
| K2p2.1 I110D | 0.287 ± 0.054 | 3 | ||
| K2p2.1 I110E | 13.6 ± 2.7 | 3 | ||
| K2p2.1 I110K | >100 | |||
| K2p2.1 I110D/G137I | 0.154 ± 0.023 | 3 | ||
| K2p2.1 I110D + 50 μM ML335 |
0.173 ± 0.021 | 3 | ||
| K2p2.1 N147D | 47.7 ± 6.3** | 2 | ||
| K2p2.1 N147E | 0.0733 ± 0.0165 | 3 | ||
| K2p2.1 I110D/N147D | 0.0127 ± 0.0023 | 3 | ||
| K2p2.1 I110D/N147E | 0.0126 ± 0.0034 | 3 | ||
| Other K2Ps | K2p10.1 (TREK-2) [D135]* | 0.23 ± 0.06 μM (Braun et al., 2015) | ||
| K2p9.1 (TASK-3) [E70]* | 0.114 ± 0.021 μM | 3 | ||
| Ru360 | K2P2.1 (TREK-1) | K2p2.1 (TREK-1) | >100 | 3 |
| K2p2.1 I110D | 11.3 ± 1.8 | 3 | ||
| Other K2Ps | K2p10.1 (TREK-2) [D135]* | 2.8 ± 1.2 | 3 | |
| K2p9.1 (TASK-3) [E70]* | 15.6 ± 2.7 | 3 | ||
n number of oocytes
Errors are SEM.
residue at the K2P2.1 (TREK-1) I110D equivalent position
Lower bound constrained to 0.2
Because another natively-RuR sensitive K2P, K2P9.1 (TASK-3), has a glutamate at the K2P2.1 I110D equivalent site (Figure S1A) that is essential for its RuR response (Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006), we asked whether I110E would also render K2P2.1 (TREK-1) sensitive to RuR. Indeed, TEVC measurements showed that RuR inhibited K2P2.1 I110E (IC50 =13.6 ± 2.7 μM, Table 1) (Figures 1D and S1B, Table 1). RuR inhibition was ~50 fold weaker than that observed for K2P2.1 I110D, indicating that the I110D and I110E changes are not equivalent even though both bear similar negative charges. Further, introduction of a positively charged residue, K2P2.1 I110K, yielded channels as insensitive to RuR as K2P2.1 (TREK-1) (Figures 1D and S1C). RuR inhibition was essentially independent of voltage for K2P2.1 I110D and K2P2.1 I110E (Figures S1D–G), consistent with prior reports of RuR inhibition of other K2Ps (Czirjak and Enyedi, 2002). Together, these results provide key support for the idea that a negative charge at the K2P CAP domain base is a crucial determinant of RuR inhibition of K2Ps (Braun et al., 2015; Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006). Importantly, the observation of the ~50 fold difference between the RuR sensitivity of two essentially equivalently negatively charged residues in the same structural context, K2P2.1 I110D and K2P2.1 I110E (Figure 1D, Table 1) indicates that electrostatics is not the sole factor contributing to the RuR:channel interaction and points to a role for the detailed geometry of the interaction of the negatively charged residues with RuR.
Structural definition of the K2P RuR binding site
To understand how RuR inhibits K2P channels, we determined the X-ray crystal structures of K2P2.1 I110D alone and bound to RuR at resolutions of 3.40Å and 3.40Å, respectively (Table S1) on the background of the previously crystallized construct K2P2.1 (TREK-1)cryst (Lolicato et al., 2017). Apart from the I110D change, the overall structure of K2P2.1 I110D was essentially identical to K2P2.1 (TREK-1)cryst (RMSDCα = 0.575Å) (Table S2). Importantly, K2P2.1 I110D was structurally similar to the natively-RuR sensitive K2P10.1 (TREK-2) (Dong et al., 2015) (RMSDCα= 0.938Å), especially in the neighborhood of K2P10.1 (TREK-2) Asp140 (Figure S2A, Table S2), the residue that is fundamental to K2P10.1 (TREK-2) RuR sensitivity (Braun et al., 2015). Hence, when taken together with the functional similarity to K2P10.1 (TREK-2), the K2P2.1 I110D:RuR complex should capture the essential elements that contribute to the RuR response of natively RuR-sensitive K2Ps.
The K2P2.1 I110D:RuR complex structure shows that RuR binds under the CAP domain archway directly above the selectivity filter at a site we term the ‘Keystone inhibitor site’ due to the location of the I110D residues at the peak of the CAP archway ceiling (Figures 1E–F and S2B–C). RuR binds with a 1:1 stoichiometry to the channel that matches expectations from functional studies (Table 1) (Braun et al., 2015; Czirjak and Enyedi, 2002; 2003). To facilitate description of this and other RuR complexes, we designate the three RuR ruthenium amine centers as RuA, RuB, and RuC. RuR binds at ~45° angle relative to the CAP and selectivity filter in a pose that places one of the terminal ruthenium-amine moieties, RuA, directly above the column of selectivity filter ions in a position that overlaps with the S0 ion site from the K2P2.1 I110D structure (Figures 1G and S2B). The RuB and RuC moieties block one EIP arm (Figures 1E and G). Notably, the observed pose is very different from the previously proposed horizontal RuR binding pose in which the RuB moiety sits above the column of selectivity filter ions (Gonzalez et al., 2013). The I110D carboxylates coordinate RuR directly through electrostatic interactions with all three ruthenium-amine moieties using a multipronged set of interactions (Figure 1G and H). Such direct coordination of RuR by multiple elements of the I110D ligands suggests why K2P2.1 I110D and K2P2.1 I110E have different magnitude RuR responses, as the extra methylene groups in K2P2.1 I110E would not allow the same type of direct coordination observed for the smaller aspartate pair (Figure 1H). Besides direct electrostatic interactions, there are van der Waals contacts between RuC and the side chain of CAP residue Val107 from chain A, RuA and RuB with Asn111 from each chain of the dimer, RuA with Asp256 from chain B, and RuB with Gly255 from chain B. Apart from a slight reorientation of the I110D sidechains and CAP to accommodate RuR (Figure S2D, Movie S1), there are only minor conformational changes with respect to the unbound K2P2.1 I110D (RMSDCα= 0.688Å)(Table S2). Hence, RuR binds to an essentially pre-organized electronegative binding site at the CAP base.
K2P RuR binding is independent of C-type gate activation
Activation of the selectivity filter, ‘C-type’ gate is central to K2P function (Bagriantsev et al., 2012; Bagriantsev et al., 2011; Lolicato et al., 2017; Piechotta et al., 2011; Schewe et al., 2016) and diverse types of activators including intracellular pH (Schewe et al., 2016), phosphatidylinositol 4,5-bisphosphate (PIP2) (Schewe et al., 2016), pressure (Schewe et al., 2016), arachidonic acid (Schewe et al., 2016), small molecule activators that target the K2P modulator pocket (Lolicato et al., 2017), the activator BL-1249 (Pope et al., 2018), and the gain-of-function mutant G137I (Lolicato et al., 2017) have equivalent effects on C-type gate activation. Because RuR binds directly above the selectivity filter and overlaps with the S0 ion, we asked whether C-type gate activation by two representative means, the G137I mutation (Bagriantsev et al., 2012; Lolicato et al., 2017; Lolicato et al., 2014) or by a small molecule activator, ML335 (Lolicato et al., 2017), would impact RuR inhibition of K2P2.1 I110D (Figure 2A–B). TEVC experiments showed that RuR inhibits K2P2.1 I110D/G137I with an IC50 (IC50 = 0.154 ± 0.023 μM) that is very similar to that for K2P2. I110D, indicating that C-type gate activation does not influence RuR block of the channel (Figure 2C, Table 1).
Figure 2. Functional and structural analysis of C-type gate activated K2P2.1 I110D:RuR complexes.
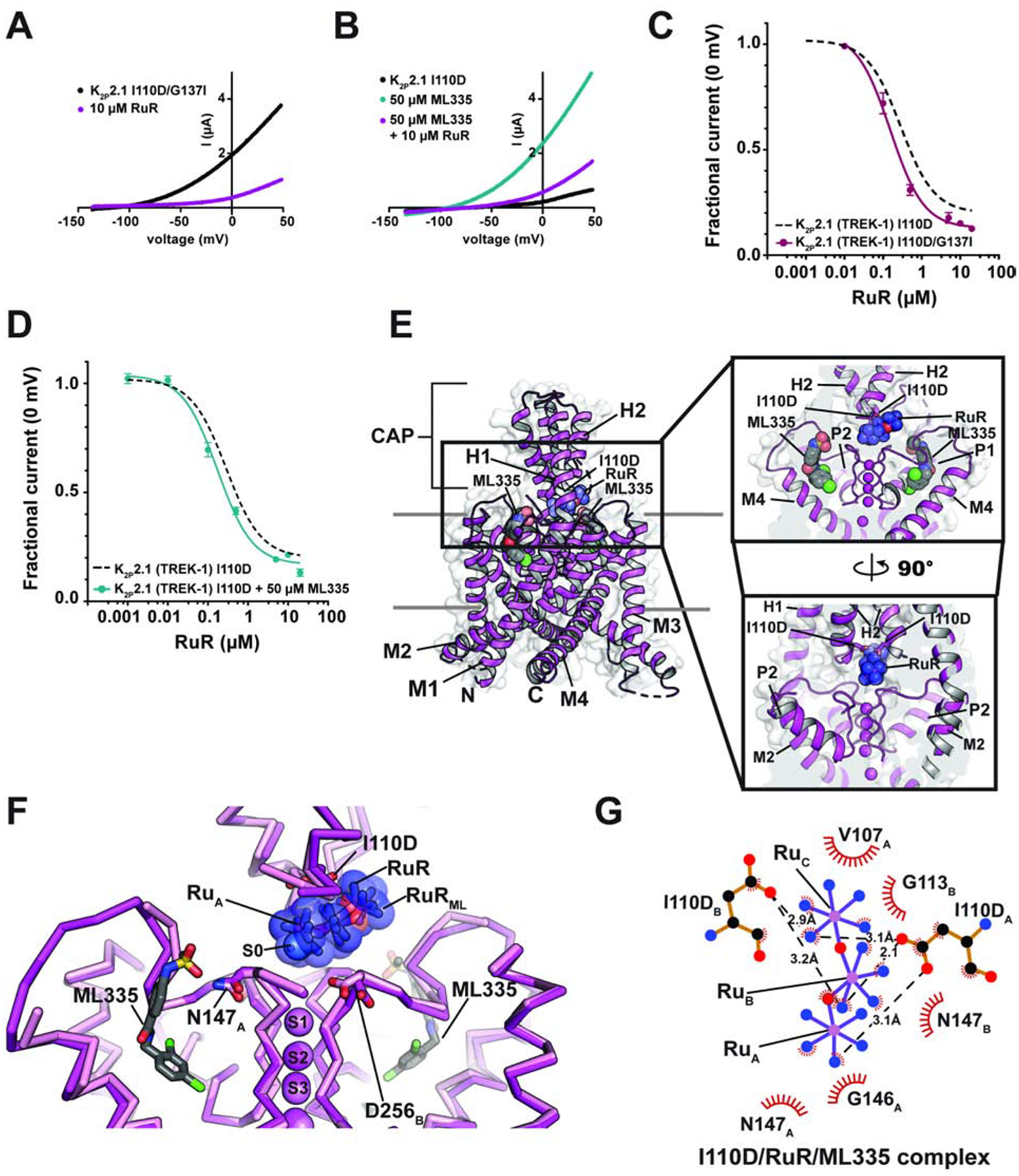
(A) and (B) Exemplar TEVC recordings of: (A) K2P2.1 I110D/G137I (black) and in the presence of 10 μM RuR (magenta), and (B) K2P2.1 I110D alone (black) in the presence of 50 μM ML335 (cyan), and in the presence of 50 μM ML335 +10 μM RuR (magenta). (C) and (D) RuR dose-response curves for (C) K2P2.1 I110D/G137I (magenta), and (D) K2P2.1 I110D in the presence of 50 μM ML335 (cyan). Dashed lines show RuR response of K2P2.1 I110D from Figure 1D. (E) Structure of the K2P2.1 I110D:ML335 RuR complex. Inset shows the location of the RuR binding site. I110D is shown as sticks. ML335 (grey) is shown as space filling. (F) Superposition of RuR binding site from the K2P2.1 I110D:RuR (pink) and K2P2.1 I110D:ML335:RuR (magenta) RuR and RuRML indicate RuR from the K2P2.1 (TREK-1) I110D:RuR and K2P2.1 I110D:ML335:RuR structures, respectively. ML335 is shown as sticks. Subscripts indicate Chain A and Chain B residues. (G) LigPLOT (Wallace et al., 1995) diagram of K2P2.1 I110D:RuR interactions from the K2P2.1 I110D:ML335:RuR complex showing ionic interactions (dashed lines) and van der Waals contacts (red) ≤5Å. See also Figure S2 and Tables S1 and S2.
Before assessing whether RuR inhibition was influenced by ML335 activation, we first measured the activation of K2P2.1 I110D by ML335. The I110D change is >15Å from the K2P modulator pocket that forms the ML335 binding site and is not expected to impact ML335 activation. In line with these expectations, there was no difference in the response of K2P2.1 I110D to ML335 activation relative to K2P2.1 (TREK-1) (EC50 (ML335) = 11.8 ± 2.3 and 14.3 ± 2.7 μM for K2P2.1 I110D and K2P2.1 (TREK-1) (Lolicato et al., 2017), respectively) (Figures S3A–B). Importantly, similar to the observations with the G137I C-type gate activation mutant, pharmacological C-type gate activation by saturating amounts of ML335 had minimal impact on the RuR response relative to K2P2.1 I110D (IC50 = 0.173 ± 0.021 μM) (Figure 2D, Table 1). Together, these data demonstrate that RuR inhibition is essentially independent of C-type gate activation (Figures 2C–D, Table 1) and are consistent with the observed position of RuR above the selectivity filter.
To see whether there might be structural differences in the interaction of RuR with an activated C-type gate, we determined the structure of the K2P2.1 I110D:RuR:ML335 complex at 2.80Å resolution (Figures 2E and S3C–D, Table S1). This structure is very similar to the K2P2.1 I110D:RuR complex (Figures S3C–E) (RMSDCα = 0.507Å) and to the previously determined K2P2.1:ML335 complex (Lolicato et al., 2017) (RMSDCα = 0.480Å) (Table S2). The structure shows that RuR binds to the Keystone inhibitor site using a pose that is very similar to that in the K2P2.1 I110D:RuR complex. Both I110D sidechains coordinate multiple Ru centers though direct interactions to RuR (Figure 2F–G) like those in the K2P2.1 I110D:RuR complex and there are van der Waals contacts with residues in the CAP and selectivity filter outer mouth. These fundamental similarities in binding to the Keystone inhibitor site are consistent with the similar IC50s measured with or without C-type gate activation (Figure 2C–D, Table 1).
Polynuclear ruthenium compounds inhibit K2Ps at a common site
The dinuclear oxo-bridged ruthenium compound, Ru360 (Figure 3A) (Ying et al., 1991), has many characteristics in common with RuR (cf. Figure 1A). Ru360 is best known as an inhibitor of the mitochondrial calcium uniporter (MCU) (Baughman et al., 2011; Kirichok et al., 2004; Oxenoid et al., 2016), a property it shares with RuR (Gunter and Pfeiffer, 1990). Yet, despite its structural similarity to RuR and the fact that both Ru360 and RuR inhibit MCU, Ru360 has not been reported to block K2P channels. To ask whether Ru360 might inhibit RuR-sensitive K2P channels, we used TEVC to measure the Ru360 responses of K2P2.1 (TREK-1), K2P2.1 I110D, and the natively RuR sensitive K2Ps, K2P10.1 (TREK-2) (Braun et al., 2015) and K2P9.1 (TASK-3) (Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006). Application of 100 μM Ru360 to K2P2.1 (TREK-1) had no effect, consistent with the insensitivity of this channel to RuR (Figures 3B and F). However, in stark contrast, Ru360 inhibited all of the RuR sensitive K2Ps with micromolar potency (IC50 = 11.3 ± 1.8, 2.8 ± 1.2, and 15.6 ± 2.7 μM for K2P2.1 I110D, K2P10.1 (TREK-2), and K2P9.1 (TASK-3), respectively) (Figures 3C–F, Table 1). Similar to RuR inhibition, Ru360 block of K2Ps was independent of voltage (Figures S4A–D). For both K2P2.1 I110D and K2P10.1 (TREK-2) the Ru360 IC50s are >10-fold weaker than those for RuR. Because there is a reported 30-fold discrepancy in the IC50 of RuR for K2P9.1 (TASK-3) in the literature (0.35 μM (Czirjak and Enyedi, 2003) vs.10 μM (Gonzalez et al., 2013)) that precluded a direct comparison with our Ru360 data, we measured inhibition of K2P9.1 (TASK-3) by RuR to resolve whether RuR and Ru360 had similar or different IC50s for K2P9.1 (TASK-3). Our TEVC experiments measured a sub-micromolar IC50 for RuR inhibition of K2P9.1 (TASK-3) (IC50 = 0.114 ± 0.021 μM) (Figures S4E–F, Table 1) that agrees with other Xenopus oocyte TEVC studies (Czirjak and Enyedi, 2003). These data establish that, as with the other polynuclear ruthenium-sensitive K2Ps we studied, Ru360 is a weaker inhibitor of K2P9.1 (TASK-3) than RuR. The uniformly weaker potency of Ru360 versus RuR against K2Ps correlates with the fact that Ru360 carries half the positive charge of RuR (+3 versus +6) and underscores the important role that electrostatics plays in the binding of these polycations.
Figure 3. Functional and structural analysis of K2P:Ru360: interactions.
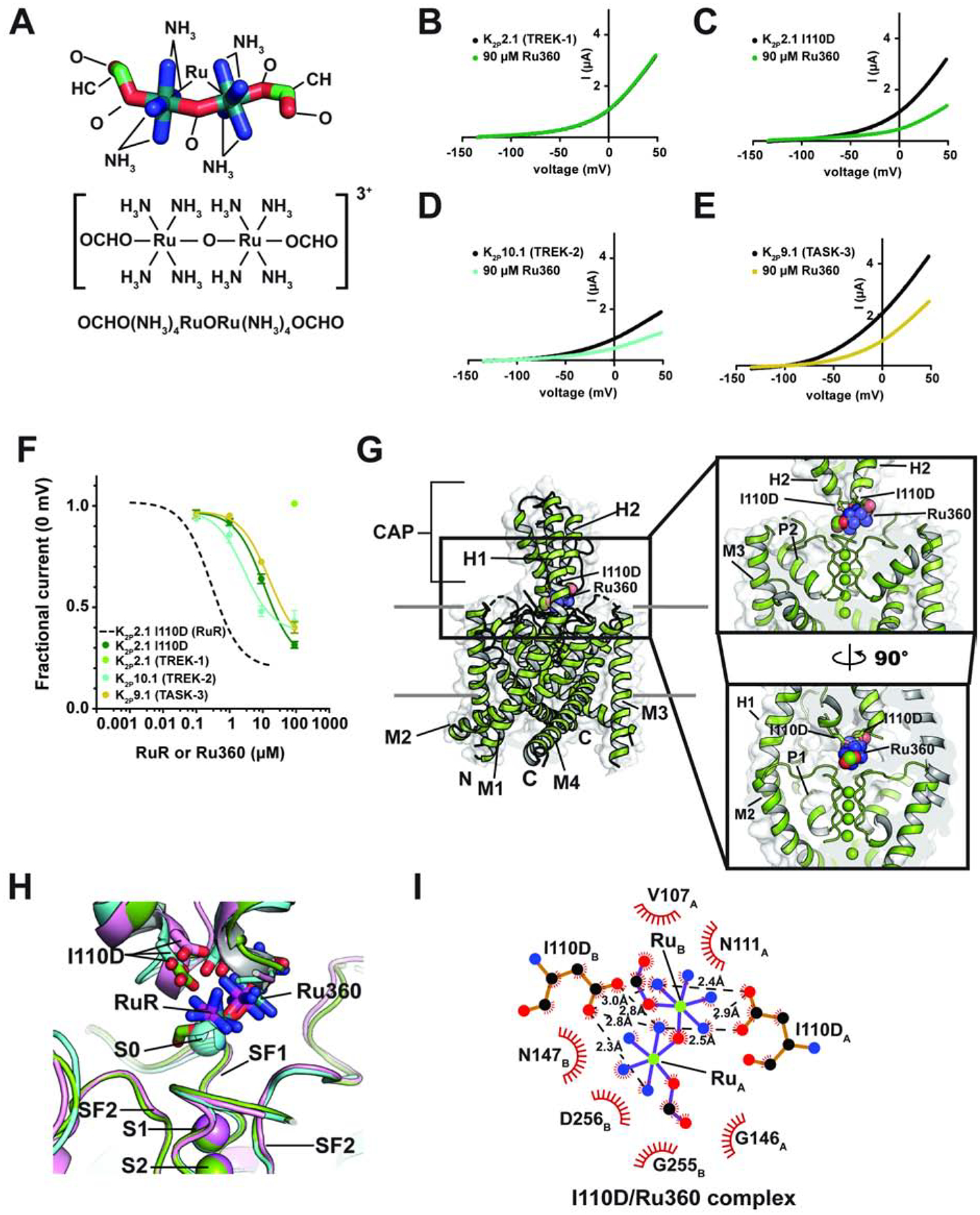
(A) Ru360 structure. (B–D) Exemplar TEVC recordings of (B) K2P2.1 (TREK-1), (C) K2P2.1 I110D, (D) K2P10.1 (TREK-2), and (E) K2P9.1 (TASK-3) alone (black) and in the presence of 90 μM Ru360 (green, teal, and olive, respectively). (F) Ru360 dose-response curves for K2P2.1 I110D (green), K2P2.1 (TREK-1) (light green), K2P10.1 (TREK-2) (teal), and K2P9.1 (TASK-3) (olive). Dashed line shows RuR dose-response for K2P2.1 I110D from Figure 1D. (G) Structure of the K2P2.1 I110D:Ru360 complex. Inset shows the location of the Ru360 binding site. I110D is shown as sticks. (H) Close up view of K2P2.1 I110D (cyan), K2P2.1 I110D:RuR (pink), and K2P2.1 I110D:Ru360 Keystone inhibitor sites. RuR and Ru360 are shown as sticks. S0 ion from the K2P2.1 I110D structure is shown as a sphere. (I) LigPLOT (Wallace et al., 1995) diagram of K2P2.1 I110D:Ru360 interactions showing ionic interactions (dashed lines) and van der Waals contacts (red) ≤5Å. Subscripts indicate Chain A and Chain B residues. See also Figure S4 and Tables S1 and S2.
To understand the details of how Ru360 inhibits K2Ps, we determined of a 3.51Å resolution X-ray crystal structure of the K2P2.1 I110D:Ru360 complex (Figures 3G–H and S4G–H, Table S1). As with the RuR complexes, the K2P2.1 I110D:Ru360 complex has a channel structure that is overall very similar to the structure of the K2P2.1 I110D in the absence of the inhibitor (RMSDCα = 0.665Å) and to the K2P2.1 I110D:RuR complex (RMSDCα = 0.561Å) (Figure S4G, Table S2). Ru360 binds to the Keystone inhibitor site in a pose that matches RuR (Figures 3H and S4G–I). Notably, even though Ru360 has one fewer ruthenium atoms than RuR, one of the Ru360 ruthenium-amine moieties, denoted RuA, occupies essentially the same site as the RuR RuA moiety and overlaps with the S0 ion site (Figure 3H), while the other ruthenium amine, RuB, overlaps the position of the RuR RuB moiety and blocks one EIP arm. There are essentially no conformational changes between K2P2.1 I110D and the K2P2.1 I110D:Ru360 complex except for a change near the base of the CAP helices similar to that seen in the RuR complexes (Figure S4I). The Keystone inhibitor site acidic sidechains directly coordinate the Ru360 RuA and RuB centers (Figure 3I). Ru360 also makes van der Waals contacts to the upper part of the selectivity filter and part of the CAP from chain A similar to those made by RuR (cf. Figures 1H, 2G, and 3I). Together, these data demonstrate that Ru360 inhibits RuR-sensitive K2Ps and reveals the common mode by which polynuclear ruthenium amines affect K2P channels by binding to the Keystone inhibitor site, blocking ion exit from the selectivity filter, and obstructing one EIP arm.
Protein engineering creates RuR super-responders
Because the electronegative nature of the Keystone inhibitor site is a key determinant of K2P channel sensitivity to polynuclear ruthenium compounds, we wanted to test whether increasing the electronegative character of surrounding portions of the EIP would also affect RuR block. To identify candidate sites, we looked for elements on the floor of the RuR and Ru360 binding site that made close contacts with the inhibitors. This analysis identified the backbone atoms of the selectivity filter outer mouth residues Asn147 and Asp256 as the nearest neighbors (Figures 4A–B). As we cannot easily change the backbone atoms, we considered changing the properties of the sidechains from these positions. Sequence comparison of representatives from each K2P subtype (Figure S5A) shows that the two sites have very different conservation patterns. The Asn147 site shows a range of amino acid types. This variability contrasts with the strict conservation at the Asp256 site. Because all K2Ps have aspartate at the 256 position regardless of whether or not they are RuR-sensitive, we reasoned that the negatively charged sidechain at this site has no influence on RuR binding. By contrast, the amino acid diversity at the Asn147 site indicated that this site might have different properties than the Asp256 site. Hence, we tested whether replacing Asn147 with a negatively charged residue would impact K2P2.1 (TREK-1) RuR sensitivity. TEVC experiments showed that RuR inhibited both K2P2.1 N147D and K2P2.1 N147E (Figures 4C–F), demonstrating that the presence of an acidic residue at the top of the Keystone inhibitor site is not the only means by which a K2P channel can acquire RuR sensitivity. Notably, there was a marked difference in the IC50s between the two mutants with N147E having a much greater susceptibility to RuR inhibition than N147D (IC50 = 0.0733 ± 0.0165 and 47.7 ± 6.3 μM for K2P2.1 N147E and K2P2.1 N147D, respectively) (Figure 4F, Table 1). Similar to RuR inhibition of other K2Ps, RuR block of K2P2.1 N147E was essentially voltage independent, whereas K2P2.1 N147D showed a mild voltage-dependence (Figures S5B–E).
Figure 4. Engineering K2P RuR super-responders.
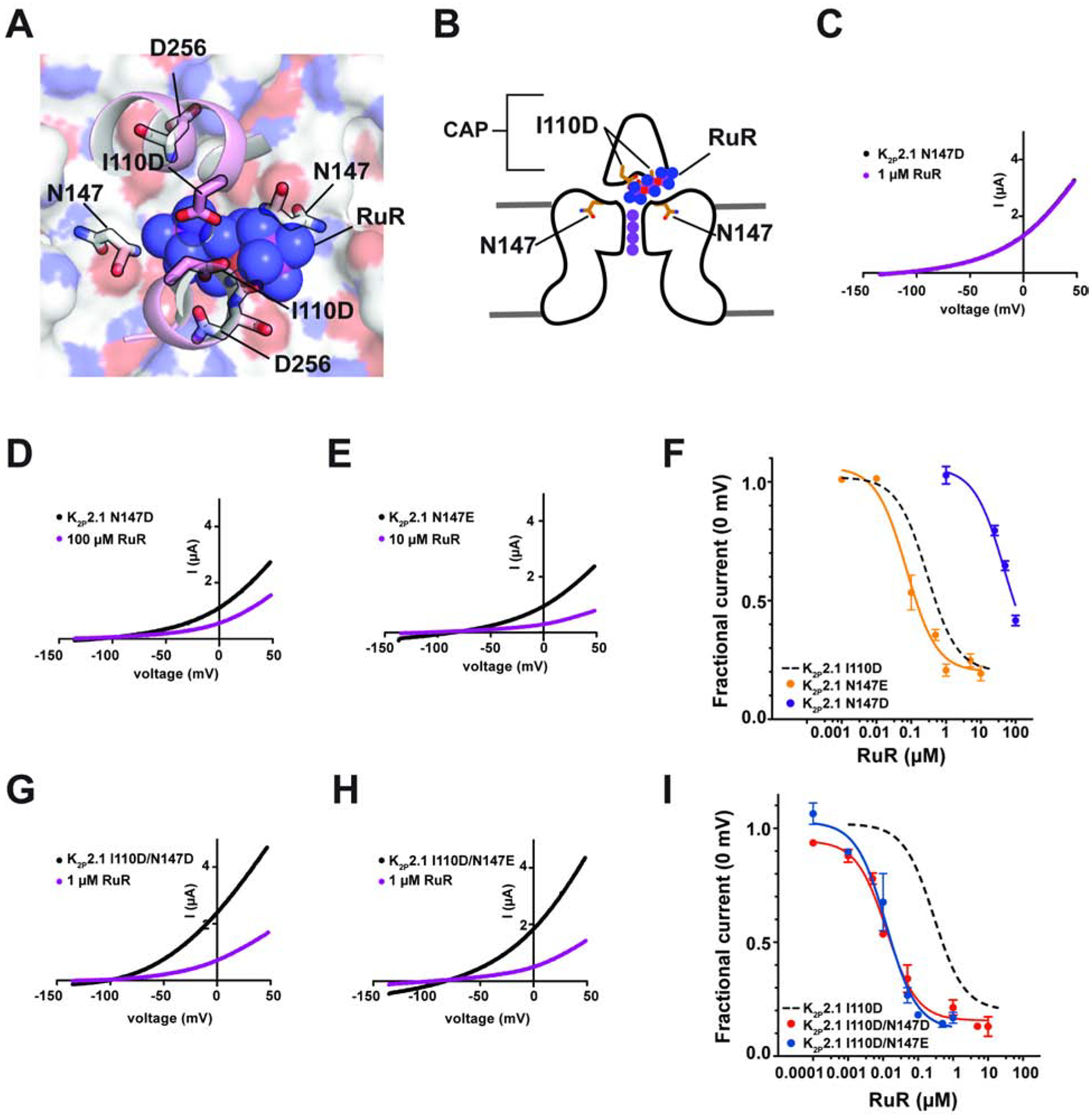
(A) View through the K2P2.1 (TREK-1) CAP to the floor of the Keystone inhibitor site. CAP H2 helix (pink) is shown as a cartoon. RuR is shown as semi-transparent spheres. Surface (white) shows the top of the selectivity filter. I110D, N147, and D256 are shown as sticks. (B) Cartoon depiction of the elements framing the Keystone inhibitor site. Locations of CAP, I110D, and N147 are indicated. Selectivity filter potassium ions are shown as purple circles. (C–E) TEVC recordings of (C) K2P2.1 N147D alone (black) and in the presence of 1 μM RuR (magenta), (D) K2P2.1 N147D alone (black) and in the presence of 100 μM RuR (magenta) and (E) K2P2.1 N147E alone (black) and in the presence of 10 μM RuR (magenta). (F) RuR response of K2P2.1 N147D (purple) and K2P2.1 N147E (orange). Dashed line shows K2P2.1 I110D response to RuR, from Figure 1D. (G) and (H) TEVC recordings of (G) K2P2.1 I110D/N147D alone (black) and in the presence of 1 μM RuR (magenta), and (H) K2P2.1 I110D/N147E alone (black) and in the presence of 1 μM RuR (magenta). (I) RuR dose-response of K2P2.1 I110D/N147D (red) and K2P2.1 I110D/N147E (blue). Dashed line shows K2P2.1 I110D response to RuR, from Figure 1D. See also Figures S5 and S6
Because both N147D and N147E changes were able to confer RuR sensitivity to K2P2.1 (TREK-1) (Figure 4F, Table 1), we asked whether having negatively charged residues on both the ceiling (residue 110) and floor (residue 147) of the RuR binding site would result in enhanced RuR inhibition. TEVC measurements of the RuR responses of K2P2.1 I110D/N147D and K2P2.1 I110D/N147E revealed that both double mutants had similar IC50s (IC50= 0.0127 ± 0.0023 and 0.0126 ± 0.0034 μM for K2P2.1 I110D/N147D and K2P2.1 I110D/N147E, respectively) (Figures 4G–I and S5F–G, Table 1). Similar to the I110D and N147E mutants, the response of both double mutant channels to RuR was essentially independent of voltage (Figures S5H–J).
The IC50s values of the double mutants were an order of magnitude better than that of K2P2.1 I110D alone and three orders of magnitude better than K2P2.1 N147D. Hence, we turned to double mutant cycle analysis (Carter et al., 1984; Hidalgo and MacKinnon, 1995) to assess the extent of synergy between the two sites with respect to RuR inhibition. This analysis uncovered a strong positive cooperativity for the I110D/N147D pair with respect to RuR binding (ΔΔG = −4.1 kcal mol−1) (Figure S6A). By contrast, the enhanced RuR response of the I110D/N147E combination resulted from essentially additive contributions of the two negatively charged residues (ΔΔG = −0.3 kcal mol−1) (Figure S6B). The fact that the two double mutant pairs do not behave equivalently even though both comprise two sets of acidic sidechains, together with the observation that there is a substantial difference in the impact of I110D versus I110E alone on the RuR sensitivity of K2P2.1 (TREK-1) (Figure 1D, Table 1) reinforces the idea that RuR molecular recognition requires both general electrostatic interactions and the direct coordination from the acidic sidechains. Taken together, our results indicate that details of these two factors tune the strength of the RuR interaction with the channel. Because of the largely conserved nature of the K2P architecture in the region of the CAP and selectivity filter (Brohawn et al., 2012; Dong et al., 2015; Lolicato et al., 2017; Miller and Long, 2012), installing acidic residues simultaneously at the equivalents of the K2P2.1 (TREK-1) Ile110 and Asn147 positions should endow any K2P of interest sensitive to nanomolar concentrations of RuR.
Discussion
Polynuclear ruthenium compounds have been used for nearly 50 years to control the function of various ion channels (Arif Pavel et al., 2016; Braun et al., 2015; Caterina et al., 1999; Caterina et al., 1997; Chaudhuri et al., 2013; Coste et al., 2012; Czirjak and Enyedi, 2003; Dreses-Werringloer et al., 2013; Gonzalez et al., 2013; Guler et al., 2002; Kirichok et al., 2004; Ma, 1993; Ma et al., 2012; Moore, 1971; Musset et al., 2006; Rahamimoff and Alnaes, 1973; Smith et al., 1988; Story et al., 2003; Strotmann et al., 2000; Voets et al., 2004; Voets et al., 2002; Zhao et al., 2016). Yet, despite this widespread application in the study of a multitude of diverse ion channels, there is only limited visualization of how such compounds might interact with and affect the function of their targets (Choi et al., 2019). The K2P:RuR and K2P:Ru360 complexes presented here provide the first detailed structural views of how this class of inorganic polycations can inhibit ion channel function. The structures demonstrate general molecular recognition principles in which a channel uses acidic sidechains to coordinate both trinuclear, RuR, and dinuclear, Ru360, ruthenium amines through direct, multipronged electrostatic interactions. Both compounds block the flow of ions through the channel using a ‘finger in the dam’ mechanism that exploits the unique archway architecture that the K2P CAP domain creates above the K2P channel mouth in which the terminal end of the polyruthenium compound occupies a site directly over the source of ion flow, the column of ions in the channel selectivity filter (Figure 5A).
Figure 5. Mechanisms of small molecule K2P modulation.
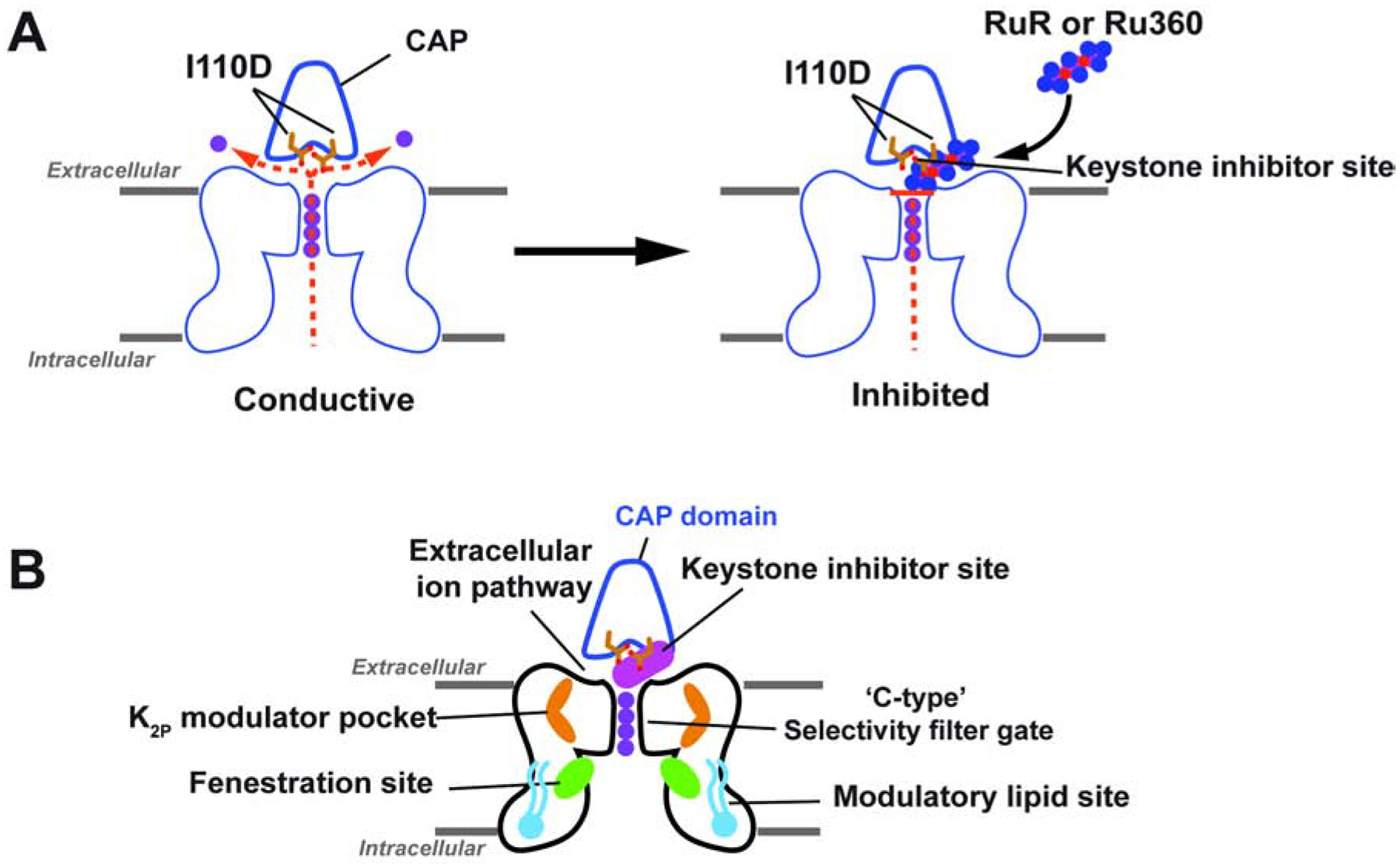
(A) Cartoon diagram depicting the ‘finger in the dam’ mechanism of K2P inhibition by polynuclear ruthenium amines. Ion flow is indicated by the dashed orange lines. Potassium ions are shown as purple circles. (B) Polysite model of K2P modulation. Diagram shows structurally defined sites for K2P modulators: Keystone inhibitor site (magenta), K2P modulator pocket (orange) (Lolicato et al., 2017), fenestration site (green) (Dong et al., 2015; Schewe et al., 2019), and modulatory lipid site (cyan) (Lolicato et al., 2017). Extracellular ion pathway and ‘C-type’ selectivity filter gate are indicated. CAP is outlined in blue and shows the position of the negatively charged residues required for the Keystone inhibitor site (sticks).
K2Ps are the only potassium channel family that bears a CAP domain, an extracellular dimerization domain positioned directly above the channel pore (Brohawn et al., 2012; Miller and Long, 2012). The structures presented here show that a pair of aspartic acids located on the underside of the CAP archway at a site that controls the RuR response of natively-RuR sensitive K2Ps, K2P10.1 (TREK-2) and K2P9.1 (TASK-3) (Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006) and of the RuR-sensitive mutant K2P2.1 I110D (Braun et al., 2015) create a polycation binding site, the Keystone inhibitor site (Figures 1E–F). This site forms the primary point of interaction with a single RuR or Ru360 that plugs one arm of the bifurcated EIP created by the CAP domain archway (Figures 1E–G, 2E–F, 3G–H). This structural observation defines an unambiguous mechanism of action for how polyruthenium amines inhibit K2Ps, as holding a large polycation above the channel pore would both physically block ion exit as well as provide an electrostatic barrier to permeant ion movement (Figure 5A). Ru360 a dinuclear oxo-bridged ruthenium amine inhibitor of the mitochondrial calcium uniporter (Baughman et al., 2011; Kirichok et al., 2004; Oxenoid et al., 2016) that had not previously been reported to affect potassium channels also inhibits engineered, K2P2.1 I110D, and natively-RuR sensitive K2Ps, K2P10.1 (TREK-2), and K2P9.1 (TASK-3), in a manner similar to RuR (Figure 3C–F). Hence, even though RuR and Ru360 bind to a largely pre-formed binding site, there is sufficient plasticity to permit the binding of different types of polyruthenium cations. Consistent with a binding site positioned above the selectivity filter and outside of the transmembrane electric field, polynuclear ruthenium amine inhibition of K2Ps is independent of voltage (Figures S1D–G, S4A–D, S5B–E and H–J) and the C-type gate activation (Figure 2C–D). Together, the data define a mechanism of action in which polynuclear ruthenium amines inhibit K2P function by preventing ion flow out of the K2P selectivity filter and through the EIP (Figure 5A).
Although many ion channels lack the extracellular archway made by the K2P CAP domain, the structures of the K2P:polyruthenium amine complexes reveal general molecular recognition principles that are likely to be shared with RuR and Ru360 sensitive ion channels. The requirement to have a negatively charged residue at the Keystone inhibitor site for RuR (Figure 1D) (Braun et al., 2015; Czirjak and Enyedi, 2003; Gonzalez et al., 2013; Musset et al., 2006) and Ru360 (Figure 3F) responses, together with the observation that inhibitor potency is proportional to the total charge (Table 1) highlights the importance of electrostatic interactions for recognizing ruthenium polycations. The multipronged direct coordination of the RuR and Ru360 ruthenium amine moieties (Figure 1H, 2G, and 3I) shows the important role that direct coordination by acidic sidechains plays in binding both compounds. The observation that equivalently charged residues having different sidechain geometries, aspartate and glutamate, have differential effects on RuR potency at the Keystone inhibitor site (Figures 1D and Table 1) highlights the importance of direct ligand coordination for tuning the strength of the interaction with polynuclear ruthenium amines. It should be noted that K2P4.1 (TRAAK) is inhibited by RuR but lacks a negatively charged residue in the Keystone inhibitor site (Figure S1A) and binds RuR with a higher stoichiometry than observed for the K2P channels studied here (Table 1) (Braun et al., 2015; Czirjak and Enyedi, 2002). These differences suggest that RuR inhibition of K2P4.1 (TRAAK) occurs using a mechanism different from the ‘finger in the dam’ mechanism. Given the importance of direct interactions between RuR and Ru360 and the acidic sidechains in the target channels studied here and the fact that acidic residues are key to the RuR sensitivity of other channels (Zhao et al., 2018; Zhao et al., 2016), we expect that similar types of multipronged coordination by sidechain carboxylates are likely to contribute to the RuR and Ru360 block of other polynuclear ruthenium amine sensitive channels such as K2P4.1 (TRAAK), TRPs (Arif Pavel et al., 2016; Caterina et al., 1999; Caterina et al., 1997; Guler et al., 2002; Story et al., 2003; Strotmann et al., 2000; Voets et al., 2004; Voets et al., 2002), the mitochondrial calcium uniporter (MCU) (Chaudhuri et al., 2013; Kirichok et al., 2004; Moore, 1971; Rahamimoff and Alnaes, 1973), CALHM calcium channels (Choi et al., 2019; Dreses-Werringloer et al., 2013; Ma et al., 2012), ryanodine receptors (Ma, 1993; Smith et al., 1988), and Piezo channels (Coste et al., 2012; Zhao et al., 2016), even if the details of where the inhibitor binds to the channel differ.
K2P channels can be modulated by a number of different small molecules and lipids (Sterbuleac, 2019). When placed in the context of previous structural studies of K2P modulator interactions (Dong et al., 2015; Lolicato et al., 2017; Schewe et al., 2019), our studies highlight an emerging picture of the complex, multisite structural pharmacology that contributes to the control of K2P function. The discovery of the Keystone inhibitor site reveals that there are at least four control sites that span from the inner leaflet of the bilayer to the extracellular parts of the channel through which exogenous molecules affect channel function (Figure 5B). These include a modulatory lipid binding site in the bilayer inner leaflet (Lolicato et al., 2017), the fenestration site residing at the intersection of the movable M4 transmembrane helix and the lower part of the selectivity filter that can be targeted by both small molecule activators and inhibitors (Dong et al., 2015; Schewe et al., 2019), the K2P modulator pocket site (Lolicato et al., 2017), and the Keystone inhibitor site. Exploring the degree of conformational coupling among these modulatory sites will be important for understanding the extent of synergistic or antagonistic actions within the various classes of K2P modulators. Acquiring this type of knowledge will be crucial for creating new interventions that could offer exquisite control of K2P function.
The ‘finger in the dam’ inhibitory mechanism defined here provides a blueprint for the development of small molecule or protein-based K2P modulators that could reach through the EIP to the Keystone inhibitor site. In this regard, exploiting the renewed interest in synthesizing novel polyruthenium amine derivatives (Woods and Wilson, 2019) or designing compounds having moieties that interact with the Keystone inhibitor site but that also make contacts to non-conserved features of CAP exterior could yield subtype-selective modulators. Biologics, such as nanobodies, may be particularly suited to this type of molecular recognition mode. Further, given the highly conserved nature of the K2P channel architecture in the region of the CAP and selectivity filter (Brohawn et al., 2012; Dong et al., 2015; Lolicato et al., 2017; Miller and Long, 2012), the strategies we used to develop RuR super-responders should be applicable to other K2P subfamily members to create subtypes endowed with RuR sensitivity. Because of the observed non-additivity with respect to RuR binding (Figure S6A), introduction of acidic residues at the Asn147 site to make super-responders seems likely to act through direct interactions with the inhibitor; however, complete understanding of how the enhanced affinity arises will require further structural studies. Regardless of the mechanism, such RuR-sensitive channels could be used to dissect the roles of various K2Ps in their native physiological settings.
Significance
Ruthenium Red (RuR) is a trinuclear, oxo-bridged ruthenium amine polycation that has many biological applications, including a ~50 year legacy as an inhibitor of diverse classes of ion channels. RuR inhibits select members of the K2P (KCNK) family, numerous TRP channels, the mitochondrial calcium uniporter, CALHM calcium channels, ryanodine receptors, and Piezo channels. Despite this remarkably wide range of ion channel targets, there are extremely limited structural data describing how RuR binds to any ion channel target. Our studies show how two polyruthenium compounds, RuR and Ru360, inhibit K2P channels through a ‘finger in the dam’ mechanism in which these polycations bind at a novel site, the ‘Keystone inhibitor site’, formed by acidic residue pair under the K2P CAP domain archway above the channel pore. This series of structures, together with functional studies, outlines the molecular recognition principles that govern how RuR and Ru360 bind to specific sites of proteins using a mixture of electrostatics and polyvalent coordination by acidic sidechains. These principles are likely to control RuR and Ru360 binding to a wide range of diverse ion channel targets. Moreover, we show that we can use knowledge of these factors to engineer RuR ‘super-responder’ K2Ps that have RuR sensitivity in the low nanomolar range. The protein engineering strategy we define should be generally applicable to any K2P of interest and provide a new method for dissecting the function of specific K2Ps in complex settings such as neurons, the brain, and the cardiovascular system. Together, the data define a ‘finger in the dam’ inhibition mechanism acting at a novel K2P inhibitor binding site. These findings highlight the polysite nature of K2P pharmacology and provide a new framework for K2P inhibitor development.
LEAD CONTACT AND MATERIALS AVAILABILTY
Further information and request for resources and reagents should be directed to and will be fulfilled by the lead contact, Daniel L. Minor, Jr. (daniel.minor@ucsf.edu)
MATERIALS AVAILABILITY STATEMENT
All unique reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Pichia pastoris strain SMD1163H was used for protein expression.
Xenopus laevis oocytes were harvested from female X. laevis according to UCSF IACUC Protocol AN129690.
METHOD DETAILS
Molecular Biology
Murine K2P2.1 (TREK-1) (Gene ID 16526), K2P10.1 (TREK-2) (Gene ID: 72258), and K2P9.1 (TASK-3) (Gene ID: 223604) were each expressed from in a pGEMHE/pMO vector for two-electrode voltage clamp (TEVC) experiments as described previously (Lolicato et al., 2017; Pope et al., 2018). A previously described version of murine K2P2.1 (TREK-1) in a Pichia pastoris pPicZ plasmid, K2P2.1(TREK-1)CRYST (Lolicato et al., 2017), encoding residues 21–322 and bearing the following mutations: K84R, Q85E, T86K, I88L, A89R, Q90A, A92P, N95S, S96D, T97Q, N119A, S300C, E306A, was used for structural studies. Mutants of K2P2.1 (TREK-1) and K2P2.1(TREK-1)CRYST were generated using site-directed mutagenesis (PFU Turbo AD, Agilent) and verified by sequencing of the complete gene.
Two-electrode voltage clamp (TEVC) electrophysiology
Xenopus laevis oocytes were harvested according to UCSF IACUC Protocol AN129690 and digested using collagenase (Worthington Biochemical Corporation, #LS004183, 0.7–0.8 mg mL−1) in Ca2+-free ND96 (96 mM NaCl, 2 mM KCl, 3.8 mM MgCl2, 5 mM HEPES pH 7.4) immediately post-harvest, as previously reported (Lolicato et al., 2017; Pope et al., 2018). Oocytes were maintained at 18°C in ND9 6 (96 mM NaCl, 2 mM KCl, 1.8 mM CaCl2, 2 mM MgCl2, 5 mM HEPES pH 7.4) supplemented with antibiotics (100 units mL−1 penicillin, 100 μg mL−1 streptomycin, 50 μg mL−1 gentimycin) and used for experiments within one week of harvest. mRNA for oocyte injection was prepared from plasmid DNA using mMessage Machine T7 Transcription Kit (Thermo Fisher Scientific), purified using RNEasy kit (Qiagen), and stored as stocks and dilutions in RNAse-free water at −80°C.
Defolliculated stage V-VI oocytes were microinjected with 50 nL of 0.1–6 ng mRNA and currents were recorded within 24–48 hours of injection. Oocytes were impaled by two standard microelectrodes (0.3–3.0 MΩ), filled with 3M KCl and subjected to constant perfusion of ND96. Currents were elicited from a −80 mV holding potential using a 500 ms ramp ranging from −140 to +50 mV.
Recording solutions containing Ruthenium red (RuR) (Millipore-Sigma, R2751) and Ru360 (Millipore-Sigma, Calbiochem – 557440) were prepared immediately prior to use. RuR was weighed and dissolved directly into ND96 at 200 μM in ND96 and then diluted into ND96 for tested experimental concentrations. The pH of the stock solution was checked to ensure no change occurred. Due to its instability in aqueous solutions, Ru360 solutions were covered with aluminum foil to minimize exposure to light and to avoid degradation. RuR and Ru360 were determined to be stable in recording solutions for duration of typical experiment length by measuring UV absorbance at 536 nm and 363 nm, respectively, before and after length of the recording session. ML335 was synthesized as described previously (Lolicato et al., 2017). ML335 recording solutions were prepared from a DMSO stock stored at −20°C (final DMSO concentration was 0.1%).
Data were recorded using a GeneClamp 500B (MDS Analytical Technologies) amplifier controlled by pClamp software (Molecular Devices), and digitized at 1 kHz using Digidata 1332A (MDS Analytical Technologies). For each recording, control solution (ND96) was perfused over a single oocyte until current was stable before switching to solutions containing the test compounds at various concentrations and again allowed to stabilize before recording final, stabilized trace. Fractional block at the potential of interest was determined as in which I is the measured current, I0 is the current in the absence of the test compound, and IB is the basal current derived from an average of uninjected oocytes (n=14). For dose-response curves, each point is an average of at least three oocytes recorded from at least two independent batches of oocytes. Representative traces and dose response plots were generated in Graphpad Prism Version 5 (GraphPad Software, San Diego California USA, www.graphpad.com). Inhibition IC50s were estimated using an auto-fitted Hill equation with a Hill coefficient = −1.0.
Protein expression
K2P2.1CRYST I110D was expressed as a fusion protein having in series from the channel C-terminus a 3C protease site, green fluorescent protein (GFP), and His10 tag as described previously for K2P2.1cryst (Lolicato et al., 2017). Linearized plasmid DNA (PmeI) was introduced into Pichia pastoris strain SMD1163H via electroporation. Strains with highest incorporation were selected for on YPD plates containing 1–2 mg mL−1 zeocin. Individual colonies were screened using fluorescence size exclusion chromatography (FSEC) (Kawate and Gouaux, 2006) to identify strain with highest expression level as described previously (Lolicato et al., 2017). The best FSEC candidate as judged by FSEC fluorescence intensity was used to inoculate a starter culture (60–120 mL) in minimal media (1% glycerol, 100 mM potassium phosphate pH 6.0, 0.4 mg L−1 biotin, 1X YNB from Invitrogen) supplemented with 1 mg mL−1 zeocin and cultured in shaker flask for 2 days at 29°C. The starter culture was then used to inoculate a large scale (6–12L) culture in shaker flasks containing minimal media without zeocin. Cells were grown at 29°C over two days in minimal media containing 1% glycerol. Cells were centrifuged at 3000g (6 min, 20°C) and pellet was resuspended in minimal induction media (100 mM potassium phosphate pH 6.0, 0.4 mg L−1 biotin, 1X YNB) containing 0.5% methanol. After 24 hrs, 0.5% methanol was added to each flask. Cells were harvested (6000g, 20 min, 4°C) two days after induction, snap-frozen in liquid nitrogen, and stored at −80°C.
Protein Purification
Purified K2P2.1cryst I110D was obtained from preparations using 100–200g of cell mass cryo-milled (Retsch, MM400) in liquid nitrogen (5 × 3 min, 25 Hz). All purification steps were carried out at 4°C and purification conditions were similar to those previously reported for K2P2.1cryst (Lolicato et al., 2017). Cell powder was solubilized at a ratio of 3 grams of cells per mL of lysis buffer containing 200 mM KCl, 21 mM octyl glucose neopentyl glycol (OGNG, Anatrace), 30 mM n-heptyl-β-D-thioglucopyranoside (HTG, Anatrace), 0.1% cholesterol hemisuccinate (CHS, Anatrace), 100 mM Tris-Cl, pH 8.2, 1 mM PMSF and 0.1 mg/mL DNaseI. Following 3 hour membrane solubilization, the sample was centrifuged at 40,000g for 45 min at 4°C. After centrifugation, supernatant wa s incubated with anti-GFP nanobodies immobilized on CNBr-activated sepharose resin (GE Healthcare, 17-0430-01) at a ratio of 1 mL resin per 10 g of cells and gently rotated on an orbital rocker for 3 hours. Resin was collected in a gravity column (Econo-Pac, 1.5 × 12 cm, BioRad) and washed with 10 CV each, buffers A-C (A-C: 200 mM KCl, 50 mM Tris-Cl pH 8.0, 15 mM HTG; A: 10 mM OGNG, 0.018% CHS; B: 5 mM OGNG, 0.018% CHS; C: 3.85 mM OGNG, 0.0156% CHS), applied in series to reduce the detergent concentration and wash away cell debris (30 CV total). The GFP-affinity tag was cleaved overnight on column using two CV of buffer C supplemented with 350 mM KCl, 1 mM EDTA and 3C protease (Shaya et al., 2011). Cleaved protein was eluted from resin with two CV of size-exclusion buffer (SEC: 200 mM KCl, 20 mM Tris-Cl pH 8.0, 2.1 mM OGNG, 15 mM HTG, 0.012% CHS), concentrated and applied to a Superdex 200 (GE, 10/300) pre-equilibrated with SEC buffer. Peak fractions were evaluated by SDS-PAGE (15% acrylamide) for purity, pooled and concentrated for crystallization.
Crystallization, structure determination, and refinement
Purified K2P2.1cryst-I110D was concentrated to 6 mg mL−1 before crystallization using hanging-drop vapor diffusion at 4°C using a mixture of 0.2 μL protein to 0.1 μL reservoir solution, over 100 μL reservoir of 20–25% PEG400, 200 mM KCl, 100 mM HEPES pH 8.0 or 7.1 and 1–2 mM CdCl2. Crystals appeared within 1–2 days and grew to full size within 2 weeks. Crystals were cryoprotected in solution containing 200 mM KCl, 0.2% OGNG, 15 mM HTG, 0.02% CHS, 100 mM HEPES pH 8.0 or 7.1 and 1–2 mM CdCl2, with 5% increase in PEG400 up to final concentration of 38% before flash freezing in liquid nitrogen. For compound bound structures, cryoprotected crystals were also soaked in final cryoprotection solution containing 1 mM each RuRed, RuRed+ML335 or Ru360, sourced as described in TEVC methods, for at least 1 hour prior to flash freezing in liquid nitrogen.
Datasets were collected at 100 K using synchrotron radiation at APS GM/CAT beamline 23-IDB/D Chicago, Illinois using a wavelength of 1.0332 Å, processed with XDS (Kabsch, 2010) and scaled and merged with Aimless (Evans and Murshudov, 2013). Highest resolution structures were obtained from crystals that were soaked with the ruthenium compounds. Structure determination of low resolution datasets from complexes obtained by co-crystallization indicated that there was no difference in the RuR position in complexes made by either soaking or co-crystallization. Final resolution cutoff was 3.40 Å, 3.40 Å, 2.80 Å and 3.51 Å for K2P2.1cryst I110D, K2P2.1cryst I110D:RuR, K2P2.1cryst I110D:RuR:ML335 and K2P2.1cryst I110D:Ru360 structures, respectively, using the CC1/2 criterion (Diederichs and Karplus, 2013). K2P2.1cryst-I110D was solved by molecular replacement utilizing K2P4.1 (G124I) structure (PDB: 4RUE) as search model. For compound bound structures, the K2P2.1cryst-I110D model was used as the molecular replacement search model. Electron density maps were improved through several cycles of manual model rebuilding, using COOT (Emsley and Cowtan, 2004), REFMAC (Murshudov et al., 1997), and PHENIX (Adams et al., 2010).
Mutant cycle analysis
Double mutant cycle analysis (Carter et al., 1984; Hidalgo and MacKinnon, 1995) was carried out using the equation in which Ω is the coupling factor (Hidalgo and MacKinnon, 1995) and IC50WT, IC50X’, IC50Y’ and IC50X’Y’ are the IC50 values for the wild-type, each single mutant of the X-Y pair, and the double mutant, respectively. As wild type K2P2.1 (TREK-1) is unaffected by RuR, the free energy of the interaction is zero and hence, Ka and Kd = 1. Coupling energy, ΔΔGΩ, was calculated as ΔΔGΩ = RTLnΩ where R=1.987 cal mol−1 deg−1 and T= 298K.
QUANTIFICATION AND STATISTICAL ANALYSIS
As outlined in the TEVC electrophysiology methods section, each dose-response curve point is an average of at least three oocytes (n=number of oocytes, indicated in Table 1) recorded from at least two independent batches of oocytes. Specific statistical details of experiments are found in figure legends. Graphpad Prism (Version 5) was used to estimate inhibition IC50 values (listed in Table 1) determined by fitting a standard Hill equation with Hill coefficient fixed to −1.0. Standard error (SEM) was calculated in Prism and is indicated in figure legends and in Table 1.
DATA AND CODE AVAILABILITY
Coordinates and structures factors for the structures of K2P2.1 I110D (6V36), K2P2.1 I110D:RuR (6V3I), K2P2.1 I110D:RuR:ML335 (6V37), and K2P2.1 I110D:Ru360 (6V3C) are available from the RCSB.
Supplementary Material
Movie S1. RuR block of K2P channels, Related to Figure 1. Morph between the K2P2.1 I110D and K2P2.1 I110D:RuR complex structures. I110D sidechains are shown as sticks. RuR is shown in space filling.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase | Worthington Biochemical Corporation | Cat#LS004183 |
| Ruthenium Red | Millipore-Sigma | Cat#R2751 |
| Ru360 | Millipore-Sigma, Calbiochem | Cat#557440 |
| ML335 | Lolicato et al., 2017 | N/A |
| Yeast Nitrogen Base (YNB) | Invitrogen | Cat#Q30007 |
| Anti-GFP Nanobody | Lolicato et al., 2017 | N/A |
| 3C Protease | Shaya et al., 2011 | N/A |
| Octyl glucose neopentyl glycol (OGNG) | Anatrace | Cat#NG31125GM |
| n-heptyl-β-D-thioglucopyranoside (HTG) | Anatrace | Cat#H30125GM |
| Cholesterol hemisuccinate (CHS) | Anatrace | Cat#CH21025GM |
| Phenylmethylsulfonyl fluoride (PMSF) | Millipore-Sigma | Cat#P7626 |
| DNase I | Roche | Cat#1010415900 |
| mMessage Machine T7 transcription kit | ThemoFisher | Cat# AM1344 |
| RNEasy kit | Qiagen | Cat# 74104 |
| Deposited Data | ||
| K2P2.1 (TREK-1) I110D (apo) structure | This paper | PDB: 6V36 |
| K2P2.1 (TREK-1) I110D bound to RuR | This paper | PDB: 6V3I |
| K2P2.1 (TREK-1) I110D co-bound to RuR and ML335 | This paper | PDB: 6V37 |
| K2P2.1 (TREK-1) I110D bound to Ru360 | This paper | PDB: 6V3C |
| K2P4.1 (TRAAK) G124I | Lolicato et al., 2014 | PDB: 4RUE |
| K2P2.1 (TREK-1) (apo) | Lolicato et al., 2017 | PDB: 6CQ6 |
| K2P2.1 (TREK-1) bound to ML335 | Lolicato et al., 2017 | PDB: 6CQ8 |
| K2P10.1 (TREK-2) (apo) | Dong et al., 2014 | PDB: 4BW5 |
| Experimental Models: Organisms/Strains | ||
| Xenopus laevis (frogs for oocyte removal) | Nasco | Female, 9+CM |
| Pichia pastoris (SMD1163H) | Lolicato et al., 2017 | N/A |
| Recombinant DNA | ||
| Murine K2P2.1 (TREK-1) (pGEM) | Bagriantsev et al., 2013 | N/A |
| K2P2.1 (TREK-1) I110D (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) I110E (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) I110K (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) I110D/G137I (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) N147D (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) N147E (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) I110D/N147D (pGEM) | This paper | N/A |
| K2P2.1 (TREK-1) I110D/N147E (pGEM) | This paper | N/A |
| Murine K2P10.1 (TREK-2) (pGEM) | Bagriantsev et al., 2013 | N/A |
| Murine K2P9.1 (TASK-3) (pGEM) | Bagriantsev et al., 2013 | N/A |
| K2P2.1 (TREK-1)CRYST (pPicZ) | Lolicato et al., 2017 | N/A |
| K2P2.1 (TREK-1)CRYST I110D (pPicZ) | This paper | N/A |
| Software and Algorithms | ||
| pClamp, Version 8 | Molecular Devices | N/A |
| Clampfit 8.1 | Molecular Devices | N/A |
| Graphpad Prism, Version 5 | Graphpad Software | https://www.graphpad.com/scientific-software/prism/ |
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| Aimless | Evans and Murshudov, 2013 | http://www.ccp4.ac.uk/ |
| PHENIX | Adams et al., 2010 | https://www.phenix-online.org/ |
| COOT | Emsley and Cowtan, 2004 | http://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| REFMAC | Murshudov et al., 1997 | http://www.ccp4.ac.uk/ |
| Other | ||
| CNBr-activated sepharose resin | GE Healthcare | Cat#17-0430-01 |
| Superdex 200 column, 10/300 | GE Healthcare | Cat#17-5175-01 |
| Econo-Pac gravity column, 1.5 × 12 cm | Bio-Rad | Cat#7321010 |
| GeneClamp 500B | MDS Analytical Technologies | N/A |
| Synchrotron radiation at APS GM/CAT Beamline 23-IDB/D | APS, Chicago | N/A |
Highlights.
Polynuclear ruthenium amines bind the CAP domain ‘Keystone inhibitor site’.
Electrostatics and direct sidechain coordination contribute to RuR and Ru360 binding.
RuR and Ru360 prevent ion flow by a ‘finger in the dam’ blockade.
Structure-based design creates RuR ‘super responder’ K2Ps.
Acknowledgements
We thank V. Nguyen for help in the initial stages of this project, Z. Wong for technical assistance, A. Natale for help with structure comparisons, J. Du Bois and M. Grabe for helpful discussions, and Y. Kirichok and members of the Minor lab for comments on the manuscript. This work was supported by grant NIH-R01-MH093603 to D.L.M.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The other authors declare no competing interests.
References
- Abraham DM, Lee TE, Watson LJ, Mao L, Chandok G, Wang HG, Frangakis S, Pitt GS, Shah SH, Wolf MJ, et al. (2018). The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J Clin Invest 128, 4843–4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta crystallographica Section D, Biological crystallography 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, et al. (2006). TREK-1, a K+ channel involved in polymodal pain perception. Embo J 25, 2368–2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif Pavel M, Lv C, Ng C, Yang L, Kashyap P, Lam C, Valentino V, Fung HY, Campbell T, Moller SG, et al. (2016). Function and regulation of TRPP2 ion channel revealed by a gain-of-function mutant. Proc Natl Acad Sci U S A 113, E2363–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Ang KH, Gallardo-Godoy A, Clark KA, Arkin MR, Renslo AR, and Minor DL Jr. (2013). A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS chemical biology 8, 1841–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Clark KA, and Minor DL Jr. (2012). Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J 31, 3297–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Peyronnet R, Clark KA, Honore E, and Minor DL Jr. (2011). Multiple modalities converge on a common gate to control K2P channel function. EMBO J 30, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, et al. (2011). Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun G, Lengyel M, Enyedi P, and Czirjak G (2015). Differential sensitivity of TREK-1, TREK-2 and TRAAK background potassium channels to the polycationic dye ruthenium red. British journal of pharmacology 172, 1728–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, del Marmol J, and MacKinnon R (2012). Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335, 436–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brohawn SG, Wang W, Handler A, Campbell EB, Schwarz JR, and MacKinnon R (2019). The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. eLife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter PJ, Winter G, Wilkinson AJ, and Fersht AR (1984). The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus). Cell 38, 835–840. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Rosen TA, Tominaga M, Brake AJ, and Julius D (1999). A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398, 436–441. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, and Julius D (1997). The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389, 816–824. [DOI] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, and Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Clemente N, Sun W, Du J, and Lu W (2019). The structures and gating mechanism of human calcium homeostasis modulator 2. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MJ (2002). Ruthenium metallopharmaceuticals. Coordin Chem Rev 232, 69–93. [Google Scholar]
- Coste B, Xiao B, Santos JS, Syeda R, Grandl J, Spencer KS, Kim SE, Schmidt M, Mathur J, Dubin AE, et al. (2012). Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483, 176–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czirjak G, and Enyedi P (2002). Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem 277, 5426–5432. [DOI] [PubMed] [Google Scholar]
- Czirjak G, and Enyedi P (2003). Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Molecular pharmacology 63, 646–652. [DOI] [PubMed] [Google Scholar]
- Decher N, Ortiz-Bonnin B, Friedrich C, Schewe M, Kiper AK, Rinne S, Seemann G, Peyronnet R, Zumhagen S, Bustos D, et al. (2017). Sodium permeable and “hypersensitive” TREK-1 channels cause ventricular tachycardia. EMBO molecular medicine 9, 403–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devilliers M, Busserolles J, Lolignier S, Deval E, Pereira V, Alloui A, Christin M, Mazet B, Delmas P, Noel J, et al. (2013). Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat Commun 4, 2941. [DOI] [PubMed] [Google Scholar]
- Diederichs K, and Karplus PA (2013). Better models by discarding data? Acta crystallographica Section D, Biological crystallography 69, 1215–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YY, Pike AC, Mackenzie A, McClenaghan C, Aryal P, Dong L, Quigley A, Grieben M, Goubin S, Mukhopadhyay S, et al. (2015). K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 347, 1256–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douguet D, and Honore E (2019). Mammalian Mechanoelectrical Transduction: Structure and Function of Force-Gated Ion Channels. Cell 179, 340–354. [DOI] [PubMed] [Google Scholar]
- Dreses-Werringloer U, Vingtdeux V, Zhao H, Chandakkar P, Davies P, and Marambaud P (2013). CALHM1 controls the Ca(2)(+)-dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J Cell Sci 126, 1199–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta crystallographica Section D, Biological crystallography 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Enyedi P, and Czirjak G (2010). Molecular background of leak K+ currents: two-pore domain potassium channels. Physiological reviews 90, 559–605. [DOI] [PubMed] [Google Scholar]
- Evans PR, and Murshudov GN (2013). How good are my data and what is the resolution? Acta crystallographica Section D, Biological crystallography 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliciangeli S, Chatelain FC, Bichet D, and Lesage F (2014). The family of K channels: salient structural and functional properties. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher JM, Greenfield BF, Hardy CJ, Scargill D, and Woodhead JL (1961). Ruthenium Red. J Chem Soc, 2000–2006. [Google Scholar]
- Gonzalez W, Zuniga L, Cid LP, Arevalo B, Niemeyer MI, and Sepulveda FV (2013). An extracellular ion pathway plays a central role in the cooperative gating of a K(2P) K+ channel by extracellular pH. J Biol Chem 288, 5984–5991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guler AD, Lee H, Iida T, Shimizu I, Tominaga M, and Caterina M (2002). Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22, 6408–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, and Pfeiffer DR (1990). Mechanisms by which mitochondria transport calcium. Am J Physiol 258, C755–786. [DOI] [PubMed] [Google Scholar]
- Heurteaux C, Guy N, Laigle C, Blondeau N, Duprat F, Mazzuca M, Lang-Lazdunski L, Widmann C, Zanzouri M, Romey G, et al. (2004). TREK-1, a K+ channel involved in neuroprotection and general anesthesia. Embo J 23, 2684–2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thummler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, et al. (2006). Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci 9, 1134–1141. [DOI] [PubMed] [Google Scholar]
- Hidalgo P, and MacKinnon R (1995). Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science 268, 307–310. [DOI] [PubMed] [Google Scholar]
- Kabsch W (2010). Xds. Acta crystallographica Section D, Biological crystallography 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanda H, Ling J, Tonomura S, Noguchi K, Matalon S, and Gu JG (2019). TREK-1 and TRAAK Are Principal K(+) Channels at the Nodes of Ranvier for Rapid Action Potential Conduction on Mammalian Myelinated Afferent Nerves. Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawate T, and Gouaux E (2006). Fluorescence-detection size-exclusion chromatography for precrystallization screening of integral membrane proteins. Structure 14, 673–681. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Laigle C, Confort-Gouny S, Le Fur Y, Cozzone PJ, and Viola A (2012). Deletion of TRAAK potassium channel affects brain metabolism and protects against ischemia. PloS one 7, e53266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert M, Boet A, Rucker-Martin C, Mendes-Ferreira P, Capuano V, Hatem S, Adao R, Bras-Silva C, Hautefort A, Michel JB, et al. (2018). Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc Res 114, 880–893. [DOI] [PubMed] [Google Scholar]
- Lazarenko RM, Fortuna MG, Shi Y, Mulkey DK, Takakura AC, Moreira TS, Guyenet PG, and Bayliss DA (2010). Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K(+) current. J Neurosci 30, 9324–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, and Minor DL Jr. (2017). K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolicato M, Riegelhaupt PM, Arrigoni C, Clark KA, and Minor DL Jr. (2014). Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K(2P) channels. Neuron 84, 1198–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J (1993). Block by ruthenium red of the ryanodine-activated calcium release channel of skeletal muscle. J Gen Physiol 102, 1031–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z, Siebert AP, Cheung KH, Lee RJ, Johnson B, Cohen AS, Vingtdeux V, Marambaud P, and Foskett JK (2012). Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc Natl Acad Sci U S A 109, E1963–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madry C, Kyrargyri V, Arancibia-Carcamo IL, Jolivet R, Kohsaka S, Bryan RM, and Attwell D (2018). Microglial Ramification, Surveillance, and Interleukin-1beta Release Are Regulated by the Two-Pore Domain K(+) Channel THIK-1. Neuron 97, 299–312 e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaud-Agrawal N, Denning EJ, Woolf TB, and Beckstein O (2011). MDAnalysis: a toolkit for the analysis of molecular dynamics simulations. J Comput Chem 32, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AN, and Long SB (2012). Crystal structure of the human two-pore domain potassium channel K2P1. Science 335, 432–436. [DOI] [PubMed] [Google Scholar]
- Moore CL (1971). Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun 42, 298–305. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, and Dodson EJ (1997). Refinement of macromolecular structures by the maximum-likelihood method. Acta crystallographica Section D, Biological crystallography 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Musset B, Meuth SG, Liu GX, Derst C, Wegner S, Pape HC, Budde T, Preisig-Muller R, and Daut J (2006). Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J Physiol 572, 639–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, et al. (2016). Architecture of the mitochondrial calcium uniporter. Nature 533, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Ehrlich G, Andres-Enguix I, Fritzenschaft H, Decher N, Sansom MS, Tucker SJ, et al. (2011). The pore structure and gating mechanism of K2P channels. EMBO J 30, 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope L, Arrigoni C, Lou H, Bryant C, Gallardo-Godoy A, Renslo AR, and Minor DL Jr. (2018). Protein and Chemical Determinants of BL-1249 Action and Selectivity for K2P Channels. ACS chemical neuroscience 9, 3153–3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahamimoff R, and Alnaes E (1973). Inhibitory action of Ruthenium red on neuromuscular transmission. Proc Natl Acad Sci U S A 70, 3613–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renigunta V, Schlichthorl G, and Daut J (2015). Much more than a leak: structure and function of K(2)p-channels. Pflugers Arch 467, 867–894. [DOI] [PubMed] [Google Scholar]
- Rödström KEJ, Kiper AK, Zhang W, Rinné S, Pike ACW, Goldstein M, Conrad L, Delbeck M, Hahn M, Meier H, et al. (2019). A unique lower X-gate in TASK channels traps inhibitors within the vestibule. bioRxiv, 706168. [DOI] [PubMed] [Google Scholar]
- Royal P, Andres-Bilbe A, Avalos Prado P, Verkest C, Wdziekonski B, Schaub S, Baron A, Lesage F, Gasull X, Levitz J, et al. (2019). Migraine-Associated TRESK Mutations Increase Neuronal Excitability through Alternative Translation Initiation and Inhibition of TREK. Neuron 101, 232–245 e236. [DOI] [PubMed] [Google Scholar]
- Schewe M, Nematian-Ardestani E, Sun H, Musinszki M, Cordeiro S, Bucci G, de Groot BL, Tucker SJ, Rapedius M, and Baukrowitz T (2016). A Non-canonical Voltage-Sensing Mechanism Controls Gating in K2P K(+) Channels. Cell 164, 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schewe M, Sun H, Mert U, Mackenzie A, Pike ACW, Schulz F, Constantin C, Vowinkel KS, Conrad LJ, Kiper AK, et al. (2019). A pharmacological master key mechanism that unlocks the selectivity filter gate in K(+) channels. Science 363, 875–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaya D, Kreir M, Robbins RA, Wong S, Hammon J, Bruggemann A, and Minor DL Jr. (2011). Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc Natl Acad Sci U S A 108, 12313–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JS, Imagawa T, Ma J, Fill M, Campbell KP, and Coronado R (1988). Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J Gen Physiol 92, 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterbuleac D (2019). Molecular determinants of chemical modulation of two-pore domain potassium channels. Chem Biol Drug Des 94, 1596–1614. [DOI] [PubMed] [Google Scholar]
- Story GM, Peier AM, Reeve AJ, Eid SR, Mosbacher J, Hricik TR, Earley TJ, Hergarden AC, Andersson DA, Hwang SW, et al. (2003). ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112, 819–829. [DOI] [PubMed] [Google Scholar]
- Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, and Plant TD (2000). OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2, 695–702. [DOI] [PubMed] [Google Scholar]
- Su ZW, Brown EC, Wang WW, and MacKinnon R (2016). Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. P Natl Acad Sci USA 113, 5748–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian F, Qiu Y, Lan X, Li M, Yang H, and Gao Z (2019). A Small-Molecule Compound Selectively Activates K2P Channel TASK-3 by Acting at Two Distant Clusters of Residues. Molecular pharmacology 96, 26–35. [DOI] [PubMed] [Google Scholar]
- Vivier D, Soussia IB, Rodrigues N, Lolignier S, Devilliers M, Chatelain FC, Prival L, Chapuy E, Bourdier G, Bennis K, et al. (2017). Development of the first Two-Pore Domain Potassium Channel TREK-1 (TWIK-Related K+ Channel 1)-selective agonist possessing in vivo anti-nociceptive activity. Journal of medicinal chemistry. [DOI] [PubMed] [Google Scholar]
- Voets T, Nilius B, Hoefs S, van der Kemp AW, Droogmans G, Bindels RJ, and Hoenderop JG (2004). TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 279, 19–25. [DOI] [PubMed] [Google Scholar]
- Voets T, Prenen J, Vriens J, Watanabe H, Janssens A, Wissenbach U, Bodding M, Droogmans G, and Nilius B (2002). Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem 277, 33704–33710. [DOI] [PubMed] [Google Scholar]
- Wallace AC, Laskowski RA, and Thornton JM (1995). LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng 8, 127–134. [DOI] [PubMed] [Google Scholar]
- Woods JJ, and Wilson JJ (2019). Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr Opin Chem Biol 55, 9–18. [DOI] [PubMed] [Google Scholar]
- Wright PD, McCoull D, Walsh Y, Large JM, Hadrys BW, Gaurilcikaite E, Byrom L, Veale EL, Jerman J, and Mathie A (2019). Pranlukast is a novel small molecule activator of the two-pore domain potassium channel TREK2. Biochem Biophys Res Commun. [DOI] [PubMed] [Google Scholar]
- Wu X, Liu Y, Chen X, Sun Q, Tang R, Wang W, Yu Z, and Xie M (2013). Involvement of TREK-1 activity in astrocyte function and neuroprotection under simulated ischemia conditions. Journal of molecular neuroscience : MN 49, 499–506. [DOI] [PubMed] [Google Scholar]
- Yarishkin O, Phuong TTT, Bretz CA, Olsen KW, Baumann JM, Lakk M, Crandall A, Heurteaux C, Hartnett ME, and Krizaj D (2018). TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells. J Gen Physiol 150, 1660–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying WL, Emerson J, Clarke MJ, and Sanadi DR (1991). Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry 30, 4949–4952. [DOI] [PubMed] [Google Scholar]
- Yoshida K, Shi S, Ukai-Tadenuma M, Fujishima H, Ohno RI, and Ueda HR (2018). Leak potassium channels regulate sleep duration. Proc Natl Acad Sci U S A 115, E9459–E9468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q, Zhou H, Chi S, Wang Y, Wang J, Geng J, Wu K, Liu W, Zhang T, Dong MQ, et al. (2018). Structure and mechanogating mechanism of the Piezo1 channel. Nature 554, 487–492. [DOI] [PubMed] [Google Scholar]
- Zhao QC, Wu K, Geng J, Chi SP, Wang YF, Zhi P, Zhang MM, and Xiao BL (2016). Ion Permeation and Mechanotransduction Mechanisms of Mechanosensitive Piezo Channels. Neuron 89, 1248–1263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1. RuR block of K2P channels, Related to Figure 1. Morph between the K2P2.1 I110D and K2P2.1 I110D:RuR complex structures. I110D sidechains are shown as sticks. RuR is shown in space filling.
Data Availability Statement
Coordinates and structures factors for the structures of K2P2.1 I110D (6V36), K2P2.1 I110D:RuR (6V3I), K2P2.1 I110D:RuR:ML335 (6V37), and K2P2.1 I110D:Ru360 (6V3C) are available from the RCSB.